
Abstract
RNA is a key regulator of almost every cellular process, and the structures adopted by RNA molecules are thought to be central to their functions. The recent fast-paced evolution of high-throughput sequencing-based RNA structure mapping methods has enabled the rapid in vivo structural interrogation of entire cellular transcriptomes. Collectively, these studies are shedding new light on the long underestimated complexity of the structural organization of the transcriptome — the RNA structurome. Moreover, recent analyses are challenging the view that the RNA structurome is a static entity by revealing how RNA molecules establish intricate networks of alternative intramolecular and intermolecular interactions and that these ensembles of RNA structures are dynamically regulated to finely tune RNA functions in living cells. This new understanding of how RNA can shape cell phenotypes has important implications for the development of RNA-targeted therapeutic strategies.
Similar content being viewed by others

Introduction
RNA is a jack-of-all-trades. Believed for too long to only represent a carrier of genetic information, a mere intermediate between DNA and protein, RNA has now emerged as a master regulator of most cellular processes, under both physiological and pathological conditions. Importantly, the regulatory functions of RNA are largely independent of its ability to encode proteins. Non-coding regions of mRNAs, such as the untranslated regions (UTRs), contribute substantially to the post-transcriptional regulation of gene expression, for example by providing binding sites for RNA binding proteins (RBPs) and microRNAs, or by hosting regulatory RNA structure elements such as G-quadruplexes1,2. The expanding repertoire of transcribed non-coding RNAs (ncRNAs)3 includes both constitutive structural ncRNAs (such as ribosomal RNAs (rRNAs), small nuclear RNAs (snRNAs) and small nucleolar RNAs (snoRNAs)) and dynamically regulated ncRNAs (such as microRNAs, piwi RNAs and long ncRNAs), which can control and orchestrate, among other functions, transcriptional and post-transcriptional regulation of gene expression, splicing, assembly of large multiprotein complexes and translation4.
Many of the non-coding functions of RNA rely on its ability to fold back on itself to create stable structures. Despite their stability, RNA structures are far from static. For a given RNA, multiple alternative structural conformations can coexist as part of a heterogeneous and dynamic ensemble. The ability to dynamically redistribute the relative abundance of specific conformations within the ensemble in response to environmental cues is crucial to the regulatory functions of RNA structures5,6 and the biological importance of RNA ensemble dynamics is widely acknowledged. Although the existence of alternative structural configurations for an RNA might simply be an evolutionary bystander, several examples of RNA structural switches with clear regulatory roles have been reported to date. Two prominent and well-characterized examples of such RNA elements are riboswitches and RNA thermometers, which are able to respond to the presence of specific metabolites or to temperature changes, respectively, to regulate gene expression, either transcriptionally or post-transcriptionally7,8. However, the true extent of RNA structural heterogeneity in living cells, the way ensemble redistribution is regulated and how it, in turn, regulates a cell’s phenotype are still largely unknown.
Determining the structure of RNA molecules is crucial for elucidating their mechanisms of action. However, the study of RNA structure has long been tedious and extremely challenging. Methods such as X-ray crystallography, nuclear magnetic resonance and cryogenic electron microscopy can provide atomistic resolution of RNA structures, but are very time-consuming, have limited throughput and are typically not suited for in vivo analyses. Conversely, biochemical RNA structure probing methods using enzymatic or chemical probes do not provide atomistic resolution but have rapidly gained popularity because of their simplicity and their potential for studying RNA structures in living cells9. More recently, the advent of high-throughput sequencing (HTS) technologies has enabled these RNA structure probing methods to be adapted to interrogate thousands of RNAs, and even whole transcriptomes, in a single experiment10. These studies have contributed greatly to an improved understanding of the regulatory principles of the RNA structurome11.
In this Review, we discuss the latest advances in HTS-based methods for the transcriptome-scale determination of RNA structures (of both mRNAs and ncRNAs) in living cells, with particular emphasis on the biological insights these methods have revealed. In particular, we outline how features of the intracellular environment are critical to the unique structural state of RNA in vivo, and hence to its biological functions. We discuss possible sources of structural heterogeneity of RNA molecules, and how the recent combination of HTS-based structure mapping and computational methods is enabling the exploration of RNA structure ensembles and the reconstruction of coexisting alternative RNA conformations. In this context, we consider how the ability of RNA molecules to interconvert between alternative structural states, through engaging in both intramolecular and intermolecular interactions, might regulate different cellular processes. Finally, we summarize open challenges concerning the study of RNA structure ensembles in living cells in the context of RNA structure as an emergent novel therapeutic target, and the role of HTS methods in informing the development of RNA-targeted therapies.
High-throughput RNA structure analyses
Obtaining direct data on RNA structural states is key to developing an understanding of how RNA structure contributes to RNA function. To date, numerous complementary experimental approaches have been developed that use chemical probes to interrogate specific structural features of RNA molecules in the cell, including base-pairing, structure flexibility and solvent accessibility. Moreover, both chemical and biochemical methods have been developed to characterize the contribution of RNA structure to RNA–protein interactions and binding selectivity. These approaches provide information on either the structural state of individual nucleotides or the structural relationship between pairs of distal nucleotides within the same or different RNA molecules. Importantly, each probe is designed to obtain structural information based on its chemical reactivity and can be interfaced with HTS technologies to analyse the entire RNA structurome.
Probing the structural state of individual nucleotides
Structure probing, more accurately viewed as chemical probing of specific functional groups, is perhaps the most widely used approach to determine RNA structure. In these methods, the reactivity towards a chemical probe (which is dependent on the chemical environment or accessibility of certain functional groups) is used to either measure or infer the base-pairing status of the probed nucleotide(s) (Fig. 1a).

a, Targets of different chemical probes on RNA, including dimethyl sulfate (DMS), α-ketoaldehydes (such as Glyoxal and N3-kethoxal), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), selective 2′-hydroxyl acylation analysed by primer extension (SHAPE) reagents, hydroxyl radicals and nicotinoyl azide (NAz). Sites of chemical modification by probes measuring the pairing status of nucleobases (circles), the solvent accessibility of RNA residues (stars) and the flexibility of the RNA backbone (pentagons) are marked. b, Psoralen interacts with uridines on opposite strands of an RNA duplex and mediates cross-linking of the two strands upon long-wave UV irradiation (365 nm). Cross-linking can occur both intramolecularly and intermolecularly. c, The reaction of bifunctional acylating compounds, such as trans-bis-isatoic anhydride (TBIA) and spatial 2′-hydroxyl acylation reversible cross-linking (SHARC) reagents, results in cross-links between structurally flexible nucleotides that are spatially proximal to each other. Cross-linking can occur both intramolecularly and intermolecularly. d, Upon long-wave UV irradiation, NHS-diazirine cross-links RNA nucleotides and amino acids (usually lysine) of interacting proteins at the RNA–protein interaction interface.
Numerous specific chemicals have been identified that can be used to measure base-pairing owing to their ability to react with unpaired residues with nucleobase specificity. Their reactivity relies on the accessibility of functional groups involved in base-pairing, such that high reactivity is associated with single-stranded (that is, unpaired) RNA. Dimethyl sulfate (DMS) is the most commonly used reagent for measuring base-pairing because its chemical reactivity is robust, the relationship between reactivity and structure read-out is well established and it can very quickly pass through cell membranes to react with RNA12. It readily reacts with the Watson–Crick faces of unpaired adenines and cytosines, but, under mildly basic conditions, it can also react at a much lower rate with unpaired uracils and guanines13. Other probes include glyoxal and other α-ketoaldehydes, which react with unpaired guanines14, and carbodiimide reagents such as 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), which display selectivity for unpaired guanines and uracils, or for G:U wobble pairs15,16.
By contrast, chemical probing by selective 2′-hydroxyl acylation analysed by primer extension (SHAPE) measures the flexibility of the RNA backbone17, which is generally used as a proxy for base-pairing: when the SHAPE reagent forms an adduct with the 2′-OH of a structurally flexible nucleotide, the position is assumed to be unpaired. Moreover, specific structural states have been shown to promote SHAPE reactivity through intramolecular catalysis18. A large-scale analysis of SHAPE reactivity with generic RNAs suggests that, owing to conformational constraints, nucleotides in small loops, bulges and internal loops have enhanced reactivity towards SHAPE probes compared with large single-stranded regions19. Although early SHAPE probes20,21 (such as N-methylisatoic anhydride (NMIA), 1-methyl-6-nitroisatoic anhydride (1M6) and benzoyl cyanide (BzCN)) were not suitable for in-cell applications, recent advances in probe development, a better understanding of reaction chemistry and design of reagent functional groups have resulted in numerous robust SHAPE probes optimized for measuring RNA flexibility in living systems. These probes include 5-nitroisatoic anhydride22 (5NIA), as well as acyl imidazoles such as 2-methylnicotinic acid imidazolide23 (NAI) and its azido-functionalized derivative 2-(azidomethyl)nicotinic acid imidazolide24 (NAI-N3), 2-methyl-3-furoic acid imidazolide23 (FAI) and the recently developed 2-aminopyridine-3-carboxylic acid imidazolide25 (2A3).
Other approaches report on the solvent accessibility of specific functional groups on the RNA molecule. Solvent accessibility has traditionally been measured through the use of Fenton reagent-generated hydroxyl (OH) radicals. OH radicals are high-energy intermediates that target accessible C3′ or C4′ positions on the ribose ring for hydrogen abstraction, resulting in strand cleavage26,27. A newer method, termed light activated structural examination of RNA (LASER), takes advantage of aroyl azide probes such as nicotinoyl azide (NAz), which, when activated with long-wavelength UV light (365 nm), form stable C8 amidation products28. In addition to examining solvent-accessible regions of RNA in living cells, both OH radicals and LASER can also be used to investigate RNA–protein interactions in their native cellular environment29 (see RNA structure of RNA–protein interactions below).
