
Abstract
During severe inflammatory and infectious diseases, various mediators modulate the equilibrium of vascular tone, inflammation, coagulation and thrombosis. This Review describes the interactive roles of the renin–angiotensin system, the complement system, and the closely linked kallikrein–kinin and contact systems in cell biological functions such as vascular tone and leakage, inflammation, chemotaxis, thrombosis and cell proliferation. Specific attention is given to the role of these systems in systemic inflammation in the vasculature and tissues during hereditary angioedema, cardiovascular and renal glomerular disease, vasculitides and COVID-19. Moreover, we discuss the therapeutic implications of these complex interactions, given that modulation of one system may affect the other systems, with beneficial or deleterious consequences.
Similar content being viewed by others
Introduction
The renin–angiotensin system (RAS), the complement system and the kallikrein–kinin system (KKS) each consists of a large number of distinct plasma and membrane-bound proteins and receptors that can become activated through proteolytic cascades. Notably, all three systems are excessively activated during inflammation, have potent pro-inflammatory and prothrombotic effects and increase vascular permeability, leading to oedema. The RAS has a major role in the control of vascular tone, blood pressure, fluid and salt balance, cell proliferation, fibrosis and inflammation1. The complement system, which can be activated via the classical, alternative and lectin pathways, forms an integral part of innate immunity by eliciting local inflammation and clearance mechanisms, including opsonization, chemotaxis, phagocytosis, anaphylaxis, cytolysis and autophagy, and by interacting with pathways of adaptive immunity in response to pathogens, altered and apoptotic cells, immune complexes or polymeric forms of immunoglobulin as well as pentraxins2. The KKS releases vasoactive kinins, such as bradykinin, which induce vasodilation, vascular leakage and pain. The KKS can be induced both in plasma, where it is referred to as the ‘plasma KKS’, and in tissue, where it is known as the ‘tissue KKS’. These systems not only differ in location; they also have different enzymes and release distinct kinins. The plasma KKS is tightly linked with the contact system, which is activated by factor XII (FXII; also called ‘Hageman factor’). In addition to kinin release, FXII also initiates the intrinsic pathway of coagulation3, leading to thrombus formation. Here we refer to the plasma KKS and the contact system as the ‘plasma KKS/contact system’.
In this Review, we describe the RAS, the complement system and the plasma KKS/contact system and their role in inflammation, with a particular focus on the mechanisms of crosstalk between the three systems. We elaborate on the role of these interactions in inflammatory disorders such as hereditary angioedema (HAE), cardiac injury, glomerulonephritis, vasculitis and severe COVID-19, and discuss the therapeutic implications of the interactions between the three systems.
The RAS
The RAS is composed of proteins, peptides, enzymes and receptors, and the main physiological functions of the RAS are determined by the balance of two counter-regulatory pathways, the canonical and non-canonical RAS pathways (Fig. 1; Supplementary Table 1). These allow the regulation of blood pressure, fluid and salt homeostasis, vasoconstriction or vasodilation, vascular permeability, oxidative stress, cell death or proliferation, inflammatory modulation (both pro-inflammatory and anti-inflammatory effects), angiogenesis, fibrosis and coagulation/impaired fibrinolysis4. Whereas the immediate effects of the RAS are related to maintaining blood flow and adequate blood pressure as well as electrolyte balance, prolonged stimulation contributes to the development of hypertension and cardiovascular and renal diseases as well as inflammatory diseases and inflammatory complications in infectious diseases, but may also yield protective countereffects as described later.

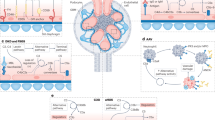
a | The renin–angiotensin system (RAS) is initiated by renin, which is secreted by the juxtaglomerular cells of the afferent renal arterioles in response to decreased perfusion, in response to low sodium levels, upon β1-adrenergic receptor stimulation or upon stimulation by nitric oxide (NO) or prostaglandin259,260. The renin precursor prorenin and renin both bind to prorenin receptor (PRR)261. In the circulation, renin cleaves angiotensinogen into angiotensin I (Ang I). Prorenin can also cleave angiotensinogen into Ang I, to a certain extent, when bound to PRR. Ang I can also be generated through the cleavage of Ang 1–12 by angiotensin-converting enzyme (ACE)240 or by chymase in cardiac tissue262. The ensuing proteolytic cascade can be divided into the canonical and non-canonical pathways. The canonical pathway is initiated when ACE, or chymase in the heart, cleaves Ang I into Ang II. Binding of Ang II or its cleavage products Ang III or Ang A to the G-protein-coupled receptor Ang II receptor type 1 (AT1R) leads to vasoconstriction, an increase in blood pressure and other effects4,221. The non-canonical pathway is initiated by Ang I cleavage by ACE2 to Ang 1–9, which is further cleaved into Ang 1–7 (mainly by ACE). In the non-canonical pathway, binding of Ang II, Ang III, Ang 1–9, Ang 1–7 and Ang A to AT2R counteracts effects mediated by AT1R. Ang IV (generated from Ang III when cleaved by aminopeptidase N (APN)) is involved in both the canonical pathway and the non-canonical pathway and binds to angiotensin 4 receptor (AT4R), also known as insulin-regulated aminopeptidase (IRAP). Binding of Ang IV to AT4R has antiapoptotic and both pro-inflammatory and anti-inflammatory effects, as it regulates responses by competitive inhibition of the enzyme/receptor218,263,264. Ang 1–7 binds to the receptors MAS and MAS-related G-protein-coupled receptor (MRGD), and alamandine, which is generated from Ang A or Ang 1–7, also binds to MRGD. Both Ang 1–7 and alamandine have antihypertensive, anti-inflammatory and antifibrotic effects265. The non-canonical and canonical RAS pathways can counterbalance each other218,263,264,265. For example, Ang II binds to both AT2R (non-canonical pathway) and AT1R (canonical pathway) but exerts a more robust effect on AT1R99,266. AT2R can, however, be upregulated during pathological conditions99. Effectors are depicted in circles and the enzymes catalysing their cleavage are listed next to blue arrows. Effectors that predominantly participate in the canonical pathway are coloured pink and those that predominantly participate in the non-canonical pathway are coloured blue. Effectors that participate in both pathways are coloured in both pink and blue, with the predominant colour indicating the more prominent pathway. Black arrows indicate which molecule binds to which receptor, with dashed lines depicting weak receptor binding. b | Amino acid sequences of angiotensin peptides. The cleavage sites of ACE and renin are indicated by blue arrows. AD, aspartate decarboxylase; ADH, antidiuretic hormone; APA, aminopeptidase A; NEP, neprilysin; PEP, prolylendopeptidase; PRCP, prolylcarboxypeptidase; THOP, thimet oligopeptidase.
A central component of the RAS is angiotensin-converting enzyme 2 (ACE2), which directs the RAS catalytic process towards the non-canonical pathway (Fig. 1). ACE2 is an integral membrane protein and, as such, is also the cellular receptor for the spike proteins of severe acute respiratory syndrome coronavirus (SARS-CoV) and SARS-CoV-2 (refs5,6). However, its soluble form also acts as an enzyme and can bind the viral spike protein7. ACE2 shares 42% homology with the enzyme ACE8 and is present in/on various types of cells in the heart, arteries (endothelial and smooth muscle cells), intestine, kidneys, lungs and brain9. ACE2 decreases angiotensin II (Ang II) levels by directly generating Ang 1–7 from Ang II (Fig. 1) and by degrading Ang I, the precursor of Ang II, to Ang 1–9. Ang 1–7 exerts inhibitory effects on inflammation, fibrosis and cell proliferation via its receptor MAS. Ace2-knockout mice develop enhanced pulmonary vascular permeability and oedema in a lung injury model10,11, an effect that might be due to excess cleavage of Ang I to Ang II by ACE and lack of Ang II degradation (Fig. 1). This mechanism has been suggested to occur in patients with COVID-19, where binding of SARS-CoV-2 to ACE2 downregulates ACE2 on cells12. ACE2 can be cleaved from the cell membrane by the enzyme ADAM17. Ang II can promote ADAM17 activity and ACE2 shedding13, which leads to reduced ACE2 tissue expression and elevated circulating levels of ACE2, with pathological effects in conditions such as cardiac failure and hypertension1,14.
The main mechanism of RAS inhibition occurs through a negative-feedback loop mediated via Ang II on renin. This feedback does not appear to be direct, as the receptors for Ang II, the Ang II type 1 receptors (AT1Rs) on renin-producing cells, are not involved15. Instead, changes in blood pressure and sodium retention induced by angiotensins regulate renin levels16. Blockade of the canonical RAS pathway (for example, by use of an ACE inhibitor) increases renin levels17 and promotes the catalytic breakdown of Ang II through the non-canonical pathway, as well as Ang 1–7 stimulation of MAS18, leading to vasodilation, decreased vascular tone and natriuresis (Fig. 1).
An interesting aspect of receptor specificity in the RAS is biased agonism4, whereby the position of the ligand determines the nature of the intracellular signalling cascades. For example, both Ang II and Ang 1–7 can bind to AT1R, but they induce different intracellular responses19. Moreover, Ang 1–7 blocks the effects of Ang II on the same receptor19. Biased agonists may accelerate receptor internalization, and this phenomenon could potentially be exploited therapeutically. Furthermore, certain G-protein-coupled receptors (GPCRs) have been found to form heterodimers (for example, AT1R and AT2R20 as well as AT2R and MAS21), adding additional complexity to the signal interaction.
The complement system
When stimulated by pathogens, apoptotic cells, immune complexes or immunoglobulin polymers, the complement system elicits local inflammation and clearance mechanisms, including opsonization, chemotaxis, phagocytosis, anaphylaxis, cytolysis and autophagy. Moreover, components of the complement system interact with pathways and cells of both innate and adaptive immunity2. The complement system may be activated on cell membranes, in the fluid phase and even intracellularly22. The three pathways of activation (the classical, lectin and alternative pathways, depicted in Fig. 2) respond to different stimuli but all ultimately generate C3 and C5 convertases and the membrane attack complex. Cleavage products of complement proteins are biologically active and can induce anaphylaxis (C3a and C5a), chemotaxis and phagocytosis (C3a and C5a) and opsonization (C3b, iC3b and C3dg). This robust defence system has multiple inhibitors, both soluble and expressed on cell surfaces, to prevent excessive activation (Supplementary Table 1). For a more comprehensive overview of the complement system, the reader is referred to ref.2. Here we focus on the components of the complement system that are important for inflammation and coagulation or thrombosis and are known to interact with the RAS or the plasma KKS/contact system (Table 1).

The complement system is a network that can be divided into the classical, lectin and alternative pathways. The classical pathway is activated when C1s within the C1qr2s2 complex cleaves C2 into C2a and C2b and cleaves C4 into C4a and C4b. Likewise, in the lectin pathway, mannose-binding lectin (MBL) serine protease 1 (MASP1) and MASP2 can also cleave C2 and C4. The classical and lectin pathways converge at the level of C2 and C4 cleavage, where a complex between C2a and C4b forms the C3 convertase. The alternative pathway exhibits a constant low-grade activation on biological surfaces whereby hydrolysed C3, C3(H2O), binds to factor B (FB), which is cleaved by factor D (FD) into the Bb and Ba fragments. The Bb fragment contributes to the formation of the initial convertase C3(H2O)Bb. This convertase cleaves C3 into C3a and C3b. C3b forms a complex with FB, which is again cleaved by FD into Bb, to form C3bBb, the C3 convertase. This convertase is stabilized by properdin (P). The formation of C3 convertases (C4b2a in the classical and lectin pathways and C3bBb in the alternative pathway) is amplified by the C3b-generation amplification loop (centre). By addition of extra C3bs to the C3 convertases, these are transformed into C5 convertases. C5 convertases cleave C5 into C5a and C5b, which promotes the assembly of the pore-forming unit C5b-9 within the cell membrane; this process is called the ‘terminal lytic complement pathway’. Binding of C3a to the receptor C3aR and of C5a to the C5a receptors C5aR1 and C5aR2 induces potent inflammatory responses. Blue lines depict enzymatic activity. Black lines depict binding or conversion into cleavage products. gC1q, globular head of C1q.
The classical pathway of complement activation is initiated when the complement component C1q binds to the Fc domains of IgM or IgG. Together with two molecules of the serine protease C1r and two molecules of the serine protease C1s, C1q forms a complex called ‘C1qr2s2’, and within this complex, C1s possesses the enzymatic activity that cleaves C2 and C4. C1q itself is composed of 18 polypeptides and has a hexameric structure, with heterotrimeric polypeptide chains forming a total of six subunits. Each heterotrimer has a collagen-like domain at the amino terminus (N terminus) and a globular head domain termed ‘gC1q’ at the carboxy terminus (C terminus). The gC1q domain can bind both to its receptor and to immunoglobulins. The multifunctional gC1q receptor (gC1qR)23 is expressed by endothelial cells and platelets24 and also binds thrombin, vitronectin25, fibrinogen, high-molecular-weight kininogen and FXII, in addition to C1q26. Of note, high-molecular-weight kininogen and FXII are components of the KKS. gC1qR can initiate the classical pathway of complement activation, even in the absence of immunoglobulins27, and can also activate the plasma KKS/contact system (Figs 3,4a).

