
Abstract
Retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) are key sensors of virus infection, mediating the transcriptional induction of type I interferons and other genes that collectively establish an antiviral host response. Recent studies have revealed that both viral and host-derived RNAs can trigger RLR activation; this can lead to an effective antiviral response but also immunopathology if RLR activities are uncontrolled. In this Review, we discuss recent advances in our understanding of the types of RNA sensed by RLRs in the contexts of viral infection, malignancies and autoimmune diseases. We further describe how the activity of RLRs is controlled by host regulatory mechanisms, including RLR-interacting proteins, post-translational modifications and non-coding RNAs. Finally, we discuss key outstanding questions in the RLR field, including how our knowledge of RLR biology could be translated into new therapeutics.
Similar content being viewed by others

Introduction
Type I interferons are highly potent cytokines that were initially identified for their essential role in antiviral defence1,2. They shape both innate and adaptive immune responses and induce the expression of restriction factors, which are proteins that directly interfere with a step in the life cycle of a virus3. Both beneficial and detrimental roles for type I interferons have been described in the context of bacterial infections and cancer2,4,5,6, and these cytokines are also linked to the development of several autoinflammatory and autoimmune diseases7 and metabolic syndromes8. Therefore, the study of the biology of type I interferons has many implications for human health and disease.
The human genome encodes 17 different type I interferons, including 13 types of interferon-α (IFNα), as well as IFNβ, IFNε, IFNκ and IFNω. Type I interferons are secreted and then signal in an autocrine and paracrine manner by binding to the type I interferon receptor (interferon-α/β receptor (IFNAR)) that activates the intracellular Janus kinase–signal transducer and activator of transcription (JAK–STAT) pathway, particularly STAT1 and STAT2 (refs9,10). Upon phosphorylation, STAT1 and STAT2 combine with interferon regulatory factor 9 (IRF9) to form the interferon-stimulated gene factor 3 (ISGF3) transcriptional complex11. ISGF3 binds to interferon-stimulated response elements in type I interferon-dependent gene promoters and thereby drives the expression of hundreds of type I interferon-stimulated genes (ISGs). The proteins encoded by ISGs mediate the effects of type I interferons; for example, some ISGs encode virus restriction factors.
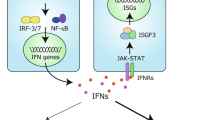
Under homeostatic conditions, type I interferons are expressed at very low and often undetectable levels. Their expression is rapidly induced by several innate immune signalling pathways. These signalling events are typically initiated by proteins called nucleic acid sensors that monitor cells for unusual nucleic acids. For example, it is an indispensable step in the life cycle of any virus to introduce a genome consisting of DNA or RNA into the infected cell. Nucleic acid sensors survey different subcellular compartments and include the endosomal Toll-like receptors (TLRs) TLR3, TLR7, TLR8 and TLR9; the cytosolic DNA sensor cyclic GMP–AMP synthase (cGAS); and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs)12,13. Once activated, these and other sensors engage converging signalling cascades that lead to transcriptional induction of the genes encoding type I interferons and other immune genes (Fig. 1).

Nucleic acid sensors of the innate immune system recognize unusual DNA and RNA molecules, for example, viral genomes. This results in the triggering of an intracellular signalling cascade that transcriptionally induces the genes encoding type I interferons, which are subsequently secreted. Type I interferons act in an autocrine (not shown) and paracrine manner by binding to their receptor, interferon-α/β receptor (IFNAR), which activates the Janus kinases (JAKs). These in turn activate the transcription factors signal transducer and activator of transcription 1 (STAT1) and STAT2, leading to expression of interferon-stimulated genes (ISGs). Several ISGs have direct antiviral effects. Type I interferons and ISGs also play important roles in bacterial infections and cancer; these effects can be both beneficial and detrimental, depending on the setting. Finally, production of type I interferons and ISGs over extended periods of time can lead to autoinflammatory and autoimmune diseases.
RLRs are RNA sensors localized in the cytosol12,13. This protein family encompasses three members: RIG-I, melanoma differentiation-associated protein 5 (MDA5) and laboratory of genetics and physiology 2 (LGP2). All RLRs have a central helicase domain and a so-called carboxy-terminal domain (CTD) (Fig. 2a). These two domains work together to detect immunostimulatory RNAs. RIG-I and MDA5 additionally have two amino-terminal caspase activation and recruitment domains (CARDs), which mediate downstream signal transduction. LGP2 lacks the CARDs and is widely believed to regulate RIG-I and MDA5 (Box 1). It is now over 15 years since RIG-I and MDA5 were discovered in overexpression studies to induce type I interferons14,15. Soon after their description, knockout studies in mice and mouse-derived cells established that RIG-I and MDA5 are essential for antiviral defence and type I interferon induction in virus infection models16,17. It is now clear that infections with almost all major virus families are recognized by RLRs (Table 1).

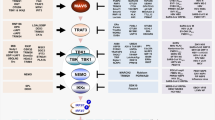
a | Domain architecture of retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) and mitochondrial antiviral-signalling protein (MAVS). The three members of the RLR family have a central helicase domain and form a subfamily of SF2 helicases. An additional RNA-binding fold, called the carboxy-terminal domain (CTD), is also found in all RLRs. RIG-I and melanoma differentiation-associated protein 5 (MDA5) contain two caspase activation and recruitment domains (CARDs) in tandem orientation at their N termini. The signalling adaptor protein MAVS also has a CARD as well as a C-terminal transmembrane domain (TM). b | RLR signalling pathway. RIG-I and MDA5 are activated by immunostimulatory RNA, for example, viral RNAs. They then undergo conformational changes that expose and multimerize their CARDs (not shown for simplicity), which allows homotypic CARD–CARD interactions with MAVS. MAVS is anchored with its TM into mitochondria, mitochondrial-associated membranes (MAMs) and peroxisomes, and relays the signal to TANK-binding kinase 1 (TBK1) and IκB kinase-ε (IKKε). These in turn activate interferon regulatory factor 3 (IRF3) and IRF7, which together with the transcription factor nuclear factor-κB (NF-κB) induce the expression of type I interferons and other genes. c | RIG-I is shown in its active conformation, bound to an immunostimulatory RNA. The molecular features of RIG-I-stimulatory RNAs are also illustrated; please see text for details. B, nucleobase; IFNα/β, interferon-α/β; LGP2, laboratory of genetics and physiology 2; P, phosphate; PP, diphosphate; PPP, triphosphate; R, ribose.
Much work has also been done to delineate the structural biology of RLR activation. The structure of full-length RIG-I without RNA ligand showed that RIG-I is in a signalling-repressed, closed conformation that is mediated by specific contacts of CARD2 with an inserting domain located between the two RecA-like helicase domains of RIG-I18. When bound to RNA ligand, the helicase domain of RIG-I tightly wraps around the RNA in a C-clamp-like fashion. ‘Pincer’ helical elements found in the helicase of RIG-I connect to the CTD and are believed to trigger RNA binding-dependent conformational changes in RIG-I that expose the CARDs for signalling. The crystal structure of 2CARD-deleted RIG-I, or the isolated CTD, bound to RNA revealed specific contacts between the CTD and the 5′-terminal region of the RNA18,19,20,21. The current model is that RIG-I binds to RNA in its monomeric form; however, signalling of RIG-I via its CARDs requires RIG-I oligomers that are nucleated and stabilized by non-degradative K63-polyubiquitin chains (as discussed in detail below). In contrast to RIG-I, MDA5 is believed to adopt a more open conformation in the absence of RNA ligand. In the presence of long double-stranded RNA (dsRNA), MDA5 assembles into helical ‘filaments’, which are MDA5 polymers22,23,24.
Upon RNA binding and oligomerization24,25,26, RLRs interact with the CARD domain found in mitochondrial antiviral-signalling protein (MAVS), which serves as the essential adaptor protein for RLR signal transduction. MAVS then activates TANK-binding kinase 1 (TBK1) and IκB kinase-ε (IKKε), which in turn activate IRF3 and IRF7; these, together with nuclear factor-κB (NF-κB), then induce transcription of the genes encoding type I interferons and other antiviral or immunoregulatory genes12,13 (Fig. 2b).
In this Review, we focus on recent developments in our understanding of the mechanisms by which RLRs are activated. First, we discuss the types and properties of RNA molecules that trigger RIG-I and MDA5 activation. Interestingly, these proteins not only detect viral RNA but also cellular RNAs that are unusual, mislocalized or misprocessed. Such disturbances of cellular RNA homeostasis can be an indirect molecular signature of infection or occur in sterile inflammatory pathologies. In the second part of the article, we highlight the regulation of RLR activity by host mechanisms such as post-translational modifications (PTMs), interacting proteins and cellular non-coding RNAs. Tight control of RLRs is important both to prevent detrimental type I interferon responses7 in the absence of infection and to allow rapid and sensitive recognition of pathogens.
RLR recognition of immunostimulatory RNA
RLRs are expressed in most cell types and are primarily located in the cytosol, although recent studies showed that RIG-I may also localize to the cell nucleus27,28. They induce type I interferon responses upon recognition of RNA molecules that are usually not present in mammalian cells, for example, the genomes of RNA viruses. In this section, we discuss the properties that mark RNA molecules as immunostimulatory and the principles explaining why RLRs are not activated by the abundant RNA content of the cytosol of healthy cells. The mechanisms by which RLRs distinguish between normal RNAs found in cells under homeostatic conditions and immunostimulatory RNAs are important for our understanding of how type I interferon responses are initiated during virus infection and in sterile pathologies. Furthermore, fundamental insights into RLR activation have led to the development of synthetic RLR agonists, which may hold therapeutic value for viral and malignant diseases, as well as vaccination (Box 2).
Activation of RIG-I by viral RNAs
Since the discovery of RIG-I, much research has been done to unravel the molecular and structural requirements for RIG-I activation by immunostimulatory RNAs. Collectively, this work shows that RIG-I monitors the 5′ ends of RNA molecules for several biochemical features, a process involving both the CTD and helicase domain (Fig. 2c). The first breakthrough was the finding that RNA molecules bearing a triphosphate (PPP) group at their 5′ end activate RIG-I29,30. This observation was subsequently extended to include additional properties of RNA 5′ ends that determine RIG-I activation. First, base pairing of the 5′-PPP-containing RNA molecule to a complementary strand of RNA is required for RIG-I activation, with the most potent RNA agonists forming a blunt end31,32. Second, RNAs with uncapped diphosphate (PP) groups at the 5′ end activate RIG-I, much like RNAs with 5′-PPP moieties33. Third, the 5′-terminal nucleotide needs to be unmethylated at its 2′-O position to allow RNA recognition by RIG-I34. These functional data are nicely underpinned by structural studies of RIG-I. The CTD has a pocket binding specifically to 5′-PPP or 5′-PP groups and also contacts the unmethylated 2′-O group of the first nucleotide20,35,36,37. The base-paired portion of the RNA forms contacts with the helicase domain in RIG-I18,19,21. The helicase domain has ATPase activity, and ATP hydrolysis determines the dissociation rate of RIG-I from RNA37,38,39,40. Stable RIG-I–RNA interaction displaces the CARDs, which in the inactive RIG-I conformation interact with parts of the helicase domain18. As a consequence, the CARDs from multiple RIG-I proteins oligomerize and become accessible for interaction with MAVS, resulting in downstream signalling (reviewed elsewhere41).