The read-out of all these methods typically relies on traditional reverse transcription experiments (reviewed elsewhere10) (Fig. 2a). Originally, these experiments were performed on one RNA at a time, whereby an RNA of interest was incubated with a particular RNA structure probe. The RNA–adduct complex was then isolated and reverse-transcribed with reverse transcriptase (RT) enzymes using a radiolabelled primer. The modification introduced by the structural probe hampers the ability of the RT to incorporate the complementary nucleotide, either by preventing the formation of hydrogen bonds with the modified base (as is the case for DMS-induced alkylations) or owing to the bulkiness of the chemical adduct (as is the case for SHAPE reagents), leading to truncation of the resulting cDNA. The truncation points (referred to here as RT drop-off sites) of the resulting cDNA molecules were then mapped to the full-length RNA to identify the sites of chemical modification. With the advent of HTS technologies, these experiments have been extended to allow mapping of RT drop-off sites on a transcriptome-wide scale. More recently, it has been demonstrated that by either using specific RT enzymes or by altering the reverse transcription conditions, it is possible to avoid termination of reverse transcription at sites of chemical probing-induced modification, incorporating instead an incorrect DNA base, leading to mutations in the cDNA sequence30,31,32,33. These mutations can be used to identify the sites of chemical modification, with the number of mutations captured related to the number of probe-induced modifications on the RNA. This methodology is referred to as mutational profiling (MaP) and is rapidly superseding traditional RT drop-off-based read-out strategies owing to its robustness and reproducibility.

a, In chemical probing experiments, RNA undergoes reverse transcription following treatment with the chemical probe. When drop-off-based read-outs are used, the reverse transcriptase (RT) drops off the template at sites that have reacted with the probe, resulting in a pool of truncated cDNA molecules that terminate at the nucleotide prior to the modified site. Alternatively, in mutational profiling (MaP) experiments, reverse transcription conditions are adjusted so that the RT reads through the chemically modified sites but incorporates incorrect bases, resulting in (possibly full-length) cDNAs containing mutations at modification sites. In both cases, cDNA fragments are ligated to adapters, converted to double-stranded DNA libraries and sequenced. Sequencing reads (corresponding to cDNA fragments) are mapped back to the reference transcriptome. For RT drop-off-based experiments, each position i along the RNA is assigned a count corresponding to the number of reads whose 5′ ends mapped one nucleotide downstream (i + 1). For MaP-based experiments, the mutation frequency at each position of the RNA is calculated as the ratio between the number of mutated reads and the total number of reads covering that position. These raw reactivity profiles are then normalized to yield reactivities ranging between 0 (unreactive) and, depending on the normalization method, ≥1 (highly reactive). b, In direct RNA–RNA interaction capture experiments, RNA duplexes are cross-linked (for example by psoralen), RNA is fragmented and the two strands of the cross-linked duplexes are intramolecularly ligated, after which cross-linking is reversed. These chimeric RNA fragments are then reverse-transcribed and the resulting cDNA fragments are ligated to adapters, converted to double-stranded DNA libraries and sequenced. Sequencing reads are then mapped back to the reference transcriptome. As these reads are derived from RNA chimeras, the two halves of these reads will map to distinct locations of the same transcript in the case of intramolecular duplexes, or distinct transcripts in the case of intermolecular duplexes. Figure 2 is adapted from ref.68, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
In summary, in the context of these methods, the function of the chemical reagents is to react with nucleobases or the RNA backbone in a way that infers their structural state. Reagents that react with the Watson–Crick face of nucleobases are used to identify unpaired residues, whereas SHAPE reagents are designed to identify positions that are structurally flexible and, by proxy, unpaired. Reagents that measure groove or backbone accessibility identify solvent-exposed positions. Lack of reactivity to any of these reagents can arise for numerous reasons. For example, interactions of the RNA with proteins or other molecules (and in the case of nucleobase-specific and SHAPE reagents, intramolecular or intermolecular base-pairing) might reduce reactivity or even shield the RNA from reacting with the probe.
Mapping RNA–RNA base-pairing and through-space interactions
There has recently been a substantial focus on developing methods that provide a better understanding of both intramolecular and intermolecular RNA–RNA interactions. These methods can be classified as either indirect or direct.
Indirect methods such as RNA interaction groups measured by mutational profiling (RING-MaP)30 and pairing ascertained from interacting RNA strands measured by mutational profiling (PAIR-MaP)13 rely on chemical probing with reagents typically used to query the pairing state of individual nucleotides, such as DMS. The main assumption of these methods is that both secondary and tertiary interactions in RNA molecules can be captured owing to equilibrium fluctuations that transiently expose interacting bases, making them temporarily accessible for modification by the structural probe. Upon modification of one of the bases, its partner becomes permanently unpaired and accessible for modification. These events can then be detected as correlated modification signals via MaP analysis. The main limitation of these approaches is that these correlated modification events are extremely rare. As such, extremely high sequencing depths are required to robustly detect them, hence making the analysis of entire transcriptomes experimentally demanding.
Direct methods, instead, rely on chemical probes whose structure can be used to impose distance constraints. Two main types of probes have emerged: those that cross-link base-paired regions and those that cross-link spatially close RNA functional groups (referred to as ‘through-space’ interactions). Base-paired regions can be captured using chemical probes that are capable of intercalating and undergoing photo-catalysed cross-linking reactions with the nucleobases. Psoralen and its derivatives have been employed to capture nucleic acid–nucleic acid interactions for decades34. These compounds are reactive upon irradiation with long-wavelength UV light, which causes the two main functional units of the psoralen core (the pyrone and the furan ring) to cross-link two adjacent pyrimidine nucleobases residing on opposite strands of an RNA duplex (Fig. 1b). Traditionally, psoralen cross-links are mapped by denaturing gel electrophoresis35. More recently, these experiments have been coupled to HTS to enable mapping of psoralen cross-links transcriptome-wide (Fig. 2b). RNA proximity ligation (RPL)36, psoralen analysis of RNA interactions and structures37 (PARIS), sequencing of psoralen cross-linked, ligated and selected hybrids38 (SPLASH), ligation of interacting RNA followed by high-throughput sequencing39 (LIGR-seq), mapping RNA interactome in vivo40 (MARIO) and cross-linking of matched RNAs and deep sequencing41 (COMRADES) were all developed at similar times to capture cross-linked RNA species for transcriptome-scale analyses. As psoralen cross-linking is characterized by low efficiency, different protocols adopted different strategies to enrich cross-linked RNA duplexes. For example, PARIS took advantage of two-dimensional gel electrophoresis37, whereas SPLASH used a biotinylated psoralen derivative to enable the direct streptavidin-mediated capture of cross-linked RNA duplexes38. After enrichment of cross-link sites, proximity ligation is performed between the two strands of the duplex, generating a chimeric RNA molecule. The cross-links are then reversed with shorter wavelength UV light (254 nm), and cDNA sequencing libraries generated. Putative duplexes are then inferred from the gapped alignment of the chimeric reads to the transcriptome (Fig. 2b). Two main caveats exist with these approaches. Firstly, these strategies are not quantitative and certain interactions, even very rare or artefactual ones, can be over-represented because they are the result of optimized base-pair interactions that enable highly efficient psoralen cross-linking. For example, capture of uracil-rich stems tends to be favoured because psoralen cross-links two uracil residues across strands of an RNA helix. Secondly, because virtually any two RNA fragments can be ligated, these strategies result in high levels of background signal, leading to reproducibility issues. The COMRADES approach partly addresses this issue by generating control libraries in which cross-linking is reverted and RNA duplexes are melted prior to intramolecular ligation, hence enabling the estimation of background ligation events41.
Chemical probes capable of directly capturing through-space interactions are a very exciting recent development in HTS-based structure probing. By extending SHAPE chemistry, bifunctional acylation reagents have been generated that capture two 2′-hydroxyl residues that are in close spatial proximity42,43,44 (Fig. 1c). For example, spatial 2′-hydroxyl acylation reversible cross-linking (SHARC) uses bifunctional acylation reagents with flexible linkers to cross-link spatially proximal nucleotides43. In this case, the linker length is assumed to set the cross-linking distance and, hence, the structural distance between two sites of SHAPE adduct formation. Cross-links are enriched using bidimensional electrophoresis, followed by intramolecular ligation and cross-link reversal using a novel base-catalysed de-acylating protocol. SHARC has been shown to improve RNA three-dimensional structure modelling to near-nanometre resolution, and is the first approach that merges 2′-hydroxyl acylation and computational predictions to directly capture tertiary contacts and alternative conformations of RNAs in their native cellular context. The resolution achievable with these probes is constrained by the distance between the two acylation-reactive functional groups. Additional SHAPE-based probes have been developed that minimize this distance to enable higher-resolution structure mapping. Such probes have been used in selective 2′-hydroxyl acylation analysed by primer extension and juxtaposed merged pairs44 (SHAPE-JuMP). Unlike SHARC, SHAPE-JuMP identifies cross-linked nucleotides using an engineered RT enzyme that ‘jumps’ across cross-linked sites, resulting in a deletion in the cDNA that is detected using HTS. SHAPE-JuMP accurately identifies close-in-space interactions at near-nucleotide resolution, as demonstrated by comparing sequencing-based data with high-resolution X-ray crystallography RNA structures44. Although, at present, these approaches have not yet been applied transcriptome-wide, they are poised to extend the capabilities and precision of two-dimensional and three-dimensional RNA structure probing experiments.
