
Abstract
Immune responses are governed by signals from the tissue microenvironment, and in addition to biochemical signals, mechanical cues and forces arising from the tissue, its extracellular matrix and its constituent cells shape immune cell function. Indeed, changes in biophysical properties of tissue alter the mechanical signals experienced by cells in many disease conditions, in inflammatory states and in the context of ageing. These mechanical cues are converted into biochemical signals through the process of mechanotransduction, and multiple pathways of mechanotransduction have been identified in immune cells. Such pathways impact important cellular functions including cell activation, cytokine production, metabolism, proliferation and trafficking. Changes in tissue mechanics may also represent a new form of ‘danger signal’ that alerts the innate and adaptive immune systems to the possibility of injury or infection. Tissue mechanics can change temporally during an infection or inflammatory response, offering a novel layer of dynamic immune regulation. Here, we review the emerging field of mechanoimmunology, focusing on how mechanical cues at the scale of the tissue environment regulate immune cell behaviours to initiate, propagate and resolve the immune response.
Similar content being viewed by others

Introduction
Mechanotransduction is the process by which cells convert mechanical cues to biochemical signals, activating cellular pathways and influencing their function. Although fundamental roles for mechanical stimuli have long been recognized in developmental biology and in organ systems that receive significant mechanical stress, such as the cardiovascular and skeletal systems, these stimuli are now emerging as important orchestrators of inflammation and immune responses1,2,3,4,5. In pathophysiological environments where tissue mechanics are altered, profound changes in mechanotransduction components occur at the cellular level, such as in adhesion molecules, ion channels and cytoskeletal proteins, and in their associated signalling pathways. Dysregulated tissue mechanics are seen in many diseases. In atherosclerosis, lung fibrosis and the peri-tumoural environment6,7,8,9 there is enhanced tissue rigidity. By contrast, other diseased tissues, such as abscesses and the necrotic core of tumours10,11, are associated with mechanically softer tissue than in the healthy state. Understanding how these tissue-scale biophysical cues impact immune cell activation, metabolic reprogramming and downstream effector functions may provide new insights to treat these disorders.
Even under homeostatic conditions, immune cells face numerous forces as they circulate the body and encounter various tissue types. Cells that become resident in tissues experience stretching and shearing forces over time, as they are mechanically influenced by the extracellular matrix (ECM), interstitial fluids and neighbouring cells. These tissue-level forces tune subsequent immune responses. Previous reviews have focused on the role of mechanical forces in immune cell trafficking12 and molecular-level forces in triggering immune cell receptor activation13,14. In this Review, we discuss the tissue-level mechanical cues and forces that intersect with immune cell activation and effector functions, including danger sensing and cytokine production. We detail how such signals tune immune cell responses and provide an integrated picture of immunity through the lens of mechanobiology to better highlight the emerging field of mechanoimmunology.
Mechanosensing pathways
We begin by examining how immune cells sense exogenous forces. Cells within tissues experience multiple types of mechanical force, including tension, compression, shear stress, interstitial flow and hydrostatic pressure (Box 1 and reviewed in refs.15,16,17). In addition, the physical properties of the tissue surrounding the cells, including matrix rigidity, topography or architecture, also provide isometric mechanical cues that impact cellular function (Box 1 and reviewed elsewhere18,19,20). As we detail in the sections below, evolutionarily conserved pathways sense changes in these physical aspects of the ECM and can trigger mechanosensors in immune cells. It is important to note that immune cells do not just passively experience these exogenous cues but can also contribute to them, generating either reciprocal or additive forces upon the matrix and upon neighbouring cells.
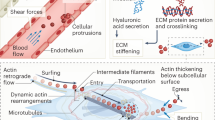
Immune cells share with other cells conserved pathways by which they can sense and respond to these mechanical cues (Fig. 1). Major mechanosensors include adhesion molecules (such as integrins), ion channels (including transient receptor potential vanilloid type 4 (TRPV4) and the PIEZO family of mechanically activated cation ion channels) and cytoskeletal components21,22,23. These mechanosensors transduce cues leading to Ca2+ fluxes, cytoskeletal reorganization and transcriptional regulation. In addition to cell-surface mechanosensors, the cytoskeletal network of filaments and tubules further allows cells to sense, withstand and respond to mechanical signals by transducing extracellular cues to the nucleus for dynamic rearrangement of chromatin following mechanical stimulation24,25. Actin filaments also form scaffolding at focal adhesions to support multiprotein signalling hubs while executing morphological changes of cells through polymerization-driven expansion, depolymerization or contraction with myosins26,27. External stress on cells results in reorientation of actin–myosin fibres (into ‘stress’ fibres) in parallel orientation to the force28. Cytoskeletal mechanotransduction is further propagated to the nuclear envelope through direct connections with the nuclear lamina via the linker of nucleoskeleton and cytoskeleton (LINC) complex, which comprises transmembrane proteins from the nesprin and SUN families29,30. These components mediate nuclear morphology and cytoskeletal force transmission with subsequent chromatin organization and gene regulation31,32,33.

a | Classical Hippo pathway activation induces phosphorylation of kinases MST1 and/or MST2, as well as LATS1 and/or LATS2, which in turn phosphorylate yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ). Phosphorylated YAP and TAZ are sequestered in the cytoplasm. Upon Hippo pathway inhibition, such as in response to mechanical force (denoted by F), YAP and TAZ translocate to the nucleus and drive gene expression that is key to immune function, including genes involved in cellular metabolic reprogramming, proliferation and responsiveness to inflammatory stimuli, through the TEA domain (TEAD) transcription factor family. b | Integrins anchor cells to the extracellular matrix (ECM), cluster and form focal adhesions, which act as multiprotein signalling hubs. Focal adhesion kinase (FAK) and SRC are key downstream effectors of focal adhesions, orchestrating various immune cell functions. Focal adhesions execute cell morphological changes either by actin polymerization-driven expansion or depolymerization and contraction with myosin. Polymerization of actin filaments (F-actin) subsequently reduces availability of G-actin (depicted by heavy arrow), decreasing its association with myocardin-related transcription factor A (MRTFA), which causes nuclear translocation of MRTFA, association with serum response factor (SRF) and gene transcription responsible for metabolic reprogramming, proliferation and cell responses to inflammatory stimuli. c | Mechanotransduction is also propagated directly from the cytoskeleton to the nuclear envelope via the linker of nucleoskeleton and cytoskeleton (LINC) complex, which includes the transmembrane proteins SUN and nesprin. LINC mediates nuclear morphology, chromatin organization, gene regulation and nuclear pore permeability, which also facilitates the nuclear translocation of YAP and TAZ and MRTFA. d | Transient receptor potential vanilloid type 4 (TRPV4) and PIEZO1 cation channels are activated by mechanical stimuli inflicted on the plasma membrane. Their activation promotes Ca2+ influx, impacting the regulation of many transcription factors, inflammatory signalling pathways and cytoskeletal reorganization.
Actin dynamics are largely regulated by a family of Rho GTPases, which themselves are controlled by many proteins and a diverse range of post-translational modifications that regulate subcellular localization and GTP exchange34,35,36,37,38,39. Molecules further downstream that influence actin dynamics promote the bundling of filaments40, the severing of filaments41,42, capping of actin43, branching of filaments44,45 and more. Important secondary mediators of force transduction include serum response factor (SRF), its coactivator myocardin-related transcription factor A (MRTFA) and yes-associated protein (YAP) or its homologue, transcriptional co-activator with PDZ-binding motif (TAZ). The role of these factors in the context of immune cells will be reviewed in the following sections.

Mechanotransduction in immune cells
A central question in immunology is how cells discern danger in the tissue environment. Traditionally, research has focused on components derived from invading pathogens (for example, lipopolysaccharide (LPS)) or damaged cells as the initiating danger signals that induce immune responses. However, emerging evidence suggests that biophysical cues may also regulate immune cell function and effector responses. For thousands of years, it has been known that ‘tumour’ (rigidity) is an early, cardinal sign of tissue inflammation (in addition to ‘calor’ (warmth), ‘rubor’ (redness), and ‘dolour’ (pain)). We now understand that the earliest changes in tissue mechanics are prompted before the activation of immune responses, by epithelial and stromal cells themselves in response to Toll-like receptor (TLR) ligands such as LPS. These signals prompt dramatic increases in the production of high molecular weight hyaluronic acid, which traps water and swells tissue (that is, causes oedema) over the course of minutes to hours. Endothelial cells respond to TLR signals by increasing the permeability of the endothelium, leading to fluid and protein leakage into the tissue. Immune cells experience these mechanical changes as they traverse these tissue environments, and these mechanical cues play an active role in the early recognition of danger and in the initiation of immune responses.
Many innate immune cells are adherent and contact-dependent, making them exquisitely sensitive to mechanical stimuli such as changes in ECM stiffness. Immune cells are frequently studied using engineered systems for their responses to mechanical cues (Box 2), and such studies have illuminated roles for mechanotransduction in immune cells. Mechanosensing of substrate rigidity by macrophages has been shown to influence phagocytosis46,47, the mode of migration46, cellular reactive oxygen species (ROS) production48, healing ability49, cell morphology50 and secretion of both anti-inflammatory and pro-inflammatory cytokines47,49,51,52. As a general trend, compared with macrophages grown on softer materials, macrophages cultured on a stiffer substrate show enhanced effector functions and produce more pro-inflammatory cytokines when exposed to TLR ligands. For instance, macrophages cultured on rigid polystyrene make more tumour necrosis factor (TNF) and interleukin-6 (IL-6), but less IL-10, in response to LPS stimulation when compared with the same cells cultured on soft fibrin hydrogels51. For TNF, this trend held across a range of LPS doses and with different TLR agonists, including Pam3CSK4 (a TLR1/TLR2 agonist) and FSL1 (a TLR2/TLR6 agonist)47,49,51. Consistent with these findings in macrophages, bone marrow-derived dendritic cells grown on a stiff substrate (50 kPa, mimicking fibro-inflammatory disease) produce increased levels of pro-inflammatory cytokines — including IL-1α, IL-1β, IL-6, IL-12 and TNF — following LPS stimulation and show increased activation and proliferation compared with dendritic cells cultured on soft hydrogel (2 kPa, mimicking the physiological stiffness of most soft tissues)53. Therefore, the mechanical signal transduced by a stiff ECM into innate immune cells tunes the inflammatory phenotype as they respond to pattern recognition receptor (PRR) ligands. Mast cells are also sensitive to mechanical stretching, which enhances their degranulation and secretion of histamine, and their release of active transforming growth factor-β1(TGFβ1)54,55.
