
Abstract
Numerous mitochondrial constituents and metabolic products can function as damage-associated molecular patterns (DAMPs) and promote inflammation when released into the cytosol or extracellular milieu. Several safeguards are normally in place to prevent mitochondria from eliciting detrimental inflammatory reactions, including the autophagic disposal of permeabilized mitochondria. However, when the homeostatic capacity of such systems is exceeded or when such systems are defective, inflammatory reactions elicited by mitochondria can become pathogenic and contribute to the aetiology of human disorders linked to autoreactivity. In addition, inefficient inflammatory pathways induced by mitochondrial DAMPs can be pathogenic as they enable the establishment or progression of infectious and neoplastic disorders. Here we discuss the molecular mechanisms through which mitochondria control inflammatory responses, the cellular pathways that are in place to control mitochondria-driven inflammation and the pathological consequences of dysregulated inflammatory reactions elicited by mitochondrial DAMPs.
Similar content being viewed by others
Introduction
Deregulated inflammatory responses are involved in numerous human disorders, encompassing not only infectious and autoimmune disorders but also neurological, cardiovascular, renal, hepatic and neoplastic conditions1,2,3,4. On the one hand, disproportionate, unwarranted or unresolving inflammation can act as a bona fide disease driver, as in the case of chronic inflammatory bowel disease1. On the other hand, uncontrolled inflammatory responses may aggravate the course of conditions that originate from non-inflammatory cues, such as myocardial infarction3. Moreover, inefficient inflammatory reactions facilitate the persistence of infectious pathogens5 and enable the emergence and progression of malignant lesions in the context of failing cancer immunosurveillance6. Of note, inflammatory reactions may affect the course of specific diseases in opposing manners, largely depending on the intensity and duration of inflammation. For example, whereas indolent, chronic inflammation has been associated with oncogenesis and accelerated tumour progression in various settings7, potent inflammatory responses culminating in the engagement of adaptive immunity underlie the beneficial effects of numerous cancer therapies, including conventional chemotherapeutics8, targeted anticancer agents9 and radiotherapy10. Moreover, recent findings indicate that numerous components of the molecular cascades underlying inflammation are key for normal embryonic and postembryonic development, at least in specific settings such as neurodevelopment11. These examples highlight the crucial requirement for regulated inflammatory responses in organismal development and homeostasis.
Inflammation is generally initiated by the activation of pattern recognition receptors (PRRs) that are expressed by both immune and non-immune cells12. Importantly, PRRs can be activated not only by viral and bacterial molecules associated with infection — so-called microorganism-associated molecular patterns or pathogen-associated molecular patterns — but also by endogenous molecules that are commonly referred to as damage-associated molecular patterns (DAMPs)12. In physiological conditions, DAMPs — which include nucleic acids, small metabolites such as ATP and proteins such as calreticulin — are generally unable to drive PRR signalling because they cannot gain physical access to PRR-containing subcellular compartments13. However, cellular stress and death can be accompanied by considerable alterations in the permeability of various cellular compartments, which enables PRR activation by DAMPs and the consequent initiation of inflammatory responses12. For example, ATP functions as a DAMP only upon release into the extracellular environment when it can bind to cognate receptors expressed on myeloid cells, such as the purinergic receptors P2RY2 and P2RX7 (refs.14,15).
On the basis of these considerations, it would seem likely that mitochondria have an important role in the control of inflammatory responses, for at least three reasons16. First, mitochondria are widely considered as the evolutionary remnants of ancestral Alphaproteobacteria (the ancestors of modern Gram-negative bacteria)17, and some mitochondrial components have considerable similarity with bacterial molecules, suggesting that they might function as PRR ligands. For example, in contrast to nuclear DNA (but similarly to bacterial genomes), the mitochondrial genome is circular and not associated with histones18. Second, mitochondria have two membranes — the inner mitochondrial membrane (IMM) and the outer mitochondrial membrane (OMM) — which together offer a dual layer of control segregating mitochondrial DAMPs (mtDAMPs) from their cognate PRRs18. Third, mitochondria have a major role in the control of apoptotic and necrotic forms of regulated cell death (RCD) (Box 1), which ultimately involves irreversible mitochondrial permeabilization (and hence loss of mitochondrial compartmentalization)19. Thus, mitochondria offer a unique platform for DAMP redistribution, PRR signalling and inflammation in the context of failing adaptation to cellular stress (which is linked to RCD initiation), the ultimate goal being to elicit innate and adaptive immune responses in support of organismal homeostasis (despite the irreversible loss of cellular fitness)20. Indeed, as we discuss herein, mitochondria are master regulators of inflammatory responses, not only as they contain several bona fide DAMPs but also as they provide a physical scaffold for the activation of some PRRs21. Moreover, various cellular responses elicited by RCD-associated mitochondrial outer membrane permeabilization (MOMP), including autophagy and caspase activation, directly affect the regulation of inflammatory processes22,23.
Here we discuss the molecular mechanisms through which mtDAMPs elicit inflammation (especially, but not exclusively, intracellular mechanisms), the signal transduction cascades through which mitochondria control inflammatory processes and the relevance of mitochondria-regulated inflammation in human disease. This is particularly relevant not only as multiple fields of biomedical research have begun to realize the impact of dysregulated inflammation in disease but also as mitochondria-targeting agents are now being used for the clinical management of specific neoplasms, raising considerable expectations for further development in the context of cancer immunotherapy24.

mtDAMP signalling pathways
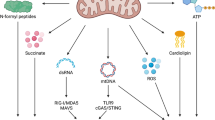
Findings from multiple, independent research teams have identified several signal transduction cascades through which mitochondrial dysfunction elicits inflammatory reactions, including (but not limited to) intracellular signalling through cyclic GMP–AMP synthase (cGAS) and stimulator of interferon response cGAMP interactor 1 (STING1), as activated by mitochondrial DNA (mtDNA), and through the inflammasome, as induced by mtDNA and reactive oxygen species (ROS) (Fig. 1).

Various mitochondrial components and products that are released as a consequence of mitochondrial dysfunction (and potentially cell death) drive inflammatory responses upon accumulation in the cytosol or the extracellular environment. Mitochondrial DNA (mtDNA), which can exit mitochondria via BCL-2-associated X, apoptosis regulator (BAX) and BCL-2 antagonist/killer 1 (BAK1) pores or via the permeability transition pore complex (PTPC), is a potent activator of cyclic GMP–AMP synthase (cGAS), resulting in stimulator of interferon response cGAMP interactor 1 (STING1) signalling and consequent synthesis of cytokines such as interferon-β1 (IFNβ1), IL-6 and tumour necrosis factor (TNF). Cytosolic mitochondrial RNA (mtRNA) has similar effects, although they depend on retinoic acid-inducible gene I protein (RIG-I), melanoma differentiation-associated protein 5 (MDA5) and mitochondrial antiviral signalling protein (MAVS). This pathway is promoted by the BAX–BAK1-dependent release of Era-like 12S mitochondrial rRNA chaperone 1 (ERAL1), which favours MAVS stabilization at the mitochondrial surface. Moreover, upon release from dysfunctional mitochondria, both mtDNA and reactive oxygen species (ROS) can drive IL-1β and IL-18 secretion as a consequence of inflammasome signalling. Electron transport chain (ETC) functions also seem to affect inflammasome activation independently of ROS as they preserve intracellular ATP availability through phosphocreatine (PCr). ATP can be released by dying cells through lysosomal secretion and pannexin 1 (PANX1) channels, mediating both chemotactic and immunostimulatory effects on antigen-presenting cells (APCs) by binding to the purinergic receptors P2RX7 and P2RY2. Upon BAX–BAK1 oligomerization during apoptosis, diablo IAP-binding mitochondrial protein (DIABLO; best known as SMAC) release not only favours caspase activation (not shown) but also rewires NF-κB signalling from canonical to non-canonical programmes. Along similar lines, mtDNA (be it naked or complexed with the mitochondrial transcription factor TFAM) and N-formyl peptides (other mitochondrial components), which accumulate in the extracellular milieu upon regulated cell death, cause neutrophil activation via Toll-like receptor 9 (TLR9) or advanced glycosylation end product-specific receptor (AGER) and formyl peptide receptor 1 (FPR1), respectively. Finally, extracellular cardiolipin (which is normally restricted to the inner mitochondrial membrane) can be presented by dendritic cells (DCs) on the MHC class I-like molecule CD1d, resulting in the activation of γδ T cells. DAMP, damage-associated molecular pattern; ER, endoplasmic reticulum; IKK, IκB kinase; IRF, interferon regulatory factor; NIK (official name MAP3K14), mitogen-activated protein kinase kinase kinase 14; TBK1, TANK-binding kinase 1; TCR, T cell receptor.
cGAS–STING1 signalling
cGAS is a nuclear and cytosolic protein that responds to cytosolic double-stranded DNA (dsDNA) molecules by catalysing the formation of cyclic GMP–AMP (cGAMP), a second messenger that initiates an inflammatory response via STING1 (ref.25) (Box 2). Although initial studies focused on exogenous26 and nuclear27 dsDNA species as key cGAS activators, it rapidly became clear that mtDNA gaining access to the cytosol as a consequence of MOMP (Box 1) or other forms of mitochondrial dysfunction can also promote cGAS signalling, although this is tightly inhibited by apoptotic caspases28,29,30 (see later). Subsequent studies showed that whereas mtDNA, naked dsDNA and dsDNA bound to proteins that introduce a specific curvature (such as TFAM, a mitochondrial transcription factor, and high mobility group box 1 (HMGB1), a nuclear non-histone DNA-binding protein) potently activate cGAS, histone-bound nuclear dsDNA (in other words, chromatin) is a poor cGAS activator and inhibits cGAS signalling driven by naked dsDNA31,32. At least in part, this reflects the strong physical interactions between cGAS and histones, which result in structural reconfigurations that conceal the cGAS DNA-binding site, prevent cGAS dimerization and favour cGAS inactivation by mitotic kinases33,34,35. Taken together, these observations delineate a system that prevents unwarranted cGAS activation in physiological settings (for example, during mitosis when chromatin becomes exposed to cytosolic cGAS, or during programmed cell death, which often involves widespread and irreversible MOMP but is accompanied by robust caspase activation)35,36, but preserves the ability to initiate inflammatory responses in situations of danger. This is particularly (but not exclusively) true for pathogen infection, because multiple intracellular pathogens, including Mycobacterium tuberculosis, type 1 herpes simplex virus (HSV-1), dengue virus, influenza virus, encephalomyocarditis virus and severe acute respiratory syndrome coronavirus 2 drive mitochondrial dysfunction and/or MOMP, culminating in mtDNA release30,37,38,39,40,41. Furthermore, the apoptotic caspases that would otherwise inhibit cGAS signalling after MOMP are often disabled by proteins encoded by intracellular pathogens as a strategy to extend the lifespan of host cells during infection42.
