
Abstract
In celebration of the 20th anniversary of Nature Reviews Genetics, we asked 12 leading researchers to reflect on the key challenges and opportunities faced by the field of genetics and genomics. Keeping their particular research area in mind, they take stock of the current state of play and emphasize the work that remains to be done over the next few years so that, ultimately, the benefits of genetic and genomic research can be felt by everyone.
The contributors
Amy L. McGuire is the Leon Jaworski Professor of Biomedical Ethics and Director of the Center for Medical Ethics and Health Policy at Baylor College of Medicine. She has received numerous teaching awards at Baylor College of Medicine, was recognized by the Texas Executive Women as a Woman on the Move in 2016 and was invited to give a TedMed talk titled “There is No Genome for the Human Spirit” in 2014. In 2020, she was elected as a Hastings Center Fellow. Her research focuses on ethical and policy issues related to emerging technologies, with a particular focus on genomic research, personalized medicine and the clinical integration of novel neurotechnologies.
Stacey Gabriel is the Senior Director of the Genomics Platform at the Broad Institute since 2012 and has led platform development, execution and operation since its founding. She is Chair of Institute Scientists and serves on the institute’s executive leadership team. She is widely recognized as a leader in genomic technology and project execution. She has led the Broad’s contributions to numerous flagship projects in human genetics, including the International HapMap Project, the 1000 Genomes Project, The Cancer Genome Atlas, the National Heart, Lung, and Blood Institute’s Exome Sequencing Project and the TOPMed programme. She is Principal Investigator of the Broad’s All of Us (AoU) Genomics Center and serves on the AoU Program Steering Committee.
Sarah A. Tishkoff is the David and Lyn Silfen University Associate Professor in Genetics and Biology at the University of Pennsylvania, Philadelphia, USA, and holds appointments in the School of Medicine and the School of Arts and Sciences. She is a member of the US National Academy of Sciences and a recipient of an NIH Pioneer Award, a David and Lucile Packard Career Award, a Burroughs/Wellcome Fund Career Award and an American Society of Human Genetics Curt Stern Award. Her work focuses on genomic variation in Africa, human evolutionary history, the genetic basis of adaptation and phenotypic variation in Africa, and the genetic basis of susceptibility to infectious disease in Africa.
Ambroise Wonkam is Professor of Medical Genetics, Director of GeneMAP (Genetic Medicine of African Populations Research Centre) and Deputy Dean Research in the Faculty of Health Sciences, University of Cape Town, South Africa. He has successfully led numerous NIH- and Wellcome Trust-funded projects over the past decade to investigate clinical variability in sickle cell disease, hearing impairment genetics and the return of individual findings in genetic research in Africa. He won the competitive Clinical Genetics Society International Award for 2014 from the British Society of Genetic Medicine. He is president of the African Society of Human Genetics.
Aravinda Chakravarti is Director of the Center for Human Genetics and Genomics, the Muriel G. and George W. Singer Professor of Neuroscience and Physiology, and Professor of Medicine at New York University School of Medicine. He is an elected member of the US National Academy of Sciences, the US National Academy of Medicine and the Indian National Science Academy. He has been a key participant in the Human Genome Project, the International HapMap Project and the 1000 Genomes Project. His research attempts to understand the molecular basis of multifactorial disease. He was awarded the 2013 William Allan Award by the American Society of Human Genetics and the 2018 Chen Award by the Human Genome Organization.
Eileen E. M. Furlong is Head of the Genome Biology Department at the European Molecular Biology Laboratory (EMBL) and a member of the EMBL Directorate. She is an elected member of the European Molecular Biology Organization (EMBO) and the Academia Europaea, and a European Research Council (ERC) advanced investigator. Her group dissects fundamental principles of how the genome is regulated and how it drives cell fate decisions during embryonic development, including how developmental enhancers are organized and function within the 3D nucleus. Her work combines genetics, (single-cell) genomics, imaging and computational approaches to understand these processes. Her research has advanced the development of genomic methods for use in complex multicellular organisms.
Barbara Treutlein is Associate Professor of Quantitative Developmental Biology in the Department of Biosystems Science and Engineering of ETH Zurich in Basel, Switzerland. Her group uses and develops single-cell genomics approaches in combination with stem cell-based 2D and 3D culture systems to study how human organs develop and regenerate and how cell fate is regulated. For her work, Barbara has received multiple awards, including the Friedmund Neumann Prize of the Schering Foundation, the Dr. Susan Lim Award for Outstanding Young Investigator of the International Society of Stem Cell Research and the EMBO Young Investigator Award.
Alexander Meissner is a scientific member of the Max Planck Society and currently Managing Director of the Max Planck Institute (MPI) for Molecular Genetics in Berlin, Germany. He heads the Department of Genome Regulation and is a visiting scientist in the Department of Stem Cell and Regenerative Biology at Harvard University. Before his move to the MPI, he was a tenured professor at Harvard University and a senior associate member of the Broad Institute, where he co-directed the epigenomics programme. In 2018, he was elected as an EMBO member. His laboratory uses genomic tools to study developmental and disease biology with a particular focus on epigenetic regulation.
Howard Y. Chang is the Virginia and D. K. Ludwig Professor of Cancer Genomics at Stanford University and an investigator at the Howard Hughes Medical Institute. He is a physician–scientist who has focused on deciphering the hidden information in the non-coding genome. His laboratory is best known for studies of long non-coding RNAs in gene regulation and development of new epigenomic technologies. He is an elected member of the US National Academy of Sciences, the US National Academy of Medicine, and the American Academy of Arts and Sciences.
Núria López-Bigas is ICREA research Professor at the Institute for Research in Biomedicine and Associate Professor at the University Pompeu Fabra. She obtained an ERC Consolidator Grant in 2015 and was elected as an EMBO member in 2016. Her work has been recognized with the prestigious Banc de Sabadell Award for Research in Biomedicine, the Catalan National Award for Young Research Talent and the Career Development Award from the Human Frontier Science Program. Her research focuses on the identification of cancer driver mutations, genes and pathways across tumour types and in understanding the mutational processes that lead to the accumulation of mutations in cancer cells.
Eran Segal is Professor in the Department of Computer Science and Applied Mathematics at the Weizmann Institute of Science, heading a multidisciplinary laboratory with extensive experience in machine learning, computational biology and analysis of heterogeneous high-throughput genomic data. His research focuses on the microbiome, nutrition and genetics, and their effect on health and disease and aims to develop personalized medicine based on big data from human cohorts. He has published more than 150 publications and received several awards and honours for his work, including the Overton and the Michael Bruno awards. He was recently elected as an EMBO member and as a member of the Israel Young Academy.
Jin-Soo Kim is Director of the Center for Genome Engineering in the Institute for Basic Science in Daejon, South Korea. He has received numerous awards, including the 2017 Asan Award in Medicine, the 2017 Yumin Award in Science and the 2019 Research Excellence Award (Federation of Asian and Oceanian Biochemists and Molecular Biologists). He was featured as one of ten Science Stars of East Asia in Nature (558, 502–510 (2018)) and has been recognized as a highly cited researcher by Clarivate Analytics since 2018. His work focuses on developing tools for genome editing in biomedical research.
Similar content being viewed by others

Making genomics truly equitable
Amy McGuire. For the field of genetics and genomics, the first decade of the twenty-first century was a time of rapid discovery, transformative technological development and plummeting costs. We moved from mapping the human genome, an international endeavour that took more than a decade and cost billions of dollars, to sequencing individual genomes for a mere fraction of the cost in a relatively short time.
During the subsequent decade, the field turned towards making sense of the vast amount of genomic information being generated and situating it in the context of one’s environment, lifestyle and other non-genetic factors. Much of the hype that characterized the previous decade was tempered as we were reminded of the exquisite complexity of human biology. A vision of medicine driven by genetically determined risk predictions was replaced with a vision of precision in which genetics, environment and lifestyle all converge to deliver the right treatment to the right patient at the right time1.
As we embark on the third decade of this century, we are now faced with the prospect of being able not only to more accurately predict disease risk and tailor existing treatments on the basis of genetic and non-genetic factors but also to potentially cure or even eliminate some diseases entirely with gene-editing technologies.
These advancements raise many ethical and policy issues, including concerns about privacy and discrimination, the right of access to research findings and direct-to-consumer genetic testing, and informed consent. Significant investment has been made to better understand the risks and benefits of clinical genomic testing, and there has been vigorous debate about the ethics of human gene editing, with many prominent scientists and bioethicists calling for a moratorium on human germline editing until it is proven to be safe and effective and there is broad societal consensus on its appropriate application2.
These are all important issues that we need to continue to explore, but as the technologies that have been developed and tested at warp speed over the past two decades begin to be integrated into routine clinical care, it is imperative that we also confront one of the most difficult and fundamental challenges in genomics, in medicine and in society — rectifying structural inequities and addressing factors that privilege some while disadvantaging others. The genomics of the future must be a genomics for all, regardless of ethnicity, geography or ability to pay.
This audacious goal of making genomics truly equitable requires multifaceted solutions. The disproportionate burden of illness and death among racial and ethnic minorities associated with the global COVID-19 pandemic3 and recent protests against police brutality towards African American citizens4 have strengthened the antiracism movement and amplified demands for racial equity.
To be part of this movement and effect change will require humility. We must actively listen and learn from each other, especially when it is uncomfortable and our own complicity may be implicated. It will require solidarity and a recognition that we are all connected through our common humanity. And it will require courage. It may seem like a platitude, but it is true that nothing will change unless actual change is made. If we continue to do things as they have always been done, we will end up where we have always been. It is time to step into the discomfort and dare to do something different.
So what can we do differently to make genomics more equitable? I propose three areas where we should focus attention to address this important question. First, we must ensure equitable representation in genomic research. Examining 2,511 studies involving nearly 35 million samples from the GWAS Catalog in 2016, Popejoy and Fullerton found that the vast majority (81%) come from individuals of European descent, with only 5% coming from non-Asian minority populations5. This has created an ‘information disparity’ that has an impact on the reliability of clinical genomic interpretation for under-represented minorities6. The US National Institutes of Health (NIH) has invested in efforts to increase diversity in genomic research, but to be successful these efforts must be accompanied by serious attention to earning the trust of disadvantaged and historically mistreated populations. This will require, at a minimum, more meaningful engagement, improved transparency, robust systems of accountability, and a commitment to creating opportunities that promote and support a genomics workforce that includes scientists and clinicians from under-represented populations.
It is insufficient to achieve diverse representation in genomic research; however, there must also be equitable access to the fruits of that research. An analysis of the US Centers for Disease Control and Prevention’s 2018 Behavioural Risk Factor Surveillance System found that non-elderly adults from self-identified racial or ethnic minority groups are significantly less likely to see a doctor because of cost than non-elderly white adults7. This finding reflects how the structure and financing of health care in the United States perpetuates inequities and contributes to the larger web of social injustice that is at the heart of the problem. Even when socio-economic factors are controlled for, racial disparities in access to genetic services persist8. Large-scale, sustained research is needed to better understand and actively address the multitude of factors that contribute to this, including issues related to structural racism, mistrust, implicit and explicit bias, a lack of knowledge of genetic testing, and concerns about misuse of genetic information.