RNA structure of RNA–protein interactions
Throughout its lifetime, RNA encounters a multitude of RBPs, which have critical regulatory roles. As such, understanding the structural interactions at the RNA–protein interface is important for gaining a mechanistic understanding of RNA function. Adapting traditional in vitro RNA structure probing techniques used for characterizing protein binding, which rely on traditional reverse transcription measured by gel electrophoresis, to in-cell, transcriptome-wide applications has been challenging, but has been met with very recent success.
The RNA–protein interface can be assessed indirectly by measuring changes in probe reactivity between free RNA and protein-bound RNA. For example, footprinting SHAPE (fSHAPE) uses differential SHAPE probing between in vivo (‘+ protein’) and ex vivo deproteinized (‘– protein’) conditions to identify RNA–protein interaction footprints45. Furthermore, by integrating SHAPE and fSHAPE with cross-linking and immunoprecipitation (CLIP) of desired RBPs, it is possible to interrogate specific RNA–protein complexes and to map which nucleotides hydrogen-bond with proteins45. Similarly, combinatorial probing of backbone flexibility using SHAPE and solvent accessibility using LASER has been shown to efficiently map protein–RNA interactions transcriptome-wide when comparing probe reactivity in cells versus in vitro refolded RNA46. Further integration of CLIP, RNA decay and polyA sequencing data sets with the SHAPE/LASER-derived protein footprinting data enabled accurate measurement of protein occupancy and prediction of RNA processing events46.
There has also been recent interest in developing chemical tools to better capture direct RNA–protein interactions. RNP network analysis by mutational profiling (RNP-MaP) employs a hetero-bifunctional cross-linker consisting of an NHS ester and diazirine to cross-link RBPs to RNAs47. The NHS ester reacts with surface-exposed lysine residues, which are known to be enriched at the RNA binding interface of RBPs. When diazirine molecules are exposed to long-wavelength UV light they react with surface-exposed functional groups on RNA through the formation of carbenes. In this way, the distance between the NHS ester and diazirine sets the distance between the NHS ester-reactive protein functional group and the cross-linked RNA (Fig. 1d). Sites of RNA reactivity are identified at single-nucleotide resolution using the same principles as SHAPE-MaP and other MaP techniques. Although this approach has so far only been applied to the targeted analysis of a subset of human ncRNAs in the cell, it is, in principle, suitable for transcriptome-wide analyses. Chemical reagents that enable direct mapping of RNA–protein interfaces can be merged with the structure probing methods detailed above to provide a more holistic approach to characterizing how proteins recognize RNA molecules and how binding of RBPs changes RNA structure.
Architecture of RNA–RNA interactomes
Although the co-transcriptional nature of RNA folding would suggest that locally stable folds mediated by short-range interactions would be generally preferred, recent studies have revealed the existence of an intricate network of both intramolecular long-range and intermolecular RNA structure interactions, particularly in the context of mRNAs and viral RNA genomes37,38,39,40,41,48,49,50,51,52. Detecting such long-range interactions is extremely challenging when relying solely on chemical probing, as structure modelling from probing data is typically constrained to limit the maximum base-pairing distance, although exceptions exist53. In this context and despite their limitations, the recent introduction of methods that allow the direct capture of RNA–RNA interactions in cells (such as PARIS, SPLASH, LIGR-seq, MARIO and COMRADES; see Mapping RNA–RNA base-pairing and through-space interactions) has been a real game changer37,38,39,40,41.
Dynamics of long-range RNA interactions in living cells
Mapping of RNA duplexes in human and mouse cells using PARIS has shown that approximately 30–40% of the duplexes occur between regions separated by more than 200 nucleotides, with 4–11% separated by more than 1,000 nucleotides37. Similarly, duplex mapping across the ZIKV RNA genome using COMRADES indicates a general preference for locally stable structures, with less than 20% of the duplexes involving distances greater than 1,000 nucleotides41. By contrast, SPLASH analysis of the SARS-CoV-2 genome revealed a high prevalence of long-range interactions, which accounted for just under half of all detected RNA duplexes51. These long-range interactions tend to have a lower read support, suggesting that they might be highly dynamic and form only transiently. In this regard, the cellular environment seems to have a major role. Comparative SPLASH analysis of RNA duplexes in virio and in vivo for ZIKV and DENV genomes revealed that nearly 80% of the interactions inside virions involve distances greater than 500 nucleotides, compared with less than 35% within the cell, and that nearly twice as many short-range duplexes are shared between in virio and in vivo conditions than long-range duplexes, suggesting that long-range interactions might be actively disrupted within the cell49.
In general, between 20 and 50% of the RNA duplexes in cellular mRNAs and roughly half of the duplexes in ZIKV, DENV and SARS-CoV-2 genomes have been reported to be mutually exclusive, confirming the existence of substantial structural heterogeneity within the cell37,49,51 (Fig. 3a). Although techniques for direct RNA–RNA interaction mapping do not preserve any information regarding the relationship between the individual duplexes, hence making it impossible to determine how many conformations were originally present within the ensemble, combined duplex clustering and structure modelling analyses of the ZIKV genome suggest that a set of as few as five structures would be sufficient to explain up to 90% of the detected RNA duplexes41. The cellular environment also seems to play a key part in regulating the structural diversity within viral genome ensembles. Comparative in virio and in vivo analysis of ZIKV and DENV genomes showed that nearly twice as many alternative interactions are formed in virions, suggesting that viral genomes are less structurally heterogeneous in the cell than they are inside viral particles49. In general, it is conceivable that both short-range and long-range interactions might be actively unwound by the helicase activity of translating ribosomes54,55, or by other host factors56, hence contributing to the overall lower level of structuring and heterogeneity of viral genomes inside host cells. In the context of virions, however, long-range interactions might have a crucial role in promoting genome compaction to ensure proper packaging. Although these studies indicate that long-range RNA interactions are more dynamic in the cellular context, it is worth pointing out that these are very preliminary investigations and that further evidence, possibly from orthogonal approaches, will be needed before solid conclusions can be reached.

a, The SARS-CoV-2 genome establishes a wide range of mutually exclusive long-range interactions, many of which involve the untranslated regions (UTRs). Four possible structural configurations, observed to coexist in the context of infected host cells, are depicted (from top-left proceeding clockwise): the linear genome; the partially circularized genome owing to an interaction between ORF1a and the 3′ UTR; the partially circularized genome owing to an interaction between ORF1a and the 5′ UTR; and the fully circularized genome owing to an interaction between the 5′ and 3′ UTRs. It is unknown what different functions these conformations play, nor which of the conformations can mutually convert one into another (represented by question marks over arrows). Regions coloured in red can form alternative, mutually exclusive, short-range and long-range RNA–RNA interactions. b, In human cells, the orphan C/D-box small nucleolar RNA (snoRNA) SNORD83B forms intermolecular interactions with the SRSF3, RPS5 and NOP14 mRNAs. The functional relevance of these interactions, which have been shown to modulate the steady-state levels of these mRNAs, is still unknown. c, The ZIKV genome can circularize owing to a long-range interaction between the 5′ and 3′ cyclization sequences (CSs) located at the termini of the genome. Genome cyclization promotes viral replication, whilst hampering translation. In its linear form, the 5′ CS region of the genome has been reported to establish an intermolecular interaction with the host hsa-miR-21 microRNA (in complex with AGO2). Although the mechanistic details of this interaction are still unknown, depletion of hsa-miR-21 potently reduces the cellular levels of viral RNA. BSL, bulged stem-loop; cHP, capsid hairpin; DAR, downstream of AUG region; DCS-PK, downstream of 5′ CS pseudoknot; HVR, hypervariable region; s2m, stem-loop II-like motif; SL, stem-loop; UAR, upstream of AUG region.
RNA establishes a network of regulatory intermolecular interactions
In addition to intramolecular interactions, mapping of RNA–RNA interactions has begun to unravel an intricate network of intermolecular interactions, which further complicates the architecture of in vivo RNA structural ensembles. Whereas mRNA–mRNA trans interactions seem to be quite rare, analysis of cellular transcriptomes by SPLASH, LIGR-seq and MARIO identified highly abundant snRNA–snRNA and rRNA–rRNA trans interactions and a large number of snoRNA–mRNA and snoRNA–ncRNA interactions, often involving orphan snoRNAs38,39,40. Although the functional importance of many of these novel interactions is still unclear, targeted antisense oligonucleotide-mediated depletion of the orphan C/D-box snoRNA SNORD83B was shown to cause a strong increase in the levels of its target mRNAs, suggesting a potential role for snoRNA–mRNA interactions in controlling steady-state RNA levels39 (Fig. 3b). Both COMRADES and SPLASH analyses detected strong interaction of cellular snRNAs, mitochondrial RNAs and snoRNAs with the SARS-CoV-2 RNA genome in infected host cells50,51. SNORD27, a C/D-box snoRNA involved in the 2′-O-methylation of adenine 27 on the 18S rRNA, establishes one of the strongest trans interactions with the SARS-CoV-2 genome51. This interaction requires the partial disruption of a secondary structure element located within ORF1A and it is proposed to drive 2′-O-methylation of the SARS-CoV-2 genome, possibly to increase its stability. Similarly, in ZIKV-infected cells the viral RNA genome establishes several interactions with both cellular tRNAs and microRNAs, as shown by COMRADES analysis41. In particular, the interaction between the ZIKV 5′ cyclization sequence (5′ CS) and miR-21 seems to have a strong proviral effect, as depletion of miR-21 reduces the cellular levels of viral RNA (Fig. 3c). This region is also involved in genome cyclization via interaction with the 3′ CS. Genome cyclization of flaviviral genomes has been recently shown to inhibit translation initiation and, possibly, promote genome replication by preventing collisions between the translating ribosome and the viral RNA polymerase, which proceed in opposite directions57. An intriguing possibility is that the interaction of the 5′ CS with miR-21 might be required to drive the switch from genome cyclization (and possibly replication) to genome translation.