Release of mtDNA in the course of MOMP generally involves the proapoptotic pore-forming proteins BCL-2-associated X, apoptosis regulator (BAX) and BCL-2 antagonist/killer 1 (BAK1)43,44 (Box 1). Mechanistically, MOMP as initiated by BAX and BAK1 rapidly impairs mitochondrial respiration owing to the loss of soluble components of the electron transport chain, such as cytochrome c, hence compromising the ability of mitochondria to preserve metabolic homeostasis and ionic equilibrium across the IMM45. In this setting, BAX–BAK1 pores in the OMM enable the extrusion of the IMM into the cytosol (driven by increasing osmotic pressure in the mitochondrial matrix), culminating in IMM breakdown and mtDNA spillage43,44. Intriguingly, it seems that a large fraction of mtDNA exiting mitochondria does not diffuse freely in the cytosol but instead remains associated with the permeabilized organelles43 (which explains, at least in part, the ability of mitophagy to inhibit cGAS signalling driven by MOMP)46,47 (see later). Recent data indicate that the relative levels of BAX and BAK1 have a key role in determining the speed of mtDNA release in the course of MOMP, largely reflecting structural and kinetic aspects of pore assembly by either of these proteins in the absence of its counterpart, with BAK1 accelerating and BAX decelerating the response in the context of negligible effects on the release of proapoptotic factors normally confined within the intermembrane space, such as diablo IAP-binding mitochondrial protein (DIABLO; best known as SMAC)48.
Of note, although MOMP was initially conceived as an ‘all-or-nothing’ phenomenon that would necessarily result in cell death45, sublethal instances of MOMP that affect only a fraction of the mitochondrial pool and do not engage RCD — known as ‘minority MOMP’ — have been described in various physiological and pathological settings49,50. In the context of sublethal stress conditions, minority MOMP, which is actively prevented by mitochondrial fusion as a consequence of BCL-2 redistribution51, not only enables inflammatory reactions driven by mtDAMPs52,53 but also favours DNA damage upon activation of DNA fragmentation factor subunit-β (DFFB)49, which can further activate inflammatory pathways.
Intriguingly, various examples of BAX-independent and BAK1-independent mtDNA release have recently been described. For example, proteolytically activated BH3-interacting domain death agonist (BID) has been shown to function as a bona fide mitochondrial pore-forming protein (rather than as an activator of BAX and BAK1) in human cells responding to Shigella infection54. Along similar lines, mild mitochondrial stress that does not result in MOMP and RCD has been associated with mtDNA release via a voltage-dependent anion channel (VDAC)-dependent mechanism37,55. This is particularly intriguing as various isoforms of VDAC are involved in the regulation (but are not required for the execution) of mitochondrial permeability transition (MPT)-driven regulated necrosis56 (Box 1), suggesting that MPT may also drive mtDNA leakage. In line with this possibility, pharmacological inhibitors of the permeability transition pore complex, which is commonly viewed as the core mediator of MPT, have been shown to limit cytosolic mtDNA accumulation and expression of interferon-stimulated genes55,57. Moreover, mutant TAR DNA-binding protein (TARDBP) has been reported to accumulate at mitochondria and initiate an MPT-like response that involves VDAC oligomerization and culminates in cGAS activation in models of amyotrophic lateral sclerosis58. As MPT generally does not cause robust caspase activation (see later), it would be tempting to suggest that mtDNA release by MPT leads to greater cGAS activation than mtDNA release by MOMP. However, the permeability transition pore complex forms pores of 1.5–3-nm diameter59, whereas dsDNA requires pores with a diameter of 3 nm or greater to diffuse60. Moreover, MPT seems to proceed efficiently with only one to nine open permeability transition pore complexes per mitochondrion, which is less than the number of BAX–BAK1 oligomers believed to underlie MOMP (more than 20)48,61. That said, both MPT and MOMP can ultimately result in the complete breakdown of mitochondrial membranes to enable mtDNA release. Molecular studies elucidating the possibility that MPT may be superior to MOMP at enabling cGAS activation by cytosolic mtDNA are urgently awaited.
In summary, despite several unknowns, an abundant literature shows the ability of mtDNA to drive potent inflammatory responses upon engagement of cGAS and STING1, especially in conditions of limited apoptotic caspase activation (for example, upon MPT, or upon MOMP in the presence of viral caspase inhibitors)42 (Fig. 1).
Inflammasome signalling
In addition to being a potent cGAS agonist, cytosolic mtDNA can also drive the activation of inflammasomes, which are supramolecular platforms for the activation of caspase 1 (CASP1), CASP4 and CASP5 (as well as CASP11, the mouse homologue of human CASP4 and CASP5)62. Initial work on inflammasomes, specifically the inflammasome that contains NLR family pyrin domain-containing 3 (NLRP3) as a sensing component, revealed that various microorganism-associated molecular patterns elicit robust CASP1 activation and consequent proteolytic maturation of IL-1β and IL-18 (refs.63,64). Subsequent work identified oxidized mtDNA released into the cytosol upon mitochondrial dysfunction as a potent NLRP3 inflammasome activator65 and also delineated a feedforward loop through which inflammasome activation facilitates mtDNA release via a ROS-dependent mechanism that connects downstream NLRP3 activation to increased upstream MPT66,67. In line with the observation that a significant fraction of mtDNA accessing the cytosol remains associated with permeabilized mitochondria43, both NLRP3 and the inflammasome adaptor PYD and CARD domain containing (PYCARD; best known as ASC) relocalize to the mitochondria-associated endoplasmic reticulum upon MOMP, via a process that depends on mitochondrial ROS68. Moreover, optimal NLRP3 inflammasome signalling seems to involve a physical interaction between NLRP3 and thioredoxin-interacting protein (TXNIP), a nuclear protein that relocalizes to mitochondria during oxidative stress69,70, as well as cardiolipin, an IMM-restricted tetraacylated phospholipid71. Taken together, these observations delineate a close association between mitochondrial dysfunction and NLRP3 inflammasome activation.
Interestingly, although various ROS-dependent mechanisms have been invoked to sustain NLRP3 inflammasome signalling downstream of mitochondrial dysfunction72, ROS inhibitors seem to disrupt inflammasome priming (the synthesis of inflammasome components), but not activation (the acquisition of proteolytic activity)73. In line with this notion, recent data suggest that oxidative phosphorylation is involved in NLRP3 signalling driven by acute exposure to bacterial lipopolysaccharide (LPS) plus ATP74 through a ROS-independent mechanism linked to preserved intracellular ATP availability via phosphocreatine75, although ROS seem to be necessary for long-term inflammasome activation upon prolonged exposure to β-amyloid. Moreover, conventional inflammasome activators such as LPS plus ATP seem to (elicit and) require mtDNA neosynthesis for optimal NLRP3 inflammasome signalling76. Indeed, NLRP3 inflammasome activation by LPS can be impaired not only by mtDNA depletion66,77 but also by the deletion of Tfam76, which is required for mtDNA replication and maintenance78, the deletion of Cmpk2 (ref.76), which encodes a rate-limiting enzyme that supplies deoxyribonucleotides for mtDNA synthesis79, or the inhibition of mtDNA synthesis with the antidiabetic agent metformin80. That said, experimental strategies that deplete mtDNA also impair oxidative phosphorylation as they limit the abundance of specific subunits of the electron transport chain, suggesting that — at least in some settings — inhibition of NLRP3 signalling downstream of mtDNA depletion may result from, or at least be aggravated by, intracellular ATP shortage. These observations suggest that the relative importance of cGAS signalling versus NLRP3 signalling induced by cytosolic mtDNA may be largely influenced by cellular bioenergetics.
Of note, NLRP3 inflammasome activation by LPS plus ATP was initially proposed to occur independently of BAX and BAK1, the key MPT regulator peptidylprolyl isomerase F (PPIF), mitochondrial antiviral signalling protein (MAVS; a signal transducer in the molecular cascade detecting foreign, altered or ectopic RNA) and the mitophagy protein parkin RBR E3 ubiquitin protein ligase (PRKN)81. Rather, this was shown to involve CASP8 and components of the necroptotic machinery, notably receptor-interacting serine/threonine kinase 3 (RIPK3)81. More recently, BAX activation and BAK1 activation have been shown to promote CASP8-dependent NLRP3 inflammasome activation as a consequence of inhibitor of apoptosis protein (IAP) degradation, and to elicit an NLRP3-independent pathway culminating in IL-1β maturation82,83, most likely upon activation of a supramolecular complex commonly referred to as the ripoptosome83. The reasons underlying these apparently discrepant findings as to the roles of BAX, BAK1 and MOMP in conventional inflammasome activation remain to be fully elucidated. At least potentially, BAX-independent and BAK1-independent inflammasome activation may involve some degree of gasdermin cleavage (which can be catalysed by inflammasome-activated CASP1 as well as by MOMP-driven CASP3 activation)84, as both gasdermin D (GSDMD) and GSDME have been shown to permeabilize mitochondria and favour mtDNA release coupled to ROS production85,86. As an alternative, the MPT has been implicated in at least some instances of NLRP3 inflammasome signalling in a recent preprint (not peer reviewed)87.
mtDNA also activates inflammasomes that use absent in melanoma 2 (AIM2) as a sensing component88, which overall resemble their NLRP3-containing counterparts in terms of their capacity to elicit CASP1-dependent IL-1β and IL-18 maturation in response to cytosolic dsDNA of various origins89, including both foreign dsDNA90 and endogenous dsDNA91. Interestingly, whereas NLRP3 inflammasomes seem to preferentially respond to oxidized DNA65, their AIM2-containing counterparts have been suggested to preferentially recognize non-oxidized DNA76. That said, both Francisella tularensis infection and non-alcoholic fatty liver disease drive mitochondrial damage coupled to AIM2 inflammasome activation upon mitochondrial ROS generation and cytosolic accumulation of oxidized mtDNA92,93. Along similar lines, Pseudomonas aeruginosa infection triggers the activation of NLR family CARD domain-containing 4 (NLRC4) inflammasomes via a process that involves ROS generation and oxidized mtDNA release by mitochondria94. Taken together, these observations highlight the importance of oxidation for robust inflammasome signalling elicited by mtDNA.
In summary, mtDNA and mitochondrial ROS function as major DAMPs for inflammasome activation at the core of a complex pathway that intersects at multiple nodes with the molecular machinery for regulation of RCD (Fig. 1).
