
Abstract
The cGAS–STING signalling pathway has emerged as a key mediator of inflammation in the settings of infection, cellular stress and tissue damage. Underlying this broad involvement of the cGAS–STING pathway is its capacity to sense and regulate the cellular response towards microbial and host-derived DNAs, which serve as ubiquitous danger-associated molecules. Insights into the structural and molecular biology of the cGAS–STING pathway have enabled the development of selective small-molecule inhibitors with the potential to target the cGAS–STING axis in a number of inflammatory diseases in humans. Here, we outline the principal elements of the cGAS–STING signalling cascade and discuss the general mechanisms underlying the association of cGAS–STING activity with various autoinflammatory, autoimmune and degenerative diseases. Finally, we outline the chemical nature of recently developed cGAS and STING antagonists and summarize their potential clinical applications.
Similar content being viewed by others
Introduction
The detection of foreign DNA serves as a crucial element of immunity in many organisms. In mammalian cells, this task is contributed in large part by the cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway, which has emerged as a critical mechanism for coupling the sensing of DNA to the induction of powerful innate immune defence programmes1. Within this pathway, the binding of cGAS to double-stranded DNA (dsDNA) allosterically activates its catalytic activity and leads to the production of 2′3′ cyclic GMP–AMP (cGAMP), a second messenger molecule and potent agonist of STING2,3,4,5,6,7,8. A salient feature of the cGAS–STING pathway, which sets it apart from several other innate immune signalling mechanisms, is that its activation is triggered by a fundamental element of life (namely DNA) and, therefore, lacks any pathogen-specific attributes9. For this reason, cGAS recognizes a broad repertoire of DNA species of both foreign and self origin. Today, our understanding of the diverse functions of the cGAS–STING pathway in host immunity has become clearer, and multiple examples highlight the protective effects of this pathway during infection. Recent studies showing that the cGAS–STING system arose from an ancient bacterial anti-phage mechanism underscore this notion10,11. Growing evidence has indicated, however, that dysregulation of this highly versatile innate immune sensing system can disrupt cellular and organismal homeostasis by fuelling aberrant innate immune responses associated with a number of pathologies1. The parameters that dictate host-protective versus pathogenic activity are still being unfolded, but it appears that the intensity and chronicity of cGAS–STING signalling are major determinants in most cases. In light of this, efforts have been undertaken or are still under way to define strategies that allow selective modulation of cGAS–STING activity in various disease settings12,13.
We note that there are major drug discovery efforts currently under way to identify and translate agonists of the cGAS–STING pathway as vaccine adjuvants or as anticancer immunostimulatory agents, and these topics have been extensively covered in recent reviews14,15. By contrast, in this Review, we particularly focus on new developments concerning the role of the cGAS–STING pathway as a major driver of inflammatory diseases. We briefly summarize the current state of understanding of signalling through the cGAS–STING pathway, outline molecular mechanisms that trigger cGAS–STING pathway activity in distinct pathophysiological contexts, and analyse its involvement in preclinical models of disease. Drawing on recent advances in understanding of the cGAS–STING pathway structure and regulation, we examine pharmacological intervention strategies that target this pathway and discuss their therapeutic potential in the treatment of inflammatory and autoinflammatory diseases.
Overview of the cGAS–STING pathway
The synthesis of cGAMP is considered the crucial first step that sets in motion cGAS-mediated antiviral effects in various species3,4,5,6,7,8. In mammals, cGAS catalytic activity is triggered through interactions with dsDNA, a process that through extensive structural and biochemical studies is now understood in detail5,16,17,18,19,20. In brief, the C-terminal part of human cGAS (h-cGAS) harbouring the nucleotidyltransferase domain (the catalytic part) contains positively charged DNA-binding sites, one primary site and two additional sites, which bind the sugar–phosphate backbone of DNA. DNA binding to the primary site, the so-called A-site, induces conformational changes in the protein that rearrange the catalytic pocket of the enzyme to allow for an optimal interaction with the substrates ATP and GTP, whereas DNA binding to the juxtaposed B-site is crucial for the formation of the core 2:2 cGAS DNA complex — the minimal active enzymatic unit. On long dsDNA molecules, cGAS dimers arrange themselves into ladder-like networks and phase-separated organelles21,22. The formation of these spatially restricted higher-order cGAS–DNA assemblies on longer stretches of DNA is considered critical for triggering biological effects23. This mode of activation offers an important inbuilt safeguard mechanism for living cells: namely, that cGAS signalling can only be initiated when longer stretches of dsDNA are available to surpass a certain signalling threshold. By contrast, spurious activation of cGAS on limited and short dsDNA is prevented.
cGAMP is then detected by the endoplasmic reticulum (ER) membrane protein STING2,24 (Fig. 1). STING is composed of a short cytosolic N-terminal segment, a four-span transmembrane domain, a connector region and a cytosolic ligand-binding domain (LBD) on which a C-terminal tail (CTT) is appended. Already in the apo-form (that is, in the absence of a ligand), STING forms a domain-swapped homodimer, which on binding to cGAMP undergoes extensive conformational rearrangements, including an untwisting (180° rotation) of the LBD, an inward rotation of the LBD and the formation of an ordered β-sheet that covers the LBD25,26,27,28,29,30. Together, these changes assist in side-by-side oligomerization of STING dimers29,30. Oligomerization is further fostered by disulfide bridges spanning separate STING dimers as well as by palmitoylation of cysteine residues, C88 and C91 (refs13,29,31). Thus, the oligomerized STING dimers form the activated STING unit capable of initiating effector functions. It is interesting to note that canonical cyclic dinucleotides — as well as a certain class of small-molecule STING agonists — confer stimulatory activity without the rearrangement of the lid region, but still promote oligomerization29,32.

A schematic detailing double-stranded DNA (dsDNA)-induced activation of cytosolic cyclic GMP–AMP synthase (cGAS), which can occur through pathogen infection or cellular stress. On binding dsDNA, cGAS dimers assemble on dsDNA resulting in enzymatic activation of cGAS and synthesis of 2′3′ cyclic GMP–AMP (cGAMP). cGAMP binds to stimulator of interferon genes (STING) dimers localized at the endoplasmic reticulum (ER) membrane, which leads to profound conformational changes that trigger STING oligomerization, liberation from anchoring factors (such as STIM1), interaction with trafficking factors (for example, SEC24/23, STEEP, not depicted in figure) and, finally, incorporation into coatomer protein complex II (COPII) vesicles. On passing through the ER–Golgi intermediate compartment (ERGIC) and Golgi, STING recruits TANK-binding kinase 1 (TBK1), promoting TBK1 autophosphorylation, STING phosphorylation at Ser366 and recruitment of interferon regulatory factor 3 (IRF3). The phosphorylation of IRF3 by TBK1 enables IRF3 dimerization and translocation to the nucleus to induce gene expression of type I interferons, interferon-stimulated genes (ISGs), and several other inflammatory mediators, pro-apoptotic genes and chemokines. Activation of STING also leads to NF-κB activation and the formation of LC3+ vesicles (autophagosomes) by a non-canonical mechanism of autophagy. In the end, both STING within autophagosomes and STING from the Golgi traffic to the lysosome, where STING degradation occurs. Steady-state STING translocation through the secretory pathway in the absence of robust cGAMP stimulation is counteracted by continuous retrograde transport to the ER, a step that is mediated by COPI vesicles and facilitated by an interaction of STING with SURF4.
At the cellular level, a prerequisite for initiation of downstream signalling is the translocation of STING from the ER through the ER–Golgi intermediate compartment (ERGIC) to the Golgi33. As yet, the precise molecular steps and interrelationship between putative players involved in this trafficking process are incompletely resolved. What is known is that trafficking of STING is facilitated by the canonical ER-to-Golgi transport machinery composed of coatomer protein complex II (COPII) vesicles, relying on the GTPase SAR1A and the COPII complex components, including SEC24C as well as the ARF-GTPase ARF1 (ref.34). Various other factors have been proposed to facilitate the regulated, activation-dependent export of STING or, alternatively, to anchor STING at the ER under non-stimulated conditions35,36. On reaching the ERGIC and Golgi compartments, STING recruits TANK-binding kinase 1 (TBK1) to initiate downstream signalling — as discussed later in detail. At the same time, STING’s movement out of the ER also promotes the formation of LC3+ autophagic vesicles34. Meanwhile, recent insights from human genetic studies (see below) have unfolded a mechanism that operates at steady state and actively retrieves STING from the Golgi back to the ER to suppress cellular activation. Specifically, the adaptor protein SURF4 interacts with STING at the Golgi to facilitate STING’s encapsulation into COPI vesicles for retrograde transport37,38. Although future work needs to refine the molecular coordination of the intracellular destinations of STING, they appear to ultimately converge with STING’s degradation in lysosomes39.
Effects of cGAS–STING pathway activation
Throughout evolutionary history, activation of the cGAS–STING pathway appears to have served to protect against viral infection and is associated with an antiviral cellular programme40. In human and mouse cells, the most prominent downstream effector function of cGAMP signalling is the de novo synthesis of antiviral type I interferons and related gene products, which are generally assumed to account for most of the pathway’s antiviral functions40. However, the cGAS–STING pathway is also present in prokaryotes and in species such as the sea anemone Nematostella vectensis and the fruit fly Drosophila melanogaster, which do not encode type I interferons10,11,41,42. Thus, cGAS–STING originally relied on more evolutionarily ancient antiviral defence strategies, which are still functional within human cells and are highly significant for various biological consequences of the cGAS–STING pathway (Fig. 2).

a | Stimulator of interferon genes (STING) mediates inflammatory and antiviral cellular programmes by engaging the transcription factors interferon regulatory factor 3 (IRF3) and canonical NF-κB (RELA–p50). IRF3 activation is strictly controlled by TANK-binding kinase 1 (TBK1), which binds to the C-terminal tail (CTT) of STING through a conserved PLPLRT/SD binding motif to then phosphorylate Ser366 within the CTT (phospho-site). In certain cells, activation of NF-κB is less dependent on the CTT and TBK1. In addition, non-canonical NF-κB (RELB–p52) is activated downstream of STING and counteracts IRF3 and canonical NF-κB signalling. STING impacts cell survival and cellular proliferation in diverse ways and is associated with the induction of apoptosis, necroptosis, pyroptosis and cellular senescence. Both necroptosis and senescence can be triggered by STING-induced cytokines. Apoptosis induction can proceed through the upregulation of pro-apoptotic genes (PUMA, NOXA) or, alternatively, directly engage BAX and BAK at the mitochondrial membrane. STING-mediated lysosomal damage has been shown to activate NLRP3 inflammasome-dependent pyroptosis. b | Intercellular 2′3′ cyclic GMP–AMP (cGAMP) transfer can be mediated by connexin proteins that form gap junctions between neighbouring cells, which can be homotypic or heterotypic. Additionally, various cell types use the voltage-dependent anion channel (VDAC) composed of LRCC8 subunits to import and export cGAMP. In the extracellular space, cGAMP is subject to degradation by the enzyme ectonucleotide pyrophosphatase/phosphodiesterase family member 1 (ENPP1). Tumour cells have also been shown to export cGAMP whereas myeloid cells use SLC19A1 or P2XR7 to import cGAMP. Connexin channels also mediate heterotypic cellular cGAMP transfer. APC, antigen-presenting cell; cGAS, cyclic GMP–AMP synthase; IFN, interferon; ISG, interferon-stimulated gene.