Finally, and perhaps most daunting, we must strive to achieve more equitable outcomes from genomic medicine. Many racial and ethnic minorities disproportionately experience chronic disease and premature death compared with white individuals. Disparities also exist by gender, sexual orientation, age, disability status, socio-economic status and geographical location. Health outcomes are heavily influenced by social, economic and environmental factors. Thus, although providing more equitable access to genomic services and ensuring more equitable representation in genomic research are necessary first steps, they are not enough9. Genomics can only be part of the solution if it is integrated with broader social, economic and political efforts aimed at addressing disparities in health outcomes. For genomics to be truly equitable, it must operate within a just health-care system and a just society.
we must strive to achieve more equitable outcomes from genomic medicine
Genome sequencing at population scale
Stacey Gabriel. Twenty years ago, I finished a PhD project that involved laboriously sequencing one gene — a rather complicated one, RET — in a couple of hundred people to catalogue pathogenic variants for Hirschsprung disease. This work required designing primers on the basis of genome sequence data as they were gradually released, amplifying the gene exon by exon (all 20!), running sequencing gels and manually scoring sequence changes. The notion of sequencing the whole genome to catalogue sequence changes was something to wish for in our wildest dreams.
Thanks to great strides in technology and the hard work of geneticists, engineers, epidemiologists and clinicians, much progress has been made; huge numbers of genomes (and exomes) have been sequenced across the world. Disease gene-finding projects such as my graduate work are now done routinely, rather than one gene at a time, using whole-exome or whole-genome sequencing (WGS) in families and affected individuals, enabling the identification of genes and causative mutations in thousands of Mendelian diseases and some complex diseases.
But the real promise of genome sequencing lies in true population-scale sequencing, ultimately at the scale of tens of millions of individuals, whereby genome sequencing of unselected people enables the unbiased, comprehensive study of our genome and the variation therein. It provides a ‘lookup table’ to catalogue disease-causing and benign variants (our ‘allelic series’). The genome sequence should become part of the electronic health record; it is a stable, persistent source of information about a person akin to physical measurements such as weight or blood pressure, exposures such as smoking or alcohol use, and (in many ways better than) self-reported family history.
the real promise of genome sequencing lies in true population-scale sequencing, ultimately at the scale of tens of millions of individuals
What can we learn? What needs to be solved? Even fairly small numbers of genomes aggregated in a consistent and searchable form have enabled a new way to use and interpret genomic data, just in the past couple of years providing a glimpse at the future. Efforts such as gnomAD10 are a start — this database contains data from more than 15,000 genomes and 125,0000 exomes. With this resource, the frequency of genetic variants within populations is readily available. A clinician interpreting the genome of a patient can ask whether a variant has been observed before. The data provide a starting point for assessing the functional impact of classes of genetic variation and the ability to ask questions about ‘missing’ genetic variation where there is constraint.
Coupled with clinical data, building up population-scale databases of genomic plus clinical information will fuel the application of better risk interpretation using polygenic risk scores (PRSs)11. More routine WGS will shorten the ‘diagnostic odyssey’, in which patients suffer through rounds of testing and parents are left uncertain about future reproductive planning. More efficient clinical trials might be built using genomic information. With existing genomic information on all individuals in a health system, trials could be designed in a way that selects individuals most likely to have an event. This enrichment could provide more promising, shorter, smaller and cheaper trial design.
These databases must also rapidly be built in such a way that is representative of the population, representing the actual racial and ethnic diversity, not just what was available as banked sample collections. These are well known to be predominantly European-descent samples and thus preclude application of risk prediction tools in non-white individuals and have limited the ability to find population-specific genetic associations, such as those that have been demonstrated in type 2 diabetes mellitus (T2DM)12.
We have to solve important issues — data sharing, privacy and getting the data to scale. Sharing genomic and clinical data is of key importance to drive forward discovery and our understanding of how to use these data in the health-care setting. To do this well and responsibly, trust must be built and maintained through adherence to the rights of privacy, protection and non-discrimination. Progress is being made through the creation of data platforms and the development of frameworks for data protection and sharing; for example, by the work of the Global Alliance for Genomics and Health (GA4GH).
Several large biobanks are already being established to launch population-scale efforts. The UK Biobank is a vanguard programme that contains genotype data, questionnaire-based health and physical measurements on 500,000 individuals and some linkage to their medical records. Other efforts such as the All of Us research programme have been launched with goals directed at true population-based representation, and biobanks that link genomic data to comprehensive medical records in specific health-care systems (for example, Geisinger) or in specific countries or regions (for example, Estonia and Iceland) are also under way.
A big piece of this puzzle is generating comprehensive genome sequence data in these programmes and far beyond. For this aim, large-scale, affordable sequencing is key. No problem, right? Is sequencing not always getting cheaper? The problem is that this assumption is no longer true. We have got to where we are today because for a long time, from 2008 to 2013, sequencing costs dropped exponentially. However, in recent years, the sequencing cost curve has flattened, as is apparent in publicly reported cost estimates provided by the US National Human Genome Research Institute13. The cost per megabase of sequence data has remained largely unchanged since around 2016, hovering around a list price of US$0.01 per megabase, which translates to a US$1,000 genome. Gone are the days of our field touting the impressive decrease of cost in comparison with Moore’s law, and this development is worrying.
Some discounting does happen at considerable volume, and whole genomes can be priced in the range of US$500 to US$700. However, large projects (more than 500,000 samples) sequenced at these prices are few and far between, and are generally dependent on pharmaceutical or biotech funding, which can bring with it restrictions on data sharing. It is my belief that a fivefold to sevenfold reduction in total costs is needed to unlock more sequencing at the population scale and, ultimately, for genome sequencing to be more widely applied in the health-care setting. At US$100 per genome, the cost represents less than 1% of the annual average health-care expenditure per person in the United States, and a genome sequence is a one-time investment that can be referenced again and again over the entire lifespan of a person. Getting that cost curve down will be important to inspire health-care systems to adopt genome sequencing routinely.
I see three main drivers that will get us to US$100 per genome: innovation, scale and competition.
-
1.
Innovation. Generating sequence data requires multiple components, and there are multiple areas ripe for innovation. Sample preparation can be improved through more efficient methods that decrease the labour required, or miniaturization can decrease the cost of the reagents used in library preparation. Developments to decrease data processing costs are also ripe for innovation. Recently, we showed that processing using optimized computing power lowered the time and cost of creating a sequence file by ~50% (S.G., unpublished observations). While decreases in the costs of sample preparation and data processing are important, they represent a small component of the total cost. Roughly 70% of the cost of sequencing a human genome is the sequencing reagent (flow cell) and the instrument. Appreciable cost decrease is made possible only by decreasing these marginal costs, as was demonstrated in the period from 2010 to 2014, when flow-cell densities doubled and sequencing cost dropped by an order of magnitude (US$100 per gigabase to US$10 per gigabase).
-
2.
Scale. One component of cost is the fixed cost borne by the sequencing centre or the sequencing vendor. With high scale, centres can become more efficient and offset costs such as the costs of personnel, equipment and facilities. Scale can also result in volume discounting of the reagents, although this process is tightly controlled and approached cautiously depending on overall market dynamics.
-
3.
Competition. Innovation and scale can only achieve so much. The cost of generating the data (the cost per gigabase) dominates and thus must come down considerably. The current market requires alternative options to drive this advance. Presently, the market for short-read sequencing is lacking viable, proven competition that would force flow-cell densities and machine yield to be increased and put pressures on volume discounting. While options for long-read sequencing exist and play a role in particular applications, such as de novo sequencing and structural variant resolution, they are at present far from cost competitive and, therefore, do not apply pressure to bring down the cost of routine WGS.
We need innovation, great economies of scale and/or real competition to come to play in the marketplace. When it comes to sequencing technology, particularly at a large scale, we cannot be complacent and work around the current barriers to realize small gains and one-off wins. This might involve specific types of investment beyond just financial ones; adopting and vetting new technology requires time, creativity, commitment and patience. It is a challenge for our community to take on now. In 5 years’ time, I hope we can look back at the era of the US$100 genome and progress towards real population-scale databases that fuel discovery, enriching our knowledge of the human allelic series and, importantly, the routine use of genomic data in the health-care setting.
A global view of human evolution
Sarah Tishkoff. The past 10 years saw an exponential increase in SNP array and high-coverage WGS data owing to innovations in genomic technologies. It is now possible to generate WGS data from tens of thousands of individuals (for example, GenomeAsia 100K14 and NIH TOPMed15). An increase in medical biobanks with access to electronic health records (for example, the UK Biobank16, the Million Veteran Project17 and BioBank Japan18) is enabling the mapping of hundreds of genetic associations with complex traits and diseases, as well as phenome-wide association studies19 to map pleiotropic associations of phenotypes with genes. The genetic associations identified in these and other studies have been used to calculate PRSs for predicting complex phenotypes and risk of diseases.
Yet despite these advances, as of 2019, nearly 80% of individuals in genome-wide association studies (GWAS) were of European ancestries, ~10% were of East Asian ancestries, ~2% were of African ancestries, ~1.5% were of Hispanic ancestries and less than 1% were of other ancestries20. There is also a strong European bias in genomic reference databases, such as gnomAD and GTEx. These biases limit our knowledge of genetic risk factors for disease in ethnically diverse populations and could exacerbate health inequities20. Furthermore, PRSs that were estimated using European data do not accurately predict phenotypes and disease risk in non-European populations, performing worst in individuals with African ancestry21. The lack of transportability of PRSs across ethnic groups is likely due to differences in patterns of linkage disequilibrium and haplotype structure (resulting in different SNPs tagging causal variants), differences in allele frequencies, gene × gene effects and gene × environment effects. It is also possible that the genetic architecture of complex traits and diseases may differ across ethnic groups owing to different demographic histories and adaptation to diverse environments.
Although there have been initiatives to increase inclusion of ethnically diverse populations in human genomics research (for example, the NIH TOPMed15 and H3Africa consortia), Indigenous populations remain under-represented. Great care must be taken to ensure that genomic research of minority and Indigenous populations is conducted in an ethical manner. This involves establishing partnerships with local research scientists, being sensitive to local customs and cultural concerns, obtaining both community and individual consent, and returning results to communities that participated when possible. In addition, there should be training and capacity building so that genomic research can be conducted locally, where feasible.
A particular area of focus in the future should be developing tools and resources that make genomic data and analyses accessible in low- and middle-income countries. We have to ensure that all people benefit from the genomics revolution and advances in precision medicine and gene editing. Thus, several of the biggest challenges in the next decade will be (1) to increase inclusion of ethnically diverse populations in human genomics research; (2) the generation of more diverse reference genomes using methods that generate long sequencing reads, and haplotype phasing, to account for the large amount of structural variation that likely exists within and between populations; (3) the training of a more diverse community of genomic research scientists; and (4) the development of better methods for accurately predicting phenotypes and genetic risk across ethnically diverse populations and for distinguishing gene × environment effects.
The inclusion of ethnically diverse populations, including Indigenous populations, is also critical for reconstructing human evolutionary history and understanding the genetic basis of adaptation to diverse environments and diets. While there have been a number of success stories for identifying genes of large effect that play a role in local adaptation (for example, lactose tolerance and sickle cell disease (SCD) associated with malaria resistance), identifying signatures of polygenic selection has been considerably more challenging22. Genomic signatures of polygenic adaptation are based on the ability to detect subtle shifts in allele frequencies at hundreds or thousands of loci with minor effect on the phenotype of a complex trait and to determine whether that shift is a result of demography or natural selection. A more daunting challenge arises from the same issues of portability of PRSs described earlier — variants associated with a complex trait may not tag well across ethnic groups and/or the genetic architecture of a trait may differ in different populations. Furthermore, it has recently been shown that uncorrected population stratification can result in a false signal of polygenic selection23. For example, several studies have identified signatures of polygenic adaptation for height across European populations (selection for increased height in northern Europeans and for decreased height in southern Europeans). However, it was recently shown that these results were influenced by population structure that could not be easily corrected using standard approaches, particularly for SNPs below genome-wide levels of significance23. When this analysis was repeated with variants identified in a more homogenous set of individuals of European ancestry from the UK Biobank, these signatures of polygenic adaptation were erased23. Thus, methods for detecting polygenic adaptation that are less biased by population structure and by population ascertainment bias will need to be developed in the future. These studies will also benefit from inclusion of more ethnically diverse populations in GWAS and identification of better tag SNPs as described earlier. A challenge of inclusion of minority populations in GWAS is that sample sizes are often small relative to majority populations. However, the high levels of genetic diversity and extremes of phenotypic diversity observed in some populations, particularly those from Africa, make them particularly informative for GWAS. For example, a GWAS of skin pigmentation in fewer than 1,600 Africans was informative for identifying novel genetic variants that affect skin colour, including a previously uncharacterized gene, MFSD12 (ref.24). Thus, genomic studies in the future must make inclusion of minority populations a priority.
