
Abstract
Natural killer (NK) cells play an important role in innate immune responses to viral infections. Here, we review recent insights into the role of NK cells in viral infections, with particular emphasis on human studies. We first discuss NK cells in the context of acute viral infections, with flavivirus and influenza virus infections as examples. Questions related to activation of NK cells, homing to infected tissues and the role of tissue-resident NK cells in acute viral infections are also addressed. Next, we discuss NK cells in the context of chronic viral infections with hepatitis C virus and HIV-1. Also covered is the role of adaptive-like NK cell expansions as well as the appearance of CD56− NK cells in the course of chronic infection. Specific emphasis is then placed in viral infections in patients with primary immunodeficiencies affecting NK cells. Not least, studies in this area have revealed an important role for NK cells in controlling several herpesvirus infections. Finally, we address new data with respect to the activation of NK cells and NK cell function in humans infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) giving rise to coronavirus disease 2019 (COVID-19).
Similar content being viewed by others


Introduction
Almost 50 years ago, a small number of research groups started to observe unexpected spontaneous cytotoxic activities among lymphocytes. While these were at first considered to be merely annoying background phenomena, subsequent studies led to the realization that a new subpopulation of lymphocytes was responsible for this activity1. The cells were termed ‘natural killer (NK) cells’. Over the years, insights into the molecular specificity of NK cells in terms of target cell recognition2,3,4 and into their role in controlling viral and intracellular bacterial infections and tumours, as well as insights into their ability to regulate other immune cells, have put NK cells at the forefront of modern immunology5. Today, they are used as therapeutic agents in the treatment of human malignant diseases6, and strategies for using them therapeutically against viral diseases, including coronavirus disease 2019 (COVID-19), are currently being explored7.
NK cells are best characterized by their cytotoxic function and their ability to produce cytokines upon stimulation5. With respect to their cytotoxic potential, they target infected, transformed and stressed cells. In this way, they not only eliminate unwanted cells but also contribute to cellular homeostasis8. The conventional known anatomical site of NK cell production is the bone marrow, where interactions with other cellular components, cytokines and soluble molecules support and drive NK cell development (reviewed in ref.9) (Box 1). Most of the current knowledge of human NK cells has arisen from studies of NK cells in peripheral blood and secondary lymphoid organs. Recently, however, there has been increased interest in NK cells residing in specific tissues10 (so-called tissue-resident NK cells) (Box 2). In humans, mature NK cells are traditionally identified as CD3−CD56+ lymphocytes. They were long thought to represent a homogeneous population of cells. However, in line with adaptive lymphocytes, we now know that NK cells undergo a well-defined differentiation process involving distinct phenotypic and functional changes (Box 1).
NK cell function is regulated by a large number of germline-encoded receptors11,12. Upon viral infection, host cells may become susceptible to NK cell-mediated recognition through a variety of mechanisms that may include upregulation of self-encoded molecules induced by the infection and/or a concomitant cellular stress response that binds activating NK cell receptors such as natural cytotoxicity receptors (NKp30, NKp44 and NKp46)13, C-type lectin-like receptors (for example, NKG2D and NKp80)14 and co-activating receptors (for example, DNAM1 and CD2)12. In parallel, contributing to increased target cell susceptibility is the often observed downregulation of MHC class I ligands for inhibitory receptors15. Importantly, NK cells can also eliminate virus-infected cells via CD16-mediated antibody-dependent cellular cytotoxicity (ADCC)16. Finally, NK cell activity is modulated by cytokines, including, but not limited to, the activating cytokines IL-2, IL-12, IL-15, IL-18 and type I interferons, which can be produced by virally infected cells or activated antigen-presenting cells17. Many of these cytokines, alone or in combination, promote NK cell survival, proliferation, cytotoxicity and cytokine production, including interferon-γ (IFNγ) production18. Through these mechanisms, NK cells are uniquely equipped to sense and quickly respond to viral infections7. In this respect, they often act in concert with other host immune responses in mediating antiviral immunity.
Here, we review recently obtained knowledge with regard to human NK cells in antiviral immunity. We address NK cell responses to typical acute viral infections. In many situations, these trigger activation of circulating NK cells and their homing to affected tissues. In parallel, tissue-resident NK cells may also become activated. With respect to chronic viral infections, NK cells contribute to viral control. We describe how such infections may lead to ‘adaptations’ in the NK cell repertoire, likely contributing to their maintenance of viral control but possibly also representing a consequence of exhaustion in some cases. Impairment in NK cell-mediated viral control is seen in patients with inborn deficiencies affecting NK cell development and/or function. Finally, we pay specific attention to the very recently developing knowledge with respect to vigorous NK cell activation in human severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and COVID-19.
NK cells in acute viral infections
NK cells respond vigorously to several acute viral infections, exemplified in the following subsections by acute infections with flavivirus and influenza virus (both RNA viruses). In many situations, this triggers activation of circulating NK cells and their homing to affected tissues. In parallel, specific tissue-resident NK cells may also become activated. Critical to all host responses to acute viral infections is the balance between possible acute antiviral mechanisms and responses contributing to tissue damage and immunopathology.
Flavivirus infections
Flaviviruses, including dengue virus (DENV), West Nile virus, Japanese encephalitis virus, tick-borne encephalitis virus, yellow fever virus and Zika virus, are major emerging human pathogens, affecting millions of individuals worldwide. Flaviviruses elicit haemorrhagic fever-like syndromes and central nervous system-associated diseases in infected hosts19. Effective vaccines exist for some of these viral infections. With a few exceptions, there is a lack of knowledge with respect to which host cells are infected by flaviviruses in humans and whether they are targets of NK cell-mediated responses. Innate immune responses elicited by flaviviruses have been studied extensively in animal models, in particular DENV and Zika virus infections20. However, fewer studies have focused specifically on NK cells in these acute viral infections, with influenza virus infections being an exception, as reviewed in the next subsection. In humans, NK cell responses to acute flavivirus infection have been addressed in clinical studies, particularly in DENV infections21,22. While clinical studies performed to date have differed in their set-up, some common observations have been made with respect to NK cell responses in different flavivirus infections. In several studies, both CD56bright NK cells and, in some studies, subpopulations of less differentiated CD56dim NK cells (Box 1) have been reported to respond vigorously to infection22,23,24,25. Type I and type III interferons likely contribute to the NK cell responses observed in several flavivirus infections, including yellow fever virus infection23. Responses occur irrespective of NK cell education, and no evidence of specific adaptive-like NK cell responses has been observed22,23. Concomitant with the increase in NK cell activation, increased levels of various cytokines, including IL-12, IL-15, IL-18, IFNγ and tumour necrosis factor (TNF), have been detected in flavivirus-infected patients22,23,25. In DENV infections, increased IL-18 levels in plasma and in induced skin blisters, as well as concomitant signalling downstream of IL-18 receptor in NK cells, have suggested an IL-18-dependent mechanism in driving the proliferative NK cell response22. This proliferation might occur via nuclear factor-κB (NF-κB), AKT, activating transcription factor 2 (ATF2) and forkhead box protein O3 (FOXO3) or indirectly via IL-18-induced upregulation of CD98 and consequently amino acid-induced activation of mechanistic target of rapamycin (mTOR)22. NK cell-produced IFNγ, on the other hand, has in other studies been suggested to be important for control of DENV infection26. These and other related studies illustrate the dynamics and specifics of the early NK cell response during prototype acute flavivirus infections in humans. How NK cells contribute to viral clearance in experimental and human flavivirus infections remains to be explored in further detail.