Importantly, the molecular features of RIG-I-stimulatory RNAs (5′-(P)PP, duplex structure, no ribose 2′-O-methylation) are found in several viral RNA molecules but are typically absent in the vast majority of host cytosolic RNAs. For example, cellular mRNAs acquire a cap structure involving 2′-O-methylation (that is, cap 1 or cap 2) in the cell nucleus following transcription, before export to the cytoplasm. Similarly, 28S and 18S ribosomal RNAs (rRNAs) and tRNAs are processed in the cell nucleus and their mature, cytosolic forms typically only have a single phosphate at the 5′ end. The 5S rRNA retains a 5′-PPP moiety but is methylated and extensively complexed with proteins. Therefore, such RNAs do not activate RIG-I; in fact, 5′-monophosphate RNAs have recently been suggested to block RIG-I42.
In contrast to cytosolic cellular RNAs, several viral RNAs have features characteristic of RIG-I agonists. The genomes of many negative-sense RNA viruses contain partially self-complementary 5′ and 3′ ends, which base pair to form duplex structures called ‘pan-handles’31. In addition, such viral RNAs often have an unmethylated 5′ end that bears a PPP group, derived from the ribonucleoside triphosphate used by viral polymerases during initiation of genome replication. Probably the best studied example of this is influenza A virus (IAV), and work by several groups has shown that RIG-I detects IAV genomes in infected cells43,44,45,46. More recently, small aberrant viral RNAs produced by the IAV polymerase were shown to also activate RIG-I; these RNAs — much like full-length genomes — have a 5′-PPP group and form a duplex structure47. In addition to negative-sense RNA viruses, infections with some positive-sense RNA viruses are also detected by RIG-I. For example, these infections include dengue and Zika viruses of the Flaviviridae family, and involve recognition of nascent viral genomes containing 5′-PPP groups prior to capping48,49. Activation of RIG-I by viral RNAs not containing the typical features of RIG-I agonists has also been proposed; for example, a uridine-rich stretch in the hepatitis C virus (HCV) RNA genome located at a distance from the 5′ end was shown to activate RIG-I50. It is noteworthy that the presence of a 5′-PPP group on viral RNAs not only makes these ‘visible’ to RIG-I but also allows restriction factors, including IFIT family proteins, to target these RNAs (reviewed elsewhere51).
Activation of RIG-I by host RNAs
Recent work has revealed that virus infection can also result in RIG-I activation by host cellular RNAs, instead of viral RNAs52,53. These studies set out to explain earlier observations that cells infected with herpesviruses, a family of double-stranded DNA viruses, induce type I interferon and pro-inflammatory cytokine responses partially dependent on RLRs. RIG-I immunoprecipitation from cells infected with herpes simplex virus type 1 (HSV-1) or Kaposi’s sarcoma-associated herpesvirus (KSHV), followed by high-throughput sequencing of co-purifying RNAs, revealed the association of cellular non-coding RNAs with RIG-I52,53. In the case of HSV-1, 5S rRNA pseudogene transcripts, and particularly RNA5SP141, were most highly enriched in RIG-I immunoprecipitations52. In contrast to the parental 5S rRNA, the biological functions of these 5S rRNA pseudogene transcripts, which have 5′-PPP groups and self-complementary regions, are poorly understood.
HSV-1 infection unmasks these cellular non-coding RNAs for RIG-I recognition in two ways. First, 5S rRNA pseudogene transcripts mislocalize to the cytoplasm in HSV-1-infected cells. Second, HSV-1-induced host shut-off leads to downregulation of the mRNAs that encode specific binding proteins of RNA5SP141; these are TST and MRPL18 (ref.52). Silencing of RNA5SP141 by small interfering RNAs or antisense oligonucleotides diminished cytokine responses during infection by HSV-1 and also upon reactivation of Epstein–Barr virus, another herpesvirus. It is noteworthy that the type I interferon response to IAV, which replicates in the cell nucleus, is partially dependent on RNA5SP141 (ref.52), suggesting that both viral RNA and cellular RNA5SP141 contribute to full RIG-I activation during IAV infection.
RNA5SP141 is transcribed by the cellular RNA polymerase III (Pol III) and an independent set of studies reported that Pol III also transcribes AT-rich viral DNA sequences into 5′-PPP-containing, RIG-I-stimulatory RNAs54,55,56. In the case of lytic KSHV infection, vault RNAs (vtRNAs) were highly enriched in RIG-I precipitates53. vtRNAs are conserved and participate in the formation of cytoplasmic ribonucleoprotein particles called vaults57. vtRNAs are highly self-complementary, are transcribed in the cell nucleus and have a 5′-PPP group from the initiating ribonucleotide. However, dual specificity phosphatase 11 (DUSP11) usually removes two phosphates from the 5′ end of vtRNAs, creating 5′-monophosphate ends58. Lytic KSHV infection results in reduced levels of DUSP11 and in the accumulation of 5′-PPP-bearing vtRNAs that then activate RIG-I53. Interestingly, DUSP11 levels are not affected during latent infection53. Given that RIG-I activation prevents lytic KSHV infection53, it is possible that a pathway involving DUSP11, vtRNAs, RIG-I and type I interferons determines the balance between lytic and latent infection. It is noteworthy that HIV-1 may also downregulate DUSP11 by means of the viral Vpr protein59. Concomitantly, non-coding cellular Y-RNAs were found to accumulate in their 5′-PPP-containing form and to bind to RIG-I in HIV-1-infected cells59. Moreover, endogenous RNAs lacking the canonical features of RIG-I agonists, including RNAs cleaved by RNase L60 and microRNAs (miRNAs)61, have been suggested to activate RIG-I in virus-infected cells. Finally, endogenous retroviral elements were shown to activate RIG-I via de-SUMOylation of the host transcriptional repressor tripartite motif-containing 28 (TRIM28; also known as KAP1)62.
Activation of MDA5 during viral infection
How MDA5 is activated during virus infection is less well understood and is only briefly described here in light of a recent review on the subject63. Infection with many different types of virus, including RNA viruses and DNA viruses, results in the triggering of MDA5 signalling. MDA5 is essential for type I interferon induction during infection with picornaviruses16,17, a family of positive-sense RNA viruses. For many other viruses, MDA5 appears either partially redundant with other sensors of infection or acts in a temporally distinct manner63. In many instances, infection with viruses that activate MDA5 results in the accumulation of dsRNA in the infected cell, which has been proposed to activate MDA5. However, studies analysing RNAs bound by MDA5 in infected cells reported that MDA5 interacts predominantly with one RNA strand of viral RNA64,65. Additionally, surrogate LGP2 immunoprecipitation identified encephalomyocarditis virus (EMCV) L-region antisense RNA as an MDA5 ligand66. Future work, perhaps using iCLIP (individual-nucleotide resolution ultraviolet crosslinking and immunoprecipitation) and other techniques to identify MDA5-associated RNAs, will be required to understand whether virus infection generates a specific ‘RNA molecular pattern’ for MDA5 recognition, as seen for RIG-I.
Activation of RLRs in sterile conditions
RLR activation has been reported in several autoinflammatory and autoimmune diseases, as well as in cancer. This activation can occur via two distinct mechanisms. In one mechanism, RLR mutations result in spontaneous signalling in the absence of virus infection. Alternatively, alterations in RNA metabolism can generate RLR-stimulatory RNAs; for example, mislocalization or defects in RNA processing can result in the detection of endogenous RNAs.
We start by discussing the first setting, which is illustrated by two rare genetic diseases: Singleton–Merten syndrome and Aicardi–Goutières syndrome. Patients affected by these diseases produce type I interferon over extended periods of time7. Activating mutations in the genes encoding RIG-I and MDA5 (DDX58 and IFIH1, respectively) have been identified in some patients with Singleton–Merten syndrome or Aicardi–Goutières syndrome67,68,69,70. Some of these mutations may change the conformation of RLRs in such a way that spontaneous signalling to MAVS occurs in an RNA-independent manner69,71. Furthermore, RLR mutations may result in aberrant detection of normal cellular RNAs38,39,70,72. For example, long hairpin RNAs derived from inverted repeat Alu elements have been suggested to activate mutant forms of MDA5 found in Aicardi–Goutières syndrome72. In addition to RLR mutations in rare diseases, IFIH1 single-nucleotide polymorphisms have been associated with many common, inflammatory diseases, including type 1 diabetes mellitus73,74,75,76,77,78,79. These IFIH1 single-nucleotide polymorphisms are likely to induce heightened type I interferon levels, but whether this involves constitutive activation or increased responses to infections remains to be determined.
Much progress has been made over the past few years connecting changes in RNA metabolism to RLR activation. For example, mislocalization of mitochondrial RNA (mtRNA) into the cytosol leads to MDA5 activation80. This occurs when the function of SUV3 and PNPase, mitochondrial enzymes involved in the degradation of mtRNA, is compromised and results in chronic type I interferon production, including in patients with mutations in the gene encoding PNPase80. The mechanism by which mtRNA escapes the mitochondria and gains access to the cytosol in this setting remains to be determined. In addition to mislocalization of cellular RNAs, defects in A-to-I RNA editing result in activation of MDA5. Indeed, adenosine deaminase acting on RNA 1 (ADAR1) deficiency in humans and mice causes chronic type I interferon production and inflammatory pathology81,82. These effects require MDA5, suggesting that editing of endogenous RNAs by ADAR1 prevents engagement of MDA5 (refs83,84,85). Unedited duplex RNAs derived from inverted repeat Alu elements have been proposed to be a major MDA5 stimulus in ADAR1-deficient cells72. Interestingly, some types of tumour are dependent on ADAR1: loss of ADAR1 in transformed cells can sensitize to immunogenic cell death, which involves MDA5-driven type I interferon production and protein kinase R (PKR)-mediated effects on cell viability86,87,88. Finally, in contrast to the cell-autonomous scenarios discussed above, RLR activation by endogenous RNAs can also occur in a cell non-autonomous manner. In a breast cancer model, the tumour microenvironment results in increased production of the non-coding, 5′-PPP-containing RN7SL1 RNA in stromal cells, followed by transfer of this RNA via exosomes to cancer cells, resulting in RIG-I activation89. Of note, in this specific setting, RIG-I activation and type I interferon induction were shown to promote cancer progression89.
RLR regulation
Accurate recognition of immunostimulatory RNA and rapid signal activation by RLRs are crucial steps in the initiation of innate immunity. As such, the sensing and signalling activities of RIG-I and MDA5 need to be tightly controlled to allow for an effective antiviral defence when needed, while maintaining immune homeostasis under normal physiological conditions. The activity of RLRs is controlled by several mechanisms that include PTMs mediated by regulatory enzymes; regulation by interacting proteins (for example, co-receptor molecules); post-transcriptional mechanisms, in particular cellular non-coding RNAs; and autophagy.
Regulation by PTMs
Over the past decade it has become clear that the activities of RIG-I and MDA5 are dynamically controlled by several types of PTM, including ubiquitylation (both non-degradative and degradative ubiquitin-linkage types), conjugation of ubiquitin-like (UBL) proteins, phosphorylation, acetylation and deamidation (Fig. 3). Accordingly, several enzymes catalysing these PTMs have been identified and their roles in RLR-mediated innate immunity have been characterized in vitro and, for many, also in vivo.