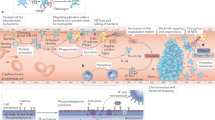
The plasma kallikrein–kinin and contact systems and their activation on endothelial cells. The zymogen factor XII (FXII) binds to urokinase plasminogen activator receptor (uPAR) in complex with cytokeratin 1 (CK1) or the globular head of the C1q receptor (gC1qR) alone or in complex with CK1 (ref.37) and is autoactivated, or is activated by plasma kallikrein, into the enzymatically active FXIIa. Alternatively, in the contact system, FXII is activated to FXIIa when it encounters negatively charged surfaces. Plasma prekallikrein (PPK) is cleaved into plasma kallikrein, its active form, by soluble or bound FXIIa267. On the surface of endothelial cells, PPK, when bound to high-molecular-weight kininogen (HK), can also be activated to plasma kallikrein by heat shock protein 90 (HSP90) or by prolylcarboxyeptidase (PRCP) (HSP90 and PRCP act as cofactors rather than enzymes). Thus, prekallikrein can be converted to plasma kallikrein both by FXIIa and by autoactivation in the presence of cofactors. Plasma kallikrein cleaves HK to release the nonapeptide bradykinin, and FXIIa activates FXI by cleaving it to form FXIa, thereby initiating the intrinsic coagulation cascade3. Bradykinin exerts inflammatory effects via the B2 receptor (B2R). Bradykinin is degraded to desArg9-bradykinin by carboxypeptidase M (CPM) on the cell surface or by carboxypeptidase N (CPN) in the fluid phase. DesArg9-bradykinin binds preferentially to the B1 receptor (B1R). Both kinins are further degraded by angiotensin-converting enzyme (ACE). Green arrows show that autoactivation of a protein into its activated form can occur, blue arrows indicate enzymatic activity, black arrows point to cleavage products and grey arrows indicate cofactors.

a | Joint pathways of the renin–angiotensin system (RAS), complement system and kallikrein–kinin system (KKS) on the endothelium. Certain components of the complement system and the KKS bind to the same surface receptor, the globular head of C1q receptor (gC1qR). It can bind C1q or, in complex with cytokeratin 1 (CK1), it binds high-molecular-weight kininogen (HK) and plasma prekallikrein (PPK). C1 inhibitor is a common inhibitor of plasma kallikrein, activated factor XII (FXIIa), mannose-binding lectin (MBL) serine proteases (MASPs), C1s and C1r. Plasma kallikrein cleaves HK, releasing bradykinin. In vitro it was also shown to cleave complement factor B (FB) and C3; however, this has not been confirmed in vivo. Renin cleaves both angiotensinogen into angiotensin I (Ang I) and C3 into C3a and C3b allowing formation of more C3bBb. Angiotensin-converting enzyme (ACE) degrades bradykinin to metabolites and also converts Ang I to Ang II. Red lines depict inhibition, green arrows indicate that autoactivation of a protein can occur, blue arrows indicate enzymatic activity (dashed if shown only in vitro) and black arrows point to cleavage products. b | Mechanisms of vascular permeability and leakage induced by the RAS, the KKS and the complement system. Vascular permeability via the RAS is induced by Ang II signalling via Ang II type 1 receptor (AT1R). In the KKS, vasopermeability is associated with bradykinin and desArg9-bradykinin signalling via the receptors B2R and B1R, respectively, and in the complement system with C5a signalling via the C5a receptor (C5aR1) and deposition of C5b-9 within the cell membrane. c | Mechanisms of endothelial cell injury, thrombosis, vasopermeability/oedema and inflammation induced by the RAS, the KKS/contact system and the complement system during severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. After binding to pulmonary epithelial cells via surface-expressed ACE2, SARS-CoV-2 undergoes endocytosis, thereby reducing the amount of ACE2 on cell surfaces. This creates an imbalance in the RAS with increased activation of the canonical pathway. ACE2 inactivates desArg9-bradykinin170. Decreased levels of ACE2 therefore increase desArg9-bradykinin levels and signalling via B1R in the KKS, leading to inflammation171. SARS-CoV-2 interacts with C1 inhibitor184, the level of which was found to be reduced in bronchoalveolar lavage fluid from patients with COVID-19 (ref.176). Downregulation of C1 inhibitor will allow activation of the classical and lectin pathways of the complement system, and the nucleocapsid proteins of SARS-CoV and SARS-CoV-2 were shown to bind to MASP2 and initiate the lectin pathway of the complement system195. The KKS/contact system is also activated because C1 inhibitor inhibits activated factor XII (FXIIa). Furthermore, activated FXII cleaves C1r254, activating the classical pathway of the complement system. Endothelial cells become injured through excess activation of the three pathways. Endothelial cell activation leads to increased vascular permeability (as shown in part b), resulting in pulmonary oedema. Ang II, C3a and C5a as well as the kinin effectors bradykinin and desArg9-bradykinin promote neutrophil recruitment and inflammation. Neutrophil proteases can further activate the KKS by cleavage of HK. As listed in Table 1, effectors of the KKS and FXII, effectors of the RAS canonical pathway and of the complement system induce thrombosis. uPAR, urokinase plasminogen activator receptor.
The lectin pathway of complement activation is initiated by mannan-binding lectin serine proteases (MASPs), which also cleave complement components C2 and C4 (ref.28). MASP1 and MASP2 can also cleave prothrombin into thrombin29,30, and MASP1 activates thrombin-activatable fibrinolysis inhibitor (TAFI), suggesting that activation of the lectin pathway may promote thrombosis by stabilizing fibrin and prolonging fibrinolysis29. MASP1 also contributes to thrombus formation in vivo31. MASPs are inhibited by C1 inhibitor; however, HAE, which is associated with C1 inhibitor deficiency, is not associated with excessive clotting32. MASP1 was also reported to cleave high-molecular-weight kininogen in vitro, thereby releasing bradykinin, whereas MASP2 was shown to cleave high-molecular-weight kininogen but bradykinin is not released33. Furthermore, MASP1 was recently found to upregulate the kinin B2 receptor34 described later.
The main inhibitor of both the classical pathway and the lectin pathway of the complement system is C1 inhibitor35, which directly inhibits the enzymatic activity of C1r/C1s in the classical pathway and MASP1/MASP2 in the lectin pathway. C1 inhibitor can also inhibit components of the KKS, such as activated FXII (FXIIa), FXI and plasma kallikrein, as well as plasmin, thereby affecting the fibrinolytic system35. Active plasma kallikrein is inhibited by both C1 inhibitor and the large glycoprotein α2-macroglobulin36. In the absence of sufficient levels of C1 inhibitor, FXII will autoactivate to form FXIIa and convert plasma prekallikrein (also called ‘Fletcher factor’) to kallikrein and FXI to FXIa37 and also allow uninhibited activation of the classical and lectin pathways of complement, ultimately leading to increased vascular leakage and inflammation.
The KKS/contact system
The plasma KKS and the tissue KKS are independent systems that both release kinins as effector peptides from kininogens through cleavage by two different kallikreins. The plasma KKS is tightly interlinked with the contact system, which is integral to the intrinsic pathway of coagulation and thrombus formation. Both involve plasma prekallikrein and FXII; the latter is activated upon contact with charged natural or artificial surfaces3,38, followed by rapid reciprocal activation of plasma prekallikrein into plasma kallikrein and FXII into FXIIa (Fig. 3). Plasma kallikrein releases bradykinin from high-molecular-weight kininogen, and FXIIa initiates blood coagulation by activating FXI. FXIIa also activates plasma kallikrein. The tissue KKS is initiated by the serine protease tissue kallikrein, which is activated, probably intracellularly, by an unknown protease, liberating kallidin from low-molecular-weight kininogen. Although there are many tissue kallikreins, only one is involved in kinin generation39,40 (Supplementary Table 1).
The only two precursor proteins for kinins in humans, high-molecular-weight kininogen and low-molecular-weight kininogen, are synthesized mainly in the liver (Supplementary Table 1). The mRNAs encoding these proteins are generated by alternative splicing. In the blood, high-molecular-weight kininogen circulates in complex with plasma prekallikrein or FXI of the intrinsic coagulation system. Plasma prekallikrein is inactive in this complex, but when the complex binds to the surface of endothelial cells, plasma prekallikrein can be activated by prolylcarboxypeptidase (PRCP) or heat shock protein 90 (HSP90) to plasma kallikrein41,42 (Fig. 3). More important, however, is its activation by FXII, which can also bind to endothelial cells via three different factors: heparan sulfate proteoglycans43, urokinase plasminogen activator receptor (uPAR)44 and gC1qR45. gC1qR and uPAR are each complexed with cytokeratin 1 (ref.46). FXII autoactivates to form FXIIa when it encounters negatively charged surfaces. These can be artificial material such as glass or plastic, or natural factors such as the subendothelial matrix of vessels after injury, or DNA, polyphosphates and heparin released from activated neutrophils, platelets and mast cells, respectively47,48. In addition to activating FXI to FXIa and thereby the intrinsic coagulation system, FXIIa also cleaves plasma prekallikrein into plasma kallikrein as mentioned earlier. This initiates a feedforward loop that activates more FXII (Fig. 3). Since plasma kallikrein releases the nonapeptide bradykinin from high-molecular-weight kininogen, its activation leads to a rapid burst in local bradykinin production, with marked effects on the surrounding tissue. Bradykinin mediates inflammation by inducing vasodilation and fluid extravasation, resulting in redness (rubor), heat (calor) and swelling (tumour), three of the four cardinal signs of inflammation. The fourth sign, pain (dolour), is mediated by kinin receptors on nociceptive nerve endings49. In addition, bradykinin induces the release of nitric oxide (NO), prostacyclin and tissue plasminogen activator, and has thrombin-inhibitory effects3,38,50 (Table 1).
In addition to kallikrein, other proteases, which do not belong to the KKS, have been described as releasing kinin peptides from high-molecular-weight kininogen. These peptides can activate the kinin receptors B1R and B2R51 (Box 1). One example of such a peptide is PR3-kinin, which has two additional amino acids at both ends of the bradykinin sequence and is released by neutrophil-derived proteinase 3 (ref.52).
Kinins have a very short half-life (less than 1 minute) in tissues and blood as they are quickly metabolized by various proteases39,53. Removal of the N-terminal lysine by aminopeptidases (such as aminopeptidase N) transforms kallidin to bradykinin. However, since both kallidin and bradykinin activate B2R, this does not alter signalling54. Carboxypeptidase M, on cell surfaces, and carboxypeptidase N (CPN; also called ‘kininase I’), in plasma, release the C-terminal arginine from bradykinin and kallidin, generating desArg9-bradykinin (DABK) and desArg10-kallidin (DAKD), respectively, which are both B1R agonists (Box 1). Various peptidases degrade kinins further. Neprilysin and ACE (also called kininase II) are two prominent peptidases, and both liberate dipeptides from the C terminus of kallidin and bradykinin, thereby abolishing their biological activity39 (Table 2). ACE inhibition therefore increases bradykinin levels55.
Interactions in inflammation
The interactions between the RAS, the complement system and the KKS/contact system (Fig. 4a; Table 2) play an important role in maintaining vasotonus and blood pressure, vasodilation and initiating thrombosis, as well as the removal of microorganisms, or their components, and dying cells. These interactions, when excessive, can lead to disease-related pathology due to increased vasopermeability, vasodilation, leukocyte chemotaxis, inflammation, thrombosis or fibrinolysis (Table 1).
Vascular tone, inflammation and permeability
Kinin activation in the vasculature induces inflammation, neutrophil chemotaxis, vasodilation and generally an increase in vasopermeability (Table 1). The KKS regulates vascular tone but has different effects depending on the affected cell type. In endothelial cells, kinins increase intracellular Ca2+ levels, leading to vasodilation via the activation of endothelial NO synthase, the production of NO and the generation of prostacyclin56. In smooth muscle cells, NO activates guanylate cyclase and thereby increases the levels of the nucleotide cGMP, and prostacyclin upregulates the nucleotide cAMP, both inducing smooth muscle relaxation. Moreover, kinins induce vasodilation via the release of endothelial-derived hyperpolarizing factor57. By contrast, the increase in intracellular Ca2+ levels induced by kinins can also lead to the constriction of smooth muscle cells via Ca2+/calmodulin-induced myosin light chain phosphorylation. Therefore, kinins can both contract or dilate vessels depending on the strength of signalling of kinin receptors, their level of expression on endothelial and smooth muscle cells and the amount of peptides that reach these cell types. In most arteries, kinins cause endothelium-dependent relaxation, resulting in a decrease in blood pressure when bradykinin is injected intravenously. However, kinins can also induce endothelium-dependent contractions in veins58.
Ang II increases vasopermeability and vasotonus via AT1R59 but may also decrease vasotonus via AT2R (Fig. 4b; Table 1). Ang II induces the expression of vascular endothelial growth factor (VEGF) in human vascular smooth muscle cells60. Ang 1–7 and bradykinin induce a synergistic vasodilatory effect that is abolished by either of their respective antagonists61 and involves AT2R signalling62, which stimulates bradykinin production63. This vasodilatory effect is reduced in kininogen-deficient rats, demonstrating the interaction between the KKS and the RAS64. The combined effect of RAS signalling and bradykinin release affects natriuresis and blood pressure. In renal proximal tubules, AT2R activation by its agonist, compound 21, induces natriuresis and lowers blood pressure. In Ang II-mediated hypertension associated with stimulation of AT1R via the canonical pathway of the RAS, AT2R counter-regulates blood pressure elevation by increasing renal production of bradykinin and promoting renal NO release65. The natriuretic effect involves the bradykinin–NO–cGMP pathway; this can be blocked by the small-molecule B2R antagonist icatibant66.