Mechanosensing in innate immune cells is not limited to sensing changes in tissue stiffness or tension. Circulating monocytes are also affected by fluid shear stress, where high shear stress promotes adhesion, phagocytosis and pro-inflammatory cytokine secretion56. Similarly, neutrophils that endure shear force show enhanced phagocytosis, greater platelet–neutrophil aggregation and amplified cytosolic Ca2+ levels leading to increased cell activation57,58. Macrophages also become pro-inflammatory when exposed to cyclical hydrostatic pressure59. These data suggest that a multitude of mechanical stimuli can influence effector functions in innate immune cells, tuning their inflammatory response.
Adaptive immune cells also respond to mechanical cues. T cells and B cells recognize cognate antigens either as peptides bound to the MHC molecules on the surface of antigen-presenting cells (APCs) or as immobilized whole proteins, respectively. Forces generated from this contact modulates their behaviour14, making the physical properties of antigen-presenting surfaces influential to adaptive immune cell responses. For example, T cells that are stimulated by ‘stiff’ dendritic cells require a lower antigen concentration for activation than do T cells stimulated by ‘soft’ dendritic cells60. In fact, dendritic cells actively elevate cortical stiffness during maturation, suggesting that increased cytoskeletal stiffness is a biophysical mechanism central to proper priming of T cells60. Substrate stiffness is also a modulator of adaptive immune cell behaviour, and has been shown to regulate T cell spreading61,62, migration, gene expression, cytokine secretion62, proliferation63 and cytotoxic function64. When tissue stiffness was specifically increased from 4 kPa (similar to what is encountered in a resting lymph node) to 40 kPa (resembling a swollen lymph node), it promoted T cell proliferation, activation and metabolism, while lowering the antigen dose needed to elicit effector responses65. T cells crawling through an artificial, mechanically tuned environment will alter their threshold for activation in a manner that depends more on the substrate stiffness than the stiffness of the APCs offering antigenic stimulation66. Thus, T cells can separately sense both environmental cues as well as the mechanics of the APCs with which they interact. For B cells, the rigidity of antigen-presenting substrates regulates B cell receptor (BCR) accumulation67, antigen uptake68, proliferation, class switching and antibody production69. B cells appear to sense the mechanics of follicular dendritic cells in the lymph node germinal centre and the forces required to extract antigens from the surface of follicular dendritic cells as a way of promoting the preferential uptake of high-affinity antigens over low-affinity antigens from the environment68. A recent publication also reported that different biomaterial implants with essentially distinct mechanical properties affect the B cell response to muscle injury70. For instance, synthetic polyester implants induced certain B cell pathways including strengthening their presentation of antigens and expressing inflammation-related genes.
Given that biophysical cues and forces in the environment are emerging as important modulators of immunity, it is important to understand the mechanisms and signalling processes by which mechanical cues influence immune cell activation and effector function. Below, we outline key studies linking mechanosensing pathways to immune cell effector outputs (see Supplementary Table 1 for a summary). In particular, we focus on two mechanosensing pathways — mechano-gated ion channels and Hippo signalling — that have been linked to inflammation.
Ion channels: PIEZO1 and TRPV4
Exposure of physical stimuli to the plasma membrane can trigger ion influx through channels and transporters71. Two of these mechano-gated ion channels have been well characterized in immune cells: PIEZO1, a member of the Piezo family of mechanically activated cation channels72, and TRPV4, a member of the TRP superfamily of non-selective cation channels73. Mechanical deformations of the plasma membrane directly open the channels of PIEZO1 and TRPV4. TRPV4 can also open in response to osmotic stress and temperature changes, and indirectly through inflammatory mediators such as arachidonic acid or histamine74,75. Both PIEZO1 and TRPV4 facilitate influx of extracellular Ca2+, leading to activation of Ca2+-regulated signalling pathways that influence cellular metabolism and effector functions. One example of signalling modulated by Ca2+ in immune cells is the calcineurin–NFAT transcription factor pathway, which is essential for T cell activation76,77. Elevation of intracellular Ca2+ triggers calcineurin, a protein phosphatase, to dephosphorylate NFAT and allows it to translocate into the nucleus78. Studies have shown that PIEZO1-mediated Ca2+ influx can activate NFAT in cells such as osteoblasts79, but this has yet to be verified in immune cells. PIEZO1 expression is particularly abundant in mechano-active tissues such as the lungs, bladder and skin80, and PIEZO1 is transcriptionally expressed across a wide range of immune cell subsets, whereas TRPV4 expression appears more limited to a few myeloid subsets81.
During macrophage activation, PIEZO1 co-localizes and coordinates with TLR4 to enhance ROS production via a calmodulin-dependent protein kinase II (CAMKII)–MST1/MST2–Rac axis, increasing pathogen ingestion and killing48. Stiff substrates were shown to activate PIEZO1, triggering Ca2+ influx that enhanced NF-κB activation and F-actin formation in bone marrow-derived macrophages stimulated with LPS and IFNγ, leading to a more prominent ‘M1-like’ inflammatory macrophage phenotype49. Intriguingly, although bone marrow-derived macrophages cultured on a stiff substrate also have Arginase 1 (ARG1) expression in response to IL-4 and IL-13, PIEZO1 activity independent of a stiff substrate suppresses the IL-4/IL-13-induced expression of ARG1, demonstrating that mechanotransductive signals can override one another49. Moreover, ECM stiffness was shown to govern population expansion and pathogenic cytokine release by myeloid-derived suppressor cells in a PIEZO1-dependent manner through the silencing of the tumour suppressor protein retinoblastoma 1 (RB1) by histone deacetylation82.
In response to cyclical hydrostatic pressure, PIEZO1 activation in macrophages leads to the production of endothelin 1 (EDN1), which stabilizes hypoxia-inducible factor 1α (HIF1α) to optimize antibacterial defence against Pseudomonas aeruginosa59. Of note, HIF1α is a major inducer of glycolysis, bridging PIEZO1 activity with glycolysis-driven inflammatory responses, highlighting mechanical regulation of immune cell metabolism83. Consistent with this concept, the addition of the PIEZO1 agonist, YODA1, to mouse dendritic cells in vitro induced expression of the major glycolytic genes MYC and hexokinase 2 (HK2), as well as SLC2A1 (which encodes the glucose transporter GLUT1), and also boosted TNF production53. Furthermore, PIEZO1 plays an important role in shear stress-induced inflammation, where it contributes to adhesion of circulating monocytes, activation of CD11b integrin (also known as Mac1), increased phagocytosis and release of pro-inflammatory cytokines56. These findings indicate that various mechanical signals can mediate innate inflammatory responses. It will be interesting to assess how PIEZO1 impacts immune cell function across different tissue types and disease states.
Studies have only recently begun to shed light on a role for PIEZO1 in regulating adaptive immunity. In culture, (partial) small interfering RNA-mediated knockdown of PIEZO1 in human CD4+ T cells reduced T cell receptor (TCR) activation and proliferation by weakening calpain-dependent immunological synapse stabilization84. Notably, unlike in osteoblasts, the Ca2+–calcineurin–NFAT pathway was not activated by agonist-induced opening of PIEZO1 in that system. By contrast, another study showed that CD4+ T cell-specific deletion of Piezo1 in mice did not impair T cell proliferation or TCR-evoked Ca2+ signalling85. Genetic deletion of Piezo1 instead enhanced expansion of regulatory T (Treg) cell populations by promoting TGFβ signalling. The conflicting requirement for PIEZO1 in T cell activation in these two systems might be the consequence of the experimental approaches used. Furthermore, PIEZO1 is responsible for maintaining a peri-arteriolar niche of lymphoid progenitors in the bone marrow; depletion of PIEZO1 from the niche caused a decline of lymphoid cells and a compromised immune state86. These initial studies support the hypothesis that PIEZO1 tunes effector functions of adaptive immune cells. However, more work is needed to better elucidate the role of PIEZO1 in adaptive immunity, especially in showing its mechanical role.
Similarly to PIEZO1, the role of TRPV4 has been largely studied in innate and tissue-resident immune cells, especially macrophages. TRPV4 facilitates skin-resident macrophage acquisition of a pro-inflammatory phenotype in conditions of high stiffness (50 kPa)47. Stiff matrices had a synergistic effect on IFNγ and LPS-induced polarization during macrophage activation, and the effect was reliant on functional TRPV4. TRPV4-deficient mice also had a reduced ability to clear bacteria during Mycobacterium tuberculosis infection owing to delayed phagosome acidification and reduced generation of reactive nitrogen species87. Similarly, during infection with P. aeruginosa, the presence of TRPV4 enhanced pathogen clearance by macrophages88. Specifically, TRPV4-mediated TLR activation of p38 via dual-specificity phosphatase 1 (DUSP1) increased macrophage phagocytosis on stiff matrices88. Interestingly, p38 activity was shown to attenuate inflammatory cytokine production through inhibition of JNK, which reduced infection-associated tissue damage. Resident cardiac macrophages also stimulate myocardial tissue remodelling and coronary angiogenesis through TRPV4-mediated sensing of mechanical stress associated with heart failure, suggesting a role for TRPV4 in promoting tissue regeneration89. By contrast, another study demonstrated that TRPV4 enhanced the release of cytokines such as IL-1α, IL-1β, IL-6, IL-8 and CC-chemokine ligand 2 (CCL2) in response to equi-biaxial stretch, a form of tension exerted by mechanical ventilators90. Conflicting results show there is still much to learn about TRPV4 in immune cells. Contradictory findings may arise from differences in cell sources, differentiation protocols, agonist dosing and adherent substrates. Still, TRPV4 has emerged as an important link between mechanosensing and pathogen clearance.
