
Abstract
Nearly all genetic variants that influence disease risk have human-specific origins; however, the systems they influence have ancient roots that often trace back to evolutionary events long before the origin of humans. Here, we review how advances in our understanding of the genetic architectures of diseases, recent human evolution and deep evolutionary history can help explain how and why humans in modern environments become ill. Human populations exhibit differences in the prevalence of many common and rare genetic diseases. These differences are largely the result of the diverse environmental, cultural, demographic and genetic histories of modern human populations. Synthesizing our growing knowledge of evolutionary history with genetic medicine, while accounting for environmental and social factors, will help to achieve the promise of personalized genomics and realize the potential hidden in an individual’s DNA sequence to guide clinical decisions. In short, precision medicine is fundamentally evolutionary medicine, and integration of evolutionary perspectives into the clinic will support the realization of its full potential.
Similar content being viewed by others

Introduction
Genetic disease is a necessary product of evolution (Box 1). Fundamental biological systems, such as DNA replication, transcription and translation, evolved very early in the history of life. Although these ancient evolutionary innovations gave rise to cellular life, they also created the potential for disease. Subsequent innovations along life’s long evolutionary history have similarly enabled both adaptation and the potential for dysfunction. Against this ancient background, young genetic variants specific to the human lineage interact with modern environments to produce human disease phenotypes. Consequently, the substrates for genetic disease in modern humans are often far older than the human lineage itself, but the genetic variants that cause them are usually unique to humans.
The advent of high-throughput genomic technologies has enabled the sequencing of the genomes of diverse species from across the tree of life1. Analysis of these genomes has, in turn, revealed the striking conservation of many of the molecular pathways that underlie the function of biological systems that are essential for cellular life2. The same technologies have also spearheaded a revolution in human genomics3; currently, more than 120,000 individual whole human genome sequences are publicly available, and genome-scale data from hundreds of thousands more have been generated by consumer genomics companies4. Huge nationwide biobanks are also characterizing the genotypes and phenotypes of millions of people from around the world5,6,7. These studies are radically changing our understanding of the genetic architecture of disease8. It is also now possible to extract and sequence ancient DNA from remains of organisms that are thousands of years old, enabling scientists to reconstruct the history of recent human adaptation with unprecedented resolution9,10. These breakthroughs have revealed the recent, often complicated, history of our species and how it influences the genetic architecture of disease8,11. With the expansion of clinical whole-genome sequencing and personalized medicine, the influence of our evolutionary past and its implications for understanding human disease can no longer remain overlooked by medical practice; evolutionary perspectives must inform medicine12,13.
Much like a family’s medical history over generations, the genome is fundamentally a historical record. Decoding the evolution of the human genome provides valuable context for interpreting and modelling disease. This context is not limited to recent human evolution but also includes more ancient events that span life’s history. In this Review, we trace the 4 billion-year interplay between evolution and disease by illustrating how innovations during the course of life’s history have established the potential for, and inevitability of, disease. Beginning with events in the very deep past, where most genes and pathways involved in human disease originate, we explain how ancient biological systems, recent genetic variants and dynamic environments interact to produce both adaptation and disease risk in human populations. Given this scope, we cannot provide a comprehensive account of all evolutionary events relevant to human disease. Instead, our goal is to illustrate through examples the relevance of both deep and recent evolution to the study and treatment of genetic disease. Many of these key insights stem from recent discoveries, which have yet to be integrated into the broader canvas of evolutionary biomedicine (Box 2).

Macroevolutionary imprints on human disease
Systems involved in disease have ancient origins
Many of cellular life’s essential biological systems and processes, such as DNA replication, transcription and translation, represent ancient evolutionary innovations shared by all living organisms. Although essential, each of these ancient innovations generated the conditions for modern disease (Fig. 1). In this section, we provide examples of how several ancient innovations have created substrates for dysfunction and disease, and how considering these histories contributes to understanding the biology of disease and extrapolating results from model systems to humans.