Influenza virus infections
Influenza viruses cause yearly global epidemics, with up to a billion infections annually27. While vaccines limit their spread, they do not prevent all infections. Typically, the virus infects lung epithelial cells. Like the situation in many viral infections, including SARS-CoV-2 infection (discussed in more detail below), infection with influenza virus can lead to excessive inflammation and tissue damage. Although NK cells are part of this response, their contribution to host defence against influenza A virus (IAV) is not fully understood. Upon IAV infection in mice, increased accumulation of NK cells is observed in the lungs28. While local proliferation of pulmonary NK cells may account for part of this response, most of the NK cell accumulation in the lungs and airways during IAV infection has been suggested to be due to NK cell recruitment that is partly dependent on CXC-chemokine receptor 3 (CXCR3) and CC-chemokine receptor 5 (CCR5), respectively28. Several NK cell natural cytotoxicity receptors have been implicated in functional activation of NK cells following influenza virus infection by binding to influenza virus haemagglutinin29. However, the degree to which NK cells contribute (if at all) to influenza virus elimination in this context is less clear. In humans, peripheral blood CD56bright and CD56dim NK cells have been shown to be primed during acute IAV infection30. Furthermore, in peripheral blood, a small subset of CD16−CD49a+CXCR3+ NK cells has been identified, with CD49a and CXCR3 potentially promoting homing to and tissue retention in the lungs during acute infection. At the primary site of infection, it is not fully clear how circulating and tissue-resident NK cells contribute to possible antiviral immunity or to pathology, such as by killing bystander cells30. Finally, studies in humans and macaques have demonstrated that cross-reactive antibodies elicited during seasonal influenza virus infections facilitate NK cell-mediated ADCC with regard to infected target cells31,32. This ADCC-mediated response has been correlated with reduced disease severity. In infections such as influenza virus infection, more research is clearly needed to better delineate and discriminate between possible protective antiviral responses and those leading to excessive inflammation and tissue damage.
NK cell activation in acute infections
The acute NK cell response to infection with flaviviruses, including tick-borne encephalitis virus, DENV and yellow fever virus, as well as SARS-CoV-2 (discussed in detail below), is to a large degree confined to less differentiated CD56bright and CD56dim NK cells22,23,25,33 (Box 1). Since these cells are more responsive to cytokines such as IL-12, IL-15, IL-18 and type I interferons, probably in part because of higher expression of receptors for these cytokines22,34,35, it is likely that early NK cell activation is cytokine driven and less reliant on receptor–ligand interactions (Fig. 1). This results in stronger downstream signalling via signal transducer and activator of transcription 1 (STAT1), STAT4 and the PI3K–AKT–mTOR pathways in less differentiated NK cells22,36,37, in which mTOR, via the IRE1α–XBP1–MYC axis, is crucial for NK cell proliferation following acute activation38. Cytokine signalling via JAK–STAT pathways in NK cells was recently comprehensively reviewed elsewhere39. For induction of early NK cell activation, type I interferon signalling via STAT1 and IL-12 plus IL-18 signalling via STAT4 result in strikingly different interactions with the NK cell epigenetic landscape40. More specifically, STAT1 and STAT4 can exhibit antagonistic functions on their downstream networks early after NK cell activation, including STAT4 actively suppressing STAT1 expression40. Furthermore, whereas STAT4 binding resulted in increased chromatin accessibility, STAT1 interacted more with promoter regions40. Although more long-lasting effects of these alterations in the chromatin landscape remain elusive, it is possible that remodelling via IL-12 plus IL-18 and STAT4 could have more permanent effects as STAT4 signalling is important for the generation of both adaptive-like and cytokine-induced memory-like NK cells41,42.

The differentiation status of human natural killer (NK) cells determines their functional response in viral infections. CD56bright NK cells and less differentiated CD56dim NK cells express high levels of NKG2A, natural cytotoxicity receptors (NCRs; NKp30, NKp44 and NKp46) and proinflammatory and antiviral cytokine receptors (IL-12 receptor (IL-12R), IL-18R and type I interferon receptor (IFNAR)). During acute viral infections, with ensuing production of IL-12, IL-18 and type I interferons by other immune cells, less differentiated NK cells are the main NK cell subset to respond. As indicated by black arrows, cytokine stimulation leads to signalling via signal transducer and activator of transcription 1 (STAT1) and STAT4 and PI3K–AKT–mTOR pathways to rapidly induce NK cell cytokine production and proliferation. With increased maturation, circulating CD56bright NK cells undergo a phenotypic shift and their responsiveness during infection becomes altered. As a consequence, more differentiated NK cells are more prone to respond to cytomegalovirus, leading to the expansion of adaptive-like NK cells that express high levels of killer cell immunoglobulin-like receptors (KIRs) and NKG2C. Circulating CD56bright NK cells might also home to tissues, giving rise to tissue-resident NK cells. Their role in the early response to infection remains elusive. However, liver-resident NK cells have been shown to mediate antigen-specific antiviral responses. Alterations in transcription factor expression during NK cell differentiation are indicated within the respective cell nuclei, and a summary of phenotypic changes during NK cell differentiation is depicted in the table. For some receptors, the expression depends on the tissue microenvironment and specific subsets studied and will deviate from what is indicated here (denoted by the asterisk). CLA, cutaneous lymphocyte-associated antigen; HAV, hepatitis A virus; HBV, hepatitis B virus; ISG, interferon-stimulated gene; NA, not available.
A recent study by Sciumè and colleagues looked into the molecular mechanism of early NK cell activation in response to cytokines43. Analysis was focused on regions surrounding genes highly induced on early NK cell activation and revealed de novo chromatin and enhancer remodelling mediated by STAT4. These de novo enhancers were further repurposed to be used not only by STATs but also by the lineage-defining transcription factor T-bet43. Thus, although T-bet and other lineage-defining transcription factors were once considered pioneers for determining the shape of the epigenetic landscape that signal-regulated transcription factors act on, this instead suggests that in acute activation of NK cells STATs will remodel chromatin accessibility and invite lineage-defining transcription factors to participate in novel ways in the acute response.
NK cell homing in acute infections
Since many viruses have narrow tropisms, infections often originate at the site of entry before spreading locally and/or systematically. Alternatively, they may be selectively restricted to a specific organ due to tissue tropism (for example, the liver in the case of hepatitis virus infections). From considerable work in mouse experimental models, it is known that recruitment of circulating NK cells to the site of infection is important for the early inflammatory response and viral containment44,45,46,47. Chemokine–chemokine receptor interactions are necessary for mouse NK cell homing to tissues early in infection, and certain combinations come into action depending on the affected organ45,46,47. In a similar way, circulating NK cells migrate into lymph nodes in a CXCR5-dependent manner in macaques and help to control simian immunodeficiency virus (SIV) infection48. In humans, however, there is a relative scarcity of studies assessing NK cell homing to tissues during acute viral infections because of the inherent challenges in performing such studies and lack of easy access to infected tissue samples. Nevertheless, in acute DENV infection, responding peripheral blood NK cells show a distinct chemokine receptor imprint, including high expression of the skin-homing marker cutaneous lymphocyte antigen. Because of this, high numbers of NK cells are present in the skin of patients early after infection22. Similarly, skin challenge with varicella zoster virus antigens in previously vaccinated individuals led to rapid recruitment of NK cells to the site of challenge49. However, studies in humans are warranted where NK cell homing to tissues during acute infections can be studied.
Tissue-resident NK cells in infection
Besides homing of NK cells to tissues in response to acute viral infections, we have in recent years learned that most peripheral organs contain tissue-resident CD56bright NK cells (in humans) and/or group 1 innate lymphoid cells (ILC1s) (in mice) in the steady state (reviewed in ref.10) (Box 2). These cells are often strategically positioned at the site of viral infection in organs, such as the liver, female genital tract, salivary glands, kidneys and intestine, poised to respond rapidly10,50. Indeed, in mouse models it has been shown that tissue-resident ILC1s confer early host protection51. Their human counterpart is most likely tissue-resident CD56bright NK cells (hereafter referred to as tissue-resident NK cells), given their functional similarities, enrichment in tissues and shared expression of tissue-residency markers (Box 2). Since tissue-resident NK cells resemble circulating CD56bright NK cells, cytokine and chemokine production is presumably the main function exhibited by these cells in early stages of infection (Fig. 1). Tissue-resident NK cells can also respond with local proliferation in acute and chronic viral infection52. Tissue-resident NK cells have also been shown to more directly influence the magnitude of T cell responses via expression of the inhibitory ligand PDL1, leading to viral persistence but more limited immunopathology after lymphocytic choriomeningitis virus infection52. In humans, a similar regulation of antiviral T cell immunity has been shown for liver-resident NK cells in chronic hepatitis B virus (HBV) infection53. However, compared with what has been reported for conventional NK cell responses to viral infections, much less is known regarding the role of tissue-resident NK cell responses. In this regard, it is striking to note that antigen-specific NK cell responses to vaccination and/or infection appear to be largely confined to tissue-resident NK cells in both humans and mice49,54,55,56 (Box 3). Future work should aim at disentangling the relative contributions of migrating and tissue-resident NK cells in antiviral responses occurring in peripheral organs.