A challenge in both GWAS and selection scans has been the identification of causal genetic variants that directly have an impact on variable traits. Most of these variants are in non-coding regions of the genome. The development of high-throughput approaches, such as massively parallel luciferase expression assays to identify gene regulatory regions and high-throughput CRISPR screens in vitro and in vivo to identify functional variants influencing the trait of interest, will be useful25. There is also a need to better understand cell type-specific variation and gene regulation at the single-cell level, including response to stimuli such as immune, pharmacological and nutrient challenges, in ethnically diverse populations. However, these approaches are still limited by the need to have informative cell lines. This can be particularly challenging to obtain for Indigenous populations living in remote regions. Improvements in the differentiation of induced pluripotent stem cells (iPS cells) into assorted cell types and into organoids will be important for facilitating functional genomic studies. Establishment of iPS cells and organoids from diverse non-human primate species will also be informative for comparative genomic studies to identify the evolution of human-specific traits such as brain development and cognition. However, iPS cell-derived cells may not accurately reflect the impact of mutations acting on developmental phenotypes, which will require development of more efficient in vivo approaches in model organisms.
Perhaps the biggest revolution in the study of recent human evolutionary history has been the development of methods that make it feasible to sequence and/or obtain targeted genotypes from ancient DNA samples. The generation of high-coverage reference genomes for archaic hominid species such as Neanderthals and Denisovans, located in Eurasia, has made it feasible to identify archaic introgressed segments within the genomes of non-Africans. Some of these regions have been shown to play a role in adaptive traits such as adaptation to high altitude and immune response26. Furthermore, there has been an explosion of studies of ancient genetic variation in Europeans within the past 30,000 years that has demonstrated a much more complex model of the peopling of Europe, and the recent evolution of adaptive traits, than previously known from the archaeological record or from studies of modern populations27. The biggest challenge has been the inability to get high-quality ancient DNA from regions with a tropical climate, such as Africa and Asia. While there has been success in analysing DNA samples as old as 15,000 years in Africa, which has been informative for tracing recent migration and admixture events28, the lack of a more ancient African reference genome makes it very challenging to detect archaic introgression, which currently relies on statistical modelling approaches. Thus, the biggest challenge in the next 10 years will be the successful sequencing of ancient DNA more than 20,000 years old from all regions of the world, so that we may have a better understanding of the complex web of population histories from across the globe.
African genomics — the next frontier
Ambroise Wonkam. To fully meet the potential of global genetic medicine, research into African genomic variation is a scientific imperative, with equitable access being a major challenge to be addressed. Studying African genomic variation represents the next frontier of genetic medicine for three major reasons: ancestry, ecology and equity.
On the basis of a ‘pan-genome’ generated from 910 individuals of African descent, at least 300 million DNA variants (10%) are not found in the current human reference genome29, and 2–19% of the genome of ancestral Africans derives from poorly investigated archaic populations that diverged before the split of Neanderthals and modern humans30. Neanderthal genome contributions make up ~2% of the genome in present-day Europeans and are enriched for variations in genes involved in dermatological phenotypes, neuropsychiatric disorders and immunological functions31. Once technical challenges in sequencing poor-quality DNA have been overcome and approaches to investigate the genomic contribution of African archaic populations have been refined, it is likely that associations between variants in ancient African DNA and human traits or diseases will be found, providing insights that can benefit modern-day humans.
As a consequence of the 300,000–500,000 years of genomic history of modern humans in Africa, ancestral African populations are the most genetically diverse in the world. By contrast, there is an extreme genetic bottleneck, resulting in much less variation, in all non-African populations who evolved from the thousands of humans who migrated out of Africa approximately 70,000 years ago. Current PRSs, which aim to predict the risk for an individual of a specific disease on the basis of the genetic variants that individual harbours, exhibit a bias regarding usability and transferability across populations, as most PRSs do not account for multiple alleles that are either limited or of high frequency among Africans. A GWAS on the genetic susceptibility to T2DM identified a previously unreported African-specific significant locus, while showing transferability of 32 established T2DM loci32. In addition, nonsense mutations found commonly among Africans in PCSK9, which are rare in Europeans33, are associated with a 40% reduction in plasma levels of low-density lipoprotein, supporting PCSK9 as a target for dyslipidaemia therapeutics. In the largest GWAS meta-analysis for 34 complex traits, conducted in 14,345 Africans, several loci had limited transferability among cohorts34, further illustrating that genomic variation is highest among Africans compared with other populations. As a consequence, linkage disequilibrium is lower in Africans, which improves fine mapping and identification of causative variants. Indeed, while only 2.4% of participants in large GWAS are African individuals, they account for 7% of all associations35. Moreover, whole-exome sequencing of nearly 1,000 African study participants of Xhosa ancestry with schizophrenia found very rare damaging mutations in multiple genes36, a finding that could be replicated in a Swedish cohort of 5,000 individuals. In comparison, results for the Xhosa cohort yielded larger effect sizes, which shows that for the same number of cases and controls, the greater genetic variation in African populations provides more power to detect genotype–phenotype relationships. Therefore, millions of African genomes must be sequenced, with genotyping and analysis tools optimized for their interrogation.
Greater availability of African genomes will improve our understanding of genomic variation and complex trait associations in all populations but will also support research into common monogenic diseases. The discovery of a single African origin of the SCD mutation, about 5,000–7,000 years ago, not only suggested recent migration and admixture events between Africans and Mediterranean and/or Middle Eastern populations but also enhanced our understanding of genetic variation in general as well as its potential impact on haemoglobinopathies37. For example, variants in the HBB-like gene cluster linked with high levels of fetal haemoglobin have been associated with less severe SCD; because the level of fetal haemoglobin is under genetic control, it is amenable to therapeutic manipulation by gene editing38. Moreover, knowledge of an individual’s genetic variants can have an impact on secondary prevention of and treatment strategies for SCD. For example, variants in APOL1 and HMOX1 and co-inheritance of α-thalassaemia are associated with kidney dysfunctions39; stroke in SCD is associated with targeted genetic variants used in a Bayesian model; and overall SCD mortality has been associated with circulating transcriptomic profiles. It is estimated that 75% of the 305,800 babies with SCD born each year are born in Africa; SCD in Africa will serve as a model for understanding the impact of genetic variation on common monogenic traits and help to illustrate the multiple layers of genomic medicine implementation.
Greater availability of African genomes will improve our understanding of genomic variation and complex trait associations in all populations
Exploring African genomic diversity will also increase discovery of novel variants and genes for rare monogenic conditions. Indeed, allelic and locus heterogeneity display important differences in African individuals compared with other populations; for example, mutations in GJB2 account for nearly 50% of cases of congenital non-syndromic hearing impairment among Eurasians but are nearly non-existent in Africans, and there is evidence that novel variants in hearing impairment-associated genes are more likely to be found in Africans than in populations of European or Asian ancestries40. Higher fertility rate, consanguinity practices and regional genetic bottlenecks will improve novel gene discovery for monogenic diseases in Africa, as well as disease–gene pair curation, and will address existing challenges surrounding database biases and inference of variant deleteriousness, which have led to the misclassification of variants.
Differential population genomic variant frequencies are shaped by natural evolutionary selection as an adaptation to environmental pressures. The African continent follows a North–South axis, which is associated with variable climates and biodiversity, both motors of natural selection. This specific African ecology has shaped genetic variation accordingly, which can have a detrimental or positive impact on health. Obvious examples are variants that cause SCD but confer resistance to malaria37, APOL1 variants that are protective against trypanosomes (the parasites that cause sleeping sickness)41 and variants of OSBPL10 and RXRA that protect against dengue fever42. Unfortunately, APOL1 variants also increase susceptibility to chronic kidney disease in populations of African ancestry39,41. A better understanding of the functional impact of genetic variants specific to African populations, particularly those that have been selected under environmental pressure, and the way they interact with each other is needed and will have a positive impact on genetic medicine practice. Moreover, immunogenetic studies among Africans will further our understanding of natural selection and responses to emerging infectious diseases, such as COVID-19.
The scientific imperative of genomic research of African populations is expected to enhance genetic medicine knowledge and practice in Africa but will face the challenges of overburdened and under-resourced public health-care systems, and often absent ethical, legal and social implication frameworks43, requiring international collaboration to be managed. Developing an African genomics workforce will be necessary to meet the major need for research across the lifespan for cohorts of millions of individuals with complex or monogenic diseases. Such endeavours can thrive on the foundation of recently established initiatives such as H3Africa. Indeed, equitable access for Africans is essential if African genomics is to reach its full potential as the next frontier of global genetic medicine.
Decoding multifactorial phenotypes
Aravinda Chakravarti. We live in a time of great technological progress in genomics and computing. And we live in a time when ‘genetics’ is a household word, with a public increasingly adept at understanding its relevance to their own lives. Not surprisingly, the study of genetics is being reinvented, rediscovered and reshaped, and we are beginning to understand the science of human heredity at a resolution that was impossible before.
The most significant genetics puzzle today, in my view, is the dissection of ‘family resemblance’ of complex phenotypes, both for intellectual (raison d'être of genetics) and practical (disease diagnosis and therapy) reasons. We have long known that family resemblance arises from shared alleles, declining as genetic relationship wanes, but the precise molecular components and composition of this resemblance are still poorly understood. At the turn of the twentieth century, the components were a matter of bitter and acrimonious debate44 between the ‘Mendelians’ and the ‘Biometricians’, until the opposing views were reconciled by Ronald Fisher’s 1918 analysis45 that complex inheritance could be explained through segregation of many genes, each individually Mendelian. In 1920, its publication delayed by World War I, this notion was elegantly demonstrated by the experimental studies of Altenburg and Muller using truncate wing, an “inconstant and modifiable character”46 in Drosophila.
Fisher’s model assumed an infinite number of genes additively contributing to a trait, with common genetic variation at each component locus comprising two alleles that differ only slightly in their genetic effects45; these genetic assumptions were quite contrary to what was then known44. Throughout the past century, this view matured, as segregation analyses of human phenotypes taught us that — beyond the effects of some major genes — most trait variation was polygenic, modulated by family-specific and random environmental factors47. Today, we have empirical evidence from GWAS, which use dense maps of genetic variants on hundreds of thousands of individuals measured for many traits and diseases, that the genetic architecture of most multifactorial traits is from common sequence variants with small allelic differences at thousands of sites across the genome48. This replacement of a pan-Mendelian view with a pan-polygenic view of traits is one of the most important contributions of genomics to genetics. Unfortunately, this mapping success has not clarified the number of genes involved, the identity of those genes or how those genes specify the phenotype. Indeed, some have concluded that many of the mapped GWAS loci are unrelated to the core biology of each phenotype49. Thus, for a deeper understanding, we need radically different approaches to understand complex trait biology in contrast to merely expanding GWAS in larger and larger samples.
for a deeper understanding, we need radically different approaches to understand complex trait biology
Yet, the most significant biology to emerge from GWAS is that most of the likely trait-causing variants fall outside coding sequences, in regulatory elements, most frequently enhancers50,51. This important finding has uncovered four new genetic puzzles. First, the non-coding regulatory machinery is vast; how much of this regulation is compromised, and how does it affect phenotypes? Second, regulatory changes affect RNA expression at many genes and protein expression at others; how does a cell ‘read’ these numerous changes as specific signals? Third, how is this coordinated expression response translated into cellular responses affecting phenotypes? Fourth, if specific environmental factors affect the same phenotype, which components do they dysregulate? In my opinion, we need to answer these questions for specific traits and diseases to truly understand their polygenic biology. Finally, these explanations must also answer the question of why some traits are decidedly Mendelian whereas others are not.