A meta-analysis of RNA duplex mapping data generated using the aforementioned methods has also identified numerous homotypic trans RNA interactions in both cellular and viral RNAs58. Although substantially rarer than heteroduplexes, these homoduplexes are enriched in specific cellular RNAs, including the U1 and U2 snRNAs, which mediate RNA splicing; the U3 and U8 snoRNAs, which mediate the cleavage and maturation of rRNAs; tRNAs; and numerous mitochondrial mRNAs. Among these, homoduplexes of the U8 snoRNA occur with substantially higher frequency. Mutations in the U8 snoRNA that are known to drive pathogenesis of leukoencephalopathy with calcifications and cysts, a rare autosomal recessive disease, are predicted to disrupt these homotypic U8–U8 interactions without affecting base-pairing of U8 to pre-rRNA or any other known U8 target. Importantly, central nervous system developmental defects in a U8–/– zebrafish model of leukoencephalopathy with calcifications and cysts can be complemented by injection of the wild-type U8 snoRNA, but not by mutant U8 snoRNAs predicted to disrupt U8 homoduplexes.
Deconvolving RNA structural heterogeneity
RNA structures are intrinsically dynamic and heterogeneous5,6. Defining a single native structural conformation for an RNA sequence that is strongly favoured over competing ones is not only extremely difficult but, in most cases, biologically incorrect. Inside the cell, multiple copies of the same RNA can fold into different conformations. Moreover, the conformation of each RNA molecule is not static over time5. Rather, each molecule can interconvert between alternative conformations, at a rate that depends on the energetic barrier separating the different conformations. It is crucial to point out that whereas the interconversion between structures involving alternative tertiary interactions can freely occur at physiological temperatures, the interconversion between alternative secondary structures is energetically very expensive because it involves the disruption of multiple base pairs5,6,59. Although alternative secondary structures can be formed upon folding of RNA molecules, these typically populate local minima of the energy landscape and are therefore separated by large energy barriers that cannot be spontaneously overcome at physiological temperatures; interconversion likely requires the contribution of proteins with RNA chaperone activity60. This heterogeneous and dynamic set of RNA structures is commonly referred to as an ensemble. Within the ensemble, each possible conformation for a given RNA is associated with a certain probability of being formed (or sampled). This probability is determined by a multitude of factors, including temperature, concentration of ions, post-transcriptional RNA modifications, RNA editing, small-molecule binding and interaction with proteins or other RNAs6 (Fig. 4 and Box 1). The traditional HTS-based chemical probing experiments described above simultaneously probe all the possible conformations making up the ensemble and, as a consequence, the reactivity profile derived from these experiments represents a weighted average of all the coexisting RNA conformations. Indeed, numerous studies have reported that a large fraction of bases in the transcriptomes of higher metazoans show intermediate reactivities24,56,61,62, hinting at an underlying structural heterogeneity arising from these bases existing in at least two distinct structural states (or conformations) in vivo. Deconvolving the individual conformations making up the ensemble from bulk structure probing data is a non-trivial task. This problem can be tackled either by using specialized RNA structure probing assays or via numerous computational approaches.

Under cellular conditions, the folding landscape of an RNA molecule is constantly changing and RNA molecules can undergo numerous structural rearrangements (Box 1). RNA molecules fold as they get transcribed, and the structures they adopt will change as transcription proceeds. Co-transcriptional events, such as the deposition of RNA post-transcriptional modifications (PTMs) or alternative splicing, can affect varying proportions of the RNA molecules and result in structurally diverse subpopulations. Differential binding of RNA binding proteins (RBPs) can further lead to substantial structural heterogeneity within and across cellular compartments. In the cytoplasm, translation (which itself can be regulated by RNA structure) can also shape the structure of RNA molecules because of the intrinsic helicase activity of the ribosome. Alternative RNA structures are coloured red. These alternative conformations may coexist in the cell, resulting in a heterogeneous ensemble.
Experimental approaches for deconvolving RNA structure ensembles
In addition to methods for direct mapping of RNA–RNA interactions that intrinsically enable the capture of RNA duplexes belonging to alternative RNA conformations (see Architecture of RNA–RNA interactomes), three experimental methods based on chemical probing have been devised to expose otherwise invisible short-lived structure intermediates and lowly populated conformations within RNA ensembles: co-transcriptional SHAPE followed by sequencing (SHAPE-seq), structural probing of elongating transcripts followed by sequencing (SPET-seq) and mutate and map (M2). Co-transcriptional SHAPE-seq and SPET-seq capture the structure of individual transcription intermediates, thereby enabling the reconstruction of RNA co-transcriptional folding pathways63,64 (Fig. 5a). Co-transcriptional SHAPE-seq relies on the generation of a pool of DNA templates for in vitro transcription that each causes the RNA polymerase to pause at a different position, thereby collectively generating all the possible transcription intermediates. SPET-seq instead relies on the assumption that, at any given time in a pool of cells, the RNA polymerase would occupy different positions on the template DNA in each cell, hence enabling the sampling of virtually any transcription intermediate. As such, SPET-seq can be readily applied to in-cell transcriptome-wide analyses, whereas co-transcriptional SHAPE-seq has so far been applied only to individual RNAs in vitro. By contrast, M2 is based on the assumption that certain lowly populated conformations can be stabilized by specific mutations that reweight the underlying structure ensemble65,66 (Fig. 5b) and, consequently, is not restricted to the analysis of co-transcriptional structure changes. Therefore, in M2 the structures of a large pool of randomly generated single-nucleotide sequence variants of an RNA of interest are queried by chemical probing and changes in the reactivity pattern across mutants indirectly inform on the presence of an alternative RNA conformation67. Although powerful, the main limitation of the M2 approach is that it cannot be scaled up to analyse the entire transcriptome.

a, Assays such as co-transcriptional selective 2′-hydroxyl acylation analysed by primer extension (SHAPE) followed by sequencing (SHAPE-seq) and structural probing of elongating transcripts followed by sequencing (SPET-seq) allow RNA co-transcriptional structure folding pathways to be deconvolved by first probing the entire population of transcription intermediates, followed by the computational reconstruction of the individual reactivity profiles. Plotting these reactivity profiles in the form of a heatmap, with the rows corresponding to distinct transcription intermediates sorted by increasing length, provides intuitive visualization of RNA structural rearrangements occurring as transcription proceeds (top to bottom). The example shows two transcription intermediates, each represented by the rows denoted in yellow. During the transition from the first to the second intermediate, the reactivity of the unpaired regions (coloured purple and green on the structures) progressively drops (purple and green boxes on the heatmap) as they begin to undergo base-pairing, resulting in a pseudoknot (purple region) and a stem-loop (SL) (green region). b, Mutate and map (M2) provides an indirect way to deconvolve RNA structure ensembles by randomly generating a large number of single-nucleotide substitution mutants of an RNA of interest, followed by structure probing analysis. Mutations capable of disrupting base-pairing interactions in the wild-type structure, whilst stabilizing alternative folds, will cause a redistribution of the relative abundance of the structures within the ensemble, leading to reactivity changes. The reactivity profiles of these mutants can then be used to infer the structure of these alternative conformations. c, The first group of computational methods for ensemble deconvolution exploits thermodynamics-guided RNA structure prediction software to sample a large number of structures from the theoretical ensemble the RNA of interest can form, and then uses the experimental data to select the smallest possible subset of structures that can explain the data. Typically, structures are then clustered together by similarity and a single representative structure is returned for each cluster. This class of approaches is suitable for the analysis of both reverse transcriptase (RT)-stop and mutational profiling (MaP) RNA structure probing data. d, The second group of computational methods for ensemble deconvolution involves direct read clustering. These methods take sequencing reads from MaP experiments and attempt to define clusters of reads with correlated patterns of mutations, corresponding to alternative RNA conformations. Clustered reads can be processed into reactivity profiles that can then be used to inform structure modelling.
Computational approaches for deconvolving RNA structure ensembles
There are two classes of computational methods available for ensemble deconvolution from bulk structure probing data, which have been recently extensively reviewed elsewhere68 and will be only briefly discussed here. The first class of methods heavily relies on thermodynamics because they attempt to predict a parsimonious set of structures for a given RNA that can justify the experimental data (Fig. 5c). These approaches typically involve sampling a large number (usually 1,000–10,000) of possible structures for the RNA of interest, followed by the identification of a smaller subset that better explains the data. This kind of approach can be applied to both structure probing and RNA–RNA interaction capture experiments69,70,71. Structures are typically sampled from the theoretical ensemble of structures that RNA can form, which is commonly referred to as a Boltzmann ensemble because structures follow a Boltzmann distribution6,72. As such, each structure within the ensemble is associated with a probability of being observed that is only dependent on its free energy and on the temperature of the system. Thus, the biggest limitation of this class of approaches is that the most stable structures will have the highest chance of being sampled; however, physiologically occurring conformations might be substantially less stable and, therefore, might (almost) never be sampled. Indeed, multiple studies showed that certain RNAs tend to be significantly less structured in the cell than they are in vitro24,56, suggesting that in silico predictions might better approximate in vitro conditions, under which thermodynamics alone can be expected to constitute the main driving force of RNA folding, in contrast to in vivo conditions. Additionally, inaccuracies with the energy potentials of the thermodynamic model might further hamper the effective sampling of physiologically occurring structures.
