
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of the ongoing coronavirus disease 2019 (COVID-19) pandemic. Alongside investigations into the virology of SARS-CoV-2, understanding the fundamental physiological and immunological processes underlying the clinical manifestations of COVID-19 is vital for the identification and rational design of effective therapies. Here, we provide an overview of the pathophysiology of SARS-CoV-2 infection. We describe the interaction of SARS-CoV-2 with the immune system and the subsequent contribution of dysfunctional immune responses to disease progression. From nascent reports describing SARS-CoV-2, we make inferences on the basis of the parallel pathophysiological and immunological features of the other human coronaviruses targeting the lower respiratory tract — severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV). Finally, we highlight the implications of these approaches for potential therapeutic interventions that target viral infection and/or immunoregulation.
Similar content being viewed by others


Introduction
The first cases of coronavirus disease 2019 (COVID-19) likely occurred from a zoonotic transmission in China in December 2019, linked to a large seafood market that also traded in live wild animals. The causative virus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is capable of human-to-human transmission and spread rapidly to other parts of China and then to other locations. By 24 March 2020, SARS-CoV-2 had infected more than 381,000 people across 195 countries/regions and killed more than 16,000: a pandemic as declared by the World Health Organization1. Daily reports of sharp rises in the number of new cases continue to emerge from many countries/regions, but efforts to overcome the virus are hampered by a lack of knowledge of several important aspects of SARS-CoV-2 infection, ranging from pathogen biology to host response and treatment options. Therefore, there is an urgent need to better understand the host–pathogen biology of COVID-19 as this will offer important insights into treatment and management of the disease, including identification of new therapies. Here, we review the literature on SARS-CoV-2 pathophysiology, its interaction with target cells and the immune response to the virus, including the contribution of dysfunctional immune responses to disease progression. Specifically, we highlight the implications of specific features of the infection for promising therapeutic interventions that could target the virus or the dysfunctional immune response. Moreover, we discuss how studies focused on the adaptive immune response will be crucial in informing the development of vaccines and therapeutic monoclonal antibodies.
Pathogenesis of COVID-19
Coronaviruses are known to cause disease in humans and animals. Among these, four (human coronaviruses 229E, NL63, OC43 and HKU1) typically infect only the upper respiratory tract and cause relatively minor symptoms2. However, there are three coronaviruses (severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS-CoV-2) that can replicate in the lower respiratory tract and cause pneumonia, which can be fatal. SARS-CoV-2 belongs to the betacoronavirus genus. Its closest relative among human coronaviruses is SARS-CoV, with 79% genetic similarity3. However, among all known coronavirus sequences, SARS-CoV-2 is most similar to bat coronavirus RaTG13, with 98% similarity4, and coronavirus sequences in the pangolin (a scaly anteater) also share high similarity5.
Like the other respiratory coronaviruses, SARS-CoV-2 is transmitted primarily via respiratory droplets, with a possible, but unproven, faecal–oral transmission route. On infection, the median incubation period is approximately 4–5 days before symptom onset6,7,8,9, with 97.5% of symptomatic patients developing symptoms within 11.5 days8. At the point of hospital admission, patients with COVID-19 typically exhibit a fever and dry cough; less commonly, patients also experience difficulty in breathing, muscle and/or joint pain, headache/dizziness, diarrhoea, nausea and the coughing up of blood6,10,11,12,13,14,15. Within 5–6 days of symptom onset, SARS-CoV-2 viral load reaches its peak — significantly earlier than that of the related SARS-CoV, where viral load peaks at about 10 days after symptom onset16,17,18,19. Severe COVID-19 cases progress to acute respiratory distress syndrome (ARDS), on average around 8–9 days after symptom onset11,20.
The pathophysiology of SARS-CoV-2 infection closely resembles that of SARS-CoV infection, with aggressive inflammatory responses strongly implicated in the resulting damage to the airways21. Therefore, disease severity in patients is due to not only the viral infection but also the host response. The pattern of increasing severity with age is also broadly consistent with the epidemiology of SARS-CoV and MERS-CoV6,11,14.
ARDS seen in severe COVID-19 is characterized by difficulty in breathing and low blood oxygen level22. As a result, some patients may succumb to secondary bacterial and fungal infections14. ARDS may lead directly to respiratory failure, which is the cause of death in 70% of fatal COVID-19 cases22. In addition, the vast release of cytokines by the immune system in response to the viral infection and/or secondary infections can result in a cytokine storm and symptoms of sepsis that are the cause of death in 28% of fatal COVID-19 cases22. In these cases, uncontrolled inflammation inflicts multi-organ damage leading to organ failure, especially of the cardiac, hepatic and renal systems (Fig. 1). Most patients with SARS-CoV infection who progressed to renal failure eventually died23.

When severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infects cells expressing the surface receptors angiotensin-converting enzyme 2 (ACE2) and TMPRSS2, the active replication and release of the virus cause the host cell to undergo pyroptosis and release damage-associated molecular patterns, including ATP, nucleic acids and ASC oligomers. These are recognized by neighbouring epithelial cells, endothelial cells and alveolar macrophages, triggering the generation of pro-inflammatory cytokines and chemokines (including IL-6, IP-10, macrophage inflammatory protein 1α (MIP1α), MIP1β and MCP1). These proteins attract monocytes, macrophages and T cells to the site of infection, promoting further inflammation (with the addition of IFNγ produced by T cells) and establishing a pro-inflammatory feedback loop. In a defective immune response (left side) this may lead to further accumulation of immune cells in the lungs, causing overproduction of pro-inflammatory cytokines, which eventually damages the lung infrastructure. The resulting cytokine storm circulates to other organs, leading to multi-organ damage. In addition, non-neutralizing antibodies produced by B cells may enhance SARS-CoV-2 infection through antibody-dependent enhancement (ADE), further exacerbating organ damage. Alternatively, in a healthy immune response (right side), the initial inflammation attracts virus-specific T cells to the site of infection, where they can eliminate the infected cells before the virus spreads. Neutralizing antibodies in these individuals can block viral infection, and alveolar macrophages recognize neutralized viruses and apoptotic cells and clear them by phagocytosis. Altogether, these processes lead to clearance of the virus and minimal lung damage, resulting in recovery. G-CSF, granulocyte colony-stimulating factor; TNF, tumour necrosis factor.