A timeline of evolutionary events (top) in the deep evolutionary past and on the human lineage that are relevant to patterns of human disease risk (bottom). The ancient innovations on this timeline (left) formed biological systems that are essential, but are also foundations for disease. During recent human evolution (right), the development of new traits and recent rapid demographic and environmental changes have created the potential for mismatches between genotypes and modern environments that can cause disease. The timeline is schematic and not shown to scale. bya, billion years ago; kya, thousand years ago; mya, million years ago.
As a foundational (if obvious) example, the origin of self-replicating molecules 4 billion years ago formed the basis of life, but also the root of genetic diseases12,14,15. Similarly, asymmetric cell division may have evolved as an efficient way to handle cellular damage, but it also established the basis for ageing in multicellular organisms16,17. Myriad age-related diseases in humans, and many other multicellular organisms, are a manifestation of this first evolutionary trade-off.
The evolution of multicellularity, which has occurred many times across the tree of life, illustrates the interplay between evolutionary innovation and disease18. The origin of multicellularity enabled complex body plans with trillions of cells, involving innovations associated with the ability of cells to regulate their cell cycles, modulate their growth and form intricate networks of communication. But multicellularity also established the foundation for cancer19,20. Genes that regulate cell cycle control are often divided into two groups: caretakers and gatekeepers21,22. The caretakers are involved in basic control of the cell cycle and DNA repair, and mutations in these genes often lead to increased mutation rates or genomic instability, both of which increase cancer risk. Caretaker genes are enriched for functions with origins dating back to the first cells23. The gatekeepers appeared later, at the genesis of metazoan multicellularity23. The gatekeepers are directly linked to tumorigenesis through their roles in regulating cell growth, death and communication. The progression of individual tumours in a given patient is likewise informed by an evolutionary perspective. Designing treatments that account for the evolution of drug resistance and heterogeneity in tumours is a tenet of modern cancer therapy24,25,26,27,28,29.
Like multicellularity, the evolution of immune systems also set the stage for dysregulation and disease. Mammalian innate and adaptive immune systems are both ancient. Components of the innate immune system are present across metazoans and even some plants30,31, whereas the adaptive immune system is present across jawed vertebrates32. These systems provided molecular mechanisms for self-/non-self-recognition and response to pathogens, but they evolved in a piecemeal fashion, using many different, pre-existing genes and processes. For example, co-option of endogenous retroviruses provided novel regulatory elements for interferon response33. As well, it is clear that the human immune system has co-evolved with parasites, such as helminths, over millions of years. Helminth infection both induces and modulates an immune response in humans34.
Evolutionary analyses of development have revealed that new anatomical structures often arise by co-opting existing structures and molecular pathways that were established earlier in the history of life. For example, animal eyes, limb structure in tetrapods and pregnancy in mammals (Box 3) each evolved by adapting and integrating ancient genes and regulatory circuits in new ways35,36,37,38. This integration of novel traits into the existing network of biological systems gives rise to links between diverse traits via the shared genes that underlie their development and function36. As a result, many genes are pleiotropic — they have effects on multiple, seemingly unrelated, traits. We do not have space here to cover the full evolutionary scope of these innovations and their legacies, but just as in each of the cases described above, innovations and adaptations spanning from the origin of metazoans to modern human populations shape the substrate upon which disease appears.
Medical implications
Although ancient macroevolutionary innovations may seem far removed from modern human phenotypes, their imprint remains on the human body and genome. Understanding the constraints they impose can provide insight into mechanisms of disease.
Mapping the origins and evolution of traits and identifying the genetic networks that underlie them are critical to the accurate selection of model systems and extrapolation to human populations. Failure to consider the evolutionary history of homologous systems, their phylogenetic relationships and their functional contexts in different organisms can lead to inaccurate generalization. Instead, when considering a model system, key evolutionary questions about both the organism and the trait of interest can indicate how translatable the research will be to humans39,40. For example, is the similarity between the trait in humans and the trait in the model system due to shared ancestry, that is, homology? The presence of homology in a human gene or system of study suggests potential as a model system; however, homology alone is not sufficient justification. Environmental and life history factors shape traits, and divergence between species complicates the simple assumption that homology provides genetic or mechanistic similarity. Thus, homology must be supplemented by understanding of whether the evolutionary divergence between humans and the proposed model led to functional divergence. For example, the rapid evolution of the placenta and variation in reproductive strategy across mammals have made it challenging to extrapolate results about the regulation of birth timing from model organisms, such as mouse, to humans (Box 3). More broadly, differences in genetic networks that underlie the development of homologous traits across mammals explain why the majority of successful animal trials fail to translate to human clinical trials41,42. Molecular mechanisms of ancient systems, such as DNA replication, can be studied using phylogenetically distant species; however, ‘humanizing’ these models to research human-specific aspects of traits may not be possible and comparative studies of closely related species may be required40.