Other inflammatory pathways
mtDNA and other mitochondrial components can also elicit inflammatory reactions via various PRRs distinct from cGAS and inflammasomes95.
Naked as well as protein-bound mtDNA molecules are potent activators of Toll-like receptor 9 (TLR9) and advanced glycosylation end product-specific receptor (AGER; also known as RAGE)96,97, two PRRs that are abundantly expressed in the endosomal compartment of myeloid cells (notably neutrophils)13. Naked mtDNA largely functions as a TLR9 agonist96, reflecting the limited methylation of CpG islands found in the mitochondrial genome98 and hence its considerable similarity to bacterial DNA (the prototypical TLR9 activator)99. Conversely, mtDNA complexed with TFAM or HMGB1, which relocalizes from the nucleus to the cytosol in the course of various stress responses (including inflammasome activation)100, has immunostimulatory effects upon binding to TLR9 or AGER97,101. Recombinant TFAM has also been shown to drive cytokine secretion by cultured human monocytes and primary microglial cells102, but the underlying mechanisms and pathophysiological relevance of this process remain to be clarified.
Importantly, activation of these inflammatory pathways most often requires mtDNA (be it naked or protein bound) to be released into the extracellular microenvironment as a consequence of RCD, where mtDNA functions as an autocrine, paracrine or endocrine immunostimulatory cue12. However, cells with increased vesicular trafficking (such as plasmacytoid dendritic cells) have been shown to engage endosomal TLR9 signalling upon mild mitochondrial dysfunction as a consequence of cytosolic mtDNA accumulation coupled to autophagic uptake and endosomal shuttling101,103,104. Along similar lines, signalling through glutamate receptors as a consequence of metabolic changes culminating in extracellular glutamate accumulation has recently been shown to promote the release of mtDNA-loaded mitochondria-derived vesicles (MDVs) by breast cancer cells independently of RCD. This is a PTEN-induced putative kinase 1 (PINK1)-dependent process that results in autocrine and/or paracrine TLR9 activation105. At odds with that study105, PRKN (a PINK1 interactor) has been proposed to actively divert mtDAMPs from inclusion in MDVs that are secreted as part of mitochondrial quality control106,107. The reasons underlying this apparent discrepancy remain to be fully elucidated. Of note, mtDAMP-containing MDVs seem to be actively produced by monocytes responding to inflammatory cues during sepsis, potentially resulting in limited neutrophil chemotaxis108. Moreover, although unrelated to inflammatory responses, the horizontal transfer of mtDNA-containing MDVs from live cancer-associated fibroblasts to tumour cells has been observed in various models of hormone-resistant breast cancer109. These studies exemplify settings in which mtDNA can be released (and potentially function as a DAMP) by cells that are not succumbing to RCD.
Another inflammatory pathway that can be stimulated by mitochondria involves the PRRs that respond to foreign, altered or ectopic RNA — known as RIG-I-like receptors (RLRs)110. In this setting, mitochondria can not only function as a source of RLR-activating RNA species as a consequence of mitochondrial dysfunction and MOMP53,111 but can also provide a cellular scaffold for optimal RLR signalling by hosting the key signal transducer MAVS on the OMM112. Specifically, mitochondria have been shown to release mitochondrial RNA (mtRNA) species that activate the RLR melanoma differentiation-associated protein 5 (MDA5; also known as IFIH1) upon depletion of polyribonucleotide nucleotidyltransferase 1 (PNPT1; a key component of the supramolecular complex responsible for mtRNA degradation), via a mechanism that involves BAX–BAK1-driven MOMP111. Moreover, mtDNA breaks have been reported to drive the BAX–BAK1-dependent release of mtRNA into the cytosol, which activates retinoic acid-inducible gene I (RIG-I; also known as DDX58) but not MDA5 (ref.53). Why some mtRNA species preferentially activate MDA5 versus RIG-I remains to be elucidated. At least potentially, this may reflect cell type-specific differences in the expression of specific PRRs or signal transducers thereof, as suggested by recent work on PNPT1 silencing in human pancreatic β-cells113. Finally, release of the mitochondrial matrix protein Era-like 12S mitochondrial rRNA chaperone 1 (ERAL1) via BAX–BAK1 pores formed in response to viral infection has recently been shown to sustain antiviral responses by promoting MAVS polymerization at the OMM, which is crucial for optimal inflammatory responses driven by RLR-activating viral RNA114. Intriguingly, MAVS resembles many other PRRs in their ability to control both cellular fate and inflammatory responses115. Thus, MAVS can promote RCD as part of its cell-intrinsic homeostatic function, but this is also connected to immune signalling via TANK-binding kinase 1 (TBK1), IκB kinase (IKK) and NLRP3 inflammasome activation116. These observations are in line with an abundant literature demonstrating that the molecular regulation of RCD may have evolved to control the kinetics and immunological manifestations of the process rather than the occurrence of cell death itself23,117,118.
At least four other mitochondrial components, namely SMAC, N-formyl peptides, cardiolipin and cytochrome c, have been shown to promote inflammatory responses. SMAC release downstream of MOMP is transduced in both proapoptotic and pro-inflammatory signalling pathways via members of the IAP family119. In addition to unleashing caspase activity, inhibition of IAPs by cytosolic SMAC (as well as by pharmacological agents commonly known as SMAC mimetics) shifts NF-κB signalling from the canonical to the non-canonical pathways upon stabilization of mitogen-activated protein kinase kinase kinase 14 (MAP3K14; best known as NIK)120,121, a process that is orchestrated by BAX–BAK1 oligomers122. Accordingly, loss of the IAP-encoding genes Birc2 and Birc3 in adult mice causes aberrant cell death and inflammation, which are fully rescued by Casp8 deletion plus pharmacological inhibition of NIK123. Moreover, SMAC mimetics have been shown to mediate anticancer effects that at least in some models involve the activation of antitumour immune responses upon macrophage repolarization124. Intriguingly, SMAC mimetics can also have direct immunomodulatory effects on T cells, for example by reprogramming CD4+ T cell differentiation from a T helper 17 (TH17)-type phenotype to a TH2-type phenotype125. Thus, whereas cytosolic mtDNA or mtRNA generally elicits a multipronged TBK1-dependent inflammatory response, SMAC-driven inflammation seems to involve mainly altered NF-κB signalling.
N-formyl peptides and cardiolipin normally reside in the mitochondrial matrix and IMM, respectively, but the structural disruption that accompanies late-stage RCD generates cellular fragments containing mitochondrial remnants that — unless taken up by professional phagocytes126 — continue to degrade in the extracellular microenvironment. Extracellular N-formyl peptides function as potent neutrophil activators upon binding to formyl peptide receptor 1 (ref.96). Extracellular cardiolipin not only promotes overexpression of the non-polymorphic MHC class I-like molecule CD1d on antigen-presenting cells (APCs)127 but also can be found associated with CD1d on the APC surface, resulting in activation of a cardiolipin-specific population of T cells expressing the unconventional γδ T cell receptor128. Whether cardiolipin is loaded on CD1d directly at the APC surface or intracellularly remains to be clarified. Extracellular cytochrome c has been proposed to have TLR4-dependent immunostimulatory effects in cultured cell models of human microglia129, but whether this pathway is activated as a response to RCD in vivo has not been specifically addressed. Along similar lines, the intra-articular injection of recombinant cytochrome c has been suggested to promote arthritis in mice via a mechanism that involves neutrophils and monocytes130, but whether cytochrome c accumulating in the synovial microenvironment downstream of RCD in vivo aetiologically contributes to arthritis remains to be formally demonstrated.
Finally, although ATP and haem are not mitochondrial components in the strictest sense, they are produced by mitochondria and mediate robust immunomodulatory effects upon release into the extracellular microenvironment131,132. Specifically, ATP released by cancer cells undergoing immunogenic cell death12, via a mechanism that involves lysosomal secretion and pannexin 1 channels133, has been shown to recruit APC precursors to the tumour microenvironment134 through a P2RY2-dependent mechanism14, and to stimulate them through P2RX7, culminating in inflammasome activation and IL-1β synthesis in support of immunosurveillance15. Extracellular haem, which functions as a mixed TLR4 and AGER agonist135,136, has been associated with multiple immunomodulatory effects, including endothelial cell and microglial activation135,137, as well as IL-1β release by macrophages downstream of NLRP3-dependent CASP1 activation plus NLRP3-independent CASP4 and CASP5 activation coupled to RCD132. The mechanistic involvement of TLR4 or AGER in this latter pathway has not been clarified.
In summary, numerous mitochondrial components and products can promote inflammation through various mechanisms (Fig. 1). Whether such a preferential position in the control of inflammatory reactions reflects the evolutionary origin of mitochondria remains to be formally established. Irrespective of this possibility, eukaryotic cells have evolved a wide range of mechanisms to control inflammatory responses elicited by mitochondria, as discussed next.
Regulation of mtDAMP signalling
As MOMP participates in multiple physiological processes, including cellular differentiation, embryonic and postembryonic development, and the maintenance of adult tissue homeostasis36,50 (Box 3), numerous safeguards have evolved to prevent unwarranted mitochondria-driven inflammation (Fig. 2). Here we describe the mechanisms through which apoptosis and autophagy suppress inflammatory responses potentially driven by mitochondria in physiological settings.

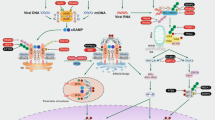
Intrinsic apoptosis proceeds with the formation of BCL-2-associated X, apoptosis regulator (BAX) and BCL-2 antagonist/killer 1 (BAK1) pores at the outer mitochondrial membrane, resulting in the cytosolic accumulation of cytochrome c and consequent activation of apoptotic caspases. Caspase 9 (CASP9)-driven CASP3 activation inhibits inflammatory responses elicited by dysfunctional mitochondria by catalysing the cleavage of cyclic GMP–AMP synthase (cGAS), mitochondrial antiviral signalling protein (MAVS) and interferon regulatory factor 3 (IRF3). A similar inhibitory effect results from the disposal of dysfunctional mitochondria via PTEN-induced putative kinase 1 (PINK1)- and parkin (PRKN)-dependent mitophagy. This is promoted (at least initially) by the capacity of TANK-binding kinase 1 (TBK1) — which is activated by phosphorylation (P) following cGAS-driven stimulator of interferon response cGAMP interactor 1 (STING1) signalling and retinoic acid-inducible gene I (RIG-I)-driven MAVS signalling downstream of the cytosolic accumulation of mitochondrial DNA (mtDNA) or mitochondrial RNA (mtRNA) — to phosphorylate optineurin (OPTN) and hence improve the ability of OPTN to recruit sequestosome 1 (SQSTM1; best known as p62) to ubiquitylated proteins at the outer mitochondrial membrane such as mitofusin 2 (MFN2). Engagement of p62 culminates in the recruitment of forming autophagosomes via lipidated microtubule-associated protein 1 light chain 3 beta (MAP1LC3B; best known as LC3-II in its lipidated form). Of note, PRKN also mediates the ubiquitylation (Ub)-dependent inactivation of BAK1. Moreover, PRKN-dependent mitophagy seems to prevent mitochondrial damage-associated molecular patterns (mtDAMPs) from being incorporated in the mitochondria-derived vesicles (MDVs) that are normally released as part of mitochondrial quality control in a PINK1-dependent manner. The underlying mechanisms, however, remain to be fully elucidated. General autophagy can likewise suppress inflammatory responses driven by mitochondrial dysfunction, at least in part reflecting its ability to degrade NLR family pyrin domain-containing 3 (NLRP3) inflammasomes. ATG, autophagy-related protein; IFNβ1, interferon-β1; ROS, reactive oxygen species; TNF, tumour necrosis factor.