Control of transcriptional responses by cGAS–STING
The activation of interferon regulatory factor 3 (IRF3) and its induction of type I interferons and other interferon-stimulated genes (ISGs), is by far the best-understood process controlled by cGAS–STING activation. The CTT of STING first recruits TBK1 through a conserved PLPLRT/SD amino acid binding motif, which promotes dimerization-mediated TBK1 autophosphorylation43,44. Activated TBK1, in turn, phosphorylates STING at Ser366 (unless otherwise indicated, we refer to the human protein sequence) in the CTT, which is part of a pLxIS motif (in which p represents the hydrophilic residue, x represents any residue and S represents the phosphorylation site) and which allows the STING–TBK1 complex to recruit IRF3 (ref.45). TBK1 then phosphorylates IRF3, enabling its dimerization, nuclear translocation and target gene induction. In addition to type I interferons and ISGs, IRF3 activity is also required for the induction of many other target genes, including genes encoding inflammatory cytokines (such as IL6 and IL12)46. Not surprisingly, the IRF3-controlled transcriptional output varies considerably depending on the cell type under study, the STING trigger used and the exact in vivo context. Crucially, the previously mentioned oligomerization of STING is highly significant for these events to occur, because owing to sterical constraints, a given STING dimer cannot serve simultaneously as the docking site for TBK1 and its substrate43. Consistently, interfering with STING oligomerization significantly reduces its ability to promote type I interferon upregulation.
Undoubtedly, type I interferons are critical for STING’s antiviral activity. However, STING’s role in immunity is not solely governed by type I interferon signalling. A case in point has been made recently by the demonstration that mice harbouring a mutation in STING that selectively impacts its type I interferon-inducing capacity are still protected against herpes simplex virus 1 (HSV-1) infection, whereas mice with complete deficiency for STING are susceptible46,47. Another major signalling module used by STING is NF-κB-mediated transcriptional activation. This STING-dependent activity does not fully depend on the CTT48,49. As such, although TBK1 itself can promote NF-κB activation, it is not required and can be compensated for by IKKε upstream of TAK1 and IKK complexes, at least in certain cell types50. Consistent with this observation, ancestral STING homologues in insects and early metazoans completely lack the CTT signalling element, but can still function in host defence by promoting NF-κB responses, arguing that this constitutes a possible archetypal STING-dependent antimicrobial mechanism. Interestingly, certain vertebrate fish species, such as zebrafish and other ray-finned fish species, have acquired additional motifs appended to the CTT to further boost NF-κB activation via the recruitment of TRAF6 (ref.49). Although STING-mediated NF-κB transcription appears to be a highly conserved and relevant downstream activity, how STING interacts with NF-κB pathway components at a molecular resolution still remains obscure. Stimulation of cGAS–STING also promotes non-canonical NF-κB responses through the triggering of p52–RELB nuclear translocation51,52. This signalling output, which limits type I interferons and canonical NF-κB, emerges as a critical negative regulator of STING effector mechanisms, which can have important biological consequences with regard to cancer immune escape and metastasis (see below). In certain contexts, cGAS–STING signalling may additionally impact p53, MAPK p38 and STAT3 signalling, suggesting yet alternative routes through which STING may influence cell states and cytokine production.
cGAS–STING and autophagy
The discussion so far has focused on STING-mediated control of gene transcription, but STING also promotes more immediate (antiviral) cellular processes, with autophagy being a key effector activity. Indeed, cGAS–STING signalling is intimately tied to autophagy — not only does cGAS–STING induce autophagy, as a cell-autonomous defence mechanism, but conversely it is also subject to regulation by autophagy components. Several earlier studies investigated the connection between STING and autophagy with some unresolved differences in the exact mechanism of autophagy induction, potentially reflecting a strong context-dependent regulatory component. Although canonical autophagy, relying on the ULK complex and TBK1, has been implicated in the control of STING-mediated autophagic vesicle formation, it appears that activation of STING can also trigger a non-canonical autophagy response requiring only selective autophagy machinery components, including the PI3P effector WIPI2 and the ATG5-12-16L1 complex34,53,54,55,56. Befitting its critical role in cell autonomous antiviral immunity, autophagy induced by STING restricts viral propagation and could have served as a major host defence programme in certain species42. For humans, the STING–autophagy connection appears to serve an additional benefit to the host. As such, a recent study reported that during replicative crisis, cells engage the STING–autophagy pathway to drive an autophagic cell death programme, thereby preventing tumour cell outgrowth57. Of note, autophagy components also feed back on the regulation of STING activity by assisting STING intracellular trafficking capabilities as well as its lysosomal degradation34,39,53. Accordingly, cells deficient in autophagy proteins or treated with drugs that inhibit lysosomal acidification, display enhanced type I interferon production39. Interfering with autophagy pathway components in tumours augments the efficacy of antitumour strategies in a STING-dependent manner, consistent with heightened cGAS–STING pathway activity39. Autophagy also prevents STING activation by delivering cytosolic DNA to the lysosome where it is degraded by DNase II34. Altogether, autophagy is a key downstream activity of STING, which is not only important as a downstream effector module, but also as a critical coordinator to restore homeostasis after pathway engagement.
Anti-proliferative and cell death functions
Activation of STING initiates cascades that support anti-proliferative cell states, including cellular senescence, and ultimately cell death (a more detailed discussion of senescence is provided further below). To that end, STING promotes induction of cell-cycle inhibitors (such as p21) and pro-apoptotic BH3-only proteins58,59. Alternatively, and in a more direct mode of action, phosphorylated IRF3 can also interact with the pro-apoptotic proteins BAX and BAK and thereby lead to transcription-independent induction of apoptosis downstream of STING60,61. Under conditions in which apoptosis is inhibited, STING activation can also promote RIPK3-dependent necroptosis by engaging type I interferon and TNF signalling pathways62. How cells balance these distinct outcomes of STING activation remains a question of ongoing research. But it appears that whether cells commit or not to death is context dependent and influenced by both the cell type and the specific cellular context (mitosis versus interphase)61. Both lymphocytes and monocytes appear to preferentially die following STING activation, a phenotype that is also observed in the context of hyperactive STING mutations in humans (see below)59,63,64,65. Given that cell death is a key initiator and amplifier of inflammatory conditions, it will be important to keep in mind these effector functions when considering the origins and/or consequences of STING-mediated inflammatory disease processes.
Intercellular cGAMP signalling networks
The finding that cGAS and STING connect through a transmittable second messenger molecule raised early on the idea that the pathway may also coordinate multicellular immune responses through the intercellular exchange of cGAMP66. Indeed, although different modes of cell-extrinisic cGAMP signalling continue to be uncovered, we now recognize that cGAMP-induced immunity engages in substantial intercellular communication, greatly amplifying the overall immune response with important biological consequences.
The phenomenon of cGAMP in trans signalling was first shown to be mediated by gap junctions, intercellular conduits built of connexin proteins that directly connect the cytosol of adjacent cells66. Following DNA transfection or virus infection of cells, cGAMP spreads from the producer cell to surrounding bystander cells causing prominent activation of STING and boosting antiviral immunity. Gap junctions not only form between identical cell types but are also established in more heterogeneous populations of cells that interact in the context of physiological immune responses. As such, gap junction-mediated cGAMP transfer has been found to amplify inflammatory responses in the setting of cancer, with cancer cells serving as the cGAMP producers and astrocytes or dendritic cells (DCs) as the cGAMP recipients67,68. Depending on the context this can have either beneficial or maladaptive effects on tumour progression. In addition, interhepatic propagation of cGAMP was shown to fuel liver injury caused by alcohol69. Evidently, this propagation mechanism is spatially restricted to layers of surrounding cells in the immediate vicinity. By contrast, vesicles, dying tumour cells and viruses have also been shown to serve carrier functions for cGAMP, which could bridge significantly longer distances70,71,72. Both transmission modes have in common that they will avoid exposure of cGAMP to the extracellular space. This could be significant, as this is where cells express ectonucleotide pyrophosphatase/phosphodiesterase family member 1 (ENPP1), an ambiguous enzyme capable of degrading cGAMP and thus counteracting cGAMP-driven intercellular communications73. However, more recent studies describe a variety of non-specific transmembrane carriers capable of shuttling cGAMP and its derivatives across cellular membranes, including members of the solute carrier family and volume-gated anion channels74,75. For example, LRCC8 facilitates cellular entry of cGAMP, a function that if genetically ablated compromises defence against HSV-1 infection76,77. In preclinical models of cancer, the transmembrane folate receptor SLC19A1-mediated cGAMP internalization is important for the efficacy of immunotherapies involving intratumoural administration of cGAMP74. Similarly, ATP-gated P2XR7-mediated cGAMP uptake promoted antitumour immunity78. Operating on the basis of diffusion along physicochemical gradients, these dedicated importers may also invert their function and promote cGAMP export in certain scenarios. More generally, the activity of intercellular transport systems dramatically depends on cell type and activation state and, thus, their contribution to cGAMP-mediated immune responses may dramatically depend on the (local) cellular composition and the ‘activation’ state of the tissue. Future work will need to expand on their contributions to inflammatory disease states, which may shed light on intercellular amplificatory loops as new targets for aberrant cGAS–STING pathway activation.
Sensing of cellular perturbation by cGAS–STING
Instead of being activated by pathogen-specific structural patterns, cGAS–STING-mediated immunity in bacteria appears to have relied on an indirect means of danger sensing, that is, overcoming a constitutive, perhaps homeostatic, mechanism of self-inhibition79. Although the acquisition of a DNA ligand-mediated mode of allosteric activation has allowed the cGAS–STING pathway to operate as a classical pattern recognition receptor, mammalian cells also benefit from danger sensing that is fuelled by cGAS-mediated recognition of out-of-context self-DNA. Similarly, several sterile disease states feature loss of cellular DNA homeostasis with distinct mechanistic scenarios of cGAS–STING pathway activation having been developed, as presented in detail below and highlighted in Fig. 3. Importantly, these different models of activation are not mutually exclusive, but instead may trigger cGAS activity in a redundant manner in certain cases and in a synergistic fashion in other cases.

Corpses of dying or apoptotic cells can become engulfed by phagocytic cells and are targeted to lysosomes (where DNase II is active) via LC3-associated phagocytosis (LAP). The efficient disposal of extracellular DNA through this route restricts cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) activity. Within mitochondria, transcription factor A mitochondrial (TFAM) plays a crucial role in stabilizing mitochondrial (mt) DNA through the formation of nucleoids, which is important to counteract mtDNA stress and aberrant cGAS activation. Exogenous stress signals, including chemotherapy or infection, may also cause mitochondrial dysfunction — in the most severe cases leading to apoptosis — which can promote loss of mtDNA compartmentalization. mtDNA either herniates through the inner mitochondrial membrane to then be released through BAX/BAK macropores or can be released via voltage-dependent anion channel (VDAC) pores in the outer mitochondrial membrane. Alternatively, mitochondrial permeability transition pore (MPTP) may present an alternative route for inner mitochondrial membrane traversal. Mitophagy of stressed mitochondria, which is executed by Parkin and PINK1, limits mtDNA-mediated stimulation of cGAS. Nuclear DNA may become accessible to cytosolic cGAS through the following mechanisms: first, the derepression of retroelements, second, DNA damage or replication stress may lead to direct nuclear DNA leakage, third, in senescence, chromatin herniations can bud off the main nucleus leading to accumulation of cytoplasmic chromatin fragments (CCFs) or, alternatively, in the context of genomic instability nuclear DNA may be encapsulated in micronuclei after mitosis. Both of these latter two events can promote cGAS activity on rupture of the aberrant micro-organelle membrane. All these instances of nuclear-derived cytosolic DNA recognition are efficiently antagonized by the exonuclease TREX1. Within the main nucleus, both nucleosomes and architectural chromatin proteins, including histone H1 and barrier-to-autointegration factor (BAF) act to limit aberrant cGAS activity. ER, endoplasmic reticulum.