Kinins mediate a drastic increase in vascular permeability (Fig. 4b). The increase in cytosolic levels of Ca2+ results in a shortening of contractile proteins within the endothelial cells, the destabilization of adherens junctions, the formation of fenestrations in the walls of microvascular tissue and the extravasation of plasma constituents and leukocytes67. Similar effects are mediated by the complement system components C3 and C5 (Table 1). C3 and C5 cleavage generates the anaphylatoxins C3a (77 amino acids) and C5a (74 amino acids, with a C-terminal receptor-binding octapeptide), respectively. These bind to GPCRs on the endothelium and trigger intracellular activation pathways such as mitogen-activated protein kinase ERK1/2 signalling68,69, cytokine release68,70 and recruitment of leukocytes71, thereby inducing inflammation. C5a induces vascular permeability (Fig. 4b) through endothelial cell retraction72, tissue factor expression, and basophil and mast cell degranulation73,74. C5a binds to two GPCRs, C5aR1 (also known as CD88) and C5aR2 (also known as C5L2 or GPR77). Binding to C5aR1 induces the aforementioned pro-inflammatory response, whereas signalling through C5aR2, which also binds desArg-C5a, has an opposing effect on C5a–C5aR1 activity69 but also plays a role in vascular inflammation. C5aR2 was recently shown to transport tissue-derived C5a into the vascular lumen and thereby induce neutrophil recruitment and adhesion75. Overall, excess complement activation on the vascular endothelium promotes inflammation by inducing leukocyte recruitment and extravasation. The enhanced vascular permeability can lead to oedema. These mechanisms are crucial in the development of sepsis76.
In a hamster model of Chagas disease, plasma leakage occurs in the cheek pouch. Targeting C5aR1 and B2R reduced vascular leakage77, suggesting that both C5a and bradykinin contribute to vascular permeability. Patients with severe capillary leakage after bone marrow transplantation improved in response to treatment with C1 inhibitor and exhibited reduced plasma levels of C5a, suggesting it may contribute to capillary leakage78. C5b-9 can also increase vascular permeability and leakage79 as demonstrated in human umbilical vein endothelial cells and rat mesenteric endothelium. C5b-9 induced the accumulation of kinins in the culture medium, suggesting that kinins mediated the response, and indeed a B2R antagonist or kallikrein inhibitor partially blocked C5b-9-induced vascular leakage79. This indicates an interaction between the terminal complement pathway and KKS activation in mediating vascular permeability.
Hereditary angioedema
Dysfunctional or deficient C1 inhibitor or dysfunctional FXII can result in angioedema. HAE is associated with mutations in the C1 inhibitor gene. In type 1 HAE, the mutation leads to lower levels of C1 inhibitor, whereas in type 2 HAE, the levels of C1 inhibitor may be normal, but the protein is dysfunctional. Both types are autosomal dominant. In patients with normal C1 inhibitor function, mutations have been found in genes encoding FXII, plasminogen, angiopoietin 1, myoferlin and kininogen80,81. Acquired C1 inhibitor dysfunction or deficiency is due to the presence of circulating autoantibodies37.
HAE is characterized by the activation of the plasma KKS/contact system due to C1 inhibitor dysfunction, leading to the release of bradykinin82. Bradykinin then binds to B2R, which increases vascular permeability and thereby causes local extravasation of plasma and tissue swelling83. The swelling may be local or generalized and can be life-threatening when airways are affected. Patients with C1 inhibitor dysfunction have high levels of circulating bradykinin84. In addition to the plasma KKS/contact system, the classical pathway of the complement system is activated in these patients, who generally have low levels of C4 and high levels of soluble C5b-9 (ref.32). However, complement activation is not the primary mediator of the swelling85. The fibrinolytic pathway is also activated and increased levels of plasmin–α2-antiplasmin complexes are observed32. Overall, C1 inhibitor dysfunction leads to excess KKS activation and bradykinin-mediated oedema as well as complement activation with release of the anaphylatoxins C3a and C5a37.
HAE can be treated with purified C1 inhibitor, with the B2R antagonist icatibant or with kallikrein inhibitors such as the small molecule ecallantide86, the monoclonal antibody ranadelumab87 or the oral inhibitor berotralstat88.
Cardiovascular injury and protection
The RAS, the complement system and the KKS interact in the induction of cardiovascular injury and hypertension as well as in cardioprotection. The pro-inflammatory and profibrotic cellular effects of the canonical RAS pathway can contribute to the development of vascular inflammation, resulting in hypertension, atherosclerosis, cardiovascular disease and renal disease, whereas the non-canonical RAS pathway generally has a protective role. Ang II plays a central role in the inflammatory response by signalling via AT1R and exerting a pro-inflammatory effect on endothelial cells, vascular smooth-muscle cells and leukocytes89. This includes promoting the release of chemokines and cytokines, the expression of cellular adhesion molecules, the induction of dendritic cell migration and endothelial cell proliferation, as well as promoting monocyte adhesion to endothelial cells90,91.
The activation of the non-canonical pathway of the RAS leads to the generation of anti-inflammatory peptides with cardioprotective effects, which can reduce the size of myocardial infarcts, improve cardiac function, decrease arrythmias, prevent the development of hypertrophy and modulate the release of pro-inflammatory cytokines92. Ang 1–9 was shown to reduce cardiac inflammation in a rat model93. Additionally, Ang 1–7 can inhibit ACE and induce vascular relaxation94. Ang 1–9 and Ang 1–7 (ref.95) can potentiate the effects of bradykinin on B2R, and all three peptides are degraded by ACE96 (Table 2). The RAS peptides do not engage the B2R itself but increase the release of arachidonic acid and NO that is induced by bradykinin97. Importantly, neprilysin activity and bradykinin levels are elevated in infarcted heart tissue98.
The activation of the bradykinin–NO–cGMP pathway through AT2R has a cardioprotective effect99. In a porcine model of myocardial ischaemia, an AT1R blocker reduced myocardial infarct size, and this cardioprotective effect was abrogated by an AT2R antagonist or by the B2R antagonist icatibant100. Similar findings were demonstrated in B2R-deficient mice, where the cardioprotective effect of ACE inhibition or AT1R blockade was diminished relative to that in wild-type mice101, suggesting that B2R has an important cardioprotective role. In Agtr2−/− mice (Agtr2 encodes AT2R), AT1R expression is upregulated102 and an imbalance between AT1R and AT2R expression favours enhanced inflammatory responses.
The effectors in the non-canonical RAS pathway, including the angiotensin family member alamandine103, have anti-inflammatory and antiproliferative effects104. Several therapeutics for heart failure, left ventricular hypertrophy, hypertension, proteinuria, renal fibrosis and inflammation99,105 target the canonical RAS pathway, thereby diverting peptide ligands to the non-canonical pathway and providing agonists for the stimulation of AT2R and MAS (Box 2, Supplementary Box 1). These therapeutics include renin and ACE inhibitors, AT1R antagonists and neprilysin inhibitors in combination with AT1R blockers, termed ‘ARN inhibitors’106. Furthermore, a number of investigational therapeutics are currently in preclinical and clinical development; these include aminopeptidase A inhibitors, MAS agonists, NPA7 (a peptide combining particulate guanylyl cyclase A activation with MAS agonism), the AT2R agonist compound 21, recombinant ACE2 (ref.107), Ang 1–9 (ref.93), angiotensin 4 receptor (AT4R) inhibitors and angiotensinogen antisense oligonucleotides104,108.
During cardiovascular inflammation and RAS activation, the complement system also becomes activated. For example, mice infused with Ang II exhibit an increase in plasma levels of C3a and C5a and have perivascular deposition of C3b in cardiac tissue. C5aR-deficient mice as well as mice treated with anti-C5 or anti-C5aR antibodies were shown to be protected from cardiac inflammation induced by Ang II (ref.109). Moreover, C3aR-deficient and C5aR-deficient mice had lower blood pressure than wild-type mice and were protected from renal and vascular damage caused by Ang II infusion110. This was found to be partly due to the presence of C3aR and C5aR on regulatory T cells110, but the precise mechanisms by which protection occurs remain to be elucidated. Thus, the blockade of complement receptors might attenuate Ang II-mediated target organ damage.
Complement-associated glomerular inflammation
Activation of the alternative pathway of the complement system can be initiated by renin, an enzyme secreted solely in the kidney. In in vitro experiments, renin was shown to cleave both angiotensinogen and C3. Renin cleavage of C3 into C3a and C3b111 enables C3b to bind to factor B, which can then be cleaved into Ba and Bb by factor D112. This results in the formation of the C3bBb complex, which is the C3 convertase (Fig. 4a). Such renin-mediated C3 cleavage also occurs in the presence of serum111. Renin levels are higher in the kidney than in the circulation113. In patients with the complement-mediated kidney disease C3 glomerulopathy, the direct renin inhibitor aliskiren decreased complement activation systemically (as measured by higher levels of circulating C3 as well as decreased C3a and C5a levels) and in renal tissue (as evidenced by reduced C3 and C5b-9 deposition in glomeruli and reduced glomerular basement membrane thickness). Furthermore, patients treated with aliskiren exhibited stabilized or improved renal function and reduced proteinuria over a long follow-up period (Box 2). The effects of aliskiren could not be evaluated in a mouse model as murine renin does not cleave C3 (ref.111). However, a study using aliskiren in double transgenic rats that express human renin and angiotensinogen genes showed that aliskiren reduced both C3 deposition and C5b-9 deposition in renal sections114.
Renin-mediated C3 cleavage is sufficient to trigger activation of the complement system via the alternative pathway (Fig. 4a), but further activation of this pathway and amplification would, under physiological conditions, be inhibited by functional complement regulators in the fluid phase and on cell membranes. Furthermore, as renin has more than one substrate, it is plausible that the presence of angiotensinogen may limit the degree of C3 cleavage. In complement-mediated renal diseases such as C3 glomerulopathy, complement regulators have been found to be dysfunctional due to mutations, or the C3 convertase may be stabilized by the circulating autoantibody C3 nephritic factor115. This will allow uninhibited complement activation once renin-mediated C3 cleavage is triggered.
Thromboinflammation
Thromboinflammation is a condition that arises from an interaction between thrombosis and inflammation involving the endothelium, leukocytes and platelets as well as the complement system, the KKS/contact system, the coagulation system and the fibrinolytic system116,117. Thromboinflammation is a serious complication of severe infection, deep vein thrombosis, stroke and other inflammatory disorders116,118. FXIIa can promote inflammation by activating plasma kallikrein and inducing the KKS/contact system. The zymogen FXII is also involved in neutrophil migration as demonstrated in a mouse model of wound healing119 and, via intracellular AKT2 signalling, in neutrophil extracellular trap formation120. Furthermore, FXII can autoactivate to form FXIIa on DNA in neutrophil extracellular traps121. FXIIa is also involved in thrombosis by its role in activating the intrinsic pathway of coagulation (Fig. 3). Accordingly, mice that lack FXII are protected from collagen–adrenaline-induced or polyphosphate-induced thromboembolism and lethality48,122,123. Both FXII-deficient and plasma prekallikrein-deficient mice show reduced thrombosis after endothelial injury123,124.
Plasma prekallikrein-deficient mice exhibit lower levels of bradykinin, as less is generated from high-molecular-weight kininogen, as well as lower expression of B2R. These mice overexpress MAS to compensate for the reduced levels of B2R. The interaction of Ang 1–7 with MAS increases plasma levels of prostacyclin125, thereby increasing the expression of the transcription factors SIRT1 and KLF4 and reducing the expression of tissue factor, which lowers the risk of thrombosis123. B2R-deficient mice also show partial protection in models of endothelial injury-induced thrombosis due to AT2R and MAS overexpression125.
Even animals that lack high-molecular-weight kininogen are partially protected from thrombosis induced by endothelial cell injury124,126. This implies that for the initial induction of thrombus formation at subendothelial surfaces, complexes of high-molecular-weight kininogen with plasma prekallikrein or with FXI are important127,128.
In mouse models of stroke and other brain injuries, B1R deficiency or inhibition of B1R was shown to be beneficial129,130. Plasma kallikrein and FXIIa have thus become targets for antithrombotic treatment strategies without the risk of bleeding episodes121. Accordingly, the reduction or inhibition of plasma kallikrein or FXIIa by small molecules, peptides, antibodies, small interfering RNAs or antisense oligonucleotides has been shown to be protective in animal models of thrombosis and is currently being tested in clinical trials131,132. In addition to their antithrombotic action, such drugs would also have anti-inflammatory effects by inhibiting kinin generation. This may be particularly important in diseases characterized by thromboinflammation, a combination of neutrophil migration and activation that includes the release of neutrophil extracellular traps containing DNA, followed by platelet activation. Neutrophil migration can be enhanced by kinins133,134, and platelet activation by extracellular DNA is FXII dependent135. Thus, FXIIa or plasma kallikrein inhibition can reduce both neutrophil migration and platelet activation and thereby potentially ameliorate thromboinflammation118,120,136.