Apoptosis
In the context of apoptosis, widespread MOMP results in the assembly of a cytosolic supramolecular CASP9-containing platform that culminates in the activation of executioner caspases such as CASP3, CASP6 and CASP719 (Box 1). Although executioner caspases have long been considered as the causative factors of apoptotic RCD, accumulating evidence suggests that they might instead have a major role in controlling its kinetics and immunological manifestations23,117,118. In line with this notion, pharmacological or genetic inhibition of CASP9 or executioner caspases (most often CASP3) is required for MOMP-inducing agents, including BH3 mimetics and radiotherapy, to drive robust synthesis of interferon-stimulated genes but does not quantitatively affect the cytotoxic response to these agents28,29,122,138,139. At least in part, this reflects the ability of executioner caspases to cleave and hence inactivate not only cGAS but also interferon regulatory factor 3 (IRF3), which signals downstream of STING1, and MAVS140. Moreover, executioner caspases promote the exposure of phosphatidylserine on the surface of dying cells141, which rapidly recruits phagocytes for the immunologically silent uptake and degradation of dying cells142. Finally, even in the absence of a phagocytic system, when executioner caspases are inactive, the transition between MOMP (when cells retain plasma membrane integrity and at least some metabolic functions) and cell death (which is characterized by plasma membrane permeabilization and complete metabolic shutdown) is delayed, potentially allowing an extended temporal window for the synthesis and secretion of pro-inflammatory factors downstream of MOMP143.
Autophagy
Sublethal (or prelethal) MOMP activates robust mitophagic responses that dispose of permeabilized or otherwise dysfunctional mitochondria via lysosomes144,145, which limits the availability of mitochondrial components, including mtDNA and ROS, for PRR signalling or release into the extracellular microenvironment upon RCD146. In line with this, pharmacological or genetic strategies for autophagy inhibition or impaired lysosomal degradation have been consistently associated with increased signalling through cGAS46, the inflammasome66 and TLR9 (ref.103) as a consequence of increased availability of mtDAMPs in the cytosol. Similar results have been obtained with experimental approaches for the selective inhibition of mitophagy (rather than autophagy in general), including deletion of the key mitophagy genes Pink1 and Prkn147,148, administration of palmitic acid149 and stimulation with tumour necrosis factor47. Furthermore, active inflammasome signalling has been shown to engage an NF-κB-dependent response that promotes the PRKN-dependent mitophagic clearance of permeabilized mitochondria77 as well as the autophagic clearance of inflammasomes themselves150, most likely as part of an adaptative pathway aimed at the restoration of cellular and inflammatory homeostasis. A similar mechanism is elicited (at least initially) by cGAS signalling, which reportedly promotes mitophagy upon activation of TBK1 or IKKα downstream of STING1 (refs.151,152). PRKN has also been shown to promote the recovery of cellular homeostasis in the context of sublethal MOMP by catalysing the ubiquitylation-dependent inactivation of BAK1 (ref.144). Conversely, mitophagy inactivation has been shown to accompany full-blown inflammatory responses driven by unrecoverable mitochondrial dysfunction, at least in part reflecting the CASP1-dependent cleavage of PRKN elicited by robust inflammasome activity153. Of note, PRKN-dependent mitophagy may also prevent the packaging of mtDAMPs into MDVs that are secreted as part of mitochondrial quality control107, which would otherwise drive inflammatory reactions in neighbouring cells106. Taken together, these observations delineate a refined system whereby autophagy (in particular, mitophagy) prevents the initiation and favours the inhibition of inflammatory responses driven by mitochondria, which require mitophagy inhibition (potentially involving CASP1-dependent PRKN degradation plus a feedforward loop linking robust inflammasome activation back to aggravated mitochondrial dysfunction)66,153 to proceed unrestricted (Fig. 3). By contrast, general autophagy is required for optimal ATP secretion (and consequent immunostimulation) in the course of immunogenic cell death, largely reflecting the ability of proficient autophagic responses to preserve intracellular ATP levels (rather than a direct effect of autophagy on mitochondria)154.

a | When only a small number of mitochondria are permeabilized, limited signalling via cyclic GMP–AMP synthase (cGAS) and the NLR family pyrin domain-containing 3 (NLRP3) inflammasome promotes mitophagy associated with the recruitment of parkin (PRKN) to dysfunctional mitochondria, TANK-binding kinase 1 (TBK1)-dependent optineurin (OPTN) phosphorylation (P) and consequent engulfment of mitochondria by forming autophagosomes. This engages a feedback mechanism that enables the restoration of cellular homeostasis in the absence of robust inflammatory responses. b | Conversely, when the functions and integrity of a large number of mitochondria are compromised, robust cGAS and NLRP3 inflammasome signalling is accompanied by mitophagy inhibition — as a consequence of caspase 1 (CASP1)-dependent cleavage of PRKN, despite OPTN phosphorylation — and increased NLRP3-dependent mitochondrial dysfunction, resulting in a feedforward loop that maximizes inflammation in the context of lost cellular homeostasis. Taken together, these mechanisms identify a rheostat that determines a threshold for recovered cellular homeostasis in the context of suppressed inflammation versus compromised cellular homeostasis in the context of acute inflammatory responses. MOMP, mitochondrial outer membrane permeabilization; mtDNA, mitochondrial DNA; ROS, reactive oxygen species; STING1, stimulator of interferon response cGAMP interactor 1.
Although these are not the only mechanisms through which eukaryotic cells fine-tune inflammatory reactions elicited by mitochondria (for example, cGAS and inflammasomes seem to inhibit each other, at least in some settings)155,156, apoptosis and autophagy exemplify molecular systems that have enabled the preservation of otherwise potentially detrimental inflammatory cues throughout the co-evolution of mitochondria and their host cells.
mtDAMP signalling in disease
Dysregulated mtDAMP signalling can be pathogenic and actively contribute to the aetiology of human disease in two opposing ways (Supplementary Table 1): inflammatory reactions driven by mtDAMPs may become disproportionate, thus fostering disorders with an (obvious or less obvious) inflammatory component; or such inflammatory reactions may be highly inefficient, ultimately enabling the emergence or persistence of infectious or neoplastic conditions.
Overactive mtDAMP signalling
Human disorders with an overtly inflammatory component that is mechanistically promoted by dysfunctional mitochondria include systemic lupus erythematosus (SLE), Crohn’s disease and multiple pulmonary and renal conditions1,4. Patients with SLE have increased circulating amounts of oxidized mtDNA as a consequence of platelet degranulation157 and impaired mitophagic responses to mitochondrial dysfunction in neutrophils (as well as autoantibodies to oxidized mtDNA), which culminate in pathogenic type I interferon responses158. Moreover, SLE is accompanied by defects in erythroid maturation that prevent the autophagic removal of mitochondria, which is a feature of normal mammalian erythropoiesis159. Therefore, mitochondria-containing erythrocytes are no longer recycled in an immunologically silent manner by the reticuloendothelial system, but instead promote potent cGAS activation and consequent type I interferon release159. In line with these findings, pharmacological inhibition of VDAC-dependent mtDNA release ameliorates lupus-like inflammation and symptoms in mice55. Finally, oxidized mtDNA is abundant within neutrophil extracellular traps, which also contribute to the aetiology of SLE via type I interferon production160.
Mutations in the autophagy-related 16-like 1 (ATG16L1) gene, which encodes a key component of the autophagy machinery22, are associated with an increased risk of Crohn’s disease161, correlating with the role of ATG16L1 in restricting accumulation of dysfunctional mitochondria and consequent inflammation in Paneth cells162. Paneth cells are functionally impaired in the ileal tissue of patients with Crohn’s disease163, a phenotype that can be recapitulated in mice by impairing mitochondrial homeostasis by deletion of the gene encoding the mitochondrial chaperone heat shock protein 1 (Hspd1)163 or of the gene encoding the mitophagy mediator prohibitin 1 (Phb1)164.
Lung biopsy samples from patients with interstitial lung disease or silicosis have increased STING1 levels, phosphorylation of its signal transducers TBK1 and IRF3, and/or higher than normal amounts of CXC-chemokine ligand 10 (CXCL10), which is elicited by type I interferon signalling165. Consistently, the sputum of patients with silicosis contains increased amounts of CXCL10 and dsDNA, which in mice exposed to silica originates from mitochondrial dysfunction165. Along similar lines, chronic obstructive pulmonary disease in humans has been associated with pulmonary mitochondrial dysfunction coupled to local ROS overgeneration166 and increased circulating levels of mtDNA and inflammatory cytokines167.
Evidence of STING1 hyperactivation has also been detected in the renal tubules of patients with acute kidney injury168. Moreover, CGAS and STING1 upregulation correlates with increased levels of inflammatory cytokines and fibrosis in patients with chronic kidney disease, who generally have low levels of TFAM expression (required for mtDNA replication and maintenance) in the kidney169. Optineurin, a component of mitophagy signalling, is often downregulated in biopsy samples from individuals with diabetic kidney disease170. Consistent with a prominent role for mitochondrial dysfunction in kidney disease, abrogation of mitophagy by Pink1 and Prkn co-deletion aggravates ischaemic acute kidney injury in mice171. Moreover, deletion of Tfam in mouse kidney tubule cells favours aberrant mtDNA packaging, cytosolic mtDNA accumulation and ultimately pathogenic cGAS-dependent inflammation169.