Although evolutionary divergence in homologous traits is an impediment to the direct translation of findings from a model system to humans, understanding how these evolutionary differences came about can also yield insights into disease mechanisms. For example, intuition would suggest that large animals (many cells and cell divisions) with long lifespans (many ageing cells), such as elephants and whales, would be at increased risk for developing cancer. However, size and lifespan are not significantly correlated with cancer risk across species; despite their large size, elephants and whales do not have a higher risk of developing cancer43,44. Why is this so? Recent studies of the evolution of genes involved in the DNA damage response in elephants have revealed mechanisms that may contribute to cancer resistance. An ancient leukaemia inhibiting factor pseudogene (LIF6) regained its function in the ancestor of modern elephants. This gene works in conjunction with the tumour suppressor gene TP53, which has increased in copy number in elephants, to reduce elephants’ risk for cancer despite their large body size45,46. This illustrates a basic life history trade-off: selection has created mechanisms for cancer suppression and somatic maintenance in large vertebrates that are not needed in small short-lived vertebrates. Studying such seeming paradoxes, especially those with clear contrasts to human disease risk, will shed light on broader disease mechanisms and suggest targets for functional interventions with translation potential.
Human-specific evolution
Human adaptation, trade-offs and disease
The macroevolutionary events described above created the foundation of genetic disease, but considering the more recent changes that occurred during the evolutionary history of the human lineage is necessary to illuminate the full context of human disease. Comparisons between humans and their closest living primate relatives, such as chimpanzees, have revealed diseases that either do not appear in other species or take very different courses47. We are beginning to understand the genetic differences underlying some of these human-specific conditions, with particular insights into infectious diseases.
The last common ancestors of humans and chimpanzees underwent a complex speciation event that is likely to have involved multiple rounds of gene flow between ~12 and 6 million years ago (mya)48. Over the millions of years after this divergence, climatic, demographic and social pressures drove the evolution of many physical and behavioural traits unique to the human lineage, including bipedalism (~7 mya), lack of body hair (~2–3 mya) and larger brain volume relative to body size (~2 mya)12,47. These traits evolved in a diverse array of hominin groups, mainly in Africa, although some of these species, such as Homo erectus, ventured into Europe and Asia.
These human adaptations developed on the substrate of tightly integrated systems shaped by billions of years of evolution, and thus beneficial adaptations with respect to one system often incurred trade-offs in the form of costs on other linked systems49. The trade-off concept derives from a branch of evolutionary biology known as life history theory. It is based on the observation that organisms contain combinations of traits that cannot be simultaneously optimized by natural selection50,51. For example, many fitness-related traits draw on common energetic reserves, and investment in one comes at the expense of another52. Large body size may improve survival in certain environments, but it comes at the expense of longer development and lower numerical investment in reproduction.