YAP and TAZ of the Hippo signalling pathway
Recent studies directly link mechanotransduction via the Hippo signalling pathway to immune cell responses. Classically, this pathway is modulated by cell contact and density91. These mechanical cues activate the kinases MST1/MST2 and LATS1/LATS2, which in turn phosphorylate the two homologous effector proteins YAP and TAZ92. Phosphorylated YAP and TAZ are sequestered in the cytoplasm, and depending on the kinase and phosphorylation site, become a substrate for degradation in the proteasome93. Upon inhibition of the Hippo pathway kinases, YAP and TAZ move into the nucleus to drive gene expression through their binding with transcription factors, particularly members of the TEA domain (TEAD) family94. YAP and TAZ can also translocate in response to ECM stiffness and cell patterning95. In immune cells, this tunes effector fates through transcription of genes integral to metabolic reprogramming, proliferation and activation-related functions. For example, nuclear YAP and TAZ direct LPS and IFNγ-triggered M1-like macrophage activation and IL-6 production, while inhibiting macrophage polarization towards an IL-4 and IL-13-induced M2-like anti-inflammatory phenotype96. Similarly, stiff substrates drove nuclear localization of YAP in macrophages and potentiated an inflammatory response after stimulation with LPS, as determined by elevated levels of NF-κB phosphorylation and pro-inflammatory cytokine production51. YAP and TAZ can also be immunomodulatory when sequestered in the cytosol (as in softer environments), where they can inhibit antiviral defence by impairing type I interferon production through interaction with the protein kinase TBK1 and interferon regulatory factor 3 (IRF3)97,98. This cytoplasmic interaction blocks IRF3 translocation to the nucleus, limiting the transcription of interferon-related genes.
Indeed, stiffness-mediated nuclear localization of YAP and TAZ has profound effects on innate immune cell signalling and metabolism. YAP–TEAD and TAZ–TEAD complexes govern the expression of genes involved in glycolysis, in glutamine metabolism and in glucose and amino acid uptake via both directly induced transcription or indirect induction. The importance of YAP and TAZ in regulating metabolism has been reviewed elsewhere99. Notably, altered metabolic reprogramming is a fundamental step in immune cell activation, effector function and maintenance100. Linking mechanotransduction-mediated Hippo signalling to metabolism in immune cells, upon contact with stiff substrates, TAZ translocates to the nucleus in bone marrow-derived dendritic cells and splenic dendritic cells and induces the transcription of genes that shape metabolic processes in these cells in response to PRR signals53. Such genes include the master regulator of glycolysis MYC, the glycolysis gene HK2, the glucose transporter SLC2A1 and the pentose phosphate pathway gene PGLS. Because of activated TAZ, dendritic cells cultured on stiff ECM have amplified activity in glycolysis, the tricarboxylic acid cycle (TCA) and the pentose phosphate pathway, which facilitates dendritic cell activation and pro-inflammatory function. Tension-mediated priming of dendritic cell metabolism occurred even without PRR input, although to a lesser extent, raising the possibility that tension acutely primes immune cell metabolism prior to activation, potentially to ready the cell in a stiff environment for optimal effector response.
In adaptive immune cells, there is similar stiffness-mediated subcellular localization of YAP and TAZ (Fig. 2). In T cells, phosphorylated YAP modulates activation by regulating the binding affinity between NFAT1, a transcription factor regulating T cell activation and metabolism, and its scaffolding protein IQGAP1, based on matrix stiffness65. YAP stabilizes NFAT1–IQGAP1 interactions in the cytoplasm in response to soft ECM contact, attenuating metabolic reprogramming and T cell activation. However, when T cells sense a stiff microenvironment YAP translocates to the nucleus, freeing NFAT1 from the NFAT1–IQGAP1 binding complex and enabling induction of downstream metabolic genes. Thus, as the mechanical environment goes from inflamed (stiff) to basal (soft), YAP mediates downregulation of T cell responses. This softness-driven inhibition also protects bystander tissues from T cell-mediated autoimmunity, as evidenced by accelerated development of type 1 diabetes in mice models when T cells lack YAP65. YAP-suppressed metabolic pathways include glycolysis, mitochondrial respiration and amino acid uptake. Consistent with these findings, a recent study reported that YAP knockout boosts CD8+ T cell production of cytotoxic cytokines and degranulation factors101, which may be related to the loss of the cytosolic functions of YAP. Knockout of YAP also perturbs Treg cell function, resulting in reduced TGFβ–SMAD signalling102. Some functions of YAP and TAZ may also occur independently of mechanical signals. For example, when interacting with the transcription factor RORγt instead of TEAD1, TAZ decreases acetylation of FOXP3, leading to attenuation of Treg cell differentiation103. Nonetheless, YAP and TAZ remain key intermediates of mechanical signal tuning of innate and adaptive immunity.

When T cells are in tissues with a soft extracellular matrix (ECM) microenvironment, yes-associated protein (YAP) is phosphorylated (likely by LATS1 or LATS2) and localized in the cytoplasm, where it interacts with IQGAP1. Owing to YAP and IQGAP1 interaction, NFAT1 is preferentially sequestered in the cytoplasm, and this results in attenuated cellular metabolism and proliferation. Conversely, when T cells encounter a stiff ECM microenvironment, YAP is dephosphorylated and migrates to the nucleus. Additionally, NFAT1 is no longer sequestered in the cytoplasm through IQGAP1–YAP interaction. Thus, with the help of calcium release-activated channels (CRAC), calcineurin and calmodulin, NFAT1 can be dephosphorylated and is free to translocate to the nucleus to induce T cell activation-related gene expression.
Integrins
Many cells are physically anchored to their extracellular environment through integrins. This family of transmembrane heterodimeric receptors is composed of α-subunits and β-subunits that bind to ECM proteins104. Upon binding to the matrix, integrins cluster to form focal adhesions, or multiprotein hubs that are connected to the cytoskeleton. Physical deformation of cells leads to cytoskeletal rearrangements, including actin bundling and stress fibre formation, which directly activates intracellular signalling pathways105. Focal adhesion kinase (FAK) and Src, two non-receptor tyrosine kinases, are important downstream effectors of focal adhesions. Under mechanical engagement, this complex directs a network of factors, such as RhoA, Rac1 and Cdc42, that modulate morphological changes, cell motility, adhesion, development, proliferation, survival and inflammation106,107.
The role of integrins is well studied in leukocyte migration; however, integrins are also mechanosensitive and actively regulate immune cell effector functions. For example, LFA1 (also known as αL integrin and CD11a–CD18) undergoes a conformational change in response to high substrate rigidity that increases its affinity for immobilized intercellular adhesion molecule 1 (ICAM1)108. This process occurs during dendritic cell maturation109. Higher affinity binding between LFA1 and ICAM1 also promotes the activation of immune cells, including T cells and natural killer cells110,111,112. Stiff matrices have been shown to enhance integrin CD11b-controlled phagocytosis in macrophages113 and LFA1-mediated actin cytoskeletal polymerization in T cells21, which promotes MRTFA translocation and consequent gene expression114,115. In addition, strong FAK activation downstream of integrins elicited by high substrate stiffness facilitates B cell spreading and adhesion, enhancing B cell activation response67. Despite these important functions of integrins in the immune system, some immune cells do not require integrins to fulfil certain effector functions in the presence of mechanical cues that occur during cell migration, or in response to infection or autoimmunity. For instance, studies have reported that integrins may be dispensable for some aspects of dendritic cell migration from tissue to lymph node, natural killer cell migration for protection against viral infection, Treg cell migration to the central nervous system during experimental autoimmune encephalomyelitis as well as T cell priming by dendritic cells during Trichuris muris infection116,117,118,119. Nonetheless, integrins still have important roles in transducing mechanical cues into effector responses in many contexts.
The MRTFA–SRF pathway
SRF and its coactivator MRTFA also link mechanotransduction, cytoskeleton dynamics and cell function. Cytoplasmic MRTFA is predominantly bound to G-actin. Mechanical stimuli induce polymerization of actin filaments, thereby reducing G-actin and causing the release of MRTFA. This allows MRTFA to translocate to the nucleus, where together with SRF it induces gene transcription115,120,121. In myeloid cells, this transcriptional activity affects migration, phagocytosis and cytoskeletal gene expression122. In dendritic cells, MRTFA–SRF also modulates cell cycle progression, adhesion and lipid metabolism114. RNA sequencing of Mrtfa–/– dendritic cells showed a reduction in cholesterol metabolism when compared with wild-type cells114, possibly connecting mechanotransduction and cholesterol metabolism. Lipid and cholesterol metabolism are critical for dendritic cell function123. Thus, attenuated cholesterol metabolism in Mrtfa–/– cells will most likely impair their immune functions, although this has yet to be shown. In macrophages, MRTFA and SRF integrate signals of spatial confinement into immunomodulating pathways124. This is especially relevant for tissue-resident macrophages, which can be tightly packed in tissues and increase in size upon activation, causing spatial confinement. The spatial confinement of macrophages suppresses the expression of late LPS-activated genes — such as those encoding IL-6, CXCL9 and inducible nitric oxide synthase (iNOS) — by downregulating histone deacetylase 3 (HDAC3), inhibiting the removal of dimethylated histone H3 K36 (H3K36me2) marks, and reducing inflammatory tone125. The transcription of such late-responsive genes is further modulated by MRTFA and SRF, which are generally induced by LPS, but reduced by spatial confinement, thus providing another link between mechanical cues and cytokine production.
Another member of the myocardin-related transcription factor family, MRTFB, has been shown to have some redundant functions to MRTFA in certain contexts, such as neuronal migration, cardiac function and haematopoietic progenitor cell chemotaxis126,127,128. Both proteins also play important roles in cancer cell immunosurveillance, and their overexpression elevates tumour cell rigidity resulting in increased killing by cytotoxic T lymphocytes and natural killer cells129. However, whether MRTFB directly responds to force induction to modulate downstream expression in immune cells themselves still requires further investigation.
LINC complex and nuclear lamins
The LINC complex and nuclear lamins are major modulators of mechanotransduction in the nucleus. Their expression influences many immune effector functions including chemotaxis, inflammation, proliferation and activation130,131. A-type lamins and the LINC complex were found to mediate T cell immunological synapse formation by promoting actin skeleton polymerization and downstream gene expression related to T cell activation, as well as ERK1 and ERK2 phosphorylation132. Indeed, T cells that lack lamins or have disconnected LINC–lamin complexes have impaired activation and proliferation. Similar to many mechanosensitive proteins in immune cells, A-type lamins together with the LINC complex have the ability to bridge actin remodelling and transcription in the nucleus, therefore hinting to their indispensable role in immune mechanotransduction. As research on the LINC complex and nuclear lamins has mainly been performed in T cells, more work is needed to better elucidate their roles across other cell types, and their link with other mechanotransduction pathways.