NK cells in chronic viral infections
NK cells adapt to chronic viral infection in ways that are only partially understood. These adaptations likely benefit viral control over time. Impairment of the ability to handle chronic viral infections, such as herpesvirus infections, is exemplified by the not seldom lethal consequences of severe inborn deficiencies affecting NK cell development and/or function.
Hepatitis C virus infection
Although hepatitis C virus (HCV) belongs to the family Flaviviridae, it only rarely causes acute symptomatic disease. Instead, HCV is successful in evading the initial immune response and establishing chronic infection. There is strong evidence suggesting that NK cells contribute to HCV infection outcome, as the combination of KIR2DL3 with homozygosity for the killer cell immunoglobulin-like receptor (KIR) ligand gene HLA-C1 has been associated with significantly elevated viral clearance57. Furthermore, certain NK cell phenotypes are linked to reduced acquisition of chronic infection in exposed individuals as well as to the outcome of acute HCV infection58,59. Yet once the virus persists and the infection becomes chronic, the rate of spontaneous clearance is low. In this phase, the immune response against the virus is hampered at several levels. T cells from patients with chronic HCV infection display reduced proliferation and cytokine production60, along with upregulation of inhibitory receptors61,62, a combination of features referred to as ‘exhaustion’. From the lymphocytic choriomeningitis virus infection model, it is known that NK cells can contribute to sustaining the chronic infection by targeting activated CD4+ T cells, which in turn leads to CD8+ T cell exhaustion63, and that NK cell depletion promotes antiviral CD8+ T cell immunity64. A similar mechanism has also been described in chronic HBV infection, in which NK cells mediate depletion of HBV-specific T cells53.
In chronic HCV infection, NK cells display an altered phenotype and functionality. Yet the impact of chronic infection on NK cells appears to be, at least in part, unique to each individual. In assessment of the expression of activating receptors, both elevated and reduced frequencies have been reported59,65,66. Along the same lines, also for NK cell degranulation both reduced levels65,67 and elevated levels68,69 have been reported. By contrast, a consistent effect of chronic HCV infection on NK cells appears to be reduced production of proinflammatory cytokines68,69,70. Thus, although the NK cell compartment is indeed affected in chronic HCV infection, certain aspects of this appear to rely on other factors, such as the duration of infection or the underlying state of liver disease. This effect of chronic HCV infection on the NK cell compartment is likely to be caused by a plethora of mechanisms, but it has been shown that the direct interaction of NK cells with HCV-infected cells leads to reduced expression of NKG2D and decreased functionality71. In line with this, NS5A-mediated production of transforming growth factor-β could also be linked to reduction of NKG2D expression67. In recent years, the development of direct-acting antivirals has revolutionized HCV infection therapy, as these drugs lead to successful viral clearance in nearly all patients, with negligible side effects. This has also made it possible to assess the capacity for reinvigoration of NK cell function after rapid clearance of a chronic infection (Box 4).
HIV-1 infection
The effector capacity of NK cells includes cytotoxic elimination of HIV-1-infected target cells, as well as secretion of IFNγ, TNF and CCL4, influencing antiviral responses and limiting viral spread72,73. Notably, however, a differential role for cytokines, including TNF and IFNγ, has been observed in chronic states of the infection74,75. NK cells also produce β-chemokines, CCL3, CCL4 and CCL5, which function as natural ligands of CCR5, hampering HIV infectivity of target cells76. Additionally, population-level genetic associations between NK cell receptor expression and HIV-1 infection outcome and evolution have clearly revealed the impact of NK cells on HIV-1 disease progression. In HIV, KIR–HLA combinations have been associated with the pace of disease progression77 and protection against disease acquisition78. In detail, HLA-B Bw4-80I in combination with KIR3DL1*h but also with KIR3DS1 could be linked to improved viral control77,79. The mechanisms conferring this protection may include both NK cell education through inhibitory receptor ligation80 and the direct interaction of KIRs with HIV-1-derived peptide motifs presented on HLA molecules81,82. Indeed, for KIR3DS1+ NK cells, an HLA-B Bw4-80I-dependent suppression of viral replication has been shown83, further supporting a direct role for NK cells in mediating the observed protective effect. HIV-1-mediated downregulation of HLA molecules, which prevents infected cells from lysis by CD8+ T cells84, may also facilitate NK cell recognition of infected cells. However, this is limited by preservation of expression of HLA-C and HLA-E85. HIV-1 infection has also been shown to trigger the upregulation of ligands for NKG2D and other activating receptors86. However, HIV-1 has enabled mechanisms to limit the recognition of these ligands through the expression of viral accessory proteins87,88. Furthermore, responses characterized by coordinated antibody function, including NK cell-mediated ADCC, are associated with viral controller phenotypes89.
Like for many other viral infections, changes in NK cell receptor repertoire and functionality are observed during HIV-1 infection. For example, treatment-naive HIV-1-infected individuals have a significant loss of CCR7+CD56bright NK cells, and this change has been associated with higher viral loads90. Loss of the CD56dim NK cell subset during HIV-1 infection has also been observed90. Animal models of SIV infection have shown accumulation of CD56−CD16+ NK cells in lymph nodes91. Since HIV-1 infection is often subjected to pharmacological intervention, the effects of therapies on NK cells have been studied. Initiation of antiretroviral therapy has been associated with alterations in the CD56bright and CD56dim NK cell populations, with the former increasing after therapy92.
Adaptive-like NK cell expansions
Studies of human peripheral blood NK cells have revealed receptor expression-associated changes throughout life93 that have been linked to an individual’s infection history94. Some of these changes are evident as the appearance of adaptive-like NK cell expansions (reviewed in ref.95) that have been extensively studied in the mouse cytomegalovirus (MCMV) infection model96. In this infection, Ly49H+ NK cells that specifically recognized the MCMV-encoded glycoprotein m157 were expanded on primary infection96. These NK cells were shown to confer protection against MCMV following rechallenge with the virus97. Adaptive-like NK cell expansions have also been identified in humans. The very first indications of adaptive-like NK cells came from observations on the expansion and persistence of CD94+NKG2C+ NK cells in humans infected with human cytomegalovirus (HCMV)98. Adaptive-like NK cell expansions have also been observed following diverse viral infections in humans, including hantavirus infection99, chikungunya virus infection100 and HIV-1 infection101. Notably, all these responses correlated with HCMV co-infection, suggesting that subclinical reactivation of HCMV could be one mechanism underlying the observed responses. By contrast, several other infections, including recurrent herpes simplex virus 2 infection102 and acute Epstein–Barr virus (EBV) infection103, have not been associated with an expansion of NKG2C+ NK cells. Furthermore, adaptive-like NK cell expansions observed in individuals infected with HBV, HCV and/or hepatitis delta virus were shown not to be linked to the chronic hepatitis infections per se but rather to be consequences of an underlying HCMV infection104. Distinct from adaptive-like NK cell expansions characterized by a reshaping of the NK cell repertoire towards certain subpopulations, additional animal models of herpes simplex virus, influenza virus and SIV infection and more recently also work in the human setting assessing vaccination responses have revealed the existence of antigen-specific NK cell responses (Box 3).
In more in-depth recent work in humans, several characteristics of adaptive-like NK cells have been identified, including clonal expansion, persistence and recall responses105,106. Furthermore, these cells are characterized by low expression of the transcription factor zinc-finger protein PLZF (also known as ZBTB16), silencing of intracellular signalling molecules and commonly also expression of CD57 (refs107,108). Human adaptive-like NK cell expansion depends on expression of the NKG2C ligand HLA-E109. Adaptive-like NKG2C+ NK cells differentially recognize distinct HCMV strains encoding variable UL40 peptides that, in combination with proinflammatory signals, control expansion and differentiation of adaptive-like NKG2C+ NK cells110. Interestingly, HCMV-seropositive individuals possessing a homozygous null allele of KLRC2 (encoding NKG2C) remain healthy. In these individuals, the adaptive-like NK cell expansions instead expressed elevated levels of CD2, which synergized with CD16 to activate NK cells in HCMV infection111. It is not unlikely that there are additional environmental factors that similarly shape NK cell phenotype and function that are even less well understood. Finally, since adaptive-like NK cell expansions decrease NK cell repertoire diversity, the long-term consequences of this for the individual remain elusive.