Host cell infection and its prevention
The first step in infection is virus binding to a host cell through its target receptor. Earlier work on SARS-CoV demonstrated that this virus principally targets airway epithelial cells, alveolar epithelial cells, vascular endothelial cells and macrophages in the lung, all of which express the angiotensin-converting enzyme 2 (ACE2) host target receptor used by SARS-CoV24,25,26 (Fig. 2). As SARS-CoV-2 uses the same entry receptor, these cell subsets are likely targeted by this virus4,27,28. SARS-CoV infection reduces ACE2 expression in lung cells. Because loss of pulmonary ACE2 function is associated with acute lung injury, virus-induced ACE2 downregulation may be important for disease pathology29,30,31,32. ACE2 has been shown to regulate the renin–angiotensin system (RAS)32. Therefore, a reduction in ACE2 function after viral infection could result in a dysfunction of the RAS, which influences blood pressure and fluid/electrolyte balance, and enhance inflammation and vascular permeability in the airways.

COVID-19 shows a difference in fatality rate between males (2.8%) and females (1.7%)33. As ACE2 is located on the X chromosome, there may be alleles that confer resistance to COVID-19, explaining the lower fatality rate in females. Alternatively, the oestrogen and testosterone sex hormones have different immunoregulatory functions, which could influence immune protection or disease severity34.
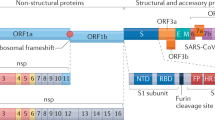
SARS-CoV-2 shares 79% genome sequence identity with SARS-CoV4. The spike (S) protein is expressed on the surface of the virus particles, giving the characteristic ‘crown’ appearance. The S protein comprises two subunits: S1 and S2. The S1 subunit consists of an amino-terminal domain and a receptor-binding domain (RBD), which in SARS-CoV spans from amino acid residue 318 to amino acid residue 510 (refs35,36,37). The RBD binds to ACE2 as its host cell target receptor, which starts the infection process4. RBD binding to ACE2 triggers endocytosis of the SARS-CoV-2 virion and exposes it to endosomal proteases38. The S2 subunit consists of a fusion peptide (FP) region and two heptad repeat regions: HR1 and HR2 (refs39,40). Within the endosome, the S1 subunit is cleaved away, exposing the fusion peptide, which inserts into the host membrane. The S2 region then folds in on itself to bring the HR1 and HR2 regions together. This leads to membrane fusion and releases the viral package into the host cytoplasm.
There is 72% similarity in the amino acid sequence of the RBDs of SARS-CoV and SARS-CoV-2, with highly similar tertiary structures. Computational modelling and biophysical measurements indicate that the SARS-CoV-2 RBD binds to ACE2 with higher affinity than that of SARS-CoV41,42. In addition, the SARS-CoV-2 S protein contains a furin-like cleavage site, similarly to MERS-CoV and human coronavirus OC43, which is not found in SARS-CoV43. These characteristics could contribute to the increased infectivity of SARS-CoV-2 relative to SARS-CoV. In addition to furin precleavage, the cellular serine protease TMPRSS2 is also required to properly process the SARS-CoV-2 spike protein and facilitate host cell entry44.
One pathway for the development of therapeutics against SARS-CoV-2 is to block the host target ACE2 receptor or TMPRSS2 (Fig. 3). Currently, there are compounds that target these molecules that have been clinically approved for other indications. For example, machine learning algorithms predicted that baricitinib, a Janus kinase (JAK) inhibitor approved for treatment of rheumatoid arthritis, could inhibit ACE2-mediated endocytosis45. Another JAK inhibitor, ruxolitinib, will be tested in clinical trials for treatment of COVID-19 later this year46. An alternative strategy is to deliver high concentrations of a soluble form of ACE2 that could potentially reduce virus entry into target host cells. This principle is being tested with APN01, a recombinant form of ACE2 developed by APEIRON that is currently in clinical trials47. Monoclonal antibodies targeting the S protein may also inhibit virus entry or fusion and are further discussed in the section entitled B cell immunity. Nafamostat mesylate48,49 and camostat mesylate44 are known inhibitors of TMPRSS2 and are currently approved in several countries/regions to treat other conditions. While there are no clinical trials specifically testing these drugs against COVID-19 at the time of writing, when camostat mesylate was tested on SARS-CoV-2 isolated from a patient, it prevented entry of the virus into lung cells44,50. If this approach is validated, rapid repurposing of these drugs will be effective and timely in the fight against COVID-19.

(1) Antibodies against the spike protein (raised through vaccination or by adoptive transfer) could block severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) from interacting with the angiotensin-converting enzyme 2 (ACE2) receptor on host cells. (2) Protease inhibitors against the serine protease TMPRSS2 can prevent spike protein cleavage, which is necessary for viral fusion into the host cell. Blocking either ACE2 interaction or viral fusion could prevent the virus from infecting the host cell. (3) Virus-specific memory CD8+ T cells from a previous vaccination or infection can differentiate into effector cells during rechallenge. When they identify infected cells presenting virus-specific epitopes, they degranulate and kill infected cells before they can produce mature virions. (4) In a novel treatment method that targets the cytokine storm symptoms, the blood of patients with coronavirus disease 2019 (COVID-19) can be passed through customized columns that are specially designed to trap pro-inflammatory cytokines, before the purified blood is passed back into patients.
Inflammatory immunopathogenesis
SARS-CoV-2 infection and the destruction of lung cells triggers a local immune response, recruiting macrophages and monocytes that respond to the infection, release cytokines and prime adaptive T and B cell immune responses. In most cases, this process is capable of resolving the infection. However, in some cases, a dysfunctional immune response occurs, which can cause severe lung and even systemic pathology.
Cytopathic viruses, including SARS-CoV-2 (ref.51), induce death and injury of virus-infected cells and tissues as part of the virus replicative cycle. Viral infection and replication in airway epithelial cells52 could cause high levels of virus-linked pyroptosis with associated vascular leakage, as seen in patients with SARS-CoV53. Pyroptosis is a highly inflammatory form of programmed cell death that is commonly seen with cytopathic viruses54. This is a likely trigger for the subsequent inflammatory response55. IL-1β, an important cytokine released during pyroptosis, is elevated during SARS-CoV-2 infection11. Using a variety of pattern-recognition receptors (PRRs), alveolar epithelial cells and alveolar macrophages detect the released pathogen-associated molecular patterns (PAMPs), such as viral RNA, and damage-associated molecular patterns (DAMPs), including ATP, DNA and ASC oligomers. A wave of local inflammation ensues, involving increased secretion of the pro-inflammatory cytokines and chemokines IL-6, IFNγ, MCP1 and IP-10 into the blood of afflicted patients11,22. These cytokines are indicators of a T helper 1 (TH1) cell-polarized response, which parallels observations made for SARS-CoV and MERS-CoV56. Secretion of such cytokines and chemokines attracts immune cells, notably monocytes and T lymphocytes, but not neutrophils, from the blood into the infected site57,58. Pulmonary recruitment of immune cells from the blood and the infiltration of lymphocytes into the airways may explain the lymphopenia and increased neutrophil–lymphocyte ratio seen in around 80% of patients with SARS-CoV-2 infection6,59.