Model linking mechanotransduction and immune cell function
Innate immune cells probe the tissue environment for danger signals to alert them for activation. Tissue-level mechanical stresses that occur in disease states, such as acute oedema during infection or increased stiffness with fibrosis, can activate mechanosensors in immune cells, allowing their signalling pathways to synergize with inputs from PRRs to boost immune effector functions (Fig. 3). When tension forces are reduced, homeostasis is restored and the inactivity of mechanosensors raises the threshold for danger signalling, and immune activation is subsequently reduced. Thus, we favour an integrated model by which tissue-level mechanotransduction directly tunes innate signalling pathways involved in danger sensing.

Pattern recognition receptor (PRR) stimulation accompanied by mechanical force causes PIEZO1 co-localization with some Toll-like receptors (TLRs). Force-triggered opening of transient receptor potential vanilloid type 4 (TRPV4) and PIEZO1 leads to Ca2+ influx. Elevated cytosolic Ca2+ levels induce actin polymerization and activate Rho GTPase, boosting phagocytosis. Concurrently, F-actin formation depletes G-actin availability, and without G-actin to bind to, myocardin-related transcription factor A (MRTFA) is shuttled into the nucleus where it forms a complex with serum response factor (SRF). MRTFA–SRF enables transcription of immune effector genes, such as those encoding interleukin-6 (IL-6) and CXC-chemokine ligand 9 (CXCL9), generally skewing the cell towards an enhanced inflammatory phenotype. Ca2+ also enforces TLR signalling by promoting the expression and activation of endothelin 1 (EDN1) and NF-κB. EDN1 stabilizes hypoxia-inducible factor 1α (HIF1α), promoting TLR-induced HIF1α accumulation. Active NF-κB and HIF1α translocate to the nucleus and drive metabolic and inflammatory gene expression. Additionally, TRPV4-dependent Ca2+ influx prompts dual-specificity phosphatase 1 (DUSP1) to activate p38 and inhibit JNK activity, further enhancing phagocytosis. The mechanosensitive molecules yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) also translocate to the nucleus upon activation, bind TEA domains (TEADs) and induce transcription of genes that boost glycolysis, the pentose phosphate pathway and the tricarboxylic acid (TCA) cycle. This immunometabolic regulation through multiple mechanosensitive inputs mediates robust cell proliferation and increases the production of inflammatory cytokines, such as IL-1α, IL-1β, IL-6 and CC-chemokine ligand 2 (CCL2). PIEZO1 also activates histone deacetylases (HDACs) to further induce inflammatory cytokine output. Dashed lines represent pathways where there are still major gaps in understanding the transduction mechanisms, and solid lines depict more well-described pathways. Linker of nucleoskeleton and cytoskeleton (LINC) complex modulation of immune activation has been shown in adaptive cells only, but we hypothesize it also plays a role in innate immune cell regulation. ECM, extracellular matrix; LPS, lipopolysaccharide; TNF, tumour necrosis factor.
This ability to tune responses to danger signals in distinct physical environments probably evolved as an additional layer of regulation atop biochemical cues. The integrated signals promote immune activity during early immune responses, which is often dynamically associated with transient increases in tissue rigidity (for example, those seen in oedematous inflamed tissues, inside proliferating lymph nodes or following cell trafficking from the blood into crowded or stiffer tissue spaces). These signals also reinforce resolution of immune responses and quiescence when stiffness is reduced by homeostatic mechanisms such as degradation of accumulated glycosaminoglycans (GAGs) and apoptosis of crowded cellular environments65. According to this model, the threshold for immune cell activation would be high when cells encounter danger signals within a mechanically soft environment. Such a mechanism might serve to reduce autoimmunity and inadvertent immunopathology in precious, irreplaceable tissues such as the brain. Likewise, according to this model, fibrotic and ageing tissues that become mechanically stiffer over time may be more likely to promote inflammation and autoimmunity. Supporting this viewpoint, older patients and patients with interstitial lung diseases or restrictive lung physiology have worse outcomes after respiratory viral infections including influenza and COVID-19 (refs.133,134,135). However, whether a threshold ultimately exists during chronic disease or ageing when immune cells become unresponsive, exhausted or potentially show resolving phenotypes by long-standing continuous exposure to biophysical cues and/or environmental forces remains to be seen. Similarly, it will also be important to better understand potential inflammatory implications of insufficient mechanotransduction in immune cells.
Although we have focused on direct mechanotransduction in immune cells, it is important to note that mechanosensing pathways can impact immunity indirectly through mesenchymal cells, such as fibroblasts, stromal cells and endothelial cells. These cells express a similar constellation of mechanosensing pathways, often to a greater extent than in immune cells. Indeed, stromal cells such as fibroblasts sense ECM mechanics and, consequently, recruit immune cells such as macrophages136,137. In both mouse and human endothelial cells, the activity of YAP and TAZ is suppressed by unidirectional blood flow with low shear stress9. YAP and TAZ inhibition dampens pro-inflammatory gene expression, and decreases the local infiltration of monocytes. Similarly, TRPV4 governs stretching-induced inflammation and expression of pro-inflammatory factors, including IL-6, IL-8, cyclooxygenase 2 (COX2), matrix metalloproteinase 1 (MMP1) and MMP3, which contributes to tissue degradation in human annulus fibrosus cells138. Given the critical crosstalk that occurs between the stroma and the immune system, a better understanding of how mechanical inputs link these two cellular compartments represents an important avenue for future research. This topic will be especially important in understanding acute and chronic integrated disease processes, such as responses to tissue injury, bacterial infections, inflammatory diseases and fibrosis.
Mechanoimmunology in health and disease
The acute and chronic rigidification of tissue following inflammatory stimuli has long been recognized139, but the physiological or pathophysiological roles of tissue mechanics in regulating immune responses have not been appreciated until recently. Here, we discuss how mechanical signals associated with altered tissue mechanics shape immunity within physiological and pathophysiological tissue environments. Unfortunately, we lack many comprehensive, in vivo studies of the impacts of mechanosensing by immune cells in pathophysiological states. Although in vitro studies have yielded important, reductionistic insights into the responses of immune cells to biophysical cues, these studies often fail to fully recapitulate integrative cell–cell responses, as well as true three-dimensional viscoelastic properties of tissues. Therefore, we offer here a perspective synthesizing the best available in vitro and in vivo studies to date.
Acute pathogen challenge
During an acute infection, early activation of PRRs induces stromal cells to produce copious amounts of hyaluronic acid, and the resulting oedema and inflammation drive extensive changes in tissue mechanics140. Surveillance of affected tissues is increased by neutrophils, which are recruited from the blood to the site of oedema. In the blood, circulating neutrophils must endure shear stresses to create and maintain a catch bond with the endothelium. The tethering catch bond is mediated by selectin and glycosylated ligand interactions, initially P-selectin and P-selectin glycoprotein ligand 1 (PSGL1)141. Only with adequate shear stresses from blood flow do high-affinity catch bonds form and neutrophils extravasate. Upon successful diapedesis, the neutrophil continues to experience forces, including compression, traction and tension, as its plasma membrane deforms to allow for extravasation into the tissue. Migration of neutrophils through dense tissue drives mechanical changes upon the nucleus, and the resulting chromatin changes promote inflammatory transcripts142. These rapidly recruited innate immune cells migrate into dense tissues, probing the tissue ahead of them by means of a nuclear-mechanical gauge143. They produce cytokines, inflammatory lipids and exosomes to increase blood flow, induce capillary leakage and augment pain sensation.
At the site of infection, tissue swelling applies mechanical forces, such as tensile stress and hydrostatic pressure, onto resident innate immune cells. Data from in vitro studies suggest that such forces are important in priming innate immune cell metabolism to ready these cells for the bioenergetic demands of activation, as well as in providing the cells with a mechano-dependent co-stimulatory signal that can reduce the signalling threshold necessary for optimal TLR stimulation48,51,53,144. Thus, pathogen-mediated triggering of PRRs elicits an acute effector response with a level of magnitude that is tuned by signals from the physical environment. Many minor, local bacterial infections are cleared by the resulting deluge of innate effectors.
Over hours, antigenic debris and hyaluronic acids are trafficked to the draining lymph node where they coat APCs140, which increases their ability to interact with T cells145. As circulating T cells and B cells enter a lymph node through high endothelial venules, they also experience shear forces upon catch-bond formation, and extravasation induces forces onto these lymphocytes similar to those encountered by neutrophils entering sites of infection. T cells within a secondary lymphoid organ can be subsequently activated by an APC, such as a dendritic cell, a process that is also influenced by mechanical forces. During migration to the secondary lymphoid organ, activated dendritic cells go through a maturation process that improves their ability to stimulate T cells. Stromal cells in the lymph node, especially fibroblastic reticular cells, undergo mechanical changes upon interacting with activated dendritic cells, further driving changes in lymph node mechanics during acute infections146.
Activation of T cells and B cells within encapsulated spaces, that is, lymph nodes, leads to robust proliferation and crowding, driving further deviation from ‘normal’ tissue mechanics. Lymph node tissue mechanics affects T cell activation. In particular, in vivo mouse models have shown that at the height of an infection with lymphocytic choriomeningitis virus the lymph node reaches 40 kPa, and decreases to 4 kPa when the infection resolves65. When T cells crawl through matrices of 40 kPa stiffness, they show increased activation regardless of the stiffness of the APC owing to increased contact area of the T cell–APC conjugate66. This mechanosensing by T cells involves YAP. T cells also bind to hyaluronic acids through CD44, a mechanosensitive receptor that is important for T cell extravasation, although future studies are needed to determine whether this interaction promotes T cell suppression or activation in the context of mechanics. Late remodelling of the lymph node architecture may occur in the days to weeks following acute inflammation146, which can have profound effects on tissue stiffness, such as in fibrosis or scarring, due to enhanced deposition of collagen and remodelling of the ECM. The integrative processing of mechanical signals into immune responses to infection is shown in Fig. 4.

