
Abstract
The COVID-19 pandemic caused by infection with SARS-CoV-2 has led to more than 200,000 deaths worldwide. Several studies have now established that the hyperinflammatory response induced by SARS-CoV-2 is a major cause of disease severity and death in infected patients. Macrophages are a population of innate immune cells that sense and respond to microbial threats by producing inflammatory molecules that eliminate pathogens and promote tissue repair. However, a dysregulated macrophage response can be damaging to the host, as is seen in the macrophage activation syndrome induced by severe infections, including in infections with the related virus SARS-CoV. Here we describe the potentially pathological roles of macrophages during SARS-CoV-2 infection and discuss ongoing and prospective therapeutic strategies to modulate macrophage activation in patients with COVID-19.
Similar content being viewed by others
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel enveloped RNA betacoronavirus that emerged in December 2019 in Wuhan, China, and is the cause of coronavirus disease 2019 (COVID-19). The most frequent clinical presentation of severe COVID-19 is pneumonia with fever, cough and dyspnoea. The Chinese Center for Disease Control and Prevention reported that of 44,500 confirmed infections the majority of infected patients (80%) experienced mild disease (with either no or only mild pneumonia), 14% developed severe disease (with dyspnoea, hypoxia or greater than 50% lung involvement on imaging tests) and 5% developed critical disease (characterized by respiratory failure, systemic shock or multi-organ failure)1. Approximately 20–30% of patients who have been hospitalized with COVID-19-associated pneumonia have required intensive care for respiratory support2,3. Acute respiratory distress syndrome (ARDS) is a common complication of severe viral pneumonia, including pneumonia caused by highly pathogenic coronaviruses such as SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV)4. In a group of 1,099 patients hospitalized with COVID-19, ARDS was reported to occur in 15.6% of patients with severe pneumonia5. A recent study from the New York area, which has emerged as the epicentre of the pandemic in the United States, reported that of 2,634 patients who were hospitalized with confirmed COVID-19 between 1 March and 4 April 2020, 14.2% of patients were treated in intensive care units (ICUs), 12.2% of patients required invasive mechanical ventilation and 21% of patients died. Mortality for those requiring mechanical ventilation was 88.1%6.
The factors that trigger severe illness in individuals infected with SARS-CoV-2 are not completely understood and the development of severe disease does not seem to be solely related to viral load and could involve a defective interferon response7. An excessive inflammatory response to SARS-CoV-2 is thought to be a major cause of disease severity and death in patients with COVID-19 (ref.8) and is associated with high levels of circulating cytokines, profound lymphopenia and substantial mononuclear cell infiltration in the lungs, heart9, spleen, lymph nodes and kidney10,11, as observed in post-mortem analysis.
Given that the morbidity and mortality seen in COVID-19 is associated with excessive inflammation, a better understanding of the immunological underpinnings of the differential responses seen in patients infected with SARS-CoV-2 is necessary to better identify therapeutic targets. Dozens of immunomodulatory agents are rapidly going into clinical trials as well as already being used routinely in the clinic in an off-label manner. Without a thorough understanding of which particular inflammatory pathways and cell types represent the best targets, some of these strategies could have detrimental effects in certain patients and/or at different stages of COVID-19 disease. In this Progress article, we discuss the nature of the inflammatory responses that have been detected so far in patients with COVID-19; in particular, we highlight the emerging roles of monocytes and macrophages in the pathology of COVID-19.