RIG-I (retinoic acid-inducible gene I) and MDA5 (melanoma differentiation-associated protein 5) are regulated by acetylation, deamidation, phosphorylation, SUMOylation, modification with FAT10 and several types of polyubiquitylation, in particular K63-linked and K48-linked polyubiquitin-linkage types (illustrated in green and red, respectively). Activating regulatory post-translational modifications (PTMs) are illustrated on the left, and those that inhibit RIG-I-like receptor (RLR) activation are depicted on the right, as well as the respective enzymes that synthesize, or remove, these PTMs. Enzymes that ultimately lead to RLR activation are shown in green; those that lead to RLR signal inhibition are shown in red. Furthermore, key modified amino acid residues in human RIG-I and MDA5 are illustrated, if known. Ac, acetylation; CARD, caspase activation and recruitment domain; CKII, casein kinase II; CTD, carboxy-terminal domain; CYLD, CYLD lysine 63 deubiquitinase; HDAC6, histone deacetylase 6; MEX3C, Mex-3 RNA binding family member C; P, phosphorylation; PFAS, phosphoribosyl-formylglycinamide synthase; PKC, protein kinase C; PP1, protein phosphatase 1; RIOK3, RIO kinase 3; RNF, ring finger protein; SENP2, sentrin/SUMO-specific protease 2; Su, SUMOylation; TRIM, tripartite motif; Ub, ubiquitin; USP, ubiquitin specific peptidase.
The first PTM identified for RLRs was covalent K63-linked polyubiquitylation of RIG-I (at K172, the major site, as well as five other residues located in the tandem CARD)90. Biochemical and functional studies have established that K63-linked ubiquitylation of RIG-I at the CARDs is important for MAVS binding and antiviral cytokine responses90. Structural analysis showed that K63-linked polyubiquitin stabilizes the RIG-I CARDs in a signalling-active, oligomeric, ‘lock-washer’ conformation in which the ubiquitin chains are bound along the outer rim of the 2CARD tetramer. Both covalently attached and unanchored (meaning, non-covalently bound) K63-linked ubiquitin chains contribute to effective RIG-I oligomerization and signal transduction91,92. TRIM25, from the tripartite motif (TRIM) protein family (Box 3), has been shown to mediate RIG-I CARD K63-linked ubiquitylation, and studies in cells in which TRIM25 was silenced or knocked out showed impaired cytokine responses to diverse stimuli (for example, infection with Sendai virus, IAV or human papillomavirus 16, or transfection with poly(I:C) or RNA from HCV)90,93,94,95,96,97. Given the important role of TRIM25 in RIG-I-mediated antiviral gene induction, several viral pathogens have evolved antagonists that directly inhibit TRIM25 activity, including NS1 from IAV96,98, NS1 from respiratory syncytial virus99, E6 from human papilloma virus97 and the BPLF1 protein from Epstein–Barr virus100, or the subgenomic RNA of dengue virus101 (reviewed in detail elsewhere102,103). Moreover, in recent years several cellular proteins (for example, nuclear dbf2-related 2 (NDR2), zinc finger CCHC-type-containing 3 (ZCCHC3), NLR family pyrin domain-containing 12 (NLRP12) and caspase 12) that specifically regulate TRIM25’s function in RIG-I K63-linked ubiquitylation have been identified (as described below). Structural studies revealed the molecular details of RIG-I signalosome assembly through a mechanism of TRIM25 dimerization104,105.
Given that K63-linked ubiquitylation is a key activation step in RIG-I signalling, it is not surprising that partial redundancy exists. Besides TRIM25, Riplet (also known as REUL or RNF135) has been shown to mediate RIG-I K63-linked ubiquitylation, thereby facilitating RIG-I signal transduction106. Of note, although several studies showed that Riplet modifies the CTD of RIG-I and thereby acts cooperatively with TRIM25 (refs106,107,108), other studies suggested that Riplet can also modify the RIG-I CARDs109,110. Interestingly, Riplet has recently been implicated in an IRF3-mediated antiviral innate response that is independent of the RIG-I–MAVS signalling pathway111, which raises the question of whether the effect of Riplet deficiency on antiviral cytokine induction is primarily due to Riplet’s role in RIG-I activation or its independent signalling function downstream of it. Finally, the ubiquitin ligases TRIM4 and Mex-3 RNA binding family member C (MEX3C) also reportedly promote RIG-I signal activation through K63-linked polyubiquitylation of multiple residues in the CARDs112, and thus a model of sequential and hierarchical RIG-I CARD ubiquitylation has been proposed113. Conversely, RIG-I stability is regulated by classical degradative K48-linked polyubiquitylation mediated by at least three E3 ligases — Cbl, ring finger protein 122 (RNF122) and RNF125 — whereas ubiquitylation mediated by linear ubiquitin chain assembly complex (LUBAC) negatively affects the stability of TRIM25 and its interaction with RIG-I114,115,116,117 (Fig. 3). Ubiquitylation events are reversible processes, and, accordingly, several deubiquitylating enzymes, in particular ubiquitin specific peptidase 3 (USP3), USP21 and CYLD lysine 63 deubiquitinase (CYLD), modulate RIG-I signalling by removing K63-polyubiquitin chains, although with unique kinetics (reviewed elsewhere112,118). On the other hand, USP4 and USP15 enhance the stability of RIG-I and TRIM25, respectively, by proteolytically cleaving K48-linked ubiquitylation from these molecules119,120.
Besides multisite ubiquitylation, phosphorylation, acetylation and deamidation are critical for regulating RIG-I signalling or sensing activity. Phosphorylation of RIG-I at both the CARDs (at S8 and T170) and the CTD (at T770 and S854/855) keeps the sensor in an autorepressed state, preventing premature signalling before infection121,122,123. Phosphomimetic mutants of RIG-I at the CARD sites exhibited abolished TRIM25 binding, K63-linked ubiquitylation and downstream signalling ability, whereas mutants of RIG-I, which cannot be phosphorylated, showed higher levels of K63-linked ubiquitylation and, accordingly, increased signalling activity compared with wild-type RIG-I. Conventional isoenzymes of PKC (PKCα and PKCβ), as well as casein kinase II (CKII), were shown to phosphorylate RIG-I at the CARDs and the CTD, respectively123,124, whereas protein phosphatase 1 (PP1) is responsible for removing the phosphorylations in the CARDs following virus infection125.
Similarly to RIG-I, aberrant activation of MDA5 in uninfected cells is prevented by phosphorylation (at S88 and S828)125,126. Upon virus infection, MDA5 is activated by removal of these phosphorylation events by PP1α/γ125,127. In contrast to RIG-I, MDA5 regulation by K63-polyubiquitin is less well established. Although studies on MDA5 CARD K63-linked ubiquitylation are conflicting90,92,94,128, it was recently shown that the helicase domain of MDA5 (specifically K743) is modified with K63-linked ubiquitin chains synthesized by TRIM65, which activates the sensor molecule by promoting its oligomerization129.
Recently, SUMOylation of MDA5 and also RIG-I (at several residues in their CARDs and CTD) has emerged as a critical regulatory mechanism of their activity130. SUMOylation of RIG-I and MDA5 by TRIM38 in uninfected cells, or at early times after viral infection, prevents their K48-polyubiquitin-dependent degradation, stabilizing the sensor proteins. During the late phase of infection, de-SUMOylation of RLRs by sentrin/SUMO-specific protease 2 (SENP2) triggers their proteasomal degradation, aiding in termination of the pro-inflammatory response once the virus has been cleared130.
Non-covalent binding of RIG-I to FAT10, a less well-characterized UBL protein, was reported to dampen the RIG-I-mediated antiviral response by sequestering signalling-active RIG-I from mitochondria131. Moreover, it was recently shown that RIG-I is regulated by deamidation, which is an emerging PTM in innate immunity132. Deamidation of RIG-I (at residues Q10, N245 and N445) by cellular phosphoribosyl-formylglycinamide synthase (PFAS) triggers RIG-I activation specifically during herpesvirus infection, and, intriguingly, this activation event is usurped by these viruses for their own replication132.
Whereas all the above described PTMs regulate the signal-transducing activity of RLRs, reversible acetylation was shown to modulate the ability of RIG-I to bind to viral RNA133,134 (Fig. 3). Acetylation of RIG-I in its CTD (at K909 and also other lysines) prevents RIG-I from binding viral RNA and assembling into a signalling-competent oligomeric form. Inversely, CTD deacetylation induced by histone deacetylase 6 (HDAC6) allows for efficient RIG-I sensing and signalling. Consistent with this observation, HDAC6-deficient cells and mice were more permissive to several RNA viruses, such as IAV, vesicular stomatitis virus (VSV) and certain flaviviruses133,134. It is noteworthy that, in contrast to the extensive characterization of PTMs regulating RIG-I and MDA5, regulation of LGP2 by PTMs remains elusive.
Regulation by interacting proteins
Recent studies identified several RLR-binding proteins that regulate RLR activity by modulating several processes: dsRNA binding to RLRs (for example, co-receptor proteins); RLR oligomerization; TRIM25-mediated K63-linked ubiquitylation of RLRs; or subcellular localization of RLRs (Fig. 4).

The activities of retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5) are regulated by interacting proteins that modulate their RNA-binding ability, oligomerization, tripartite motif-containing 25 (TRIM25)-mediated K63-linked ubiquitylation or subcellular localization. Furthermore, several cellular long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) regulate RIG-I-mediated and MDA5-mediated innate immune signalling. Conceptually, these RNAs promote or diminish RIG-I-like receptor (RLR) signalling by regulating the gene expression of these sensors or regulatory proteins in the RLR pathway, or they modulate the activity of RLRs or TRIM25 through a direct physical interaction. CARD, caspase activation and recruitment domain; CTD, carboxy-terminal domain; CYLD, CYLD lysine 63 deubiquitinase; DDX60, DExD/H-box helicase 60; DHX15, DEAH-box helicase 15; dsRNA, double-stranded RNA; IKKε, IκB kinase-ε; IRF, interferon regulatory factor; ISG, interferon-stimulated gene; K48-(Ub)n, K48-linked ubiquitylation; K63-(Ub)n, K63-linked ubiquitylation; LGP2, laboratory of genetics and physiology 2; lncRNA, long non-coding RNA; MAVS, mitochondrial antiviral-signalling protein; NDR2, nuclear dbf2-related 2; NLRP12, NLR family pyrin domain-containing 12; OASL, oligoadenylate synthetase-like; P, phosphorylation; (P)PP, (tri)diphosphate; RNF, ring finger protein; SFPQ, splicing factor proline and glutamine rich; TBK1, TANK-binding kinase 1; Ub, ubiquitin; ZCCHC3, zinc finger CCHC-type-containing 3.
Numerous dsRNA-binding proteins, which are involved in many aspects of RNA biology, have been shown to regulate RLRs. One of the first dsRNA-binding proteins reported to regulate RLRs was PACT. PACT was shown to interact with the CTD of RIG-I and to stimulate the ATPase activity of RIG-I135,136. Interestingly, recent studies showed that PACT also promotes MDA5 activation by facilitating its ability to oligomerize137. In addition to PACT, the third RLR member, LGP2, positively and negatively regulates RLR activities through multiple mechanisms (Box 1).