a | Danger signals in the skin (such as those arising from bacteria or tissue damage) activate Langerhans cells and stromal cells to produce inflammatory cytokines and/or hyaluronic acid to promote oedema and attract immune cells to the infection site. b | Within minutes to hours, diapedesis of cells from the vasculature into inflamed tissues occurs. This requires force-sensing at the molecular level (shear force-dependent catch bonds) and at the cellular level (cells squeezing through endothelium and extracellular matrix (ECM)). Forces exerted by diapedesis prime circulating immune cells to acquire an enhanced pro-inflammatory phenotype. Simultaneously, local oedema occurs. Mechanosensation of oedema further drives a pro-inflammatory phenotype in tissue-resident cells that encounter pathogens. c | Eventually, antigenic debris, hyaluronic acids and activated dendritic cells traffic to the lymph node. There, fibroblastic reticular cells sense activated dendritic cells and change their shape to allow for imminent increases in lymph node size. Migrating T cells and B cells encounter mechanical forces as they squeeze through high endothelial venules and into interfollicular and follicular spaces. After activation, T cells and B cells proliferate in the lymph node, causing lymph node swelling and stiffening, which propagates mechanical forces onto cells. T cells crawling through a stiff lymph node increase the contact area for T cell–antigen-presenting cell (APC) conjugates, which may also impact T follicular helper (TFH) cell interactions with germinal centre B cells. Activated T cells and B cells eventually migrate to the infection site via the blood, continuing to experience multiple mechanical stimuli at the inflamed tissue. Resolution of the infection leads to breakdown of hyaluronic acid, decreasing oedema and leading to the refractoriness of immune cells to danger signals. The resulting diminishment of inflammatory cytokines reduces immune cell trafficking and promotes their apoptosis through cytokine withdrawal-induced cell death and other mechanisms. Under the influence of ageing and other factors, tissue may not return to a ‘healthy’ baseline and might, instead, adopt remodelling changes including collagen deposition (scarring) and elastin degradation, leaving a lasting mechanical imprint at the inflammatory response site. ILC, innate lymphoid cell; IL, interleukin; PRR, pattern recognition receptor; TNF, tumour necrosis factor.
Wound healing and fibrosis
Immune cells play an important role in wound healing, and recent studies suggest mechanosensing may drive immune cell-dependent responses involved in aberrant healing, such as scarring and fibrosis147. There are four stages in wound healing: haemostasis, inflammation, proliferation and remodelling. During haemostasis in the skin, tissue-resident cells such as fibroblasts, myofibroblasts and macrophages endure tensive forces, which are generated by the skin’s resistance to breaking. If an injury breaks the skin, the haemostatic phase transitions into the inflammatory phase, and fibroblasts, macrophages and neutrophils will travel to the site of the wound where they recognize tissue damage and create a pro-inflammatory environment. There is a subsequent generation of vascularized ECM, known as granulation tissue, through which epithelial cells migrate to close the wound. Fibroblasts then differentiate into myofibroblasts, which synthesize collagen and ECM components including hyaluronic acid to generate a scar148. Higher mechanical tension in the healing wound can clinically worsen the healing response and lead to hyperplastic scars149. There is speculation that some of this mechanism is mediated through tension-induced FAK activity in fibroblasts150. These tension-primed fibroblasts release high levels of CCL2 to recruit immune cells (that is, CCR2+ inflammatory macrophages), which ultimately help orchestrate inflammation and potentiate scar formation149. Using a high-tension scar model, global CCL2-knockout mice showed a 70% reduction in scar formation at 10 days post injury. This study demonstrates a direct link between mechanotransduction, recruitment of macrophages and aberrant wound responses149. Moreover, another study using a similar high-tension model of scarring showed that treatment of skin wounds with verteporfin, an inhibitor of YAP and TAZ, prevented an engrailed 1-expressing subset of fibroblasts from generating a fibrotic response151. Further insight into the roles of mechanosensing in scar formation is needed, particularly to assess the direct impacts of different mechanosensing pathways in immune cells during wound healing.
Cancer
One of the hallmarks of the solid tumour microenvironment is that it is often stiffer than healthy tissue, which typically results from abnormal physical properties of the ECM. Numerous works have demonstrated the co-progression of tumorigenesis and mechanical stiffness152,153. The stiffness of the ECM can forestall immune responses by acting as a physical barrier that blocks motility. The collagen-rich ECM around tumours includes features such as alignment of fibres in the perivascular regions and around nests of tumour epithelial cells. This ECM has been shown to alter the migration of T cells and restrict them away from tumour cell nests154. Furthermore, breast cancer tissue samples that show a high collagen density in the ECM surrounding the tumour have fewer infiltrating T cells155. Beyond this stiff barrier of solid tumours, the tumour cells themselves are often mechanically softer and more heterogeneous than neighbouring healthy cells10. As tumours hit a mechanical barrier owing to the constraint offered by nearby tissues, they undergo a change from a proliferative phenotype to a metastatic phenotype. The resulting metastatic cells are alternatively said to be mechanically stiff because they crawl on basement membranes129 or mechanically soft because they have to crawl through small pores created by their own ECM ‘fence’156. How tumour-infiltrating immune cells respond to the heterogeneous mechanical environment of tumours has barely been studied. However, tumour killing by cytotoxic T cells was shown to be potentiated when the tumour cells are mechanically rigid because stretched tumour membranes are more susceptible to T cell-delivered perforin157. The impact of this differential capability to kill tumour cells may promote the survival of tumour cells that are able to repopulate the tumour64. Much more work is needed to study whether therapeutic alterations of the ECM could impact tumour stiffness and the associated immune response to improve clinical outcomes. For example, collagenases, lysyl-oxidase inhibitors (which block the cross-linking of collagen) or hyaluronidases could be employed to modify the tumour ECM.
Conclusions and outlook
Mechanical changes in tissues occur with the natural process of ageing and in disease states such as infection, tissue injury and cancer. These mechanical changes influence immune cells, tuning their effector functions, with the potential to promote both adaptive and maladaptive responses. The alterations in ECM that underlie tissue changes and the mechanotransducing pathways that sense tissue mechanics offer therapeutic targets for diseases such as fibrosis and cancer. Regardless of the advances presented in this Review, the field of mechanoimmunology is still in its infancy; improved means to measure tissue mechanics in vivo are needed. Consistently, we lack in vivo models that specifically test the impact of mechanical forces on immune cells in various pathophysiological states. Thus, many findings inferred from in vitro testing may need to be revised as the field matures. Furthermore, measurements of tissue mechanics, as well as constructive interpretation of the findings, have hardly made their way into clinical practice, and thus offer ripe potential for exploiting MRI, ultrasound and other modalities to probe mechanics and guide therapeutic algorithms158,159. Further study into the regulation of immune responses by tissue mechanics will no doubt provide new insights with direct applications into boosting or resolving inflammation, treating autoimmunity and promoting regenerative healing after tissue injury or infection.
References
Garoffolo, G. & Pesce, M. Mechanotransduction in the cardiovascular system: from developmental origins to homeostasis and pathology. Cells 8, 1607 (2019).
Hsieh, J. Y. et al. Differential regulation of macrophage inflammatory activation by fibrin and fibrinogen. Acta Biomater. 47, 14–24 (2017).
Meli, V. S. et al. Biophysical regulation of macrophages in health and disease. J. Leukoc. Biol. 106, 283–299 (2019).
Rowley, A. T., Nagalla, R. R., Wang, S.-W. & Liu, W. F. Extracellular matrix-based strategies for immunomodulatory biomaterials engineering. Adv. Healthc. Mater. 8, e1801578 (2019).
Smith, T. D., Nagalla, R. R., Chen, E. Y. & Liu, W. F. Harnessing macrophage plasticity for tissue regeneration. Adv. Drug Deliv. Rev. 114, 193–205 (2017).
Casal, J. I. & Bartolomé, R. A. RGD cadherins and α2β1 integrin in cancer metastasis: a dangerous liaison. Biochim. Biophys. Acta Rev. Cancer 1869, 321–332 (2018).
Feng, Y. et al. The signaling protein Wnt5a promotes TGFβ1-mediated macrophage polarization and kidney fibrosis by inducing the transcriptional regulators Yap/Taz. J. Biol. Chem. 293, 19290–19302 (2018).
Jansson, L. & Larsson, J. Normal hematopoietic stem cell function in mice with enforced expression of the Hippo signaling effector YAP1. PLoS ONE 7, e32013 (2012).
Wang, L. et al. Integrin–YAP/TAZ–JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540, 579–582 (2016). This report identifies that atheroprone-disturbed blood flow increases shear stress, thereby promoting endothelial YAP/TAZ activity; in addition, YAP/TAZ inhibition downregulates pro-inflammatory gene expression, which reduces monocyte attachment and infiltration.
Plodinec, M. et al. The nanomechanical signature of breast cancer. Nat. Nanotechnol. 7, 757–765 (2012).
Gaspari, R. et al. Use of ultrasound elastography for skin and subcutaneous abscesses. J. Ultrasound Med. 28, 855–860 (2009).
Huse, M. Mechanical forces in the immune system. Nat. Rev. Immunol. 17, 679–690 (2017).
Pageon, S. V., Govendir, M. A., Kempe, D. & Biro, M. Mechanoimmunology: molecular-scale forces govern immune cell functions. Mol. Biol. Cell 29, 1919–1926 (2018).
Zhu, C., Chen, W., Lou, J., Rittase, W. & Li, K. Mechanosensing through immunoreceptors. Nat. Immunol. 20, 1269–1278 (2019).
Wells, R. G. Tissue mechanics and fibrosis. Biochim. Biophys. Acta 1832, 884–890 (2013).
Orr, A. W., Helmke, B. P., Blackman, B. R. & Schwartz, M. A. Mechanisms of mechanotransduction. Dev. Cell 10, 11–20 (2006).
Sugimura, K., Lenne, P.-F. & Graner, F. Measuring forces and stresses in situ in living tissues. Development 143, 186–196 (2016).
Vogel, V. Unraveling the mechanobiology of extracellular matrix. Annu. Rev. Physiol. 80, 353–387 (2018).
Guimarães, C. F., Gasperini, L., Marques, A. P. & Reis, R. L. The stiffness of living tissues and its implications for tissue engineering. Nat. Rev. Mater. https://doi.org/10.1038/s41578-019-0169-1 (2020).
Paul, C. D., Hung, W.-C., Wirtz, D. & Konstantopoulos, K. Engineered models of confined cell migration. Annu. Rev. Biomed. Eng. 18, 159–180 (2016).
Roy, N. H., MacKay, J. L., Robertson, T. F., Hammer, D. A. & Burkhardt, J. K. Crk adaptor proteins mediate actin-dependent T cell migration and mechanosensing induced by the integrin LFA-1. Sci. Signal. 11, eaat3178 (2018).
Köhler, R. et al. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler. Thromb. Vasc. Biol. 26, 1495–1502 (2006).