Hyperinflammation in severe COVID-19
Almost all patients with COVID-19 present with lung involvement, as evidenced by chest radiography, whereas severe complications — such as ARDS and death — are only observed in a subgroup of patients. Although observational studies reported older age and the presence of comorbidities as risk factors for increased disease severity in patients with COVID-19, it rapidly became clear that severe disease can also occur in younger patients with no pre-existing medical conditions. Higher levels of inflammatory markers in blood (including C-reactive protein, ferritin, and D-dimers), an increased neutrophil-to-lymphocyte ratio1,12,13 and increased serum levels of several inflammatory cytokines and chemokines3,14,15,16,17 have been associated with disease severity and death. The systemic cytokine profiles observed in patients with severe COVID-19 show similarities to those observed in cytokine release syndromes, such as macrophage activation syndrome, with increased production of cytokines such as IL-6, IL-7 and tumour necrosis factor (TNF) and also of inflammatory chemokines including CC-chemokine ligand 2 (CCL2), CCL3 and CXC-chemokine ligand 10 (CXCL10), as well as of the soluble form of the α-chain of the IL-2 receptor. This has led to the hypothesis that dysregulated activation of the mononuclear phagocyte (MNP) compartment contributes to COVID-19-associated hyperinflammation8,18. Levels of IL-6 are often increased in the sera of patients with severe disease, but most studies (including unpublished observations from our own laboratories; M.M and J.C.M.) have not seen increased serum IL-1β levels in patients with COVID-19, including in patients with severe disease. Whether the discrepancy between IL-6 and IL-1β levels seen in patients with COVID-19 reflects technical limitations or localized tissue production of cytokines or indicates the absence of inflammasome activation requires further clarification. Measurement of systemic IL-18 as well as local measurement of IL-1β and IL-18 should better assess the role of inflammasome activation in COVID-19 pathophysiology
As is seen in infections with other highly pathogenic human coronaviruses, a common feature in many patients with COVID-19 is the presence of a global T cell lymphopenia, which is more pronounced in the CD8+ T cell compartment and is particularly prominent in patients with severe disease. Although it is possible that the reduced levels of circulating T cells in patients may reflect their massive recruitment to inflamed tissues or the use of steroid treatment to mitigate inflammation, some studies have reported significant T cell depletion from the secondary lymphoid organs of patients infected with SARS-CoV and SARS-CoV-2 (refs10,19). The potential mechanisms responsible for T cell depletion are still poorly understood but are not likely to be related to direct infection of T cells by the virus, at least in the case of patients with COVID-19 for which no evidence supports possible T cell infection by SARS-CoV-2 (ref.10). Serum levels of TNF, IL-6 and IL-10 negatively correlated with T cell numbers20. In situ terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining revealed high levels of lymphocyte apoptosis in the spleens and lymph nodes of patients who had died from COVID-19, and this was associated with increased expression of the death receptor FAS, suggesting that activation-induced cell death (AICD) may cause T cell lymphopenia10. Because T cells exert major functions in viral containment and clearance, further exploration to clarify whether T cell AICD is antigen-induced and/or increased by macrophage-derived cytokines such as IL-6 will be important.
Trials to block IL-6 signalling have been launched across the world and some clinical benefits have been seen in a subset of patients. A study from China reported a clear benefit in 15 of 20 (75%) patients treated with IL-6 blockade21, leading the FDA to approve trials of IL-6 blockade in patients with COVID-19. A more recent study from Italy reported a benefit in only 7 of 21 (33%) patients22. Neither of these studies had control arms and very few data are available on the exact clinical and inflammatory status of patients who responded versus those who did not. Several groups across the world have now initiated clinical trials combining IL-6 receptor and IL-1β blockade in patients with COVID-19. Hopefully, careful immune monitoring of responders and non-responders will reveal immune correlates of response or resistance to cytokine blockade and shed light on the pathophysiology of the COVID-19-associated inflammatory response. Measuring the prevalence and severity of disease seen in patients with COVID-19 who are on immunomodulatory biologics for other comorbidities will also help us to understand whether blockade of particular immune pathways can modulate COVID-19 disease outcomes; examples include patients on anti-TNF, anti-IL-4 receptor, anti-IL-12/23, anti-IL-17A and Janus kinase (JAK) inhibitors, as well as those receiving drugs that interfere with leukocyte migration (for example, α4β1 integrin, α4β7 integrin and S1PR inhibitors) and T cell co-stimulation. These patients will provide important additional insights on the disease pathophysiology of COVID-19.
Monocytes/macrophages in COVID-19
Bronchoalveolar fluid (BALF) from patients with severe COVID-19 was shown to be enriched in CCL2 and CCL7, two chemokines that are most potent at the recruitment of CC-chemokine receptor 2-positive (CCR2+) monocytes23. Accordingly, single-cell RNA sequencing (scRNA-seq) analysis of BALF collected from patients with severe or mild COVID-19 revealed increased proportions of MNPs, with these cells accounting for as much as 80% of total BALF cells in patients with severe COVID-19 compared with only approximately 60% and 40%, respectively, of total BALF cells in patients with mild disease or healthy controls24. MNP composition was further characterized by a depletion of tissue-resident alveolar macrophages and an abundance of inflammatory monocyte-derived macrophages in patients with severe disease24. Inflammatory programmes seen in these macrophages24 were consistent with findings from bulk RNA-seq analysis of BALF23 from patients with severe COVID-19, and notably both studies identified a strong interferon gene signature. In addition, a subset of macrophages has been described in patients with COVID-19 that is enriched in genes associated with tissue repair and promotes fibrosis generation, such as is seen in liver cirrhosis25. This suggests that the pathogenicity of infiltrating macrophages could extend beyond the promotion of acute inflammation and is also in line with the fibrotic complications observed in patients under mechanical ventilation26.