In most individuals, recruited cells clear the infection in the lung, the immune response recedes and patients recover. However, in some patients, a dysfunctional immune response occurs, which triggers a cytokine storm that mediates widespread lung inflammation. It was observed that patients with severe COVID-19, requiring intensive care in hospitals, exhibited higher blood plasma levels of IL-2, IL-7, IL-10, granulocyte colony-stimulating factor (G-CSF), IP-10, MCP1, macrophage inflammatory protein 1α (MIP1α) and tumour necrosis factor (TNF)11. IL-6 levels in these patients continue to increase over time and are relatively more elevated in non-survivors than survivors60. Notably, there exists a highly inflammatory monocyte-derived FCN1+ macrophage population in the bronchoalveolar lavage fluid of patients with severe but not mild COVID-19 (ref.61). Also, patients with severe disease show a significantly higher percentage of CD14+CD16+ inflammatory monocytes in peripheral blood than patients with mild disease62. These cells secrete inflammatory cytokines that contribute to the cytokine storm, including MCP1, IP-10 and MIP1α (Fig. 1).
The mechanisms by which SARS-CoV-2 subverts the body’s innate antiviral cytokine responses are yet to be studied, but research on SARS-CoV shows that multiple viral structural and non-structural proteins antagonize interferon responses. Antagonism occurs at various stages of the interferon signalling pathway, including by preventing PRR recognition of viral RNA63,64,65, by preventing PRR signalling through TBK1/inhibitor of nuclear factor-κB kinase subunit-ε (IKKε), TRAF3 and IRF3 (refs63,66), by preventing downstream interferon signalling through STAT1 (ref.67) and by promoting host mRNA degradation and inhibiting host protein translation68. It is very likely that at least some of these pathways are conserved in SARS-CoV-2. Antagonism of the interferon response aids viral replication, resulting in increased release of pyroptosis products that can further induce aberrant inflammatory responses.
Unrestrained inflammatory cell infiltration can itself mediate damage in the lung through excessive secretion of proteases and reactive oxygen species, in addition to the direct damage resulting from the virus. Together, these result in diffuse alveolar damage, including desquamation of alveolar cells, hyaline membrane formation and pulmonary oedema57,58. This limits the efficiency of gas exchange in the lung, causing difficulty in breathing and low blood oxygen levels. The lung also becomes more vulnerable to secondary infections.
In addition to local damage, cytokine storm also has ripple effects across the body. Elevated levels of cytokines such as TNF can cause septic shock and multi-organ failure. These may result in myocardial damage and circulatory failure observed in some patients69. Older people (those aged over 60 years) and people with co-morbidities are more likely to develop such a dysfunctional immune response that causes pathology and also fails to successfully eradicate the pathogen. The exact reasons for this are unclear, although one reason may be an ageing lung microenvironment causing altered dendritic cell maturation and migration to the lymphoid organs70, and thereby defective T cell activation. In contrast, children tend not to develop severe disease despite being capable of experiencing high viral titres71. Across all age groups younger than 18 years, more than 50% of children experienced mild symptoms or were asymptomatic, with less than 6% of children developing severe symptoms72. Thus, while the aforementioned studies represent important inroads, a full picture of the critical host immune factors that underlie the development of severer inflammatory responses in some patients remains poorly defined.
It remains controversial whether virus persistence is necessary to drive the ongoing damage. The peak of viral titres in respiratory tract samples might occur even before symptom onset of pneumonia in SARS-CoV and SARS-CoV-2 infections17,19. However, a large retrospective cohort study showed that viral RNA was detectable in non-survivors up until the point of death, suggesting a correlation between virus persistence and poor disease outcome60. As viral RNA may linger even after active infection, and is not representative of the infectivity of the virus, whether the poor disease outcome is directly due to large amounts of infectious particles is speculative at this moment. Furthermore, earlier studies of SARS-CoV found that the virus may infect other targets besides lung cells. Notably, virus was found in T lymphocytes73, macrophages74,75,76 and monocyte-derived dendritic cells77. Direct virus killing of lymphocytes could contribute to the observed lymphopenia in patients73. Viral infection in immune cells such as monocytes and macrophages can result in aberrant cytokine production, even if viral infection is not productive74,75,76,77. The degree to which SARS-CoV-2 targets these cells remains poorly defined. Understanding the precise drivers of immune dysfunction is crucial to guide the application of appropriate immunomodulatory treatments.
Several immunosuppressive therapies aimed at limiting immunomediated damage in COVID-19 are at various phases of development and are listed in Table 1. Currently, trials of corticosteroids for treatment of COVID-19 are under way78, although this class of treatment was not recommended during the 2003 SARS epidemic79,80. A clinical trial of the IL-6 antagonist tocilizumab is also under way to test its efficacy81, and sarilumab is also being explored82. Other clinical trials are also testing the effects of targeting granulocyte–macrophage colony-stimulating factor (GM-CSF), including the use of gimsilumab83, lenzilumab84 and namilumab85. Another novel adjunctive therapy is cytosorb86, which acts by absorbing a broad spectrum of cytokines, DAMPs and PAMPs in order to reduce their circulating levels and ameliorate immunopathology. Thalidomide, an agent with immunomodulatory properties, has also been successfully administered to a single patient with COVID-19 (ref.87). As a result, two clinical trials have now been initiated to test its potential to reduce lung injury88,89. TNF antagonism was suggested but not tested in the context of SARS-CoV infection, and it has not yet been tested in patients with COVID-19 (ref.90). A small open-label, non-randomized study suggested that a combination of hydroxychloroquine (a known antimalarial agent) and azithromycin (a common antibiotic) may be beneficial for treating patients with severe COVID-19 (ref.91). Although hydroxychloroquine’s effect on direct inhibition of the virus92 and its anti-inflammatory and immunomodulatory activities are known93, whether these mechanisms play a role against COVID-19 remains to be determined94.
T cell immunity
Both T and B cell responses against SARS-CoV-2 are detected in the blood around 1 week after the onset of COVID-19 symptoms. CD8+ T cells are important for directly attacking and killing virus-infected cells, whereas CD4+ T cells are crucial to prime both CD8+ T cells and B cells. CD4+ T cells are also responsible for cytokine production to drive immune cell recruitment. The first autopsy of a patient with COVID-19 revealed an accumulation of mononuclear cells (likely monocytes and T cells) in the lungs, coupled with low levels of hyperactive T cells in the peripheral blood57. Together with reports of lymphopenia and reduced peripheral T cell levels in patients 6,95,96,97, these findings suggest that T cells are attracted away from the blood and into the infected site to control the viral infection. In patients with COVID-19, increased T cell exhaustion and reduced functional diversity predicted severe disease98. Despite the impaired response, patients who recovered from SARS-CoV infection developed coronavirus-specific memory T cells, which were found up to 2 years after recovery99,100.