The questions of tomorrow will need to focus on four areas: the biology of enhancers and the transcription factors that bind them51; the effect of genetic variation in enhancers50; gene regulatory networks (GRNs) that regulate expression of multiple genes52; and how GRN changes lead to specific cellular responses53. Despite many advances, the number of enhancers regulating expression of a specific gene remains unknown. How many enhancers are cell type specific versus ubiquitous? How many are constitutive rather than stage specific? And do they act additively or synergistically in gene expression? Additionally, which cognate transcription factors bind these enhancers, with what dynamics and how are they regulated54? These details of a gene’s ‘enhancer code’ are critical for assessing its relative effect on a trait. Next, how does enhancer sequence variation affect a gene’s activity? Does such variation affect transcription factor binding only or its interaction with the promoter? Is the enhancer variant’s effect evident in all cellular states or only some? Is variation in only one enhancer sufficient to alter gene expression, or are multiple changes in multiple elements necessary?
Additional critical questions include which genes are involved in the core pathway underlying a trait, and how do we identify them49? Elegant work has shown how genes are regulated within integrated modular GRNs, whereby one gene’s product is required in a subsequent step by another gene, with feedback interactions52. These GRNs comprise elements from the genome, transcriptome and proteome, with rate-limiting steps that require regulation. As our work on Hirschsprung disease has shown50,53, a GRN is composed of core genes, is the logic diagram of regulation of a major rate-limiting cellular step, is enriched in coding and enhancer disease variants with disease susceptibility scaling with increasing number of variants, and with disease resulting from effects on its rate-limiting gene product53. That is, the GRN integrates the expression of multiple genes. Finally, we need to understand how GRN changes alter cell properties and behaviour. I speculate that rate-limiting steps in GRNs are major regulators of broad cell properties, be they differentiation, migration, proliferation or apoptosis, the cellular integrator of GRN variation. Thus, genetic variation across the genome affects enhancers dysregulating many genes, but only when they dysregulate GRNs through rate-limiting steps do they affect cell and tissue biology55. This offers the promise of a mechanistic understanding of human polygenic disease.
The way forward for complex trait biology, including disease, is to shift our approach from reverse to forward genetics, using genome-wide approaches to cell type-specific gene perturbation. I believe we can construct cell-type GRNs en masse, inclusive of their enhancers, transcription factors and feedback or feedforward interactions, to then assay functionally defined variation in phenotypes. But, even this approach will be insufficient. We need to test our success by solving at least a few complex traits completely and demonstrating their veracity using a synthetic biology approach to recapitulate the phenotype in a model system; similarly to the field of chemistry, analysis has to be followed by de novo synthesis. Our genomic technologies are getting up to the task to enable this advance; as geneticists, are we?
Enhancers and embryonic development
Eileen Furlong. The work of my group sits at the interface of genome regulation and animal development, and there have been many exciting advances in both during the past decade. Developmental biology studies fundamental processes such as tissue and organ development and how complexity emerges through the combined action of cell communication, movement and mechanical forces. After the discovery that differentiated cells could be reprogrammed to a naive embryonic stem cell-like state, the past decade has witnessed an explosion in in vitro cellular reprogramming and differentiation studies. Organoids are a very exciting extension of this. The extent to which these fairly simple systems can self-organize and generate complexity56 is one of the unexpected surprises of the past 5–10 years. The buzz around stem cells has also renewed interest in cellular plasticity in vivo and has uncovered an unexpected degree of transdifferentiation and dedifferentiation57. In the mouse heart, for example, cardiomyocytes dedifferentiate and proliferate to regenerate heart tissue when damaged within the first week after birth58.
Our understanding of the molecular changes that accompany differentiation has hugely advanced owing to the jump in scale, resolution and sensitivity of next-generation sequencing technologies over the past decade. This has led to a flood of studies in embryonic stem cells, iPS cells and embryos that revealed new concepts underlying genome regulation by measuring transcript diversity, transcription factor occupancy, chromatin accessibility and conformation, and chromatin, DNA and RNA modifications. The future challenge will be to connect this information to the physical characteristics of cells and how they form complex tissues. New technologies that solve many challenges of working with embryos will help, including CRISPR to engineer genomes, optogenetics to perturb proteins, lattice light-sheet and selective plane illumination microscopy to image processes in vivo, and low-input methods to overcome issues with scarce material. Particularly exciting to me are recent advances in single-cell genomics, which, although they are in their early days, will dramatically change the way we study embryogenesis. Many new insights have already emerged, including the discovery of unknown cell types and new developmental trajectories for well-established cell types. Even the concept of ‘cell identity’ has come into question.
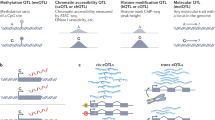
Cell identities are largely driven by transcription factors, which act through cis-regulatory elements called ‘enhancers.’ One of the most exciting unsolved mysteries, in my opinion, is how enhancers relay information to their target genes. The textbook view of enhancers is of elements with exclusive function that regulate a specific target gene through direct promoter interactions, which occur sequentially if multiple enhancers are involved. However, emerging concepts in the past decade question many of these ‘dogmas’. Some enhancers have dual functions, whereas others may even regulate two genes. Enhancer–promoter communication is now viewed in the light of spatial genome organization, including topologically associating domains (TADs) and membraneless nuclear microcompartments (that is, hubs or condensates)59. Being present within the same TAD likely increases the frequency of enhancer–promoter interactions, but how a specific enhancer finds its correct promoter within a TAD, or when TADs are rearranged60,61, remains a mystery. Hubs or condensates are dynamic microcompartments62 that contain high local concentrations of proteins, including transcription factors and the transcriptional machinery. One potential implication of condensates is that enhancers may not need to ‘directly’ touch a gene’s promoter to regulate transcription — rather, it may be sufficient to come in close proximity within the same condensate. Presumably, once proteins reach a critical concentration, transcription will be initiated. While this model fits a lot of emerging data, there are still many open questions. What is the required distance between an enhancer and a promoter to trigger transcription? Does this distance differ for different enhancers63 depending on their transcription factor–DNA affinities? Do different chromatin environments64 influence the process? At some loci, mutation of a single transcription factor-binding site in a single enhancer can have dramatic effects on gene expression and development. It is difficult to reconcile such cases with a shared condensate model, as other proteins bound to the enhancers and promoter should still phase separate. By contrast, there are many examples where mutation of a single transcription factor-binding site, or even an entire enhancer, has minimal impact on the expression of a gene. These observations suggest that there may be different types of loci, with requirements for different types of chromatin topologies and local nuclear environments, which will be important to tease apart in the coming years.
The genetic dissection of model loci in the 1990s and the first decade of the twenty-first century led to much of our understanding of how genes are regulated. The power of genomics in the past few decades has captured regulatory information for all genes genome-wide, providing more unbiased views of regulatory signatures, leading to new models of gene regulation. What is missing is empirical testing at a large scale. A major challenge is to move to more systematic in vivo functional dissection in organisms. CRISPR-based pooled screens have advanced the interrogation of genomic regions in cell culture systems. However, scaling functional assays in embryos remains a huge challenge. The task is enormous — even long-standing model organisms, such as Drosophila and mice, lack knockout strains for all protein-coding genes, and the number of regulatory elements is at least an order of magnitude higher. There has been little progress in developing scalable methods to quantify the contribution of a transcription factor’s input to an enhancer’s activity, and gene expression, in embryos. More systematic unbiased data will uncover more generalizable regulatory principles, increase our predictive abilities of gene regulation and developmental programmes, and enhance our understanding of the impact of genetic variation.
A major challenge is to move to more systematic in vivo functional dissection in organisms
Perhaps the most promising and exciting prospects in the coming years are to use single-cell genomics, imaging and the integration of the two to dissect the amazing complexity of embryonic development. Single-cell genomics can reveal information about developmental transitions in a way that was unfeasible before. When combined with temporal information, such data can reconstruct developmental trajectories65,66 and identify the regulatory regions and transcription factors likely responsible for each transition67. The scale and unbiased nature of the data, profiling tens to hundreds of thousands of cells, provides much richer information than anyone envisaged just 5 years ago, bringing a new level of inference and causal modelling. The ability to measure single-cell parameters in situ (called ‘spatial omics’) will be transformative in the context of developing embryos to reveal the functional impact of spatial gradients, inductive signals and cell–cell interactions, and to move to digital 4D embryos. Combining these approaches with genetic perturbations holds promise to decode developmental programmes as they unfold. Will this bring us to a predictive understanding of the regulatory networks driving embryonic development during the next decade? ‘Simple’ model organisms are a fantastic test case to determine the types and scale of data required and to develop the computational framework to build predictive networks. The systematic functional dissection of gene regulation and true integration of single-cell genomics with single-cell imaging will bring many exciting advances in our understanding of the programmes driving embryonic development in the coming years.
Spatial multi-omics in single cells
Barbara Treutlein. Incredibly, the first single-cell transcriptome was sequenced just over a decade ago68! Since this milestone, transcriptomes of millions of cells have been sequenced and analysed from diverse organisms, tissues and other cellular biosystems, and these maps of cell states are revolutionizing the life sciences. The technologies and associated computational methods have matured and been democratized to such an extent that nearly all laboratories can apply the approach to their particular system or question.
Of course, the transcriptome is not enough, and protocols have already been developed to measure chromatin accessibility, histone modifications, protein abundances, cell lineages and other features linked to genome activity in single cells69. Currently, many studies use dissociation-based single-cell genomics methods, where the spatial context is disrupted to facilitate the capture of single cells for downstream processing. Methods are improving to measure genomic features in situ70, as well as to computationally map features to spatial contexts71,72. The stage is set for the next phase of single-cell genomics, where spatial registration of multimodal genome activity across molecular, cellular and tissue or ecosystem scales will enable virtual reconstructions with extraordinary resolution and predictive capacity. These virtual maps will rely on multi-omic profiling of healthy and perturbed tissues and organisms, which presents major challenges and opportunities for innovation.
Cell throughput remains a challenge, and it is unclear what role dissociation-based single-cell sequencing protocols will play in the future. These protocols are fairly easy to implement, and laboratories around the world can execute projects with tens of thousands of cells analysed per experiment. However, there are scenarios in which measuring millions of cells per experiment would be desired, such as in perturbation screens. Combinatorial barcoding methods push cell-throughput boundaries73; however, it is unclear how to scale full transcriptome sequencing economically to millions of cells using current sequencing technologies. ‘Compressed sensing’ modalities — whereby a limited, selected and/or random number of features are measured per cell, and high-dimensional feature levels are recovered through inference or similarity to a known reference — provide an interesting possibility to increasing cell throughput74.