Activation by extracellular self-DNA sources
Non-apoptotic cell death can lead to unprogrammed extracellular release of DNA, and several conditions associated with accrual of dying cells have been linked to activation of cGAS and/or STING. For example, irradiation of the tumour bed promotes internalization of tumour-cell-derived DNA in DCs, which elicits cGAS–STING activation80. Likewise, synchronized cell death arising from acute ischaemic events, including occlusion of the coronary arteries, promotes the recruitment and activation of monocytes, an inflammatory event depending upon STING81. Adjuvant-mediated cell death has been shown to promote immune activation via STING82. Additionally, neutrophil extracellular trap (NET) formation — a process implicated in various sterile inflammatory diseases — results in cell death and extrusion of neutrophil DNA–protein complexes into the extracellular space and in turn activates STING83.
What these distinct mechanisms appear to have in common is that the dominant responding cell type is of myeloid origin, suggesting a correlation with the cellular capacity to engulf extracellular material. Whether internalized DNA simply overloads the homeostatic disposal mechanism or whether there are dedicated host factors involved in facilitating extracellular DNA recognition by cGAS remains unknown. What is known, however, is that impairment of the regulated turnover of apoptotic cell corpses can also trigger cGAS–STING activity. The best studied example of this is deficiency of DNase II, a lysosomal DNase crucial for the degradation of apoptotic-cell-derived DNA84. Mechanistically, this phenotype has been associated with accumulation of dying erythrocytes in liver macrophages. More recent studies have illuminated further upstream factors that could play a role in the regulated degradation of DNA in apoptotic cells and prevention of cGAS activity. LC3-associated phagocytosis (LAP) is a non-canonical form of autophagy, which crucially functions in phagosome maturation and the regulation of downstream (immune) signalling events in macrophages and other cell types85. It has been shown that deficiency of LAP components can promote STING activation within myeloid cells86. The additional observation that TIM4 deficiency mimics this phenotype may suggest that dedicated apoptotic receptor pathways fuel extracellular DNA into an immunologically silent degradation process. However, if this route is overwhelmed and DNA internalization is mediated by other mechanisms, cGAS–STING activation may follow. Given that extracellular DNA is a major factor underlying many inflammatory phenotypes, a better understanding of the molecular routes taken by extracellular DNA will be of importance.
Activation by cell-intrinsic mitochondrial DNA
Release of mitochondrial (mt) DNA into the cytosol has emerged as a prominent trigger of cGAS–STING pathway activation in multiple contexts. Pathogens are well known to produce various signals capable of perturbing mitochondrial integrity, suggesting that the recognition of mtDNA via cGAS–STING could serve as an important means to signal the presence of infection. In support of this notion, antiviral immunity to HSV-1 and Dengue virus has been found to rely — at least to a certain degree — on cGAS recognition of mtDNA87,88. In addition, certain strains of Mycobacterium tuberculosis were found to drive cGAS activity via the release of mtDNA, although in this particular case the evoked type I interferon response may favour the pathogen rather than the host89. Mitochondrial dysfunction and an accompanying rise in mtDNA levels can also accompany sterile inflammatory processes through the effects of cytokine signalling. For example, IL-1β, a hallmark inflammatory cytokine, and IFNα both compromise mitochondrial homeostasis and trigger an amplificatory immune reaction through the activity of mtDNA engaging cGAS90. It is possible that, through such positive regulatory mechanisms, diverse cytokine signalling programmes can cause potentiation of inflammation involving mtDNA recognition via cGAS. Defects in the clearance of damaged mitochondria due to mutations in PINK1 and Parkin, have also been linked to STING activity, implicating mitophagy as an important constitutive cellular process that limits aberrant immune activation91.
Similar to what is seen with genomic nuclear DNA, disruption of processes surrounding mtDNA intactness, replication and repair can result in cGAS–STING signalling. Among the best studied examples, is that defective packaging of mtDNA into nucleoids following the depletion of transcription factor A mitochondrial (TFAM) is a prominent signal for cGAS activity in diverse cells87. In addition, mtDNA stress invoked by lack of mitochondrial endonuclease G or by treatment with chemotherapeutic drugs activates cells via cGAS92,93. In these and other instances, mtDNA may also be modified by mitochondrial reactive oxygen species that can render mtDNAs more resistant to processing by the nuclease TREX1 and hence more stimulatory94.
A most prominent cellular event of mitochondrial disintegration is apoptosis — a fundamental non-inflammatory mechanism of cell death. A compelling case has been made in support of an essential function of apoptotic caspases to impair cGAS sensing of mtDNA and, hence, maintain the immunologically silent mechanism of apoptotic cell demise95,96. How exactly caspases exert this anti-inflammatory function is not fully elucidated, but one potential strategy involves the regulated cleavage of cGAS and its downstream transcription factor IRF3 (ref.97).
Compared with the importance to mtDNA-mediated activation of cGAS the mechanisms allowing mtDNA to translocate into the cytosol have only more recently been developed. To reach the cytosol, mtDNA must overcome two barriers — the inner mitochondrial membrane (IMM) and the outer mitochondrial membrane (OMM). In the context of apoptosis, BAX/BAK macropores located in the OMM have been shown to serve as gateways for mtDNA herniating through inner nuclear membrane (INM) ruptures and, in turn, its escape into the cytosol98,99. Importantly, beyond apoptosis, which is characterized by synchronized and complete mitochondrial outer membrane permeabilization (MOMP), cells may also experience more limited forms of MOMP affecting only a minority of mitochondria100. Such minor MOMP could provide one explanation of how MOMP could connect with the cGAS–STING pathway in the context of more subtle (mitochondrial) stress responses. In addition, the oligomerization of the voltage-dependent anion channel proteins VDAC1 and VDAC3 leads to the formation of pores in the OMM that also allow mtDNA fragments to reach the cytosol92. How the IMM permeabilizes is not yet fully understood, but it may involve the mitochondrial permeability transition pore (MPTP), which spans the IMM and forms as a consequences of diverse cellular stresses101.
Activation by cell-intrinsic genomic DNA
Molecularly distinct disease-associated processes have been identified that underlie the cell-intrinsic activation of the cGAS–STING pathway by genomic DNA. As of today, most of these processes are characterized by the accumulation of nuclear DNA inside the cytoplasm providing a straightforward mechanism of cGAS activation by out-of-context DNA. However, the localization of cGAS within the nucleus raises the possibility that under conditions of disease, aberrant cGAS activity may also arise directly from sensing genomic DNA inside the nucleus (Box 1).
Early studies recognized that the cytosolic enrichment of retrotransposable elements serves as a potent trigger of type I interferon responses102. One could well argue that the sensing of retroelements may in fact represent a primary function of cGAS in the context of retrovirus infection. Retrotransposon sequences occupy a considerable fraction of the human genome, but most of these sequences are severely truncated and, thus, considered non-functional. Whereas so-called LINE-1 (L1) elements can still undergo retrotransposition, various actions counteract the propagation of L1 elements in living cells under homeostatic conditions. However, imbalances in the surveillance strategies for endogenous retroelements can trigger cGAS activity, owing to improper accumulation of cDNA intermediates. The best studied example of the ability to directly recognize overt retrotransposon activity by cGAS is loss of the 3′ repair exonuclease TREX1, which naturally antagonizes this process by cleavage of DNA intermediates. Deficiency of TREX1 causes accumulation of L1 elements in mouse tissue and human neural stem cells103,104. Further upstream derepression of retroelements via epigenetic alterations occurs naturally during ageing and this gives rise to cytoplasmic L1 and cGAS–STING-dependent inflammation105. Given that retro-transcription of L1 elements is believed to occur within the nucleus it remains to be fully elucidated how exactly these cDNA intermediates are exported to the cytosol.
Another obvious category of cell-intrinsic stimulatory DNA products are those emerging from DNA damage and defective replication and repair. Mutations in or depletion of a range of genes that control these processes have served as important experimental paradigms that link DNA damage with activation of cGAS–STING signalling. Likewise, various exogenous genotoxic treatments have been linked to a cell-intrinsic cGAS-dependent type I interferon response. Importantly, however, there are profound differences with regard to the mechanisms of DNA accrual and subsequent cGAS engagement. First, under certain conditions, free DNA-damage products have been reported to reach the cytosol directly, although how these products traverse the nuclear membrane remains to be resolved106,107. Alternatively, unrepaired or profoundly damaged genomic DNA promotes chromosome missegregation and micronuclei formation or the generation of chromatin bridges in mitosis. Upon rupture, cytosolic cGAS gains access to the damaged chromatin inside micronuclei leading to immune activation in several contexts of genome instability108,109. Alternatively, enveloped chromatin fragments can also bud off the main nucleus in senescent cells, an event that is believed to be due to increased fragility of the nuclear lamina110.
Activation of cGAS–STING in disease contexts
The remarkable complexity that is inherent in the proper regulation of DNA abundance, sequestration and clearance at both the cellular and organismal level makes it vulnerable to perturbations. Although the inducers of pathological processes are disease-specific, there is evidence for considerable convergence on the involvement of the cGAS–STING pathway as a driver of both (hyper-)acute and chronic, low-grade inflammatory conditions associated with diverse pathologies. Owing to its central role as a ubiquitous and sensitive mechanism of out-of-context DNA recognition, this may not be too surprising. In addition, first evidence indicates that imbalances in intracellular trafficking routes can have profound repercussions on STING activity and contribute to improper immune activation. Below we highlight implications of the cGAS–STING pathway in distinct disease processes and provide a summary of the additional literature surrounding this aspect in Table 1.
Implications of cGAS–STING in monogenic diseases
Rare monogenic autoinflammatory diseases are associated with mutations in genes that participate in innate immunity or that control innate immune homeostasis. Here, we briefly highlight a selection of conditions as they relate to a cGAS-STING-driven pathogenesis implying the opportunity for interventions predicated on the inhibition of cGAS and STING. For more comprehensive reviews on type I interferonopathies, particularly Aicardi–Goutières syndrome (AGS), we refer to excellent recent reviews on that topic111,112.
Gain-of-function mutations in STING
Rare mutations in STING1 (formerly known as TMEM173) can lead to paediatric onset of a severe autoinflammatory syndrome named STING-associated vasculopathy with onset in infancy (SAVI). SAVI is characterized by recurrent fevers, ulcerative skin lesions, vasculitis and interstitial lung disease and is for the most part driven by autosomal dominant mutations in STING1, although an autosomal recessive mutation that drives SAVI was recently identified113,114,115. The mutated residues localize around two separate regions on STING, namely the dimerization interface, more precisely the connector helix loop (N154S, V155M and V147L), and the polymerization interface (G207E, R281Q, R284G and R284S)30,113,114,116,117,118,119. Mechanistically, it is thought that the disease-causing substitutions trigger ligand-independent activation of STING either by inducing spontaneous rotation of the LBD along the connector helix loop or by promoting STING polymerization, possibly by relieving an inhibitory interaction29,30. At the cellular level, these mutations cause spontaneous trafficking of STING to the Golgi and activation of downstream signalling leading to heightened type I interferon signatures in primary patient cells. In mice, the introduction of SAVI mutations (N153S or V154M knock-in; mouse residues) causes lung disease, cytokine production, skin ulcerations and premature death120,121,122,123. A salient feature of murine SAVI models is the development of severe lymphocytopenia and immunodeficiency owing to abnormal lymphocyte development and intrinsic T cell abnormalities. Reduced numbers and anti-proliferative effects in T cells have also been observed in patients with SAVI, although these features are less prominent compared with what is seen in mice48,114. Notably, ablation of type I interferon signalling or disabling necroptosis had no effect on the disease pathogenesis in the N153S mouse model of SAVI, whereas depletion of T cells protected N153S mice from developing lung disease120,122,123,124. The extent to which the clinical manifestations seen in humans rely on type I interferon signalling or on T cell-mediated immunopathology is not yet clear.