CD56− NK cells in chronic infections
A particular hallmark of chronic viral infections such as HIV-1 and HCV infections is the expansion of a subset of CD56− NK cells (reviewed in ref.112). More recently, CD56− NK cells have also been observed to be expanded in HCMV-seropositive and EBV-seropositive elderly individuals113. In healthy individuals, the CD56− NK cell subset encompasses only a small proportion of the NK cell compartment in peripheral blood114. In chronic infections, however, it may constitute up to half of all peripheral NK cells, largely at the expense of the CD56dim NK cell subset115,116,117. The CD56− NK cell population displays an exhausted phenotype, with reduced levels of perforin, granzyme B, IFNγ and TNF, and lower cellular cytotoxicity than CD56dim NK cells117,118. It has remained unclear to what extent these expanded CD56− NK cells represent a similar or distinct phenotype as compared with the CD56bright or CD56dim NK cell populations in healthy individuals. However, more recently, an unbiased surface receptor screen and a global proteome analysis were performed to obtain detailed molecular and phenotypic information of the CD56− NK cell subset119. It was revealed that CD56− NK cells have a surface proteome and a total proteome related to, but not identical to, those of CD56dim NK cells. Functional investigations in that study examining degranulation, cytotoxicity and IFNγ production of CD56− NK cells from healthy individuals largely confirmed an, at most, moderate responsiveness of these cells119.
NK cell deficiencies and infections
Studies of patients with more than 400 different primary immunodeficiency diseases (PIDs) have provided key insights into host control of viral infections. NK cells are affected in more than 50 PIDs. At present, at least seven PIDs have an abnormality specifically affecting NK cells (reviewed in ref.120) (Fig. 2). These specific NK cell deficiency disorders include mutations affecting total NK cell numbers, NK cell subsets and/or NK cell functions120. The genes affected in these seven NK cell deficiencies are GATA2 (ref.121), MCM4 (ref.122), RTEL1 (ref.123), GINS1 (ref.124), IRF8 (ref.125), MCM10 (ref.126) and FCGR3A127. The first six genes affect NK cell development or maturation, with resulting low total NK cell numbers and specific defects affecting NK cell subsets. FCGR3A gene mutations affect NK cell function despite normal numbers of NK cells120. This is due to a homozygous mutation in FCGR3A (which encodes CD16)127,128. Intriguingly, despite CD16 functioning as an IgG Fc receptor, patients with mutations in FCGR3A have impaired natural cytotoxicity but intact ADCC function128. The clinical presentation of human NK cell deficiency varies from patient to patient; however, hallmark viral infections affecting these individuals include herpesvirus infections such as HCMV, EBV, herpes simplex virus and varicella zoster virus infections120,126 (Fig. 2). These viruses cause disease in almost 60% of reported NK cell deficiency cases120. On the basis of these findings, one may argue that host defence against herpesvirus infections might be one of the most important functions of NK cells in the context of antiviral immunity. In addition, some other viral infections have been reported in patients with NK cell deficiency, including human papillomavirus infection and some respiratory infections120. Interestingly, these observations suggest an important role for NK cells in keeping these, often latent, infections under control. Notably, of patients with reported NK cell deficiency, almost half have been reported to have died prematurely120, underscoring the severity of these diseases. Corroborating these findings, in studies of patients with a GATA2 deficiency, the frequencies of NK cells were directly associated with increasing complications of disease129.

Different genetic mutations have been linked to specific natural killer (NK) cell deficiencies. Affected genes encode either intranuclear proteins (GATA2, IRF8, RTEL1, GINS1, MCM4 and MCM10) or the surface-expressed Fc receptor for IgG CD16 (encoded by FCGR3A). Defects in the intranuclear proteins lead to disturbed NK cell development and/or differentiation. The mutation affecting CD16 causes altered NK cell functionality. A common denominator for NK cell deficiencies is an inappropriate response to viral infections, as evidenced by severe and/or recurrent herpesvirus infections (human cytomegalovirus (HCMV), Epstein–Barr virus (EBV), herpes simplex virus (HSV) and varicella zoster virus (VZV)), as well as human papillomavirus virus (HPV) infection and respiratory infections.
NK cells in COVID-19
In late 2019, a new zoonotic viral pathogen, SARS-CoV-2, emerged and rapidly spread throughout the world, causing more than 60 million infections and 1.5 million deaths during the first year of the pandemic130. SARS-CoV-2 infection can cause COVID-19, a respiratory and vascular disease which, in severe cases, can lead to acute respiratory distress syndrome, multi-organ failure and death130,131. Although the disease pathogenesis is not yet completely understood, a misdirected and hyperactivated immune system is thought to contribute to severe COVID-19, with a likely contribution from NK cells.
NK cell response to SARS-CoV-2
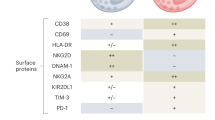
NK cells are early responders to acute SARS-CoV-2 infection, with recruitment of CD56bright and CD56dim NK cells from the circulation to the lungs, leading to reduced numbers of circulating NK cells in acute infection33,132,133,134,135 (Fig. 3). A gene module for chemotaxis is induced in lung NK cells33,135, and lungs of patients with COVID-19 contain elevated levels of chemokines such as CCL3, CCL3L1, CCL4, CXCL9, CXCL10 and CXCL11 (refs135,136). Although further work is needed in the area, this chemokine production suggests that CXCR3 and CCR5 are important for homing of NK cells to the lungs in acute SARS-CoV-2 infection137. The early depletion of NK cells from the circulation and their redistribution to the site of infection is in line with what has been observed in other severe acute infections, such as acute hantavirus infection99. Furthermore, NK cells display an activated and cycling phenotype in acute SARS-CoV-2 infection at both the protein level and the transcriptomic level, with upregulation of Ki67, CD69, HLA-DR and CD38 (refs33,132,134). In the CD56dim NK cell compartment, less differentiated NKG2A+CD62L+CD57−KIR− cells were the main responding cells, suggesting a cytokine-driven mechanism of activation33 (Box 1; Fig. 1). Paralleling the activation of NK cells, regulatory programmes appear to be initiated, as evidenced by the upregulation of inhibitory checkpoint receptors such as LAG3, TIGIT and TIM3 (refs33,132) (Fig. 3). This could possibly explain why peripheral blood NK cell function has been reported to be blunted in acute SARS-CoV-2 infection133,138.

In the circulation, natural killer (NK) cells respond strongly to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and display an altered phenotype, including elevated expression of activation markers (HLA-DR, CD69 and CD38), inhibitory molecules (TIM3, LAG3 and possibly PD1) and tissue-homing markers (CCR5, CXCR3 and CD62L). Furthermore, circulating NK cells are highly proliferative and upregulate perforin and granzyme B expression. This response is primarily confined to less differentiated NK cells, suggestive of a cytokine-driven response. NK cells likely home to the lungs, where they exhibit an inflamed transcriptional signature. In severe coronavirus disease 2019 (COVID-19), adaptive-like NK cells are found at higher frequencies in the circulation, but it remains unclear whether these cells home to the lungs and interact with infected epithelia that show increased expression of HLA-E, a ligand for the activating receptor NKG2C. Furthermore, the transcriptional profile of NK cells in the lung microenvironment of patients with severe COVID-19 is even further skewed towards inflammation. This lung microenvironment also contains high numbers of myeloid-derived suppressor cells (MDSCs) and immature neutrophils. However, details on how NK cells might interact with these cells remain elusive. Additional outstanding questions related to NK cells in COVID-19 are highlighted in the figure. CCL, CC-chemokine ligand; CCR, CC-chemokine receptor; CXCL, CXC-chemokine ligand; CXCR, CXC-chemokine receptor; KIR, killer cell immunoglobulin-like receptor; LAG3, lymphocyte activation gene 3; TIGIT, T cell immunoreceptor with immunoglobulin and ITIM domains; TIM3, T cell immunoglobulin mucin receptor 3.
Role of NK cells in COVID-19
Although NK cells appear to be robustly activated and home to the lungs in acute SARS-CoV-2 infection, from the currently available studies, the level of NK cell activation (induction of CD69 and HLA-DR expression) or proliferation is not associated strongly with COVID-19 severity. Instead, Maucourant and colleagues reported that severe/critical COVID-19 was associated with the appearance of adaptive-like NK cell expansions33 (Fig. 3). These expansions where characterized by high NKG2C and CD57 expression as well as by narrow KIR profiles33. Since the first report, similar findings have been reported in two additional studies133,139. It currently remains unclear whether the increased frequency of adaptive-like NK cells in severe COVID-19 is because of a direct effect of SARS-CoV-2 or whether this is a bystander phenomenon driven by HCMV33. Indeed, a history of HCMV infection appears more common in patients with severe COVID-19 (ref.140), and some evidence of local HCMV reactivation in the lungs of patients with COVID-19 receiving ventilator treatment has been reported141. On the other hand, both immune and parenchymal cells in the lungs of patients with COVID-19 display elevated HLA-E expression33, and it has been suggested that the SARS-CoV-2 spike protein can induce surface HLA-E expression by lung epithelial cells142. However, it remains to be determined whether upregulated HLA-E will stimulate NK cells via NKG2C or rather inhibit the NKG2A-expressing NK cells.