Li, J. et al. Piezo1 integration of vascular architecture with physiological force. Nature 515, 279–282 (2014).
Maniotis, A. J., Chen, C. S. & Ingber, D. E. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc. Natl Acad. Sci. USA 94, 849–854 (1997).
Le, H. Q. et al. Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 18, 864–875 (2016).
Théry, M. et al. Anisotropy of cell adhesive microenvironment governs cell internal organization and orientation of polarity. Proc. Natl Acad. Sci. USA 103, 19771–19776 (2006).
Even-Ram, S. et al. Myosin IIA regulates cell motility and actomyosin–microtubule crosstalk. Nat. Cell Biol. 9, 299–309 (2007).
Uyeda, T. Q. P., Iwadate, Y., Umeki, N., Nagasaki, A. & Yumura, S. Stretching actin filaments within cells enhances their affinity for the myosin II motor domain. PLoS ONE 6, e26200 (2011).
Tajik, A. et al. Transcription upregulation via force-induced direct stretching of chromatin. Nat. Mater. 15, 1287–1296 (2016).
Lombardi, M. L. et al. The interaction between nesprins and sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J. Biol. Chem. 286, 26743–26753 (2011).
Jain, N., Iyer, K. V., Kumar, A. & Shivashankar, G. V. Cell geometric constraints induce modular gene-expression patterns via redistribution of HDAC3 regulated by actomyosin contractility. Proc. Natl Acad. Sci. USA 110, 11349–11354 (2013).
Alam, S. G. et al. The mammalian LINC complex regulates genome transcriptional responses to substrate rigidity. Sci. Rep. 6, 38063 (2016).
Versaevel, M., Grevesse, T. & Gabriele, S. Spatial coordination between cell and nuclear shape within micropatterned endothelial cells. Nat. Commun. 3, 671 (2012).
Michaelson, D. et al. Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 152, 111–126 (2001).
Oberoi, T. K. et al. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. EMBO J. 31, 14–28 (2012).
Lang, P. et al. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 15, 510–519 (1996).
Adamson, P., Marshall, C. J., Hall, A. & Tilbrook, P. A. Post-translational modifications of p21rho proteins. J. Biol. Chem. 267, 20033–20038 (1992).
Katayama, M. et al. The posttranslationally modified C-terminal structure of bovine aortic smooth muscle rhoA p21. J. Biol. Chem. 266, 12639–12645 (1991).
Hodge, R. G. & Ridley, A. J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 17, 496–510 (2016).
Wang, C. et al. Actin-bundling protein L-plastin regulates T cell activation. J. Immunol. 185, 7487–7497 (2010).
Klemke, M. et al. Oxidation of cofilin mediates T cell hyporesponsiveness under oxidative stress conditions. Immunity 29, 404–413 (2008).
Thauland, T. J., Hu, K. H., Bruce, M. A. & Butte, M. J. Cytoskeletal adaptivity regulates T cell receptor signaling. Sci. Signal. 10, eaah3737 (2017).
Thauland, T. J., Khan, H. A. & Butte, M. J. The actin-capping protein α-adducin is required for T-cell costimulation. Front. Immunol. 10, 2706 (2019).
Amann, K. J. & Pollard, T. D. Direct real-time observation of actin filament branching mediated by Arp2/3 complex using total internal reflection fluorescence microscopy. Proc. Natl Acad. Sci. USA 98, 15009–15013 (2001).
Mullins, R. D., Heuser, J. A. & Pollard, T. D. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl Acad. Sci. USA 95, 6181–6186 (1998).
Sridharan, R., Cavanagh, B., Cameron, A. R., Kelly, D. J. & O’Brien, F. J. Material stiffness influences the polarization state, function and migration mode of macrophages. Acta Biomater. 89, 47–59 (2019).
Dutta, B., Goswami, R. & Rahaman, S. O. TRPV4 plays a role in matrix stiffness-induced macrophage polarization. Front. Immunol. 11, 570195 (2020).
Geng, J. et al. TLR4 signalling via Piezo1 engages and enhances the macrophage mediated host response during bacterial infection. Nat. Commun. 12, 3519 (2021). This report identifies PIEZO1 as an associated complex to TLR4 during bacterial infection or LPS activation. Thus, this work links a TLR to a mechanosensor in organizing integral components of the innate response to pathogens, including phagocytosis, ROS production and bacterial clearance.
Atcha, H. et al. Mechanically activated ion channel Piezo1 modulates macrophage polarization and stiffness sensing. Nat. Commun. 12, 3256 (2021).
Chen, M. et al. Substrate stiffness modulates bone marrow-derived macrophage polarization through NF-κB signaling pathway. Bioact. Mater. 5, 880–890 (2020).
Meli, V. S. et al. YAP-mediated mechanotransduction tunes the macrophage inflammatory response. Sci. Adv. 6, eabb8471 (2020). This study identifies nuclear localization of YAP as a key mediator in tuning inflammatory macrophage responses to enhanced substrate stiffness.
Scheraga, R. G., Southern, B. D., Grove, L. M. & Olman, M. A. The role of TRPV4 in regulating innate immune cell function in lung inflammation. Front. Immunol. 11, 1211 (2020).
Chakraborty, M. et al. Mechanical stiffness controls dendritic cell metabolism and function. Cell Rep. 34, 108609 (2021). This study connects mechanosensing via Hippo signalling and PIEZO1 to the immunometabolism of innate immune cells, by showing how substrate stiffness impacts dendritic cell metabolism, maturation and inflammatory function.
Shimbori, C. et al. Mechanical stress-induced mast cell degranulation activates TGF-β1 signalling pathway in pulmonary fibrosis. Thorax 74, 455–465 (2019).
Hu, K. K., Bruce, M. A. & Butte, M. J. Spatiotemporally and mechanically controlled triggering of mast cells using atomic force microscopy. Immunol. Res. 58, 211–217 (2014).
Baratchi, S. et al. Transcatheter aortic valve implantation represents an anti-inflammatory therapy via reduction of shear stress-induced, Piezo-1-mediated monocyte activation. Circulation 142, 1092–1105 (2020).
Morikis, V. A., Masadeh, E. & Simon, S. I. Tensile force transmitted through LFA-1 bonds mechanoregulate neutrophil inflammatory response. J. Leukoc. Biol. 108, 1815–1828 (2020).
Sun, W. et al. Neutrophil injury and function alterations induced by high mechanical shear stress with short exposure time. Artif. Organs 45, 577–586 (2021).
Solis, A. G. et al. Mechanosensation of cyclical force by PIEZO1 is essential for innate immunity. Nature 573, 69–74 (2019). This study shows that PIEZO1 acts as a sensor of cyclical pressure in macrophages and monocytes, triggering pro-inflammatory responses to bacteria and likely exacerbating inflammation in fibrotic microenvironments.
Blumenthal, D., Chandra, V., Avery, L. & Burkhardt, J. K. Mouse T cell priming is enhanced by maturation-dependent stiffening of the dendritic cell cortex. eLife 9, e55995 (2020).
Wahl, A. et al. Biphasic mechanosensitivity of T cell receptor-mediated spreading of lymphocytes. Proc. Natl Acad. Sci. USA 116, 5908–5913 (2019).
Saitakis, M. et al. Different TCR-induced T lymphocyte responses are potentiated by stiffness with variable sensitivity. eLife 6, e23190 (2017). This study demonstrates that physiological-relevant increases in substrate stiffness influence migration, transcription, cytokine production, metabolism and cell cycle progression of CD4+ T cells.
Hickey, J. W. et al. Engineering an artificial T-cell stimulating matrix for immunotherapy. Adv. Mater. 31, e1807359 (2019).
Liu, Y. et al. Cell softness prevents cytolytic T-cell killing of tumor-repopulating cells. Cancer Res. 81, 476–488 (2021).
Meng, K. P., Majedi, F. S., Thauland, T. J. & Butte, M. J. Mechanosensing through YAP controls T cell activation and metabolism. J. Exp. Med. 217, e20200053 (2020). This study ties the tissue-level mechanical changes to the optimal priming of T cell effector responses via YAP-mediated effects on NFAT1 and metabolic reprogramming.
Majedi, F. S. et al. T-cell activation is modulated by the 3D mechanical microenvironment. Biomaterials 252, 120058 (2020).
Shaheen, S. et al. Substrate stiffness governs the initiation of B cell activation by the concerted signaling of PKCβ and focal adhesion kinase. eLife 6, e23060 (2017).
Spillane, K. M. & Tolar, P. B cell antigen extraction is regulated by physical properties of antigen-presenting cells. J. Cell Biol. 216, 217–230 (2017).
Zeng, Y. et al. Substrate stiffness regulates B-cell activation, proliferation, class switch, and T-cell-independent antibody responses in vivo. Eur. J. Immunol. 45, 1621–1634 (2015).
Moore, E. M. et al. Biomaterials direct functional B cell response in a material-specific manner. Sci. Adv. 7, eabj5830 (2021).
Feske, S., Wulff, H. & Skolnik, E. Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 33, 291–353 (2015).
Coste, B. et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330, 55–60 (2010).
Zheng, J. Molecular mechanism of TRP channels. Compr. Physiol. 3, 221–242 (2013).
Michalick, L. & Kuebler, W. M. TRPV4 — a missing link between mechanosensation and immunity. Front. Immunol. 11, 413 (2020).
Sulk, M. & Steinhoff, M. Role of TRP channels in skin diseases. in TRP channels as therapeutic targets (ed. Szallasi, A.) 293–323 (Elsevier, 2015).
Loh, C., Carew, J. A., Kim, J., Hogan, P. G. & Rao, A. T-cell receptor stimulation elicits an early phase of activation and a later phase of deactivation of the transcription factor NFAT1. Mol. Cell. Biol. 16, 3945–3954 (1996).
Feske, S., Giltnane, J., Dolmetsch, R., Staudt, L. M. & Rao, A. Gene regulation mediated by calcium signals in T lymphocytes. Nat. Immunol. 2, 316–324 (2001).
Bueno, O. F., Brandt, E. B., Rothenberg, M. E. & Molkentin, J. D. Defective T cell development and function in calcineurin A β-deficient mice. Proc. Natl Acad. Sci. USA 99, 9398–9403 (2002).
Zhou, T. et al. Piezo1/2 mediate mechanotransduction essential for bone formation through concerted activation of NFAT–YAP1–β-catenin. eLife 9, e52779 (2020).
