
Abstract
Tissue-resident memory T (TRM) cells were originally identified as a tissue-sequestered population of memory T cells that show lifelong persistence in non-lymphoid organs. That definition has slowly evolved with the documentation of TRM cells having variable terms of tissue residency combined with a capacity to return to the wider circulation. Nonetheless, reductionist experiments have identified an archetypical population of TRM cells showing intrinsic permanent residency in a wide range of non-lymphoid organs, with one notable exception: the lungs. Despite the fact that memory T cells generated during a respiratory infection are maintained in the circulation, local TRM cell numbers in the lung decline concomitantly with a decay in T cell-mediated protection. This Perspective describes the mechanisms that underpin long-term T cell lodgement in non-lymphoid tissues and explains why residency is transient for select TRM cell subsets. In doing so, it highlights the unusual nature of memory T cell egress from the lungs and speculates on the broader disease implications of this process, especially during infection with SARS-CoV-2.
Similar content being viewed by others


Introduction
Memory T cells can show a range of persistence within non-lymphoid compartments. Many lymphocytes move freely through the various organs unimpeded before exiting the tissue via the draining lymphatic vessels1,2,3. Recognition of antigen leads to their transient retention4 while physical barriers may slow the return of cells to the circulation5. Finally, a subset of T cells is specialized for purely localized patterns of immune surveillance6,7 and only poorly exits the tissues, if at all8. These tissue-resident memory T (TRM) cells9 have a cell-autonomous limitation in their recirculation capacity10,11,12 and show a superior ability to control localized infections in a number of settings13,14,15. In this Perspective, I detail the transcription networks that define sequestered TRM cells, identifying CD103+CD8+ memory T cells as the key population of memory CD8+ T cells that encompasses all the hallmarks of permanent tissue residency. Finally, I describe how these archetypical TRM cells show an unusual pattern of egress from the lungs and discuss how this impacts the course of respiratory infections, including SARS-CoV-2.
Identifying tissue-resident memory T cells
TRM cells were initially identified as a distinct, sessile T cell subset that coexisted alongside tissue-emigrating T cells8. This was a break from the prevailing understanding of tissue T cells based on early lymphocyte circulation experiments5,16,17. At that time, the widely accepted view was that these T cells were simply recirculating memory cells that either happened to be found in non-lymphoid tissues in large numbers18,19,20 or, alternatively, were trapped by some sort of gating mechanisms or by structural barriers such as the basement membrane that lines epithelia5. The identification of a unique TRM cell subset meant that non-lymphoid tissues contained at least two populations of memory T cells, each with its own distinct phenotype and functional properties. One was a recirculating subset that at the time was thought to comprise effector memory T (TEM) cells17 and the other, the newly identified permanently resident TRM cell population.
A major challenge from that point onwards has been distinguishing non-migrating TRM cells from recirculating memory T cells, largely because of the phenotypic overlap between these populations. For example, TRM cells do not express CC-chemokine receptor 7 (CCR7) — a receptor required for entry into lymphoid tissues and the marker that was originally used to differentiate TEM cells (identified as CCR7-negative) from lymphoid-tissue-constrained central memory T (TCM) cells (identified as CCR7-positive)17. Separately, CD69 had been proposed to be a pan-TRM cell identifier21, yet it is upregulated by both antigen-specific and nonspecific stimuli22 and a substantial fraction of migratory T cells express this molecule once in the tissues23. Compounding the confusion is the extensive heterogeneity seen in both circulating and tissue-resident memory T cell populations24,25,26,27, expanded by a history of natural infection28. Therefore, although combinations of surface markers can cover a range of TRM-like tissue cells, it would be fair to say that to date there remains no unifying phenotypic identifier for this population.
CD103+CD8+ TRM cells: the archetypical TRM cell
Although it has proven difficult to identify TRM cells by definitive phenotypic means, therapeutic and experimental interventions can eliminate all circulating T cells from the blood, leaving long-term tissue residents as the only memory T cells remaining in non-lymphoid compartments. Two approaches have proven particularly useful in this regard. The first exploits T cell responses against a transplantation mismatch to selectively eliminate cells in the circulation29,30,31 whereas the second uses a more versatile cytolytic antibody-based technique for the same purpose27,32,33. Of additional importance is the in vivo infusion of labelling antibodies before tissue analyses to exclude cells that are simply in the vasculature34. This technique eliminates confounding contributions by blood-borne cells and is critical when examining highly vascularized organs such as the lung, although it does not identify TRM cells per se.