Interestingly, clonal expansion of subsets of CD8+ T cells with a tissue-resident memory T cell gene signature was observed in the BALF of patients with mild COVID-19, in which inflammatory monocyte infiltration was minimal. These results could suggest that pre-existing populations of tissue-resident memory T cells with potential cross-reactivity against SARS-CoV-2 enable rapid control of the virus and protect against disease progression by limiting epithelial damage, local inflammation and accumulation of pathological macrophage populations. While this hypothesis remains to be addressed, epidemiological studies have reported that prior Bacillus Calmette–Guérin vaccination may protect from COVID-19, leading several countries to launch clinical trials testing the protective benefit of intracutaneous administration of Bacillus Calmette–Guérin vaccine in health-care workers (ClincialTrials.gov identifiers NCT04328441 and NCT04327206).
A significant expansion of populations of CD14+CD16+ monocytes producing IL-6 was also observed in the peripheral blood of patients with COVID-19 in ICUs compared with those patients who did not require ICU hospitalization27,28, a finding also supported by scRNA-seq analysis of peripheral blood mononuclear cells29 and our personal observations (J.C.M. and R. Josien, unpublished observations).
In addition, immunostaining of post-mortem tissue from patients who had died from COVID-19 revealed that populations of CD169+ lymph node subcapsular and splenic marginal zone macrophages express the SARS-CoV-2 entry receptor ACE2 and that these macrophages contained SARS-CoV-2 nucleoprotein (NP)10. This finding is surprising given that scRNA-seq analysis of human tissues failed to identify ACE2 expression on most tissue-resident macrophages, including on secondary lymphoid tissue-resident macrophages (M.M., E. Kenigsberg and A. Leader, unpublished observations). However, it is possible that ACE2 expression on macrophages can be triggered by inflammatory signals such as type I interferon30. It is also possible that additional receptors such as CD147 (ref.31) are involved in virus entry. Although SARS-CoV-2 NP was detected in secondary lymphoid tissue macrophages10, it is still unclear whether the virus productively infected these cells or whether the NP positivity reflected macrophage uptake of other infected cells. Macrophages that contained SARS-CoV-2 viral particles were found to express IL-6, and the presence of IL-6+ macrophages was associated with severe depletion of lymphocytes from the spleen and lymph nodes7. CD68+NP+ macrophages were also observed in the kidneys of patients with COVID-19, and acute kidney tubular damage was associated with marked accumulation of monocytes and macrophages8. These preliminary findings in patients with COVID-19 are in line with the pathology observed in post-mortem tissues obtained from patients infected with SARS-CoV. The disease pathology seen in these patients is also characterized by damage to the lungs consistent with ARDS, focal necrosis and lymphocytic and monocytic infiltration into the lungs, heart, kidney, liver and muscles, as well as massive necrosis of the spleen and focal necrosis of lymph nodes32.
The exact drivers of monocyte and macrophage activation in COVID-19 and the contribution to COVID-19 disease pathophysiology remain to be clarified. Key questions to address include: one, the exact contribution of direct viral sensing as opposed to cytokine exposure to macrophage activation; two, the influence of prior infections and epigenetic remodelling events in shaping monocyte responsiveness33; three, the contribution of the tissue site of immune activation (that is, the inflamed tissue, blood or bone marrow) to macrophage activation state; and four, the contribution of tissue-resident macrophages as opposed to monocyte-derived macrophages to tissue damage.