Hennes, A. et al. Functional expression of the mechanosensitive PIEZO1 channel in primary endometrial epithelial cells and endometrial organoids. Sci. Rep. 9, 1779 (2019).
Heng, T. S. P. & Painter, M. W., Immunological Genome Project Consortium. The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol. 9, 1091–1094 (2008).
Aykut, B. et al. Targeting Piezo1 unleashes innate immunity against cancer and infectious disease. Sci. Immunol. 5, eabb5168 (2020).
Denko, N. C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705–713 (2008).
Liu, C. S. C. et al. Cutting edge: piezo1 mechanosensors optimize human T cell activation. J. Immunol. 200, 1255–1260 (2018).
Jairaman, A. et al. Piezo1 channels restrain regulatory T cells but are dispensable for effector CD4+ T cell responses. Sci. Adv. 7, eabg5859 (2021).
Shen, B. et al. A mechanosensitive peri-arteriolar niche for osteogenesis and lymphopoiesis. Nature 591, 438–444 (2021).
Naik, S. K. et al. Differential roles of the calcium ion channel TRPV4 in host responses to Mycobacterium tuberculosis early and late in infection. iScience 23, 101206 (2020).
Scheraga, R. G. et al. TRPV4 protects the lung from bacterial pneumonia via MAPK molecular pathway switching. J. Immunol. 204, 1310–1321 (2020).
Wong, N. R. et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity https://doi.org/10.1016/j.immuni.2021.07.003 (2021).
Pairet, N. et al. TRPV4 inhibition attenuates stretch-induced inflammatory cellular responses and lung barrier dysfunction during mechanical ventilation. PLoS ONE 13, e0196055 (2018).
Aragona, M. et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154, 1047–1059 (2013).
Halder, G., Dupont, S. & Piccolo, S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 13, 591–600 (2012).
Totaro, A., Panciera, T. & Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 20, 888–899 (2018).
Panciera, T., Azzolin, L., Cordenonsi, M. & Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 18, 758–770 (2017).
Dupont, S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011).
Zhou, X. et al. YAP aggravates inflammatory bowel disease by regulating M1/M2 macrophage polarization and gut microbial homeostasis. Cell Rep. 27, 1176–1189.e5 (2019).
Zhang, Q. et al. Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat. Cell Biol. 19, 362–374 (2017).
Wang, S. et al. YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKɛ-mediated phosphorylation. Nat. Immunol. 18, 733–743 (2017).
Koo, J. H. & Guan, K.-L. Interplay between YAP/TAZ and metabolism. Cell Metab. 28, 196–206 (2018).
O’Neill, L. A. J., Kishton, R. J. & Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565 (2016).
Lebid, A., Chung, L., Pardoll, D. M. & Pan, F. YAP attenuates CD8 T cell-mediated anti-tumor response. Front. Immunol. 11, 580 (2020).
Ni, X. et al. YAP is essential for Treg-mediated suppression of antitumor immunity. Cancer Discov. 8, 1026–1043 (2018).
Geng, J. et al. The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and Treg cells. Nat. Immunol. 18, 800–812 (2017).
Takada, Y., Ye, X. & Simon, S. The integrins. Genome Biol. 8, 215 (2007).
Wang, N. Review of cellular mechanotransduction. J. Phys. D. Appl. Phys. 50, (2017).
Sun, Z., Guo, S. S. & Fässler, R. Integrin-mediated mechanotransduction. J. Cell Biol. 215, 445–456 (2016).
Byeon, S. E. et al. The role of Src kinase in macrophage-mediated inflammatory responses. Mediators Inflamm. 2012, 512926 (2012).
Ben-Shmuel, A., Joseph, N., Sabag, B. & Barda-Saad, M. Lymphocyte mechanotransduction: the regulatory role of cytoskeletal dynamics in signaling cascades and effector functions. J. Leukoc. Biol. 105, 1261–1273 (2019).
Comrie, W. A., Li, S., Boyle, S. & Burkhardt, J. K. The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM-1 mobility. J. Cell Biol. 208, 457–473 (2015).
Li, D., Molldrem, J. J. & Ma, Q. LFA-1 regulates CD8+ T cell activation via T cell receptor-mediated and LFA-1-mediated Erk1/2 signal pathways. J. Biol. Chem. 284, 21001–21010 (2009).
Matalon, O. et al. Actin retrograde flow controls natural killer cell response by regulating the conformation state of SHP-1. EMBO J. 37, e96264 (2018).
Ni, H. T., Deeths, M. J. & Mescher, M. F. LFA-1-mediated costimulation of CD8+ T cell proliferation requires phosphatidylinositol 3-kinase activity. J. Immunol. 166, 6523–6529 (2001).
Jaumouillé, V., Cartagena-Rivera, A. X. & Waterman, C. M. Coupling of β2 integrins to actin by a mechanosensitive molecular clutch drives complement receptor-mediated phagocytosis. Nat. Cell Biol. 21, 1357–1369 (2019).
Guenther, C. et al. A β2-integrin/MRTF-A/SRF pathway regulates dendritic cell gene expression, adhesion, and traction force generation. Front. Immunol. 10, 1138 (2019).
Olson, E. N. & Nordheim, A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 11, 353–365 (2010).
Lämmermann, T. et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 453, 51–55 (2008).
Altorki, T., Muller, W., Brass, A. & Cruickshank, S. The role of β2 integrin in dendritic cell migration during infection. BMC Immunol. 22, 2 (2021).
Stotesbury, C. et al. α2β1 integrin is required for optimal NK cell proliferation during viral infection but not for acquisition of effector functions or NK cell-mediated virus control. J. Immunol. 204, 1582–1591 (2020).
Mair, I. et al. A context-dependent role for αv integrins in regulatory T cell accumulation at sites of inflammation. Front. Immunol. 9, 264 (2018).
Miralles, F., Posern, G., Zaromytidou, A.-I. & Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113, 329–342 (2003).
Vartiainen, M. K., Guettler, S., Larijani, B. & Treisman, R. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 316, 1749–1752 (2007).
Record, J. et al. Immunodeficiency and severe susceptibility to bacterial infection associated with a loss-of-function homozygous mutation of MKL1. Blood 126, 1527–1535 (2015).
Everts, B. et al. TLR-driven early glycolytic reprogramming via the kinases TBK1–IKKɛ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 15, 323–332 (2014).
Jain, N., Moeller, J. & Vogel, V. Mechanobiology of macrophages: how physical factors coregulate macrophage plasticity and phagocytosis. Annu. Rev. Biomed. Eng. 21, 267–297 (2019).
Jain, N. & Vogel, V. Spatial confinement downsizes the inflammatory response of macrophages. Nat. Mater. 17, 1134–1144 (2018). This study demonstrates mechanotransduction pathway modulation of inflammation by showing that spatial confinement of macrophages downregulates late inflammatory responses through epigenetic alterations and MRTFA–SRF-mediated transcription.
Mokalled, M. H. et al. Myocardin-related transcription factors are required for cardiac development and function. Dev. Biol. 406, 109–116 (2015).
Mokalled, M. H., Johnson, A., Kim, Y., Oh, J. & Olson, E. N. Myocardin-related transcription factors regulate the Cdk5/Pctaire1 kinase cascade to control neurite outgrowth, neuronal migration and brain development. Development 137, 2365–2374 (2010).
Costello, P. et al. MRTF–SRF signaling is required for seeding of HSC/Ps in bone marrow during development. Blood 125, 1244–1255 (2015).
Tello-Lafoz, M. et al. Cytotoxic lymphocytes target characteristic biophysical vulnerabilities in cancer. Immunity 54, 1037–1054.e7 (2021).
Saez, A. et al. Lamin A/C and the immune system: one intermediate filament, many faces. Int. J. Mol. Sci. 21, 6109 (2020).
Donahue, D. A., Porrot, F., Couespel, N. & Schwartz, O. SUN2 silencing impairs CD4 T cell proliferation and alters sensitivity to HIV-1 infection independently of cyclophilin A. J. Virol. 91, e02303-16 (2017).
González-Granado, J. M. et al. Nuclear envelope lamin-A couples actin dynamics with immunological synapse architecture and T cell activation. Sci. Signal. 7, ra37 (2014).
Esposito, A. J. et al. Increased odds of death for patients with interstitial lung disease and COVID-19: a case–control study. Am. J. Respir. Crit. Care Med. 202, 1710–1713 (2020).
Drake, T. M. et al. Outcome of hospitalization for COVID-19 in patients with interstitial lung disease. An international multicenter study. Am. J. Respir. Crit. Care Med. 202, 1656–1665 (2020).
Bartleson, J. M. et al. SARS-CoV-2, COVID-19 and the aging immune system. Nat. Aging 1, 769–782 (2021).
Pakshir, P. et al. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat. Commun. 10, 1850 (2019).
Zhou, X. et al. Microenvironmental sensing by fibroblasts controls macrophage population size. Preprint at BioRxiv https://doi.org/10.1101/2022.01.18.476683 (2022).
Cambria, E. et al. TRPV4 inhibition and CRISPR–Cas9 knockout reduce inflammation induced by hyperphysiological stretching in human annulus fibrosus cells. Cells 9, 1736 (2020).
Celsus, A. C. De medicina (Harvard Univ. Press, 1935).
Nagy, N. et al. Hyaluronan in immune dysregulation and autoimmune diseases. Matrix Biol. 78–79, 292–313 (2019).
Xiao, Z., Goldsmith, H. L., McIntosh, F. A., Shankaran, H. & Neelamegham, S. Biomechanics of P-selectin PSGL-1 bonds: shear threshold and integrin-independent cell adhesion. Biophys. J. 90, 2221–2234 (2006).
Jacobson, E. C. et al. Migration through a small pore disrupts inactive chromatin organization in neutrophil-like cells. BMC Biol. 16, 142 (2018).
Renkawitz, J. et al. Nuclear positioning facilitates amoeboid migration along the path of least resistance. Nature 568, 546–550 (2019).
Hsieh, J. Y. et al. Matrix crosslinking enhances macrophage adhesion, migration, and inflammatory activation. Apl. Bioeng. 3, 016103 (2019).
Bollyky, P. L. et al. TH1 cytokines promote T-cell binding to antigen-presenting cells via enhanced hyaluronan production and accumulation at the immune synapse. Cell. Mol. Immunol. 7, 211–220 (2010).