Whereas the concept of a receptor/co-receptor mechanism is well known in adaptive immunity, it is less well established in innate immunity. The zinc-finger protein ZCCHC3 has recently been shown to function as a co-receptor for both RIG-I and MDA5 (ref.94). Co-immunoprecipitation and cell fractionation experiments showed that ZCCHC3 binds with its C-terminal zinc finger domains to RNAs that activate RLRs (including 5′-PPP RNA and high molecular weight poly(I:C)), whereas its N-terminal domain interacts with the helicase and CTD of RIG-I and MDA5. This interaction mode enhanced RNA binding by RLRs, as well as the K63-linked ubiquitylation of RLRs by TRIM25, which was also found to be in complex with ZCCHC3 (of note, Riplet, TRIM4 and TRIM65 were not found to be associated with ZCCHC3). Functional studies in ZCCHC3-deficient cells and mice demonstrated that ZCCHC3 was required for efficient RLR-mediated cytokine responses to several RNA viruses. Of note, the phenotype of ZCCHC3 knockout was comparable with that of TRIM25 gene editing using CRISPR, which also markedly reduced Sendai virus-triggered cytokine responses94. Moreover, several DExD/H-box helicases may regulate RLR signalling (Fig. 4). For example, DEAH-box helicase 15 (DHX15) was shown to bind dsRNA and to facilitate the ability of RIG-I to hydrolyse ATP and to signal downstream138. In accord, cells depleted of DHX15 were more susceptible to infection by RNA viruses from several families, including paramyxoviruses and rhabdoviruses. DExD/H-box helicase 60 (DDX60) has been suggested to promote RIG-I-dependent (and potentially also MDA5-dependent) antiviral interferon and ISG induction, although conflicting results have been reported in DDX60-knockout cells and mice139,140,141. Riplet, which is one of the E3 ligases mediating RIG-I K63-linked ubiquitylation, was recently shown to promote RIG-I signalling in a ubiquitylation-independent manner. It was suggested that by forming a dimer and by crossbridging RIG-I filaments on longer RNA molecules, Riplet induces higher-assembly aggregates of RIG-I110.
Recently, several regulatory proteins for TRIM25 have been identified and functionally characterized. The kinase NDR2 (also known as serine/threonine-protein kinase 38-like (STK38L)) was shown to be crucial for TRIM25-mediated and RIG-I-mediated antiviral interferon responses, evidenced by the impairment of antiviral cytokine induction when NDR2 was gene-edited in cells (in particular, macrophages) and in mice. Interestingly, the kinase activity of NDR2 was shown to be dispensable for its positive effect on RIG-I signalling. Using cell-based assays and in vitro experiments with recombinant RIG-I and TRIM25 proteins, this study showed that NDR2, through interaction with both RIG-I and TRIM25, stabilized their complex formation and promoted TRIM25-induced K63-linked ubiquitylation of the RIG-I CARDs142. Ting and colleagues recently unveiled a novel regulatory mechanism that interconnects the RLR and NOD-like receptor (NLR) pathways. The pyrin-containing NLR member, NLRP12, which is expressed predominantly in dendritic cells and neutrophils, was shown to dampen cytokine responses by RIG-I following RNA virus infection or 5′-PPP RNA stimulation, evidenced by higher innate immune signalling in NLRP12-deficient bone marrow-derived dendritic cells. This study further showed that, under normal conditions, NLRP12 interacts with TRIM25, blocking its ability to ubiquitylate and activate RIG-I. At the same time, NLRP12 enhanced the K48-linked ubiquitylation of RIG-I by RNF125, thereby destabilizing RIG-I. During the course of viral infection, however, NLRP12 protein abundance is gradually reduced, relieving the TRIM25-inhibitory mechanism and, at the same time, stabilizing RIG-I143. Caspase 12, a negative regulator of inflammasome signalling, was shown to be required for type I interferon induction in response to West Nile virus infection in vitro and in vivo by interacting with RIG-I and regulating its K63-linked ubiquitylation by TRIM25 (ref.144). In addition to these TRIM25-regulatory proteins, several cellular RNAs have been discovered to bind to TRIM25 and regulate its E3 ligase activity (as described below). RIG-I reportedly can also be activated in a K63-linked polyubiquitin-independent manner by oligoadenylate synthetase-like (OASL), which is a unique member of the OAS family in that it is devoid of 2′–5′ oligoadenylate synthetase activity and possesses two UBL domains. It has been proposed that upon transcriptional induction of OASL and its binding to RIG-I, the UBL domains of OASL mimic the presence of K63-polyubiquitin chains, leading to RIG-I signalling145.
Besides viral RNA binding, oligomerization and modification by various enzymes, RLRs need to relocate from areas of RNA sensing to mitochondria and mitochondrial-associated membranes, where further downstream signalling occurs via MAVS. Members of the 14-3-3 protein family, which comprises seven isoforms in human, have emerged as critical regulators of the cytosol-to-mitochondria translocation step of RLRs. Gale and colleagues showed that 14-3-3ɛ forms a complex with endogenous RIG-I and TRIM25, stabilizing their interaction and mediating their translocation to MAVS on mitochondrial-associated membranes93. Accordingly, silencing of 14-3-3ɛ diminished TRIM25-induced and RIG-I-induced signalling to Sendai virus infection or HCV RNA stimulation. Similarly, relocalization of MDA5 was recently shown to be mediated by 14-3-3η, indicating that specific 14-3-3 isoforms function as ‘RLR translocon’ proteins146.
Regulation by non-coding RNAs
Cellular non-coding RNAs, in particular long non-coding RNAs (lncRNAs) and miRNAs, have emerged as important regulators of RLR signal transduction. In most cases, viral infection and/or interferon stimulation triggers the expression of these non-coding RNAs, often in a cell type-specific or tissue-specific manner. Upon induction, some lncRNAs and miRNAs promote or dampen RLR signalling by regulating the gene expression of RLRs or other key molecules in the RLR pathway. In addition, some lncRNAs were shown to directly bind to the RLR sensor protein (in particular RIG-I), or its activator TRIM25, which itself has RNA-binding capacity101,147,148 (Fig. 4). Lastly, miRNAs and lncRNAs modulate interferon-dependent and ISG-dependent innate immune responses by multiple pattern-recognition receptors, including RLRs, through broader mechanisms, such as chromatin remodelling (reviewed in detail elsewhere149).
Several miRNAs and lncRNAs have been shown to regulate the gene expression of RLRs, or other molecules in the interferon induction pathway, thereby creating feedback or feedforward regulatory loops. lncRNA NEAT1, which is induced upon hantavirus infection, promotes the expression of RIG-I through sequestration of the splicing factor and transcriptional repressor protein splicing factor proline and glutamine rich (SFPQ)150. Inversely, miR-485 targets the RIG-I mRNA for degradation, thereby dampening the antiviral type I interferon response151. miRNAs also regulate the expression of RLR-regulatory enzymes. miR-526a downregulates the expression of the deubiquitylating enzyme CYLD, which ultimately promotes RIG-I K63-linked ubiquitylation and interferon responses152. miR-15b negatively regulates gene expression of the E3 ligase RNF125, thereby controlling RIG-I protein stability during Japanese encephalitis virus infection153. Using an RIG-I affinity purification–RNA immunoprecipitation approach combined with ultraviolet crosslinking, Cao and colleagues identified that, in murine macrophages, several lncRNAs were enriched with RIG-I following VSV infection154. Individual silencing of RIG-I-bound lncRNAs showed that lnc-Lsm3b, when depleted, increased IFNβ production upon infection with RNA viruses (for example, Sendai virus and VSV), but not the DNA virus HSV-1. Furthermore, lnc-Lsm3b was transcriptionally upregulated during the course of RNA virus infection, suggesting that lnc-Lsm3b could function as a negative-feedback regulator of RIG-I signalling, maintaining immune homeostasis. Indeed, lnc-Lsmb3-deficient mice challenged with VSV produced higher amounts of type I interferons and, correlated to this, showed reduced viral burden and disease symptoms compared with infected control mice. Biochemical characterization showed that lnc-Lsm3b interacts with the CTD of RIG-I, competing with RNA agonists for RIG-I binding. Furthermore, lnc-Lsm3b stabilized the intramolecular bond between the N-terminal CARDs and the central helicase domain, maintaining RIG-I in an autorepressed, monomeric state. By contrast, lnc-Lsmb3 deficiency promoted the interaction of RIG-I with TRIM25, TRIM25-induced K63-linked ubiquitylation and, consequently, RIG-I oligomerization following VSV infection (Fig. 4). It is important to note that lnc-Lsm3b is specific to mice; as such, exploring human lncRNAs that act as feedback inhibitors, similarly to lnc-Lsm3b, will be an exciting new avenue of research.
Indeed, a recent study showed that lnc-ATV, which is a previously uncharacterized, human-specific lncRNA, dampens RIG-I signalling155. lnc-ATV was identified by high-throughput transcriptome analysis as one of 272 lncRNAs that were significantly upregulated in response to stimulation by type I or III interferons. Functional RNA interference screening showed that lnc-ATV promoted the replication of several RNA viruses, specifically HCV, Zika virus, Newcastle disease virus and Sendai virus, hinting at the possibility that lnc-ATV promotes an antiviral host pathway commonly triggered by these viruses. Functional studies indicated that lnc-ATV suppresses the RIG-I-mediated antiviral response as knockdown of lnc-ATV promoted TBK1-dependent and IRF3-dependent interferon responses to RNA virus infection. lnc-ATV was shown to bind to RIG-I, although more detailed studies are needed to define the precise mechanism by which lnc-ATV inhibits RIG-I activity.
Although RIG-I is well known to be a bona fide RNA sensor, its activator, TRIM25, has recently been shown to also have RNA-binding capacity101,147,148. Biochemical and functional studies showed that the ability of TRIM25 to bind RNA is important for its E3 ligase activity and, thus, its positive effect on RIG-I antiviral signalling148. However, which type (or types) of host or viral RNAs are bound by TRIM25 in physiological contexts, such as virus infection, has remained unknown until recently. Lin et al. identified that a previously uncharacterized lncRNA, called Lnczc3h7a, directly binds to TRIM25, stabilizing the interaction of the E3 ligase with RIG-I, which ultimately promotes RIG-I signalling156. Silencing or gene-editing of Lnczc3h7a significantly decreased interferon and ISG expression in peritoneal macrophages infected with viruses that predominantly trigger RIG-I (VSV and Sendai virus). However, silencing of Lnczc3h7a had no effect on pro-inflammatory responses to non-RIG-I stimuli (for example, EMCV or lipopolysaccharide (LPS)). Lnczc3h7a is transcriptionally induced by interferon stimulation or viral infection, and gene expression profiling showed that Lnczc3h7a is expressed in many immune cell types. Lncxc3h7a-knockout mice showed defective cytokine responses, enhanced virus replication and more severe pathology following VSV infection compared with control mice156. Through the iCLIP assay, the authors showed that Lnczc3h7a not only interacts with TRIM25 but also, through a short RNA stem–loop structure, with the helicase domain of RIG-I, suggesting that Lnczc3h7a acts as a molecular scaffold to enhance RIG-I–TRIM25 complex formation; this promoted TRIM25-mediated K63-linked ubiquitylation of RIG-I and, ultimately, its ability to form signalling-active oligomers early during infection. With regard to MDA5 activation, lnc-ITPRIP-1 was shown to interact with the sensor, stabilizing its binding to viral RNAs and promoting its oligomeric signalling-competent form157. Finally, besides the described lncRNAs and miRNAs that directly regulate RLR activities, others act downstream of RLRs or broadly regulate inflammatory gene or ISG expression158,159,160.