One of the striking features of the mouse TRM cells that remain after circulating T cells are depleted from the tissues is the dominance of a population of CD8+ T cells expressing the CD103 (also known as αE integrin) subunit of the αEβ7 integrin complex23,33. CD103+ TRM cells are highly enriched in the environmentally exposed epithelia of the skin, small intestine and female reproductive tract8,10,35. At these epithelial sites, interaction between αEβ7 and its abundantly expressed target ligand E-cadherin36 probably plays a role in cell adhesion and retention. However, CD103+CD8+ TRM cells are also found in non-epithelial tissues such as the brain12,37, and although CD103 has variously been implicated as being important for TRM cell development38,39,40, its expression is not ubiquitous37 and therefore not mandatory for all forms of T cell residency. Nonetheless, tissue-lodged CD103+CD8+ memory T cells are highly resistant to equilibration across parabiotic pairs41, are uniquely spared from elimination by the approaches mentioned above23,30, selectively survive for prolonged periods in transplanted tissues in mice8,33 as well as in humans42,43 and persist independently of antigen recognition15,37. Moreover, CD103+CD8+ memory T cells are usually not found in secondary lymphoid organs15,44 — with one striking exception45,46 to be described in detail below. Thus, although not all TRM cells express CD103, the balance of evidence argues that CD103+CD8+ tissue T cells are true TRM cells, making this an easily identifiable archetypical population and an ideal reductionist means for delineating tissue residency mechanisms.
RUNX3 and TGFβ in CD8+ TRM cell development
Early experiments in mice comparing the transcriptomes of CD103+CD8+ TRM cells isolated from a range of organs with those of their circulating counterparts provided some of the first insights into the transcription networks critical for TRM cell development and survival10,39. Not surprisingly, genes associated with tissue egress were found to be downregulated in TRM cells, including Ccr7 and the genes encoding the sphingosine-1-phosphate receptors S1PR1 and S1PR510,11. Without the downregulation of these receptors, the precursors of TRM cells return to the circulation, thereby dampening TRM cell development11,47. Other genes that come into play are those involved in dealing with local metabolite availability7,48,49 and those that prolong T cell survival23, with both sets of genes necessary to maintain a long-lived sequestered T cell population. Further experiments fleshed out how transcription factors control the various networks, such as the involvement of KLF2, which modulates the expression of S1PR1 and CCR711. Following this, key upstream gene regulators were identified, such as T-bet and EOMES23,50 as well as BLIMP1 and the BLIMP1 homologue HOBIT51,52; of note, BLIMP1 and HOBIT are also involved in the development of innate-like lymphocytes that permanently reside in mouse tissues, such as natural killer cells and natural killer T cells51. Most recently, an overarching transcription factor has come into focus. RUNX3 has been identified as contributing to TRM cell formation, and it directly or indirectly regulates BLIMP1 and KLF2 expression as well as modulating downstream retention components53. This contribution is particularly striking as RUNX3 is a pivotal player in CD8+ T cell development and functionality54,55.
As the network analyses evolved, one commonality to emerge was the involvement of TGFβ in TRM cell development and survival in a range of tissues and organs23,40,56,57,58. TGFβ appears to use a non-canonical signalling pathway59 that controls much of the CD8+ TRM cell gene expression signature60. It has been shown to facilitate tissue entry via selectin upregulation61 and can regulate a broad range of transcription regulators and cytokine-driven survival factors during CD8+ TRM cell development11,23,50. Combined, there is now a wealth of data regarding the tenets of transcriptional control of TRM cell formation, which largely pivots around a TGFβ–RUNX3 axis, at least in the case of the mouse CD8+ TRM cell subset.
TRM cell re-entry into the circulation
Although TRM cells were originally shown to persist in non-lymphoid organs in quasi-perpetuity8, there have been subsequent descriptions of TRM cell egress with the resultant ‘ex-TRM cells’ ultimately being incorporated into the circulation33,41,62. CD8+ TRM cell numbers show an intrinsic decline in organs such as the lung and liver30,41, but not in tissues such as the skin and small intestine, where the cells effectively remain in place for life once lodged8,41. However even for these fixed populations, TRM cells can be forced to leave using in situ antigen stimulation via peptide challenge33,44. Such active dislodgement is not universal, with CD103+CD8+ TRM cells sometimes remaining resident in the tissue even after multiple rounds of cell division initiated by local infection8,63,64. Perhaps tellingly, when CD103+CD8+ TRM cells are selectively dislodged by intervention, the resultant ex-TRM cells appear to adopt a phenotype intermediate between those of upstream resident memory T cells and conventional recirculating memory T cell populations, with a CD103 expression status that is either undefined or reported to be transient33,44. Moreover, when these same cells are directly isolated from non-lymphoid compartments, they are inferior in their capacity to enter the circulation compared to counterparts extracted from lymphoid organs33,65.