Future research should also address whether expansion of adaptive-like NK cells is needed to unleash the full antiviral capacity of NK cells against SARS-CoV-2 infection or whether these cells are instead contributing to COVID-19 pathogenesis. In this regard, it is interesting to note that a recent artificial intelligence-guided big-data approach identified a ‘severe viral pandemic core gene signature’ that highlighted IL-15–IL-15RA and NK cell senescence as important determinants for severe or fatal COVID-19 (ref.143). Corroborating this, it was recently suggested that IL-15 could be linked to NK cell dysfunction in COVID-19 and that this occurs late after symptom onset and only in the most severely ill patients144. Thus, these studies support a model in which misdirected and/or exhausted NK cell responses, rather than hyperactivated NK cells, contribute to severe COVID-19. Future work should explore the contribution of myeloid-derived suppressor cells and immature neutrophils145 in relation to NK cell dysfunction in severe COVID-19 (Fig. 3).
Concluding remarks and outlook
As is evident from studies reviewed herein, it is clear that NK cells can mount vigorous responses to several acute viral infections, including acute flavivirus and influenza virus infections as discussed here. In many situations, this triggers homing of circulating NK cells to affected tissues. In parallel, less well-characterized tissue-resident NK cells may also become activated. Critical in all host responses to acute viral infections is the balance between acute antiviral mechanisms and responses contributing to tissue damage and immunopathology. Clearly, more knowledge is needed with respect to direct NK cell-mediated interactions with viral infected cells and the consequences of possible viral clearance by such interactions. NK cells adapt to chronic viral infections in ways that are only partially understood. These adaptations likely benefit viral control over time, although the detailed mechanisms are far from completely understood. Impairment of the ability to handle chronic viral infections, such as herpesvirus infections, is exemplified by the lethal consequences of severe inborn deficiencies affecting NK cell development and/or function. As discussed, at least seven PIDs have an abnormality specifically affecting NK cells. With the recent COVID-19 pandemic, NK cells have come into the spotlight in this context as well. Although the disease pathogenesis is still being worked out, a misdirected and hyperactivated immune system is thought to contribute to severe COVID-19, with a likely significant contribution from NK cells (Fig. 3).
While many insights with respect to a possible role for NK cells in viral infections have been revealed, from studies in mouse models to studies in humans, there are several outstanding questions and unresolved issues. What is the nature of NK cell viral antigen specificity as observed in, for example, hepatitis A virus and HBV vaccination (Box 3)? Furthermore, regarding antigen-specific NK cell responses, what are specific surface receptor-mediated responses versus what might be novel forms of antigen-specificity ‘dictated’ by changes at an epigenetic level (Box 3)? On the same note, what drives adaptive-like expansions of NK cell populations? What is the function of these cells in the context of viral infection? How are NK cells driven towards sites of infection and what is the role of responses from tissue-resident NK cells in the context of acute infections? Is there a division of labour between infiltrating NK cells and tissue-resident NK cells? What distinguishes functional antiviral NK cell responses from disease-causing responses? This is a question of relevance with regard to both acute responses, like in the SARS-CoV-2 situation (Fig. 3), and chronic infections, such as HIV-1 and HCV infections. With regard to vaccination, a presently hot topic, what is the role of NK cells in the generation of efficient vaccine responses? Finally, while this has been discussed for years, is there a role for NK cells in settings of adoptive immunotherapy for viral diseases such as what we are currently witnessing in the context of human malignant diseases?
In summary, although we have gained a great deal of knowledge of NK cells in the context of human viral infections over the past 10 years, much more remains to be learned.
References
Kiessling, R., Klein, E. & Wigzell, H. ‘Natural’ killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 5, 112–117 (1975).
Kärre, K., Ljunggren, H. G., Piontek, G. & Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319, 675–678 (1986).
Ljunggren, H. G. & Kärre, K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 11, 237–244 (1990).
Lanier, L. L. Natural killer cells: from no receptors to too many. Immunity 6, 371–378 (1997).
Vivier, E., Tomasello, E., Baratin, M., Walzer, T. & Ugolini, S. Functions of natural killer cells. Nat. Immunol. 9, 503–510 (2008).
Myers, J. A. & Miller, J. S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 16, 230–16 (2020).
Market, M. et al. Flattening the COVID-19 curve with natural killer cell based immunotherapies. Front. Immunol. 11, 1512 (2020).
Cichocki, F., Sitnicka, E. & Bryceson, Y. T. NK cell development and function-plasticity and redundancy unleashed. Semin. Immunol. 26, 114–126 (2014).
Sun, J. C. & Lanier, L. L. NK cell development, homeostasis and function: parallels with CD8+ T cells. Nat. Rev. Immunol. 11, 645–657 (2011).
Björkström, N. K., Ljunggren, H.-G. & Michaëlsson, J. Emerging insights into natural killer cells in human peripheral tissues. Nat. Rev. Immunol. 16, 310–320 (2016).
Lanier, L. L. NK cell recognition. Annu. Rev. Immunol. 23, 225–274 (2005).
Bryceson, Y. T., March, M. E., Ljunggren, H.-G. & Long, E. O. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol. Rev. 214, 73–91 (2006).
Biassoni, R. et al. Human natural killer cell receptors and co-receptors. Immunol. Rev. 181, 203–214 (2001).
Bartel, Y., Bauer, B. & Steinle, A. Modulation of NK cell function by genetically coupled C-type lectin-like receptor/ligand pairs encoded in the human natural killer gene complex. Front. Immunol. 4, 362 (2013).
Halenius, A., Gerke, C. & Hengel, H. Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: so many targets — but how many arrows in the quiver? Cell. Mol. Immunol. 12, 139–153 (2015).
Ochoa, M. C. et al. Antibody-dependent cell cytotoxicity: immunotherapy strategies enhancing effector NK cells. Immunol. Cell Biol. 95, 347–355 (2017).
Wu, Y., Tian, Z. & Wei, H. Developmental and functional control of natural killer cells by cytokines. Front. Immunol. 8, 930 (2017).
Fauriat, C., Long, E. O., Ljunggren, H.-G. & Bryceson, Y. T. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 115, 2167–2176 (2010).
Gould, E. A. & Solomon, T. Pathogenic flaviviruses. Lancet 371, 500–509 (2008).
Nelemans, T. & Kikkert, M. Viral innate immune evasion and the pathogenesis of emerging RNA virus infections. Viruses 11, 961 (2019).
Mathew, A. Defining the role of NK cells during dengue virus infection. Immunology 154, 557–562 (2018).
Zimmer, C. L. et al. NK cells are activated and primed for skin-homing during acute dengue virus infection in humans. Nat. Commun. 10, 3897 (2019). A detailed assessment of the NK cell response including tissue homing in acute DENV infection.
Marquardt, N. et al. The human NK cell response to yellow fever virus 17D is primarily governed by NK cell differentiation independently of NK cell education. J. Immunol. 195, 3262–3272 (2015).
Yao, Y. et al. The natural killer cell response to West Nile virus in young and old individuals with or without a prior history of infection. PLoS ONE 12, e0172625 (2017).
Blom, K. et al. NK cell responses to human tick-borne encephalitis virus infection. J. Immunol. 197, 2762–2771 (2016).
Costa, V. V. et al. Dengue virus-infected dendritic cells, but not monocytes, activate natural killer cells through a contact-dependent mechanism involving adhesion molecules. mBio 8, e00741-17 (2017).
Iuliano, A. D. et al. Estimates of global seasonal influenza-associated respiratory mortality: a modelling study. Lancet 391, 1285–1300 (2018).
Carlin, L. E., Hemann, E. A., Zacharias, Z. R., Heusel, J. W. & Legge, K. L. Natural killer cell recruitment to the lung during influenza a virus infection is dependent on CXCR3, CCR5, and virus exposure dose. Front. Immunol. 9, 781 (2018).