The complement system also plays an important role in thromboinflammation117. Complement activation on the endothelium leads to cellular activation and induces the expression of adhesion molecules, the release of cytokines and chemokines, a decrease in NO production, an increase in shedding of complement-coated endothelial microvesicles137 and the assembly of C5b-9, ultimately leading to cell lysis138. Tissue factor activity on the endothelium will lead to a procoagulant phenotype and is dependent on the generation of C5b-9 (ref.139). Moreover, C5b-9 deposition on the endothelium promotes the assembly of the prothrombinase complex and thrombin generation140. Thrombin can cleave both C3 and C5 and perpetuate complement activation141,142. In addition, complement activation on the endothelium triggers the release of von Willebrand factor (VWF)143 onto the vessel wall, which then promotes platelet adhesion, and VWF multimers form strings on the endothelium that serve as a platform for the deposition of more C3 via the alternative complement pathway144. Accordingly, high plasma levels of C3 have been associated with a risk of venous thromboembolism145.
The complement components C3 and C4 can bind to platelets146, C3b can bind to P-selectin and propagate complement activation147 and C5b-9 assembly can activate platelets148 and promote thrombin-mediated platelet aggregation149. C5b-9 deposition on the platelet membrane induces the release of platelet-derived microvesicles and results in a prothrombotic and procoagulant platelet surface137. Platelet microvesicles promote coagulation as they express both phosphatidylserine and tissue factor150. C3a and C5a are generated in the process of complement activation on platelets24, and C3a will further promote platelet activation151. C5a induces the release of thromboxane 4, an eicosanoid that is a potent platelet aggregator, from neutrophils70. Both C5a and C5b-9 contribute to platelet and neutrophil activation during extracorporeal circulation152. In turn, platelet activation in itself can induce complement activation on the cell surface both via the classical pathway and via the alternative pathway24,147. Importantly, uninhibited complement activation on the endothelium153 or on platelets154 will promote thrombosis and microangiopathy.
Vasculitis
Vasculitides are disorders characterized by severe inflammation of the vasculature with multi-organ damage affecting the kidneys, intestines, respiratory tract, skin, joints and other organs. Tissue injury involves endothelial cell damage and neutrophil-mediated inflammation155. The KKS and the complement system can both be activated during vasculitis. In patients with anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) and other vasculitides, plasma kallikrein activity and levels of kinins are increased, whereas plasma high-molecular-weight kininogen is degraded52,156,157,158. Neutrophil-derived proteases such as proteinase 3 can generate vasoactive kinin peptides that bind to the B1R52. Blocking B1R ameliorates vascular inflammation in animal models of glomerulonephritis and vasculitis159,160. B1R was detected on leukocyte-derived and endothelial cell-derived microvesicles in the circulation of patients with vasculitis133,161. The microvesicles were capable of transferring the receptor to recipient cells to induce KKS activation and, importantly, B1R on the vesicles served as a neutrophil chemoattractant, which could, thereby, promote inflammation. C1 inhibitor reduced the release of B1R-positive endothelial microvesicles in vitro133.
In patients with AAV, complement activation occurs primarily via the alternative pathway162. Circulating complement-coated endothelial microvesicles were detected during the acute phase of vasculitis163. Blocking B1R and B2R inhibited complement activation on microvesicles in an in vitro perfusion model. In a mouse model of nephrotoxic serum-induced glomerulonephritis, which mimics vasculitis, a B1R antagonist reduced glomerular complement deposition, a finding that could have potential therapeutic implications because complement activation on the endothelium is an important feature of vasculitides163. Sera from patients with AAV have also been shown to induce the shedding of tissue factor-positive microvesicles from neutrophils in vitro. Similarly, in vitro experiments showed that C5a-primed neutrophils treated with ANCA release tissue factor-positive microvesicles, an effect that is abrogated by treatment with a C5aR antagonist164. This suggests that complement activation contributes to excess coagulability in AAV. In patients with vasculitis, the small-molecule C5aR antagonist avacopan was shown to sustain remission165.
SARS-CoV-2 infection and inflammation
Severe SARS-CoV-2 infection is associated with multi-organ failure, primarily affecting the lungs, heart, liver and kidneys. Patients present with histopathological lesions indicative of endotheliitis, diffuse thrombotic microangiopathy and vasculitis166,167. The RAS has been of particular interest in this context as ACE2, in addition to its carboxypeptidase function in the non-canonical pathway of the RAS, is also the receptor for SARS-CoV-2 and the related virus SARS-CoV168. ACE2 is expressed on lung alveolar epithelial cells, as well as in many other organs9. Upon binding of the virus spike protein to ACE2, the virus–receptor complex is taken up by endocytosis. This leads to a downregulation of ACE2 on cell surfaces11 (Fig. 4c), where ACE2 should have a protective effect10, and a reduction in the generation of Ang 1–7. This results in unhindered activation of the canonical RAS pathway, without counterbalance by the non-canonical RAS pathway, with profound deleterious pro-inflammatory and prothrombotic effects in the lungs, heart and kidneys169.
ACE2 can inactivate DABK by cleaving off its C-terminal amino acid170. Therefore, decreased levels of ACE2 will result in increased DABK activity (Fig. 4c) and promote signalling via B1R and contribute to lung inflammation171. Since the availability of ACE2 is reduced in the lungs of patients with COVID-19, DABK may be stabilized and contribute to the pathogenesis of the disease. A case report of a patient who was successfully treated with recombinant ACE2, leading to markedly reduced Ang II and increased Ang 1–7 and Ang 1–9 levels, has been published172.
Conflicting data have been reported with regard to Ang II levels in patients with COVID-19. One study showed elevated levels of Ang II, correlating with the degree of lung injury173, a second study showed that Ang II levels were higher in severely ill patients174, whereas a third study did not detect altered levels of ACE2 and Ang II in serum175. These data may, however, not fully reflect cellular and tissue levels as gene expression profiles in cells from bronchoalveolar lavage fluid from patients with COVID-19 showed upregulation of ACE2, angiotensinogen, renin, AT1R and AT2R and downregulated ACE176. Upregulation of ACE2 seemingly conflicts with data showing that ACE2 expression is decreased on the surface of cultured cell lines during SARS-CoV uptake11. However, ACE2 is physiologically upregulated in conditions in which the effects of the canonical RAS pathway and Ang II are counterbalanced by the non-canonical RAS pathway, such as in hypertension, cardiomyopathy177, heart failure and chronic kidney disease178, and ACE2 upregulation may increase the risk of severe COVID-19 (ref.177). Moreover, ACE2 levels are elevated in the plasma of severely ill patients with COVID-19 (ref.179). Downregulation of ACE will theoretically lead to increased levels of bradykinin. Gene expression of high-molecular-weight kininogen, kallikreins and B1R and B2R was upregulated in cells from bronchoalveolar lavage fluid176. Activated products of the KKS might also play a pivotal role in the inflammatory processes in COVID-19 (refs180,181). Severely ill patients exhibited reduced plasma levels of high-molecular-weight kininogen, prekallikrein and factor XII (ref.182), which could potentially lead to oedematous lung injury183. The SERPING1 gene, which encodes C1 inhibitor, was downregulated in bronchioalveolar lavage fluid from these patients176, allowing KKS/contact system activation, and SARS-CoV proteins have been shown to interact with C1 inhibitor184. The combination of a decrease in ACE level and increased levels of AT1R, AT2R, B1R and B2R creates an imbalance that results in increased vascular permeability, bradykinin-driven inflammation and RAS/KKS-mediated thrombosis (Fig. 4c). KKS-mediated thromboinflammation may also be involved in this process given that increased platelet/neutrophil aggregates and augmented FXII activity have been observed in patients with severe COVID-19, which may cause embolisms, particularly in the lung185.
ACE inhibitors do not affect ACE2 activity186. Studies of animal models and patients treated with ACE inhibitors and Ang II receptor blockers (ARBs) have produced conflicting results with regard to the effect of these drugs on ACE2 expression, although studies have not explicitly addressed its expression on lung epithelium187. ACE inhibitors and ARBs may have a protective effect with regard to the pro-inflammatory and profibrotic effects induced by the canonical RAS pathway. Indeed, in mice injected with the SARS-CoV spike protein, RAS blockade attenuated lung injury11. Although the effects of ACE inhibitors and ARBs in COVID-19 are still being evaluated, there is a general consensus that these drugs should not be withdrawn in patients being treated for cardiovascular disease, diabetic nephropathy or hypertension187,188,189 and that they do not increase the risk of hospitalization190 or death191. ACE inhibitors and ARBs were shown to reduce the severity of COVID-19 in patients with hypertension192.
The complement system plays a prominent role in the immune response to viral infections during acute lung injury193,194, where it contributes to systemic inflammation, coagulopathy, thrombosis and multi-organ dysfunction. The nucleocapsid proteins of SARS-CoV and SARS-CoV-2 bind to MASP2, thereby activating the lectin pathway of the complement system195, and the viral spike proteins activate the alternative pathway of complement196. In vitro experiments showed that SARS-CoV-2-infected cells upregulate the transcription of genes encoding C3, factor B, C1s and C1r, and the increased expression of C3 and factor B was confirmed in bronchoalveolar lavage fluid from patients with COVID-19 (ref.197). Proteomic profiles of sera from patients with severe COVID-19 revealed elevated levels of complement proteins, including C5, C6, factor B and CPN198, as well as C5a or soluble C5b-9 (refs199,200,201,202). The levels of C3a and soluble C5b-9 were higher in patients with thromboembolic events203. Deposits of C3, C4, mannose-binding lectin (MBL), MASP2 and C5b-9 were demonstrated in the lungs, and deposits of C3 and MASP2 were demonstrated in the kidneys, of patients with severe COVID-19 (refs204,205). The severe inflammatory response to SARS-CoV-2 may be attributed, in part, to elevated levels of C5a, which acts as a neutrophil chemoattractant that can also contribute to inducing a cytokine storm and increase vascular permeability166,193. C1 inhibitor has been proposed to interact with SARS-CoV-2 proteins184, causing a physiological C1 inhibitor deficiency. Such a scenario would promote unbridled activation of t he plasma KKS/contact system and complement system (Fig. 4c), which, together with activation of tissue factor and the extrinsic coagulation pathway, would contribute to extensive thromboembolism.
Treatment with C1 inhibitor, narsoplimab (a MASP2 inhibitor), C3 inhibitor (AMY-101) or eculizumab (anti-C5) was effective in uncontrolled studies and in case reports of patients with COVID-19 (refs200,201,206,207,208,209,210). A small phase II trial of the C5a-targeted monoclonal antibody IFX-1 (vilobelimab) produced promising results211. Likewise, in case–control studies, blockade of B2R with icatibant was shown to be effective in a limited number of patients212,213, and use of the plasma kallikrein inhibitor lanadelumab or FXIIa-inhibiting drugs (Supplementary Box 1) have been proposed as potential therapies180,181. The timing of intervention is likely to be crucial, as inhibition of a fulminant inflammatory process once it has occurred may be less effective.
Conclusion
Severe inflammation is a complex condition involving multiple pathways and mediators. Blocking the RAS, the complement system or the plasma KKS/contact system individually may not be sufficient in hyperinflammatory disorders such as sepsis or COVID-19. The RAS, the complement system and the plasma KKS/contact system interact and contribute to the development of shock, severe cytokine storm and capillary leakage. The three pathways may either potentiate or counterbalance each other. The large number of interactions between components of the three systems lead us to suggest that the three systems actually constitute subdivisions of a joint protein cascade. Systemic and tissue inflammation can be effectively treated by blockade of the canonical RAS pathway or potentiation of the non-canonical RAS pathway, inhibition of complement activation via its three pathways and/or inhibition of the KKS at the level of enzymatic activation or receptor signalling. Understanding the intricate interactions between the three systems is of crucial importance for the treatment of inflammatory and infectious diseases as pharmacological interference with one system may have beneficial or deleterious effects in the other systems.
References
Santos, R. A. S. et al. The renin-angiotensin system: going beyond the classical paradigms. Am. J. Physiol. Heart Circ. Physiol. 316, H958–H970 (2019).
Noris, M. & Remuzzi, G. Overview of complement activation and regulation. Semin. Nephrol. 33, 479–492 (2013).
Colman, R. W. & Schmaier, A. H. Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood 90, 3819–3843 (1997). This is a seminal review of the contact system.
Forrester, S. J. et al. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol. Rev. 98, 1627–1738 (2018).
Li, F., Li, W., Farzan, M. & Harrison, S. C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309, 1864–1868 (2005).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e278 (2020).
Yeung, M. L. et al. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell 184, 2212–2228.e12 (2021).
Donoghue, M. et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 87, E1–E9 (2000).
Hamming, I. et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203, 631–637 (2004).
Imai, Y. et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436, 112–116 (2005). This study demonstrates the role of the canonical pathway in severe lung injury and the protective role of ACE2.
Kuba, K. et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 11, 875–879 (2005). This study provides evidence for ACE2 as the receptor for the SARS-CoV spike protein.
de Maat, S., de Mast, Q., Danser, A. H. J., van de Veerdonk, F. L. & Maas, C. Impaired breakdown of bradykinin and its metabolites as a possible cause for pulmonary edema in COVID-19 infection. Semin. Thromb. Hemost. 46, 835–837 (2020).
Patel, V. B. et al. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: a positive feedback mechanism in the RAS. J. Mol. Cell Cardiol. 66, 167–176 (2014).