It remains difficult to reconcile these conflicting results, but studies on CD8+ TRM cells in the liver and recent revelations regarding the basis for CD4+ T cell residency provide valuable insight that might explain egress variability. Although much more is known about CD8+ TRM cells, there are many examples of CD4+ TRM cell-type counterparts13,27,66. Comparisons make it clear that the two are unrelated in terms of mechanistic underpinnings and they can exhibit quite distinct patterns of tissue residency even in the same organ29,67. As noted above, the archetypical CD103+CD8+ TRM cells use a set of TGFβ-driven transcriptional networks to shut down tissue egress, upregulate survival factors and tailor metabolic pathways. By contrast, few of these networks have been associated with CD4+ TRM cell residency, which instead relies on retention mechanisms variously operating via cell aggregation, antigen-specific T cell activation and chemotactic agents68,69 (Fig. 1). The reason why CD4+ and CD8+ TRM cells are likely to differ at the mechanistic level is the pivotal role RUNX3 plays in TRM cell development and survival53. This transcription factor is repressed in CD4+ T cells by the opposing gene regulator ThPOK (also known as ZBT7B), which is itself a lineage-determining factor70,71. Although natural RUNX3 upregulation can convert CD4+ T cells to an unconventional CD8αα+ intraepithelial regulatory T cell population with CD8+ TRM cell-like qualities72, the intrinsic paucity of RUNX3 expression in conventional CD4+ TRM cells results in low CD103 levels in these cells and more transient tissue residency as a direct consequence of their inability to access the RUNX3-mediated pathways downstream of TGFβ signalling53,73.

The mechanism promoting permanent residency in non-lymphoid tissues for the CD103+CD8+ tissue-resident memory T (TRM) cell population involves a RUNX3-driven transcriptional network that is downstream of TGFβ receptor signalling53,60. This transcription programme is missing in CD4+ TRM cells as a consequence of deficiencies in RUNX3 expression73. Instead, these populations use a combination of cell aggregation and extrinsic chemokine networks for tissue retention68,69. The typical CD103+CD8+ TRM cell transcription programme is also missing in CD103− liver-like TRM cells because of deficiencies in TGFβ engagement65.
Somewhat analogous to their CD4+ tissue-resident T cell counterparts, mouse liver CD8+ TRM cells are also deficient in CD103 expression74. These cells show medium-to-long-term tissue residency74, but not the almost lifelong persistence of TRM cells in organs such as skin and small intestine41,65. Although the liver T cells are fully capable of responding to TGFβ, local requirements negate this capacity, resulting in an immature or less differentiated CD103− TRM cell population (Fig. 1) with an inferior term of residency combined with more flexible reprograming compared to mature CD103+ TRM cell counterparts65. Collectively, the results show that although CD103− TRM cells can reside in tissues for a considerable period, they can exhibit a range of spontaneous egress and reprogramming capabilities because of deficiencies in TGFβ-mediated maturation. Given the heterogeneous nature of tissue-resident T cells, including variable CD103+ T cell content across different organs37 and the known recruitment of recirculating T cells by the peptide stimulation used for TRM cell dislodgement32, it is possible that less differentiated populations analogous to the liver CD103− TRM cells may preferentially contribute to the egress process. Regardless, although some TRM cells can leave the tissues and enter the circulation, the balance of data argues that for the archetypical CD103+CD8+ TRM cells, this process is not constitutive and when it does happen, it usually results in cells that do not phenocopy their direct upstream antecedents.