Most single-cell transcriptome protocols are currently limited to priming the polyadenylation track present on all cellular mRNAs; however, this approach leads to biased sampling of highly expressed mRNAs. Clever innovations for random or targeted RNA enrichment could be a way to build up composite representations of cell states. Image-based in situ sequencing methods provide a means for increasing the number ofcells measured per experiment, as millions of cells can be imaged without a substantial increase in financial cost, although imaging time is a limiting factor. There remains a lot of room for experimental and computational optimizations to measure the transcriptome, random barcodes, DNA conformations and protein abundances from the micrometre scale to the centimetre scale spatially, and it will be interesting to see how methods for spatial registration advance over the next 5 years.
Currently, most high-throughput measurements are performed on cell suspensions or on intact tissues using one modality. That said, studies are emerging that measure several features from the same cell; for example, mRNA and chromatin accessibility75 or mRNA and lineage76. To build virtual maps, independent measurements from different cells can be integrated with use of data integration tools77, although it can be difficult to align cell states across modalities in particular in developing systems. Therefore, the ultimate goal is to directly measure as many features as possible (for example, RNA, lineage, chromatin, proteins and DNA methylation) in the same cell78, ideally with spatial resolution. Furthermore, combining genetic and pharmacological perturbation screens with single-cell multi-omic measures will be informative to understand cell state landscapes and underlying regulatory networks for each cell type. The CRISPR–Cas field continues to develop creative tools for precise single-locus editing and other manipulations79, and incorporation of these toolkits with single-cell sequencing readouts will certainly bring new mechanistic insight.
Life forms are inherently dynamic, and each cell has a story to tell. Static measurements do not provide sufficient insight into the mechanisms that give rise to each cell state observed in a tissue. Computational approaches to stitch together independent measurements across time can be used to reconstruct potential histories; however, these are indirect inferences. Long-term live imaging in 2D cultures using confocal microscopy and in 3D tissues using light-sheet microscopy provides morphology, behaviour, location and, in some cases, molecular information on the history of a cell. Indeed, such long-term imaging experiments revealed that cell fates or states can be predicted from cell behaviour across many generations80. Cell tracking combined with end point single-cell genomics experiments can help to understand how cell states came to be; however, these experiments lack molecular resolution of the intermediates. There are strategies using CRISPR–Cas systems to capture highly prevalent RNAs inside cells at given times and insert these RNAs into DNA for storage and subsequent readout81. Together with live tracking and end-point single-cell genomics, such methods could provide unprecedented insight into cell histories.
My vision is that the emerging technologies described above can be applied to human 2D cell culture and 3D organoid biosystems to understand human development and disease mechanisms. My team and others are working to build virtual human organs that are based on high-throughput, multimodal single-cell genomics data. Organoid counterparts provide opportunities to perturb the system and understand lineage histories. Together, the next generation of single-cell genomics methods and human organoid technologies will provide unprecedented opportunities to develop new therapies for human disease.
the next generation of single-cell genomics methods and human organoid technologies will provide unprecedented opportunities
Unravelling the layers of the epigenome
Alexander Meissner. Around 1975, the idea that 5-methylcytosine could provide a mechanism to control gene expression gained traction, despite little knowledge of its genomic distribution or the associated enzymes82. With similarly limited genomic information or knowledge of the players involved, the histone code hypothesis was put forward in 2000 to explain how multiple different covalent modifications of chromatin may be coordinated to direct specific regulatory functions83. Tremendous progress has been made since, and the list of core epigenetic regulators that have been discovered and characterized seems largely complete84.
DNA sequencing has continued to dominate the past decade and contributed to an exponential growth of genome-wide maps of all layers of regulation. In the early days, individual CpG sites could be measured by restriction enzymes, whereas now we have generated probably well over a trillion cytosine methylation measurements. An equally astonishing number of genome-wide data sets have been collected for transcriptomes, histone modifications, transcription factor occupancy and DNA accessibility. Furthermore, the number of single-cell transcriptome and epigenome data sets continues to grow at an unprecedented pace.
On the basis of this overabundance of data across many normal and diseased cell states, for instance, we now clearly understand the non-random distribution of cytosine methylation across many different organisms. These maps have helped to refine our understanding of its relationship to gene expression, including the realization that only a few promoters are normally controlled via this modification, whereas gene bodies are actively targeted, and most dynamic changes occur at distal regulatory sites. Similar insights exist for many core histone modifications, and, in general, we have an improved appreciation of the epigenetic writers, readers and erasers involved. Over the past decade, we have seen substantially integrated and multilayered epigenomic analyses that provide a fairly comprehensive picture of epigenomic landscapes, including their dynamics across development and disease.
Additional innovation is now needed around data access and sharing. As noted, there is certainly no shortage of data, but to enable individual researchers to generate and verify hypotheses quickly improved tools are required to access and browse these data. Over the past decade, large coordinated projects such as ENCODE, the Roadmap Epigenomics Project and Blueprint Epigenome have initiated such efforts, but it remains a reality that data are not at everyone’s fingertips quite yet.
Moreover, despite decades of steady and recently accelerated progress, many important questions remain regarding the molecular coordination and developmental functions of these epigenetic modifications. For instance, cytosine methylation at gene bodies has been preserved for more than a billion years of evolution and yet its precise function is still under investigation. How and why did genomic methylation switch to a global mechanism in vertebrates compared with the selected methylation observed in invertebrates? What is the precise function of this modification in each of its regulatory contexts, and how are its ubiquitously acting enzymes recruited to specific sites in the genome? The latter is particularly timely given recent observations that enhancers, but also some repetitive elements, show ongoing recruitment of both de novo methylation and demethylation activity. Moreover, extraembryonic tissues show redirected activity that shares notable similarities with the long observed altered DNA methylation landscape found across most cancer types85. Lastly, it is abundantly clear that DNA methylation is essential for mammalian development; but despite us knowing this for nearly three decades, it is not clear how and why developing knockout embryos die. The specific developmental requirements are also largely true for many histone-modifying enzymes; however, it remains incompletely understood how exactly these modifications interact to support gene regulation.
A decade ago it seemed likely that we would answer questions such as these using newly gained sequencing power as a potent tool for generating hypotheses. However, for the most part, epigenomic analyses have expanded a highly valuable, but still largely descriptive, understanding of numerous epigenetic layers. So one may ask, what is different now and why should we expect to answer these questions in the coming years?
Technological innovation has always played a key role in biology, and some broadly applicable, recent breakthroughs will enable us to drive progress in the coming years. These include the transfer of the bacterial innate immunity CRISPR–Cas system as a universal genome-targeting tool86 as well as for base editing, epigenome editing and various genome manipulations. Similarly, new fast-acting endogenous protein degradation systems have been developed that further enhance our ability to probe for precise function87. The past decade also saw major improvements in imaging technologies as well as cell and molecular biology, moving from the 2D space into the 3D space with both organoid cell culture models88 and chromosome conformation capture approaches for exploring nuclear organization89.
Another major shift included the reappreciation that membraneless organelles are a widespread mechanism of cellular organization90. In particular, there have been many advances in our understanding of how condensates form and function, including for transcriptional regulation. Together with known properties of modified histones on DNA and the fact that many epigenetic regulators also contain intrinsically disordered regions, it is reasonable to assume that these physical properties will have a major impact on our understanding of chromatin. Importantly, changes in topology have been linked to disease91, and similar connections have been reported recently for condensates92. This will likely be an exciting area to follow in the coming years.
there have been many advances in our understanding of how condensates form and function, including for transcriptional regulation
Lastly, our research continues to be more and more reliant on multidisciplinary skills, with mathematics, physics, chemistry and computer science playing an ever-more central role in biology, which will require some rethinking in training and institutional organization to accomplish our goals. Going forward, we will need more functional integration, which in part due to the aforementioned selected discoveries is now very tractable. In particular, more refined perturbation of gene activity, which for many chromatin regulators should be separated into catalytic and regulatory functions, together with readouts at multiple levels of resolution will bring us closer to the insights needed. We recently exemplified this with a pipeline that explores epigenetic regulator mutant phenotypes at single-cell resolution93. From these studies, we may be able to understand how epigenetic regulators interact with the environment to influence or protect the organismal phenotype, connecting detailed molecular genetics to classical theories of epigenetic phenomena.
As we approach the 100-year anniversary of the detection of 5-methylcytosine in DNA94, it seems we can hope to declare at least for some layers of the epigenome that we fully understand the rules under which they operate. This may enable the exploration of more precise therapeutic interventions, for instance by redirecting chromatin modifiers rather than blocking their universal catalytic activities, which are shared between normal and diseased states. Of course, looking back at predictions made just 10 years ago95, one should expect many additional unforeseen advances that are just as difficult to predict now as they were back then.
Long non-coding RNAs: a time to build
Howard Chang. Long non-coding RNAs (lncRNAs) are the dominant transcriptional output of many eukaryotic genomes. Although studies over the past decade have revealed diverse mechanisms and disease implications for many lncRNAs, the vast majority of lncRNAs remain mysterious. The fundamental challenge is that we lack the knowledge to systematically transform lncRNA sequence into function. Progress in the next decade may come from a paradigm shift from ‘reading’ to ‘writing’ lncRNAs.
Gene regulation was once thought to be the exclusive province of proteins. Intense efforts for disease diagnosis and treatment focused almost entirely on protein-coding genes and their products, ignoring the vast majority of the genome. Even at the time of the completion of the Human Genome Project, only a handful of functional lncRNAs were known that silenced the expression of neighbouring genes. Thus, it was widely believed that the genome contained mostly ‘junk’, which sometimes made RNA as transcriptional noise.
The human genome is currently estimated to encode nearly 60,000 lncRNAs, ranging from several hundred to tens of thousands of bases, that apparently do not function by encoding proteins96. Studies over the past decade discovered that many lncRNAs act at the interface between chromatin modification machinery and the genome. Specific lncRNAs can act as guides, scaffolds or decoys to control the recruitment of specific chromatin modification enzymes or transcription factors to DNA or their dismissal from DNA97. lncRNAs can activate as well as silence genes, and these RNAs can target neighbouring genes as a function of local chromosomal folding (in cis) or at a distance throughout the genome (in trans). Detailed dissections of individual lncRNAs have revealed that lncRNAs are composed of modular RNA motifs that enable one lncRNA to connect proteins that read, write or erase specific chromatin marks. These findings have galvanized substantial excitement about lncRNAs; laboratories around the world are now investigating the roles of lncRNAs in diverse systems, ranging from control of flowering time in plants to mutations in human genetic disorders.
Nonetheless, the notable progress to date can be viewed as anecdotal — each lncRNA is its own story. When a new lncRNA sequence is recognized in a genome database or RNA profiling experiment, we are still in the dark about what may happen to the cell or organism (if anything) when the lncRNA is removed. Indeed, efforts to ‘read’ lncRNAs have been the dominant experimental strategy over the past two decades. Systematic efforts in the ENCODE, FANTOM and emerging cell atlas consortia have mapped the transcriptional landscape, transcript isoforms and, more recently, single-cell expression profiles of lncRNAs. These powerful data are now combined with genome-scale CRISPR-based methods to inactivate tens of thousands of lncRNAs, one at a time, to observe possible cell defects98,99. However, many challenges remain. Positive hits require further exploratory studies to define possible mechanisms of action, and we lack a principled strategy to combine lncRNA knockouts to address genetic redundancy and compensation.
A potentially fruitful and complementary direction is the pivot from ‘reading’ to ‘writing’ long RNA scripts. On the basis of the systematic dissection of RNA sequences and secondary structures in lncRNAs, we and others believe that the information in lncRNAs resembles that on a billboard (in which keywords and catchphrases are repeated) rather than a finely honed legal document (where every comma counts). Small units of RNA shapes are repeated within lncRNAs to build up the meaning in the lncRNA billboard, but these RNA shapes can be rearranged in different orders or locations without affecting meaning. These insights have allowed scientists to recognize lncRNA genes from different species that perform the same function even though the primary sequences bear little similarity100. Moreover, investigators were able to strip down lncRNAs to their essential ‘words’, composed of these key repeating shapes and one-tenth the size of the original lncRNA, which still functioned in vivo to control chromatin state over a whole chromosome100,101. Finally, it is now possible to successfully create synthetic lncRNAs. By adding RNA shapes to carefully chosen RNA templates, investigators are starting to create designer lncRNAs that can regulate chromatin in vivo100, suffice to partly rescue the physiological lncRNA gene knockout102, or target RNAs to specific cytotopic locations within the cell103,104.