The second class of methods are RNA structure agnostic (Fig. 5d). These methods do not make any a priori assumptions based on the reactivity data and, most importantly, do not rely on thermodynamics; rather, they attempt to directly assign the sequencing reads to distinct clusters that represent the coexisting conformations populated by the RNA30,73,74,75. Crucial to these direct read clustering analyses is the use of chemical probing data derived from MaP experiments, which record multiple sites of chemical modification as mutations within the same cDNA product. By analysing the co-mutation patterns in sequencing reads, it is possible to deconvolve the reactivity profiles of the individual conformations making up the ensemble. As such, the main limitation of this class of methods is that they are not suited for the analysis of RT drop-off-based RNA probing experiments. Although these methods can efficiently deconvolve the ensemble in a thermodynamics-independent fashion, their resolution is typically limited as they can only distinguish major structural differences, likely resulting from the presence of alternative secondary structures. However, the high sequencing depths required to sample a sufficiently high number of reads harbouring two or more mutations is the major constraint of this class of methods, making their application to the analysis of the whole transcriptome a currently unmet challenge.
Insights into RNA structure ensembles
To date, only a small number of studies have attempted to analyse RNA structure ensembles in living cells. Most have focused on viral genomes, which have long been known to carry structurally heterogeneous and dynamic structure elements76, but a couple have focused on mammalian RNAs. Although these studies analyse various different contexts (that is, in virio, in vivo and in vitro), they collectively point to the existence of a vast and previously unappreciated RNA structural heterogeneity.
RNA structural switches in the HIV-1 genome regulate viral replication
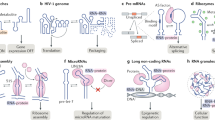
Ensemble deconvolution of the HIV-1 virus genome, probed in living infected host cells by DMS-MaPseq, revealed that more than 90% of the genomic RNA folds into at least two alternative structures73. The Rev protein recognition element (RRE) is crucial for regulating the nuclear export of the unspliced HIV-1 genome and the known minor conformation (a four-way junction) and major conformation (a five-way junction) were observed to consistently form under in vitro, in vivo and in virio conditions. The five-way junction configuration, which has previously been reported to confer a replicative advantage to the virus77, was the most prominent as expected (Fig. 6a). Similarly, the region encompassing the A3 splice site, which regulates the abundance of the transcript encoding the Tat protein (an activator of viral transcription), was shown to adopt two structures73. In the minor conformation, the splice site and the polypyrimidine tract are sequestered within a stem-loop (SL) structure, thereby preventing U2AF binding and inhibiting splicing, whereas in the major conformation the splice site and the polypyrimidine tract are exposed, hence promoting splicing and viral transcription (Fig. 6b). It thus seems that for both the RRE and A3 structural switches the major conformations promote replication and transcription, respectively, hence fostering viral spread and disease severity. The development of RNA-targeted therapeutics capable of promoting the switch from the major to the minor conformation of these structure elements might therefore provide a novel effective approach to treat HIV-1 infections. Ensemble deconvolution analyses conducted both in CD4+ primary T cells and in the human embryonic kidney cell line HEK293t show strong agreement, suggesting that the observed structure heterogeneity is largely independent of the cell type73. Moreover, in vitro analyses indicate that these RNA elements are intrinsically heterogeneous73,77. However, whereas the in vivo and in vitro ensembles of the RRE are remarkably similar, the in vitro ensemble of the A3 element does not populate the major conformation observed in infected cells, suggesting that cellular factors, such as protein binding, might be driving the selection of this conformation.

a, The structure ensemble of the HIV-1 Rev response element (RRE) populates two conformations, a four-way junction (the minor conformation) and a five-way junction (the major conformation); regions that adopt alternative structures in these two conformations are coloured red. The major conformation can interact with the viral protein, Rev, which promotes nuclear export of the viral genome. This export is crucial both for the translation of the Gag and Gag-Pol proteins and for the packaging of new virions. b, Splicing of the transcript encoding the transactivator protein Tat of HIV-1 is controlled by a switch between two alternative conformations, with consequences for transcription of the HIV-1 genome. In the minor conformation, the A3 splice site is inaccessible to binding by the U2AF splicing factor and, as a result, no functional Tat protein is produced. In the absence of Tat, transcription of the HIV-1 double-stranded DNA genome by the host RNA Polymerase II is highly inefficient. By contrast, the A3 splice site of the Tat transcript is accessible in the major conformation leading to productive splicing, and the resulting Tat protein promotes efficient transcription of the HIV-1 genome. c, In human cells, the activity of P-TEFb, a positive regulator of transcription, is controlled by the 7SK snRNA, which is capable of binding and sequestering P-TEFb. The structure ensemble of 7SK populates two major conformations: one that contains the SL1 stem-loop, which can bind to and sequester P-TEFb (P-TEFb-bound); and one that contains the SL1alt stem-loop and cannot sequester P-TEFb (P-TEFb-unbound). Thus, switching between SL1 and SL1alt stem-loop containing-structures regulates the binding of P-TEFb and, thereby, its availability for promoting transcription. A third highly dynamic minor conformation of 7SK has also been identified and hypothesized to represent an intermediate state between the two major conformations. Arrows with questions marks above indicate that it is not yet known whether those conformations can interconvert. This highly dynamic intermediate is possibly an average of multiple low-abundance conformations. Part c is adapted with permission from ref.75, Elsevier.
The SARS-CoV-2 frameshifting element samples a large folding space
Two independent studies reported the ensemble deconvolution analysis of the SARS-CoV-2 genome, probed by DMS-MaPseq under either in vivo or in vitro conditions74,78. Both studies concordantly detected extensive structural heterogeneity along the SARS-CoV-2 genome and, in particular, deconvolved two alternative conformations making up the structural ensemble of the ribosomal frameshifting element (FSE). The FSE is crucial for regulating programmed –1 ribosomal frameshifting of ORF1b, which encodes five non-structural viral proteins79. Previous in silico and in vitro analyses, focusing solely on an 88 nucleotide-long segment at the interface of ORF1a and ORF1b, proposed a 3-stemmed pseudoknot structure for the FSE80,81. However, neither of the structures identified by ensemble deconvolution analyses of the SARS-CoV-2 genome correspond to the three-stemmed pseudoknot conformation. Although the structures reported by the two studies are slightly different, both contain the same bipartite SL element, which harbours the attenuator hairpin and the slippery site that are essential for slowing down the translating ribosome to enable –1 frameshifting79. The structure model for the major conformation reported by one of the studies further involves a long-range interaction spanning ~1.1 kb that, in a dual-luciferase reporter assay, promotes an approximate 25% increase in ribosomal frameshifting compared with the previously proposed three-stemmed pseudoknot78. Importantly, the same study illustrates the dependency of the FSE structure on the surrounding context. For example, when only the 88 nucleotide-long segment encompassing the FSE is transcribed and probed in vitro, it folds consistently with the 3-stemmed pseudoknot model, whereas in vitro refolding of the entire SARS-CoV-2 genome recapitulates the in vivo FSE structure78. It is essential to note that during the COVID-19 pandemic, different studies using either chemical probing or RNA duplex capture approaches have proposed several alternative structural configurations for the FSE element50,51,74,78,82,83,84. Importantly, a recent study using direct RNA duplex capture via a simplified SPLASH approach found both in virio and in vivo RNA–RNA interactions that support most of these proposed FSE structures, including the three-stemmed pseudoknot52. The relevance of this finding is twofold. On the one hand, it reveals that the FSE is characterized by much higher structural heterogeneity than anticipated on the basis of each individual study. On the other, it suggests that different methods might be better at capturing different subsets of conformations and that a comprehensive description of the ensemble might require the combination of complementary approaches for ensemble deconvolution. These observations become particularly relevant when it comes to the development of RNA-targeted therapies aimed at inhibiting frameshifting by targeting the FSE. Indeed, most studies aimed at the development of RNA-targeted therapies have focused solely on the 88 nucleotide-long segment that folds into the 3-stemmed pseudoknot conformation, yet it does not represent the major conformation in the context of infected cells85,86.
Structural dynamics of human 7SK ncRNA regulate transcription
Unlike for the HIV-1 RNA genome, the cellular context seems to have a major role in determining the distribution of alternative conformations within the ensemble for certain cellular RNAs. In human cells, in vivo ensemble deconvolution analysis of 7SK, the RNA component of an snRNP involved in sequestering P-TEFb to downregulate transcription initiation by RNA Polymerase II, revealed the existence of two major conformations, P-TEFb-bound and P-TEFb-unbound, whose relative stoichiometries are highly cell context-dependent and state-dependent75 (Fig. 6c). Indeed, analysis of the 7SK ensemble in fast versus slowly proliferating cells revealed an increase in the relative abundance of the P-TEFb-bound conformation in the slowly proliferating cells, which was even more prominent in quiescent cells. Antisense oligonucleotide-mediated destabilization of the P-TEFb-bound conformation was shown to skew the ensemble towards the P-TEFb-unbound state and to induce transcription in cells, supporting the role of this structural switch in regulating transcription in the cell by sequestration of P-TEFb. This type of antisense oligonucleotide-based approach has great therapeutic potential and P-TEFb is an attractive therapeutic target; for example, it is an essential cellular cofactor of HIV-1 Tat-activated transcription and it is dysregulated in many human diseases, such as infectious diseases and cancer87.