TRM cells in the lungs
From the discussion above, it can be reasonably argued that because they fully engage the TGFβ–RUNX3 residency programme, mouse CD103+CD8+ tissue T cells fit the original TRM cell definition8; specifically, they form a distinct subset of memory T cells that remains lodged in peripheral compartments in virtual perpetuity. However, there is one organ where the CD103+CD8+ T cells do not abide by this rule, and its uniqueness has important disease implications. Unlike the situation in other tissues, CD103+CD8+ TRM cells in the lung do not require local antigen stimulation for dislodgement45. Also unusually, the egressing memory T cells retain cell surface expression of CD103 post-exit, meaning that the lung-draining lymph nodes are unique in having a substantive subset of memory CD8+ T cells with this marker45,46. Lung CD103+CD8+ TRM cells are fully mature and unremarkable in terms of their TGFβ requirement for development and survival58. They also express the gene signatures associated with tissue residency10,12, including a cluster of TRM cell-associated transcription factors, namely HOBIT, NR4A1, aryl hydrocarbon receptor (AhR) and BHLHE407,51,75,76. In all critical aspects, they resemble TRM cells from other tissues, meaning that their exit from the lung is probably an organ-specific feature rather than due to a cell-intrinsic programme. Such a mechanistic distinction is important, as it would suggest that the egress process would probably capture TRM cells beyond the archetypical CD103+CD8+ subset that was used to define this phenomenon and would do so regardless of where they fall on a maturation and term-of-residency continuum.
Exacting experiments by Stolley and colleagues45 proved that the resultant draining lymph node-resident memory T cells were indeed constitutively derived from upstream lung tissue counterparts, possibly dislodged as a consequence of virus-induced tissue damage77,78 or the interruption of tonic TGFβ signalling needed to retain TRM cells in tissues79. Although the resultant lymph node accumulation offers an additional avenue to maintain regional protection45,80, memory T cell exit helps to explain one of the intriguing conundrums associated with immune protection in the lungs. It has long been known that T cell immunity in the lung wanes over time, with this first reported for respiratory infections with influenza virus and Sendai virus in mice81,82. This decline in lung-based immunity occurs despite virus-specific memory cells persisting in the circulation30,82,83,84. Non-TRM cell-based mechanisms were originally proposed to describe the behaviour of lung T cell populations81,85,86,87, variously confounded by blood-borne cells that are particularly problematic when dealing with this highly vascularized organ34. More recently, it was shown that the waning local immunity correlates with declining lung TRM cell numbers in mouse after influenza virus infection83,84 and in humans after respiratory syncytial virus challenge88. Although other mechanisms have been posited to account for this TRM cell attrition, such as the selective death of lung TRM cells30,84 or the disappearance of structures associated with focal damage67, none exclude concurrent tissue egress. Once lost, lung TRM cells are difficult to replace in the absence of renewed infection owing to the strict antigen recognition requirements for effective lodgement67,83,89, which are optional in many other tissues37 including the upper respiratory tract90. Overall, a range of mechanistic overlays would imply that losing TRM cells over time is important for this organ — for example, to limit ongoing damage to its delicate oxygen-exchange architecture91.
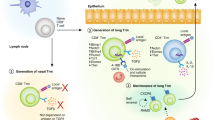
Finally, the natural decay of lung TRM cells stands in stark contrast to what is seen elsewhere in the body, where CD103+CD8+ TRM cell populations can remain tightly contained (Fig. 2). CD103+CD8+ TRM cells show long-term persistence in organs such as the brain, skin and cervicovaginal tissue8,39,92, despite the loss of their CD103− counterparts. The extent to which these spatial and temporal restrictions can operate was dramatically illustrated by experiments that lodged CD103+CD8+ TRM cells in a small patch of skin, thus confining effective protection to just that location while leaving the remainder of the torso under the inferior control of memory cells in the blood8,63. By contrast, lung TRM cell residency is unstable and transient, resulting in surveillance that is increasingly dependent on recirculating populations over time, with a concomitant decline in local T cell immunity.

Inflammation associated with infection of tissues such as skin, small intestine and reproductive tract (left panels) and lung (right panels) leads to the recruitment of a variety of CD4+ and CD8+ T cells that combat the invading pathogens (part a). These populations include effector memory T (TEM) cells that continuously recirculate between non-lymphoid organs and blood as well as tissue-resident memory T (TRM) cell precursors (not shown). Following resolution of the infection (part b), most of the recruited T cells exit or die, leaving local immunosurveillance to recirculating TEM cells and the more potent TRM cells. Over time, some TRM cell subsets selectively disappear (part c, left panel), resulting in a resident population highly enriched in long-lived CD103+CD8+ TRM cells that afford long-term local immunity against re-infection (part d, left panel). In the lungs, CD103+CD8+ TRM cells are gradually lost after the infection has resolved and instead accumulate in the proximal draining lymph nodes (part c, right panel) leaving the lower respiratory tract deficient in CD103+CD8+ TRM cells and thus susceptible to re-infection (part d, right panel).