Regulation by autophagy
Accumulating evidence indicates that autophagy regulates RLR signalling through diverse mechanisms that include autophagy-dependent degradation of RLRs or downstream innate signalling molecules; direct interaction between autophagic proteins and RLRs; and RLR signal amplification via reactive oxygen species (ROS) production regulated by autophagy (reviewed elsewhere161). In most cases, an absence of autophagy promotes the RLR-mediated cytokine responses, indicating that autophagy has a dampening effect on RLR signalling. The LRR-containing protein leucine rich repeat-containing 25 (LRRC25) recognizes RIG-I ‘earmarked’ with ISG15, inducing its degradation by p62-mediated selective autophagy162. The autophagy-related protein conjugate, ATG5–ATG12, suppresses IFN gene expression through direct binding to both RIG-I and MAVS, preventing their CARD–CARD interaction163. NLR family member X1 (NLRX1) together with its binding partner, the mitochondrial Tu translation elongation factor (TUFM), dually regulates virus-induced type I interferon induction and autophagy164. Complex formation of NLRX1, TUFM, ATG5–ATG12 and ATG16L1 negatively controlled RIG-I–MAVS binding during infection while, at the same time, promoting virus-induced autophagy164,165. Furthermore, in autophagy-defective cells, RLR signalling has been found to be more effective owing to overzealous production of ROS due to the accumulation of dysfunctional mitochondria161. Interestingly, RLR activation also induces autophagy166,167, showing an intricate relationship between these two host pathways that, on the one hand, could aid in terminating cytokine responses, while, at the same time, promoting direct virus restriction by autophagy.
Conclusions and future directions
Significant progress has been made in dissecting the mechanisms that lead to RLR activation and, equally importantly, on what keeps RLRs controlled in uninfected conditions. Although much has been learned about the molecular features of viral RNAs that induce RIG-I activation, significantly less is known concerning the circumstances in which host-derived RNAs activate RLRs and the mechanistic basis of this. Recent work highlighted the importance of mislocalized, misprocessed or unmasked cellular RNAs (mostly non-coding RNAs) in RLR-driven autoimmunity or antiviral responses, which is expected to stimulate future studies to determine the physiological relevance of such RNAs in disease contexts. In particular, the recognition of DNA virus infection by RLRs via sensing of non-coding RNAs is an emerging theme. It will be important to test whether this concept applies not only to herpesviruses but generally to DNA viruses and to other virus classes detected by RLRs (Table 1). The recent advance in the characterization of RLR-mediated immune sensing of nucleic acid ligands can now be utilized for the development of novel RLR agonist or antagonist-based therapies for autoimmunity, infectious diseases or cancer. With regard to the regulatory mechanisms of RLR function by various enzymes, it remains to be determined how these regulators themselves are activated during viral infection and what their cell type-specific and species-specific roles are. Moreover, the role of miRNAs and lncRNAs in RLR signal modulation has just begun to be elucidated, and future work may further expand the repertoire of non-coding RNAs implicated in the RLR regulatory circuit, which will be critical information for potential therapeutic purposes.
References
Isaacs, A. & Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 147, 258–267 (1957).
McNab, F., Mayer-Barber, K., Sher, A., Wack, A. & O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 15, 87–103 (2015).
Yan, N. & Chen, Z. J. Intrinsic antiviral immunity. Nat. Immunol. 13, 214–222 (2012).
Minn, A. J. Interferons and the immunogenic effects of cancer therapy. Trends Immunol. 36, 725–737 (2015).
Barber, G. N. STING: infection, inflammation and cancer. Nat. Rev. Immunol. 15, 760–770 (2015).
Zitvogel, L., Galluzzi, L., Kepp, O., Smyth, M. J. & Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 15, 405–414 (2015).
Crow, Y. J. & Manel, N. Aicardi–Goutieres syndrome and the type I interferonopathies. Nat. Rev. Immunol. 15, 429–440 (2015).
Ganguly, D. Do type I interferons link systemic autoimmunities and metabolic syndrome in a pathogenetic continuum? Trends Immunol. 39, 28–43 (2018).
Pestka, S., Krause, C. D. & Walter, M. R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202, 8–32 (2004).
Stark, G. R. & Darnell, J. E. Jr. The JAK–STAT pathway at twenty. Immunity 36, 503–514 (2012).
Schreiber, G. & Piehler, J. The molecular basis for functional plasticity in type I interferon signaling. Trends Immunol. 36, 139–149 (2015).
Hartmann, G. Nucleic acid immunity. Adv. Immunol. 133, 121–169 (2017).
Goubau, D., Deddouche, S. & Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 38, 855–869 (2013).
Andrejeva, J. et al. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-β promoter. Proc. Natl Acad. Sci. USA 101, 17264–17269 (2004).
Yoneyama, M. et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737 (2004). This manuscript reports the discovery of the role of RIG-I in antiviral immunity.
Kato, H. et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 (2006). This study demonstrates, using gene knockout in mice, non-redundant functions of RIG-I and MDA5 in virus sensing.
Gitlin, L. et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl Acad. Sci. USA 103, 8459–8464 (2006).
Kowalinski, E. et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147, 423–435 (2011). This work presents a crystal structure of full-length RIG-I revealing CARD2–helicase interactions in the inactive conformation, and also provides a structural model for RIG-I activation upon RNA binding.
Jiang, F. et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 479, 423–427 (2011).
Cui, S. et al. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol. Cell 29, 169–179 (2008).
Luo, D. et al. Structural insights into RNA recognition by RIG-I. Cell 147, 409–422 (2011).
Berke, I. C. & Modis, Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 31, 1714–1726 (2012).
Wu, B. et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 152, 276–289 (2013).
Yu, Q., Qu, K. & Modis, Y. Cryo-EM structures of MDA5–dsRNA filaments at different stages of ATP hydrolysis. Mol. Cell 72, 999–1012.e6 (2018).
Binder, M. et al. Molecular mechanism of signal perception and integration by the innate immune sensor retinoic acid-inducible gene-I (RIG-I). J. Biol. Chem. 286, 27278–27287 (2011).
Peisley, A., Wu, B., Yao, H., Walz, T. & Hur, S. RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol. Cell 51, 573–583 (2013).
Liu, G. et al. Nuclear-resident RIG-I senses viral replication inducing antiviral immunity. Nat. Commun. 9, 3199 (2018).
Li, W., Chen, H., Sutton, T., Obadan, A. & Perez, D. R. Interactions between the influenza A virus RNA polymerase components and retinoic acid-inducible gene I. J. Virol. 88, 10432–10447 (2014).
Hornung, V. et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 (2006).
Pichlmair, A. et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314, 997–1001 (2006). This study, together with the work by Hornung et al. (2006), shows that RIG-I recognizes RNA molecules with 5′-PPP groups and provides important insight into the discrimination of viral and host RNAs.
Schlee, M. et al. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31, 25–34 (2009).
Schmidt, A. et al. 5′-Triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc. Natl Acad. Sci. USA 106, 12067–12072 (2009).
Goubau, D. et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 514, 372–375 (2014).
Schuberth-Wagner, C. et al. A conserved histidine in the RNA sensor RIG-I controls immune tolerance to N1-2′O-methylated self RNA. Immunity 43, 41–51 (2015).
Lu, C. et al. The structural basis of 5′ triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure 18, 1032–1043 (2010).
Devarkar, S. C. et al. Structural basis for m7G recognition and 2′-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl Acad. Sci. USA 113, 596–601 (2016).
Zheng, J. et al. HDX-MS reveals dysregulated checkpoints that compromise discrimination against self RNA during RIG-I mediated autoimmunity. Nat. Commun. 9, 5366 (2018).
Lassig, C. et al. ATP hydrolysis by the viral RNA sensor RIG-I prevents unintentional recognition of self-RNA. eLife 4, e10859 (2015).
Lassig, C. et al. Unified mechanisms for self-RNA recognition by RIG-I Singleton–Merten syndrome variants. eLife 7, e38958 (2018).
Devarkar, S. C., Schweibenz, B., Wang, C., Marcotrigiano, J. & Patel, S. S. RIG-I uses an ATPase-powered translocation-throttling mechanism for kinetic proofreading of RNAs and oligomerization. Mol. Cell 72, 355–368.e4 (2018).
Wu, B. & Hur, S. How RIG-I like receptors activate MAVS. Curr. Opin. Virol. 12, 91–98 (2015).
Ren, X., Linehan, M. M., Iwasaki, A. & Pyle, A. M. RIG-I selectively discriminates against 5′-monophosphate RNA. Cell Rep. 26, 2019–2027.e4 (2019).
Rehwinkel, J. et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140, 397–408 (2010).
Baum, A., Sachidanandam, R. & Garcia-Sastre, A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl Acad. Sci. USA 107, 16303–16308 (2010).
Weber, M. et al. Incoming RNA virus nucleocapsids containing a 5′-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe 13, 336–346 (2013).
Liu, G., Park, H. S., Pyo, H. M., Liu, Q. & Zhou, Y. Influenza a virus panhandle structure is directly involved in RIG-I activation and interferon induction. J. Virol. 89, 6067–6079 (2015).
Te Velthuis, A. J. W. et al. Mini viral RNAs act as innate immune agonists during influenza virus infection. Nat. Microbiol. 3, 1234–1242 (2018).
Chazal, M. et al. RIG-I recognizes the 5′ region of dengue and Zika virus genomes. Cell Rep. 24, 320–328 (2018).
Hertzog, J. et al. Infection with a Brazilian isolate of Zika virus generates RIG-I stimulatory RNA and the viral NS5 protein blocks type I IFN induction and signaling. Eur. J. Immunol. 48, 1120–1136 (2018).
Saito, T., Owen, D. M., Jiang, F., Marcotrigiano, J. & Gale, M. Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454, 523–527 (2008).
Rehwinkel, J. & Reis e Sousa, C. Targeting the viral Achilles’ heel: recognition of 5′-triphosphate RNA in innate anti-viral defence. Curr. Opin. Microbiol. 16, 485–492 (2013).
Chiang, J. J. et al. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol. 19, 53–62 (2018). This study demonstrates that virus infection can lead to the mislocalization and unmasking of cellular non-coding RNAs, resulting in RIG-I activation.
Zhao, Y., Ye, X., Dunker, W., Song, Y. & Karijolich, J. RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat. Commun. 9, 4841 (2018). This manuscript reports the activation of RIG-I in KSHV-infected cells due to virus-induced defects in the normal post-transcriptional processing of cellular non-coding RNAs.
Samanta, M., Iwakiri, D., Kanda, T., Imaizumi, T. & Takada, K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 25, 4207–4214 (2006).