Luczo, J. M., Ronzulli, S. L. & Tompkins, S. M. Influenza a virus hemagglutinin and other pathogen glycoprotein interactions with nk cell natural cytotoxicity receptors NKp46, NKp44, and NKp30. Viruses 13, 156 (2021).
Scharenberg, M. et al. Influenza a virus infection induces hyperresponsiveness in human lung tissue-resident and peripheral blood NK cells. Front. Immunol. 10, 1116 (2019).
Jegaskanda, S., Weinfurter, J. T., Friedrich, T. C. & Kent, S. J. Antibody-dependent cellular cytotoxicity is associated with control of pandemic H1N1 influenza virus infection of macaques. J. Virol. 87, 5512–5522 (2013).
Jegaskanda, S. et al. Cross-reactive influenza-specific antibody-dependent cellular cytotoxicity in intravenous immunoglobulin as a potential therapeutic against emerging influenza viruses. J. Infect. Dis. 210, 1811–1822 (2014).
Maucourant, C. et al. Natural killer cell immunotypes related to COVID-19 disease severity. Sci. Immunol. 5, eabd6832 (2020). The first report on the NK cell response in COVID-19.
Björkström, N. K. et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood 116, 3853–3864 (2010). An article outlining a stepwise programme for human NK cell differentiation.
Lunemann, S. et al. Effects of HDV infection and pegylated interferon α treatment on the natural killer cell compartment in chronically infected individuals. Gut 64, 469–482 (2015).
Yu, J. et al. CD94 surface density identifies a functional intermediary between the CD56bright and CD56dim human NK-cell subsets. Blood 115, 274–281 (2010).
Wagner, J. A. et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J. Clin. Invest. 127, 4042–4058 (2017).
Dong, H. et al. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat. Immunol. 20, 865–878 (2019).
Gotthardt, D., Trifinopoulos, J., Sexl, V. & Putz, E. M. JAK/STAT cytokine signaling at the crossroad of NK cell development and maturation. Front. Immunol. 10, 2590 (2019).
Lau, C. M. et al. Epigenetic control of innate and adaptive immune memory. Nat. Immunol. 19, 963–972 (2018).
Madera, S. & Sun, J. C. Cutting edge: stage-specific requirement of IL-18 for antiviral NK cell expansion. J. Immunol. 194, 1408–1412 (2015).
Romee, R. et al. Cytokine activation induces human memory-like NK cells. Blood 120, 4751–4760 (2012).
Sciumè, G. et al. Rapid enhancer remodeling and transcription factor repurposing enable high magnitude gene induction upon acute activation of NK cells. Immunity 53, 745–758.e4 (2020). A study presenting a new mechanism for transcription factor use in acute activation of NK cells.
Salazar-Mather, T. P., Orange, J. S. & Biron, C. A. Early murine cytomegalovirus (MCMV) infection induces liver natural killer (NK) cell inflammation and protection through macrophage inflammatory protein 1alpha (MIP-1alpha)-dependent pathways. J. Exp. Med. 187, 1–14 (1998).
Trifilo, M. J. et al. CXC chemokine ligand 10 controls viral infection in the central nervous system: evidence for a role in innate immune response through recruitment and activation of natural killer cells. J. Virol. 78, 585–594 (2004).
Thapa, M., Kuziel, W. A. & Carr, D. J. J. Susceptibility of CCR5-deficient mice to genital herpes simplex virus type 2 is linked to NK cell mobilization. J. Virol. 81, 3704–3713 (2007).
Gazit, R. et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat. Immunol. 7, 517–523 (2006).
Huot, N. et al. Natural killer cells migrate into and control simian immunodeficiency virus replication in lymph node follicles in African green monkeys. Nat. Med. 23, 1277–1286 (2017). A study showing the importance of NK cell migration to tissues in control of viral infection in non-human primates.
Nikzad, R. et al. Human natural killer cells mediate adaptive immunity to viral antigens. Sci. Immunol. 4, eaat8116 (2019). The first article showing antigen-specific NK cell responses in humanized mice.
Dogra, P. et al. Tissue determinants of human NK cell development, function, and residence. Cell 180, 749–763.e13 (2020).
Weizman, O.-E. et al. ILC1 confer early host protection at initial sites of viral infection. Cell 171, 795–808.e12 (2017).
Zhou, J. et al. Liver-resident NK cells control antiviral activity of hepatic T cells via the PD-1-PD-L1 axis. Immunity 50, 403–417.e4 (2019).
Peppa, D. et al. Up-regulation of a death receptor renders antiviral T cells susceptible to NK cell-mediated deletion. J. Exp. Med. 210, 99–114 (2013).
O’Leary, J. G., Goodarzi, M., Drayton, D. L. & Andrian, von, U. H. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat. Immunol. 7, 507–516 (2006). The first report on antigen-specific NK cell responses.
Paust, S. et al. Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat. Immunol. 11, 1127–1135 (2010).
Stary, V. et al. A discrete subset of epigenetically primed human NK cells mediates antigen-specific immune responses. Sci. Immunol. 5, eaba6232 (2020). The first study reporting on antigen-specific NK cell responses in humans.
Khakoo, S. I. et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 305, 872–874 (2004).
Golden-Mason, L., Cox, A. L., Randall, J. A., Cheng, L. & Rosen, H. R. Increased natural killer cell cytotoxicity and NKp30 expression protects against hepatitis C virus infection in high-risk individuals and inhibits replication in vitro. Hepatology 52, 1581–1589 (2010).
Alter, G. et al. Reduced frequencies of NKp30+NKp46+, CD161+, and NKG2D+ NK cells in acute HCV infection may predict viral clearance. J. Hepatol. 55, 278–288 (2011).
Semmo, N. et al. Preferential loss of IL-2-secreting CD4+ T helper cells in chronic HCV infection. Hepatology 41, 1019–1028 (2005).
Shen, T. et al. PD-1 expression on peripheral CD8+ TEM/TEMRA subsets closely correlated with HCV viral load in chronic hepatitis C patients. Virol. J. 7, 310 (2010).
Bengsch, B. et al. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. 6, e1000947 (2010).
Waggoner, S. N., Cornberg, M., Selin, L. K. & Welsh, R. M. Natural killer cells act as rheostats modulating antiviral T cells. Nature 481, 394–398 (2012).
Lang, P. A. et al. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc. Natl Acad. Sci. USA 109, 1210–1215 (2012).
Nattermann, J. et al. Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis C. Gut 55, 869–877 (2006).
De Maria, A. et al. Increased natural cytotoxicity receptor expression and relevant IL-10 production in NK cells from chronically infected viremic HCV patients. Eur. J. Immunol. 37, 445–455 (2007).
Sène, D. et al. Hepatitis C virus (HCV) evades NKG2D-dependent NK cell responses through NS5A-mediated imbalance of inflammatory cytokines. PLoS Pathog. 6, e1001184 (2010).
Oliviero, B. et al. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology 137, 1151–1160 (2009).
Serti, E. et al. Successful interferon-free therapy of chronic hepatitis C virus infection normalizes natural killer cell function. Gastroenterology 149, 190–200.e2 (2015).
Lunemann, S. et al. Compromised function of natural killer cells in acute and chronic viral hepatitis. J. Infect. Dis. 209, 1362–1373 (2014).
Yoon, J. C., Lim, J.-B., Park, J. H. & Lee, J. M. Cell-to-cell contact with hepatitis C virus-infected cells reduces functional capacity of natural killer cells. J. Virol. 85, 12557–12569 (2011).
Scully, E. & Alter, G. NK cells in HIV disease. Curr. HIV AIDS Rep. 13, 85–94 (2016).
Flórez-Álvarez, L., Hernández, J. C. & Zapata, W. NK cells in HIV-1 infection: from basic science to vaccine strategies. Front. Immunol. 9, 2290 (2018).
Gallitano, S. M., McDermott, L., Brar, K. & Lowenstein, E. Use of tumor necrosis factor (TNF) inhibitors in patients with HIV/AIDS. J. Am. Acad. Dermatol. 74, 974–980 (2016).
Roff, S. R., Noon-Song, E. N. & Yamamoto, J. K. The significance of Interferon-γ in HIV-1 pathogenesis, therapy, and prophylaxis. Front. Immunol. 4, 498 (2014).
Zapata, W. et al. Influence of CCR5 and CCR2 genetic variants in the resistance/susceptibility to HIV in serodiscordant couples from Colombia. AIDS Res. Hum. Retroviruses 29, 1594–1603 (2013).