SARS-CoV-specific CD4+ T cells express IFNγ, TNF and IL-2, which suggests that patients with SARS-CoV infection exhibit a TH1 cell response and mainly use cellular immunity to control the infection101,102. Although this pro-inflammatory profile may be an aggravating factor for immunopathogenesis, CD4+ T cells have been hypothesized to control SARS, as depletion of these cells in mice resulted in slower clearance of the virus from the host and severer lung inflammation103. With the use of a mouse-adapted strain of SARS-CoV, immunization with dendritic cells bearing SARS-CoV peptides resulted in higher numbers of virus-specific CD4+ and CD8+ T cells that accumulated in the lungs and increased survival104,105. Also, transfer of SARS-CoV-specific CD4+ and CD8+ T cells into immunodeficient mice resulted in better protection against a mouse-adapted strain of SARS-CoV105.
Despite evidence for an important role of T cells in controlling infection, several vaccine formulations against SARS-CoV previously tested in animal models showed signs of immunopathology associated with TH2 cell-mediated eosinophil infiltration106,107. In particular, aged mice that were vaccinated seemed to display increased immunopathology rather than protection108. Further study of the nature of protective versus detrimental T cell responses is critically needed to determine the optimal T cell engagement strategies for vaccines109.
Coronavirus-specific T cells are clearly important in eliminating the virus and controlling disease development and should be considered in vaccine strategies. However, whether T cell responses alone are capable of preventing infection in human settings remains to be investigated. This knowledge will be important for vaccine development.
B cell immunity
B cell responses in patients with COVID-19 occur concomitantly with T follicular helper cell responses, from around 1 week after symptom onset110. In patients with SARS-CoV infection, B cell responses typically arise first against the nucleocapsid (N) protein. Within 4–8 days after symptom onset, antibody responses to S protein are found111,112. Neutralizing antibody responses, likely to the S protein, begin to develop by week 2, and most patients develop neutralizing antibodies by week 3 (refs113,114). Given that viral titres peak earlier for SARS-CoV-2 than for SARS-CoV16,17,18,19, antibody responses may also arise earlier. It seems that a subset of patients may not develop long-lasting antibodies to SARS-CoV-2 (ref.115). It remains unknown whether these patients are susceptible to reinfection, of which there are sporadic reports116,117. Antibodies are likely to be effective against SARS-CoV-2: convalescent serum samples have been applied with apparently good clinical results in COVID-19 (ref.118) and were also previously used successfully in the treatment of SARS119,120,121.
While mechanistic correlates of protection have not yet been identified in humans, neutralization of the virus is presumed to be an important mechanism of action for antibodies, although the specific titre and specificity of the antibody repertoire required (for protection) remain undefined. In SARS-CoV, the primary target of neutralizing antibodies is the RBD122, comprising a 193 amino acid region (amino acids 318–510) in the S protein, which can independently bind to the host target ACE2 receptor35,36,37. Although a few previously identified monoclonal antibodies to SARS-CoV also bind to or neutralize SARS-CoV-2 (ref.123), the majority do not124. This could be due to significant differences in the RBDs of SARS-CoV-2 and SARS-CoV (Fig. 4). In particular, of the 33 amino acids in the region (amino acids 460–492) in the SARS-CoV S protein that contains the critical residues that contact ACE2 (ref.125), less than half (15/33) are conserved in SARS-CoV-2. Nevertheless, mouse antiserum raised against SARS-CoV protein can cross-neutralize SARS-CoV-2 pseudovirus, indicating overlapping neutralizing epitopes between the two viruses28,126.

a | Sequence alignment of severe acute respiratory syndrome coronavirus (SARS-CoV) spike protein and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein, with conserved amino acid residues shown in black and non-conserved residues shown in colours. b | The 3D structure of SARS-CoV-2 (Protein Data Bank ID 6VSB42, peach ribbon) is superimposed on the SARS-CoV receptor-binding motif (RBM) complex with the neutralizing antibody (nAb; red ribbon) interfacing with the RBM (Protein Data Bank 2DD8 (ref.151), purple ribbon). Peach and purple spheres denote the RBMs of SARS-CoV-2 and SARS-CoV, respectively. Magenta spheres denote non-synonymous alterations in the SARS-CoV-2 spike protein that have been reported135.
In China, hospitals have initiated the use of convalescent plasma as a source of therapeutic polyclonal antibodies for treatment of COVID-19, and early data suggest a positive impact on respiratory viral load and mortality127,128. Efforts are under way to develop therapeutic monoclonal antibodies to SARS-CoV-2, using approaches including phage library display, traditional mouse immunization and hybridoma isolation, and cloning of B cell sequences from convalescent human patients129,130,131,132. SARS-CoV does not appear to have strong mechanisms to escape or prevent antibody neutralization, such as glycan shielding of the receptor-binding site against antibody binding133. This is further corroborated by the fact that patients with SARS-CoV infection were generally capable of developing neutralizing antibodies. A recombinant S protein fragment that included the RBD of SARS-CoV showed the highest immunogenicity relative to other recombinant S protein fragments tested, suggesting that the immune system is capable of targeting neutralizing epitopes effectively134. Thus, if SARS-CoV-2 behaves like SARS-CoV in this respect, it is likely that these efforts will be successful in developing neutralizing monoclonal antibodies.
It is possible that alterations in the S protein will render SARS-CoV-2 resistant to some monoclonal antibodies, especially as it spreads and mutates. As of now, the entire RBD remains conserved, and there are only four known rare non-synonymous alterations in the S protein: V483A, L455I, F456V and G476S135. The V483A alteration maps to a similar natural alteration found in MERS-CoV, I529T, where it reduced viral protein binding to the host receptor target and also increased resistance to antibody neutralization from serum samples from patients with MERS136. F456V and G476S alterations also map to similar alteration positions in SARS-CoV (L443R and D463G), which were found in a panel of neutralization escape mutants137.