Krishnamurty, A. T. & Turley, S. J. Lymph node stromal cells: cartographers of the immune system. Nat. Immunol. 21, 369–380 (2020).
Kuehlmann, B., Bonham, C. A., Zucal, I., Prantl, L. & Gurtner, G. C. Mechanotransduction in wound healing and fibrosis. J. Clin. Med. 9, 1423 (2020).
Ruppert, S. M., Hawn, T. R., Arrigoni, A., Wight, T. N. & Bollyky, P. L. Tissue integrity signals communicated by high-molecular weight hyaluronan and the resolution of inflammation. Immunol. Res. 58, 186–192 (2014).
Wong, V. W. et al. Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat. Med. 18, 148–152 (2011). Using a wound repair model of cutaneous injury, this study shows a direct link between mechanotransduction, immune cell recruitment and fibrosis through an inflammatory FAK–ERK–MCP1 axis.
Greenberg, R. S. et al. FAK-dependent regulation of myofibroblast differentiation. FASEB J. 20, 1006–1008 (2006).
Mascharak, S. et al. Preventing Engrailed-1 activation in fibroblasts yields wound regeneration without scarring. Science 372, eaba2374 (2021).
Cukierman, E. & Bassi, D. E. Physico-mechanical aspects of extracellular matrix influences on tumorigenic behaviors. Semin. Cancer Biol. 20, 139–145 (2010).
Acerbi, I. et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 7, 1120–1134 (2015).
Salmon, H. et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Invest. 122, 899–910 (2012).
Kuczek, D. E. et al. Collagen density regulates the activity of tumor-infiltrating T cells. J. Immunother. Cancer 7, 68 (2019).
Kim, T.-H. et al. Cancer cells become less deformable and more invasive with activation of β-adrenergic signaling. J. Cell Sci. 129, 4563–4575 (2016).
Basu, R. et al. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell 165, 100–110 (2016). This study shows that cytotoxic T lymphocytes require mechanical forces both in the form of force exertion cross the synapse and tumour cell tension to modulate perforin pore formation for optimal tumour cell killing.
Venkatesh, S. K., Yin, M. & Ehman, R. L. Magnetic resonance elastography of liver: technique, analysis, and clinical applications. J. Magn. Reson. Imaging 37, 544–555 (2013).
Kennedy, P. et al. Quantitative elastography methods in liver disease: current evidence and future directions. Radiology 286, 738–763 (2018).
Vining, K. H. & Mooney, D. J. Mechanical forces direct stem cell behaviour in development and regeneration. Nat. Rev. Mol. Cell Biol. 18, 728–742 (2017).
Hsiai, T. K. et al. Monocyte recruitment to endothelial cells in response to oscillatory shear stress. FASEB J. 17, 1648–1657 (2003).
Rutkowski, J. M. & Swartz, M. A. A driving force for change: interstitial flow as a morphoregulator. Trends Cell Biol. 17, 44–50 (2007).
Knothe Tate, M. L. “Whither flows the fluid in bone?” An osteocyte’s perspective. J. Biomech. 36, 1409–1424 (2003).
Evans, R. C. & Quinn, T. M. Dynamic compression augments interstitial transport of a glucose-like solute in articular cartilage. Biophys. J. 91, 1541–1547 (2006).
Fitzgerald, J. B., Jin, M. & Grodzinsky, A. J. Shear and compression differentially regulate clusters of functionally related temporal transcription patterns in cartilage tissue. J. Biol. Chem. 281, 24095–24103 (2006).
Stavenschi, E., Corrigan, M. A., Johnson, G. P., Riffault, M. & Hoey, D. A. Physiological cyclic hydrostatic pressure induces osteogenic lineage commitment of human bone marrow stem cells: a systematic study. Stem Cell Res. Ther. 9, 276 (2018).
Tansey, E. A., Montgomery, L. E. A., Quinn, J. G., Roe, S. M. & Johnson, C. D. Understanding basic vein physiology and venous blood pressure through simple physical assessments. Adv. Physiol. Educ. 43, 423–429 (2019).
Ogawa, R. Mechanobiology of scarring. Wound Repair. Regen. 19, s2–s9 (2011).
Darwish, A. & Lui, F. Physiology, colloid osmotic pressure. in StatPearls (StatPearls Publishing, 2022).
Sachot, N., Engel, E. & Castano, O. Hybrid organic–inorganic scaffolding biomaterials for regenerative therapies. COC 18, 2299–2314 (2014).
Acevedo-Acevedo, S. & Crone, W. C. Substrate stiffness effect and chromosome missegregation in hIPS cells. J. Negat. Results Biomed. 14, 22 (2015).
Leung, V. Y. et al. Quantitative elastography of liver fibrosis and spleen stiffness in chronic hepatitis B carriers: comparison of shear-wave elastography and transient elastography with liver biopsy correlation. Radiology 269, 910–918 (2013).
Colecchia, A. et al. Measurement of spleen stiffness to evaluate portal hypertension and the presence of esophageal varices in patients with HCV-related cirrhosis. Gastroenterology 143, 646–654 (2012).
Pawluś, A. et al. Shear wave elastography of the spleen: evaluation of spleen stiffness in healthy volunteers. Abdom. Radiol. 41, 2169–2174 (2016).
Arda, K., Ciledag, N., Aktas, E., Aribas, B. K. & Köse, K. Quantitative assessment of normal soft-tissue elasticity using shear-wave ultrasound elastography. AJR Am. J. Roentgenol. 197, 532–536 (2011).
Murphy, M. C. et al. Decreased brain stiffness in Alzheimer’s disease determined by magnetic resonance elastography. J. Magn. Reson. Imaging 34, 494–498 (2011).
Jansen, L. E., Birch, N. P., Schiffman, J. D., Crosby, A. J. & Peyton, S. R. Mechanics of intact bone marrow. J. Mech. Behav. Biomed. Mater. 50, 299–307 (2015).
Ramião, N. G. et al. Biomechanical properties of breast tissue, a state-of-the-art review. Biomech. Model. Mechanobiol. 15, 1307–1323 (2016).
Kawano, S. et al. Assessment of elasticity of colorectal cancer tissue, clinical utility, pathological and phenotypical relevance. Cancer Sci. 106, 1232–1239 (2015).
Wang, L., Yan, F., Yang, Y., Xiang, X. & Qiu, L. Quantitative assessment of skin stiffness in localized scleroderma using ultrasound shear-wave elastography. Ultrasound Med. Biol. 43, 1339–1347 (2017).
Diridollou, S. et al. Skin ageing: changes of physical properties of human skin in vivo. Int. J. Cosmet. Sci. 23, 353–362 (2001).
Sasaki, Y. & Ogura, I. Shear wave elastography in differentiating between benign and malignant cervical lymph nodes in patients with oral carcinoma. Dentomaxillofac Radiol. 48, 20180454 (2019).
Sicard, D. et al. Aging and anatomical variations in lung tissue stiffness. Am. J. Physiol. Lung Cell Mol. Physiol. 314, L946–L955 (2018).
Booth, A. J. et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am. J. Respir. Crit. Care Med. 186, 866–876 (2012).
de la Zerda, A., Kratochvil, M. J., Suhar, N. A. & Heilshorn, S. C. Review: bioengineering strategies to probe T cell mechanobiology. Apl. Bioeng. 2, 021501 (2018).
Vedadghavami, A. et al. Manufacturing of hydrogel biomaterials with controlled mechanical properties for tissue engineering applications. Acta Biomater. 62, 42–63 (2017).
Acknowledgements
J.M.B. is a recipient of a National Institutes of Health (NIH) T32 (AG000266) postdoctoral fellowship award. This work was funded, in part, through the Huiying Memorial Foundation (D.A.W.), the Canadian Institutes of Health Research PJT169175 (D.A.W.), and the National Institutes of Health National Institute of Allergy and Infectious Diseases (NIAID) R01 AI151301 (W.F.L.), National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) R21 AR077288 (W.F.L.) and the National Institute of Aging (NIA) R21 G069067 (W.F.L).
Author information
Authors and Affiliations
Contributions
All authors contributed to the design and writing of the manuscript and the generation of the figures.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Immunology thanks M. Biro and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- ‘M1-like’ inflammatory macrophage
-
‘M1’ and ‘M2’ are classifications historically used to define macrophages activated in vitro as pro-inflammatory (when ‘classically’ activated with IFNγ and lipopolysaccharide (LPS)) or anti-inflammatory (when ‘alternatively’ activated with interleukin-4 (IL-4) or IL-10), respectively. However, in vivo macrophages are highly specialized, transcriptomically dynamic and extremely heterogeneous with regards to their phenotypes and functions, which are continuously shaped by their tissue microenvironment. Therefore, the M1 or M2 classification is too simplistic to explain the true nature of in vivo macrophages, although these terms are still often used to indicate whether the macrophages in question are more pro-inflammatory or anti-inflammatory.
- Immunological synapse
-
The site of discrete contact formed between an antigen-presenting cell (APC) and a T cell. Similar synapses have been described in other immune cells such as natural killer cells or cytotoxic T cells, where the contact is formed with a target cell. It is important in establishing adhesion with the partner cell and polarization of the signalling and cytotoxic machinery. This structure is heavily influenced by the cytoskeleton and transduces controlled secretory signals, thereby allowing the directed release of cytokines or lytic granules towards the APC or target cell.
- Annulus fibrosus
-
A ring of fibrous tissue, such as that surrounding an intervertebral disc or heart valve.
Rights and permissions
About this article

Cite this article
Du, H., Bartleson, J.M., Butenko, S. et al. Tuning immunity through tissue mechanotransduction. Nat Rev Immunol 23, 174–188 (2023). https://doi.org/10.1038/s41577-022-00761-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-022-00761-w
This article is cited by
-
Matrix stiffness affects tumor-associated macrophage functional polarization and its potential in tumor therapy
Journal of Translational Medicine (2024)
-
Evidence and therapeutic implications of biomechanically regulated immunosurveillance in cancer and other diseases
Nature Nanotechnology (2024)
-
Particle elasticity influences polymeric artificial antigen presenting cell effectiveness in vivo via CD8+ T cell activation, macrophage uptake, and the protein corona
Nano Research (2024)
-
Inflammasomes as regulators of mechano-immunity
EMBO Reports (2023)
-
Modelling Keloids Dynamics: A Brief Review and New Mathematical Perspectives
Bulletin of Mathematical Biology (2023)