TRM cell lung egress and immunity to SARS-CoV-2
At the time of this writing and nearly three years since the emergence of the SARS-CoV-2 virus in late 201993,94, the COVID-19 pandemic continues to be a major challenge in many parts of the world. Despite reports showing that circulating antiviral T cell immunity can be cross-reactive against emerging variants95, long lived96,97 and associated with better disease outcomes98,99, immunity from combinations of COVID-19 vaccination and SARS-CoV-2 infection has been found to steadily decline100,101. One possible contributor may be that anti-SARS-CoV-2 tissue-resident T cells that are pivotal for immune protection show the same type of numerical decay as reported for mouse CD103+CD8+ TRM cells. Employing strategies that slow TRM cell loss102 could be advantageous, as might approaches that circumvent the lung altogether. The upper respiratory tract, especially the nasal mucosa, is a prime target with respect to the latter possibility as it does not show the TRM cell decline that is intrinsic to the lung90. Alternatively, it may be that TRM cells are actually counterproductive, leading to tissue damage. This is especially poignant because repeated antigen encounters extend the durability of CD103+CD8+ TRM cells in the lung102, yet a recent report found that experiencing successive SARS-CoV-2 infections progressively increases the risk of adverse health outcomes103. In terms of their potential to contribute to tissue damage, TRM cells have an innate immune alarm and recruitment function32,104, and the innate response has been shown to be a key mediator of COVID-19-associated lung pathology105,106.
Conclusion
Overall, TRM cells provide superior protection against tissue-localized infection, primarily because of constraints in their migration capabilities. Despite proving to be long-lived and effective in a range of different infectious diseases, lung TRM cells have an unusual propensity for tissue exit reflected in a decay in local T cell immunity. Such a feature may have evolved to protect this organ against long-term damage or may simply be a by-product of some unique anatomical feature intrinsic to lung function. Given the ability of TRM cells to respond to infection with an immediacy unmatched by the blood-based memory populations, there is a need to focus on their deposition in the different compartments of the respiratory system, especially in settings or sub-regions that support their long-term survival.
References
Gowans, J. L. & Knight, E. J. The route of re-circulation of lymphocytes in the rat. Proc. R. Soc. Lond. B Biol. Sci. 159, 257–282 (1964).
Issekutz, T. B., Chin, W. & Hay, J. B. The characterization of lymphocytes migrating through chronically inflamed tissues. Immunology 46, 59–66 (1982).
Rannie, G. H. & Ford, W. L. Recirculation of lymphocytes: its role in implementing immune responses in the skin. Lymphology 11, 193–201 (1978).
Hall, J., Scollay, R. & Smith, M. Studies on the lymphocytes of sheep. I. Recirculation of lymphocytes through peripheral lymph nodes and tissues. Eur. J. Immunol. 6, 117–120 (1976).
Klonowski, K. D. et al. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity 20, 551–562 (2004).
Ariotti, S. et al. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc. Natl Acad. Sci. USA 109, 19739–19744 (2012).
Zaid, A. et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc. Natl Acad. Sci. USA 111, 5307–5312 (2014).
Gebhardt, T. et al. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 10, 524–530 (2009).
Iwasaki, A. Local advantage: skin DCs prime; skin memory T cells protect. Nat. Immunol. 10, 451–453 (2009).
Mackay, L. K. et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 14, 1294–1301 (2013).
Skon, C. N. et al. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 14, 1285–1293 (2013).
Wakim, L. M. et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J. Immunol. 189, 3462–3471 (2012).
Glennie, N. D. et al. Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J. Exp. Med. 212, 1405–1414 (2015).
Jiang, X. et al. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature 483, 227–231 (2012).
Mackay, L. K. et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc. Natl Acad. Sci. USA 109, 7037–7042 (2012).
Mackay, C. R., Marston, W. L. & Dudler, L. Naive and memory T cells show distinct pathways of lymphocyte recirculation. J. Exp. Med. 171, 801–817 (1990).
Sallusto, F., Lenig, D., Forster, R., Lipp, M. & Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712 (1999).
Clark, R. A. et al. The vast majority of CLA+ T cells are resident in normal skin. J. Immunol. 176, 4431–4439 (2006).
Masopust, D., Vezys, V., Marzo, A. L. & Lefrancois, L. Preferential localization of effector memory cells in nonlymphoid tissue. Science 291, 2413–2417 (2001).
Reinhardt, R. L., Khoruts, A., Merica, R., Zell, T. & Jenkins, M. K. Visualizing the generation of memory CD4 T cells in the whole body. Nature 410, 101–105 (2001).
Sathaliyawala, T. et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 38, 187–197 (2013).
Shiow, L. R. et al. CD69 acts downstream of interferon-α/β to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440, 540–544 (2006).