RNA structure ensembles tend to be evolutionarily conserved
Phylogenetic analyses have revealed how certain RNA structure ensembles are highly conserved across evolution, which typically implies functionality. Ensemble deconvolution analysis of the SARS-CoV-2 genome probed by DMS-MaPseq identified numerous regions that form two alternative structures, including the 3′ UTR74. The major 3′ UTR conformation corresponds to the structure previously identified by phylogenetic analyses79, whereas the minor conformation has a rearrangement of the hypervariable region (HVR) but retains two highly conserved structure elements of betacoronaviruses: the bulged stem-loop (BSL) and the stem-loop II-like motif (s2m). The BSL has been previously shown to be crucial for viral replication in mouse hepatitis virus88 (MHV). The functional relevance of the s2m is not yet clear, but the peculiar three-dimensional geometry of this structure has been proposed to be involved in hijacking the protein synthesis machinery via molecular mimicry of an rRNA fold89. Although the biological function of this structural switch in the SARS-CoV-2 3′ UTR is still unknown, the exceptional covariation support exhibited by both conformations across thousands of coronavirus genomes supports its functionality.
In mouse cells, ensemble analysis of the extremely conserved 5′ UTR of Csde1 mRNA, which encodes an RBP that regulates cell cycle, differentiation and apoptosis and is implicated in various human diseases90, revealed the existence of three structurally distinct conformations, whose proportions are dynamically regulated by ATP-dependent RNA helicases66. Regions showing differential reactivity upon ATP depletion are characterized by significantly higher sequence conservation compared with the rest of the RNA, suggesting that the extreme conservation of 5′ UTR sequences might ensure the structural conservation of the ensemble, ultimately needed for the active structure remodelling by RNA helicases. Notably, mutations leading to a redistribution of the ensemble altered translation levels of a luciferase reporter by up to 50%, indicating that the proportions of the different conformations making up the ensemble are crucial to finely tune protein levels66.
Open challenges in RNA structuromics
Despite substantial advances in our ability to interrogate the structure of RNA molecules in vivo, several important challenges impede a full understanding of the RNA structurome. Firstly, substantial limitations remain when it comes to modelling the structure of RNA molecules. Although many orthogonal approaches have been introduced to query different aspects of RNA structures, combining the data they generate into a single coherent structure prediction is a non-trivial task. Importantly, the prediction of RNA secondary structures often disregards complex non-nested structure elements such as pseudoknots, mostly because of the associated computational cost. Furthermore, existing computational methods for predicting RNA pseudoknots with the aid of constraints from chemical probing experiments cannot model RNAs containing multiple pseudoknots91, although this limitation can be mitigated by adopting sliding window-based solutions31,92. Although techniques for direct RNA–RNA interaction mapping theoretically have the potential to capture pseudoknots in RNAs, they do not preserve information about the relationship between individual RNA duplexes, which makes it impossible to determine whether two non-nested duplexes coexist as part of a pseudoknot, or whether they belong to two mutually exclusive alternative conformations. In this regard, combining computational approaches for RNA structure ensemble deconvolution from chemical probing experiments with RNA–RNA interaction capture data might provide the means for identifying pseudoknots at scale (Fig. 7a).

a, Mapping of pseudoknots can potentially be achieved by combining direct RNA–RNA interaction capture with methods for ensemble deconvolution from chemical probing experiments. Although RNA duplex mapping does not preserve any information about the relationship between two independent helices, using ensemble deconvolution analysis to determine whether the region of the RNA encompassing these helices populates one or two conformations can help determine whether two incompatible helices coexist within the same RNA molecule, forming a pseudoknot, or whether they belong to two independent RNA molecules. b, Specialized structure probing assays can aid the analysis of RNA structure ensembles in vivo. Coupling of chemical probing with single-cell analysis (top), RNA immunoprecipitation (middle) or polysome fractionation (bottom) would increase the resolution of RNA structure analyses, possibly enabling the characterization of lowly abundant RNA conformations. c, RNA chemical probing can aid the mapping of small molecule–RNA interactions. Analysis of population-averaged reactivities can be used to identify footprints of small molecules binding to RNA. The coupling of chemical probing with ensemble deconvolution analysis can further help elucidate binding modes of small molecules, possibly enabling the identification of specific RNA conformations targeted by the small molecule.
Secondly, a thorough characterization of RNA structure ensembles and their dynamics in living cells requires that substantial technical limitations are overcome. On the one hand, better chemical probes (and RTs) are needed to achieve a higher signal-to-noise ratio in MaP-based RNA chemical probing experiments. This would, in turn, facilitate ensemble deconvolution by direct read clustering, further lowering the sequencing depth required for the detection of lowly abundant conformations. On the other hand, the timescale for the analysis of RNA structure dynamics is directly dependent on the reaction time needed for the chemical probe to efficiently permeate the cell and modify the RNA. Although chemical probing on a millisecond scale is achievable in vitro21, the shortest time frame for efficient in vivo probing of RNA is in the order of minutes12,22,23,25, thus hampering the possibility of capturing fast structural transitions and short-lived structure intermediates. In general, dissecting RNA structure ensembles in vivo remains a substantial challenge and a full description of RNA structure ensembles in a living cell has yet to be achieved. The application of methods for RNA ensemble deconvolution to full transcriptomes has the unique potential to accelerate the discovery of dynamic regulatory RNA structure elements such as riboswitches that have remained largely elusive, especially in eukaryotes92. RNA structure heterogeneity in vivo might arise as a consequence of numerous cellular determinants, which might affect only a small fraction of the RNA population (Fig. 4 and Box 1). However, at present, the resolution of methods based on direct read clustering is limited to the reconstruction of conformations with sufficiently high stoichiometries (typically 10% or higher) and such reconstructions likely represent an aggregate of highly similar, yet structurally distinct, conformations, hence providing only a coarse-grained overview of RNA ensembles73,74,75. The combination of these methods with computational approaches relying on thermodynamics might partly help address this limitation by enabling the further deconvolution and refinement of these sub-ensembles. Additionally, the development of specialized experimental assays — such as co-transcriptional SHAPE-seq, SPET-seq and M2 — could further enable the enrichment of lowly abundant subpopulations of structures. For instance, the coupling of chemical probing with RNA metabolic or proximity labelling93,94, polysome fractionation, RNA immunoprecipitation or single-cell RNA-sequencing analyses would provide the means to characterize in greater detail the RNA structure sub-ensembles generated as a consequence of RNA compartmentalization, translation, protein binding, RNA post-transcriptional modification (PTM) and editing, as well as RNA structure differences between individual cells in a heterogeneous population (Fig. 7b). However, an important caveat is that RNA structure mapping experiments are typically read out on the Illumina platform, which has a maximum achievable read length of 600 bp; this is a major limitation for the analysis of RNA structure ensembles for transcripts longer than this maximum read length. Although approaches such as DRACO74 can use tiled overlapping reads to deconvolve structurally heterogeneous regions longer than the actual read length, it is impossible to infer any relationship between distal regions in a long transcript. The use of long-read platforms such as Oxford Nanopore and PacBio provides an opportunity to tackle this problem. Indeed, a recent in vivo analysis of the long ncRNA COOLAIR (including a 795-nucleotide isoform) in Arabidopsis thaliana using a chemical probing read-out via MaP and long-read PacBio sequencing demonstrates that this approach can be used to deconvolve the structural ensemble of longer transcripts95.
Lastly, the ability to deconvolve RNA structure ensembles becomes particularly relevant when thinking of RNA structure as a target for small-molecule drugs. Alternative structures might mediate different biological functions, so knowing which conformation is responsible for a specific pathological phenotype is crucial for target identification. In this regard, the characterization of RNA structure ensembles in living cells represents a key step towards mapping the druggable transcriptome. Little is known to date about the mechanisms by which small molecules can establish productive interactions with RNA or about the features that define a good druggable pocket within an RNA structure element, with most of our knowledge coming from the study of interactions between riboswitches and their ligands. Binding of small molecules to RNA has been shown to shield certain nucleotides from chemical probing96, or to alter their reactivity97, hence allowing the sites of RNA–small molecule interactions to be directly pinpointed. Chemical probing can be potentially leveraged to obtain large-scale maps of small-molecule binding sites in RNAs. Indeed, a novel SHAPE-like approach, which exploits the functionalization of small-molecule drugs with an acylimidazole-substituted linker, has been recently reported and used to map transcriptome-wide the interaction sites of numerous US Food and Drug Administration (FDA)-approved drugs98. Combining these analyses with methods for ensemble deconvolution by direct read clustering could additionally make it possible to dissect complex small-molecule binding modes, such as induced fit or conformational selection (Fig. 7c).
Conclusions
We have just begun to scratch the surface of the complexity of the RNA structurome. A key advance has been the ability to robustly probe RNA structures in living cells and compare the data generated with in vitro refolded RNA, thereby revealing unique structural aspects of RNA molecules in the cell. The fast-paced parallel development of experimental and computational methods has enabled the analysis of RNA structure ensembles for individual transcripts, with the potential to scale these analyses to the entire transcriptome in the near future. Nonetheless, it must be pointed out that, although powerful, sequencing-based methods such as those discussed in this Review can only provide coarse-grained representations of RNA structure ensembles because they can only detect major structural rearrangements. In this regard, future efforts should be aimed at improving the sensitivity of these approaches, to enable smaller RNA structural changes to be captured. Finally, the development of novel chemical methods in combination with higher-resolution transcriptomic technologies will be needed to reveal the secrets of the RNA structurome in ever greater detail.
References
Leppek, K., Das, R. & Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 19, 158–174 (2018).
Mayr, C. Regulation by 3′-untranslated regions. Annu. Rev. Genet. 51, 171–194 (2017).
Frankish, A. et al. GENCODE 2021. Nucleic Acids Res. 49, D916–D923 (2021).
Fu, X.-D. Non-coding RNA: a new frontier in regulatory biology. Natl Sci. Rev. 1, 190–204 (2014).
Mustoe, A. M., Brooks, C. L. & Al-Hashimi, H. M. Hierarchy of RNA functional dynamics. Annu. Rev. Biochem. 83, 441–466 (2014).