In addition, it is now clear that various conditions originating from non-inflammatory insults, including neurodegenerative diseases as well as hepatic and cardiovascular diseases, ultimately involve inflammatory processes that in some settings can result from mitochondrial dysfunction2,3. For example, missense mutations in PRKN and PINK1 are associated with familial Parkinson disease172, correlating with increased circulating levels of mtDNA and inflammatory cytokines147,173. Consistent with this, old Prkn–/– mice develop Parkinson disease-like symptoms together with structural and functional abnormalities of mitochondria in the brain174. Along similar lines, Alzheimer disease is accompanied by inflammatory responses driven by microglial cells that engulf mtDNA released by dying neurons175. Moreover, post-mortem brain samples from patients with Alzheimer disease show the accumulation of damaged mitochondria and signs of mitophagy inhibition, and experimental activation of mitophagy limits disease progression in a mouse model of Alzheimer disease176. Of note, defective mitophagy in Alzheimer disease may originate from alterations in the lipid profile of microglial cells (notably cholesterol accumulation)177, perhaps explaining, at least in part, the strong link between apolipoprotein E mutations and the incidence of Alzheimer disease in humans178.
Additional disorders in which mitochondrial dysfunction upstream of inflammation may have an aetiological role include cardiac maladaptation upon myocardial infarction179, septic shock180, rheumatoid arthritis47,130, sickle cell disease137, trauma96, intracerebral haemorrhage135, non-alcoholic fatty liver disease and non-alcoholic steatohepatitis93, liver failure181 and obesity182. Discussing each of these conditions in detail is beyond the scope of this Review, but these observations suggest that various additional disorders with an inflammatory component may be linked to mitochondrial dysfunction.
Inefficient mtDAMP signalling
The deregulation of inflammatory reactions elicited by mitochondrial dysfunction has broad, context-dependent effects on the pathogenesis of viral infections and cancer. Inhibition of MOMP or inflammatory responses driven by MOMP52 — reflecting, for example, the expression of viral proteins that inhibit MOMP, mtDNA, cGAS, STING1 or TBK1 (refs.41,183,184,185) or the hyperactivation of autophagy in cancer cells22 — favours viral persistence and tumour progression by impairing immunosurveillance.
For example, co-deletion of BAX and BAK1 from human cervical cancer HeLa cells not only promotes the growth of the intracellular pathogen Chlamydia trachomatis but also suppresses the ability of infected cells to secrete pro-inflammatory cytokines that would engage antibacterial immune responses upon sublethal MOMP52. HSV-1 encodes a conserved nuclease that actively depletes mtDNA during infection184, highlighting a direct mechanism by which this human pathogen avoids immune responses elicited by mtDAMPs. A similar function is mediated by non-structural protein 1 (NS1) of influenza virus41. Specifically, NS1 sequesters mtDNA molecules that access the cytosol in a MAVS-dependent manner during infection (driven by the influenza virus protein M2), thus preventing them from activating cGAS41. Accordingly, both Cgas–/– mice and Sting1–/– mice (but not Mavs–/– mice) exhibit increased viral titres upon influenza virus infection compared with their wild-type littermates, correlating with reduced pulmonary levels of type I interferon41.
Along similar lines, the HSV-1 virulence factor ICP27 inhibits STING1 and TBK1 (ref.183), the latter of which is required for the control of infection by mice, largely reflecting its ability to elicit NF-κB (rather than IRF3) responses186. Moreover, hepatitis B virus X protein promotes the ubiquitylation-dependent degradation of cGAS in infected cells185, and defects in the cGAS–STING1 pathway have been linked to increased sensitivity to hepatitis B virus infection in both human and mouse hepatocytes187. In these cases, however, the relative contribution of mtDAMP signalling to inflammatory reactions that would promote pathogen control has not been directly quantified.
Likewise, multiple cancer cell types harness autophagy46 and MOMP-driven CASP3 activation138,139 to avoid antitumour immune responses elicited by radiotherapy via mtDNA-dependent cGAS signalling. Conversely, autophagic defects that result in compromised ATP secretion in the context of immunogenic cell death not only support the resistance of cancer cells to various chemotherapeutic agents that engage antitumour immunity, such as anthracyclines and oxaliplatin15,154, correlating with inefficient recruitment and activation of myeloid cells to the tumour microenvironment134, but also promote malignant transformation in numerous immunocompetent mouse models of early oncogenesis188, a process that is under strict control by immunosurveillance189.
That said, the pathogenesis of some viral and bacterial infections involves late-stage uncontrolled inflammation, which at least in some cases originates from mitochondrial dysfunction37,190,191,192. Similarly, chronic low-grade inflammation generally contributes to oncogenesis and tumour progression7, and at least in some scenarios this is initiated by mitochondrial dysfunction193,194,195,196. Finally, SMAC release upon minority MOMP driven by Helicobacter pylori seems to have a role in both the short-term and the long-term detrimental effects of the infection as it drives pathogenic inflammation as well as DNA damage in the absence of overt RCD, ultimately promoting malignant transformation197.
Therapeutic prospects
Mitochondria have long been considered master regulators of (at least some forms of) RCD45,56, but accumulating evidence from various experimental settings now shows that the disruption of mitochondrial functions and structural integrity that accompanies RCD is also closely associated with an inflammatory response to preserve organismal homeostasis20. In keeping with this, the deregulation of inflammatory responses elicited by mitochondrial components or products has been shown to contribute to numerous human disorders, ranging from diseases that are driven by excessive inflammation to diseases that are enabled by inefficient inflammatory reactions95.
Importantly, most (if not all) of these conditions are managed clinically with therapeutic interventions that target the effector phase of inflammation, such as cytokine-neutralizing agents198, or target PRRs and their signal transducers, such as STING1 agonists199. By contrast, little attention has been given to the possibility of modulating inflammation through mitochondria-targeted agents, which may reflect the novelty of research in this area as well as a relatively small number of pharmacological interventions to target mitochondrial functions, particularly MOMP and MPT, in patients.
As it stands, only one drug that directly targets the molecular machinery of MOMP is licensed for use in humans, the selective BCL-2 inhibitor venetoclax (which is currently used for the treatment of some haematological malignancies and is under clinical evaluation for other neoplasms, including some solid tumours)24. The development of less selective inhibitors of BCL-2-like proteins200, such as navitoclax and ABT-737, has been troublesome (with the latter being discontinued) as they both mediate on-target, dose-limiting thrombocytopenia201. Although venetoclax and navitoclax would both be expected to promote inflammatory reactions that may support anticancer immunity by blocking the MOMP-inhibiting functions of BCL-2 (ref.202), they have been developed and are currently used or being tested as cytotoxic drugs against tumours that are ‘addicted’ to BCL-2-like proteins for survival9. Similar considerations apply to the use of pharmacological BAX activators to initiate MOMP, which are also in preclinical development to overcome apoptosis resistance in cancer203,204.
BAX inhibitors are being developed as cytoprotective agents for cardiovascular disorders, including chemotherapy-driven cardiotoxicity205,206. Although these agents would be expected to limit MOMP and consequently reduce inflammatory reactions driven by mitochondria, recent findings raise the intriguing possibility that (at least in BAK1-competent cancer cells) pharmacological BAX inhibitors may accelerate mtDNA release driven by MOMP (in the absence of major effects on the kinetics of caspase activation), potentially offering a window for inducing cGAS signalling before caspase-dependent cGAS cleavage and inactivation48,207. This hypothesis remains to be experimentally tested.
Cyclosporine A, a PPIF-targeting agent with prominent MPT-inhibitory activity, is also approved for use in humans56. Intriguingly, cyclosporine A is commonly used as an immunosuppressive drug for the management of autoimmune disorders and the prophylaxis of transplant rejection, largely on the basis of its ability to bind the PPIF-like cytosolic protein PPIA, ultimately resulting in lymphocyte inhibition through blocking calcineurin208. At least potentially, however, part of the immunosuppressive effects of cyclosporine A could result from MPT inhibition and the consequent suppression of inflammatory reactions driven by permeabilized mitochondria, a possibility that has not yet been formally addressed.
In conclusion, although mitochondria are clearly master regulators of inflammation, additional research is needed to address outstanding questions and surmount existing obstacles (Box 4), with the ultimate objective of targeting mitochondrial functions as a means to control inflammatory reactions in patients. As an additional layer of complexity, specific pharmacological modulators of autophagy for clinical use remain elusive209, and the development of emricasan, a pan-caspase inhibitor introduced into clinical testing for the management of cirrhosis, seems to have stalled210. Despite these and other caveats, we surmise that an improved understanding of the molecular mechanisms linking mitochondrial dysfunction to intracellular and extracellular DAMP signalling will ultimately unlock the development of mitochondria-targeting drugs for the control of inflammation.
References
Roda, G. et al. Crohn’s disease. Nat. Rev. Dis. Prim. 6, 22 (2020).
Tansey, M. G. et al. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-022-00684-6 (2022).
Stark, K. & Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 18, 666–682 (2021).
Basso, P. J., Andrade-Oliveira, V. & Camara, N. O. S. Targeting immune cell metabolism in kidney diseases. Nat. Rev. Nephrol. 17, 465–480 (2021).
Marchi, S., Morroni, G., Pinton, P. & Galluzzi, L. Control of host mitochondria by bacterial pathogens. Trends Microbiol. 30, 452–465 (2022).
Vesely, M. D., Kershaw, M. H., Schreiber, R. D. & Smyth, M. J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 29, 235–271 (2011).
Mantovani, A., Ponzetta, A., Inforzato, A. & Jaillon, S. Innate immunity, inflammation and tumour progression: double-edged swords. J. Intern. Med. 285, 524–532 (2019).
Galluzzi, L., Humeau, J., Buqué, A., Zitvogel, L. & Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 17, 725–741 (2020).
Petroni, G., Buque, A., Coussens, L. M. & Galluzzi, L. Targeting oncogene and non-oncogene addiction to inflame the tumour microenvironment. Nat. Rev. Drug Discov. 21, 440–462 (2022).
Rodriguez-Ruiz, M. E., Vitale, I., Harrington, K. J., Melero, I. & Galluzzi, L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat. Immunol. 21, 120–134 (2020).
Zengeler, K. E. & Lukens, J. R. Innate immunity at the crossroads of healthy brain maturation and neurodevelopmental disorders. Nat. Rev. Immunol. 21, 454–468 (2021).
Kroemer, G., Galassi, C., Zitvogel, L. & Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 23, 487–500 (2022).
Vanpouille-Box, C., Hoffmann, J. A. & Galluzzi, L. Pharmacological modulation of nucleic acid sensors - therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 18, 845–867 (2019).
Elliott, M. R. et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286 (2009).