DNA-driven inflammation in Aicardi–Goutières syndrome and DNase II deficiency
AGS is a rare genetic disorder driven by various genes associated with nucleic acid sensing or metabolism111. The disease is predominantly characterized by early onset of progressive encephalopathy and skin lesions (referred to as chilblains), with the development of other features being common and considerable clinical heterogeneity reportedly existing between individual patients. Mutations in a subset of these genes — namely TREX1, genes encoding the three RNase H2 endonuclease subunits, RNASEH2A, RNASEH2C and SAMHD1 — have been associated with disturbed self-DNA metabolism and constitutive activation of the cGAS–STING pathway. As such, mice deficient for or with mutation in AGS-associated genes have been developed, and data derived from these animals have shown that co-depletion of cGAS or STING reverses the pathogenesis seen in mice125,126,127. More recently, a clinical syndrome with an elevated type I interferon signature was found to be caused by hypomorphic mutations in DNASE2 (ref.128) As distinct from AGS, the disease manifested with neonatal anaemia, kidney disease and arthropathy. Dnase2−/− mice die during embryonic development owing to severe anaemia and if crossed with Ifnar null mice, develop an arthritis phenotype84,129. In that sense, the mouse models, although more severe, overlap with the features seen in patients. Notably, depletion of either cGAS or STING protects Dnase2−/− mice from developing disease126,130. It is important to keep in mind the existence of considerable differences in the phenotypic consequences between these preclinical models and patients with mutations in DNA metabolism with regard to the overall disease severity, the variability of developing disease and the involvement of given organ systems, especially with regard to the neurological phenotype, which is absent in mouse models. Still, these genetic disease models unequivocally support the view that cGAS–STING signalling drives autoinflammatory disease features and that drugs blocking cGAS or STING could be used to treat these conditions.
COPA syndrome
COPI is critical for the retrieval of proteins from the Golgi to the ER and for intra-Golgi transport. COPA syndrome is a rare early-onset autosomal dominant disease caused by missense mutations in the COPA gene, which encodes the COPα protein subunit of the COPI complex; the mutation impairs the binding and sorting of proteins targeted for ER retrieval131. Clinically, patients typically present with inflammatory arthritis, interstitial lung disease and kidney disease and exhibit a type I interferon signature in blood — features that are to some degree reminiscent of SAVI132. Recently, four independent studies have shown that defects caused by COPA mutations promote ligand-independent activation of STING-mediated signalling and that the elevated type I interferon signature associated with COPA dysfunction can be reduced by genetic or pharmacological interference with STING37,38,133,134. In a mouse model of COPA syndrome (CopaE241K/+), type I interferon-driven inflammation was rescued by crossing these mice with STING-deficient (STINGgt/gt) animals and, moreover, the embryonic lethality of homozygous CopaE241K/E241K mice was rescued by co-deletion of STING37. Together, these studies suggest that STING may play an important role in the excessive inflammatory phenotype in COPA syndrome.
cGAS–STING in SLE and other autoimmune diseases
With both type I interferons and self-nucleic acids recognized as hallmark drivers in the pathogenesis of systemic autoimmune diseases, the involvement of the cGAS–STING pathway in these diseases is of great interest. For systemic lupus erythematosus (SLE), the prototypic chronic systemic autoimmune disease, it has been documented that ~15% of a total cohort of 41 patients displayed elevated levels of cGAMP in the serum — a strong indicator of the presence of cGAS pathway activity135. Moreover, serum collected from patients with SLE was shown to amplify type I interferon induction via STING, providing a mechanism for how cGAS–STING may amplify or accelerate disease symptoms136. Mutations of TREX1 can cause monogenic forms of cutaneous lupus, and TREX1 variants have been reported in a large cohort of patients with SLE, although these occur in only a minor fraction of patients137,138. Judging from animal models, the relevance of cGAS–STING signalling in the pathogenesis of SLE is variable and considerably dependent on the mouse model under study. Given the multitude of pathological mechanisms underlying SLE, this is not unexpected and rather reflects the considerable heterogeneity of SLE in humans. Early work found that the genetic deletion of STING in the lupus-prone MRL.Faslpr mice aggravated the disease phenotype139. By contrast, in another model driven by a deletion in the lupus susceptibility gene Fcgr2b, Sting1 null mice were protected from development of various disease manifestations, including elevated autoantibody titres and glomerulonephritis, and showed improved overall survival140. Moreover, cGAS–STING plays a pathogenic role in a variety of distinct models that promote lupus-like symptoms through a range of mechanisms. As such, defects in extracellular clearance of apoptotic cells, accumulation of extracellular NETs from dying neutrophils, defects in mtDNA replication and dysregulation of endogenous retroelements have been suggested to predispose to the development of cardinal disease features of SLE and present plausible mechanisms of aberrant cGAS activity in SLE92,141,142,143.
Implications of cGAS–STING in neurodegeneration
Inflammation is a prominent hallmark of several neurodegenerative diseases, including Alzheimer disease, Parkinson disease and amyotrophic lateral sclerosis (ALS)144. Multiple recent studies associate activation of the cGAS–STING pathway with the development of some of these neurodegenerative pathologies. As a most illustrative example, a mechanism that connects compromised mitophagy to activation of cGAS–STING signalling has recently been identified that may be relevant to the pathology of Parkinson disease91. Parkinson disease is a debilitating movement disorder resulting from selective death of dopamine-producing neurons in the substantia nigra region of the midbrain. Missense mutations in PARKIN and PINK1, which encode proteins that assist in the removal of dysfunctional mitochondria, are linked to familiar forms of Parkinson disease. In the context of in vivo mitochondrial stress, it was found that mice deficient in either Parkin or Pink1 accumulate mtDNA and display elevated cytokine levels in the bloodstream, the latter effect being fully abrogated by co-deletion of STING91. Importantly, absence of STING also rescued the motor deficit and neuronal cell loss seen in aged Parkin mutator mice, which develop neuronal abnormalities similar to those in patients with Parkinson disease91. More recently, cGAS–STING-dependent inflammatory processes driven through the recognition of mtDNA have been found to trigger neuropathological processes associated with certain forms of ALS and frontotemporal lobar degeneration (FTLD)101. Cytosolic and mitochondrial accumulation of TDP43, a DNA/RNA-binding protein normally sequestered in the nucleus, is a hallmark of many cases of ALS and FTLD. Mislocalized mitochondrial TDP43 causes mtDNA release through the opening of the MPTP and leakage via VDAC1 followed by the induction of type I interferons and inflammatory cytokines in a cGAS–STING-dependent fashion in human and mouse cells. Moreover, motorneurons derived from patients with ALS showed STING-dependent upregulation of inflammatory cytokines. In a preclinical model of ALS driven by overexpression of TDP43, co-depletion of STING dampened neuroinflammation, mitigated rapid disease progression and protected from early death. Strikingly, attenuation of signs of neurodegeneration was also observed upon deletion of only one allele of STING or by the application of a pharmacological inhibitor of STING. A link between inflammation associated with ALS and cGAS–STING activity also came from another recent report that investigated the biological consequences of diminished C9orf72 levels — mutations of which are the most common genetic causes of ALS and FTLD. Myeloid cells deficient for C9orf72 as well as cells from patients with C9-ALS-/FTLD showed STING-mediated production of type I interferons and inflammatory mediators145. Mechanistically, compromised C9orf72 expression was suggested to hamper the swift degradation of STING, consistent with a function of C9orf72 in the regulation of lysosomal vesicle formation146. Lastly, elevated levels of type I interferons and contributions of STING have also been observed in models and disease samples of Huntington disease147 or neurodegeneration due to extracellular protein aggregate formation148. Future work will be needed to further dissect the cell types involved in cGAS–STING-driven inflammation and to assess whether interfering with the cGAS–STING signalling axis may merely stop and slow down or, perhaps, even reverse molecular changes associated with neurological deterioration. In summary, these studies imply that despite occurring secondary to initial degenerative changes in the brain, aberrant activation of the cGAS–STING pathway is relevant in distinct types of neurodegeneration, and inhibiting cGAS or STING may improve certain disease states or mitigate disease progression.
Implications of protumorigenic roles of cGAS–STING
Owing to its potent immune-stimulatory effects, the cGAS–STING pathway holds particular translational appeal as a promising target for cancer immunotherapy. However, in striking contrast to their tumour suppressive roles, cGAS and STING have also been implicated in promoting tumour burden and worse disease outcomes in models of cancer. This raises the question of what dictates these fundamentally distinct outcomes of cGAS–STING activity in tumours? Valuable clues to answering this question came from studying the kinetics and strength of cGAS–STING activation. As such, whilst transient activation appears favourable, adverse effects seem to rely on the stimulation of a chronic inflammatory programme that promotes an immune-suppressive tumour environment. For example, chronic exposure to genotoxic 7,12-dimethylbenz(a)anthracene (DMBA) promotes tumour growth in a STING-dependent manner149. Similarly, STING-mediated recruitment of myeloid-derived suppressor cells promotes MC38 colon cancer growth following radiation therapy150. STING ligands can also induce the expression of inhibitory molecules, for example, PDL1 or IDO1, or activate non-canonical NF-κB signalling, which counteracts the tumour-suppressive effects of type I interferons52. The pro-apoptotic effects of STING on T cells may also contribute to limited tumour cell clearance59. Beyond shaping the immune cell composition at the primary tumour site, the cGAS–STING pathway also impacts metastatic behaviours. Indeed, by activation of STING signalling, chromosomally unstable tumours upregulate genes relevant for epithelial–mesenchymal transition and metastasis through non-canonical NF-κB signalling51. Moreover, on reaching unaffected distant sites, such as the brain, metastatic breast cancer cells also utilize cGAMP transfer to manipulate immune cells for their own growth advantage67. Although these pro-tumorigenic effects are reliant on cGAS–STING signalling, a recent report implies that nuclear cGAS can directly promote uncontrolled cancer cell proliferation independently of STING by inhibiting homologous recombination151. All in all, these aspects suggest a complex role of the cGAS–STING pathway in cancer. In the context of more advanced disease, a STING-mediated inflammatory retrenchment may allow aggressive cancer cells to adapt, and overcome and finally to further thrive and propagate — highlighting that chronic STING signalling may represent a key problem that must be taken into consideration for tumour therapies targeting this pathway.
cGAS–STING in senescence and ageing
During ageing crucial mechanisms that maintain tissue and cellular homeostasis collapse, generating the accumulation of a plethora of molecules that can fuel the heightened inflammatory state characteristic of old age in various species, including humans. Crucial to this ‘inflammageing’ process are senescent cells, which amongst other features have lost the capacity to proliferate and display prominent secretory activity, referred to as the senescence-associated secretory phenotype (SASP), which can impair tissue function152. Depletion of senescent cells prolongs lifespan and healthspan and ameliorates several ageing-associated dysfunctions153. Furthermore, transplantation of senescent cells into young mice accelerates the establishment of several ageing-associated frailty aspects, underscoring the critical role of senescent cells in regulating features of ageing154. We, and other groups, have discovered that aberrant activity of cGAS–STING is a conserved pattern across multiple types of senescent cells and that this is critical for the secretion of several SASP components58,155,156. Mechanistically, cGAS was found to be enriched on cytosolic chromatin that can accumulate within senescent cells and represent a bona fide activator for cGAS in this context (see above). As potential sources, cytosolic DNA fragments may derive from ruptured micronuclei or chromatin herniations, as a consequence of chronic DNA damage of disrupted nuclear envelope integrity, both of which are features of senescent cells108,109,110. Alternatively, a recent report proposed accumulation of cytosolic retrotransposable elements, which can become reactivated during ageing in somatic tissues, to drive cGAS-dependent type I interferon responses105. Strikingly, repressing retrotransposon transcription using the nucleoside reverse transcriptase inhibitor lamivudine dampens signs of inflammageing in multiple tissues in naturally aged mice and in a progeroid model of ageing105,157. Given that senescent cells have gained broader attention in other disease settings, such as osteoarthritis and artherosclerosis, further studies into the connection between inflammatory senescence and cGAS–STING are warranted158. Age-associated decline in mitochondrial function is also a hallmark of ageing. Interestingly, depletion of TFAM — a well-known trigger of cGAS–STING activity in multiple settings — in T cells is sufficient to trigger an accelerated ageing phenotype and multimorbidity by promoting a pro-senescent inflammatory milieu in vivo159. In light of these advances, it is tempting to speculate that aberrant cGAS–STING signalling may contribute to functional decline and ageing-related dysfunction imparted by its pro-inflammatory properties.