Ganser, L. R., Kelly, M. L., Herschlag, D. & Al-Hashimi, H. M. The roles of structural dynamics in the cellular functions of RNAs. Nat. Rev. Mol. Cell Biol. 20, 474–489 (2019).
Kortmann, J. & Narberhaus, F. Bacterial RNA thermometers: molecular zippers and switches. Nat. Rev. Microbiol. 10, 255–265 (2012).
Serganov, A. & Nudler, E. A decade of riboswitches. Cell 152, 17–24 (2013).
Kubota, M., Tran, C. & Spitale, R. C. Progress and challenges for chemical probing of RNA structure inside living cells. Nat. Chem. Biol. 11, 933–941 (2015).
Strobel, E. J., Yu, A. M. & Lucks, J. B. High-throughput determination of RNA structures. Nat. Rev. Genet. 19, 615–634 (2018).
Kwok, C. K., Tang, Y., Assmann, S. M. & Bevilacqua, P. C. The RNA structurome: transcriptome-wide structure probing with next-generation sequencing. Trends Biochem. Sci. 40, 221–232 (2015).
Wells, S. E., Hughes, J. M., Igel, A. H. & Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Methods Enzymol. 318, 479–493 (2000).
Mustoe, A. M., Lama, N. N., Irving, P. S., Olson, S. W. & Weeks, K. M. RNA base-pairing complexity in living cells visualized by correlated chemical probing. Proc. Natl Acad. Sci. USA 116, 24574–24582 (2019).
Mitchell, D. et al. Glyoxals as in vivo RNA structural probes of guanine base-pairing. RNA 24, 114–124 (2018).
Wang, P. Y., Sexton, A. N., Culligan, W. J. & Simon, M. D. Carbodiimide reagents for the chemical probing of RNA structure in cells. RNA 25, 135–146 (2019).
Mitchell, D. et al. In vivo RNA structural probing of uracil and guanine base-pairing by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC). RNA 25, 147–157 (2019).
Merino, E. J., Wilkinson, K. A., Coughlan, J. L. & Weeks, K. M. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc. 127, 4223–4231 (2005).
McGinnis, J. L., Dunkle, J. A., Cate, J. H. D. & Weeks, K. M. The mechanisms of RNA SHAPE chemistry. J. Am. Chem. Soc. 134, 6617–6624 (2012).
Xiao, L., Fang, L. & Kool, E. T. Acylation probing of “generic” RNA libraries reveals critical influence of loop constraints on reactivity. Cell Chem. Biol. 29, 1341–1352.e8 (2022).
Steen, K.-A., Rice, G. M. & Weeks, K. M. Fingerprinting noncanonical and tertiary RNA structures by differential SHAPE reactivity. J. Am. Chem. Soc. 134, 13160–13163 (2012).
Mortimer, S. A. & Weeks, K. M. Time-resolved RNA SHAPE chemistry. J. Am. Chem. Soc. 130, 16178–16180 (2008).
Busan, S., Weidmann, C. A., Sengupta, A. & Weeks, K. M. Guidelines for SHAPE reagent choice and detection strategy for RNA structure probing studies. Biochemistry 58, 2655–2664 (2019).
Spitale, R. C. et al. RNA SHAPE analysis in living cells. Nat. Chem. Biol. 9, 18–20 (2013).
Spitale, R. C. et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486–490 (2015).
Marinus, T., Fessler, A. B., Ogle, C. A. & Incarnato, D. A novel SHAPE reagent enables the analysis of RNA structure in living cells with unprecedented accuracy. Nucleic Acids Res. 49, e34 (2021).
Ingle, S., Azad, R. N., Jain, S. S. & Tullius, T. D. Chemical probing of RNA with the hydroxyl radical at single-atom resolution. Nucleic Acids Res. 42, 12758–12767 (2014).
Kielpinski, L. J. & Vinther, J. Massive parallel-sequencing-based hydroxyl radical probing of RNA accessibility. Nucleic Acids Res. 42, e70 (2014).
Feng, C. et al. Light-activated chemical probing of nucleobase solvent accessibility inside cells. Nat. Chem. Biol. 14, 276–283 (2018).
Zinshteyn, B. et al. Assaying RNA structure with LASER-Seq. Nucleic Acids Res. 47, 43–55 (2019).
Homan, P. J. et al. Single-molecule correlated chemical probing of RNA. Proc. Natl Acad. Sci. USA 111, 13858–13863 (2014).
Siegfried, N. A., Busan, S., Rice, G. M., Nelson, J. A. E. & Weeks, K. M. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods 11, 959–965 (2014).
Zubradt, M. et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 14, 75–82 (2017).
Guo, L.-T. et al. Sequencing and structure probing of long RNAs using MarathonRT: a next-generation reverse transcriptase. J. Mol. Biol. 432, 3338–3352 (2020).
Cimino, G. D., Gamper, H. B., Isaacs, S. T. & Hearst, J. E. Psoralens as photoactive probes of nucleic acid structure and function: organic chemistry, photochemistry, and biochemistry. Annu. Rev. Biochem. 54, 1151–1193 (1985).
Nilsen, T. W. Detecting RNA-RNA interactions using psoralen derivatives. Cold Spring Harb. Protoc. 2014, 996–1000 (2014).
Ramani, V., Qiu, R. & Shendure, J. High-throughput determination of RNA structure by proximity ligation. Nat. Biotechnol. 33, 980–984 (2015).
Lu, Z. et al. RNA duplex map in living cells reveals higher order transcriptome structure. Cell 165, 1267–1279 (2016).
Aw, J. G. A. et al. In vivo mapping of eukaryotic RNA interactomes reveals principles of higher-order organization and regulation. Mol. Cell 62, 603–617 (2016).
Sharma, E., Sterne-Weiler, T., O’Hanlon, D. & Blencowe, B. J. Global mapping of human RNA–RNA interactions. Mol. Cell 62, 618–626 (2016).
Nguyen, T. C. et al. Mapping RNA–RNA interactome and RNA structure in vivo by MARIO. Nat. Commun. 7, 12023 (2016).
Ziv, O. et al. COMRADES determines in vivo RNA structures and interactions. Nat. Methods 15, 785–788 (2018).
Velema, W. A., Park, H. S., Kadina, A., Orbai, L. & Kool, E. T. Trapping transient RNA complexes by chemically reversible acylation. Angew. Chem. Int. Ed. Engl. 59, 22017–22022 (2020).
Van Damme, R. et al. Chemical reversible crosslinking enables measurement of RNA 3D distances and alternative conformations in cells. Nat. Commun. 13, 911 (2022).
Christy, T. W. et al. Direct mapping of higher-order RNA interactions by SHAPE-JuMP. Biochemistry 60, 1971–1982 (2021).
Corley, M. et al. Footprinting SHAPE-eCLIP reveals transcriptome-wide hydrogen bonds at RNA–protein interfaces. Mol. Cell 80, 903–914.e8 (2020).
Chan, D. et al. Diverse functional elements in RNA predicted transcriptome-wide by orthogonal RNA structure probing. Nucleic Acids Res. 49, 11868–11882 (2021).
Weidmann, C. A., Mustoe, A. M., Jariwala, P. B., Calabrese, J. M. & Weeks, K. M. Analysis of RNA–protein networks with RNP-MaP defines functional hubs on RNA. Nat. Biotechnol. 39, 347–356 (2021).
Li, P. et al. Integrative analysis of Zika virus genome RNA structure reveals critical determinants of viral infectivity. Cell Host Microbe 24, 875–886.e5 (2018).
Huber, R. G. et al. Structure mapping of dengue and Zika viruses reveals functional long-range interactions. Nat. Commun. 10, 1408 (2019).
Ziv, O. et al. The short- and long-range RNA–RNA interactome of SARS-CoV-2. Mol. Cell 80, 1067–1077.e5 (2020).
Yang, S. L. et al. Comprehensive mapping of SARS-CoV-2 interactions in vivo reveals functional virus–host interactions. Nat. Commun. 12, 5113 (2021).
Zhang, Y. et al. In vivo structure and dynamics of the SARS-CoV-2 RNA genome. Nat. Commun. 12, 5695 (2021).
Uroda, T. et al. Conserved pseudoknots in lncRNA MEG3 are essential for stimulation of the p53 pathway. Mol. Cell 75, 982–995.e9 (2019).
Mustoe, A. M. et al. Pervasive regulatory functions of mRNA structure revealed by high-resolution SHAPE probing. Cell 173, 181–195.e18 (2018).
Beaudoin, J.-D. et al. Analyses of mRNA structure dynamics identify embryonic gene regulatory programs. Nat. Struct. Mol. Biol. 25, 677–686 (2018).
Rouskin, S., Zubradt, M., Washietl, S., Kellis, M. & Weissman, J. S. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505, 701–705 (2014).
Sanford, T. J., Mears, H. V., Fajardo, T., Locker, N. & Sweeney, T. R. Circularization of flavivirus genomic RNA inhibits de novo translation initiation. Nucleic Acids Res. 47, 9789–9802 (2019).
Gabryelska, M. M. et al. Global mapping of RNA homodimers in living cells. Genome Res. 32, 956–967 (2022).
Dethoff, E. A., Chugh, J., Mustoe, A. M. & Al-Hashimi, H. M. Functional complexity and regulation through RNA dynamics. Nature 482, 322–330 (2012).
Herschlag, D. RNA chaperones and the RNA folding problem. J. Biol. Chem. 270, 20871–20874 (1995).
Ding, Y. et al. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505, 696–700 (2014).