However, the selection of therapeutic antibody candidates should include careful consideration of potential unwanted side effects. For example, pre-existing antibodies to other coronaviruses may exacerbate SARS-CoV infections through antibody-dependent enhancement138,139,140. Also, previous studies in animal models showed that in SARS-CoV infection, neutralizing antibodies to S protein can potentially augment severe lung injury by exacerbating inflammatory responses141. In addition, a correlation has been observed where development of ARDS coincides with antiviral IgG seroconversion in 80% of patients19. Patients who developed neutralizing antibodies to S protein earlier in infection had a higher rate of disease; it took an average of only 14.7 days for patients who died of infection to reach their peak levels of neutralizing antibody activity, as opposed to 20 days for patients who went on to recover142. Similarly, for MERS, patients with severer disease appear to have higher antibody titres than those with mild disease143,144, although one study argues that it is a delay in the development of antibody responses that is associated with disease145. The binding of antibody–virus immune complexes to activating Fc receptors on alveolar macrophages could induce the expression of pro-inflammatory factors, including IL-8 and MCP1, which add to the immunostimulatory milieu146. Such complexes may also activate the complement system and lead to further unwanted inflammation141. As a result, it is important to consider engineering therapeutic antibodies with little or no pro-inflammatory activity but that retain their virus-neutralizing capacity147. For instance, alterations could be made to the Fc region and/or its glycosylation to change its binding affinity for activating Fc receptors146,148.
Conclusion
This Review has presented the various mechanisms of SARS-CoV-2 infection and COVID-19 immunopathogenesis. Controlling the inflammatory response may be as important as targeting the virus. Therapies inhibiting viral infection and regulation of dysfunctional immune responses may synergize to block pathologies at multiple steps. At the same time, the association between immune dysfunction and outcome of disease severity in patients with COVID-19 should serve as a note of caution in vaccine development and evaluation. Further studies of the host immune response to SARS-CoV-2 are necessary, including a detailed investigation of the determinants of healthy versus dysfunctional outcomes. These will also help identify biomarkers to define immune correlates of protection and disease severity for effective triage of patients.
References
World Health Organization. WHO Director-General’s opening remarks at the media briefing on COVID-19 - 11 March 2020. WHO https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (2020).
Fehr, A. R. & Perlman, S. Coronaviruses: an overview of their replication and pathogenesis. Methods. Mol. Biol. 1282, 1–23 (2015).
Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C. & Garry, R. F. The proximal origin of SARS-CoV-2. Nat. Med. https://doi.org/10.1038/s41591-020-0820-9 (2020).
Guan, W. J. et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2002032 (2020).
Pung, R. et al. Investigation of three clusters of COVID-19 in Singapore: implications for surveillance and response measures. Lancet 395, 1039–1046 (2020).
Lauer, S. A. et al. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Ann. Intern. Med. https://doi.org/10.7326/m20-0504 (2020).
Li, Q. et al. Early transmission dynamics in Wuhan, China, of Novel Coronavirus-infected pneumonia. N. Engl. J. Med. 382, 1199–1207 (2020).
Chan, J. F. et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 395, 514–523 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020). This prospective study is the earliest to include an analysis of cytokine levels in severe and mild COVID-19, showing the presence of a cytokine storm analogous to that found for SARS-CoV infection.
Chen, G. et al. Clinical and immunologic features of severe and moderate coronavirus disease 2019. J. Clin. Invest. https://doi.org/10.1172/jci137244 (2020).
Liu, Y. et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 63, 364–374 (2020).
Chen, N. et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395, 507–513 (2020).
Phan, L. T. et al. Importation and human-to-human transmission of a novel coronavirus in Vietnam. N. Engl. J. Med. 382, 872–874 (2020).
Pan, Y., Zhang, D., Yang, P., Poon, L. L. M. & Wang, Q. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect. Dis. https://doi.org/10.1016/s1473-3099(20)30113-4 (2020).
Kim, J. Y. et al. Viral load kinetics of SARS-CoV-2 infection in first two patients in Korea. J. Korean Med. Sci. 35, e86 (2020).
Zou, L. et al. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N. Engl. J. Med. 382, 1177–1179 (2020).
Peiris, J. S. et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361, 1767–1772 (2003).
Wang, D. et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 323, 1061–1069 (2020).
Wong, C. K. et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 136, 95–103 (2004).
Zhang, B. et al. Clinical characteristics of 82 death cases with COVID-19. Preprint at medRxiv https://doi.org/10.1101/2020.02.26.20028191 (2020).
Chu, K. H. et al. Acute renal impairment in coronavirus-associated severe acute respiratory syndrome. Kidney Int. 67, 698–705 (2005).
Jia, H. P. et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 79, 14614–14621 (2005).
Xu, H. et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral. Sci. 12, 8 (2020).
Hamming, I. et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203, 631–637 (2004).
Zhao, Y. et al. Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCov. Preprint at bioRxiv https://doi.org/10.1101/2020.01.26.919985 (2020).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell https://doi.org/10.1016/j.cell.2020.02.058 (2020). Together with Wrapp et al. (2020), this article presents a cryo-electron microscopy structure of the SARS-CoV-2 spike glycoprotein used for cell entry, including an analysis of its receptor-binding kinetics and antigenicity with respect to SARS-CoV.
Imai, Y. et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436, 112–116 (2005).
Imai, Y., Kuba, K. & Penninger, J. M. The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp. Physiol. 93, 543–548 (2008).
Kuba, K. et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 11, 875–879 (2005).
Kuba, K., Imai, Y. & Penninger, J. M. Angiotensin-converting enzyme 2 in lung diseases. Curr. Opin. Pharmacol. 6, 271–276 (2006).
Epidemiology Working Group for NCIP Epidemic Response. The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) in China. Chin. J. Epidemiol. 41, 145–151 (2020).
Taneja, V. Sex hormones determine immune response. Front. Immunol. 9, 1931 (2018).
Xiao, X., Chakraborti, S., Dimitrov, A. S., Gramatikoff, K. & Dimitrov, D. S. The SARS-CoV S glycoprotein: expression and functional characterization. Biochem. Biophys. Res. Commun. 312, 1159–1164 (2003).
Babcock, G. J., Esshaki, D. J., Thomas, W. D. Jr. & Ambrosino, D. M. Amino acids 270 to 510 of the severe acute respiratory syndrome coronavirus spike protein are required for interaction with receptor. J. Virol. 78, 4552–4560 (2004).
Wong, S. K., Li, W., Moore, M. J., Choe, H. & Farzan, M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 279, 3197–3201 (2004).
Simmons, G. et al. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl Acad. Sci. USA 102, 11876–11881 (2005).
Bosch, B. J. et al. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl Acad. Sci. USA 101, 8455–8460 (2004).
Liu, S. et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 363, 938–947 (2004).