Martin, M. P. et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat. Genet. 39, 733–740 (2007).
Boulet, S. et al. Increased proportion of KIR3DS1 homozygotes in HIV-exposed uninfected individuals. AIDS 22, 595–599 (2008).
Martin, M. P. et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat. Genet. 31, 429–434 (2002).
Long, E. O., Kim, H. S., Liu, D., Peterson, M. E. & Rajagopalan, S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu. Rev. Immunol. 31, 227–258 (2013).
Alter, G. et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature 476, 96–100 (2011).
Hölzemer, A. et al. Selection of an HLA-C*03:04-restricted HIV-1 p24 Gag sequence variant is associated with viral escape from KIR2DL3+natural killer cells: data from an observational cohort in South Africa. PLoS Med. 12, e1001900; discussion e1001900 (2015).
Alter, G. et al. Differential natural killer cell-mediated inhibition of HIV-1 replication based on distinct KIR/HLA subtypes. J. Exp. Med. 204, 3027–3036 (2007).
Collins, K. L., Chen, B. K., Kalams, S. A., Walker, B. D. & Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391, 397–401 (1998).
Specht, A. et al. Selective downmodulation of HLA-A and -B by Nef alleles from different groups of primate lentiviruses. Virology 373, 229–237 (2008).
Richard, J., Sindhu, S., Pham, T. N. Q., Belzile, J.-P. & Cohen, E. A. HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood 115, 1354–1363 (2010).
Norman, J. M. et al. The antiviral factor APOBEC3G enhances the recognition of HIV-infected primary T cells by natural killer cells. Nat. Immunol. 12, 975–983 (2011).
Shah, A. H. et al. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe 8, 397–409 (2010).
Ackerman, M. E. et al. Polyfunctional HIV-specific antibody responses are associated with spontaneous HIV control. PLoS Pathog. 12, e1005315 (2016).
Hong, H. S. et al. Loss of CCR7 expression on CD56bright NK cells is associated with a CD56dimCD16+ NK cell-like phenotype and correlates with HIV viral load. PLoS ONE 7, e44820 (2012).
Schafer, J. L., Li, H., Evans, T. I., Estes, J. D. & Reeves, R. K. Accumulation of cytotoxic CD16+ NK cells in simian immunodeficiency virus-infected lymph nodes associated with in situ differentiation and functional anergy. J. Virol. 89, 6887–6894 (2015).
Ripa, M. et al. Dynamics of adaptive and innate immunity in patients treated during primary human immunodeficiency virus infection: results from Maraviroc in HIV Acute Infection (MAIN) randomized clinical trial. Clin. Microbiol. Infect. 21, 876.e1–4 (2015).
Horowitz, A. et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci. Transl Med. 5, 208ra145 (2013).
Strauss-Albee, D. M. et al. Human NK cell repertoire diversity reflects immune experience and correlates with viral susceptibility. Sci. Transl Med. 7, 297ra115 (2015).
Adams, N. M., Grassmann, S. & Sun, J. C. Clonal expansion of innate and adaptive lymphocytes. Nat. Rev. Immunol. 20, 694–707 (2020).
Arase, H., Mocarski, E. S., Campbell, A. E., Hill, A. B. & Lanier, L. L. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296, 1323–1326 (2002).
Sun, J. C., Beilke, J. N. & Lanier, L. L. Adaptive immune features of natural killer cells. Nature 457, 557–561 (2009).
Gumá, M. et al. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood 107, 3624–3631 (2006).
Björkström, N. K. et al. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J. Exp. Med. 208, 13–21 (2011).
Petitdemange, C. et al. Unconventional repertoire profile is imprinted during acute chikungunya infection for natural killer cells polarization toward cytotoxicity. PLoS Pathog. 7, e1002268 (2011).
Gumá, M. et al. Human cytomegalovirus infection is associated with increased proportions of NK cells that express the CD94/NKG2C receptor in aviremic HIV-1-positive patients. J. Infect. Dis. 194, 38–41 (2006). An important article reporting on adaptive-like NK cell expansions in response to HCMV-infected cells.
Björkström, N. K., Svensson, A., Malmberg, K.-J., Eriksson, K. & Ljunggren, H.-G. Characterization of natural killer cell phenotype and function during recurrent human HSV-2 infection. PLoS ONE 6, e27664 (2011).
Hendricks, D. W. et al. Cutting edge: NKG2ChiCD57+ NK cells respond specifically to acute infection with cytomegalovirus and not Epstein-Barr virus. J. Immunol. 192, 4492–4496 (2014).
Malone, D. F. G. et al. Cytomegalovirus-driven adaptive-like natural killer cell expansions are unaffected by concurrent chronic hepatitis virus infections. Front. Immunol. 8, 14725–7 (2017).
Schlums, H. et al. Adaptive NK cells can persist in patients with GATA2 mutation depleted of stem and progenitor cells. Blood 129, 1927–1939 (2017).
Cichocki, F. et al. ARID5B regulates metabolic programming in human adaptive NK cells. J. Exp. Med. 215, 2379–2395 (2018).
Lee, J. et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 42, 431–442 (2015).
Schlums, H. et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 42, 443–456 (2015). Together with Lee et al. (2015), the first work showing epigenetic modifications of adaptive-like NK cells.
Rölle, A. et al. IL-12-producing monocytes and HLA-E control HCMV-driven NKG2C+ NK cell expansion. J. Clin. Invest. 124, 5305–5316 (2014).
Hammer, Q. et al. Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat. Immunol. 19, 453–463 (2018). An article reporting on the viral specificity for adaptive-like NK cell expansions.
Liu, L. L. et al. Critical role of CD2 co-stimulation in adaptive natural killer cell responses revealed in NKG2C-deficient humans. Cell Rep. 15, 1088–1099 (2016).
Björkström, N. K., Ljunggren, H.-G. & Sandberg, J. K. CD56 negative NK cells: origin, function, and role in chronic viral disease. Trends Immunol. 31, 401–406 (2010).
Müller-Durovic, B., Grählert, J., Devine, O. P., Akbar, A. N. & Hess, C. CD56-negative NK cells with impaired effector function expand in CMV and EBV co-infected healthy donors with age. Aging 11, 724–740 (2019).
Mavilio, D. et al. Characterization of CD56-/CD16+ natural killer (NK) cells: a highly dysfunctional NK subset expanded in HIV-infected viremic individuals. Proc. Natl Acad. Sci. USA 102, 2886–2891 (2005).
Eller, M. A. et al. Elevated natural killer cell activity despite altered functional and phenotypic profile in Ugandans with HIV-1 clade A or clade D infection. J. Acquir. Immune Defic. Syndr. 51, 380–389 (2009).
Mavilio, D. et al. Natural killer cells in HIV-1 infection: dichotomous effects of viremia on inhibitory and activating receptors and their functional correlates. Proc. Natl Acad. Sci. USA 100, 15011–15016 (2003).
Alter, G. et al. Sequential deregulation of NK cell subset distribution and function starting in acute HIV-1 infection. Blood 106, 3366–3369 (2005).
Gonzalez, V. D. et al. Expansion of functionally skewed CD56-negative NK cells in chronic hepatitis C virus infection: correlation with outcome of pegylated IFN-alpha and ribavirin treatment. J. Immunol. 183, 6612–6618 (2009).
Voigt, J. et al. Proteome analysis of human CD56neg NK cells reveals a homogeneous phenotype surprisingly similar to CD56dim NK cells. Eur. J. Immunol. 48, 1456–1469 (2018).
Mace, E. M. & Orange, J. S. Emerging insights into human health and NK cell biology from the study of NK cell deficiencies. Immunol. Rev. 287, 202–225 (2019).
Biron, C. A., Byron, K. S. & Sullivan, J. L. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 320, 1731–1735 (1989). The first article reporting on an individual with a complete NK cell deficiency.
Gineau, L. et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J. Clin. Invest. 122, 821–832 (2012).
Hanna, S., Béziat, V., Jouanguy, E., Casanova, J.-L. & Etzioni, A. A homozygous mutation of RTEL1 in a child presenting with an apparently isolated natural killer cell deficiency. J. Allergy Clin. Immunol. 136, 1113–1114 (2015).
Cottineau, J. et al. Inherited GINS1 deficiency underlies growth retardation along with neutropenia and NK cell deficiency. J. Clin. Invest. 127, 1991–2006 (2017).