Ghiringhelli, F. et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 15, 1170–1178 (2009). Elliott et al. (2009) and Ghiringhelli et al. (2009) document the potent chemotactic and immunostimulatory effects of extracellular ATP.
Wein, T. & Sorek, R. Bacterial origins of human cell-autonomous innate immune mechanisms. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-022-00705-4 (2022).
Roger, A. J., Muñoz-Gómez, S. A. & Kamikawa, R. The origin and diversification of mitochondria. Curr. Biol. 27, R1177–R1192 (2017).
Harapas, C. R. et al. Organellar homeostasis and innate immune sensing. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-022-00682-8 (2022).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541 (2018).
Galluzzi, L., Yamazaki, T. & Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 19, 731–745 (2018).
Mehta, M. M., Weinberg, S. E. & Chandel, N. S. Mitochondrial control of immunity: beyond ATP. Nat. Rev. Immunol. 17, 608–620 (2017).
Klionsky, D. J. et al. Autophagy in major human diseases. EMBO J. 40, e108863 (2021).
Galluzzi, L., López-Soto, A., Kumar, S. & Kroemer, G. Caspases connect cell-death signaling to organismal homeostasis. Immunity 44, 221–231 (2016).
Diepstraten, S. T. et al. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat. Rev. Cancer 22, 45–64 (2022).
Decout, A., Katz, J. D., Venkatraman, S. & Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 21, 548–569 (2021).
Li, X. D. et al. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394 (2013).
Civril, F. et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337 (2013).
White, M. J. et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159, 1549–1562 (2014).
Rongvaux, A. et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577 (2014). White et al. (2014) and Rongvaux et al. (2014) show that robust activation of apoptotic caspases downstream of MOMP suppresses cGAS signalling driven by mtDNA.
West, A. P. et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015).
Andreeva, L. et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 549, 394–398 (2017).
Zierhut, C. et al. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 178, 302–315.e323 (2019). This study shows that chromatin is a poor cGAS activator and suppresses cGAS signalling elicited by naked dsDNA.
Michalski, S. et al. Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature 587, 678–682 (2020).
Zhao, B. et al. The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature 587, 673–677 (2020).
Li, T. et al. Phosphorylation and chromatin tethering prevent cGAS activation during mitosis. Science 371, eabc5386 (2021).
Fuchs, Y. & Steller, H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 16, 329–344 (2015).
Domizio, J. D. et al. The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature 603, 145–151 (2022).
Wiens, K. E. & Ernst, J. D. The mechanism for type I interferon induction by mycobacterium tuberculosis is bacterial strain-dependent. PLoS Pathog. 12, e1005809 (2016).
Sun, B. et al. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 7, 3594 (2017).
Zhou, C. M. et al. Identification of cGAS as an innate immune sensor of extracellular bacterium Pseudomonas aeruginosa. iScience 24, 101928 (2021).
Moriyama, M., Koshiba, T. & Ichinohe, T. Influenza A virus M2 protein triggers mitochondrial DNA-mediated antiviral immune responses. Nat. Commun. 10, 4624 (2019).
Danthi, P. Viruses and the diversity of cell death. Annu. Rev. Virol. 3, 533–553 (2016).
McArthur, K. et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359, eaao6047 (2018). These authors used super-resolution microscopy to define the roles of BAX and BAK1 in the release of mtDNA by permeabilized mitochondria.
Riley, J. S. et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 37, e99238 (2018).
Bock, F. J. & Tait, S. W. G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 21, 85–100 (2020).
Yamazaki, T. et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat. Immunol. 21, 1160–1171 (2020). This study shows that mtDNA released by permeabilized mitochondria in the context of limited apoptotic caspase activation underlies the ability of radiotherapy to initiate acute cGAS signalling.
Willemsen, J. et al. TNF leads to mtDNA release and cGAS/STING-dependent interferon responses that support inflammatory arthritis. Cell Rep. 37, 109977 (2021).
Cosentino, K. et al. The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Mol. Cell 82, 933–949.e939 (2022). The authors elegantly dissect the relative effects of BAX and BAK1 on the kinetics of the assembly of mtDNA-releasing pores at the OMM.
Ichim, G. et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 57, 860–872 (2015).
Galluzzi, L., Kepp, O., Trojel-Hansen, C. & Kroemer, G. Non-apoptotic functions of apoptosis-regulatory proteins. EMBO Rep. 13, 322–330 (2012).
Cao, K. et al. Mitochondrial dynamics regulate genome stability via control of caspase-dependent DNA damage. Dev. Cell 57, 1211–1225.e1216 (2022).
Brokatzky, D. et al. A non-death function of the mitochondrial apoptosis apparatus in immunity. EMBO J. 38, e100907 (2019).
Tigano, M., Vargas, D. C., Tremblay-Belzile, S., Fu, Y. & Sfeir, A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 591, 477–481 (2021). This article delineates a mechanism through which breaks in mtDNA initiate inflammatory responses as a consequence of mtRNA accumulation in the cytosol and RIG-I activation.
Flores-Romero, H. et al. BCL-2-family protein tBID can act as a BAX-like effector of apoptosis. EMBO J. 41, e108690 (2022).
Kim, J. et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 366, 1531–1536 (2019). These authors suggest a role for VDACs in the pathogenic release of mtDNA that accompanies the development of a lupus-like syndrome in mice.
Bonora, M., Giorgi, C. & Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 23, 266–285 (2022).
Patrushev, M. et al. Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell. Mol. Life Sci. 61, 3100–3103 (2004).
Yu, C. H. et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 183, 636–649.e618 (2020).
Kruglov, A. G., Kharechkina, E. S., Nikiforova, A. B., Odinokova, I. V. & Kruglova, S. A. Dynamics of the permeability transition pore size in isolated mitochondria and mitoplasts. FASEB J. 35, e21764 (2021).
Heng, J. B. et al. The electromechanics of DNA in a synthetic nanopore. Biophys. J. 90, 1098–1106 (2006).
Neginskaya, M. A. et al. The very low number of calcium-induced permeability transition pores in the single mitochondrion. J. Gen. Physiol. 152, e202012631 (2020).
Ross, C. et al. Inflammatory caspases: toward a unified model for caspase activation by inflammasomes. Annu. Rev. Immunol. 40, 249–269 (2022).
Agostini, L. et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity 20, 319–325 (2004).
Muruve, D. A. et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452, 103–107 (2008).
Shimada, K. et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012).
Nakahira, K. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 (2011). This article reveals the existence of a feedforward loop linking inflammasome activation and mitochondrial dysfunction that is under tonic control by autophagy.
Renaudin, X. Reactive oxygen species and DNA damage response in cancer. Int. Rev. Cell Mol. Biol. 364, 139–161 (2021).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Zhou, R., Tardivel, A., Thorens, B., Choi, I. & Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140 (2010).
Saxena, G., Chen, J. & Shalev, A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J. Biol. Chem. 285, 3997–4005 (2010).
Iyer, S. S. et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323 (2013).
Pervaiz, S., Bellot, G. L., Lemoine, A. & Brenner, C. Redox signaling in the pathogenesis of human disease and the regulatory role of autophagy. Int. Rev. Cell Mol. Biol. 352, 189–214 (2020).
Bauernfeind, F. et al. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 187, 613–617 (2011).
Niemi, K. et al. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J. Immunol. 186, 6119–6128 (2011).
Billingham, L. K. et al. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat. Immunol. 23, 692–704 (2022).
Zhong, Z. et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 (2018). The authors elegantly reveal that mtDNA neosynthesis is required for the generation of oxidized mtDNA species that elicit NLRP3 inflammasome activation.
Zhong, Z. et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164, 896–910 (2016).
Kang, D., Kim, S. H. & Hamasaki, N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion 7, 39–44 (2007).
Xu, Y., Johansson, M. & Karlsson, A. Human UMP-CMP kinase 2, a novel nucleoside monophosphate kinase localized in mitochondria. J. Biol. Chem. 283, 1563–1571 (2008).
Xian, H. et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity 54, 1463–1477.e1411 (2021).
Allam, R. et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 15, 982–990 (2014).
Vince, J. E. et al. The mitochondrial apoptotic effectors BAX/BAK activate caspase-3 and -7 to trigger NLRP3 inflammasome and caspase-8 driven IL-1β activation. Cell Rep. 25, 2339–2353.e2334 (2018).
Chauhan, D. et al. BAX/BAK-induced apoptosis results in caspase-8-dependent IL-1β maturation in macrophages. Cell Rep. 25, 2354–2368.e2355 (2018).
Wang, Y. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017).
Rogers, C. et al. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 10, 1689 (2019).
Platnich, J. M. et al. Shiga toxin/lipopolysaccharide activates caspase-4 and gasdermin D to trigger mitochondrial reactive oxygen species upstream of the NLRP3 inflammasome. Cell Rep. 25, 1525–1536.e1527 (2018).
Guarnieri, J. W. et al. SARS-CoV-2 viroporins activate the NLRP3-inflammasome via the mitochondrial permeability transition pore. Preprint at bioRxiv https://doi.org/10.1101/2022.02.19.481139 (2022).
Dang, E. V., McDonald, J. G., Russell, D. W. & Cyster, J. G. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell 171, 1057–1071.e1011 (2017).
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J. & Alnemri, E. S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 (2009).
Rathinam, V. A. et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402 (2010).
Di Micco, A. et al. AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity. Proc. Natl Acad. Sci. USA 113, E4671–E4680 (2016).
Crane, D. D., Bauler, T. J., Wehrly, T. D. & Bosio, C. M. Mitochondrial ROS potentiates indirect activation of the AIM2 inflammasome. Front. Microbiol. 5, 438 (2014).
Xu, L. et al. Mitochondrial DNA enables AIM2 inflammasome activation and hepatocyte pyroptosis in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 320, G1034–G1044 (2021).
Jabir, M. S. et al. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy 11, 166–182 (2015).
Riley, J. S. & Tait, S. W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 21, e49799 (2020).
Zhang, Q. et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010). This study shows that mitochondrial products that are released into the circulation as a consequence of cell death, including mtDNA and formylated peptides, contribute to the aetiology of systemic inflammatory response syndrome.
Tian, J. et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 8, 487–496 (2007).
Mangelinck, A. & Mann, C. DNA methylation and histone variants in aging and cancer. Int. Rev. Cell Mol. Biol. 364, 1–110 (2021).
Hemmi, H. et al. A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 (2000).
Lamkanfi, M. et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 185, 4385–4392 (2010).
Liu, Y. et al. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J. Hepatol. 63, 114–121 (2015).