Chen, Y., Guo, Y., Pan, Y. & Zhao, Z. J. Structure analysis of the receptor binding of 2019-nCoV. Biochem. Biophys. Res. Commun. 525, 135–140 (2020).
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020). Together with Walls et al. (2020), this article presents a high-resolution cryo-electron microscopy structure of the SARS-CoV-2 spike glycoprotein used for cell entry, including an analysis of its receptor-binding kinetics with respect to SARS-CoV.
Coutard, B. et al. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 176, 104742 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell https://doi.org/10.1016/j.cell.2020.02.052 (2020).
Richardson, P. et al. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 395, e30–e31 (2020).
Chinese Clinical Trial Register. Chictr.org.cn http://www.chictr.org.cn/showprojen.aspx?proj=49088 (2020).
Pipeline Review. APEIRON’s respiratory drug product to start pilot clinical trial to treat coronavirus disease COVID-19 in China. Pipeline Review https://pipelinereview.com/index.php/2020022673884/Proteins-and-Peptides/APEIRONs-respiratory-drug-product-to-start-pilot-clinical-trial-to-treat-coronavirus-disease-COVID-19-in-China.html (2020).
Wang, M. et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 30, 269–271 (2020).
Yamamoto, M. et al. Identification of nafamostat as a potent inhibitor of Middle East respiratory syndrome coronavirus S protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob. Agents Chemother. 60, 6532–6539 (2016).
Zhang, H., Penninger, J. M., Li, Y., Zhong, N. & Slutsky, A. S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 46, 586–590 (2020).
Park, W. B. et al. Virus isolation from the first patient with SARS-CoV-2 in Korea. J. Korean Med. Sci. 35, e84 (2020).
Zhang, H. et al. Histopathologic changes and SARS-CoV-2 immunostaining in the lung of a patient with COVID-19. Ann. Intern. Med. https://doi.org/10.7326/m20-0533 (2020).
Chen, I. Y., Moriyama, M., Chang, M. F. & Ichinohe, T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 10, 50 (2019).
Fink, S. L. & Cookson, B. T. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 73, 1907–1916 (2005).
Yang, M. Cell pyroptosis, a potential pathogenic mechanism of 2019-nCoV infection. SSRN https://doi.org/10.2139/ssrn.3527420 (2020).
Huang, K. J. et al. An interferon-gamma-related cytokine storm in SARS patients. J. Med. Virol. 75, 185–194 (2005).
Xu, Z. et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 8, 420–422 (2020). This study is the first to describe pathological findings in severe COVID-19 and demonstrates the aberrant immune cell infiltrates found in the lung.
Tian, S. et al. Pulmonary pathology of early phase 2019 novel coronavirus (COVID-19) pneumonia in two patients with lung cancer. J. Thorac. Oncol. https://doi.org/10.1016/j.jtho.2020.02.010 (2020).
Qin, C. et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa248 (2020).
Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062 (2020).
Liao, M. et al. The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. Preprint at medRxiv https://doi.org/10.1101/2020.02.23.20026690 (2020).
Zhou, Y. et al. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID-19 patients. Natl Sci. Rev. https://doi.org/10.1093/nsr/nwaa041 (2020).
Siu, K. L., Chan, C. P., Kok, K. H., Chiu-Yat Woo, P. & Jin, D. Y. Suppression of innate antiviral response by severe acute respiratory syndrome coronavirus M protein is mediated through the first transmembrane domain. Cell. Mol. Immunol. 11, 141–149 (2014).
Versteeg, G. A., Bredenbeek, P. J., van den Worm, S. H. & Spaan, W. J. Group 2 coronaviruses prevent immediate early interferon induction by protection of viral RNA from host cell recognition. Virology 361, 18–26 (2007).
Sun, L. et al. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One 7, e30802 (2012).
Frieman, M., Ratia, K., Johnston, R. E., Mesecar, A. D. & Baric, R. S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 83, 6689–6705 (2009).
Frieman, M. et al. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 81, 9812–9824 (2007).
Narayanan, K. et al. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 82, 4471–4479 (2008).
Ruan, Q., Yang, K., Wang, W., Jiang, L. & Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. https://doi.org/10.1007/s00134-020-05991-x (2020).
Zhao, J., Zhao, J., Legge, K. & Perlman, S. Age-related increases in PGD2 expression impair respiratory DC migration, resulting in diminished T cell responses upon respiratory virus infection in mice. J. Clin. Invest. 121, 4921–4930 (2011).
Kam, K. Q. et al. A well infant with coronavirus disease 2019 (COVID-19) with high viral load. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa201 (2020).
Dong, Y. et al. Epidemiological characteristics of 2143 pediatric patients with 2019 coronavirus disease in China. Pediatrics https://doi.org/10.1542/peds.2020-0702 (2020).
Gu, J. et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 202, 415–424 (2005). This article describes the presence of SARS-CoV viral particles and RNA in T cells, monocytes and macrophages, suggesting that SARS-CoV and potentially SARS-CoV-2 may drive immunopathogenesis by direct infection of immune cells.
Cheung, C. Y. et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J. Virol. 79, 7819–7826 (2005).
Yilla, M. et al. SARS-coronavirus replication in human peripheral monocytes/macrophages. Virus Res. 107, 93–101 (2005).
Tseng, C. T., Perrone, L. A., Zhu, H., Makino, S. & Peters, C. J. Severe acute respiratory syndrome and the innate immune responses: modulation of effector cell function without productive infection. J. Immunol. 174, 7977–7985 (2005).
Law, H. K. et al. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood 106, 2366–2374 (2005).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04273321 (2020).
Stockman, L. J., Bellamy, R. & Garner, P. SARS: systematic review of treatment effects. PLoS Med. 3, e343 (2006).
Tai, D. Y. Pharmacologic treatment of SARS: current knowledge and recommendations. Ann. Acad. Med. Singap. 36, 438–443 (2007).
Chinese Clinical Trial Register. Chictr.org.cn http://www.chictr.org.cn/showproj.aspx?proj=49409 (2020).
Liu, A. China turns Roche arthritis drug Actemra against COVID-19 in new treatment guidelines. FiercePharma https://www.fiercepharma.com/pharma-asia/china-turns-roche-arthritis-drug-actemra-against-covid-19-new-treatment-guidelines (2020).
Roivant Sciences. Roivant announces development of anti-GM-CSF monoclonal antibody to prevent and treat acute respiratory distress syndrome (ARDS) in patients with COVID-19. Roivant Sciences https://roivant.com/roivant-announces-development-of-anti-gm-csf-monoclonal-antibody-to-prevent-and-treat-acute-respiratory-distress-syndrome-ards-in-patients-with-covid-19/ (2020).