Patel, V. B., Zhong, J. C., Grant, M. B. & Oudit, G. Y. Role of the ACE2/angiotensin 1–7 axis of the renin-angiotensin system in heart failure. Circ. Res. 118, 1313–1326 (2016).
Neubauer, B. et al. Angiotensin II short-loop feedback: is there a role of Ang II for the regulation of the renin system in vivo? Hypertension 71, 1075–1082 (2018).
Bader, M. Of mice and renin. Hypertension 70, 35–37 (2017).
Azizi, M. et al. Pharmacologic demonstration of the synergistic effects of a combination of the renin inhibitor aliskiren and the AT1 receptor antagonist valsartan on the angiotensin II-renin feedback interruption. J. Am. Soc. Nephrol. 15, 3126–3133 (2004).
Sevá Pessôa, B. et al. Key developments in renin-angiotensin-aldosterone system inhibition. Nat. Rev. Nephrol. 9, 26–36 (2013).
Galandrin, S. et al. Cardioprotective angiotensin-(1–7) peptide acts as a natural-biased ligand at the angiotensin II type 1 receptor. Hypertension 68, 1365–1374 (2016).
AbdAlla, S., Lother, H., Abdel-tawab, A. M. & Quitterer, U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J. Biol. Chem. 276, 39721–39726 (2001).
Leonhardt, J. et al. Evidence for heterodimerization and functional interaction of the angiotensin type 2 receptor and the receptor MAS. Hypertension 69, 1128–1135 (2017).
Arbore, G. et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science 352, aad1210 (2016).
Ghebrehiwet, B., Lim, B. L., Peerschke, E. I., Willis, A. C. & Reid, K. B. Isolation, cDNA cloning, and overexpression of a 33-kD cell surface glycoprotein that binds to the globular “heads” of C1q. J. Exp. Med. 179, 1809–1821 (1994).
Peerschke, E. I., Yin, W. & Ghebrehiwet, B. Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol. Immunol. 47, 2170–2175 (2010).
Lim, B. L. et al. The binding protein for globular heads of complement C1q, gC1qR. Functional expression and characterization as a novel vitronectin binding factor. J. Biol. Chem. 271, 26739–26744 (1996).
Ghebrehiwet, B., Geisbrecht, B. V., Xu, X., Savitt, A. G. & Peerschke, E. I. B. The C1q receptors: focus on gC1qR/p33 (C1qBP, p32, HABP-1)(1). Semin. Immunol. 45, 101338 (2019).
Yin, W., Ghebrehiwet, B., Weksler, B. & Peerschke, E. I. Classical pathway complement activation on human endothelial cells. Mol. Immunol. 44, 2228–2234 (2007).
Garred, P. et al. A journey through the lectin pathway of complement-MBL and beyond. Immunol. Rev. 274, 74–97 (2016).
Hess, K. et al. Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PLoS ONE 7, e35690 (2012).
Krarup, A., Wallis, R., Presanis, J. S., Gál, P. & Sim, R. B. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS ONE 2, e623 (2007).
La Bonte, L. R. et al. Mannose-binding lectin-associated serine protease-1 is a significant contributor to coagulation in a murine model of occlusive thrombosis. J. Immunol. 188, 885–891 (2012).
Nielsen, E. W. et al. Activation of the complement, coagulation, fibrinolytic and kallikrein-kinin systems during attacks of hereditary angioedema. Scand. J. Immunol. 44, 185–192 (1996).
Dobó, J. et al. Cleavage of kininogen and subsequent bradykinin release by the complement component: mannose-binding lectin-associated serine protease (MASP)-1. PLoS ONE 6, e20036 (2011).
Debreczeni, M. L. et al. MASP-1 increases endothelial permeability. Front. Immunol. 10, 991 (2019).
Ratnoff, O. D., Pensky, J., Ogston, D. & Naff, G. B. The inhibition of plasmin, plasma kallikrein, plasma permeability factor, and the C’1r subcomponent of the first component of complement by serum C’1 esterase inhibitor. J. Exp. Med. 129, 315–331 (1969).
Harpel, P. C., Lewin, M. F. & Kaplan, A. P. Distribution of plasma kallikrein between C-1 inactivator and alpha 2-macroglobulin in plasma utilizing a new assay for alpha 2-macroglobulin-kallikrein complexes. J. Biol. Chem. 260, 4257–4263 (1985).
Kaplan, A. P. & Ghebrehiwet, B. The plasma bradykinin-forming pathways and its interrelationships with complement. Mol. Immunol. 47, 2161–2169 (2010).
Hasan, A. A., Amenta, S. & Schmaier, A. H. Bradykinin and its metabolite, Arg-Pro-Pro-Gly-Phe, are selective inhibitors of alpha-thrombin-induced platelet activation. Circulation 94, 517–528 (1996).
Bhoola, K. D., Figueroa, C. D. & Worthy, K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol. Rev. 44, 1–80 (1992).
Kalinska, M., Meyer-Hoffert, U., Kantyka, T. & Potempa, J. Kallikreins - the melting pot of activity and function. Biochimie 122, 270–282 (2016).
Shariat-Madar, Z., Mahdi, F. & Schmaier, A. H. Identification and characterization of prolylcarboxypeptidase as an endothelial cell prekallikrein activator. J. Biol. Chem. 277, 17962–17969 (2002).
Joseph, K., Tholanikunnel, B. G. & Kaplan, A. P. Heat shock protein 90 catalyzes activation of the prekallikrein-kininogen complex in the absence of factor XII. Proc. Natl Acad. Sci. USA 99, 896–900 (2002).
Renné, T., Dedio, J., David, G. & Müller-Esterl, W. High molecular weight kininogen utilizes heparan sulfate proteoglycans for accumulation on endothelial cells. J. Biol. Chem. 275, 33688–33696 (2000).
Mahdi, F. et al. Mapping the interaction between high molecular mass kininogen and the urokinase plasminogen activator receptor. J. Biol. Chem. 279, 16621–16628 (2004).
Joseph, K., Ghebrehiwet, B., Peerschke, E. I., Reid, K. B. & Kaplan, A. P. Identification of the zinc-dependent endothelial cell binding protein for high molecular weight kininogen and factor XII: identity with the receptor that binds to the globular “heads” of C1q (gC1q-R). Proc. Natl Acad. Sci. USA 93, 8552–8557 (1996).
Joseph, K., Tholanikunnel, B. G., Ghebrehiwet, B. & Kaplan, A. P. Interaction of high molecular weight kininogen binding proteins on endothelial cells. Thromb. Haemost. 91, 61–70 (2004).
Maas, C. & Renné, T. Coagulation factor XII in thrombosis and inflammation. Blood 131, 1903–1909 (2018).
Müller, F. et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell 139, 1143–1156 (2009).
Dray, A. & Perkins, M. Bradykinin and inflammatory pain. Trends Neurosci. 16, 99–104 (1993).
Schmaier, A. H. The plasma kallikrein-kinin system counterbalances the renin-angiotensin system. J. Clin. Invest. 109, 1007–1009 (2002).
Stuardo, M. et al. Stimulated human neutrophils form biologically active kinin peptides from high and low molecular weight kininogens. J. Leukoc. Biol. 75, 631–640 (2004).
Kahn, R. et al. Neutrophil-derived proteinase 3 induces kallikrein-independent release of a novel vasoactive kinin. J. Immunol. 182, 7906–7915 (2009). This work shows that neutrophil-derived proteinase 3 cleaves high-molecular-weight kininogen, generating a vasoactive peptide, PR3-kinin, that binds to B1R, and this pathway circumvents kallikrein-mediated kinin release.
Regoli, D. & Barabé, J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 32, 1–46 (1980).
Marceau, F. et al. Bradykinin receptors: agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. Int. Immunopharmacol. 82, 106305 (2020).
Matthews, P. G. & Johnston, C. I. Responses of the renin-angiotensin system and kallikrein-kinin system to sodium and converting enzyme inhibitor (SQ 14,225). Adv. Exp. Med. Biol. 120b, 447–457 (1979).
Kuhr, F., Lowry, J., Zhang, Y., Brovkovych, V. & Skidgel, R. A. Differential regulation of inducible and endothelial nitric oxide synthase by kinin B1 and B2 receptors. Neuropeptides 44, 145–154 (2010).
Campbell, W. B. & Gauthier, K. M. Inducible endothelium-derived hyperpolarizing factor: role of the 15-lipoxygenase-EDHF pathway. J. Cardiovasc. Pharmacol. 61, 176–187 (2013).
Basei, F. L. et al. Endothelium dependent expression and underlying mechanisms of des-Arg9-bradykinin-induced B1R-mediated vasoconstriction in rat portal vein. Peptides 37, 216–224 (2012).
Bodor, C. et al. Angiotensin II increases the permeability and PV-1 expression of endothelial cells. Am. J. Physiol. Cell Physiol. 302, C267–C276 (2012).
Williams, B., Baker, A. Q., Gallacher, B. & Lodwick, D. Angiotensin II increases vascular permeability factor gene expression by human vascular smooth muscle cells. Hypertension 25, 913–917 (1995).
Oliveira, M. A., Fortes, Z. B., Santos, R. A., Kosla, M. C. & De Carvalho, M. H. Synergistic effect of angiotensin-(1–7) on bradykinin arteriolar dilation in vivo. Peptides 20, 1195–1201 (1999).
Gorelik, G., Carbini, L. A. & Scicli, A. G. Angiotensin 1–7 induces bradykinin-mediated relaxation in porcine coronary artery. J. Pharmacol. Exp. Ther. 286, 403–410 (1998).
Tsutsumi, Y. et al. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J. Clin. Invest. 104, 925–935 (1999).
Katada, J. & Majima, M. AT2 receptor-dependent vasodilation is mediated by activation of vascular kinin generation under flow conditions. Br. J. Pharmacol. 136, 484–491 (2002).
Siragy, H. M. & Carey, R. M. Protective role of the angiotensin AT2 receptor in a renal wrap hypertension model. Hypertension 33, 1237–1242 (1999). This study shows that in Ang II-mediated hypertension, AT2R counter-regulates blood pressure elevation via the renal bradykinin–NO–cGMP pathway.
Kemp, B. A. et al. AT2 receptor activation induces natriuresis and lowers blood pressure. Circ. Res. 115, 388–399 (2014).
Zuraw, B. L. & Christiansen, S. C. HAE pathophysiology and underlying mechanisms. Clin. Rev. Allergy Immunol. 51, 216–229 (2016).
Monsinjon, T. et al. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 17, 1003–1014 (2003).
Li, X. X., Lee, J. D., Kemper, C. & Woodruff, T. M. The complement receptor C5aR2: a powerful modulator of innate and adaptive immunity. J. Immunol. 202, 3339–3348 (2019).
Ward, P. A. The dark side of C5a in sepsis. Nat. Rev. Immunol. 4, 133–142 (2004).
Wu, F. et al. Complement component C3a plays a critical role in endothelial activation and leukocyte recruitment into the brain. J. Neuroinflammation 13, 23 (2016).
Schraufstatter, I. U., Trieu, K., Sikora, L., Sriramarao, P. & DiScipio, R. Complement C3a and C5a induce different signal transduction cascades in endothelial cells. J. Immunol. 169, 2102–2110 (2002).
Kourtzelis, I. et al. Complement anaphylatoxin C5a contributes to hemodialysis-associated thrombosis. Blood 116, 631–639 (2010).
Lim, H. W., He, D., Esquenazi-Behar, S., Yancey, K. B. & Soter, N. A. C5a, cutaneous mast cells, and inflammation: in vitro and in vivo studies in a murine model. J. Invest. Dermatol. 97, 305–311 (1991).
Miyabe, Y., Miyabe, C., Mani, V., Mempel, T. R. & Luster, A. D. Atypical complement receptor C5aR2 transports C5a to initiate neutrophil adhesion and inflammation. Sci. Immunol. 4, eaav5951 (2019).
Mollnes, T. E. & Huber-Lang, M. Complement in sepsis-when science meets clinics. FEBS Lett. 594, 2621–2632 (2020).
Schmitz, V. et al. C5a and bradykinin receptor cross-talk regulates innate and adaptive immunity in Trypanosoma cruzi infection. J. Immunol. 193, 3613–3623 (2014).
Nürnberger, W., Petrik, K., Burdach, S. & Göbel, U. C1 esterase inhibitor (C1-INH) can reduce plasma concentrations of the complement activation product C5a. Intensive Care Med. 20, 242 (1994).
Bossi, F. et al. Platelet-activating factor and kinin-dependent vascular leakage as a novel functional activity of the soluble terminal complement complex. J. Immunol. 173, 6921–6927 (2004). This work shows that both bradykinin and soluble C5b-9 induced vascular permeability and that the effect of soluble C5b-9 was inhibited by a B2R antagonist and a kallikrein inhibitor.
Bork, K. et al. Clinical features of genetically characterized types of hereditary angioedema with normal C1 inhibitor: a systematic review of qualitative evidence. Orphanet J. Rare Dis. 15, 289 (2020).
Germenis, A. E., Rijavec, M. & Veronez, C. L. Leveraging genetics for hereditary angioedema: a road map to precision medicine. Clin. Rev. Allergy Immunol. 60, 416–428 (2021).
Fields, T., Ghebrehiwet, B. & Kaplan, A. P. Kinin formation in hereditary angioedema plasma: evidence against kinin derivation from C2 and in support of “spontaneous” formation of bradykinin. J. Allergy Clin. Immunol. 72, 54–60 (1983).