The shift from reading to writing lncRNAs will challenge us on the technical front, leading to potential transformative technologies. Current technologies for massively parallel reporter gene assays are built on short sequence inserts. A plan to build tens of thousands of synthetic lncRNAs will require accurate long DNA or RNA synthesis. These designer sequences will need to be placed into the appropriate locations in the genome and controlled to have proper developmental expression, splicing pattern and RNA chemical modifications. Landmark studies using the XIST lncRNA, which normally silences the second X chromosome in female cells, to silence the ectopic chromosome 21 in Down syndrome cells highlight the biomedical promise of such an approach105.
As the field develops technologies for large-scale creation and testing of synthetic lncRNAs, we can rigorously test our understanding of the information content in the language of RNA sequences and shapes. The next decade promises to be an exciting time for building non-coding RNAs and to create entirely new tools to manipulate gene function for biology and medicine.
FAIR genomics to track tumorigenesis
Núria López-Bigas. Cancer research is one of the fields that has probably benefited the most from the technological and methodological advances of genomics. In the span of less than two decades, the field has witnessed an incredible boost in the generation of cancer genomic, epigenomic and transcriptomic data of patients’ tumours, both in bulk and more recently at the single-cell level. My dream as a cancer researcher is to have a full understanding of the path that cells follow towards tumorigenesis. Which events in the life of an individual, a tissue and a particular cell lead to the malignant transformation of some cells? Of course I do not expect to have a deterministic answer, as this is not a deterministic process. Instead we should aim for a quantitative or probabilistic understanding of the key events that drive tumorigenesis. We have solid epidemiological evidence showing that smoking increases the probability of lung cancer, exposure to the Sun raises the probability of developing melanoma and some anticancer treatments increase the probability of secondary neoplasms. But which specific mechanisms at the molecular and cellular levels influence these increases?
One first clear goal of cancer genomics is to catalogue all genes involved in tumorigenesis across different tissues. Although this is a daunting task, it is actually feasible106. By analysing the mutational patterns of genes across tumours, one can identify those with significant deviations from what is expected under neutrality, which indicates that these mutations provide a selective advantage in tumorigenesis and are thus driver mutations. We can imagine a future in which through the systematic analysis of millions of sequenced tumour genomes this catalogue or compendium moves closer and closer to completion. For this to happen, not only do we need genome sequencing to expand — this process is already in motion in research, clinical settings and the pharmaceutical industry — but more importantly the resulting data must be made FAIR (findable, accessible, interoperable and reusable)107. To this end, consortia and initiatives that promote, catalyse and facilitate the sharing of genomic data, such as the Beyond 1 Million Genomes consortium, the GA4GH or the cBioPortal for Cancer Genomics, are necessary.
Of note, cataloguing genes and mutations involved in cancer development, albeit a very important first step, is still far from the final goal of understanding how and under which conditions they drive tumorigenesis. Framing cancer development as a Darwinian evolutionary process helps me to navigate the path towards this final objective. As is true of any Darwinian process, its two key features are variation and selection. Thanks to the past 15 years of cancer genomics, we now have a much better grasp of the origin of somatic genetic variation between cells across different tissues. The study of the variability in the number, type and genomic distribution of mutations across tumours provides a window into the life history of cells across the somatic tissues of an individual108,109. In addition, recent studies sequencing the genome of healthy cells in different tissues110,111,112 have shown that mutations accumulate in hundreds and thousands in our cells in normal conditions over time. These studies have also detected positive selection in some genes across healthy tissues. Hence, positive selection is a pervasive process that operates not only in tumorigenesis but also in healthy tissues, where it is a hallmark of somatic development of skin, oesophagus, blood and other tissues. Take, for example, clonal haematopoiesis: it results from a continuous Darwinian evolutionary process in which over time (with age) some haematopoietic cells harbouring mutations in certain blood development genes, such as DNMT3A and TET2, outcompete other cells in the compartment113,114. This process is part of normal haematopoietic development. Problems arise only when this process gets out of control, leading to leukaemia in the case of blood, or a malignant tumour in solid tissues. Why is it only in rare cases that this ubiquitous interplay between variation and selection becomes uncontrollable and results in full-blown tumorigenesis? Which events, beside known tumorigenic mutations, drive this process?
we now have a much better grasp of the origin of somatic genetic variation between cells across different tissues
If we have learnt something in recent years, it is that virtually all tumours harbour driver mutations115,116,117, implying that driver genomic events are necessary. However, they are clearly not sufficient for tumorigenesis to occur. So, what are these other triggers of the tumorigenic process? What happens in the lung cells of a smoker or in the haematopoietic cells of a patient treated with chemotherapy that increases their chances to become malignant? Epigenetic modifications and changes in selective constraints, such as evolutionary bottlenecks, for example, at the time of chemotherapy, may be part of the answer.
For the near future, my dream is to see a further increase in FAIR cancer genomics data to help us disentangle the step-by-step game of variation and selection in our tissues that leads to tumorigenesis and likely other ageing-related diseases.
Integrating genomics into medicine
Eran Segal. The past 20 years in genomics have been extraordinary. We developed high-throughput sequencing and learned how to use it to efficiently sequence full genomes and measure gene expression and epigenetic marks at the genome-wide scale and even at the single-cell level118. Using these capabilities, we created unprecedented catalogues of novel genomes, functional DNA elements and non-coding RNAs from all kingdoms of life119. But — perhaps with the exception of cancer120 and gene therapy for some monogenic diseases121 — genomics has yet to deliver on its promise to have an impact on our everyday life. For example, drugs and diagnostics are still being developed in the traditional way, with screening assays to find lead compounds for targets typically arising from animal studies, without involving genomics in any of the steps. Moreover, when the global COVID-19 pandemic hit, the genome of the spreading severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was rapidly sequenced, but why some infected individuals exhibit severe disease and others do not remains unknown.
Indeed, our next challenge is to translate the incredible resources and technologies developed in genomics into an improved understanding of health and disease. This improved understanding should transform the field of medicine to use genomics in its transition to personalized medicine, which promises individualized treatment by targeting the right medication to the right person at the right time on the basis of that person’s unique profile. By continuing to focus on more and more measurements and the creation of more atlases and catalogues, we run the danger of drowning in ever-growing amounts of data and correlative findings. Walking down this path can lead to an endless endeavour, as bulk measurements can always be replaced with single-cell ones, or measures at higher temporal and spatial resolution, across more conditions and wider biological contexts.
Instead, we should use genomics to tackle big unanswered questions such as what causes the variation that we see across people in phenotypes, disease susceptibility and drug responses? What is the relative contribution of genetic, epigenetic, microbiome and environmental factors? How are their effects mediated, and what would be the effect of different interventions? Ultimately, we should strive to use genomics to generate actionable and personalized insights that lead to better health. We are now at an inflexion point in genomics that allows us for the first time to apply it to study human biology and realize these ambitious aims122.
At the cellular level, we can use iPS cells from patients to derive cellular models of multiple diseases and prioritize treatments based on measuring both their cellular and molecular response (for example, gene expression and epigenetics) to existing drugs and drug combinations. We can even use massively parallel assays to separately measure the effect of each of tens of thousands of rationally designed mutations, including patient-specific mutations, as we have done, for example, in testing the effect of all clinically identified mutations in TP53 on cellular function123. Measuring the molecular effects of directed mutations in genes encoding transcription factors and signalling molecules and in other genes can reveal the underlying pathways and regulatory networks of the disease studied and identify putative therapeutic targets. The application of such approaches to fields that are still poorly understood, such as neurodegenerative diseases, can be particularly impactful.
But we can be much more ambitious and directly profile large cohorts of human individuals using diverse ‘omics’ assays. As molecular changes typically precede clinical disease manifestations, longitudinal measurements coupled with clinical phenotyping have the potential of identifying novel disease diagnostics and therapeutic targets. Indeed, biobanks that track large samples of hundreds of thousands of individuals have recently emerged and are proving highly informative124. However, at the molecular level their focus has thus far been on genetics. Technological advances and cost reductions now allow us to obtain much deeper person-specific multi-omic profiles that include transcriptome, proteome, methylome, microbiome, immune system and metabolome measurements. Having these data on the same individual and at multiple time points can reveal which omic layer is more perturbed and informative for each disease and identify associations between molecular markers and disease.
The challenge in using such observational data from human cohorts is to identify which of the associations are causal. One way to address this is to wisely select the nature and type of the associations studied. For example, in working with microbiome data, we can move from analyses at the level of species composition to analyses at the level of SNPs in bacterial genes. Such associations are more specific and more likely to be causal, as in the case of a SNP in the dadH bacterial gene, which correlated with metabolism of the primary medication to treat Parkinson disease and the gut microbiota from patients125. Another approach is to use longitudinal measurements and separation of time to emulate target trials from observational data126. For example, we can select distinct subsets from the cohort that match on several known risk factors (for example, age or body mass index) but differ on a marker of interest (for example, expression of a gene or presence of an epigenetic mark), and compare future disease onset or progression in these two populations. Similarly, retrospective analysis of baseline multi-omic measurements from participants in randomized clinical trials may identify markers that distinguish responders from non-responders and be used for patient stratification or for identifying additional putative targets.
Ultimately, biomarkers identified from observational cohorts need to be tested in randomized clinical trials to establish causality and assess efficacy. In the case of microbial strains extracted from humans, we may be able to skip animal testing and go directly to human trials. In other cases, such as when human genes are being manipulated, we will need to start with cell culture assays and animal testing before performing clinical trials in humans. However, in all cases, tested omic targets should have already shown associations in human individuals, thus making them more likely to be relevant and succeed in trials, as is the case with drug targets for which genetic evidence links them to the disease127.
Beyond these scientific challenges, there is the challenge of engaging the public and diverse ethnic and socio-economic groups to participate in such large-scale multi-omic profiling endeavours even before we can present them with immediate benefits. We can start with incentives in the form of informational summary reports of the data measured and gradually move towards carefully and responsibly conveyed actionable insights as we learn more.
Overcoming the aforementioned challenges is not an easy task, but with the breathtaking advances that genomics has undergone in the past two decades, the time may be right to tackle them. Success can transform genomics from being applied mostly in research settings to having it become an integral and inseparable part of medicine.
CRISPR genome editing enters the clinic
Jin-Soo Kim. In the past several years, genome editing has come of age128, in particular because of the repurposing of CRISPR systems. Genomic DNA can be modified in a targeted manner in vivo or in vitro with high efficiency and precision, potentially enabling therapeutic genome editing for the treatment of both genetic and non-genetic diseases. All three types of programmable nucleases developed for genome editing, namely zinc-finger nucleases, transcription activator-like effector nucleases and CRISPR nucleases, are now under clinical investigation. In the next several years, we will be able to learn whether these genome-editing tools will be effective and safe enough to treat patients with an array of diseases, including HIV infection, leukaemia, blood disorders and hereditary blindness, heralding a new era in medicine.