Mace, E. M. et al. Biallelic mutations in IRF8 impair human NK cell maturation and function. J. Clin. Invest. 127, 306–320 (2017).
Mace, E. M. et al. Human NK cell deficiency as a result of biallelic mutations in MCM10. J. Clin. Invest. 130, 5272–5286 (2020).
Grier, J. T. et al. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J. Clin. Invest. 122, 3769–3780 (2012).
de Vries, E. et al. Identification of an unusual Fc gamma receptor IIIa (CD16) on natural killer cells in a patient with recurrent infections. Blood 88, 3022–3027 (1996).
Spinner, M. A. et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood 123, 809–821 (2014).
Guan, W.-J. et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 382, 1708–1720 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Wilk, A. J. et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 26, 1070–1076 (2020).
Varchetta, S. et al. Unique immunological profile in patients with COVID-19. Cell. Mol. Immunol. 395, 497 (2020).
Xu, G. et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discov. 6, 73–14 (2020).
Liao, M. et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 26, 842–844 (2020).
Chua, R. L. et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 38, 970–979 (2020).
Brownlie, D. et al. Distinct lung-homing receptor expression and activation profiles on NK cell and T cell subsets in COVID-19 and influenza. Preprint at bioRxiv https://doi.org/10.1101/2021.01.13.426553 (2021).
Osman, M. et al. Impaired natural killer cell counts and cytolytic activity in patients with severe COVID-19. Blood Adv. 4, 5035–5039 (2020).
Rendeiro, A. F. et al. Longitudinal immune profiling of mild and severe COVID-19 reveals innate and adaptive immune dysfunction and provides an early prediction tool for clinical progression. Preprint at medRxiv https://doi.org/10.1101/2020.09.08.20189092 (2020).
Shrock, E. et al. Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity. Science 370, eabd4250 (2020).
Le Balc’h, P. et al. Herpes simplex virus and cytomegalovirus reactivations among severe COVID-19 patients. Crit. Care 24, 530–3 (2020).
Bortolotti, D., Gentili, V., Rizzo, S., Rotola, A. & Rizzo, R. SARS-CoV-2 spike 1 protein controls natural killer cell activation via the HLA-E/NKG2A pathway. Cells 9, 1975 (2020).
Sahoo, D. et al. AI-guided discovery of the invariant host response to viral pandemics. Preprint at bioRxiv https://doi.org/10.1101/2020.09.21.305698 (2020).
Liu, C. et al. Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19. Cell 184, 1836–1857.e22 (2021).
Kvedaraite, E. et al. Major alterations in the mononuclear phagocyte landscape associated with COVID-19 severity. Proc. Natl Acad. Sci. USA 118, e2018587118 (2021).
Freud, A. G. et al. A human CD34+ subset resides in lymph nodes and differentiates into CD56bright natural killer cells. Immunity 22, 295–304 (2005).
Juelke, K. et al. CD62L expression identifies a unique subset of polyfunctional CD56dim NK cells. Blood 116, 1299–1307 (2010).
Lopez-Vergès, S. et al. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood 116, 3865–3874 (2010).
Marquardt, N. et al. Cutting edge: identification and characterization of human intrahepatic CD49a+ NK cells. J. Immunol. 194, 2467–2471 (2015).
Marquardt, N. et al. Unique transcriptional and protein-expression signature in human lung tissue-resident NK cells. Nat. Commun. 10, 3841–12 (2019).
Ivarsson, M. A. et al. Composition and dynamics of the uterine NK cell KIR repertoire in menstrual blood. Mucosal Immunol. 10, 322–331 (2017).
Snyder, M. E. et al. Generation and persistence of human tissue-resident memory T cells in lung transplantation. Sci. Immunol. 4, eaav5581 (2019).
Zuber, J. et al. Bidirectional intragraft alloreactivity drives the repopulation of human intestinal allografts and correlates with clinical outcome. Sci. Immunol. 1, eaah3732 (2016).
Lim, A. I. et al. Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell 168, 1086–1100.e10 (2017).
Seillet, C., Brossay, L. & Vivier, E. Natural killers or ILC1s? That is the question. Curr. Opin. Immunol. 68, 48–53 (2021).
Reeves, R. K. et al. Antigen-specific NK cell memory in rhesus macaques. Nat. Immunol. 16, 927–932 (2015). A study showing the presence of antigen-specific NK cell responses in non-human primates.
Wijaya, R. S. et al. HBV vaccination and HBV infection induces HBV-specific natural killer cell memory. Gut 70, 357–369 (2021).
Martin, B. et al. Restoration of HCV-specific CD8+ T cell function by interferon-free therapy. J. Hepatol. 61, 538–543 (2014).
Aregay, A. et al. Elimination of hepatitis C virus has limited impact on the functional and mitochondrial impairment of HCV-specific CD8+ T cell responses. J. Hepatol. 71, 889–899 (2019).
Hengst, J. et al. Direct-acting antiviral-induced hepatitis C virus clearance does not completely restore the altered cytokine and chemokine milieu in patients with chronic hepatitis C. J. Infect. Dis. 214, 1965–1974 (2016).
Hengst, J. et al. Nonreversible MAIT cell-dysfunction in chronic hepatitis C virus infection despite successful interferon-free therapy. Eur. J. Immunol. 46, 2204–2210 (2016).
Serti, E. et al. Rapid decrease in hepatitis C viremia by direct acting antivirals improves the natural killer cell response to IFNα. Gut 66, 724–735 (2017).
Strunz, B. et al. Chronic hepatitis C virus infection irreversibly impacts human natural killer cell repertoire diversity. Nat. Commun. 9, 2275 (2018). A report assessing the restoration of the NK cell compartment after resolution of chronic HCV infection.
Merino, A. et al. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J. Clin. Invest. 129, 3770–3785 (2019).
Acknowledgements
The authors thank present and past members of their groups for their contributions towards the understanding of human natural killer cells in health and disease. Their work was supported by the Swedish Research Council, Sweden’s Innovation Agency, the European Research Council under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 948692), the Swedish Cancer Society, the Swedish Foundation for Strategic Research, the Knut and Alice Wallenberg Foundation, Nordstjernan AB, the Center for Innovative Medicine at Karolinska Institutet, Region Stockholm, SRP Diabetes Karolinska Institutet, StratRegen Karolinska Institutet and Karolinska Institutet. The authors apologize to those colleagues whose work has not been cited owing to space constraints.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Immunology thanks C. Biron and the other, anonymous, reviewers for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Antibody-dependent cellular cytotoxicity
-
(ADCC). A mechanism by which natural killer cells, via their Fc receptor CD16, kill a target cell whose membrane-surface antigens have been bound by a specific antibody.
- NK cell education
-
The process involving acquisition of functional competence in natural killer (NK) cells, often mediated by interactions between inhibitory receptors and the corresponding ligands.
- Adaptive-like NK cell
-
A subset of expanded terminally differentiated natural killer (NK) cells that, in the context of human cytomegalovirus infection, is characterized by expression of NKG2C.
- JAK–STAT pathways
-
Signalling pathways involving Janus kinases (JAKs) and signal transducers and activators of transcription (STATs) downstream of many cytokine receptors, including those for IL-2, IL-12, IL-15 and type I interferons, but not IL-18, important in natural killer cell activation, proliferation and survival.
- Killer cell immunoglobulin-like receptor
-
(KIR). A family of highly polymorphic activating and inhibitory receptors that serve as key regulators of human natural killer cell function.
- HLA-E
-
Non-classical MHC class I molecule characterized by a limited polymorphism that normally binds a restricted subset of peptides derived from signal peptides.
Rights and permissions
About this article

Cite this article
Björkström, N.K., Strunz, B. & Ljunggren, HG. Natural killer cells in antiviral immunity. Nat Rev Immunol 22, 112–123 (2022). https://doi.org/10.1038/s41577-021-00558-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-021-00558-3
This article is cited by
-
Nanoparticles in tumor microenvironment remodeling and cancer immunotherapy
Journal of Hematology & Oncology (2024)
-
The causality between CD8+NKT cells and CD16−CD56 on NK cells with hepatocellular carcinoma: a Mendelian randomization study
Infectious Agents and Cancer (2024)
-
Cytokine-responsive T- and NK-cells portray SARS-CoV-2 vaccine-responders and infection in multiple myeloma patients
Leukemia (2024)
-
Natural killer cell therapies
Nature (2024)
-
Targeting immunogenic cell stress and death for cancer therapy
Nature Reviews Drug Discovery (2024)