Humanigen. Humanigen partners with CTI, a leading contract research organization, for planned phase III study for lenzilumab for coronavirus treatment. Humanigen https://www.humanigen.com/press/Humanigen-Partners-With-CTI%2C-A-Leading-Contract-Research-Organization%2C-For-Planned-Phase-III-Study-For-Lenzilumab-For-Coronavirus-Treatment (2020).
Izana Bioscience. Initiation of two-centre compassionate use study involving namilumab in the treatment of individual patients with rapidly worsening COVID-19 infection in Italy. Izana Bioscience https://izanabio.com/initiation- of-two-centre-compassionate-use-study-involving-namilumab-in-the-treatment-of-individual-patients-with-rapidly-worsening-covid-19-infection-in-italy/ (2020).
CytoSorbents Corportation. CytoSorb, the Wuhan coronavirus, and cytokine storm. PR Newswire https://www.prnewswire.com/news-releases/cytosorb-the-wuhan-coronavirus-and-cytokine-storm-300994196.html (2020).
Chen, C. et al. Thalidomide combined with low-dose glucocorticoid in the treatment of COVID-19 pneumonia. Preprints https://www.preprints.org/manuscript/202002.0395/v1 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04273581 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04273529 (2020).
Tobinick, E. TNF-alpha inhibition for potential therapeutic modulation of SARS coronavirus infection. Curr. Med. Res. Opin. 20, 39–40 (2004).
Gautret, P. et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents https://doi.org/10.1016/j.ijantimicag.2020.105949 (2020).
Yao, X. et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa237 (2020).
Shukla, A. M. & Wagle Shukla, A. Expanding horizons for clinical applications of chloroquine, hydroxychloroquine, and related structural analogues. Drugs Context. 8, 2019-9-1 (2019).
Cortegiani, A., Ingoglia, G., Ippolito, M., Giarratano, A. & Einav, S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J. Crit. Care https://doi.org/10.1016/j.jcrc.2020.03.005 (2020).
Wong, R. S. et al. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ 326, 1358–1362 (2003).
Cui, W. et al. Expression of lymphocytes and lymphocyte subsets in patients with severe acute respiratory syndrome. Clin. Infect. Dis. 37, 857–859 (2003).
Li, T. et al. Significant changes of peripheral T lymphocyte subsets in patients with severe acute respiratory syndrome. J. Infect. Dis. 189, 648–651 (2004).
Zheng, H.-Y. et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cell. Mol. Immunol. https://doi.org/10.1038/s41423-020-0401-3 (2020). This article examines the immune states of patients with severe or mild COVID-19 and shows reduced T cell functional diversity in severe COVID-19, supporting a role for T cell function in controlling COVID-19.
Libraty, D. H., O’Neil, K. M., Baker, L. M., Acosta, L. P. & Olveda, R. M. Human CD4+ memory T-lymphocyte responses to SARS coronavirus infection. Virology 368, 317–321 (2007).
Yang, L. T. et al. Long-lived effector/central memory T-cell responses to severe acute respiratory syndrome coronavirus (SARS-CoV) S antigen in recovered SARS patients. Clin. Immunol. 120, 171–178 (2006).
Oh, H. L. J., Gan, K.-E. S., Bertoletti, A. & Tan, Y. J. Understanding the T cell immune response in SARS coronavirus infection. Emerg. Microbes. Infect. 1, e23 (2012).
Shin, H. S. et al. Immune responses to Middle East respiratory syndrome coronavirus during the acute and convalescent phases of human infection. Clin. Infect. Dis. 68, 984–992 (2019).
Chen, J. et al. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J. Virol. 84, 1289–1301 (2010).
Roberts, A. et al. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 3, e5 (2007).
Zhao, J., Zhao, J. & Perlman, S. T cell responses are required for protection from clinical disease and for virus clearance in severe acute respiratory syndrome coronavirus-infected mice. J. Virol. 84, 9318–9325 (2010).
Deming, D. et al. Vaccine efficacy in senescent mice challenged with recombinant SARS-CoV bearing epidemic and zoonotic spike variants. PLoS Med. 3, e525 (2006).
Yasui, F. et al. Prior immunization with severe acute respiratory syndrome (SARS)-associated coronavirus (SARS-CoV) nucleocapsid protein causes severe pneumonia in mice infected with SARS-CoV. J. Immunol. 181, 6337–6348 (2008).
Bolles, M. et al. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J. Virol. 85, 12201–12215 (2011).
Lurie, N., Saville, M., Hatchett, R. & Halton, J. Developing Covid-19 vaccines at pandemic speed. N. Engl. J. Med. https://doi.org/10.1056/NEJMp2005630 (2020).
Thevarajan, I. et al. Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat. Med. https://doi.org/10.1038/s41591-020-0819-2 (2020).
Tan, Y. J. et al. Profiles of antibody responses against severe acute respiratory syndrome coronavirus recombinant proteins and their potential use as diagnostic markers. Clin. Diagn. Lab. Immunol. 11, 362–371 (2004).
Wu, H. S. et al. Early detection of antibodies against various structural proteins of the SARS-associated coronavirus in SARS patients. J. Biomed. Sci. 11, 117–126 (2004).
Nie, Y. et al. Neutralizing antibodies in patients with severe acute respiratory syndrome-associated coronavirus infection. J. Infect. Dis. 190, 1119–1126 (2004).
Temperton, N. J. et al. Longitudinally profiling neutralizing antibody response to SARS coronavirus with pseudotypes. Emerg. Infect. Dis. 11, 411–416 (2005).
CGTN. Expert: recovered coronavirus patients are still prone to reinfection. YouTube https://www.youtube.com/watch?v=GZ99J7mlaIQ (2020).
The Straits Times. Japanese woman reinfected with coronavirus weeks after initial recovery. The Straits Times https://www.straitstimes.com/asia/east-asia/japanese-woman-reinfected-with-coronavirus-weeks-after-initial-recovery (2020).
NHK World-Japan. Japanese man tests positive for coronavirus again. NHK https://www3.nhk.or.jp/nhkworld/en/news/20200315_13/ (2020).
Xinhua. China puts 245 COVID-19 patients on convalescent plasma therapy. Xinhuanet http://www.xinhuanet.com/english/2020-02/28/c_138828177.htm (2020).
Cheng, Y. et al. Use of convalescent plasma therapy in SARS patients in Hong Kong. Eur. J. Clin. Microbiol. Infect. Dis. 24, 44–46 (2005).
Soo, Y. O. et al. Retrospective comparison of convalescent plasma with continuing high-dose methylprednisolone treatment in SARS patients. Clin. Microbiol. Infect. 10, 676–678 (2004).