If the history of the development of novel drugs or treatments such as gene therapy and monoclonal antibodies is any guide, the road to therapeutic genome editing is likely to be bumpy but ultimately worth travelling. Key questions related to medical applications of programmable nucleases concern their mode of delivery, specificity, on-target activity and immunogenicity. First, in vivo delivery (or direct delivery into patients) of genes or mRNAs encoding programmable nucleases or preassembled Cas9 ribonucleoproteins can be a challenge, given the large size of these nucleases. Ex vivo (or indirect) delivery is, in general, more efficient than in vivo delivery but is limited to cells from blood or bone marrow, which can be collected with ease, edited in vitro and transfused back into patients. Ongoing developments of nanoparticles and viral vectors are expected to enhance and expand in vivo genome editing in tissues or organs not readily accessible with current delivery systems, such as the brain.
Second, programmable nucleases, including CRISPR nucleases, can cause unwanted on-target and off-target mutations, which may contribute to oncogenesis. Several cell-based and cell-free methods have been developed to identify genome-wide CRISPR off-target sites in an unbiased manner129,130,131. But it remains a challenge to validate off-target activity at sites with low mutation frequencies (less than 0.1%) in a population of cells, owing to the intrinsic error rates of current sequencing technologies. Even at on-target sites, CRISPR–Cas9 can induce unexpected outcomes such as large deletions of chromosomal segments132. It will be important to understand the mechanisms behind the unusual on-target activity and to measure and reduce the frequencies of such events.
Last but not least, Cas9 and other programmable nucleases can be immunogenic, potentially causing undesired innate and adaptive immune responses. In this regard, it makes sense that initial clinical trials have focused on ex vivo delivery of Cas9 ribonucleoproteins into T cells or in vivo gene editing in the eye, an immunologically privileged organ. Cas9 epitope engineering or novel Cas9 orthologues derived from non-pathogenic bacteria may avoid some of the immune responses, offering therapeutic modalities for in vivo genome editing in tissues or organs with little or no immune privilege.
Base editing133,134 and prime editing135 are promising new approaches that may overcome some of the limitations of nuclease-mediated genome editing. Base editors and prime editors are composed of a Cas9 nickase, rather than the wild-type Cas9 nuclease, and a nucleobase deaminase and a reverse transcriptase, respectively. Because a nickase, unlike a nuclease, produces DNA single-strand breaks or nicks, but not double-strand breaks (DSBs), base editors and prime editors are unlikely to induce large deletions at on-target sites and chromosomal rearrangements resulting from non-homologous end joining (NHEJ) repair of concurrent on-target and off-target DSBs. Furthermore, when it comes to gene correction rather than gene disruption, these new types of gene editors are much more efficient and ‘cleaner’ than DSB-producing nucleases because they neither require donor template DNA nor rely on error-prone NHEJ; in human cells, DSBs are preferentially repaired by NHEJ, leading to small insertions or deletions (indels), rather than by homologous recombination involving donor DNA.
Base editors and prime editors are also well suited for germline editing and in utero editing (that is, gene editing in the fetus), which should be done with caution, in full consideration of ethical, legal and societal issues. In principle, CRISPR–Cas9 can be used for the correction of pathogenic mutations in human embryos; however, donor DNA is seldom used as a repair template in human embryos136. Recurrent or non-recurrent de novo mutations are responsible for the vast majority of genetic diseases. Cell-free fetal DNA in the maternal blood can be used to detect these de novo mutations in fetuses, which are absent in the parents. Some de novo mutations are manifested even before birth, leading to miscarriage, disability or early death after birth; it is often too late and inefficient to attempt gene editing in newborns. These mutations could be corrected in utero using base editors or prime editors without inducing unwanted indels and without relying on inefficient homologous recombination. Compared with germline editing or preimplantation genetic diagnosis, in utero editing, if proven safe and effective in the future, should be ethically more acceptable because it does not involve the creation or destruction of human embryos.
As promising and powerful as they are, current versions of base editors and prime editors can be further optimized and improved. For instance, Cas9 evolved in microorganisms as a nuclease rather than a nickase. Current Cas9 nickases used for base editing (D10A SpCas9 variant) and prime editing (H840A variant) can be engineered to increase their activities and specificities. In parallel, deaminase and reverse transcriptase moieties in base editors and prime editors, respectively, can be engineered or replaced with appropriate orthologues to increase the efficiency and scope of genome editing. It has been shown that base editors can cause both guide RNA-dependent and guide RNA-independent DNA or RNA off-target mutations, raising concerns for their applications in medicine. Prime editors may also cause unwanted on-target and off-target mutations, which must be carefully studied before moving on to therapeutic applications.
Biomedical researchers are now equipped with powerful tools for genome editing. I expect that these tools will be developed further and applied more broadly in both research and medicine in the coming years.
References
Collins F. The director of the NIH lays out his vision of the future of medical science. Time https://time.com/5709207/medical-science-age-of-discovery (2019).
The National Academies of Sciences, Engineering, and Medicine Organizing Committee for the International Summit on Human Gene Editing. On human gene editing: international summit statement. The National Academies of Sciences, Engineering, and Medicine https://www.nationalacademies.org/news/2015/12/on-human-gene-editing-international-summit-statement (2015).
Centers for Disease Control and Prevention. COVID-19 in racial and ethnic minority groups. CDC https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/racial-ethnic-minorities.html (2020).
Edwards, F., Lee, H. & Esposito, M. Risk of being killed by police use of force in the United States by age, race–ethnicity, and sex. Proc. Natl Acad. Sci. USA 116, 16793–16798 (2019).
Popejoy, A. B. & Fullerton, S. M. Genomics is failing on diversity. Nature 538, 161–164 (2016).
Popejoy, A. B. et al. The clinical imperative for inclusivity: race, ethnicity, and ancestry (REA) in genomics. Hum. Mutat. 39, 1713–1720 (2018).
Artiga, S. & Orgera, K. Key facts on health and health care by race and ethnicity. Kaiser Family Foundation https://www.kff.org/report-section/key-facts-on-health-and-health-care-by-race-and-ethnicity-coverage-access-to-and-use-of-care/ (2019).
Armstrong, K., Micco, E., Carney, A., Stopfer, J. & Putt, M. Racial differences in the use of BRCA1/2 testing among women with a family history of breast or ovarian cancer. JAMA 293, 1729–1736 (2005).
Bonham, V. L., Callier, S. L. & Royal, C. D. Will precision medicine move us beyond race? N. Engl. J. Med. 374, 2003–2005 (2016).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
The SIGMA Type 2 Diabetes Consortium. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 506, 97–101 (2014).
Wetterstrand, K. A. DNA sequencing costs: data from the NHGRI genome sequencing program (GSP). National Human Genome Research Institute https://www.genome.gov/sequencingcostsdata (2019).
Wall, J. D. et al. The GenomeAsia 100K Project enables genetic discoveries across Asia. Nature 576, 106–111 (2019).
Kowalski, M. H. et al. Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 15, e1008500 (2019).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Gaziano, J. M. et al. Million veteran program: a mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol. 70, 214–223 (2016).
Nagai, A. et al. Overview of the BioBank Japan Project: study design and profile. J. Epidemiol. 27, S2–S8 (2017).
Denny, J. C. et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 31, 1102–1110 (2013).
Sirugo, G., Williams, S. M. & Tishkoff, S. A. The missing diversity in human genetic studies. Cell 177, 1080 (2019).
Martin, A. R. et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 51, 584–591 (2019).
McQuillan, M. A., Zhang, C., Tishkoff, S. A. & Platt, A. The importance of including ethnically diverse populations in studies of quantitative trait evolution. Curr. Opin. Genet. Dev. 62, 30–35 (2020).
Sohail, M. et al. Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. eLife 8, e39702 (2019).
Crawford, N. G. et al. Loci associated with skin pigmentation identified in African populations. Science 358, eaan8433 (2017).
Gasperini, M., Tome, J. M. & Shendure, J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. 21, 292–310 (2020).
Racimo, F., Sankararaman, S., Nielsen, R. & Huerta-Sánchez, E. Evidence for archaic adaptive introgression in humans. Nat. Rev. Genet. 16, 359–371 (2015).
Skoglund, P. & Mathieson, I. Ancient genomics of modern humans: the first decade. Annu. Rev. Genomics Hum. Genet. 19, 381–404 (2018).
Vicente, M. & Schlebusch, C. M. African population history: an ancient DNA perspective. Curr. Opin. Genet. Dev. 62, 8–15 (2020).
Sherman, R. M. et al. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet. 51, 30–35 (2019).
Durvasula, A. et al. Recovering signals of ghost archaic introgression in African populations. Sci. Adv. 12, eaax5097 (2020).
Skov, L. et al. The nature of Neanderthal introgression revealed by 27,566 Icelandic genomes. Nature 582, 78–83 (2020).
Adeyemo, A. A. et al. ZRANB3 is an African-specific type 2 diabetes locus associated with beta-cell mass and insulin response. Nat. Commun. 10, 3195 (2019).
Cohen, J. et al. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 37, 161–165 (2005).
Gurdasani, D. et al. Uganda genome resource enables insights into population history and genomic discovery in Africa. Cell 179, 984–002.e36 (2019).
Gurdasani, D. et al. Genomics of disease risk in globally diverse populations. Nat. Rev. Genet. 20, 520–535 (2019).
Gulsuner, S. et al. Genetics of schizophrenia in the South African Xhosa. Science 367, 569–573 (2020).
Shriner, D. & Rotimi, C. N. Whole-genome-sequence-based haplotypes reveal single origin of the sickle allele during the Holocene wet phase. Am. J. Hum. Genet. 102, 547–556 (2018).
Wu, Y. et al. Highly efficient therapeutic gene editing of human haematopoietic stem cells. Nat. Med. 25, 776–783 (2019).
Geard, A. et al. Clinical and genetic predictors of renal dysfunctions in sickle cell anaemia in Cameroon. Br. J. Haematol. 178, 629–639 (2017).
Lebeko, K. et al. Targeted genomic enrichment and massively parallel sequencing identifies novel nonsyndromic hearing impairment pathogenic variants in Cameroonian families. Clin. Genet. 90, 288–290 (2016).
Genovese, G. et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329, 841–845 (2010).
Sierra, B. et al. OSBPL10, RXRA and lipid metabolism confer African-ancestry protection against dengue haemorrhagic fever in admixed Cubans. PLoS Pathog. 13, e1006220 (2017).
Wonkam, A. & de Vries, J. Returning incidental findings in African genomics research. Nat. Genet. 52, 17–20 (2020).
Provine, W. B. The Origins of Theoretical Population Genetics (University of Chicago Press, 1971)
Fisher, R. A. The correlation between relatives on the supposition of Mendelian inheritance. Trans. R. Soc. Edinb. 52, 399–433 (1918).
Altenburg, E. & Muller, H. J. The genetic basis of truncate wing – an inconstant and modifiable character in Drosophila. Genetics 5, 1–59 (1920).
Morton, N. E. Analysis of family resemblance. I. Introduction. Am. J. Hum. Genet. 26, 318–330 (1974).
Visscher, P. M. et al. 10 Years of GWAS discovery: biology, function and translation. Am. J. Hum. Genet. 101, 5–22 (2017).
Boyle, E. A., Li, Y. I. & Pritchard, J. K. An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 (2017).
Emison, E. S. et al. A common, sex-dependent mutation in a putative RET enhancer underlies Hirschsprung disease susceptibility. Nature 434, 857–863 (2005).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Davidson, E. Emerging properties of animal gene regulatory networks. Nature 468, 911–920 (2010).
Chatterjee, S. et al. Enhancer variants synergistically drive dysregulation of the RET gene regulatory network in Hirschsprung disease. Cell 167, 355–368 (2016).
Segal, E., Raveh-Sadka, T., Schroeder, M., Unnerstall, U. & Gaul, U. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature 451, 535–540 (2008).