Macrophage activation in SARS
A similar disease course to that seen during SARS-CoV-2 infection has been described during infection with other highly pathogenic coronaviruses, such as SARS-CoV and MERS-CoV, with 20% of patients ultimately progressing to fatal ARDS. Extensive cellular infiltration dominated by macrophages was found in post-mortem lungs from these patients. Similar to what has been reported in SARS-CoV-2, high levels of interferon-γ (IFNγ), IL-6, IL-12, transforming growth factor-β (TGFβ), CCL2, CXCL10, CXCL9 and IL-8 were reported in patients with SARS-CoV. However, in contrast to patients infected with SARS-CoV-2, low levels of IL-10 and high levels of IL-1β were often detected in SARS-CoV. Of note, while infection of MNPs is abortive in SARS-CoV, MERS-CoV can replicate in monocytes, macrophages and dendritic cells4. As mentioned above, SARS-CoV-2 viral particles were detected in MNPs but evidence of productive infection remains to be demonstrated.
The factors that drive severe lung pathology during infection with highly pathogenic human coronaviruses are poorly understood. Potential mechanisms include high rates of viral replication, which could be responsible for enhanced host cell cytolysis and the strong production of inflammatory cytokines and chemokines by infected epithelial cells; delayed induction of antiviral interferon responses owing to virus escape mechanisms, such as production of interferon inhibitory proteins, which perpetuates virual damage, leading to excessive accumulation of monocytes, macrophages and neutrophils in the lungs4; or modulation of the ACE2-mediated epithelial protective pathway upon virus binding to its receptor34. Although viruses have evolved many mechanisms to block type I interferon production in infected cells35, it is surprising that reduced interferon activity can persist in patients with COVID-19 who show massive inflammatory responses7; this suggests that additional pathways, such as inflammasome activation, could contribute to the persistent low level of type I interferon induction, as was recently shown36. Identifying the exact mechanisms that contribute to reduced type I interferon activity will be critical for the development of targeted immunomodulatory strategies in patients with COVID-19.
Studies in animal models of virus-induced acute lung injury (ALI) suggest that several pathways drive pro-inflammatory programmes, including the infiltration of monocytes. For example, sustained activation of infiltrating monocytes and monocyte-derived macrophages could be driven by type I interferon37, oxidative stress38,39, anti-spike protein IgG immune complexes40 and NLRP3 inflammasome activation41,42. Interestingly, however, deletion of the Toll-like receptor (TLR)/IL-1 receptor adaptor MYD88 had no effect on mouse ALI38; in fact, a protective role of MYD88 was even reported, although protection was independent of IL-1 receptor engagement by IL-1β43. These experiments were conducted in mice with constitutive MYD88 deficiency and do not rule out a pathological function for MYD88-dependent TLR and IL-1β-mediated signalling in late stages of the disease. However, the temporal contribution of inflammasome-induced IL-1β to viral clearance and/or COVID-19 severity remains to be investigated.
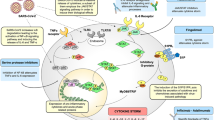
It remains to be clarified whether similar pathways are involved in SARS-CoV-2-mediated activation of inflammatory macrophage responses, the extent of their contribution to disease severity and the therapeutic benefits of targeting the MYD88, TLR and/or IL-1β pathways. It has also yet to be determined whether additional receptors trigger the direct or indirect activation of macrophages by SARS-CoV-2 and what the contribution of these putative receptors may be to COVID-19 disease stages and progression to severe forms of disease. Figure 1 considers potential pathways that may drive hyperactivation of macrophages during infection with SARS-CoV-2.

Several mechanisms likely contribute to the hyperactivation of monocyte-derived macrophages that is seen in patients with COVID-19. Delayed production of type I interferon leading to enhanced cytopathic effects and increased sensing of microbial threats promotes the enhanced release of monocyte chemoattractants by alveolar epithelial cells (and likely also by macrophages and stromal cells), leading to sustained recruitment of blood monocytes into the lungs. Monocytes differentiate into pro-inflammatory macrophages though activation of Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathways. Activated natural killer (NK) cells and T cells further promote the recruitment and activation of monocyte-derived macrophages through the production of granulocyte–macrophage colony-stimulating factor (GM-CSF), tumour necrosis factor (TNF) and interferon-γ (IFNγ). Oxidized phospholipids (OxPLs) accumulate in infected lungs and activate monocyte-derived macrophages through the Toll-like receptor 4 (TLR4)–TRAF6–NF-κB pathway. Virus sensing can trigger TLR7 activation through viral single-stranded RNA recognition. It is possible that type I interferons induce the expression of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) entry receptors, enabling the virus to gain access to the cytoplasm of macrophages and to activate the NLRP3 inflammasome, leading to the secretion of mature IL-1β and/or IL-18. IL-1β can amplify activation of monocyte-derived macrophages in an autocrine or paracrine way, but it can also reduce type I interferon production in infected lungs. The engagement of Fcγ receptors (FcγRs) by anti-spike protein IgG immune complexes can contribute to increased inflammatory activation of monocyte-derived macrophages. Activated monocyte-derived macrophages contribute to the COVID-19 cytokine storm by releasing massive amounts of pro-inflammatory cytokines. CCL, CC-chemokine ligand; CXCL10, CXC-chemokine ligand 10; ISG, interferon-stimulated gene; ITAM, immunoreceptor tyrosine-based activation motif; TRAM, TRIF-related adaptor molecule.