Chiu, Y. H., Macmillan, J. B. & Chen, Z. J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591 (2009).
Ablasser, A. et al. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 10, 1065–1072 (2009).
Steiner, E., Holzmann, K., Elbling, L., Micksche, M. & Berger, W. Cellular functions of vaults and their involvement in multidrug resistance. Curr. Drug. Targets 7, 923–934 (2006).
Burke, J. M. & Sullivan, C. S. DUSP11 — an RNA phosphatase that regulates host and viral non-coding RNAs in mammalian cells. RNA Biol. 14, 1457–1465 (2017).
Vabret, N. et al. Y-RNAs lead an endogenous program of RIG-I agonism mobilized upon RNA virus infection and targeted by HIV. Preprint at bioRxiv https://doi.org/10.1101/773820 (2019).
Malathi, K., Dong, B., Gale, M. Jr. & Silverman, R. H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448, 816–819 (2007).
Zhao, L. et al. Identification of cellular microRNA-136 as a dual regulator of RIG-I-mediated innate immunity that antagonizes H5N1 IAV replication in A549 cells. Sci. Rep. 5, 14991 (2015).
Schmidt, N. et al. An influenza virus-triggered SUMO switch orchestrates co-opted endogenous retroviruses to stimulate host antiviral immunity. Proc. Natl Acad. Sci. USA 116, 17399–17408 (2019).
Dias Junior, A. G., Sampaio, N. G. & Rehwinkel, J. A balancing act: MDA5 in antiviral immunity and autoinflammation. Trends Microbiol. 27, 75–85 (2019).
Runge, S. et al. In vivo ligands of MDA5 and RIG-I in measles virus-infected cells. PLoS Pathog. 10, e1004081 (2014).
Sanchez David, R. Y. et al. Comparative analysis of viral RNA signatures on different RIG-I-like receptors. eLife 5, e11275 (2016).
Deddouche, S. et al. Identification of an LGP2-associated MDA5 agonist in picornavirus-infected cells. eLife 3, e01535 (2014).
Jang, M. A. et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton–Merten syndrome. Am. J. Hum. Genet. 96, 266–274 (2015).
Rutsch, F. et al. A specific IFIH1 gain-of-function mutation causes Singleton–Merten syndrome. Am. J. Hum. Genet. 96, 275–282 (2015).
Oda, H. et al. Aicardi–Goutieres syndrome is caused by IFIH1 mutations. Am. J. Hum. Genet. 95, 121–125 (2014).
Rice, G. I. et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 46, 503–509 (2014). This work shows that mutations in the IFIH1 gene that encodes MDA5 cause aberrant sensor activation and disease driven by type I interferons.
Funabiki, M. et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 40, 199–212 (2014).
Ahmad, S. et al. Breaching self-tolerance to Alu duplex RNA underlies MDA5-mediated inflammation. Cell 172, 797–810.e13 (2018). This study suggests that duplex RNAs originating from Alu elements activate MDA5 in settings where either MDA5 is mutated or the RNA-editing enzyme ADAR1 is impaired.
Smyth, D. J. et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat. Genet. 38, 617–619 (2006).
Sheng, Y. et al. Sequencing-based approach identified three new susceptibility loci for psoriasis. Nat. Commun. 5, 4331 (2014).
Martinez, A. et al. Association of the IFIH1–GCA–KCNH7 chromosomal region with rheumatoid arthritis. Ann. Rheum. Dis. 67, 137–138 (2008).
Jin, Y. et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat. Genet. 44, 676–680 (2012).
Enevold, C. et al. Multiple sclerosis and polymorphisms of innate pattern recognition receptors TLR1-10, NOD1-2, DDX58, and IFIH1. J. Neuroimmunol. 212, 125–131 (2009).
Cunninghame Graham, D. S. et al. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with systemic lupus erythematosus. PLoS Genet. 7, e1002341 (2011).
Nejentsev, S., Walker, N., Riches, D., Egholm, M. & Todd, J. A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324, 387–389 (2009).
Dhir, A. et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242 (2018). This study indicates that mtRNA, when released into the cytosol, activates MDA5.
Hartner, J. C., Walkley, C. R., Lu, J. & Orkin, S. H. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 10, 109–115 (2009).
Rice, G. I. et al. Mutations in ADAR1 cause Aicardi–Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 44, 1243–1248 (2012).
Pestal, K. et al. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43, 933–944 (2015).
Liddicoat, B. J. et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120 (2015).
Chung, H. et al. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172, 811–824.e14 (2018).
Liu, H. et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat. Med. 25, 95–102 (2019).
Ishizuka, J. J. et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48 (2019).
Gannon, H. S. et al. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat. Commun. 9, 5450 (2018).
Nabet, B. Y. et al. Exosome RNA unshielding couples stromal activation to pattern recognition receptor signaling in cancer. Cell 170, 352–366.e13 (2017).
Gack, M. U. et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920 (2007).
Zeng, W. et al. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 141, 315–330 (2010).
Peisley, A., Wu, B., Xu, H., Chen, Z. J. & Hur, S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 509, 110–114 (2014).
Liu, H. M. et al. The mitochondrial targeting chaperone 14–3–3ε regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe 11, 528–537 (2012).
Lian, H. et al. The zinc-finger protein ZCCHC3 binds RNA and facilitates viral RNA sensing and activation of the RIG-I-like receptors. Immunity 49, 438–448.e5 (2018). This work identifies that ZCCHC3 functions as a co-receptor for both RIG-I and MDA5.
Castanier, C. et al. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 10, 44 (2012).
Gack, M. U. et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5, 439–449 (2009).
Chiang, C. et al. The human papillomavirus E6 oncoprotein targets USP15 and TRIM25 to suppress RIG-I-mediated innate immune signaling. J. Virol. 92, e01737-17 (2018).
Koliopoulos, M. G. et al. Molecular mechanism of influenza A NS1-mediated TRIM25 recognition and inhibition. Nat. Commun. 9, 1820 (2018).
Ban, J. et al. Human respiratory syncytial virus NS 1 targets TRIM25 to suppress RIG-I ubiquitination and subsequent RIG-I-mediated antiviral signaling. Viruses 10, 716 (2018).
Gupta, S. et al. Herpesvirus deconjugases inhibit the IFN response by promoting TRIM25 autoubiquitination and functional inactivation of the RIG-I signalosome. PLoS Pathog. 14, e1006852 (2018).
Manokaran, G. et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 350, 217–221 (2015).
Chan, Y. K. & Gack, M. U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 14, 360–373 (2016).
Martin-Vicente, M., Medrano, L. M., Resino, S., Garcia-Sastre, A. & Martinez, I. TRIM25 in the regulation of the antiviral innate immunity. Front. Immunol. 8, 1187 (2017).
Sanchez, J. G. et al. The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc. Natl Acad. Sci. USA 111, 2494–2499 (2014).
Sanchez, J. G. et al. Mechanism of TRIM25 catalytic activation in the antiviral RIG-I pathway. Cell Rep. 16, 1315–1325 (2016).
Oshiumi, H. et al. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 8, 496–509 (2010).
Oshiumi, H., Miyashita, M., Matsumoto, M. & Seya, T. A distinct role of Riplet-mediated K63-linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 9, e1003533 (2013).
Rajsbaum, R. et al. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 8, e1003059 (2012).
Gao, D. et al. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS One 4, e5760 (2009).
Cadena, C. et al. Ubiquitin-dependent and -independent roles of E3 Ligase RIPLET in innate immunity. Cell 177, 1187–1200.e16 (2019).
Vazquez, C., Tan, C. Y. & Horner, S. M. Hepatitis C virus infection is inhibited by a non-canonical antiviral signaling pathway targeted by NS3–NS4A. J. Virol. https://doi.org/10.1128/JVI.00725-19 (2019).
Davis, M. E. & Gack, M. U. Ubiquitination in the antiviral immune response. Virology 479–480, 52–65 (2015).
Xian, H. et al. Stratified ubiquitination of RIG-I creates robust immune response and induces selective gene expression. Sci. Adv. 3, e1701764 (2017).
Wang, W. et al. RNF122 suppresses antiviral type I interferon production by targeting RIG-I CARDs to mediate RIG-I degradation. Proc. Natl Acad. Sci. USA 113, 9581–9586 (2016).
Arimoto, K. et al. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl Acad. Sci. USA 104, 7500–7505 (2007).
Inn, K. S. et al. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol. Cell 41, 354–365 (2011).
Chen, W. et al. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 152, 467–478 (2013).
Brisse, M. & Ly, H. Comparative structure and function analysis of the RIG-I-like receptors: RIG-I and MDA5. Front. Immunol. 10, 1586 (2019).
Wang, L. et al. USP4 positively regulates RIG-I-mediated antiviral response through deubiquitination and stabilization of RIG-I. J. Virol. 87, 4507–4515 (2013).
Pauli, E. K. et al. The ubiquitin-specific protease USP15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci. Signal. 7, ra3 (2014).
Gack, M. U., Nistal-Villan, E., Inn, K. S., Garcia-Sastre, A. & Jung, J. U. Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J. Virol. 84, 3220–3229 (2010).
Nistal-Villan, E. et al. Negative role of RIG-I serine 8 phosphorylation in the regulation of interferon-β production. J. Biol. Chem. 285, 20252–20261 (2010).
Sun, Z., Ren, H., Liu, Y., Teeling, J. L. & Gu, J. Phosphorylation of RIG-I by casein kinase II inhibits its antiviral response. J. Virol. 85, 1036–1047 (2011).
Maharaj, N. P., Wies, E., Stoll, A. & Gack, M. U. Conventional protein kinase C-alpha (PKC-α) and PKC-β negatively regulate RIG-I antiviral signal transduction. J. Virol. 86, 1358–1371 (2012).
Wies, E. et al. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 38, 437–449 (2013).
Takashima, K., Oshiumi, H., Takaki, H., Matsumoto, M. & Seya, T. RIOK3-mediated phosphorylation of MDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep. 11, 192–200 (2015).
Davis, M. E. et al. Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host Microbe 16, 19–30 (2014).
Jiang, X. et al. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 36, 959–973 (2012).
Lang, X. et al. TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J. Exp. Med. 214, 459–473 (2017).
Hu, M. M., Liao, C. Y., Yang, Q., Xie, X. Q. & Shu, H. B. Innate immunity to RNA virus is regulated by temporal and reversible sumoylation of RIG-I and MDA5. J. Exp. Med. 214, 973–989 (2017).
Nguyen, N. T., Now, H., Kim, W. J., Kim, N. & Yoo, J. Y. Ubiquitin-like modifier FAT10 attenuates RIG-I mediated antiviral signaling by segregating activated RIG-I from its signaling platform. Sci. Rep. 6, 23377 (2016).
He, S. et al. Viral pseudo-enzymes activate RIG-I via deamidation to evade cytokine production. Mol. Cell 58, 134–146 (2015).
Choi, S. J. et al. HDAC6 regulates cellular viral RNA sensing by deacetylation of RIG-I. EMBO J. 35, 429–442 (2016).
Liu, H. M. et al. Regulation of retinoic acid inducible gene-I (RIG-I) activation by the histone deacetylase 6. EBioMedicine 9, 195–206 (2016).