The trade-off concept is clinically relevant because it dispenses with the notion of a single ‘optimal’ phenotype or fitness state for an individual49,53,54. Given the interconnected deep evolution of the human body, many diseases are tightly linked, in the sense that decreasing the risk for one increases the risk for the other. Such diametric diseases and the trade-offs that produce them are the starkest when there is competition within the body for limited resources; for example, energy used for reproduction cannot be used for growth, immune function or other energy-consuming survival processes54. The molecular basis for diametric diseases often results from antagonistic pleiotropy at the genetic level — when a variant has contrasting effects on multiple bodily systems. In extreme cases, some diseases that manifest well after reproductive age, for example, Alzheimer disease, have been less visible to selection and, thus, potentially more susceptible to trade-offs. Cancer and neurodegenerative disorders also exhibit this diametric pattern, where cancer risk is inversely associated with Alzheimer disease, Parkinson disease and Huntington disease. This association is hypothesized to be mediated by differences in the neuronal energy use and trade-offs in cell proliferation and apoptosis pathways49. Similarly, osteoarthritis (breakdown of cartilage in joints often accompanied by high bone mineral density) and osteoporosis (low bone mineral density) rarely co-occur. Their diametric pattern reflects, at least in part, different probabilities across individuals of mesenchymal stem cells within bone marrow to develop into osteoblasts versus non-bone cells such as adipocytes49,55. In another example, a history of selection for a robust immune response can now lead to an increased risk for autoimmune and inflammatory diseases, especially when coupled with new environmental mismatches49,54. Other examples of trade-offs are found throughout the human body, manifesting in risk for diverse diseases, including psychiatric and rheumatoid disorders49,56.
Just as adaptations in deep evolutionary time created new substrates for disease, evolutionary pressures exerted on the human lineage established the foundation for complex cognitive capabilities, but they also established the potential for many neuropsychiatric or neurodevelopmental diseases. For example, genomic structural variants enabled functional innovation in the brain through the emergence of novel genes57,58,59,60. Many human-specific segmental duplications influence genes that are essential to the development of the human brain, such as SRGAP2C and ARHGAP11B. Both of these genes function in cortical development and may be involved in the expansion of human brain size61,62,63. The human-specific NOTCH2NL is also hypothesized to have evolved from a partial duplication event, and is implicated in increased output during human corticogenesis, another potential key contributor to human brain size59,60. Although these structural variants were probably adaptive58, they may have also predisposed humans to neuropsychiatric diseases and developmental disorders. Copy number variation in the region flanking ARHGAP11B, specifically a microdeletion at 15q13.3, is associated with risk for intellectual disability, autism spectrum disorder (ASD), schizophrenia and epilepsy58,64. Duplications and deletions of NOTCH2NL and surrounding regions are implicated in macrocephaly and ASD or microcephaly and schizophrenia, respectively59. These trade-offs also play out at the protein domain level. For example, the Olduvai domain (previously known as DUF1220) is a 1.4-kb sequence that appears in ~300 copies in the human genome; this domain has experienced a large human-specific increase in copy number. These domains appear in tandem arrays in neuroblastoma breakpoint family (NBPF) genes, and have been associated with both increased brain size and neuropsychiatric diseases, including autism and schizophrenia65. These examples suggest that the genomic organization of these human-specific duplications may have enabled human-specific changes in brain development while also increasing the likelihood of detrimental rearrangements that cause human disease59,64. Furthermore, genomic regions associated with neuropsychiatric diseases have experienced human-specific accelerated evolution and recent positive selection, providing additional evidence for the role of recent evolutionary pressures on human disease risk66,67. Schizophrenia-associated loci, for example, are enriched near human accelerated regions (HARs) that are conserved in non-human primates68. Variation in HARs has also been associated with risk for ASD, possibly through perturbations of gene regulatory architecture69.
Human immune systems have adapted in response to changes in environment and lifestyles over the past few million years; however, the rapid evolution of the immune system may have left humans vulnerable to certain diseases, such as HIV-1 infection. A similar virus, simian immunodeficiency virus (SIV), is found in chimpanzees and other primates, and studies in the early 2000s found evidence of AIDS-like symptoms (primarily a reduction in CD4+ T cells) in chimpanzees infected with SIV. Although the effects of SIV in chimpanzees mirror some of the effects of HIV in humans70, captive chimpanzees infected with HIV-1 do not typically develop AIDS and have better clinical outcomes. The differences in outcome are influenced by human-specific immune evolution. For example, humans have lost expression of several Siglecs, cell surface proteins that binds sialic acids, in T lymphocytes compared with great apes71. In support of this hypothesis, human T cells with high Siglec-5 expression survive longer after HIV-1 infection72. Moreover, there is a possible role for the rapidly evolving Siglecs in other diseases, such as epithelial cancers, that differentially affect humans relative to closely related primates73,74.