De Maat, S., Hofman, Z. L. M. & Maas, C. Hereditary angioedema: the plasma contact system out of control. J. Thromb. Haemost. 16, 1674–1685 (2018).
Nussberger, J. et al. Plasma bradykinin in angio-oedema. Lancet 351, 1693–1697 (1998).
Kaplan, A. P. Enzymatic pathways in the pathogenesis of hereditary angioedema: the role of C1 inhibitor therapy. J. Allergy Clin. Immunol. 126, 918–925 (2010).
Martello, J. L., Woytowish, M. R. & Chambers, H. Ecallantide for treatment of acute attacks of hereditary angioedema. Am. J. Health Syst. Pharm. 69, 651–657 (2012).
Syed, Y. Y. Lanadelumab: a review in hereditary angioedema. Drugs 79, 1777–1784 (2019).
Aygören-Pürsün, E. et al. Oral plasma kallikrein inhibitor for prophylaxis in hereditary angioedema. N. Engl. J. Med. 379, 352–362 (2018).
Benigni, A., Cassis, P. & Remuzzi, G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol. Med. 2, 247–257 (2010).
Li, R. et al. Long-term stimulation of angiotensin II induced endothelial senescence and dysfunction. Exp. Gerontol. 119, 212–220 (2019).
Muller, D. N. et al. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am. J. Pathol. 161, 1679–1693 (2002).
Santos, R. A. S. et al. The ACE2/angiotensin-(1-7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1–7). Physiol. Rev. 98, 505–553 (2018). This is a comprehensive review of the protective effects of the non-canonical pathway of the RAS in tissues.
Gonzalez, L. et al. Angiotensin-(1–9) reduces cardiovascular and renal inflammation in experimental renin-independent hypertension. Biochem. Pharmacol. 156, 357–370 (2018).
Tom, B., de Vries, R., Saxena, P. R. & Danser, A. H. Bradykinin potentiation by angiotensin-(1–7) and ACE inhibitors correlates with ACE C- and N-domain blockade. Hypertension 38, 95–99 (2001).
Marques, F. D. et al. An oral formulation of angiotensin-(1–7) produces cardioprotective effects in infarcted and isoproterenol-treated rats. Hypertension 57, 477–483 (2011).
Jackman, H. L. et al. Angiotensin 1–9 and 1–7 release in human heart: role of cathepsin A. Hypertension 39, 976–981 (2002).
Marcic, B., Deddish, P. A., Jackman, H. L. & Erdös, E. G. Enhancement of bradykinin and resensitization of its B2 receptor. Hypertension 33, 835–843 (1999).
Walther, T. et al. AT1 receptor blockade increases cardiac bradykinin via neutral endopeptidase after induction of myocardial infarction in rats. FASEB J. 16, 1237–1241 (2002).
Kaschina, E., Namsolleck, P. & Unger, T. AT2 receptors in cardiovascular and renal diseases. Pharmacol. Res. 125, 39–47 (2017).
Jalowy, A., Schulz, R., Dörge, H., Behrends, M. & Heusch, G. Infarct size reduction by AT1-receptor blockade through a signal cascade of AT2-receptor activation, bradykinin and prostaglandins in pigs. J. Am. Coll. Cardiol. 32, 1787–1796 (1998).
Yang, X. P. et al. Diminished cardioprotective response to inhibition of angiotensin-converting enzyme and angiotensin II type 1 receptor in B2 kinin receptor gene knockout mice. Circ. Res. 88, 1072–1079 (2001).
Tanaka, M. et al. Vascular response to angiotensin II is exaggerated through an upregulation of AT1 receptor in AT2 knockout mice. Biochem. Biophys. Res. Commun. 258, 194–198 (1999).
de Carvalho Santuchi, M. et al. Angiotensin-(1–7) and alamandine promote anti-inflammatory response in macrophages in vitro and in vivo. Mediators Inflamm. 2019, 2401081 (2019).
Arendse, L. B. et al. Novel therapeutic approaches targeting the renin-angiotensin system and associated peptides in hypertension and heart failure. Pharmacol. Rev. 71, 539–570 (2019).
Paz Ocaranza, M. et al. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 17, 116–129 (2020).
Solomon, S. D. et al. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N. Engl. J. Med. 381, 1609–1620 (2019).
Haschke, M. et al. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin. Pharmacokinet. 52, 783–792 (2013).
Romero, C. A., Orias, M. & Weir, M. R. Novel RAAS agonists and antagonists: clinical applications and controversies. Nat. Rev. Endocrinol. 11, 242–252 (2015).
Zhang, C. et al. Complement 5a receptor mediates angiotensin II-induced cardiac inflammation and remodeling. Arterioscler. Thromb. Vasc. Biol. 34, 1240–1248 (2014).
Chen, X. H. et al. Deficiency of complement C3a and C5a receptors prevents angiotensin II-induced hypertension via regulatory T cells. Circ. Res. 122, 970–983 (2018).
Békássy, Z. D. et al. Aliskiren inhibits renin-mediated complement activation. Kidney Int. 94, 689–700 (2018). This work demonstrates for the first time that C3 is a renin substrate and that renin inhibition had a beneficial effect in complement-mediated renal disease.
Forneris, F. et al. Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science 330, 1816–1820 (2010).
Gerdts, K. G., Shah, V., Savage, J. M. & Dillon, M. J. Renal vein renin measurements in normotensive children. J. Pediatr. 95, 953–958 (1979).
Shagdarsuren, E. et al. Complement activation in angiotensin II-induced organ damage. Circ. Res. 97, 716–724 (2005).
Smith, R. J. H. et al. C3 glomerulopathy — understanding a rare complement-driven renal disease. Nat. Rev. Nephrol. 15, 129–143 (2019).
Schattner, M., Jenne, C. N., Negrotto, S. & Ho-Tin-Noe, B. Editorial: platelets and immune responses during thromboinflammation. Front. Immunol. 11, 1079 (2020).
Ekdahl, K. N. et al. Dangerous liaisons: complement, coagulation, and kallikrein/kinin cross-talk act as a linchpin in the events leading to thromboinflammation. Immunol. Rev. 274, 245–269 (2016).
Göb, E. et al. Blocking of plasma kallikrein ameliorates stroke by reducing thromboinflammation. Ann. Neurol. 77, 784–803 (2015).
Stavrou, E. X. et al. Factor XII and uPAR upregulate neutrophil functions to influence wound healing. J. Clin. Invest. 128, 944–959 (2018).
Schmaier, A. H. & Stavrou, E. X. Factor XII — what’s important but not commonly thought about. Res. Pract. Thromb. Haemost. 3, 599–606 (2019).
Renné, T. & Stavrou, E. X. Roles of factor XII in innate immunity. Front. Immunol. 10, 2011 (2019).
Nickel, K. F. et al. The polyphosphate-factor XII pathway drives coagulation in prostate cancer-associated thrombosis. Blood 126, 1379–1389 (2015).
Stavrou, E. X. et al. Reduced thrombosis in Klkb1−/− mice is mediated by increased Mas receptor, prostacyclin, Sirt1, and KLF4 and decreased tissue factor. Blood 125, 710–719 (2015). This study shows a novel mechanism of kallikrein-mediated thrombosis whereby its decrease increases expression of the receptor MAS.
Kokoye, Y. et al. A comparison of the effects of factor XII deficiency and prekallikrein deficiency on thrombus formation. Thromb. Res. 140, 118–124 (2016).
Fang, C. et al. Angiotensin 1–7 and Mas decrease thrombosis in Bdkrb2−/− mice by increasing NO and prostacyclin to reduce platelet spreading and glycoprotein VI activation. Blood 121, 3023–3032 (2013).
Merkulov, S. et al. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood 111, 1274–1281 (2008).
Wiggins, R. C., Bouma, B. N., Cochrane, C. G. & Griffin, J. H. Role of high-molecular-weight kininogen in surface-binding and activation of coagulation factor XI and prekallikrein. Proc. Natl Acad. Sci. USA 74, 4636–4640 (1977).
Thompson, R. E., Mandle, R. Jr & Kaplan, A. P. Studies of binding of prekallikrein and factor XI to high molecular weight kininogen and its light chain. Proc. Natl Acad. Sci. USA 76, 4862–4866 (1979).
Austinat, M. et al. Blockade of bradykinin receptor B1 but not bradykinin receptor B2 provides protection from cerebral infarction and brain edema. Stroke 40, 285–293 (2009).
Raslan, F. et al. Inhibition of bradykinin receptor B1 protects mice from focal brain injury by reducing blood-brain barrier leakage and inflammation. J. Cereb. Blood Flow. Metab. 30, 1477–1486 (2010).
Wilbs, J. et al. Cyclic peptide FXII inhibitor provides safe anticoagulation in a thrombosis model and in artificial lungs. Nat. Commun. 11, 3890 (2020).
DeLoughery, E. P., Olson, S. R., Puy, C., McCarty, O. J. T. & Shatzel, J. J. The safety and efficacy of novel agents targeting factors XI and XII in early phase human trials. Semin. Thromb. Hemost. 45, 502–508 (2019).
Mossberg, M. et al. C1-inhibitor decreases the release of vasculitis-like chemotactic endothelial microvesicles. J. Am. Soc. Nephrol. 28, 2472–2481 (2017). This study shows that B1R-positive endothelial microvesicles are chemotactic for neutrophils and that this effect could be reduced by C1 inhibitor.
Paegelow, I., Trzeczak, S., Böckmann, S. & Vietinghoff, G. Migratory responses of polymorphonuclear leukocytes to kinin peptides. Pharmacology 66, 153–161 (2002).
Gould, T. J. et al. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler. Thromb. Vasc. Biol. 34, 1977–1984 (2014).
Larsson, M. et al. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci. Transl. Med. 6, 222ra217 (2014).
Sims, P. J. & Wiedmer, T. Induction of cellular procoagulant activity by the membrane attack complex of complement. Semin. Cell Biol. 6, 275–282 (1995).
Karpman, D. & Tati, R. Complement activation in thrombotic microangiopathy. Hamostaseologie 33, 96–104 (2013).
Saadi, S., Holzknecht, R. A., Patte, C. P., Stern, D. M. & Platt, J. L. Complement-mediated regulation of tissue factor activity in endothelium. J. Exp. Med. 182, 1807–1814 (1995).
Hamilton, K. K., Hattori, R., Esmon, C. T. & Sims, P. J. Complement proteins C5b-9 induce vesiculation of the endothelial plasma membrane and expose catalytic surface for assembly of the prothrombinase enzyme complex. J. Biol. Chem. 265, 3809–3814 (1990).
Huber-Lang, M. et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat. Med. 12, 682–687 (2006).
Amara, U. et al. Molecular intercommunication between the complement and coagulation systems. J. Immunol. 185, 5628–5636 (2010).
Hattori, R., Hamilton, K. K., McEver, R. P. & Sims, P. J. Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J. Biol. Chem. 264, 9053–9060 (1989).
Tati, R. et al. Complement activation associated with ADAMTS13 deficiency in human and murine thrombotic microangiopathy. J. Immunol. 191, 2184–2193 (2013).
Nørgaard, I., Nielsen, S. F. & Nordestgaard, B. G. Complement C3 and high risk of venous thromboembolism: 80517 individuals from the Copenhagen General Population Study. Clin. Chem. 62, 525–534 (2016).
Karpman, D., Manea, M., Vaziri-Sani, F., Ståhl, A. L. & Kristoffersson, A. C. Platelet activation in hemolytic uremic syndrome. Semin. Thromb. Hemost. 32, 128–145 (2006).
Del Conde, I., Crúz, M. A., Zhang, H., López, J. A. & Afshar-Kharghan, V. Platelet activation leads to activation and propagation of the complement system. J. Exp. Med. 201, 871–879 (2005).
Zimmerman, T. S. & Kolb, W. P. Human platelet-initiated formation and uptake of the C5-9 complex of human complement. J. Clin. Invest. 57, 203–211 (1976).
Polley, M. J. & Nachman, R. L. Human complement in thrombin-mediated platelet function: uptake of the C5b-9 complex. J. Exp. Med. 150, 633–645 (1979).
Ståhl, A. L., Sartz, L., Nelsson, A., Békássy, Z. D. & Karpman, D. Shiga toxin and lipopolysaccharide induce platelet-leukocyte aggregates and tissue factor release, a thrombotic mechanism in hemolytic uremic syndrome. PLoS ONE 4, e6990 (2009).
Polley, M. J. & Nachman, R. L. Human platelet activation by C3a and C3a des-arg. J. Exp. Med. 158, 603–615 (1983).
Rinder, C. S. et al. Blockade of C5a and C5b-9 generation inhibits leukocyte and platelet activation during extracorporeal circulation. J. Clin. Invest. 96, 1564–1572 (1995).
Manuelian, T. et al. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J. Clin. Invest. 111, 1181–1190 (2003).
Ståhl, A. L. et al. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood 111, 5307–5315 (2008).
Michailidou, D., Mustelin, T. & Lood, C. Role of neutrophils in systemic vasculitides. Front. Immunol. 11, 619705 (2020).
Bleian, G. T., Pogosian, A. S., Stepanian, M. A. & Osipova, E. N. [Role of the blood kinin system in the pathogenesis of the hemorrhagic syndrome in hemorrhagic vasculitis]. Vopr. Med. Khim 24, 779–781 (1978).