Mackay, L. K. et al. T-box transcription factors combine with the cytokines TGF-β and IL-15 to control tissue-resident memory T cell fate. Immunity 43, 1101–1111 (2015).
Gerlach, C. et al. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity 45, 1270–1284 (2016).
Hikono, H. et al. Activation phenotype, rather than central- or effector-memory phenotype, predicts the recall efficacy of memory CD8+ T cells. J. Exp. Med. 204, 1625–1636 (2007).
Olson, J. A., McDonald-Hyman, C., Jameson, S. C. & Hamilton, S. E. Effector-like CD8+ T cells in the memory population mediate potent protective immunity. Immunity 38, 1250–1260 (2013).
Watanabe, R. et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci. Transl. Med. 7, 279ra39 (2015).
Beura, L. K. et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516 (2016).
Gebhardt, T. et al. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature 477, 216–219 (2011).
Slutter, B. et al. Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aag2031 (2017).
Takamura, S. et al. Interstitial-resident memory CD8+ T cells sustain frontline epithelial memory in the lung. J. Exp. Med. 216, 2736–2747 (2019).
Schenkel, J. M., Fraser, K. A., Vezys, V. & Masopust, D. Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol. 14, 509–513 (2013).
Fonseca, R. et al. Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat. Immunol. 21, 412–421 (2020).
Anderson, K. G. et al. Cutting edge: intravascular staining redefines lung CD8 T cell responses. J. Immunol. 189, 2702–2706 (2012).
Masopust, D. et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med. 207, 553–564 (2010).
Cepek, K. L. et al. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the αEβ7 integrin. Nature 372, 190–193 (1994).
Casey, K. A. et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 188, 4866–4875 (2012).
Schon, M. P. et al. Mucosal T lymphocyte numbers are selectively reduced in integrin αE (CD103)-deficient mice. J. Immunol. 162, 6641–6649 (1999).
Wakim, L. M., Woodward-Davis, A. & Bevan, M. J. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc. Natl Acad. Sci. USA 107, 17872–17879 (2010).
Lee, Y. T. et al. Environmental and antigen receptor-derived signals support sustained surveillance of the lungs by pathogen-specific cytotoxic T lymphocytes. J. Virol. 85, 4085–4094 (2011).
Wijeyesinghe, S. et al. Expansible residence decentralizes immune homeostasis. Nature 592, 457–462 (2021).
Lian, C. G. et al. Biomarker evaluation of face transplant rejection: association of donor T cells with target cell injury. Mod. Pathol. 27, 788–799 (2014).
Snyder, M. E. et al. Generation and persistence of human tissue-resident memory T cells in lung transplantation. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aav5581 (2019).
Beura, L. K. et al. T cells in nonlymphoid tissues give rise to lymph-node-resident memory T cells. Immunity 48, 327–338e5 (2018).
Stolley, J. M. et al. Retrograde migration supplies resident memory T cells to lung-draining LN after influenza infection. J. Exp. Med. https://doi.org/10.1084/jem.20192197 (2020).
Takamura, S. et al. The route of priming influences the ability of respiratory virus-specific memory CD8+ T cells to be activated by residual antigen. J. Exp. Med. 207, 1153–1160 (2010).
Evrard, M. et al. Sphingosine 1-phosphate receptor 5 (S1PR5) regulates the peripheral retention of tissue-resident lymphocytes. J. Exp. Med. https://doi.org/10.1084/jem.20210116 (2022).
Pan, Y. et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256 (2017).
Frizzell, H. et al. Organ-specific isoform selection of fatty acid-binding proteins in tissue-resident lymphocytes. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aay9283 (2020).
Laidlaw, B. J. et al. CD4+ T cell help guides formation of CD103+ lung-resident memory CD8+ T cells during influenza viral infection. Immunity 41, 633–645 (2014).
Mackay, L. K. et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463 (2016).
Parga-Vidal, L. et al. Hobit identifies tissue-resident memory T cell precursors that are regulated by Eomes. Sci. Immunol. https://doi.org/10.1126/sciimmunol.abg3533 (2021).
Milner, J. J. et al. Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257 (2017).
Cruz-Guilloty, F. et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J. Exp. Med. 206, 51–59 (2009).
Taniuchi, I. et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell 111, 621–633 (2002).
Sheridan, B. S. et al. Oral infection drives a distinct population of intestinal resident memory CD8+ T cells with enhanced protective function. Immunity 40, 747–757 (2014).