Kok, K. H. et al. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 9, 299–309 (2011).
Luthra, P. et al. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 14, 74–84 (2013).
Lui, P. Y. et al. PACT facilitates RNA-induced activation of MDA5 by promoting MDA5 oligomerization. J. Immunol. 199, 1846–1855 (2017).
Pattabhi, S., Knoll, M. L., Gale, M. Jr. & Loo, Y. M. DHX15 is a coreceptor for RLR signaling that promotes antiviral defense against RNA virus infection. J. Interf. Cytok. Res. 39, 331–346 (2019).
Miyashita, M., Oshiumi, H., Matsumoto, M. & Seya, T. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell Biol. 31, 3802–3819 (2011).
Oshiumi, H. et al. DDX60 is involved in RIG-I-dependent and independent antiviral responses, and its function is attenuated by virus-induced EGFR activation. Cell Rep. 11, 1193–1207 (2015).
Goubau, D. et al. Mouse superkiller-2-like helicase DDX60 is dispensable for type I IFN induction and immunity to multiple viruses. Eur. J. Immunol. 45, 3386–3403 (2015).
Liu, Z. et al. NDR2 promotes the antiviral immune response via facilitating TRIM25-mediated RIG-I activation in macrophages. Sci. Adv. 5, eaav0163 (2019).
Chen, S. T. et al. NLRP12 regulates anti-viral RIG-I activation via interaction with TRIM25. Cell Host Microbe 25, 602–616.e7 (2019). This study reveals a novel mechanism of interplay between the NLR and RLR pathways.
Wang, P. et al. Caspase-12 controls West Nile virus infection via the viral RNA receptor RIG-I. Nat. Immunol. 11, 912–919 (2010).
Zhu, J. et al. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 40, 936–948 (2014).
Lin, J. P., Fan, Y. K. & Liu, H. M. The 14–3–3η chaperone protein promotes antiviral innate immunity via facilitating MDA5 oligomerization and intracellular redistribution. PLoS Pathog. 15, e1007582 (2019).
Choudhury, N. R. et al. RNA-binding activity of TRIM25 is mediated by its PRY/SPRY domain and is required for ubiquitination. BMC Biol. 15, 105 (2017).
Sanchez, J. G. et al. TRIM25 binds RNA to modulate cellular anti-viral defense. J. Mol. Biol. 430, 5280–5293 (2018).
Zhang, Q. & Cao, X. Epigenetic regulation of the innate immune response to infection. Nat. Rev. Immunol. 19, 417–432 (2019).
Ma, H. et al. The long noncoding RNA NEAT1 exerts antihantaviral effects by acting as positive feedback for RIG-I signaling. J. Virol. 91, e02250-16 (2017).
Ingle, H. et al. The microRNA miR-485 targets host and influenza virus transcripts to regulate antiviral immunity and restrict viral replication. Sci. Signal. 8, ra126 (2015).
Xu, C. et al. Downregulation of microRNA miR-526a by enterovirus inhibits RIG-I-dependent innate immune response. J. Virol. 88, 11356–11368 (2014).
Zhu, B. et al. MicroRNA-15b modulates Japanese encephalitis virus-mediated inflammation via targeting RNF125. J. Immunol. 195, 2251–2262 (2015).
Jiang, M. et al. Self-recognition of an inducible host lncRNA by RIG-I feedback restricts innate immune response. Cell 173, 906–919.e13 (2018). This work shows that a lncRNA represses RIG-I signalling through direct interaction with the sensor.
Fan, J., Cheng, M., Chi, X., Liu, X. & Yang, W. A human long non-coding RNA LncATV promotes virus replication through restricting RIG-I-mediated innate immunity. Front. Immunol. 10, 1711 (2019).
Lin, H. et al. The long noncoding RNA Lnczc3h7a promotes a TRIM25-mediated RIG-I antiviral innate immune response. Nat. Immunol. 20, 812–823 (2019). This study provides evidence that a cellular non-coding RNA functions as a molecular scaffold to promote RIG-I–TRIM25 complex formation and RIG-I signalling.
Xie, Q. et al. Long noncoding RNA ITPRIP-1 positively regulates the innate immune response through promotion of oligomerization and activation of MDA5. J. Virol. 92, e00507-18 (2018).
Atianand, M. K., Caffrey, D. R. & Fitzgerald, K. A. Immunobiology of long noncoding RNAs. Annu. Rev. Immunol. 35, 177–198 (2017).
Valadkhan, S. & Plasek, L. M. Long non-coding RNA-mediated regulation of the interferon response: a new perspective on a familiar theme. Pathog. Immun. 3, 126–148 (2018).
Forster, S. C., Tate, M. D. & Hertzog, P. J. MicroRNA as type I interferon-regulated transcripts and modulators of the innate immune response. Front. Immunol. 6, 334 (2015).
Deretic, V., Saitoh, T. & Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 13, 722–737 (2013).
Du, Y. et al. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J. 37, 351–366 (2018).
Jounai, N. et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl Acad. Sci. USA 104, 14050–14055 (2007).
Lei, Y. et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 36, 933–946 (2012).
Allen, I. C. et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I–MAVS and TRAF6–NF-κB signaling pathways. Immunity 34, 854–865 (2011).
Sun, X. et al. MAVS maintains mitochondrial homeostasis via autophagy. Cell Discov. 2, 16024 (2016).
Lee, N. R. et al. Activation of RIG-I-mediated antiviral signaling triggers autophagy through the MAVS–TRAF6–Beclin-1 signaling axis. Front. Immunol. 9, 2096 (2018).
Rodriguez, K. R., Bruns, A. M. & Horvath, C. M. MDA5 and LGP2: accomplices and antagonists of antiviral signal transduction. J. Virol. 88, 8194–8200 (2014).
Rothenfusser, S. et al. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol. 175, 5260–5268 (2005).
Quicke, K. M., Kim, K. Y., Horvath, C. M. & Suthar, M. S. RNA helicase LGP2 negatively regulates RIG-I signaling by preventing TRIM25-mediated caspase activation and recruitment domain ubiquitination. J Interf. Cytok. Res. 39, 669–683 (2019).
Venkataraman, T. et al. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol. 178, 6444–6455 (2007).
Bruns, A. M., Leser, G. P., Lamb, R. A. & Horvath, C. M. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5–RNA interaction and filament assembly. Mol. Cell 55, 771–781 (2014).
Uchikawa, E. et al. Structural analysis of dsRNA binding to anti-viral pattern recognition receptors LGP2 and MDA5. Mol. Cell 62, 586–602 (2016).
Parisien, J. P. et al. RNA sensor LGP2 inhibits TRAF ubiquitin ligase to negatively regulate innate immune signaling. EMBO Rep. 19, e45176 (2018).
Suthar, M. S. et al. The RIG-I-like receptor LGP2 controls CD8+ T cell survival and fitness. Immunity 37, 235–248 (2012). This study identifies a prominent role for LGP2 in regulating the sensitivity of T cells to cell death.
van der Veen, A. G. et al. The RIG-I-like receptor LGP2 inhibits Dicer-dependent processing of long double-stranded RNA and blocks RNA interference in mammalian cells. EMBO J. 37, e97479 (2018).
Wu, Y., Wu, X., Wu, L., Wang, X. & Liu, Z. The anticancer functions of RIG-I-like receptors, RIG-I and MDA5, and their applications in cancer therapy. Transl Res. 190, 51–60 (2017).
Roulois, D. et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973 (2015).
Chiappinelli, K. B. et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986 (2015).
Ranoa, D. R. et al. Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget 7, 26496–26515 (2016).
Kato, H. et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205, 1601–1610 (2008).
Chiang, C. et al. Sequence-specific modifications enhance the broad-spectrum antiviral response activated by RIG-I agonists. J. Virol. 89, 8011–8025 (2015).
Dassler-Plenker, J. et al. RIG-I activation induces the release of extracellular vesicles with antitumor activity. Oncoimmunology 5, e1219827 (2016).
Dassler-Plenker, J. et al. Direct RIG-I activation in human NK cells induces TRAIL-dependent cytotoxicity toward autologous melanoma cells. Int. J. Cancer 144, 1645–1656 (2019).
Helms, M. W. et al. Utility of the RIG-I agonist triphosphate RNA for melanoma therapy. Mol. Cancer Ther. https://doi.org/10.1158/1535-7163.MCT-18-1262 (2019).
Hochheiser, K. et al. Cutting edge: the RIG-I ligand 3pRNA potently improves CTL cross-priming and facilitates antiviral vaccination. J. Immunol. 196, 2439–2443 (2016).
Coch, C. et al. RIG-I activation protects and rescues from lethal influenza virus infection and bacterial superinfection. Mol. Ther. 25, 2093–2103 (2017).
Castiello, L. et al. An optimized retinoic acid-inducible gene I agonist M8 induces immunogenic cell death markers in human cancer cells and dendritic cell activation. Cancer Immunol. Immunother. 68, 1479–1492 (2019).
Heidegger, S. et al. RIG-I activation is critical for responsiveness to checkpoint blockade. Sci. Immunol. 4, eaau8943 (2019). This study shows RIG-I activation by synthetic RNAs as a therapeutic strategy to boost antitumour immunity and reveals synergy with checkpoint blockade.
Jiang, X. et al. Intratumoral delivery of RIG-I agonist SLR14 induces robust antitumor responses. J. Exp. Med. 216, 2854–2868 (2019).
Chen, Y. G. et al. Sensing self and foreign circular RNAs by intron identity. Mol. Cell 67, 228–238.e5 (2017).
Chen, Y. G. et al. N6-methyladenosine modification controls circular RNA immunity. Mol. Cell 76, 96–109.e9 (2019).
Wesselhoeft, R. A. et al. RNA circularization diminishes immunogenicity and can extend translation duration in vivo. Mol. Cell 74, 508–520.e4 (2019).
Luke, J. M. et al. Coexpressed RIG-I agonist enhances humoral immune response to influenza virus DNA vaccine. J. Virol. 85, 1370–1383 (2011).
Lazear, H. M. et al. Pattern recognition receptor MDA5 modulates CD8+ T cell-dependent clearance of West Nile virus from the central nervous system. J. Virol. 87, 11401–11415 (2013).
Kandasamy, M. et al. RIG-I signaling is critical for efficient polyfunctional T cell responses during influenza virus infection. PLoS Pathog. 12, e1005754 (2016).
Stone, A. E. L., Green, R., Wilkins, C., Hemann, E. A. & Gale, M. Jr. RIG-I-like receptors direct inflammatory macrophage polarization against West Nile virus infection. Nat. Commun. 10, 3649 (2019).
van Gent, M., Sparrer, K. M. J. & Gack, M. U. TRIM proteins and their roles in antiviral host defenses. Annu. Rev. Virol. 5, 385–405 (2018).
Versteeg, G. A. et al. The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 38, 384–398 (2013).
Ozato, K., Shin, D. M., Chang, T. H. & Morse, H. C. III. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8, 849–860 (2008).
Heaton, S. M., Borg, N. A. & Dixit, V. M. Ubiquitin in the activation and attenuation of innate antiviral immunity. J. Exp. Med. 213, 1–13 (2016).