Yamada, T., Harber, P., Pettit, G. W., Wing, D. A. & Oster, C. N. Activation of the kallikrein-kinin system in Rocky Mountain spotted fever. Ann. Intern. Med. 88, 764–768 (1978).
Kahn, R. et al. Contact-system activation in children with vasculitis. Lancet 360, 535–541 (2002).
Klein, J. et al. Blockade of the kinin B1 receptor ameloriates glomerulonephritis. J. Am. Soc. Nephrol. 21, 1157–1164 (2010).
Hu, P. et al. Kinin B1 receptor is important in the pathogenesis of myeloperoxidase-specific ANCA GN. J. Am. Soc. Nephrol. 31, 297–307 (2020). This study demonstrates the role of B1R for inflammation in ANCA-associated glomerulonephritis.
Kahn, R. et al. Microvesicle transfer of kinin B1-receptors is a novel inflammatory mechanism in vasculitis. Kidney Int. 91, 96–105 (2017). This work shows that circulating leukocyte-derived microvesicles can transfer B1R to cells and thereby promote kinin-mediated inflammation.
Chen, M., Jayne, D. R. W. & Zhao, M. H. Complement in ANCA-associated vasculitis: mechanisms and implications for management. Nat. Rev. Nephrol. 13, 359–367 (2017).
Lopatko Fagerström, I. et al. Blockade of the kallikrein-kinin system reduces endothelial complement activation in vascular inflammation. EBioMedicine 47, 319–328 (2019). This study shows that blockade of kinin receptors blocked endothelial complement deposition in vitro and in vivo.
Huang, Y. M., Wang, H., Wang, C., Chen, M. & Zhao, M. H. Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol. 67, 2780–2790 (2015).
Jayne, D. R. W., Merkel, P. A., Schall, T. J. & Bekker, P. Avacopan for the treatment of ANCA-associated vasculitis. N. Engl. J. Med. 384, 599–609 (2021).
Noris, M., Benigni, A. & Remuzzi, G. The case of complement activation in COVID-19 multiorgan impact. Kidney Int. 98, 314–322 (2020).
Varga, Z. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 395, 1417–1418 (2020).
Yan, R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448 (2020).
Henry, B. M., Vikse, J., Benoit, S., Favaloro, E. J. & Lippi, G. Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: a novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin. Chim. Acta 507, 167–173 (2020).
Vickers, C. et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 277, 14838–14843 (2002). This article comprehensively lists ACE2 substrates.
Sodhi, C. P. et al. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell Mol. Physiol. 314, L17–L31 (2018).
Zoufaly, A. et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir. Med. 8, 1154–1158 (2020).
Liu, Y. et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 63, 364–374 (2020).
Wu, Z. et al. Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID-19 patients. Crit. Care 24, 290 (2020).
Rieder, M. et al. Serum ACE-2, angiotensin II, and aldosterone levels are unchanged in patients with COVID-19. Am. J. Hypertens 34, 278–281 (2020).
Garvin, M. R. et al. A mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. eLife 9, e591747 (2020).
Bos, J. M. et al. Marked up-regulation of ACE2 in hearts of patients with obstructive hypertrophic cardiomyopathy: implications for SARS-CoV-2-mediated COVID-19. Mayo Clin. Proc. 95, 1354–1368 (2020).
Dean, A. Q. et al. The fight against COVID-19: striking a balance in the renin-angiotensin system. Drug Discov. Today 26, 2214–2220 (2021).
Reindl-Schwaighofer, R. et al. ACE2 elevation in severe COVID-19. Am. J. Respir. Crit. Care Med. 203, 1191–1196 (2021).
Meini, S. et al. Understanding the pathophysiology of COVID-19: could the contact system be the key? Front. Immunol. 11, 2014 (2020).
van de Veerdonk, F. L. et al. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. eLife 9, e57555 (2020).
Lipcsey, M. et al. The outcome of critically ill COVID-19 patients is linked to thromboinflammation dominated by the kallikrein/kinin system. Front. Immunol. 12, 627579 (2021).
Kaplan, A. P. & Ghebrehiwet, B. Pathways for bradykinin formation and interrelationship with complement as a cause of edematous lung in COVID-19 patients. J. Allergy Clin. Immunol. 147, 507–509 (2021).
Thomson, T. M., Toscano-Guerra, E., Casis, E. & Paciucci, R. C1 esterase inhibitor and the contact system in COVID-19. Br. J. Haematol. 190, 520–524 (2020).
Taus, F. et al. Platelets promote thromboinflammation in SARS-CoV-2 pneumonia. Arterioscler. Thromb. Vasc. Biol. 40, 2975–2989 (2020).
Rice, G. I., Thomas, D. A., Grant, P. J., Turner, A. J. & Hooper, N. M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 383, 45–51 (2004).
Vaduganathan, M. et al. Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N. Engl. J. Med. 382, 1653–1659 (2020).
Chung, M. K. et al. SARS-CoV-2 and ACE2: the biology and clinical data settling the ARB and ACEI controversy. EBioMedicine 58, 102907 (2020).
Chan, C. K. et al. Renin-angiotensin-aldosterone system inhibitors and risks of severe acute respiratory syndrome coronavirus 2 infection: a systematic review and meta-analysis. Hypertension 76, 1563–1571 (2020).
Dublin, S. et al. Renin-angiotensin-aldosterone system inhibitors and COVID-19 infection or hospitalization: a cohort study. Am. J. Hypertens. 34, 339–347 (2020).
Trifirò, G. et al. Renin-angiotensin-aldosterone system inhibitors and risk of death in patients hospitalised with COVID-19: a retrospective Italian cohort study of 43,000 patients. Drug. Saf. 43, 1297–1308 (2020).
Semenzato, L. et al. Antihypertensive drugs and COVID-19 risk: a cohort study of 2 million hypertensive patients. Hypertension 77, 833–842 (2021).
Wang, R., Xiao, H., Guo, R., Li, Y. & Shen, B. The role of C5a in acute lung injury induced by highly pathogenic viral infections. Emerg. Microbes Infect. 4, e28 (2015).
Holter, J. C. et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl Acad. Sci. USA 117, 25018–25025 (2020).
Gao, T. et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv https://doi.org/10.1101/2020.03.29.20041962 (2020).
Yu, J. et al. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood 136, 2080–2089 (2020).
Yan, B. et al. SARS-CoV-2 drives JAK1/2-dependent local complement hyperactivation. Sci. Immunol. 6, eabg0833 (2021).
Shen, B. et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 182, 59–72.e15 (2020).
Cugno, M. et al. Complement activation in patients with COVID-19: a novel therapeutic target. J. Allergy Clin. Immunol. 146, 215–217 (2020).
Urwyler, P. et al. Treatment of COVID-19 with conestat alfa, a regulator of the complement, contact activation and kallikrein-kinin system. Front. Immunol. 11, 2072 (2020).
Regis Peffault de, L. et al. Complement C5 inhibition in patients with COVID-19 — a promising target? Haematologica 105, 2847–2850 (2020).
Diorio, C. et al. Evidence of thrombotic microangiopathy in children with SARS-CoV-2 across the spectrum of clinical presentations. Blood Adv. 4, 6051–6063 (2020).
de Nooijer, A. H. et al. Complement activation in the disease course of coronavirus disease 2019 and its effects on clinical outcomes. J. Infect. Dis. 223, 214–224 (2021).
Magro, C. et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl. Res. 220, 1–13 (2020).
Pfister, F. et al. Complement activation in kidneys of patients with COVID-19. Front. Immunol. 11, 594849 (2020).
Rambaldi, A. et al. Endothelial injury and thrombotic microangiopathy in COVID-19: treatment with the lectin-pathway inhibitor narsoplimab. Immunobiology 225, 152001 (2020).
Annane, D. et al. Eculizumab as an emergency treatment for adult patients with severe COVID-19 in the intensive care unit: a proof-of-concept study. EClinicalMedicine 28, 100590 (2020).
Mahajan, R., Lipton, M., Broglie, L., Jain, N. G. & Uy, N. S. Eculizumab treatment for renal failure in a pediatric patient with COVID-19. J. Nephrol. 33, 1373–1376 (2020).
Mastellos, D. C. et al. Complement C3 vs C5 inhibition in severe COVID-19: early clinical findings reveal differential biological efficacy. Clin. Immunol. 220, 108598 (2020).
Diurno, F. et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur. Rev. Med. Pharmacol. Sci. 24, 4040–4047 (2020).
Vlaar, A. P. J. et al. Anti-C5a antibody IFX-1 (vilobelimab) treatment versus best supportive care for patients with severe COVID-19 (PANAMO): an exploratory, open-label, phase 2 randomised controlled trial. Lancet Rheumatol. 2, e764–e773 (2020).
van de Veerdonk, F. L. et al. Outcomes associated with use of a kinin B2 receptor antagonist among patients with COVID-19. JAMA Netw. Open 3, e2017708 (2020).
Mansour, E. et al. Safety and outcomes associated with the pharmacological inhibition of the kinin-kallikrein system in severe COVID-19. Viruses 13, 309 (2021).
Nabah, Y. N. et al. Angiotensin II induces neutrophil accumulation in vivo through generation and release of CXC chemokines. Circulation 110, 3581–3586 (2004).
Senchenkova, E. Y., Russell, J., Almeida-Paula, L. D., Harding, J. W. & Granger, D. N. Angiotensin II-mediated microvascular thrombosis. Hypertension 56, 1089–1095 (2010).
Vaughan, D. E. Angiotensin and vascular fibrinolytic balance. Am. J. Hypertens. 15, 3s–8s (2002).
Yugandhar, V. G. & Clark, M. A. Angiotensin III: a physiological relevant peptide of the renin angiotensin system. Peptides 46, 26–32 (2013).
Ruiz-Ortega, M., Esteban, V. & Egido, J. The regulation of the inflammatory response through nuclear factor- κb pathway by angiotensin IV extends the role of the renin angiotensin system in cardiovascular diseases. Trends Cardiovasc. Med. 17, 19–25 (2007).
Wang, Q. G. et al. Angiotensin IV suppresses inflammation in the brains of rats with chronic cerebral hypoperfusion. J. Renin Angiotensin Aldosterone Syst. 19, 1470320318799587 (2018).
Ocaranza, M. P. et al. Recent insights and therapeutic perspectives of angiotensin-(1–9) in the cardiovascular system. Clin. Sci. 127, 549–557 (2014).
Jankowski, V. et al. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler. Thromb. Vasc. Biol. 27, 297–302 (2007).
Lautner, R. Q. et al. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ. Res. 112, 1104–1111 (2013).
Björk, J., Hugli, T. E. & Smedegård, G. Microvascular effects of anaphylatoxins C3a and C5a. J. Immunol. 134, 1115–1119 (1985).
Klos, A. et al. The role of the anaphylatoxins in health and disease. Mol. Immunol. 46, 2753–2766 (2009).
Propson, N. E., Roy, E. R., Litvinchuk, A., Köhl, J. & Zheng, H. Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J. Clin. Invest. 131, e140966 (2021).
Sauter, R. J. et al. Functional relevance of the anaphylatoxin receptor C3aR for platelet function and arterial thrombus formation marks an intersection point between innate immunity and thrombosis. Circulation 138, 1720–1735 (2018).
Wang, H., Ricklin, D. & Lambris, J. D. Complement-activation fragment C4a mediates effector functions by binding as untethered agonist to protease-activated receptors 1 and 4. Proc. Natl Acad. Sci. USA 114, 10948–10953 (2017).
Morgan, B. P. The membrane attack complex as an inflammatory trigger. Immunobiology 221, 747–751 (2016).
Bossi, F., Peerschke, E. I., Ghebrehiwet, B. & Tedesco, F. Cross-talk between the complement and the kinin system in vascular permeability. Immunol. Lett. 140, 7–13 (2011).
Marceau, F., Hess, J. F. & Bachvarov, D. R. The B1 receptors for kinins. Pharmacol. Rev. 50, 357–386 (1998).
Balaguer, J. M. et al. Contribution of endogenous bradykinin to fibrinolysis, inflammation, and blood product transfusion following cardiac surgery: a randomized clinical trial. Clin. Pharmacol. Ther. 93, 326–334 (2013).
Ehrenfeld, P. et al. Activation of kinin B1 receptors induces chemotaxis of human neutrophils. J. Leukoc. Biol. 80, 117–124 (2006).
Ehrenfeld, P. et al. Kinin B1 receptor activation turns on exocytosis of matrix metalloprotease-9 and myeloperoxidase in human neutrophils: involvement of mitogen-activated protein kinase family. J. Leukoc. Biol. 86, 1179–1189 (2009).
Fang, C. & Schmaier, A. H. Novel anti-thrombotic mechanisms mediated by Mas receptor as result of balanced activities between the kallikrein/kinin and the renin-angiotensin systems. Pharmacol. Res. 160, 105096 (2020).
Tidjane, N. et al. A primary role for kinin B1 receptor in inflammation, organ damage, and lethal thrombosis in a rat model of septic shock in diabetes. Eur. J. Inflamm. 13, 40–52 (2015).
Cruden, N. L. et al. B1 kinin receptor does not contribute to vascular tone or tissue plasminogen activator release in the peripheral circulation of patients with heart failure. Arterioscler. Thromb. Vasc. Biol. 25, 772–777 (2005).