Hypercoagulation and MNP activation
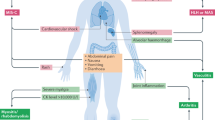
Importantly, lower platelets counts, increased levels of fibrin degradation products (known as D-dimers)44 and coagulation abnormalities are being increasingly associated with poor prognosis and could represent a main cause of organ failure and death in patients with severe COVID-19 (ref.45) (Fig. 2). Microthrombi of the lungs, lower limbs, hands, brain46, heart, liver and kidneys47 have been described in patients with COVID-19. Notably, activation of coagulation and intravascular coagulation is a hallmark of organ injury in sepsis, where it is mainly mediated by inflammatory cytokines48.

Circulating pro-inflammatory stimuli, such as viral pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs) and cytokines trigger activation of blood monocytes, which respond by inducing tissue factor membrane expression. Endothelial cells are activated by cytokines and viral particles and produce monocyte chemoattractants and adhesion molecules. Endothelial damage induced by the virus can also expose tissue factor on endothelial cells. Activated monocytes are recruited to endothelial cells. Tissue factor expressed by activated monocytes, monocyte-derived microvesicles and endothelial cells activates the extrinsic coagulation pathway, leading to fibrin deposition and blood clotting. Neutrophils are recruited by activated endothelial cells and release neutrophil extracellular traps (NETs), which activate the coagulation contact pathway and bind and activate platelets to amplify blood clotting. Major endogenous anticoagulant pathways, which include tissue factor pathway inhibitor (TFPI), antithrombin and protein C, are reduced further, supporting coagulation activation. CCL2, CC-chemokine ligand 2; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; TLR, Toll-like receptor; TNF, tumour necrosis factor; vWF, von Willebrand factor.
The initiation of inflammation-induced coagulation leading to an increased coagulation state is almost always mediated by the expression of the tissue factor (TF; also called CD142 or coagulation factor III) pathway49,50. TF is expressed on mononuclear cells in response to pro-inflammatory cytokines (mainly IL-6), but TF is also expressed on vascular endothelial cells and is known to promote the transformation of prothrombin into thrombin, which in turn converts circulating fibrinogen into fibrin leading to fibrin-based blood clots51. In addition, major natural anticoagulant pathways, such as antithrombin or TF pathway inhibitor, are almost always impaired during inflammation, contributing further to the propagation of coagulation. In the absence of vascular injury, the initiation of coagulation is completely dependent on the recruitment of TF-expressing inflammatory monocytes by activated endothelial cells52 (Fig. 2). It is also possible that oxidized phospholipids (OxPLs) — which have been detected in the lungs of patients infected with SARS-CoV38 — contribute to the process53. OxPLs are produced following oxidative stress; they promote the induction of TF expression and inflammatory programmes in monocytes and activate endothelial cells to recruit and bind to monocytes53,54. In experimental ALI, OxPLs trigger macrophage activation through the TLR4–TRIF–TRAF6–NF-κB pathway38. Interfering with monocyte and/or endothelial cell activation in response to OxPLs could help prevent thrombotic complications, especially in patients with COVID-19 who have pre-existing cardiovascular and metabolic comorbidities13. However, it is important to note that the SARS-CoV-2 entry receptor ACE2 is expressed on arterial and venous endothelial cells55,56, where it plays an anti-inflammatory protective effect. Whether the increased coagulopathy observed in patients with COVID-19 is also partly due to direct vascular damage induced by SARS-CoV-2 infection or ACE2 inhibition remains to be determined.