Yeh, K. M. et al. Experience of using convalescent plasma for severe acute respiratory syndrome among healthcare workers in a Taiwan hospital. J. Antimicrob. Chemother. 56, 919–922 (2005).
Zhu, Z. et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc. Natl Acad. Sci. USA 104, 12123–12128 (2007).
Wang, C. et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Preprint at bioRxiv https://doi.org/10.1101/2020.03.11.987958 (2020).
Tian, X. et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 9, 382–385 (2020).
Li, F., Li, W., Farzan, M. & Harrison, S. C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309, 1864–1868 (2005).
Tai, W. et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunology https://doi.org/10.1038/s41423-020-0400-4 (2020).
Li, X. Y. et al. The keypoints in treatment of the critical coronavirus disease 2019 patient. Chin. J. Tuberculosis Respir. Dis. 43, E026 (2020).
CNA. Chinese doctors ‘using plasma therapy’ on COVID-19 patients. CNA https://www.channelnewsasia.com/news/asia/chinese-doctors-using-plasma-therapy-on-covid-19-patients-12444244 (2020).
Johnson, R. F. et al. 3B11-N, a monoclonal antibody against MERS-CoV, reduces lung pathology in rhesus monkeys following intratracheal inoculation of MERS-CoV Jordan-n3/2012. Virology 490, 49–58 (2016).
VIR. Press release details: Vir Biotechnology applying multiple platforms to address public health risk from Wuhan coronavirus. VIR https://investors.vir.bio/news-releases/news-release-details/vir-biotechnology-applying-multiple-platforms-address-public (2020).
Cohen, J. Can an anti-HIV combination or other existing drugs outwit the new coronavirus? Science https://www.sciencemag.org/news/2020/01/can-anti-hiv-combination-or-other-existing-drugs-outwit-new-coronavirus (2020).
Duddu, P. Coronavirus treatment: vaccines/drugs in the pipeline for COVID-19. Clinical Trials Arena https://www.clinicaltrialsarena.com/analysis/coronavirus-mers-cov-drugs/ (2020).
Berry, J. D. et al. Neutralizing epitopes of the SARS-CoV S-protein cluster independent of repertoire, antigen structure or mAb technology. MAbs 2, 53–66 (2010). This article shows the immunodominance of neutralizing epitopes on the RBD for SARS-CoV, suggesting the feasibility of a recombinant antigen strategy focused on the RBD for vaccination against COVID-19.
Qiu, M. et al. Antibody responses to individual proteins of SARS coronavirus and their neutralization activities. Microbes Infect. 7, 882–889 (2005).
GISAID. Receptor binding surveillance for high quality genomes. GISAID https://www.gisaid.org/ (2020).
Kleine-Weber, H. et al. Mutations in the spike protein of Middle East respiratory syndrome coronavirus transmitted in Korea increase resistance to antibody-mediated neutralization. J. Virol. 93, e01381-18 (2019).
Rockx, B. et al. Escape from human monoclonal antibody neutralization affects in vitro and in vivo fitness of severe acute respiratory syndrome coronavirus. J. Infect. Dis. 201, 946–955 (2010).
Yang, Z. Y. et al. Evasion of antibody neutralization in emerging severe acute respiratory syndrome coronaviruses. Proc. Natl. Acad. Sci. USA 102, 797–801 (2005).
Ho, M. S. et al. Neutralizing antibody response and SARS severity. Emerg. Infect. Dis. 11, 1730–1737 (2005).
Tetro, J. A. Is COVID-19 receiving ADE from other coronaviruses? Microbes Infect. 22, 72–73 (2020).
Liu, L. et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight https://doi.org/10.1172/jci.insight.123158 (2019).
Zhang, L. et al. Antibody responses against SARS coronavirus are correlated with disease outcome of infected individuals. J. Med. Virol. 78, 1–8 (2006).
Arabi, Y. M. et al. Feasibility of using convalescent plasma immunotherapy for MERS-CoV infection, Saudi Arabia. Emerg. Infect. Dis. 22, 1554 (2016).
Drosten, C. et al. Transmission of MERS-coronavirus in household contacts. N. Engl. J. Med. 371, 828–835 (2014).
Park, W. B. et al. Kinetics of serologic responses to MERS coronavirus infection in humans, South Korea. Emerg. Infect. Dis. 21, 2186–2189 (2015).
Nimmerjahn, F. & Ravetch, J. V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34–47 (2008).
Bournazos, S., DiLillo, D. J. & Ravetch, J. V. The role of Fc-FcgammaR interactions in IgG-mediated microbial neutralization. J. Exp. Med. 212, 1361–1369 (2015).
Kaneko, Y., Nimmerjahn, F. & Ravetch, J. V. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 313, 670–673 (2006).
Zhang, Q., Wang, Y., Qi, C., Shen, L. & Li, J. Clinical trial analysis of 2019-nCoV therapy registered in China. J. Med. Virol. https://doi.org/10.1002/jmv.25733 (2020).
Shang, J. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature https://doi.org/10.1038/s41586-020-2179-y (2020).
Prabakaran, P. et al. Structure of severe acute respiratory syndrome coronavirus receptor-binding domain complexed with neutralizing antibody. J. Biol. Chem. 281, 15829–15836 (2006).
Acknowledgements
The authors thank Insight Editing London for its inputs on an early draft. This work was supported by core funds at the Singapore Immunology Network (SIgN) through the Biomedical Medical Research Council (BMRC), A*STAR.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Immunology thanks A. Barrett, A. Hosmalin, K. Ishii and H. Wei for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tay, M.Z., Poh, C.M., Rénia, L. et al. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol 20, 363–374 (2020). https://doi.org/10.1038/s41577-020-0311-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-020-0311-8
This article is cited by
-
Beyond prediction: unveiling the prognostic power of μ-opioid and cannabinoid receptors, alongside immune mediators, in assessing the severity of SARS-CoV-2 infection
BMC Infectious Diseases (2024)
-
Cell-free DNA methylation reveals cell-specific tissue injury and correlates with disease severity and patient outcomes in COVID-19
Clinical Epigenetics (2024)
-
Association of dietary inflammatory index and the SARS-CoV-2 infection incidence, severity and mortality of COVID-19: a systematic review and dose-response meta-analysis
Nutrition Journal (2024)
-
Identification of biomarkers and pathways for the SARS-CoV-2 infections in obstructive sleep apnea patients based on machine learning and proteomic analysis
BMC Pulmonary Medicine (2024)
-
Serum soluble toll-like receptor 4 and risk for clinical severity in COVID-19 patients
Pneumonia (2024)