Another human-specific immune change is the deletion of an exon of CMP-N-acetylneuraminic acid hydroxylase (CMAH) leading to a difference in human cell surface sialoglycans compared with other great apes75,76,77. The change in human sialic acid to an N-acetylneuraminic acid (Neu5Ac) termination, rather than N-glycolylneuraminic acid (Neu5Gc), may have been driven by pressure to escape infection by Plasmodium reichenowi, a parasite that binds Neu5Gc and causes malaria in chimpanzees. Conversely, the prevalence of Neu5Ac probably made humans more susceptible to infection by the malaria parasite Plasmodium falciparum, which binds to Neu5Ac78,79, and another human-specific pathology: typhoid fever80. Typhoid toxin binds specifically and is cytotoxic to cells expressing Neu5Ac glycans. Thus, the deletion of CMAH was likely to have been selected for by pressure from pathogens, but has in turn enabled other human-specific diseases such as malaria and typhoid fever81. The rapid evolution of the human immune system creates the potential for human-specific disease. As a result, human-specific variation in many other human immune genes influences human-specific disease risk82,83.
Medical implications
These examples from recent human evolution highlight the ongoing interplay of genetic variation, adaptation and disease. Understanding the evolutionary history of traits along with the aetiology of related diseases can help identify and evaluate risks for unintended consequences of treatments due to trade-offs. For example, ovarian steroids have pleiotropic effects stimulating both bone growth and mitosis in breast tissues to mobilize calcium stores during lactation54. However, later in life this link gives rise to a clinical trade-off. Hormone replacement therapy in postmenopausal women reduces the risk for osteoporosis and ovarian cancer, but also, as a result of its effects on breast tissue, increases the risk for breast cancer. Given the commonality of the trade-off between maintenance and proliferation, this is just one of many examples of cancer risk emerging as a result of trade-offs in immune, reproductive and metabolic systems56,84. Pregnancy is also rife with clinically relevant trade-offs given the interaction between multiple individuals and genomes (mother, father and fetus) with different objectives (Box 3). Trade-offs at the cellular level also have medical implications. For example, cellular senescence is a necessary and beneficial part of many basic bodily responses, but the accumulation of senescent cells underlies many ageing-related disorders. Thus, individuals with different solutions to this trade-off may have very different ‘molecular’ versus ‘chronological’ ages85.
Identifying such trade-offs by studying disease and treatment response is of great interest, but is challenging for several reasons: the number of possible combinations of traits to consider is large; many humans must have experienced the negative effects; and data must be available on both traits in the same individuals. Here, evolution paired with massive electronic health record (EHR)-linked biobanks5,86,87 provides a possible solution. By considering the evolutionary context and potential linkages between traits, the search space of possible trade-offs can be constrained. Then, diametric traits can be tested for among individuals in the EHRs by performing phenome-wide association studies (PheWAS) either on traits or genetic loci of interest and looking for inverse relationships88. The mechanisms underlying the observed associations could then be evaluated in model systems and, if validated, anticipated in future human treatments.
In addition to trade-offs, evolutionary analyses can help us identify therapeutic targets for uniquely human diseases. A small subset of humans infected with HIV never progress to AIDS — a resistance phenotype that has been generally attributed to host genomics89,90,91. Identifying and understanding the genes that contribute to non-progression is of great interest in the development of vaccines and treatments for HIV infection. Genome-wide association studies (GWAS) and functional studies have supported the role of the MHC class I region, specifically the HLA-B*27/B*57 molecules, in HIV non-progression92,93,94. Comparative genomics with chimpanzees identified a chimpanzee MHC class I molecule functionally analogous to that of the non-progressors that contains amino acid substitutions that change binding affinity for conserved areas of the HIV-1 and SIV viruses. Evolutionary analysis of this region suggests that these substitutions are the result of an ancient selective sweep in chimpanzee genomes that did not occur in humans95. This analysis not only helps us understand how humans are uniquely susceptible to HIV progression but also highlights functional variation in the MHC that are potential targets of medical intervention.