Incarnato, D., Neri, F., Anselmi, F. & Oliviero, S. Genome-wide profiling of mouse RNA secondary structures reveals key features of the mammalian transcriptome. Genome Biol. 15, 491 (2014).
Watters, K. E., Strobel, E. J., Yu, A. M., Lis, J. T. & Lucks, J. B. Cotranscriptional folding of a riboswitch at nucleotide resolution. Nat. Struct. Mol. Biol. 23, 1124–1131 (2016).
Incarnato, D. et al. In vivo probing of nascent RNA structures reveals principles of cotranscriptional folding. Nucleic Acids Res. 45, 9716–9725 (2017).
Cheng, C. Y., Kladwang, W., Yesselman, J. D. & Das, R. RNA structure inference through chemical mapping after accidental or intentional mutations. Proc. Natl Acad. Sci. USA 114, 9876–9881 (2017).
Byeon, G. W. et al. Functional and structural basis of extreme conservation in vertebrate 5′ untranslated regions. Nat. Genet. 53, 729–741 (2021).
Cordero, P. & Das, R. Rich RNA structure landscapes revealed by mutate-and-map analysis. PLoS Comput. Biol. 11, e1004473 (2015).
Aviran, S. & Incarnato, D. Computational approaches for RNA structure ensemble deconvolution from structure probing data. J. Mol. Biol. https://doi.org/10.1016/j.jmb.2022.167635 (2022).
Li, H. & Aviran, S. Statistical modeling of RNA structure profiling experiments enables parsimonious reconstruction of structure landscapes. Nat. Commun. 9, 606 (2018).
Spasic, A., Assmann, S. M., Bevilacqua, P. C. & Mathews, D. H. Modeling RNA secondary structure folding ensembles using SHAPE mapping data. Nucleic Acids Res. 46, 314–323 (2018).
Zhou, J. et al. IRIS: a method for predicting in vivo RNA secondary structures using PARIS data. Quant. Biol. 8, 369–381 (2020).
McCaskill, J. S. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers 29, 1105–1119 (1990).
Tomezsko, P. J. et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature 582, 438–442 (2020).
Morandi, E. et al. Genome-scale deconvolution of RNA structure ensembles. Nat. Methods 18, 249–252 (2021).
Olson, S. W. et al. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Mol. Cell 82, 1708–1723.e10 (2022).
Wu, M. T.-P. & D’Souza, V. Alternate RNA structures. Cold Spring Harb. Perspect. Biol. 12, a032425 (2020).
Sherpa, C., Rausch, J. W., Le Grice, S. F. J., Hammarskjold, M.-L. & Rekosh, D. The HIV-1 Rev response element (RRE) adopts alternative conformations that promote different rates of virus replication. Nucleic Acids Res. 43, 4676–4686 (2015).
Lan, T. C. T. et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nat. Commun. 13, 1128 (2022).
Manfredonia, I. & Incarnato, D. Structure and regulation of coronavirus genomes: state-of-the-art and novel insights from SARS-CoV-2 studies. Biochemical Soc. Trans. 49, 341–352 (2020).
Plant, E. P. et al. A three-stemmed mRNA pseudoknot in the SARS coronavirus frameshift signal. PLoS Biol. 3, e172 (2005).
Rangan, R. et al. De novo 3D models of SARS-CoV-2 RNA elements from consensus experimental secondary structures. Nucleic Acids Res. 49, 3092–3108 (2021).
Manfredonia, I. et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Res. 48, 12436–12452 (2020).
Huston, N. C. et al. Comprehensive in vivo secondary structure of the SARS-CoV-2 genome reveals novel regulatory motifs and mechanisms. Mol. Cell 81, 584–598.e5 (2021).
Schlick, T. et al. To knot or not to knot: multiple conformations of the SARS-CoV-2 frameshifting RNA element. J. Am. Chem. Soc. 143, 11404–11422 (2021).
Park, S.-J., Kim, Y.-G. & Park, H.-J. Identification of RNA pseudoknot-binding ligand that inhibits the –1 ribosomal frameshifting of SARS-coronavirus by structure-based virtual screening. J. Am. Chem. Soc. 133, 10094–10100 (2011).
Sun, Y. et al. Restriction of SARS-CoV-2 replication by targeting programmed −1 ribosomal frameshifting. Proc. Natl Acad. Sci. USA 118, e2023051118 (2021).
Fujinaga, K. P-TEFb as a promising therapeutic target. Molecules 25, E838 (2020).
Hsue, B. & Masters, P. S. A bulged stem-loop structure in the 3’ untranslated region of the genome of the coronavirus mouse hepatitis virus is essential for replication. J. Virol. 71, 7567–7578 (1997).
Robertson, M. P. et al. The structure of a rigorously conserved RNA element within the SARS virus genome. PLOS Biol. 3, e5 (2004).
Guo, A.-X., Cui, J.-J., Wang, L.-Y. & Yin, J.-Y. The role of CSDE1 in translational reprogramming and human diseases. Cell Commun. Signal. 18, 14 (2020).
Hajdin, C. E. et al. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc. Natl Acad. Sci. USA 110, 5498–5503 (2013).
Batey, R. T. Riboswitches: still a lot of undiscovered country. RNA 21, 560–563 (2015).
Fazal, F. M. et al. Atlas of subcellular RNA localization revealed by APEX-seq. Cell 178, 473–490.e26 (2019).
Singha, M., Spitalny, L., Nguyen, K., Vandewalle, A. & Spitale, R. C. Chemical methods for measuring RNA expression with metabolic labeling. Wiley Interdiscip. Rev. RNA 12, e1650 (2021).
Yang, M. et al. In vivo single-molecule analysis reveals COOLAIR RNA structural diversity. Nature https://doi.org/10.1038/s41586-022-05135-9 (2022).
Sengupta, A., Rice, G. M. & Weeks, K. M. Single-molecule correlated chemical probing reveals large-scale structural communication in the ribosome and the mechanism of the antibiotic spectinomycin in living cells. PLOS Biol. 17, e3000393 (2019).
Zeller, M. J. et al. SHAPE-enabled fragment-based ligand discovery for RNA. Proc. Natl Acad. Sci. USA 119, e2122660119 (2022).
Fang, L. et al. Pervasive transcriptome interactions of protein-targeted drugs. Preprint at https://doi.org/10.1101/2022.07.18.500496 (2022).
Bushhouse, D. Z., Choi, E. K., Hertz, L. M. & Lucks, J. B. How does RNA fold dynamically? J. Mol. Biol. 167665 https://doi.org/10.1016/j.jmb.2022.167665 (2022).
Strobel, E. J., Cheng, L., Berman, K. E., Carlson, P. D. & Lucks, J. B. A ligand-gated strand displacement mechanism for ZTP riboswitch transcription control. Nat. Chem. Biol. 15, 1067–1076 (2019).
Cheng, L. et al. Cotranscriptional RNA strand exchange underlies the gene regulation mechanism in a purine-sensing transcriptional riboswitch. Nucleic Acids Res. https://doi.org/10.1093/nar/gkac102 (2022).
Saldi, T., Riemondy, K., Erickson, B. & Bentley, D. L. Alternative RNA structures formed during transcription depend on elongation rate and modify RNA processing. Mol. Cell 81, 1789–1801.e5 (2021).
Fu, Y., Dominissini, D., Rechavi, G. & He, C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 15, 293–306 (2014).
Roost, C. et al. Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J. Am. Chem. Soc. 137, 2107–2115 (2015).
Liu, N. et al. N6-Methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature 518, 560–564 (2015).
Aw, J. G. A. et al. Determination of isoform-specific RNA structure with nanopore long reads. Nat. Biotechnol. 39, 336–346 (2021).
Sun, L. et al. RNA structure maps across mammalian cellular compartments. Nat. Struct. Mol. Biol. 26, 322–330 (2019).
Liu, Z. et al. In vivo nuclear RNA structurome reveals RNA-structure regulation of mRNA processing in plants. Genome Biol. 22, 11 (2021).
Ray, P. S. et al. A stress-responsive RNA switch regulates VEGF expression. Nature 457, 915–919 (2009).
Acknowledgements
This work was supported by funding from the Groningen Biomolecular Sciences and Biotechnology Institute (GBB, University of Groningen) to D.I.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Genetics thanks Y. Ding and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Boltzmann distribution
-
A probability distribution describing the probability that a system will be in a certain state (in this case, a certain RNA conformation) as a function of the state’s energy and of the system’s temperature.
- Covariation
-
In an RNA multiple sequence alignment, two covarying positions are those for which the sequence changes but their ability to base-pair is preserved.
- Hydrogen abstraction
-
Removal of an atom or group from a molecule by a radical.
- Pseudoknot
-
A non-nested structural RNA motif formed upon base-pairing between the loop of a secondary structure element (such as a stem-loop (SL)) and any complementary region along the RNA.
- RNA structurome
-
The full range of RNA structures formed by the transcriptome of an organism.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Spitale, R.C., Incarnato, D. Probing the dynamic RNA structurome and its functions. Nat Rev Genet 24, 178–196 (2023). https://doi.org/10.1038/s41576-022-00546-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41576-022-00546-w
This article is cited by
-
Co-transcriptional gene regulation in eukaryotes and prokaryotes
Nature Reviews Molecular Cell Biology (2024)
-
Small RNA structural biochemistry in a post-sequencing era
Nature Protocols (2024)
-
Isoform-specific RNA structure determination using Nano-DMS-MaP
Nature Protocols (2024)
-
KARR-seq maps higher-order RNA structures and RNA–RNA interactions across the transcriptome
Nature Biotechnology (2024)
-
Accurate prediction of RNA secondary structure including pseudoknots through solving minimum-cost flow with learned potentials
Communications Biology (2024)