Little, J. P. et al. Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Mol. Cell. Neurosci. 60, 88–96 (2014).
Oka, T. et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251–255 (2012). The authors show that deletion of a lysosomal nuclease-encoding gene results in inefficient mtDNA disposal by autophagy, culminating in pathogenic TLR9 activation.
De Leo, M. G. et al. Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat. Cell Biol. 18, 839–850 (2016).
Rabas, N. et al. PINK1 drives production of mtDNA-containing extracellular vesicles to promote invasiveness. J. Cell Biol. 220, e202006049 (2021).
Todkar, K. et al. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 12, 1971 (2021).
Cadete, V. J. et al. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J. Physiol. 594, 5343–5362 (2016).
Konecna, B. et al. Monocyte exocytosis of mitochondrial danger-associated molecular patterns in sepsis suppresses neutrophil chemotaxis. J. Trauma. Acute Care Surg. 90, 46–53 (2021).
Sansone, P. et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl Acad. Sci. USA 114, E9066–E9075 (2017).
Hur, S. Double-stranded RNA sensors and modulators in innate immunity. Annu. Rev. Immunol. 37, 349–375 (2019).
Dhir, A. et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242 (2018). This study shows that mtRNA can function as a DAMP and initiate MDA5-dependent inflammatory responses in eukaryotic cells.
Seth, R. B., Sun, L., Ea, C. K. & Chen, Z. J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682 (2005).
Coomans de Brachène, A. et al. Endogenous mitochondrial double-stranded RNA is not an activator of the type I interferon response in human pancreatic beta cells. Auto. Immun. Highlights 12, 6 (2021).
Li, S. et al. The mitochondrial protein ERAL1 suppresses RNA virus infection by facilitating RIG-I-like receptor signaling. Cell Rep. 34, 108631 (2021).
Vanpouille-Box, C., Demaria, S., Formenti, S. C. & Galluzzi, L. Cytosolic DNA sensing in organismal tumor control. Cancer Cell 34, 361–378 (2018).
Franchi, L. et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J. Immunol. 193, 4214–4222 (2014).
Galluzzi, L. et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell. Death Differ. 22, 58–73 (2015).
McArthur, K. & Kile, B. T. Apoptotic caspases: multiple or mistaken identities? Trends Cell Biol. 28, 475–493 (2018).
Gyrd-Hansen, M. & Meier, P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat. Rev. Cancer 10, 561–574 (2010).
Vince, J. E. et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 131, 682–693 (2007).
Varfolomeev, E. et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 131, 669–681 (2007).
Giampazolias, E. et al. Mitochondrial permeabilization engages NF-kappaB-dependent anti-tumour activity under caspase deficiency. Nat. Cell Biol. 19, 1116–1129 (2017).
Zhang, J. et al. Ubiquitin ligases cIAP1 and cIAP2 limit cell death to prevent inflammation. Cell Rep. 27, 2679–2689.e2673 (2019).
Lecis, D. et al. Smac mimetics induce inflammation and necrotic tumour cell death by modulating macrophage activity. Cell Death Dis. 4, e920 (2013).
Rizk, J. et al. SMAC mimetics promote NIK-dependent inhibition of CD4+ TH17 cell differentiation. Sci. Signal. 12, eaaw3469 (2019).
Boada-Romero, E., Martinez, J., Heckmann, B. L. & Green, D. R. The clearance of dead cells by efferocytosis. Nat. Rev. Mol. Cell Biol. 21, 398–414 (2020).
Leslie, D. S. et al. Serum lipids regulate dendritic cell CD1 expression and function. Immunology 125, 289–301 (2008).
Dieudé, M. et al. Cardiolipin binds to CD1d and stimulates CD1d-restricted γδ T cells in the normal murine repertoire. J. Immunol. 186, 4771–4781 (2011).
Gouveia, A., Bajwa, E. & Klegeris, A. Extracellular cytochrome c as an intercellular signaling molecule regulating microglial functions. Biochim. Biophys. Acta Gen. Subj. 1861, 2274–2281 (2017).
Pullerits, R., Bokarewa, M., Jonsson, I. M., Verdrengh, M. & Tarkowski, A. Extracellular cytochrome c, a mitochondrial apoptosis-related protein, induces arthritis. Rheumatology 44, 32–39 (2005).
Kepp, O. et al. ATP and cancer immunosurveillance. EMBO J. 40, e108130 (2021).
Bolivar, B. E. et al. Noncanonical roles of caspase-4 and caspase-5 in heme-driven IL-1beta release and cell death. J. Immunol. 206, 1878–1889 (2021).
Wang, Y. et al. Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy 9, 1624–1625 (2013).
Ma, Y. et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 38, 729–741 (2013).
Lin, S. et al. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J. Neuroinflamm. 9, 46 (2012).
May, O. et al. The receptor for advanced glycation end products is a sensor for cell-free heme. FEBS J. 288, 3448–3464 (2021).
Belcher, J. D. et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 123, 377–390 (2014).
Han, C. et al. Tumor cells suppress radiation-induced immunity by hijacking caspase 9 signaling. Nat. Immunol. 21, 546–554 (2020).
Rodriguez-Ruiz, M. E. et al. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology 8, e1655964 (2019).
Ning, X. et al. Apoptotic caspases suppress type I interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol. Cell 74, 19–31.e17 (2019).
Segawa, K. et al. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344, 1164–1168 (2014).
Fadok, V. A. et al. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207–2216 (1992).
Buqué, A., Rodriguez-Ruiz, M. E., Fucikova, J. & Galluzzi, L. Apoptotic caspases cut down the immunogenicity of radiation. Oncoimmunology 8, e1655364 (2019).
Bernardini, J. P. et al. Parkin inhibits BAK and BAX apoptotic function by distinct mechanisms during mitophagy. EMBO J. 38, e99916 (2019).
Lindqvist, L. M. et al. Autophagy induced during apoptosis degrades mitochondria and inhibits type I interferon secretion. Cell Death Differ. 25, 784–796 (2018).
Galluzzi, L. et al. Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836 (2017).
Sliter, D. A. et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262 (2018). This study shows that PINK1- and PRKN-dependent mitophagy has a major role in restricting STING1 signalling by clearing permeabilized, mtDNA-releasing mitochondria.
Mouton-Liger, F. et al. Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia 66, 1736–1751 (2018).
Zhang, N. P., Liu, X. J., Xie, L., Shen, X. Z. & Wu, J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Invest. 99, 749–763 (2019).
Shi, C. S. et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263 (2012).
Richter, B. et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl Acad. Sci. USA 113, 4039–4044 (2016).
Di Rita, A. et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKα. Nat. Commun. 9, 3755 (2018).
Yu, J. et al. Inflammasome activation leads to caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl Acad. Sci. USA 111, 15514–15519 (2014).
Michaud, M. et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1577 (2011).
Wang, Y. et al. Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity 46, 393–404 (2017).
Corrales, L. et al. Antagonism of the STING pathway via activation of the AIM2 inflammasome by intracellular DNA. J. Immunol. 196, 3191–3198 (2016).
Melki, I. et al. Platelets release mitochondrial antigens in systemic lupus erythematosus. Sci. Transl. Med. 13, eaav5928 (2021).
Caielli, S. et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 213, 697–713 (2016).
Caielli, S. et al. Erythroid mitochondrial retention triggers myeloid-dependent type I interferon in human SLE. Cell 184, 4464–4479.e4419 (2021). This is an elegant demonstration of the links between defective mitophagy in erythroid precursors and the formation of mitochondria-containing erythrocytes that drive pathogenic type I interferon responses in SLE.
Lood, C. et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 22, 146–153 (2016).
Cadwell, K. et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263 (2008).
Adolph, T. E. et al. Paneth cells as a site of origin for intestinal inflammation. Nature 503, 272–276 (2013).
Khaloian, S. et al. Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut 69, 1939–1951 (2020).
Jackson, D. N. et al. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut 69, 1928–1938 (2020).
Benmerzoug, S. et al. STING-dependent sensing of self-DNA drives silica-induced lung inflammation. Nat. Commun. 9, 5226 (2018).
Wiegman, C. H. et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 136, 769–780 (2015).
Giordano, L. et al. Extracellular release of mitochondrial DNA: triggered by cigarette smoke and detected in COPD. Cells 11, 369 (2022).
Maekawa, H. et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. 29, 1261–1273.e1266 (2019).
Chung, K. W. et al. Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. 30, 784–799.e785 (2019).
Chen, K. et al. Optineurin inhibits NLRP3 inflammasome activation by enhancing mitophagy of renal tubular cells in diabetic nephropathy. FASEB J. 33, 4571–4585 (2019).
Tang, C. et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 14, 880–897 (2018).
Kasten, M. et al. Genotype-phenotype relations for the Parkinson’s disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov. Disord. 33, 730–741 (2018).
Borsche, M. et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain 143, 3041–3051 (2020).
Noda, S. et al. Loss of Parkin contributes to mitochondrial turnover and dopaminergic neuronal loss in aged mice. Neurobiol. Dis. 136, 104717 (2020).
Wilkins, H. M. et al. Mitochondrial lysates induce inflammation and Alzheimer’s disease-relevant changes in microglial and neuronal cells. J. Alzheimers Dis. 45, 305–318 (2015).
Fang, E. F. et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412 (2019).
Roca-Agujetas, V. et al. Cholesterol alters mitophagy by impairing optineurin recruitment and lysosomal clearance in Alzheimer’s disease. Mol. Neurodegener. 16, 15 (2021).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
King, K. R. et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 23, 1481–1487 (2017).
Kwon, W. Y. et al. Circulating mitochondrial N-formyl peptides contribute to secondary nosocomial infection in patients with septic shock. Proc. Natl Acad. Sci. USA 118, e2018538118 (2021).
Marques, P. E. et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 56, 1971–1982 (2012).
Bai, J. et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc. Natl Acad. Sci. USA 114, 12196–12201 (2017).
Christensen, M. H. et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 35, 1385–1399 (2016).
Saffran, H. A., Pare, J. M., Corcoran, J. A., Weller, S. K. & Smiley, J. R. Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep. 8, 188–193 (2007).
Chen, H. et al. HBx inhibits DNA sensing signaling pathway via ubiquitination and autophagy of cGAS. Virol. J. 19, 55 (2022).
Yum, S., Li, M., Fang, Y. & Chen, Z. J. TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc. Natl Acad. Sci. USA 118, e2100225118 (2021).
Thomsen, M. K. et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology 64, 746–759 (2016).
Galluzzi, L. et al. Autophagy in malignant transformation and cancer progression. EMBO J. 34, 856–880 (2015).