Therapeutic perspectives
Clinical trials to assess the benefits of inflammatory cytokine blockade targeting IL-6 and IL-1β alone or in combination are in progress (Table 1). Trials examining the blockade of additional myeloid-derived inflammatory cytokines, such as TNF57 and granulocyte–macrophage colony-stimulating factor (GM–CSF) (NCT04341116), have also been considered and/or initiated. Strategies aimed at broadly interfering with cytokine signalling could also significantly reduce hyperinflammation in patients with severe COVID-19, and several trials testing the use of JAK inhibitors are ongoing. Alternatively, strategies upstream of the production of individual cytokines could represent a broader and possibly more effective way to dampen the occurrence of cytokine release syndromes. Current evidence suggests that pathological macrophages mostly derive from circulating monocytes that massively infiltrate the lungs and other organs rather than from tissue-resident macrophage populations. Circulating CD14+ monocytes require the chemokine receptor CCR2 to exit the bone marrow and to accumulate in inflamed tissues58,59. CCR2 blockade could potentially help to reduce the accumulation of pathological monocytes in inflamed tissues, although it is likely that redundant mechanisms independent of CCR2 may also contribute to monocyte accumulation in tissues during severe inflammation. Trials targeting CCR5, another chemokine receptor that regulates monocyte and T cell migration, have been initiated in patients with COVID-19 who show mild-to-moderate symptoms of respiratory illness (NCT04343651). Although the antiviral activity of hydroxychloroquine remains uncertain, its immunomodulatory activity in chronic inflammatory diseases is well established60 and this could have contributed to the reported (although still controversial) clinical benefits of this drug in patients with COVID-19.
Based on evidence that interferon responses to coronaviruses deviate from those induced by other respiratory viruses37,61, trials testing the administration of type I or type III interferons have been initiated. Type I interferon (IFNαβ) receptors are ubiquitously expressed and type I interferons can drive antiviral effects but also induce the activation of immune cells that could potentially enhance tissue pathology. By contrast, type III interferon (also known as IFNλ) mainly targets epithelial cells as well as a restricted pool of immune cells, and could therefore promote a potent antiviral effect without enhancing pathological tissue inflammation62. As discussed above, inflammatory macrophages from the lungs of patients with severe COVID-19 exhibit a strong interferon signature and it is possible that, while interferon may be protective during the early stages of the disease, persistent IFNγ production could ultimately drive macrophage hyperactivation as is seen in macrophage activation syndrome. Trials exploring IFNγ blockade in patients with COVID-19 who show respiratory distress and hyperinflammation have been initiated (NCT04324021).
Current models of COVID-19 propose three distinct immune stages that are crucial for the ultimate disease course. In the first stage, early activation of the immune system through the induction of a potent interferon response is important to control the virus. In the second stage, a delayed interferon response may lead to progressive tissue damage. This may ultimately lead to the third stage, a deleterious hyperinflammation characterized by the excessive macrophage activation and coagulation that is seen in patients with severe disease, possibly followed by dysregulation of tissue repair mechanisms and fibrosis63. The exact drivers of pathological inflammation in COVID-19 remain to be identified, but it is crucial that we establish the pathways involved in the different stages of the inflammatory response to SARS-CoV-2 and determine the contribution of these pathways to ultimate disease outcome. The results of these studies should help identify not only the most relevant immunomodulatory strategies but also the optimal timing of such immunomodulatory interventions to maximize therapeutic effect.
Conclusion
Although data characterizing the immune and inflammatory status in patients with COVID-19 are only starting to emerge, it is clear that hyperinflammation and coagulopathy contribute to disease severity and death in patients infected with SARS-CoV-2. A better understanding of which subsets of patients are at risk of pathological inflammation and which particular inflammatory pathways drive disease pathology is crucial to develop rationale-based clinical therapeutic strategies. It is critical to start measuring longitudinally and with as much granularity as possible the systemic and potentially local inflammatory responses induced by SARS-CoV-2 and how these pathways are modulated by the different therapies currently used as standard of care or in clinical trials. Although it is difficult to engage in research in the middle of a pandemic, clinical centres that have the means to perform deep-monitoring studies now have the responsibility to conduct such studies and to share their strategies and results with the scientific community.
Change history
02 June 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41577-020-0353-y
References
Wu, C. et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. https://doi.org/10.1001/jamainternmed.2020.0994 (2020).
Chen, N. et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395, 507–513 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Channappanavar, R. & Perlman, S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 39, 529–539 (2017).
Guan, W.-J. et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2002032 (2020).