Chakravarti, A. & Turner, T. N. Revealing rate-limiting steps in complex disease biology: The crucial importance of studying rare, extreme-phenotype families. Bioessays 38, 578–586 (2016).
Lancaster, M. A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013).
Rothman, J. & Jarriault, S. Developmental plasticity and cellular reprogramming in caenorhabditis elegans. Genetics 213, 723–757 (2019).
Porrello, E. R. et al. Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080 (2011).
Mir, M., Bickmore, W., Furlong, E. E. M. & Narlikar, G. Chromatin topology, condensates and gene regulation: shifting paradigms or just a phase? Development 146, dev182766 (2019).
Ghavi-Helm, Y. et al. Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat. Genet. 51, 1272–1282 (2019).
Despang, A. et al. Functional dissection of the Sox9-Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nat. Genet. 51, 1263–1271 (2019).
Hnisz, D., Shrinivas, K., Young, R. A., Chakraborty, A. K. & Sharp, P. A. A phase separation model for transcriptional control. Cell 169, 13–23 (2017).
Shrinivas, K. et al. Enhancer features that drive formation of transcriptional condensates. Mol. Cell 75, 549–561 e547 (2019).
Narlikar, G. J. Phase-separation in chromatin organization. J. Biosci. 45, 5 (2020).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Farrell, J. A. et al. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 360, eaar3131 (2018).
Cusanovich, D. A. et al. The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature 555, 538–542 (2018).
Tang, F. et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009).
Camp, J. G., Platt, R. & Treutlein, B. Mapping human cell phenotypes to genotypes with single-cell genomics. Science 365, 1401–1405 (2019).
Lein, E., Borm, L. E. & Linnarsson, S. The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science 358, 64–69 (2017).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
Achim, K. et al. High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat. Biotechnol. 33, 503–509 (2015).
Cao, J. et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 (2017).
Cleary, B., Cong, L., Cheung, A., Lander, E. S. & Regev, A. Efficient generation of transcriptomic profiles by random composite measurements. Cell 171, 1424–1436 e1418 (2017).
Cao, J. et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018).
Kester, L. & van Oudenaarden, A. Single-cell transcriptomics meets lineage tracing. Cell Stem Cell 23, 166–179 (2018).
Stuart, T. & Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 20, 257–272 (2019).
Zhu, C., Preissl, S. & Ren, B. Single-cell multimodal omics: the power of many. Nat. Methods 17, 11–14 (2020).
Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. (2020).
Loeffler, D. et al. Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature 573, 426–429 (2019).
Schmidt, F., Cherepkova, M. Y. & Platt, R. J. Transcriptional recording by CRISPR spacer acquisition from RNA. Nature 562, 380–385 (2018).
Holliday, R. & Pugh, J. E. DNA modification mechanisms and gene activity during development. Science 187, 226–232 (1975).
Strahl, B. D. & Allis, C. D. The language of covalent histone modifications. Nature 403, 41–45 (2000).
Jambhekar, A., Dhall, A. & Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 20, 625–641 (2019).
Smith, Z. D. et al. Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature 549, 543–547 (2017).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Nabet, B. et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 14, 431–441 (2018).
Clevers, H. Modeling development and disease with organoids. Cell 165, 1586–1597 (2016).
Dekker, J., Marti-Renom, M. A. & Mirny, L. A. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat. Rev. Genet. 14, 390–403 (2013).
Banani, S. F., Lee, H. O., Hyman, A. A. & Rosen, M. K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017).
Lupianez, D. G. et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025 (2015).
Basu, S. et al. Unblending of transcriptional condensates in human repeat expansion disease. Cell 181, 1062–1079 e1030 (2020).
Grosswendt, S. et al. Epigenetic regulator function through mouse gastrulation. Nature 584, 102–108 (2020).
Johnson, T. B. & Coghill, R. D. Researches on pyrimidines. C111. The discovery of 5-methyl-cytosine in tuberculinic acid, the nucleic acid of the tubercle bacillus. J. Am. Chem. Soc. 47, 2838–2844,47 (1925).
Heard, E. et al. Ten years of genetics and genomics: what have we achieved and where are we heading? Nat. Rev. Genet. 11, 723–733 (2010).
Quinn, J. J. & Chang, H. Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 17, 47–62 (2016).
Kopp, F. & Mendell, J. T. Functional classification and experimental dissection of long noncoding RNAs. Cell 172, 393–407 (2018).
Liu, S. J. et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 355, eaah7111 (2017).
Rubin, A. J. et al. Coupled single-cell CRISPR screening and epigenomic profiling reveals causal gene regulatory networks. Cell 176, 361–376.e17 (2019).
Quinn, J. J. et al. Rapid evolutionary turnover underlies conserved lncRNA-genome interactions. Genes. Dev. 30, 191–207 (2016).
Kirk, J. M. et al. Functional classification of long non-coding RNAs by k-mer content. Nat. Genet. 50, 1474–1482 (2018).
Carter, A. C. et al. Spen links RNA-mediated endogenous retrovirus silencing and X chromosome inactivation. eLife 9, e54508 (2020).
Lubelsky, Y. & Ulitsky, I. Sequences enriched in Alu repeats drive nuclear localization of long RNAs in human cells. Nature 555, 107–111 (2018).
Shukla, C. J. et al. High-throughput identification of RNA nuclear enrichment sequences. EMBO J. 37, e98452 (2018).
Czerminski, J. T. & Lawrence, J. B. Silencing Trisomy 21 with XIST in neural stem cells promotes neuronal differentiation. Dev. Cell 52, 294–308 e3 (2020).
Martínez-Jiménez, F. et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer https://doi.org/10.1038/s41568-020-0290-x (2020).
Wilkinson, M. et al. The FAIR guiding principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Gonzalez-Perez, A., Radhakrishnan, S. & Lopez-Bigas, N. Local determinants of the mutational landscape of the human genome. Cell 177, 101–114 (2019).
Martincorena, I. et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886 (2015).
Martincorena, I. et al. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917 (2018).
Yokoyama, A. et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 565, 312–317 (2019).
Genovese, G. et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014).
Jaiswal, S. et al. Age related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498 (2014).
Sabarinathan, R. et al. The whole-genome panorama of cancer drivers. Preprint at bioRxiv https://doi.org/10.1101/190330 (2017).
Pich, O. et al. The mutational footprints of cancer therapies. Nat. Genet. 51, 1732–1740 (2019).
Campbell, P. J. et al. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
Rozenblatt-Rosen, O., Stubbington, M. J. T., Regev, A. & Teichmann, S. A. The Human Cell Atlas: from vision to reality. Nature 550, 451–453 (2017).
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Damodaran, S. et al. Cancer Driver Log (CanDL): catalog of potentially actionable cancer mutations. J. Mol. Diagn. 17, 554–559 (2015).
High, K. A. & Roncarolo, M. G. Gene therapy. N. Engl. J. Med. 381, 455–464 (2019).
Shilo, S., Rossman, H. & Segal, E. Axes of a revolution: challenges and promises of big data in healthcare. Nat. Med. 26, 29–38 (2020).
Kotler, E. et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol. Cell 71, 873 (2018).
Swanson, J. M. The UK Biobank and selection bias. Lancet 380, 110 (2012).
Maini Rekdal, V., Bess, E. N., Bisanz, J. E., Turnbaugh, P. J. & Balskus, E. P. Discovery and inhibition of an interspecies gut bacterial pathway for levodopa metabolism. Science 364, eaau6323 (2019).
Hernán, M. A. & Robins, J. M. Using big data to emulate a target trial when a randomized trial is not available. Am. J. Epidemiol. 183, 758–764 (2016).
Nelson, M. R. et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860 (2015).
Kim, J.-S. Genome editing comes of age. Nat. Protoc. 11, 1573–1578 (2016).
Kim, D. et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 12, 237–243 (2015).
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015).
Wienert, B. et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 364, 286–289 (2019).
Kosicki, M., Tomberg, K. & Bradley, A. et al. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771 (2018).
Komor, A. C. et al. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Nishida, K. et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353, aaf8729 (2016).
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
Ma, H. et al. Correction of a pathogenic gene mutation in human embryos. Nature 548, 413–419 (2017).
Acknowledgements
A.C. acknowledges that the ideas in his contribution were developed through studies on Hirschsprung disease and thanks the many trainees who have contributed to this work over the past 5 years. A.L.M. acknowledges A. Gutierrez, K. Kostick, G. Lazaro, M. Majumder, K. Munoz, S. Pereira, H. Smith and P. Zuk for feedback. A.M. thanks D. Hnisz, Z. D. Smith, J. Charlton and H. Kretzmer for feedback and the Max Planck Society for funding. A.W. is supported by NIH awards U54HG009790, U01HG009716, U01HG007459 and U24HL135600, and Wellcome Trust award H3A/18/001, and states that the funders had no role in study design, and analysis, decision to publish or preparation of the manuscript. B.T. acknowledges J. G. Camp for helpful discussions. E.E.M.F. is very grateful to A. Ephrussi, M. Mir, M. Perino, Y. Kherdjemil, T. Pollex and S. Secchia for useful comments. E. E. M. F is supported by European Research Council (Advanced Grant) agreement no. 787611 (DeCRyPT). E.S. is supported by grants from the European Research Council and the Israel Science Foundation. H.Y.C. is supported by NIH RM1-HG007735 and R35-CA209919. H.Y.C. is an investigator of the Howard Hughes Medical Institute. J.-S.K. is supported by the Institute for Basic Science (IBS-R021-D1). N.L-B. acknowledges funding from the European Research Council (Consolidator Grant 682398), the Spanish Ministry of Economy and Competitiveness (SAF2015-66084-R, European Regional Development Fund) and the Asociación Española Contra el Cáncer (GC16173697BIGA). S.A.T. is funded by NIH grants R35 GM134957-01 and NIAMS R01AR076241-01A1 and American Diabetes Association Pathway to Stop Diabetes grant #1-19-VSN-02.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
H.Y.C. is a co-founder of Accent Therapeutics and Boundless Bio and an advisor of 10x Genomics, Arsenal Biosciences and Spring Discovery. J.-S.K. is a co-founder of and holds stock in ToolGen Inc. A.C., A.L.M., A.M., A.W., B.T., E.E.M.F., E.S., N.L.-B., S.G. and S.A.T. declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Beyond 1 Million Genomes: https://b1mg-project.eu/
Blueprint Epigenome: https://www.blueprint-epigenome.eu/
cBioPortal for Cancer Genomics: https://www.cbioportal.org/
ENCODE: https://www.encodeproject.org/
Global Alliance for Genomics and Health: https://www.ga4gh.org/
gnomAD: https://gnomad.broadinstitute.org/
GTEx: https://www.gtexportal.org/home/
GWAS Catalog: https://www.ebi.ac.uk/gwas
H3Africa: https://h3africa.org
Roadmap Epigenomics Project: http://www.roadmapepigenomics.org/
Rights and permissions
About this article

Cite this article
McGuire, A.L., Gabriel, S., Tishkoff, S.A. et al. The road ahead in genetics and genomics. Nat Rev Genet 21, 581–596 (2020). https://doi.org/10.1038/s41576-020-0272-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41576-020-0272-6
This article is cited by
-
Overcoming barriers to single-cell RNA sequencing adoption in low- and middle-income countries
European Journal of Human Genetics (2024)
-
An overview of artificial intelligence in the field of genomics
Discover Artificial Intelligence (2024)
-
Current trends, limitations and future research in the fungi?
Fungal Diversity (2024)
-
Improving variant calling using population data and deep learning
BMC Bioinformatics (2023)
-
Methods in molecular biology and genetics: looking to the future
BMC Research Notes (2023)