Buque, A. et al. Immunoprophylactic and immunotherapeutic control of hormone receptor-positive breast cancer. Nat. Commun. 11, 3819 (2020).
Li, S. et al. SFTSV infection induces BAK/BAX-dependent mitochondrial DNA release to trigger NLRP3 inflammasome activation. Cell Rep. 30, 4370–4385.e4377 (2020).
Costa, T. J. et al. Mitochondrial DNA and TLR9 activation contribute to SARS-CoV-2-induced endothelial cell damage. Vasc. Pharmacol. 142, 106946 (2022).
Neufeldt, C. J. et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-κB. Commun. Biol. 5, 45 (2022).
Kuo, C. L. et al. Mitochondrial oxidative stress by Lon-PYCR1 maintains an immunosuppressive tumor microenvironment that promotes cancer progression and metastasis. Cancer Lett. 474, 138–150 (2020).
Singel, K. L. et al. Mitochondrial DNA in the tumour microenvironment activates neutrophils and is associated with worse outcomes in patients with advanced epithelial ovarian cancer. Br. J. Cancer 120, 207–217 (2019).
Bao, D. et al. Mitochondrial fission-induced mtDNA stress promotes tumor-associated macrophage infiltration and HCC progression. Oncogene 38, 5007–5020 (2019).
Li, C. et al. PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron-mediated immunometabolism. Dev. Cell 46, 441–455.e448 (2018).
Dorflinger, B. et al. Mitochondria supply sub-lethal signals for cytokine secretion and DNA-damage in H. pylori infection. Cell Death Differ. https://doi.org/10.1038/s41418-022-01009-9 (2022).
Schmidts, A., Wehrli, M. & Maus, M. V. Toward better understanding and management of CAR-T cell-associated toxicity. Annu. Rev. Med. 72, 365–382 (2021).
Le Naour, J., Zitvogel, L., Galluzzi, L., Vacchelli, E. & Kroemer, G. Trial watch: STING agonists in cancer therapy. Oncoimmunology 9, 1777624 (2020).
Glab, J. A., Cao, Z. & Puthalakath, H. Bcl-2 family proteins, beyond the veil. Int. Rev. Cell Mol. Biol. 351, 1–22 (2020).
Mason, K. D. et al. Programmed anuclear cell death delimits platelet life span. Cell 128, 1173–1186 (2007).
Galluzzi, L., Zitvogel, L. & Kroemer, G. Immunological mechanisms underneath the efficacy of cancer therapy. Cancer Immunol. Res. 4, 895–902 (2016).
Lopez, A. et al. Co-targeting of BAX and BCL-XL proteins broadly overcomes resistance to apoptosis in cancer. Nat. Commun. 13, 1199 (2022).
Reyna, D. E. et al. Direct activation of BAX by BTSA1 overcomes apoptosis resistance in acute myeloid leukemia. Cancer Cell 32, 490–505.e410 (2017).
Spitz, A. Z., Zacharioudakis, E., Reyna, D. E., Garner, T. P. & Gavathiotis, E. Eltrombopag directly inhibits BAX and prevents cell death. Nat. Commun. 12, 1134 (2021).
Amgalan, D. et al. A small-molecule allosteric inhibitor of BAX protects against doxorubicin-induced cardiomyopathy. Nat. Cancer 1, 315–328 (2020).
Yamazaki, T. & Galluzzi, L. BAX and BAK dynamics control mitochondrial DNA release during apoptosis. Cell Death Differ. 29, 1296–1298 (2022).
Swanson, S. K. et al. Cyclosporin-mediated inhibition of bovine calcineurin by cyclophilins A and B. Proc. Natl Acad. Sci. USA 89, 3741–3745 (1992).
Galluzzi, L., Bravo-San Pedro, J. M., Levine, B., Green, D. R. & Kroemer, G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 16, 487–511 (2017).
Lekakis, V. & Cholongitas, E. The impact of emricasan on chronic liver diseases: current data. Clin. J. Gastroenterol. 15, 271–285 (2022).
Galluzzi, L., Vanpouille-Box, C., Bakhoum, S. F. & Demaria, S. SnapShot: CGAS-STING signaling. Cell 173, 276–276.e271 (2018).
Yatim, N. et al. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science 350, 328–334 (2015).
Bakhoum, S. F. et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018).
Li, H., Zimmerman, S. E. & Weyemi, U. Genomic instability and metabolism in cancer. Int. Rev. Cell Mol. Biol. 364, 241–265 (2021).
Acknowledgements
S.M. is supported by the Italian Ministry of Health (GR-2016-02364602), Nanoblend and local funds from Marche Polytechnic University (Ancona, Italy). S.W.G.T. is supported by funding from Cancer Research UK, Prostate Cancer UK, Tenovus Scotland and the Swiss National Science Foundation. The laboratory of L.G. (as a principal investigator unless otherwise indicated) is or has been supported by two Breakthrough Level 2 grants from the US Department of Defense Breast Cancer Research Program (BC180476P1 and BC210945), by a Transformative Breast Cancer Consortium Grant from the US Department of Defense Breast Cancer Research Program (W81XWH2120034; principal investigator Formenti), by a U54 grant from the NCI of the NIH (CA274291; principal investigators Deasy, Formenti and Weichselbaum), by the 2019 Laura Ziskin Prize in Translational Research (ZP-6177; principal investigator Formenti) from Stand Up to Cancer, by a Mantle Cell Lymphoma Research Initiative grant from the Leukaemia and Lymphoma Society (principal investigator Chen-Kiang), by a Rapid Response Grant from the Functional Genomics Initiative (New York, USA), by start-up funds from the Department of Radiation Oncology at Weill Cornell Medicine (New York, USA), by industrial collaborations with Lytix Biopharma (Oslo, Norway) and Promontory (New York, USA), and by donations from Promontory (New York, USA), the Luke Heller TECPR2 Foundation (Boston, USA), Sotio a.s. (Prague, Czech Republic), Lytix Biopharma (Oslo, Norway), Onxeo (Paris, France), Ricerchiamo (Brescia, Italy) and Noxopharm (Chatswood, Australia).
Author information
Authors and Affiliations
Contributions
T.Y. and L.G. conceived the idea for the Review. S.M. and E.G. prepared the first version of the manuscript, with constructive input from S.W.G.T. and under the supervision of T.Y. and L.G. E.G. prepared display items under the supervision of T.Y. and L.G. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
L.G. has held research contracts with Lytix Biopharma and Promontory, has received consulting/advisory honoraria from Boehringer Ingelheim, AstraZeneca, OmniSEQ, Onxeo, The Longevity Labs, Inzen, Sotio, Promontory, Noxopharm, EduCom and the Luke Heller TECPR2 Foundation, and holds Promontory stock options. All other authors declare no conflicts of interest.
Peer review
Peer review information
Nature Reviews Immunology thanks N. Chandel, C. Hauser and A. García-Sáez for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- Cancer immunosurveillance
-
A process through which the immune system recognizes and eliminates the majority of newly formed cancer cell precursors, hence suppressing oncogenesis.
- Type II cells
-
Mammalian cells that (in contrast to type I cells) require mitochondrial outer membrane permeabilization to enable death receptors to induce full activation of executioner caspases during apoptosis.
- Regulated cell death
-
(RCD). A type of cell death that depends on a genetically encoded machinery and hence can be modulated by pharmacological or genetic interventions.
- Mitochondrial outer membrane permeabilization
-
(MOMP). A key event in apoptotic cell death, culminating in the release of mitochondrial components into the cytosol and activation of proapoptotic caspases.
- Autophagy
-
An evolutionarily conserved, lysosome-dependent mechanism through which eukaryotic cells clear the cytoplasm of potentially cytotoxic or superfluous material to preserve homeostasis.
- Caspase
-
A member of a family of cysteine-dependent proteases that regulate the timing and immunological effects of various forms of cell death.
- Programmed cell death
-
A variant of regulated cell death that is triggered as a part of physiological programmes (such as embryonic development or preservation of adult tissue homeostasis) and not as a consequence of failing adaptation to stress.
- Mitophagy
-
A type of autophagy response that preferentially degrades permeabilized or otherwise dysfunctional mitochondria.
- Mitochondrial permeability transition
-
(MPT). A regulated process resulting in the abrupt loss of the impermeability of the inner mitochondrial membrane to solutes and water, resulting in osmotic swelling of the mitochondrial matrix and, ultimately, cell death.
- Inhibitor of apoptosis protein
-
(IAP). A member of a protein family that inhibits apoptosis by antagonizing the catalytic activity of caspases and functioning as a ubiquitin ligase to control upstream apoptotic signal transduction.
- Ripoptosome
-
A supramolecular complex containing receptor-interacting serine/threonine kinase 1 (RIPK1) and RIPK3 that promotes cell death coupled to inflammatory responses in various stress conditions (for example, exposure to genotoxins and allergens).
- Non-canonical NF-κB signalling
-
A transcriptional response generally initiated by the NIK-driven activation of NF-κB heterodimers composed of RELB and p52.
- Canonical NF-κB signalling
-
A transcriptional response generally initiated by the TAK1-driven activation of NF-κB heterodimers composed of RELA and p50.
- Immunogenic cell death
-
A variant of regulated cell death that is sufficient, in immunocompetent and syngeneic settings, to elicit an adaptive immune response to dead cell-associated antigens.
- BH3 mimetics
-
Pharmacological agents that mimic the ability of natural BH3-only proteins to directly or indirectly promote mitochondrial outer membrane permeabilization.
- Neutrophil extracellular traps
-
Networks of extracellular fibres enriched in DNA and proteins that are released by neutrophils in response to activating stimuli (such as pathogens).
Rights and permissions
About this article

Cite this article
Marchi, S., Guilbaud, E., Tait, S.W.G. et al. Mitochondrial control of inflammation. Nat Rev Immunol 23, 159–173 (2023). https://doi.org/10.1038/s41577-022-00760-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-022-00760-x
This article is cited by
-
RNA sequencing reveals differential long noncoding RNA expression profiles in bacterial and viral meningitis in children
BMC Medical Genomics (2024)
-
Mcl-1 mediates intrinsic resistance to RAF inhibitors in mutant BRAF papillary thyroid carcinoma
Cell Death Discovery (2024)
-
Mitochondrial outer membrane integrity regulates a ubiquitin-dependent and NF-κB-mediated inflammatory response
The EMBO Journal (2024)
-
A break in mitochondrial endosymbiosis as a basis for inflammatory diseases
Nature (2024)
-
Inflammation and mitophagy are mitochondrial checkpoints to aging
Nature Communications (2024)