Recent human demographic history
Most genetic variants are young, but have diverse histories
The complex demographic history of modern humans in the past 200,000 years has created differences in the genetic architecture of and risk for specific diseases among human populations. With genomic sequences of thousands of humans from diverse locations, we can compare genetic information over time and geography to better understand the origins and evolution of both individual genetic variants and human populations96,97,98. The vast majority of human genetic variants are not shared with other species99. Demographic events such as bottlenecks, introgression and population expansion shaped the genetic composition of human populations, whereas rapid introduction of humans into new environments and the subsequent adaptations created potential for evolutionary mismatches (Figs 2,3).

Representative genes that have experienced local adaptive evolution over the past 100,000 years as humans moved across the globe. We focus on adaptations that also produced the potential for disease due to trade-offs or mismatches with modern environments. For each, we list the evolutionary pressure, the trait(s) influenced and the associated disease(s). The approximate regions where the adaptations occurred are indicated by blue circles. Arrows represent the expansion of human populations, and purple shading represents introgression events with archaic hominins. Supplementary Table S1 presents more details and references. COVID-19, coronavirus disease 2019; G6PD, glucose-6-phosphate dehydrogenase; UV, ultraviolet.

Ancient human migrations, introgression events with other archaic hominins and recent population expansions have all contributed to the introduction of variants associated with human disease. Schematic of human evolutionary history, where the branches represent different human populations and the branch widths represent population size (top left). Letter labels refer to the processes illustrated in parts a–d. a | Human populations migrating out of Africa maintained only a subset of genetic diversity present in African populations. The resulting out-of-Africa bottleneck is likely to have increased the fraction of deleterious, disease-associated variants in non-African populations. Coloured circles represent different genetic variants. Circles marked with X denote deleterious, disease-associated variants. b | When anatomically modern humans left Africa, they encountered other archaic hominin populations. Haplotypes introduced by archaic introgression events (illustrated in grey) contained Neanderthal-derived variants (denoted by red circles) associated with increased disease risk in modern populations. c | In the last 10,000 years, the burden of rare disease-associated variants (denoted by yellow circles) has increased due to rapid population expansion. d | Modern human individuals with admixture in their recent ancestry, such as African Americans, can have differences in genetic risk for disease, because of each individual’s unique mix of genomic regions with African and European evolutionary ancestry. For example, each of the three admixed individuals depicted have the same proportions of African and European ancestry, but do not all carry the disease-associated variant found at higher frequency in European populations (illustrated by yellow circles). Summarizing clinical risk for a patient requires a higher resolution view of evolutionary ancestry along the genome and improved representation of genetic variation from diverse human populations.
Approximately 200,000 years ago, ‘anatomically modern humans’ (AMHs) first appeared in Africa. This group had the key physical characteristics of modern human groups and exhibited unique behavioural and cognitive abilities that enabled rapid improvements in tool development, art and material culture. Approximately 100,000 years ago, AMH groups began to migrate out of Africa. The populations ancestral to all modern Eurasians are likely to have left Africa tens of thousands of years later98, but quickly spread across Eurasia. Expansions into the Americas and further bottlenecks are thought to have occurred between 35,000 and 15,000 years ago. The details and uncertainties surrounding these origin and migration events are more extensively reviewed elsewhere98.
Populations that experience bottlenecks and founder effects have a higher mutation load than populations that do not, largely due to their lower effective population sizes reducing the efficacy of selection100 (Fig. 3a). During this dispersal, the migrant human populations harboured less genetic variation than was present in Africa. The reduction in diversity caused by the out-of-Africa and subsequent bottlenecks shaped the genetic landscape of all populations outside Africa.
AMHs did not live in isolation after migration out of Africa. Instead, there is evidence of multiple admixture events with other archaic hominin groups, namely Neanderthals and Denisovans101,102. Modern non-African populations derive approximately 2% of their ancestry from Neanderthals, with some Asian populations having an even higher proportion of archaic hominin ancestry (Fig. 3b). African populations have only a small amount of Neanderthal and Denisovan ancestry, largely from back migration from European populations with archaic ancestry103. However, there is evidence of admixture with other, as yet unknown, archaic hominins in the genomes of modern African populations104,105,106.