Therapeutic targeting of cGAS–STING
The findings discussed above on the involvement of cGAS–STING in a number of pathologies with so far unsatisfactory or unmet therapeutic options portend its high potential as a drug target. The emergence of first drug-like compounds selectively targeting cGAS or STING has opened the door for the development of first clinical candidates. Here, we discuss emerging small-molecule-based strategies to therapeutically target cGAS–STING signalling in models of disease. The chemical structures of specific agents targeting cGAS and STING are summarized in Table 2 and Table 3, respectively. An overview of their mechanisms of action is provided in Fig. 4. Considerations on human STING allelic variants are presented in Box 2.

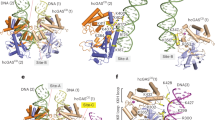
a | Inhibitory molecules targeting the catalytic pocket of cyclic GMP–AMP synthase (cGAS) are shown on the left, and those reported to impede DNA binding are shown on the right. b | Inhibitory compounds affecting stimulator of interferon genes (STING) can either occupy the ligand-binding domain (LBD) or covalently bind to C91 of both human and mouse STING. In the 2′3′ cyclic GMP–AMP (cGAMP)-bound state, STING undergoes conformational rearrangements leading to its oligomerization. The inset highlights the position of C88 and C91, residues of STING that are both subject to palmitoylation. Structures shown: cGAS catalytic domain (CD-cGAS) apo form (Homo sapiens PDB: 4O68)18; CD-cGAS DNA-bound (H. sapiens PDB: 6CT9)191; STING full-length apo form (H. sapiens PDB: 6NT5); STING full-length cGAMP-bound (H. sapiens PDB: 6NT5, modelled with Gallus gallus PDB: 6NT7 (front view) or G. gallus PDB: 6NT8 (tetramer side-view)30.

cGAS inhibitors
Given the amenability of cGAS to crystallography, a number of reports have detailed the structure-based design of cGAS inhibitors12,160,161,162,163. Most of the cGAS antagonists discovered in this manner bind to the active site and are thus competitive with the ATP or GTP substrates or the product cGAMP. The other main class of cGAS antagonists described in the literature compete with DNA for binding to cGAS, thus interfering with the initial activation step of cGAS164,165,166,167,168. A brief summary of these cGAS inhibitors is presented in the following section.
Catalytic site inhibitors
PF-06928125
Hall et al.160 conducted a saturation transfer difference 1H NMR screen of the Pfizer fragment library against a truncated cGAS construct. In concert with surface plasmon resonance (SPR) and fluorescence polarization (FP) assays, they identified a tetrazolo[1,5-a]pyrimidine fragment with a Kd of 171 µM (SPR). X-ray crystallography (3.1 Å resolution structure) confirmed that this compound binds to the active site of cGAS at a region typically occupied by the adenosine base of ATP or cGAMP. Using this information and together with other published crystal structures, the team modified the core and created additional interactions within the active site to identify pyrazolopyrimidine PF-06928125, with a Kd of 0.2 µM by SPR and Kd of 4.9 µM in the FP assay. Although this compound was shown to bind to cGAS in biochemical assays, PF-06928125 lacked inhibitory activity in cellular assays. The authors speculated that inhibiting the active site might require more potent compounds, owing to high intracellular levels of ATP and GTP. In addition, they proposed that additional changes to the core to replace the carboxylic acid with a motif that would have a higher pKa, lower polar surface area and fewer H-bonds could improve resultant cellular potency.
RU.521
Vincent et al.12 conducted a high-throughput screen using recombinant mouse cGAS (m-cGAS) and a RapidFire mass spectrometry system (RF-MS) to quantify levels of ATP, GTP and cGAMP as readout. Screening 123,306 compounds led to the identification of four compounds that survived both in silico filters for PAINS motifs and counter-screening to remove DNA intercalators. A 2.13 Å resolution crystal structure of one of these hits (RU.365) bound to cGAS with dsDNA confirmed that the compound occupied the active site of cGAS. Subsequent structure-guided chemical synthesis of analogues led to the identification of RU.521, with a Kd of 36 nM as assessed by isothermal titration calorimetry. This compound was also the most potent compound of this series in a cellular assay (IC50 = 0.70 µM) that measured the inhibition of dsDNA-induced cGAS signalling in RAW macrophage cells. The inhibitory effect of RU.521 was selective for cGAS versus other innate immune signalling pathways and was shown to reduce expression levels of Ifnb1 mRNA in bone marrow-derived macrophages (BMDMs) from Trex1−/− mice, demonstrating the potential for this compound to be effective in a constitutively activated system.
G150
Lama et al.161 recently reported the identification of a new class of active site-targeted inhibitors of cGAS. In this communication, the group used a new luminescence-based assay system that allowed them to screen h-cGAS. This screening campaign of 281,348 compounds was triaged in a manner similar to that used to identify RU.521. This screen identified two main chemotypes (J and G), of which chemotype G, with a pyridoindole tricyclic core, was used as a starting point to ultimately derive G150 (RF-MS IC50 = 0.0102 µM for h-cGAS). Using an IFNB1 mRNA readout, G150 had an IC50 of 1.96 µM in THP-1 cells and an IC50 of 0.62 µM in primary human macrophages. G150 also showed no off-target effects in a series of assays testing inhibition of other innate immune sensing pathways. The X-ray structures of G150 and a number of other analogues confirmed their binding of the cGAS active site. However, the authors note that they have not been able to rationalize all of the structure–activity relationships (SARs) based on this information, and more work will be needed to design and develop next-generation inhibitors.
Compound S3
Zhao et al.163 solved a 1.8 Å resolution crystal structure of a h-cGAS structure bound to PF-06928125, which they then used to conduct a virtual screen of about 1.5 million compounds in the ChemDiv database. Following up on 59 compounds for biological testing in thermal shift assay provided four positive hits. Co-crystal structures of these hits revealed that they bound the active site of cGAS in a manner that correlated well with their docking models from the virtual screen. However, subsequent assessment of the compounds in a PPiase-coupled assay measuring cGAS catalytic activity revealed that they did not inhibit cGAS enzymatic activity. Instead, while screening the initial virtual screen hits in the PPiase-coupled assay, they found that one of the original 59 hits had an IC50 of 29.9 µM even though it did not have any activity in the thermal shift assay. An expanded similarity search around this compound, combined with docking assessment, identified 21 new compounds for purchase. From this endeavour, they identified compound S3, with an IC50 of 4.9 µM in the PPiase-coupled assay (for comparison, PF-06928125 has an IC50 of 2.1 µM and RU.521 has an IC50 of 5.7 µM in this assay). The authors compared the binding modes of compound S3 with those of PF-06928125 and RU.521 and recommended the virtual screening approach as a complementary method to identify new starting points for cGAS inhibitors.
Inhibitors that disrupt DNA binding
Antimalarial drugs
Although inhibition of the active site of cGAS is an approach many research groups have pursued, another strategy would be to disrupt the binding of cGAS to dsDNA, which is the activating trigger for cGAS. An et al.164,165 reported that a series of antimalarial drugs, such as hydroxychloroquine and quinacrine, could be used as potential treatments for SLE and that these drugs inhibited IFNβ production by selectively blocking the cGAS–dsDNA interaction. They went on to describe the development of a second-generation molecule X6, with improved in vitro and in vivo activity166. It has been shown that these classes of molecule can interact with DNA by both intercalation and association of the positively charged amino side chain within the minor groove of DNA169. As such, the authors and others168 postulated that these molecules inhibit cGAS indirectly by binding to dsDNA and preventing the formation of the cGAS–dsDNA complex.
Suramin
Wang et al.168 screened a small 268-compound library and identified suramin as an inhibitor of cGAS activity. A series of experiments revealed that suramin likely inhibits cGAS activity by binding at the dsDNA-binding site, thus disrupting the formation of the cGAS–dsDNA complex. The authors also reported that the observed inhibition of cGAS is selective, with no effect on the TLR1/TLR2 or TLR4 pathways. Although the molecule is highly charged, suramin has been shown to have cellular activy. One potential mechanism of action suggested in this publication is that the anionic sulfates of suramin act as phosphate mimics and bind to the positive regions on cGAS.
Suppressive oligodeoxynucleotides
A151 is a suppressive oligodeoxynucleotide containing four TTAGGG motif repeats (5′-tt agg gtt agg gtt agg gtt agg g-3′). Initial reports described its inhibitory effects on both TLR9 signalling170 and AIM2 (ref.171). In a recent report, Steinhagen et al.167 demonstrated that A151 can also function as a competitive inhibitor of cGAS by interacting with the dsDNA-binding domain. The exact binding sites of A151 within the DNA-binding domain are not known, but the authors did determine that binding is both sequence- and backbone-specific. In this study, A151 was also able to inhibit a type I interferon response in TREX1-deficient cells, raising the possibility of this approach to treat dsDNA-driven autoimmune diseases.
Compounds with undetermined mechanism of action
Padilla-Salinas et al.162 also used the Hall et al. crystal structure as a starting point to conduct a virtual screen. In particular, they targeted a potential druggable pocket around Lys347 and Lys394 in h-cGAS (Lys335/Lys382 in m-cGAS) near the dsDNA interface, with the aim of developing a protein–protein interface inhibitor of the cGAS dimer itself. They conducted a virtual screen of both the Maybridge (53,000 compounds) and Enamine (1.7 million compounds) collections, using reported co-crystal structures of m-cGAS, to identify 10 compounds that met their screening criteria. Only one compound from this effort inhibited h-cGAS (1,3,5-triazine Z918) with an IC50 of 100 µM in a KinaseGlo cGAS in vitro assay. They initiated a dedicated synthetic effort about this core and, through a series of optimizations, identified CU-76 as a potent cGAS inhibitor (IC50 = 0.24 µM). The compounds were determined to be selective for cGAS versus other pathways, using a suite of assays similar to those described in the RU.251 and G150 work. The authors were not able to obtain a crystal structure of CU-76 bound to cGAS, although they did confirm that it does not disrupt formation of the cGAS–dsDNA complex, as originally hypothesized. As such, it is not known whether CU-76 binds to the active site or inhibits cGAS via another mechanism.
STING inhibitors
To date, two main approaches have been reported in the literature to identify STING inhibitors. The first is to design molecules that occupy the cyclic dinucleotide (CDN)-binding site, thus acting as competitive antagonists of STING activators. The second approach has been to identify inhibitors that bind to either the Cys88 or the Cys91 residue near the transmembrane domain of the STING protein, each of which is subject to palmitoylation. We briefly describe these approaches and representative STING inhibitors in the following sections.
STING antagonists targeting the CDN-binding site
Tetrahydroisoquinolines
Biophysical and X-ray crystallographic data show that the STING C-terminal domain exists as a symmetrical dimer, with the ligand-binding site located at the interface between the two monomers. Siu et al.172 took advantage of the symmetrical nature of the CDN-binding domain to design small-molecule antagonists that bind two identical molecules in the larger CDN-binding site. Using a mass spectrometry-based ligand screening technique, they identified a low-affinity hit (compound 1, HAQ STING IC50 = 7.5 μM). X-ray structure determination demonstrated that two molecules occupied the CDN site and that STING adopted the inactive, open conformation. This group identified several key polar and hydrophobic contacts, both between the two molecules occupying the binding site, and the protein itself. SAR studies probing these contacts, including modification of the pKa of the acid, led to identification of compound 18 (HAQ STING IC50 = 0.08 μM). This molecule bound to STING in a similar manner (two molecules per STING dimer) and inhibited cGAMP-induced IFNβ secretion with an IC50 of 11 μM in THP-1 cells.