Werle, E. & Schievelbein, H. Activity of nicotine and inactivity of kallikrein and kallidin in aggregation of blood platelets. Nature 207, 871–872 (1965).
Muenstermann, M. et al. Distinct roles of the anaphylatoxin receptors C3aR, C5aR1 and C5aR2 in experimental meningococcal infections. Virulence 10, 677–694 (2019).
Barnum, S. R. C4a: an anaphylatoxin in name only. J. Innate Immun. 7, 333–339 (2015).
Moniwa, N. et al. Primacy of angiotensin converting enzyme in angiotensin-(1–12) metabolism. Am. J. Physiol. Heart Circ. Physiol. 305, H644–H650 (2013).
Reilly, C. F., Schechter, N. B. & Travis, J. Inactivation of bradykinin and kallidin by cathepsin G and mast cell chymase. Biochem. Biophys. Res. Commun. 127, 443–449 (1985).
Ahmad, S. et al. Chymase-dependent generation of angiotensin II from angiotensin-(1–12) in human atrial tissue. PLoS ONE 6, e28501 (2011).
Velez, J. C. et al. Angiotensin I is largely converted to angiotensin (1–7) and angiotensin (2–10) by isolated rat glomeruli. Hypertension 53, 790–797 (2009).
Kokkonen, J. O., Kuoppala, A., Saarinen, J., Lindstedt, K. A. & Kovanen, P. T. Kallidin- and bradykinin-degrading pathways in human heart: degradation of kallidin by aminopeptidase M-like activity and bradykinin by neutral endopeptidase. Circulation 99, 1984–1990 (1999).
Pelorosso, F. G., Brodsky, P. T., Zold, C. L. & Rothlin, R. P. Potentiation of des-Arg9-kallidin-induced vasoconstrictor responses by metallopeptidase inhibition in isolated human umbilical artery. J. Pharmacol. Exp. Ther. 313, 1355–1360 (2005).
Sandén, C., Enquist, J., Bengtson, S. H., Herwald, H. & Leeb-Lundberg, L. M. Kinin B2 receptor-mediated bradykinin internalization and metalloendopeptidase EP24.15-dependent intracellular bradykinin degradation. J. Pharmacol. Exp. Ther. 326, 24–32 (2008).
Odya, C. E., Marinkovic, D. V., Hammon, K. J., Stewart, T. A. & Erdös, E. G. Purification and properties of prolylcarboxypeptidase (angiotensinase C) from human kidney. J. Biol. Chem. 253, 5927–5931 (1978).
Chajkowski, S. M. et al. Highly selective hydrolysis of kinins by recombinant prolylcarboxypeptidase. Biochem. Biophys. Res. Commun. 405, 338–343 (2011).
Ideishi, M., Ikeda, M. & Arakawa, K. Direct angiotensin II formation by rat submandibular gland kallikrein. J. Biochem. 102, 859–868 (1987).
Sasaguri, M. et al. Purification and characterization of a kinin- and angiotensin II-forming enzyme in the dog heart. J. Hypertens. 15, 675–682 (1997).
Oikonomopoulou, K. et al. Induction of complement C3a receptor responses by kallikrein-related peptidase 14. J. Immunol. 191, 3858–3866 (2013).
Irmscher, S. et al. Kallikrein cleaves C3 and activates complement. J. Innate Immun. 10, 94–105 (2018).
DiScipio, R. G. The activation of the alternative pathway C3 convertase by human plasma kallikrein. Immunology 45, 587–595 (1982).
Ghebrehiwet, B., Randazzo, B. P., Dunn, J. T., Silverberg, M. & Kaplan, A. P. Mechanisms of activation of the classical pathway of complement by Hageman factor fragment. J. Clin. Invest. 71, 1450–1456 (1983).
Skidgel, R. A., Davis, R. M. & Tan, F. Human carboxypeptidase M. Purification and characterization of a membrane-bound carboxypeptidase that cleaves peptide hormones. J. Biol. Chem. 264, 2236–2241 (1989).
Bokisch, V. A. & Müller-Eberhard, H. J. Anaphylatoxin inactivator of human plasma: its isolation and characterization as a carboxypeptidase. J. Clin. Invest. 49, 2427–2436 (1970).
Campbell, W. D., Lazoura, E., Okada, N. & Okada, H. Inactivation of C3a and C5a octapeptides by carboxypeptidase R and carboxypeptidase N. Microbiol. Immunol. 46, 131–134 (2002).
Plug, T. & Meijers, J. C. Structure-function relationships in thrombin-activatable fibrinolysis inhibitor. J. Thromb. Haemost. 14, 633–644 (2016).
Krop, M. & Danser, A. H. Circulating versus tissue renin-angiotensin system: on the origin of (pro)renin. Curr. Hypertens. Rep. 10, 112–118 (2008).
Kurtz, A. Control of renin synthesis and secretion. Am. J. Hypertens. 25, 839–847 (2012).
Ichihara, A. & Yatabe, M. S. The (pro)renin receptor in health and disease. Nat. Rev. Nephrol. 15, 693–712 (2019).
Ahmad, S. et al. Angiotensin-(1–12): a chymase-mediated cellular angiotensin II substrate. Curr. Hypertens. Rep. 16, 429 (2014).
Park, B. M., Cha, S. A., Lee, S. H. & Kim, S. H. Angiotensin IV protects cardiac reperfusion injury by inhibiting apoptosis and inflammation via AT4R in rats. Peptides 79, 66–74 (2016).
Kramár, E. A., Krishnan, R., Harding, J. W. & Wright, J. W. Role of nitric oxide in angiotensin IV-induced increases in cerebral blood flow. Regul. Pept. 74, 185–192 (1998).
Tetzner, A. et al. G-protein-coupled receptor MrgD is a receptor for angiotensin-(1–7) involving adenylyl cyclase, cAMP, and phosphokinase A. Hypertension 68, 185–194 (2016).
de Gasparo, M., Catt, K. J., Inagami, T., Wright, J. W. & Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 52, 415–472 (2000).
Revak, S. D., Cochrane, C. G., Bouma, B. N. & Griffin, J. H. Surface and fluid phase activities of two forms of activated Hageman factor produced during contact activation of plasma. J. Exp. Med. 147, 719–729 (1978).
Leeb-Lundberg, L. M., Marceau, F., Müller-Esterl, W., Pettibone, D. J. & Zuraw, B. L. International Union of Pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol. Rev. 57, 27–77 (2005). This is a comprehensive review of kinin receptors.
Yosipiv, I. V., Dipp, S. & El-Dahr, S. S. Targeted disruption of the bradykinin B2 receptor gene in mice alters the ontogeny of the renin-angiotensin system. Am. J. Physiol. Ren. Physiol. 281, F795–F801 (2001).
Abadir, P. M., Periasamy, A., Carey, R. M. & Siragy, H. M. Angiotensin II type 2 receptor-bradykinin B2 receptor functional heterodimerization. Hypertension 48, 316–322 (2006).
Abadir, P. M., Carey, R. M. & Siragy, H. M. Angiotensin AT2 receptors directly stimulate renal nitric oxide in bradykinin B2-receptor-null mice. Hypertension 42, 600–604 (2003).
Cerrato, B. D., Carretero, O. A., Janic, B., Grecco, H. E. & Gironacci, M. M. Heteromerization between the bradykinin B2 receptor and the angiotensin-(1–7) mas receptor: functional consequences. Hypertension 68, 1039–1048 (2016).
AbdAlla, S., Lother, H. & Quitterer, U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature 407, 94–98 (2000).
Qadri, F. & Bader, M. Kinin B1 receptors as a therapeutic target for inflammation. Expert Opin. Ther. Targets 22, 31–44 (2018).
Menke, J. G. et al. Expression cloning of a human B1 bradykinin receptor. J. Biol. Chem. 269, 21583–21586 (1994).
Merino, V. F. et al. Molecular structure and transcriptional regulation by nuclear factor- κB of the mouse kinin B1 receptor gene. Biol. Chem. 386, 515–522 (2005).
Duchene, J. et al. A novel inflammatory pathway involved in leukocyte recruitment: role for the kinin B1 receptor and the chemokine CXCL5. J. Immunol. 179, 4849–4856 (2007).
Linz, W. & Schölkens, B. A. Role of bradykinin in the cardiac effects of angiotensin-converting enzyme inhibitors. J. Cardiovasc. Pharmacol. 20 (Suppl 9), S83–S90 (1992).
Marin-Castaño, M. E. et al. Induction of functional bradykinin B1-receptors in normotensive rats and mice under chronic angiotensin-converting enzyme inhibitor treatment. Circulation 105, 627–632 (2002).
Marcic, B. M. & Erdös, E. G. Protein kinase C and phosphatase inhibitors block the ability of angiotensin I-converting enzyme inhibitors to resensitize the receptor to bradykinin without altering the primary effects of bradykinin. J. Pharmacol. Exp. Ther. 294, 605–612 (2000).
Montinaro, V. & Cicardi, M. ACE inhibitor-mediated angioedema. Int. Immunopharmacol. 78, 106081 (2020).
Campbell, D. J. Neprilysin inhibitors and bradykinin. Front. Med. 5, 257 (2018).
Remuzzi, G., Perico, N., Macia, M. & Ruggenenti, P. The role of renin-angiotensin-aldosterone system in the progression of chronic kidney disease. Kidney Int. Suppl. 68, S57–S65 (2005).
Nakano, D. & Nishiyama, A. A novel role of renin inhibitor in the complement cascade. Kidney Int. 94, 650–652 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04183101 (2020).
Acknowledgements
Z.B. is supported by the Crafoord Foundation, the Swedish Kidney Foundation and the Maggie Stephens Foundation. M.B. is supported by the German Research Foundation (SFB1365). D.K. is supported by the Swedish Research Council (2017-01920), the Knut and Alice Wallenberg Foundation (Wallenberg Clinical Scholar 2015.0320), the Skåne Centre of Excellence in Health, the IngaBritt and Arne Lundberg’s Research Foundation and the Olle Engkvist Byggmästare Foundation.
Author information
Authors and Affiliations
Contributions
All authors researched data for the article. Z.B., M.B. and D.K. contributed substantially to discussion of the content. All authors wrote the article and reviewed the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Immunology thanks Johannes Stegbauer, Allen P. Kaplan, Berhane Gherbehiwet and Claudia Kemper for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- Angiotensin
-
A group of peptide hormones that are effectors of the renin–angiotensin system and are involved in control of vascular tone, blood pressure, fluid and salt balance, cell proliferation, fibrosis and inflammation.
- Kinin
-
A group of peptides that are effectors of the kallikrein–kinin system and induce vasodilation, vascular leakage and pain, cardinal signs of inflammation.
- Pentraxins
-
A family of innate pattern recognition proteins, of which C-reactive protein (CRP), serum amyloid P and pentraxin 3 (PTX3) activate the classical pathway of the complement system.
- Intrinsic pathway of coagulation
-
A cascade of serial activation of the coagulation factors. Factor XII (FXII; in activated form FXIIa), FXI and FIX generate a complex between FVIIIa and FIXa that activates FX. FXa with FVa form the prothrombinase complex that cleaves prothrombin into thrombin.
- Membrane attack complex
-
An assembly of complement proteins C5b, C6, C7 and C8 and a number of C9 molecules forming a cytolytic pore in the cell membrane.
- Chagas disease
-
A tropical infection caused by the intracellular protozoan Trypanosoma cruzi.
- Extracorporeal circulation
-
Temporary circulation and oxygenation of blood through an instrument that takes over bodily functions. For example, a cardiopulmonary bypass is a form of extracorporeal circulation.
- Microangiopathy
-
A pathological lesion affecting small vessels, primarily capillaries, characterized by endothelial cell injury, detachment of endothelial cells, accumulation of amorphous material in the subendothelial space, platelet aggregation, vascular luminal narrowing and partial or complete vessel occlusion.
- Endotheliitis
-
Generalized inflammation and damage to the endothelium with leukocyte infiltration into the perivascular space.
- Cytokine storm
-
A systemic inflammatory disorder triggered by pathogens, genetic or autoimmune diseases or iatrogenic treatments leading to hyperactivation of immune cells, such as T cells, macrophages and dendritic cells, and extremely high levels of circulating pro-inflammatory cytokines resulting in multi-organ failure and a high risk of death.
Rights and permissions
About this article

Cite this article
Bekassy, Z., Lopatko Fagerström, I., Bader, M. et al. Crosstalk between the renin–angiotensin, complement and kallikrein–kinin systems in inflammation. Nat Rev Immunol 22, 411–428 (2022). https://doi.org/10.1038/s41577-021-00634-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-021-00634-8
This article is cited by
-
Impact of ACE I gene insertion/deletion, A-240T polymorphisms and the renin–angiotensin–aldosterone system on COVID-19 disease
Virology Journal (2024)
-
The use of angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers may relate to the survival and walking ability in geriatric patients with hip fractures: a 1-year follow-up study
BMC Musculoskeletal Disorders (2023)
-
Defining blood-induced microglia functions in neurodegeneration through multiomic profiling
Nature Immunology (2023)
-
The Role of Complement in HSCT-TMA: Basic Science to Clinical Practice
Advances in Therapy (2022)
-
What have we learnt from the COVID-19 epidemic? Considerations of a troubled editor after two troubled years
Journal of Nephrology (2022)