Richardson, S. et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA https://doi.org/10.1001/jama.2020.6775 (2020).
Hadjadj, J. et al. Impaired type I interferon activity and exacerbated inflammatory responses in severe COVID-19 patients. Preprint at medRxiv https://doi.org/10.1101/2020.04.19.20068015 (2020).
Mehta, P. et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395, 1033–1034 (2020).
Xu, Z. et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 8, 420–422 (2020).
Chen, Y. et al. The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) directly decimates human spleens and lymph nodes. Preprint at medRxiv https://doi.org/10.1101/2020.03.27.20045427 (2020).
Diao, B. et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Preprint at medRxiv https://doi.org/10.1101/2020.03.04.20031120 (2020).
Ruan, Q., Yang, K., Wang, W., Jiang, L. & Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. https://doi.org/10.1007/s00134-020-05991-x (2020).
Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062 (2020).
Chen, G. et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 130, 2620–2629 (2020).
Gong, J. et al. Correlation analysis between disease severity and inflammation-related parameters in patients with COVID-19 pneumonia. Preprint at medRxiv https://doi.org/10.1101/2020.02.25.20025643 (2020).
Qin, C. et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa248 (2020).
Yang, Y. et al. Exuberant elevation of IP-10, MCP-3 and IL-1ra during SARS-CoV-2 infection is associated with disease severity and fatal outcome. Preprint at medRxiv https://doi.org/10.1101/2020.03.02.20029975 (2020).
Schulert, G. S. & Grom, A. A. Pathogenesis of macrophage activation syndrome and potential for cytokine-directed therapies. Annu. Rev. Med. 66, 145–159 (2015).
Gu, J. et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 202, 415–424 (2005).
Diao, B. et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Preprint at medRxiv https://doi.org/10.1101/2020.02.18.20024364 (2020).
Xu, X. et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl Acad. Sci. USA https://doi.org/10.1073/pnas.2005615117 (2020).
Gritti, G. et al. Use of siltuximab in patients with COVID-19 pneumonia requiring ventilatory support. Preprint at medRxiv https://doi.org/10.1101/2020.04.01.20048561 (2020).
Zhou, Z. et al. Overly exuberant innate immune response SARS-CoV-2 infect. Cell Host Microbe https://doi.org/10.2139/ssrn.3551623 (2020).
Liao, M. et al. The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. Preprint at medRxiv https://doi.org/10.1101/2020.02.23.20026690 (2020).
Ramachandran, P. et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518 (2019).
Cabrera-Benitez, N. E. et al. Mechanical ventilation-associated lung fibrosis in acute respiratory distress syndrome: a significant contributor to poor outcome. Anesthesiology 121, 189–198 (2014).
Zhou, Y. et al. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID-19 patients. Natl Sci. Rev. https://doi.org/10.1093/nsr/nwaa041 (2020).
Zhang, D. et al. COVID-19 infection induces readily detectable morphological and inflammation-related phenotypic changes in peripheral blood monocytes, the severity of which correlate with patient outcome. Preprint at medRxiv https://doi.org/10.1101/2020.03.24.20042655 (2020).
Wen, W. et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Preprint at medRxiv https://doi.org/10.1101/2020.03.23.20039362 (2020).
Ziegler, C. et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is enriched in specific cell subsets across tissues. Cell https://doi.org/10.2139/ssrn.3555145 (2020).
Wang, K. et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. Preprint at bioRxiv https://doi.org/10.1101/2020.03.14.988345 (2020).
Ding, Y. et al. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J. Pathol. 200, 282–289 (2003).
Netea, M. G. et al. Trained immunity: a program of innate immune memory in health and disease. Science 352, aaf1098 (2016).
Kuba, K. et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 11, 875–879 (2005).
García-Sastre, A. Ten strategies of interferon evasion by viruses. Cell Host Microbe 22, 176–184 (2017).
Mayer-Barber, K. D. et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 511, 99–103 (2014).
Channappanavar, R. et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 19, 181–193 (2016).
Imai, Y. et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133, 235–249 (2008).
Vijay, R. et al. Critical role of phospholipase A2 group IID in age-related susceptibility to severe acute respiratory syndrome-CoV infection. J. Exp. Med. 212, 1851–1868 (2015).
Liu, L. et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 4, e123158 (2019).