Astin C
Li et al.173 identified the natural product astin C from a reporter gene-based screen of a cyclopeptide library. Pull-down experiments using biotinylated astin C and human STING demonstrated that astin C binds competitively to the CDN site. In addition, adding an excess of unlabelled free astin C (10-fold) abolished the binding of biotinylated astin C to STING. Additional mechanistic studies revealed that astin C blocks the recruitment of IRF3 to the STING signalosome, thus preventing downstream signalling through the pathway. In vitro, astin C inhibited the expression of type I interferon in Trex1−/− BMDMs. The investigators also explored the in vivo activity of astin C, administered as a C-HP-β-CD inclusion complex to Trex1−/− mice, where it strongly inhibited the expression of Ifnb, Cxcl10, Isg15, Isg56 and Tnf mRNA in the heart, muscle, stomach and tongue of the mice, thus demonstrating in vivo modulation of STING.
Targeting STING palmitoylation sites
Nitrofurans (C-176 and C-178)
We identified a series of nitrofuran derivatives from a cell-based screening campaign (HEK293T cells expressing murine STING)13. Using protein mass spectroscopy and an alanine scan we demonstrated that these inhibitors specifically and irreversibly bind to Cys91 of STING, but not the nearby Cys88, both of which are palmitoylated after CDN-stimulated activation. Using an azide-based clickable C-176 probe (C-176-AZ) and a gel-based protein profiling approach, we showed that C-176 was very selective for STING against a panel of proteins that contain hyper-reactive cysteine residues. Furthermore, pre-treatment of mice with C-176 markedly reduced STING agonist-mediated elevation of serum levels of type I interferons and IL-6. In addition, treatment of Trex1−/− mice with C-176 resulted in a significant reduction in serum levels of type I interferons and in strong suppression of inflammatory parameters in the heart. A small set of analogues designed around C-176 and C-178 revealed that both the nitro group and the furan moiety were essential for the activity of the compounds. Compounds C-176 and C-178 were found to be specific for mouse STING; however, introduction of butyl and hexyl alkyl groups at the 4-position of the phenyl ring (C-170 and C-171, respectively) showed activity against both human and mouse STING.
Indole ureas (H-151)
A second screen of a 30,000-member library using HEK293T cells expressing human STING led us to identify the 3-acylamino indole derivative H-151 (ref.13). We showed that H-151 is active against human and mouse STING, as evidenced by reduction of type I interferon responses, inhibition of TBK1 phosphorylation and suppression of STING Cys91 palmitoylation in various cellular assays. Similar to the nitrofurans discussed above, protein mass spectroscopy studies demonstrated that H-151 also forms a covalent bond to Cys91. When H-151 was dosed by intraperitoneal administration followed by a STING agonist challenge, systemic cytokine responses were markedly inhibited. In addition, treatment of Trex1−/− mice expressing a bioluminescent IFNβ-responsive reporter showed a reduction in IFNβ reporter levels after dosing for 1 week. We speculated on a mechanism by which the indole series exemplified by H-151 covalently inactivates STING.
Nitro fatty acids
Hansen et al.174 have shown that nitro fatty acids are formed after HSV infection. They showed that endogenous nitro fatty acids (NO2-FA) such as nitrolinoleic acid (NO2 -cLA) and nitrooleic acid (NO2-OA) covalently modify STING via a Michael addition into the nitro alkene of the fatty acids. Cell-based studies with NO2-FA led to reduction of type I interferons in response to HSV-2 infection in both THP-1 cells and in BMDMs. The authors showed that NO2-OA alkylated both Cys88 and Cys91, in addition to alkylation of His16, in a non-selective fashion. Additional studies in fibroblasts from patients with SAVI, all bearing the gain-of-function STING-N154S alteration, showed that the increase in pTBK1 was almost completely inhibited by NO2-OA treatment.
Acrylamides
Vinogradova et al.175 used chemical proteomic activity-based protein profiling to map 3,000 cysteines in T cells that were reactive with a library of electrophilic small molecules. Mass-spectroscopic analysis showed that acrylamides BPK-21 and BPK-25 formed adducts with Cys91 of STING, as well as with cysteines of other immune-related proteins. Furthermore, this group showed that BPK-25 inhibited cGAMP-mediated STING in peripheral blood mononuclear cells. The authors presented a model for the binding of these acrylamides to Cys91 of STING.
Inhibitors of unknown mechanism
A recent report by Huffman et al.176 detailed a mechanistic study into the synthesis of a library of butanolide heterodimers. A cell-based phenotypic screen of ~250,000 compounds seeking cGAS–STING pathway inhibitors conducted by this group identified four butanolide heterodimers as hits, including compound 13. THP-1 cells treated with compound 13 showed a reduction in CXCL10 mRNA levels following stimulation with dsDNA, which is indicative of reduced type I interferon induction. Although it was demonstrated that compound 13 could modulate the cGAS–STING pathway, the exact mechanism of action was not described.
Outlook and future perspective
The past few years have seen an explosion of interest in and knowledge about the cGAS–STING pathway, which, as we have discussed above, is a central danger-sensing mechanism of innate immunity. Although much remains to be learned, compelling evidence suggests that the sensing of out-of-context DNA drives activation of cGAS–STING signalling in a broad range of inflammatory disease settings, thereby rationalizing the blockade of this pathway for therapeutic benefit. Undoubtedly, continuing efforts will need to carefully validate clinical implications drawn from animal models to human diseases and to provide more precise knowledge on the causative roles of the cGAS–STING pathway in disease pathogenesis. These efforts will go hand in hand with the advancement of solid biomarkers as well as with the use of conditional mouse models or selective drug-like compounds to monitor and inhibit cGAS–STING pathway activity in mice in vivo or in ex vivo human samples, respectively. Concerning the first aspect, measuring the levels of cGAMP and phosphorylation of Ser366 STING are pathway-specific read-outs that can serve as useful, reactive clinical biomarkers, faithfully reflecting cGAS–STING pathway engagement. As for the second aspect, screening approaches have yielded first compounds with drug-like properties that will be of immense value to complement genetic studies by intervening at different stages along the disease process and allowing us to track efficacy in primary human samples. At the same time, the existing chemical matter can serve as a valuable basis to develop clinical candidates. A unique aspect of the pathway is that it offers two druggable candidates that operate in series — at least as far as the transduction of DNA-initiated signals is concerned. Whether targeting one over the other may be preferred in terms of improving efficacy or safety profiles is still unclear.
A critical aspect for the future will be to better understand the minimal level of inhibition required for therapeutic benefit. It is possible that strong reduction of the pathway provokes adverse effects in humans by increasing susceptibility to infection. This may be particularly relevant for the treatment of chronic conditions that require repetitive or continuous treatment regimens. Still, the direct targeting of cGAS–STING bears potential benefits over more non-specific and broadly acting anti-cytokine antibodies or compounds targeting key signalling molecules, such as JAK inhibitors or TBK1 inhibitors, as it leaves intact essential compensatory innate immune recognition pathways, most critically the TLR, RIG-like receptor and inflammasome pathways. Moreover, insight from fundamental biology as well as preclinical disease models demonstrates that activation of the cGAS–STING pathway occurs in a highly cooperative manner. Therefore, partial — instead of complete — blockade appears to suffice to confer anti-inflammatory effects, while still allowing for cGAS–STING activity in the context of overt infection. If such a balance could be achieved then the breadth and essentiality of cGAS–STING in inflammation is no longer a liability, but in fact may be its most powerful advantage as it could serve as a therapeutic target of broad utility.
References
Ablasser, A. & Chen, Z. J. cGAS in action: expanding roles in immunity and inflammation. Science 363, eaat8657 (2019).
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009).
Ablasser, A. et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384 (2013).
Diner, E. J. et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 3, 1355–1361 (2013).
Gao, P. et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107 (2013).
Zhang, X. et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 51, 226–235 (2013). Together with references 3–5, this study demonstrates that cGAS produces a non-canonical cyclic dinucleotide second messenger molecule.
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Wu, J. et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013). Together with reference 7, this study identifies cGAS as a cytosolic DNA sensor inducing type I interferon signalling by producing the second messenger cGAMP.
Ablasser, A. & Hur, S. Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat. Immunol. 21, 17–29 (2020).
Cohen, D. et al. Cyclic GMP-AMP signalling protects bacteria against viral infection. Nature 574, 691–695 (2019).
Morehouse, B. R. et al. STING cyclic dinucleotide sensing originated in bacteria. Nature 586, 429–433 (2020). Together with reference 10, this study identifies a role for cGAMP signalling and STING in anti-phage immunity in bacteria, indicating that the eukaryotic cGAS–STING system arose from an ancient bacterial defence mechanism against phages.
Vincent, J. et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat. Commun. 8, 750 (2017). This study identifies the first class of small-molecule inhibitors of cGAS.
Haag, S. M. et al. Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273 (2018). This study identifies the first covalent small-molecule inhibitors of STING.
Yum, S., Li, M., Frankel, A. E. & Chen, Z. J. Roles of the cGAS-STING pathway in cancer immunosurveillance and immunotherapy. Annu. Rev. Cancer Biol. 3, 323–344 (2019).
Motedayen Aval, L., Pease, J. E., Sharma, R. & Pinato, D. J. Challenges and opportunities in the clinical development of STING agonists for cancer immunotherapy. J. Clin. Med. 9, 3323 (2020).
Civril, F. et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337 (2013).
Kranzusch, P. J., Lee, A. S., Berger, J. M. & Doudna, J. A. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 3, 1362–1368 (2013).
Li, X. et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39, 1019–1031 (2013).
Zhang, X. et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 6, 421–430 (2014).
Xie, W. et al. Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc. Natl Acad. Sci. USA 116, 11946–11955 (2019).
Andreeva, L. et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 549, 394–398 (2017).
Du, M. & Chen, Z. J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709 (2018).
Luecke, S. et al. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 18, 1707–1715 (2017).
Ishikawa, H. & Barber, G. N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008). This work identifies STING as a key mediator of DNA sensing and antiviral immunity.
Huang, Y. H., Liu, X. Y., Du, X. X., Jiang, Z. F. & Su, X. D. The structural basis for the sensing and binding of cyclic di-GMP by STING. Nat. Struct. Mol. Biol. 19, 728–730 (2012).
Shang, G. et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat. Struct. Mol. Biol. 19, 725–727 (2012).
Shu, C., Yi, G., Watts, T., Kao, C. C. & Li, P. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat. Struct. Mol. Biol. 19, 722–724 (2012).
Gao, P. et al. Structure-function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell 154, 748–762 (2013).
Ergun, S. L., Fernandez, D., Weiss, T. M. & Li, L. STING polymer structure reveals mechanisms for activation, hyperactivation, and inhibition. Cell 178, 290–301 e210 (2019).
Shang, G., Zhang, C., Chen, Z. J., Bai, X. C. & Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 567, 389–393 (2019). This study provides the first cryo-EM structure of full-length STING.
Mukai, K. et al. Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 7, 11932 (2016).
Ramanjulu, J. M. et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 564, 439–443 (2018).
Dobbs, N. et al. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18, 157–168 (2015).
Gui, X. et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266 (2019).
Srikanth, S. et al. The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat. Immunol. 20, 152–162 (2019).
Zhang, B.-C. et al. STEEP mediates STING ER exit and activation of signaling. Nat. Immunol. 21, 868–879 (2020).
Deng, Z. et al. A defect in COPI-mediated transport of STING causes immune dysregulation in COPA syndrome. J. Exp. Med. 217, e20201045 (2020).
Mukai, K. et al. Homeostatic regulation of STING by Golgi-to-ER membrane traffic. Preprint at bioRxiv https://doi.org/10.1101/2020.05.20.107664 (2020).