Zhang, N. & Bevan, M. J. Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39, 687–696 (2013).
Wakim, L. M., Smith, J., Caminschi, I., Lahoud, M. H. & Villadangos, J. A. Antibody-targeted vaccination to lung dendritic cells generates tissue-resident memory CD8 T cells that are highly protective against influenza virus infection. Mucosal Immunol. 8, 1060–1071 (2015).
Hu, Y., Lee, Y. T., Kaech, S. M., Garvy, B. & Cauley, L. S. Smad4 promotes differentiation of effector and circulating memory CD8 T cells but is dispensable for tissue-resident memory CD8 T cells. J. Immunol. 194, 2407–2414 (2015).
Nath, A. P. et al. Comparative analysis reveals a role for TGF-β in shaping the residency-related transcriptional signature in tissue-resident memory CD8+ T cells. PLoS ONE 14, e0210495 (2019).
Ma, C., Mishra, S., Demel, E. L., Liu, Y. & Zhang, N. TGF-β controls the formation of kidney-resident T cells via promoting effector T cell extravasation. J. Immunol. 198, 749–756 (2017).
Klicznik, M. M. et al. Human CD4+CD103+ cutaneous resident memory T cells are found in the circulation of healthy individuals. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aav8995 (2019).
Park, S. L. et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol. 19, 183–191 (2018).
Wakim, L. M., Waithman, J., van Rooijen, N., Heath, W. R. & Carbone, F. R. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science 319, 198–202 (2008).
Christo, S. N. et al. Discrete tissue microenvironments instruct diversity in resident memory T cell function and plasticity. Nat. Immunol. 22, 1140–1151 (2021).
Beura, L. K. et al. CD4+ resident memory T cells dominate immunosurveillance and orchestrate local recall responses. J. Exp. Med. 216, 1214–1229 (2019).
Takamura, S. et al. Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J. Exp. Med. 213, 3057–3073 (2016).
Iijima, N. & Iwasaki, A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science 346, 93–98 (2014).
Collins, N. et al. Skin CD4+ memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat. Commun. 7, 11514 (2016).
Egawa, T. & Littman, D. R. ThPOK acts late in specification of the helper T cell lineage and suppresses Runx-mediated commitment to the cytotoxic T cell lineage. Nat. Immunol. 9, 1131–1139 (2008).
He, X. et al. The zinc finger transcription factor Th-POK regulates CD4 versus CD8 T-cell lineage commitment. Nature 433, 826–833 (2005).
Reis, B. S., Rogoz, A., Costa-Pinto, F. A., Taniuchi, I. & Mucida, D. Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4+ T cell immunity. Nat. Immunol. 14, 271–280 (2013).
Fonseca, R. et al. Runx3 drives a CD8+ T cell tissue residency program that is absent in CD4+ T cells. Nat. Immunol. 23, 1236–1245 (2022).
Fernandez-Ruiz, D. et al. Liver-resident memory CD8+ T cells form a front-line defense against malaria liver-stage infection. Immunity 45, 889–902 (2016).
Boddupalli, C. S. et al. ABC transporters and NR4A1 identify a quiescent subset of tissue-resident memory T cells. J. Clin. Invest. 126, 3905–3916 (2016).
Li, C. et al. The transcription factor Bhlhe40 programs mitochondrial regulation of resident CD8+ T cell fitness and functionality. Immunity 51, 491–507.e7 (2019).
Quantius, J. et al. Influenza virus infects epithelial stem/progenitor cells of the distal lung: impact on Fgfr2b-driven epithelial repair. PLoS Pathog. 12, e1005544 (2016).
Ray, S. et al. Rare SOX2+ airway progenitor cells generate KRT5+ cells that repopulate damaged alveolar parenchyma following influenza virus infection. Stem Cell Rep. 7, 817–825 (2016).
Hirai, T. et al. Competition for active TGFβ cytokine allows for selective retention of antigen-specific tissue-resident memory T cells in the epidermal niche. Immunity 54, 84–98.e5 (2021).
Anthony, S. M. et al. Protective function and durability of mouse lymph node-resident memory CD8+ T cells. eLife https://doi.org/10.7554/eLife.68662 (2021).
Hogan, R. J. et al. Activated antigen-specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J. Immunol. 166, 1813–1822 (2001).
Liang, S., Mozdzanowska, K., Palladino, G. & Gerhard, W. Heterosubtypic immunity to influenza type A virus in mice. Effector mechanisms and their longevity. J. Immunol. 152, 1653–1661 (1994).