Carthagena, L. et al. Human TRIM gene expression in response to interferons. PLoS One 4, e4894 (2009).
Mandell, M. A. et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev. Cell 30, 394–409 (2014).
Sparrer, K. M. J. et al. TRIM23 mediates virus-induced autophagy via activation of TBK1. Nat. Microbiol. 2, 1543–1557 (2017).
van Tol, S., Hage, A., Giraldo, M. I., Bharaj, P. & Rajsbaum, R. The TRIMendous role of TRIMs in virus–host interactions. Vaccines 5, 23 (2017).
Melchjorsen, J. et al. Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J. Virol. 84, 11350–11358 (2010).
Nguyen, T. A. et al. SIDT2 transports extracellular dsRNA into the cytoplasm for innate immune recognition. Immunity 47, 498–509.e6 (2017).
Zhao, J. et al. A viral deamidase targets the helicase domain of RIG-I to block RNA-induced activation. Cell Host Microbe 20, 770–784 (2016).
Duan, Y. et al. Nasopharyngeal carcinoma progression is mediated by EBER-triggered inflammation via the RIG-I pathway. Cancer Lett. 361, 67–74 (2015).
Samanta, M., Iwakiri, D. & Takada, K. Epstein–Barr virus-encoded small RNA induces IL-10 through RIG-I-mediated IRF-3 signaling. Oncogene 27, 4150–4160 (2008).
Wang, F. et al. RIG-I mediates the co-induction of tumor necrosis factor and type I interferon elicited by myxoma virus in primary human macrophages. PLoS Pathog. 4, e1000099 (2008).
Pham, A. M. et al. PKR transduces MDA5-dependent signals for type I IFN induction. PLoS Pathog. 12, e1005489 (2016).
Pichlmair, A. et al. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 83, 10761–10769 (2009).
Minamitani, T., Iwakiri, D. & Takada, K. Adenovirus virus-associated RNAs induce type I interferon expression through a RIG-I-mediated pathway. J. Virol. 85, 4035–4040 (2011).
Watkinson, R. E., McEwan, W. A., Tam, J. C., Vaysburd, M. & James, L. C. TRIM21 promotes cGAS and RIG-I sensing of viral genomes during infection by antibody-opsonized virus. PLoS Pathog. 11, e1005253 (2015).
Dou, Y., Yim, H. C., Kirkwood, C. D., Williams, B. R. & Sadler, A. J. The innate immune receptor MDA5 limits rotavirus infection but promotes cell death and pancreatic inflammation. EMBO J. 36, 2742–2757 (2017).
Uzri, D. & Greenberg, H. B. Characterization of rotavirus RNAs that activate innate immune signaling through the RIG-I-like receptors. PLoS One 8, e69825 (2013).
Broquet, A. H., Hirata, Y., McAllister, C. S. & Kagnoff, M. F. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J. Immunol. 186, 1618–1626 (2011).
Feng, Q. et al. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. 2, 1187–1196 (2012).
Wang, J. P. et al. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J. Virol. 84, 254–260 (2010).
Slater, L. et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 6, e1001178 (2010).
Cao, X. et al. MDA5 plays a critical role in interferon response during hepatitis C virus infection. J. Hepatol. 62, 771–778 (2015).
Fredericksen, B. L., Keller, B. C., Fornek, J., Katze, M. G. & Gale, M. Jr. Establishment and maintenance of the innate antiviral response to West Nile Virus involves both RIG-I and MDA5 signaling through IPS-1. J. Virol. 82, 609–616 (2008).
Riedl, W. et al. Zika virus NS3 mimics a cellular 14–3–3-binding motif to antagonize RIG-I- and MDA5-mediated innate immunity. Cell Host Microbe 26, 493–503.e6 (2019).
Zust, R. et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 12, 137–143 (2011).
Zalinger, Z. B., Elliott, R., Rose, K. M. & Weiss, S. R. MDA5 is critical to host defense during infection with murine coronavirus. J. Virol. 89, 12330–12340 (2015).
Li, J., Liu, Y. & Zhang, X. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J. Virol. 84, 6472–6482 (2010).
Ikegame, S. et al. Both RIG-I and MDA5 RNA helicases contribute to the induction of α/β interferon in measles virus-infected human cells. J. Virol. 84, 372–379 (2010).
Gitlin, L. et al. Melanoma differentiation-associated gene 5 (MDA5) is involved in the innate immune response to Paramyxoviridae infection in vivo. PLoS Pathog. 6, e1000734 (2010).
Banos-Lara Mdel, R., Ghosh, A. & Guerrero-Plata, A. Critical role of MDA5 in the interferon response induced by human metapneumovirus infection in dendritic cells and in vivo. J. Virol. 87, 1242–1251 (2013).
Dutta, M. et al. A systems approach reveals MAVS signaling in myeloid cells as critical for resistance to Ebola virus in murine models of infection. Cell Rep. 18, 816–829 (2017).
Cardenas, W. B. et al. Ebola virus VP35 protein binds double-stranded RNA and inhibits α/β interferon production induced by RIG-I signaling. J. Virol. 80, 5168–5178 (2006).
Solis, M. et al. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol. 85, 1224–1236 (2011).
Ringeard, M., Marchand, V., Decroly, E., Motorin, Y. & Bennasser, Y. FTSJ3 is an RNA 2′-O-methyltransferase recruited by HIV to avoid innate immune sensing. Nature 565, 500–504 (2019).
Berg, R. K. et al. Genomic HIV RNA induces innate immune responses through RIG-I-dependent sensing of secondary-structured RNA. PLoS One 7, e29291 (2012).
Lu, H. L. & Liao, F. Melanoma differentiation-associated gene 5 senses hepatitis B virus and activates innate immune signaling to suppress virus replication. J. Immunol. 191, 3264–3276 (2013).
Sato, S. et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42, 123–132 (2015).
Acknowledgements
The authors apologize to all colleagues whose important contributions could not be discussed and cited owing to space constraints. Current research in the Gack laboratory is supported by US National Institutes of Health (NIH) grants (R01 AI087846, R01 AI127774 and R21 AI148082) and an award from the ClayCo Foundation. J.R. acknowledges funding by the UK Medical Research Council (core funding of the MRC Human Immunology Unit).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Immunology thanks J. Hiscott, J. Jung and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Toll-like receptors
-
(TLRs). A family of membrane-bound innate immune receptors that recognize various bacterial or virus-derived pathogen-associated molecular patterns. These sensors are crucial for eliciting an early innate immune response and for modulating adaptive immunity.
- Cyclic GMP–AMP synthase
-
(cGAS). A key intracellular innate immune receptor that recognizes DNA from invading pathogens or cellular DNA that is mislocalized inside the cell. When activated, cGAS enzymatically synthesizes cyclic GMP–AMP, which acts as a second messenger to trigger type I interferon induction via stimulator of interferon genes (STING).
- Negative-sense RNA viruses
-
Viruses containing single-stranded RNA in negative polarity as their genomes. Viral RNA-dependent RNA polymerases copy these genomes into reverse-complementary, positive-sense RNA, which is then translated into viral proteins.
- Host shut-off
-
A process by which viruses prevent expression of cellular proteins. This may involve degradation of cellular transcripts or disruption of protein translation by viral factors. Host shut-off is believed to facilitate viral gene expression.
- Latent infection
-
An often lifelong form of herpesvirus infection during which no infectious virus is produced. Viral DNA is maintained in the nucleus of infected cells and a small set of viral RNAs and proteins may be expressed.
- Y-RNAs
-
Small non-coding, structured RNAs that are components of the Ro60 ribonucleoprotein complex and play roles in DNA replication.
- MicroRNAs
-
(miRNAs). Cellular non-coding, single-stranded RNAs of 17–24 nucleotides that are found in most organisms and that silence gene expression via direct targeting of complementary messenger RNAs.
- iCLIP
-
(Individual-nucleotide resolution ultraviolet crosslinking and immunoprecipitation). A method used to identify and map RNA–protein interactions in which ultraviolet light induces covalent bonds between the protein and RNA binding partners.
- Alu elements
-
Endogenous retroelements found in more than 1 million copies in the human genome. Alu elements rarely retrotranspose but can be transcribed. Adjacent Alu elements in inverted orientation can give rise to long duplex RNA structures.
- Single-nucleotide polymorphisms
-
Substitutions of a single nucleotide at a specific position compared with the reference genome. Single-nucleotide polymorphisms may affect the expression level or amino acid sequence of a protein.
- A-to-I RNA editing
-
A process that converts adenosine to inosine (A-to-I) in RNA. A-to-I editing is catalysed by adenosine deaminases acting on RNA (ADARs) and can change RNA structure and stability. A-to-I editing can also introduce mutations into proteins as ‘I’ bases are read as guanosines instead of adenosines during translation.
- Autophagy
-
A lysosome-dependent recycling and degradation process used to remove unnecessary or dysfunctional cellular material, such as long-lived proteins or organelles. Autophagy is also used by the cell to eliminate microorganisms, such as certain bacterial and viral pathogens.
- Deamidation
-
A process catalysed by aminotransferases in which an amide functional moiety is removed from a glutamine or asparagine residue in the substrate protein. Deamidation can have diverse outcomes for the target protein, including degradation and activation of its signal-transducing activity.
- Tripartite motif (TRIM) protein family
-
A family of ~80 proteins in humans that is structurally defined by the tripartite motif (also known as the RBCC motif) comprising a RING-finger E3 ligase, B-box and coiled-coil domain. In addition, TRIM proteins encode numerous different carboxy-terminal domains (for example, SPRY) that allow for the recruitment of specific proteins or have other functions.
- Deubiquitylating enzymes
-
Proteases that cleave the (iso)peptide bond between ubiquitin, or ubiquitin-like molecules, and the modified substrate. The human genome encodes ~100 deubiquitinating enzymes, some of which regulate pathogen recognition and signalling in innate immunity.
- Long non-coding RNAs
-
(lncRNAs). A class of host RNAs longer than 200 nucleotides that do not encode a protein. lncRNAs, which are often only expressed in certain cell types or tissues, regulate many cellular processes through modulation of gene expression or post-transcriptional mechanisms.
- RNA interference
-
A process in which long double-stranded RNA is cut into RNA fragments of ~21 nucleotides long by the enzyme Dicer. These RNA fragments, when further processed, can bind to complementary messenger RNA target sequences and, thereby, silence gene expression.
Rights and permissions
About this article

Cite this article
Rehwinkel, J., Gack, M.U. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol 20, 537–551 (2020). https://doi.org/10.1038/s41577-020-0288-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-020-0288-3
This article is cited by
-
Emerging roles of the Protein Phosphatase 1 (PP1) in the context of viral infections
Cell Communication and Signaling (2024)
-
Novel dual inhibitors of PARP and HDAC induce intratumoral STING-mediated antitumor immunity in triple-negative breast cancer
Cell Death & Disease (2024)
-
Longitudinal analysis of influenza vaccination implicates regulation of RIG-I signaling by DNA methylation
Scientific Reports (2024)
-
Harnessing innate immune pathways for therapeutic advancement in cancer
Signal Transduction and Targeted Therapy (2024)
-
Cytosolic DNA sensors in neurodegenerative diseases: from physiological defenders to pathological culprits
EMBO Molecular Medicine (2024)