Gonugunta, V. K. et al. Trafficking-mediated STING degradation requires sorting to acidified endolysosomes and can be targeted to enhance anti-tumor response. Cell Rep. 21, 3234–3242 (2017).
Schoggins, J. W. et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011).
Kranzusch, P. J. et al. Ancient origin of cGAS-STING reveals mechanism of universal 2′,3′ cGAMP signaling. Mol. Cell 59, 891–903 (2015).
Margolis, S. R., Wilson, S. C. & Vance, R. E. Evolutionary origins of cGAS-STING signaling. Trends Immunol. 38, 733–743 (2017).
Zhang, C. et al. Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398 (2019).
Zhao, B. et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 569, 718–722 (2019).
Liu, S. et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 (2015).
Wu, J., Dobbs, N., Yang, K. & Yan, N. Interferon-independent activities of mammalian STING mediate antiviral response and tumor immune evasion. Immunity 53, 115–126.e115 (2020).
Yamashiro, L. H. et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat. Commun. 11, 3382 (2020).
Cerboni, S. et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J. Exp. Med. 214, 1769–1785 (2017).
de Oliveira Mann, C. C. et al. Modular architecture of the STING C-terminal tail allows interferon and NF-κB signaling adaptation. Cell Rep. 27, 1165–1175.e1165 (2019).
Balka, K. R. et al. TBK1 and IKKepsilon act redundantly to mediate STING-induced NF-kappaB responses in myeloid cells. Cell Rep. 31, 107492 (2020).
Bakhoum, S. F. et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018). This study describes how tumour cells can subvert the cGAS–STING-mediated immune response to promote tumour metastasis and cancer spread to distant organs.
Hou, Y. et al. Non-canonical NF-kappaB antagonizes STING sensor-mediated DNA sensing in radiotherapy. Immunity 49, 490–503.e494 (2018).
Saitoh, T. et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl Acad. Sci. USA 106, 20842–20846 (2009).
Konno, H., Konno, K. & Barber, G. N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155, 688–698 (2013).
Prabakaran, T. et al. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM 1-dependent autophagy pathway activated by TBK1. EMBO J. 37, e97858 (2018).
Liu, D. et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 26, 1735–1749 (2019).
Nassour, J. et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 565, 659–663 (2019).
Glück, S. et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 19, 1061–1070 (2017). Together with references 155 and 156, this study delineates the cGAS-dependent mechanisms underlying cellular senescence and senescence-associated secretory phenotype.
Gulen, M. F. et al. Signalling strength determines proapoptotic functions of STING. Nat. Commun. 8, 427 (2017).
Petrasek, J. et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl Acad. Sci. USA 110, 16544–16549 (2013).
Zierhut, C. et al. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 178, 302–315 e323 (2019). This study describes how prolonged mitotic arrest induces cGAS-mediated cell death independently of transcription induction.
Brault, M., Olsen, T. M., Martinez, J., Stetson, D. B. & Oberst, A. Intracellular nucleic acid sensing triggers necroptosis through synergistic type I IFN and TNF signaling. J. Immunol. 200, 2748–2756 (2018).
Tang, C. H. et al. Agonist-mediated activation of STING induces apoptosis in malignant B cells. Cancer Res. 76, 2137–2152 (2016).
Gaidt, M. M. et al. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell 171, 1110–1124 e1118 (2017).
Larkin, B. et al. Cutting edge: activation of STING in T cells induces type I IFN responses and cell death. J. Immunol. 199, 397–402 (2017).
Ablasser, A. et al. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503, 530–534 (2013). This study establishes that cGAMP can be transferred between cells to promote in trans STING activation and antiviral immunity.
Chen, Q. et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498 (2016).
Schadt, L. et al. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep. 29, 1236–1248 e1237 (2019).
Luther, J. et al. Hepatic gap junctions amplify alcohol liver injury by propagating cGAS-mediated IRF3 activation. Proc. Natl Acad. Sci. USA 117, 11667–11673 (2020).
Bridgeman, A. et al. Viruses transfer the antiviral second messenger cGAMP between cells. Science 349, 1228–1232 (2015).
Gentili, M. et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science 349, 1232–1236 (2015).
Ahn, J., Xia, T., Rabasa Capote, A., Betancourt, D. & Barber, G. N. Extrinsic phagocyte-dependent STING signaling dictates the immunogenicity of dying cells. Cancer Cell 33, 862–873.e865 (2018).
Li, L. et al. Hydrolysis of 2′3′-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol. 10, 1043–1048 (2014).
Luteijn, R. D. et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 573, 434–438 (2019).
Ritchie, C., Cordova, A. F., Hess, G. T., Bassik, M. C. & Li, L. SLC19A1 is an importer of the immunotransmitter cGAMP. Mol. Cell 75, 372–381 e375 (2019).
Lahey, L. J. et al. The LRRC8A:C heteromeric channel is a cGAMP transporter and the dominant cGAMP importer in human vasculature cells. Preprint at bioRxiv https://doi.org/10.1101/2020.02.13.948273 (2020).
Zhou, C. et al. Transfer of cGAMP into bystander cells via LRRC8 volume-regulated anion channels augments STING-mediated interferon responses and anti-viral immunity. Immunity 52, 767–781 e766 (2020).
Zhou, Y. et al. Blockade of the phagocytic receptor MerTK on tumor-associated macrophages enhances P2X7R-dependent STING activation by tumor-derived cGAMP. Immunity 52, 357–373.e359 (2020).
Kranzusch, P. J. cGAS and CD-NTase enzymes: structure, mechanism, and evolution. Curr. Opin. Struct. Biol. 59, 178–187 (2019).
Deng, L. et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41, 843–852 (2014).
King, K. R. et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 23, 1481–1487 (2017).
Marichal, T. et al. DNA released from dying host cells mediates aluminum adjuvant activity. Nat. Med. 17, 996–1002 (2011).
Lood, C. et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 22, 146–153 (2016).
Kawane, K. et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443, 998–1002 (2006).
Martinez, J. et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl Acad. Sci. USA 108, 17396–17401 (2011).
Cunha, L. D. et al. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell 175, 429–441 e416 (2018).
West, A. P. et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015). This study establishes mitochondrial DNA release as a cell-intrinsic trigger of antiviral signalling via cGAS–STING signalling.
Aguirre, S. et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2, 17037 (2017).
Wiens, K. E. & Ernst, J. D. The mechanism for type I interferon induction by Mycobacterium tuberculosis is bacterial strain-dependent. PLoS Pathog. 12, e1005809 (2016).
Aarreberg, L. D. et al. Interleukin-1β induces mtDNA release to activate innate immune signaling via cGAS-STING. Mol. Cell 74, 801–815.e806 (2019).
Sliter, D. A. et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262 (2018).
Kim, J. et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 366, 1531–1536 (2019).
Wu, Z. et al. Mitochondrial DNA stress signalling protects the nuclear genome. Nat. Metab. 1, 1209–1218 (2019).
Gehrke, N. et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 39, 482–495 (2013).
Rongvaux, A. et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577 (2014).
White, M. J. et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159, 1549–1562 (2014).
Ning, X. et al. Apoptotic caspases suppress type I interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol. Cell 74, 19–31.e17 (2019).
McArthur, K. et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359, eaao6047 (2018).
Riley, J. S. et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 37, e99238 (2018).
Ichim, G. et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 57, 860–872 (2015).
Yu, C. H. et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 183, 636–649 (2020).
Stetson, D. B. & Medzhitov, R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103 (2006). This study links the presence of cytosolic DNA to the production of type I interferons.
Stetson, D. B., Ko, J. S., Heidmann, T. & Medzhitov, R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598 (2008). This study identifies TREX1 as a key regulator of cytosolic DNA levels through degradation of accumulating retroelements in the cytosol and reveals a mechanistic link to the autoinflammatory AGS.
Thomas, C. A. et al. Modeling of TREX1-dependent autoimmune disease using human stem cells highlights L1 accumulation as a source of neuroinflammation. Cell Stem Cell 21, 319–331.e318 (2017).
De Cecco, M. et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78 (2019).
Hartlova, A. et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 42, 332–343 (2015).
Coquel, F. et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557, 57–61 (2018).
Harding, S. M. et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 (2017).
Mackenzie, K. J. et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 (2017). These two studies unveil the role of micronuclei in cell-intrinsic immune surveillance by cGAS to detect genomic instability.
Ivanov, A. et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 202, 129–143 (2013).
Crow, Y. J. & Manel, N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat. Rev. Immunol. 15, 429–440 (2015).
Uggenti, C., Lepelley, A. & Crow, Y. J. Self-awareness: nucleic acid-driven inflammation and the type I interferonopathies. Annu. Rev. Immunol. 37, 247–267 (2019).
Jeremiah, N. et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J. Clin. Invest. 124, 5516–5520 (2014).
Liu, Y. et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 371, 507–518 (2014).
Lin, B. et al. A novel STING1 variant causes a recessive form of STING-associated vasculopathy with onset in infancy (SAVI). J. Allergy Clin. Immunol. 146, 1204–1208 e6 (2020).
Melki, I. et al. Disease-associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J. Allergy Clin. Immunol. 140, 543–552 e545 (2017).
Konno, H. et al. Pro-inflammation associated with a gain-of-function mutation (R284S) in the innate immune sensor STING. Cell Rep. 23, 1112–1123 (2018).
Saldanha, R. G. et al. A mutation outside the dimerization domain causing atypical STING-associated vasculopathy with onset in infancy. Front. Immunol. 9, 1535 (2018).
Keskitalo, S. et al. Novel TMEM173 mutation and the role of disease modifying alleles. Front. Immunol. 10, 2770 (2019).
Warner, J. D. et al. STING-associated vasculopathy develops independently of IRF3 in mice. J. Exp. Med. 214, 3279–3292 (2017).
Bouis, D. et al. Severe combined immunodeficiency in stimulator of interferon genes (STING) V154M/wild-type mice. J. Allergy Clin. Immunol. 143, 712–725 e715 (2019).
Motwani, M. et al. Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models. Proc. Natl Acad. Sci. USA 116, 7941–7950 (2019).
Siedel, H., Roers, A., Rosen-Wolff, A. & Luksch, H. Type I interferon-independent T cell impairment in a Tmem173 N153S/WT mouse model of STING associated vasculopathy with onset in infancy (SAVI). Clin. Immunol. 216, 108466 (2020).
Bennion, B. G. et al. STING gain-of-function disrupts lymph node organogenesis and innate lymphoid cell development in mice. Cell Rep. 31, 107771 (2020).
Gall, A. et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 36, 120–131 (2012).
Gao, D. et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl Acad. Sci. USA 112, E5699–E5705 (2015).
Gray, E. E., Treuting, P. M., Woodward, J. J. & Stetson, D. B. Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of Aicardi-Goutieres syndrome. J. Immunol. 195, 1939–1943 (2015).
Rodero, M. P. et al. Type I interferon-mediated autoinflammation due to DNase II deficiency. Nat. Commun. 8, 2176 (2017).
Yoshida, H., Okabe, Y., Kawane, K., Fukuyama, H. & Nagata, S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat. Immunol. 6, 49–56 (2005).
Ahn, J., Gutman, D., Saijo, S. & Barber, G. N. STING manifests self DNA-dependent inflammatory disease. Proc. Natl Acad. Sci. USA 109, 19386–19391 (2012).
Watkin, L. B. et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat. Genet. 47, 654–660 (2015).
Vece, T. J. et al. Copa syndrome: a novel autosomal dominant immune dysregulatory disease. J. Clin. Immunol. 36, 377–387 (2016).
Lepelley, A. et al. Mutations in COPA lead to abnormal trafficking of STING to the Golgi and interferon signaling. J. Exp. Med. 217, e20200600 (2020).