Wu, T. et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J. Leukoc. Biol. 95, 215–224 (2014).
Hayward, S. L. et al. Environmental cues regulate epigenetic reprogramming of airway-resident memory CD8+ T cells. Nat. Immunol. 21, 309–320 (2020).
Ely, K. H. et al. Nonspecific recruitment of memory CD8+ T cells to the lung airways during respiratory virus infections. J. Immunol. 170, 1423–1429 (2003).
Tripp, R. A., Hou, S. & Doherty, P. C. Temporal loss of the activated L-selectin-low phenotype for virus-specific CD8+ memory T cells. J. Immunol. 154, 5870–5875 (1995).
Roberts, A. D., Ely, K. H. & Woodland, D. L. Differential contributions of central and effector memory T cells to recall responses. J. Exp. Med. 202, 123–133 (2005).
Jozwik, A. et al. RSV-specific airway resident memory CD8+ T cells and differential disease severity after experimental human infection. Nat. Commun. 6, 10224 (2015).
Wakim, L. M., Gupta, N., Mintern, J. D. & Villadangos, J. A. Enhanced survival of lung tissue-resident memory CD8+ T cells during infection with influenza virus due to selective expression of IFITM3. Nat. Immunol. 14, 238–245 (2013).
Pizzolla, A. et al. Resident memory CD8+ T cells in the upper respiratory tract prevent pulmonary influenza virus infection. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aam6970 (2017).
Van Braeckel-Budimir, N. & Harty, J. T. Influenza-induced lung Trm: not all memories last forever. Immunol. Cell Biol. 95, 651–655 (2017).
Dave, V. A. et al. Cervicovaginal tissue residence confers a distinct differentiation program upon memory CD8 T cells. J. Immunol. 206, 2937–2948 (2021).
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Tarke, A. et al. SARS-CoV-2 vaccination induces immunological T cell memory able to cross-recognize variants from Alpha to Omicron. Cell 185, 847–859.e11 (2022).
Cohen, K. W. et al. Longitudinal analysis shows durable and broad immune memory after SARS-CoV-2 infection with persisting antibody responses and memory B and T cells. Cell Rep. Med. 2, 100354 (2021).
Dan, J. M. et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science https://doi.org/10.1126/science.abf4063 (2021).
Sekine, T. et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell 183, 158–168.e14 (2020).
Rydyznski Moderbacher, C. et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell 183, 996–1012.e19 (2020).
Gazit, S. et al. SARS-CoV-2 naturally acquired immunity vs. vaccine-induced immunity, reinfections versus breakthrough infections: a retrospective cohort study. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciac262 (2022).
Goldberg, Y. et al. Protection and waning of natural and hybrid immunity to SARS-CoV-2. N. Engl. J. Med. 386, 2201–2212 (2022).
Van Braeckel-Budimir, N., Varga, S. M., Badovinac, V. P. & Harty, J. T. Repeated antigen exposure extends the durability of influenza-specific lung-resident memory CD8+ T cells and heterosubtypic immunity. Cell Rep. 24, 3374–3382.e3 (2018).
Bowe, B., Xie, T. & Al-Aly, Z. Acute and postacute sequelae associated with SARS-CoV-2 reinfection. Nat. Med. https://doi.org/10.1038/s41591-022-02051-3 (2022).
Ariotti, S. et al. T cell memory. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 346, 101–105 (2014).
Merad, M. & Martin, J. C. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat. Rev. Immunol. 20, 355–362 (2020).
Hu, B., Huang, S. & Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 93, 250–256 (2021).
Acknowledgements
The author wishes to thank L. Mackay, L. Wakim and T. Gebhardt for insightful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Peer review
Peer review information
Nature Reviews Immunology thanks J. Harty, M. Hassert and J. Schenkel for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Carbone, F.R. Unique properties of tissue-resident memory T cells in the lungs: implications for COVID-19 and other respiratory diseases. Nat Rev Immunol 23, 329–335 (2023). https://doi.org/10.1038/s41577-022-00815-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-022-00815-z
This article is cited by
-
CD28/PD1 co-expression: dual impact on CD8+ T cells in peripheral blood and tumor tissue, and its significance in NSCLC patients' survival and ICB response
Journal of Experimental & Clinical Cancer Research (2023)
-
Assessing the generation of tissue resident memory T cells by vaccines
Nature Reviews Immunology (2023)
-
Cationic crosslinked carbon dots-adjuvanted intranasal vaccine induces protective immunity against Omicron-included SARS-CoV-2 variants
Nature Communications (2023)