Chen, I.-Y., Moriyama, M., Chang, M.-F. & Ichinohe, T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 10, 50 (2019).
Shi, C.-S., Nabar, N. R., Huang, N.-N. & Kehrl, J. H. SARS-coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 5, 101 (2019).
Sheahan, T. et al. MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathog. 4, e1000240 (2008).
Tang, N. et al. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. https://doi.org/10.1111/jth.14817 (2020).
Xiang-Hua, Y. et al. Severe acute respiratory syndrome and venous thromboembolism in multiple organs. Am. J. Respir. Crit. Care Med. 182, 436–437 (2010).
Zhang, Y. et al. Coagulopathy and antiphospholipid antibodies in patients with COVID-19. N. Engl. J. Med. 382, e38 (2020).
Liu, P. P., Blet, A., Smyth, D. & Li, H. The science underlying COVID-19: implications for the cardiovascular system. Circulation https://doi.org/10.1161/CIRCULATIONAHA.120.047549 (2020).
Levi, M., Nieuwdorp, M., van der Poll, T. & Stroes, E. Metabolic modulation of inflammation-induced activation of coagulation. Semin. Thromb. Hemost. 34, 26–32 (2008).
Simmons, J. & Pittet, J.-F. The coagulopathy of acute sepsis. Curr. Opin. Anaesthesiol. 28, 227–236 (2015).
Iba, T., Levy, J. H., Raj, A. & Warkentin, T. E. Advance in the management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J. Clin. Med. 8, 728 (2019).
van der Poll, T., van de Veerdonk, F. L., Scicluna, B. P. & Netea, M. G. The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 17, 407–420 (2017).
von Brühl, M.-L. et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 209, 819–835 (2012).
Berliner, J. A. & Watson, A. D. A role for oxidized phospholipids in atherosclerosis. N. Engl. J. Med. 353, 9–11 (2005).
Owens, A. P. et al. Monocyte tissue factor-dependent activation of coagulation in hypercholesterolemic mice and monkeys is inhibited by simvastatin. J. Clin. Invest. 122, 558–568 (2012).
Hamming, I. et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203, 631–637 (2004).
Chen, L., Li, X., Chen, M., Feng, Y. & Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 116, 1097–1100 (2020).
Feldmann, M. et al. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet https://doi.org/10.1016/S0140-6736(20)30858-8 (2020).
Serbina, N. V. & Pamer, E. G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7, 311–317 (2006).
Shi, C. et al. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating Toll-like receptor ligands. Immunity 34, 590–601 (2011).
Schrezenmeier, E. & Dörner, T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat. Rev. Rheumatol. 16, 155–166 (2020).
Blanco-Melo, D. et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell https://doi.org/10.1016/j.cell.2020.04.026 (2020).
Broggi, A., Granucci, F. & Zanoni, I. Type III interferons: balancing tissue tolerance and resistance to pathogen invasion. J. Exp. Med. 217, e20190295 (2020).
Siddiqi, H. K. & Mehra, M. R. COVID-19 illness in native and immunosuppressed states: a clinical–therapeutic staging proposal. J. Heart Lung Transplant. 39, 405–407 (2020).
Acknowledgements
The authors thank C. Gianarelli, A. Leader (ISMMS), R. Josien, A. Roquilly, C. Bressollette, B. Gaborit, J. Poschmann and C. Braudeau (Université de Nantes, Inserm, CHU Nantes) for important discussion and insights.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Immunology thanks F. Tacke, M. Travis and T. A. Wynn for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ClinicalTrials.gov: https://clinicaltrials.gov/
Rights and permissions
About this article

Cite this article
Merad, M., Martin, J.C. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol 20, 355–362 (2020). https://doi.org/10.1038/s41577-020-0331-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-020-0331-4
This article is cited by
-
Role of epinephrine in attenuating cytokine storm, decreasing ferritin, and inhibiting ferroptosis in SARS-CoV-2
The Egyptian Heart Journal (2024)
-
Degree of COVID-19 severity and mortality in stroke: correlation of clinical and laboratory parameters
BMC Neuroscience (2024)
-
A new DNA aptamer which binds to SARS-CoV-2 spike protein and reduces pro-inflammatory response
Scientific Reports (2024)
-
Targeted therapies of inflammatory diseases with intracellularly gelated macrophages in mice and rats
Nature Communications (2024)
-
Organoids in virology
npj Viruses (2024)