Steiner, A. et al. Activation of STING due to COPI-deficiency. Preprint at bioRxiv https://doi.org/10.1101/2020.07.09.194399 (2020).
An, J. et al. Expression of cyclic GMP-AMP synthase in patients with systemic lupus erythematosus. Arthritis Rheumatol. 69, 800–807 (2017).
Kato, Y. et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann. Rheum. Dis. 77, 1507–1515 (2018).
Lee-Kirsch, M. A. et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 39, 1065–1067 (2007).
Namjou, B. et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 12, 270–279 (2011).
Sharma, S. et al. Suppression of systemic autoimmunity by the innate immune adaptor STING. Proc. Natl Acad. Sci. USA 112, E710–E717 (2015).
Thim-uam, A. et al. STING mediates lupus via the activation of conventional dendritic cell maturation and plasmacytoid dendritic cell differentiation. iScience 23, 101530 (2020).
Martinez, J. et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 533, 115–119 (2016).
Mavragani, C. P. et al. Expression of long interspersed nuclear element 1 retroelements and induction of type I interferon in patients with systemic autoimmune disease. Arthritis Rheumatol. 68, 2686–2696 (2016).
Gestermann, N. et al. Netting neutrophils activate autoreactive B cells in lupus. J. Immunol. 200, 3364–3371 (2018).
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C. & Gage, F. H. Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934 (2010).
McCauley, M. E. et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature 585, 96–101 (2020).
Su, M.-Y., Fromm, S. A., Zoncu, R. & Hurley, J. H. Structure of the C9orf72 ARF GAP complex that is haploinsufficient in ALS and FTD. Nature 585, 251–255 (2020).
Sharma, M., Rajendrarao, S., Shahani, N., Ramírez-Jarquín, U. N. & Subramaniam, S. Cyclic GMP-AMP synthase promotes the inflammatory and autophagy responses in Huntington disease. Proc. Natl Acad. Sci. USA 117, 15989–15999 (2020).
Nazmi, A. et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia 67, 1254–1276 (2019).
Ahn, J. et al. Inflammation-driven carcinogenesis is mediated through STING. Nat. Commun. 5, 5166 (2014).
Liang, H. et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun. 8, 1736 (2017).
Liu, H. et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563, 131–136 (2018).
van Deursen, J. M. The role of senescent cells in ageing. Nature 509, 439–446 (2014).
Baker, D. J. et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Xu, M. et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256 (2018).
Dou, Z. et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 (2017).
Yang, H., Wang, H., Ren, J., Chen, Q. & Chen, Z. J. cGAS is essential for cellular senescence. Proc. Natl Acad. Sci. USA 114, E4612–E4620 (2017).
Simon, M. et al. LINE1 derepression in aged wild-type and SIRT6-deficient mice drives inflammation. Cell Metab. 29, 871–885 e875 (2019).
Childs, B. G. et al. Senescent cells: an emerging target for diseases of ageing. Nat. Rev. Drug Discov. 16, 718–735 (2017).
Desdín-Micó, G. et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 368, 1371–1376 (2020).
Hall, J. et al. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS ONE 12, e0184843 (2017).
Lama, L. et al. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat. Commun. 10, 2261 (2019).
Padilla-Salinas, R. et al. Discovery of small-molecule cyclic GMP-AMP synthase inhibitors. J. Org. Chem. 85, 1579–1600 (2020).
Zhao, W. et al. In silico screening-based discovery of novel inhibitors of human cyclic GMP–AMP synthase: a cross-validation study of molecular docking and experimental testing. J. Chem. Inf. Model. 60, 3265–3276 (2020).
An, J., Woodward, J. J., Sasaki, T., Minie, M. & Elkon, K. B. Cutting edge: antimalarial drugs inhibit IFN-β production through blockade of cyclic GMP-AMP synthase-DNA interaction. J. Immunol. 194, 4089–4093 (2015).
An, J., Minie, M., Sasaki, T., Woodward, J. J. & Elkon, K. B. Antimalarial drugs as immune modulators: new mechanisms for old drugs. Annu. Rev. Med. 68, 317–330 (2017).
An, J. et al. Inhibition of cyclic GMP-AMP synthase using a novel antimalarial drug derivative in trex1-deficient mice. Arthritis Rheumatol. 70, 1807–1819 (2018).
Steinhagen, F. et al. Suppressive oligodeoxynucleotides containing TTAGGG motifs inhibit cGAS activation in human monocytes. Eur. J. Immunol. 48, 605–611 (2018).
Wang, M., Sooreshjani, M. A., Mikek, C., Opoku-Temeng, C. & Sintim, H. O. Suramin potently inhibits cGAMP synthase, cGAS, in THP1 cells to modulate IFN-β levels. Future Med. Chem. 10, 1301–1317 (2018).
Ehsanian, R., Van Waes, C. & Feller, S. M. Beyond DNA binding - a review of the potential mechanisms mediating quinacrine’s therapeutic activities in parasitic infections, inflammation, and cancers. Cell Commun. Signal. 9, 13 (2011).
Gursel, I. et al. Repetitive elements in mammalian telomeres suppress bacterial DNA-induced immune activation. J. Immunol. 171, 1393–1400 (2003).
Kaminski, J. J. et al. Synthetic oligodeoxynucleotides containing suppressive TTAGGG motifs inhibit AIM2 inflammasome activation. J. Immunol. 191, 3876–3883 (2013).
Siu, T. et al. Discovery of a novel cGAMP competitive ligand of the inactive form of sting. ACS Med. Chem. Lett. 10, 92–97 (2019).
Li, S. et al. The cyclopeptide astin C specifically inhibits the innate immune CDN sensor STING. Cell Rep. 25, 3405–3421.e3407 (2018).
Hansen, A. L. et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc. Natl Acad. Sci. USA 115, E7768–E7775 (2018).
Vinogradova, E. V. et al. An activity-guided map of electrophile-cysteine interactions in primary human T cells. Cell 182, 1009–1026.e1029 (2020).
Huffman, B. J. et al. Electronic complementarity permits hindered butenolide heterodimerization and discovery of novel cGAS/STING pathway antagonists. Nat. Chem. 12, 310–317 (2020).
Orzalli, M. H. et al. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc. Natl Acad. Sci. USA 112, E1773–E1781 (2015).
Gentili, M. et al. The N-terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep. 26, 3798 (2019).
Volkman, H. E., Cambier, S., Gray, E. E. & Stetson, D. B. Tight nuclear tethering of cGAS is essential for preventing autoreactivity. eLife 8, e47491 (2019). This study shows that cGAS is predominantly a nuclear protein and that cGAS is tethered to chromatin.
Guey, B. et al. BAF restricts cGAS on nuclear DNA to prevent innate immune activation. Science 369, 823–828 (2020).
Boyer, J. A. et al. Structural basis of nucleosome-dependent cGAS inhibition. Science 370, 450–454 (2020).
Kujirai, T. et al. Structural basis for the inhibition of cGAS by nucleosomes. Science 370, 455–458 (2020).
Michalski, S. et al. Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature 587, 678–682 (2020).
Pathare, G. R. et al. Structural mechanism of cGAS inhibition by the nucleosome. Nature 587, 668–672 (2020).
Zhao, B. et al. The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature 587, 673–677 (2020). These five studies report the cryo-EM structure of cGAS tethered to nucleosomes and identify how cGAS is inactivated when bound to the nucleosome.
Uggenti, C. et al. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat. Genet. 52, 1364–1372 (2020).
Yi, G. et al. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLoS ONE 8, e77846 (2013).
Patel, S. et al. The common R71H-G230A-R293Q human TMEM173 is a null allele. J. Immunol. 198, 776–787 (2017).
Sivick, K. E. et al. Comment on “The common R71H-G230A-R293Q human TMEM173 is a null allele”. J. Immunol. 198, 4183–4185 (2017).
Patel, S. & Jin, L. TMEM173 variants and potential importance to human biology and disease. Genes. Immun. 20, 82–89 (2019).
Zhou, W. et al. Structure of the human cGAS-DNA complex reveals enhanced control of immune surveillance. Cell 174, 300–311 e311 (2018).
Rice, G. I. et al. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 12, 1159–1169 (2013).
Mackenzie, K. J. et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 35, 831–844 (2016).
Pokatayev, V. et al. RNase H2 catalytic core Aicardi-Goutieres syndrome-related mutant invokes cGAS- STING innate immune-sensing pathway in mice. J. Exp. Med. 213, 329–336 (2016).
Konig, N. et al. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann. Rheum. Dis. 76, 468–472 (2017).
Wang, J. et al. Accumulation of cytosolic dsDNA contributes to fibroblast-like synoviocytes-mediated rheumatoid arthritis synovial inflammation. Int. Immunopharmacol. 76, 105791 (2019).
Li, Q. et al. Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol. Med. 12, e11002 (2020).
Kerur, N. et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med. 24, 50–61 (2018).
Yu, Y. et al. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Invest. 129, 546–555 (2018).
Zhao, Q., Wei, Y., Pandol, S. J., Li, L. & Habtezion, A. STING signaling promotes inflammation in experimental acute pancreatitis. Gastroenterology 154, 1822–1835 e1822 (2018).
Benmerzoug, S. et al. STING-dependent sensing of self-DNA drives silica-induced lung inflammation. Nat. Commun. 9, 1–19 (2018).
Heipertz, E. L., Harper, J. & Walker, W. E. STING and TRIF contribute to mouse sepsis, depending on severity of the disease model. Shock 47, 621–631 (2017).
Zeng, L. et al. ALK is a therapeutic target for lethal sepsis. Sci. Transl Med. 9, eaan5689 (2017).
Cao, D. et al. Cytosolic DNA sensing promotes macrophage transformation and governs myocardial ischemic injury. Circulation 137, 2613–2634 (2018).
Hu, D. et al. Cytosolic DNA sensor cGAS plays an essential pathogenetic role in pressure overload-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 318, H1525–H1537 (2020).
Zhu, Q. et al. Cutting edge: STING mediates protection against colorectal tumorigenesis by governing the magnitude of intestinal inflammation. J. Immunol. 193, 4779–4782 (2014).
Abdullah, A. et al. STING-mediated type-I interferons contribute to the neuroinflammatory process and detrimental effects following traumatic brain injury. J. Neuroinflammation 15, 323 (2018).
Acknowledgements
The authors thank W. R. Roush and H. M. Seidel for advice during the preparation of this document. A.A. acknowledges support from the SNF (31003A-159836) and the European Research Council (ERC-StG: 804933).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
A.A. is a member of the scientific advisory board of IFM Therapeutics and scientific co-founder of IFM Due. J.D.K. and S.V. are employees of IFM Therapeutics. A.D. declares no competing interests.
Additional information
Peer review information
Nature Reviews Immunology thanks P. Li and N. Yan for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Decout, A., Katz, J.D., Venkatraman, S. et al. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol 21, 548–569 (2021). https://doi.org/10.1038/s41577-021-00524-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-021-00524-z
This article is cited by
-
Dual deficiency of melatonin and dihydrotestosterone promotes stromal cell damage and mediates prostatitis via the cGAS-STING pathway in sleep-deprived mice
Cell Communication and Signaling (2024)
-
Recent research advances in pain mechanisms in McCune–Albright syndrome thinking about the pain mechanism of FD/MAS
Journal of Orthopaedic Surgery and Research (2024)
-
Silencing PinX1 enhances radiosensitivity and antitumor-immunity of radiotherapy in non-small cell lung cancer
Journal of Translational Medicine (2024)
-
A new physiological medium uncovers biochemical and cellular alterations in Lesch-Nyhan disease fibroblasts
Molecular Medicine (2024)
-
The contribution of pattern recognition receptor signalling in the development of age related macular degeneration: the role of toll-like-receptors and the NLRP3-inflammasome
Journal of Neuroinflammation (2024)