Following their expansion around the globe, humans have experienced explosive growth over the past 10,000 years, in particular in modern Eurasian populations107,108 (Fig. 3c). Growth in population size modifies the genetic architecture of traits by increasing the efficacy of selection and generating many more low-frequency genetic variants. Although the impact of rare alleles is not completely understood, they often have a deleterious role in variation in traits in modern populations109. Although there is still debate about the combined effects of these recent demographic differences, a consensus is emerging that they are likely to have only minor effects on the efficacy of selection and the mutation load between human populations100,110,111,112,113,114. Nonetheless, there are substantial differences in allele frequency between populations that are relevant to disease risk115.
The exposure of humans to new environments and major lifestyle shifts, such as agriculture and urbanization, created the opportunity for adaptation96,116. Ancient DNA sequencing efforts coupled with recent statistical advances are beginning to enable the linking of human adaptations to specific environmental shifts in the recent past96,117,118. However, these rapid environmental changes also created new patterns of complex disease. Mismatch between our biological suitability for ancestral environments and modern environments accounts for the prevalence of many common diseases, such as obesity, diabetes or heart disease that derive from sedentary lifestyles and poor nutrition. The ancestral susceptibility model proposes that ancestral alleles that were adapted to ancient environments can, in modern populations, increase the risk for disease119,120. Supporting this hypothesis, both ancestral and derived alleles increase disease risk in modern humans121,122. However, underscoring the importance of recent demographic history, patterns of risk for ancestral and derived alleles differ in African and European populations, with ancestral risk alleles at higher frequencies in African populations115.
Medical implications
The different evolutionary histories of modern human individuals and populations described in the previous section influence disease susceptibilities and outcomes. Perhaps most striking are the mismatches and trade-offs resulting from recent immune system adaptations. Classic examples include genetic variants conferring resistance to malaria also causing sickle cell-related diseases in homozygotes96,123, or the predominantly African G1 and G2 variants in APOL1 protecting against trypanosomes and ‘sleeping sickness’ but leading to chronic kidney disease in individuals with these genotypes96. Similarly, a variant in CREBRF that is thought to have improved survival for people in times of starvation is now linked to obesity and type 2 diabetes124. In a study of ancient European populations, a variant in SLC22A4, the ergothioneine transporter, that may have been selected for to protect against deficiency of ergothioneine (an antioxidant) is also associated with gastrointestinal problems such as coeliac disease, ulcerative colitis and irritable bowel syndrome118. The variant responsible did not reach high frequency in European populations until relatively recently, and current disease associations are likely to be new, perhaps as a result of mismatches with the current environment118. The possibility of mismatch is further supported by the varying prevalence of coeliac disease between human populations related to population-specific selection for several risk alleles82. Indeed, recent studies suggest that there is a relationship between ancestry and immune response, with individuals of African ancestry demonstrating stronger responses. This could be the result of selective processes in response to new environments for European populations, or a larger pathogen burden in Africa now leading to a higher instance of inflammatory and autoimmune disorders. This is still an open area of research, and more evidence is needed before strong conclusions can be drawn125.
In modern human environments, there is also a mismatch between the current low parasite infection levels and the immune system that evolved under higher parasite load. This mismatch is hypothesized to contribute to the increase in inflammatory and autoimmune diseases seen in modern humans34. For example, loci associated with ten different inflammatory diseases, including Crohn’s disease and multiple sclerosis, show evidence of selection consistent with the hygiene hypothesis126. Furthermore, recent positive selection on variants in the type 2 immune response pathway favoured alleles associated with susceptibility to asthma127. This suggests that recent evolutionary processes may have led to elevated or altered immune responses at the expense of increased susceptibility to inflammatory and autoimmune diseases. This insight has broad clinical implications, including the potential targeted use of helminths and natural products for immune modulation in patients with chronic inflammatory disease128,129.
Archaic introgression is relevant to modern medicine because alleles introduced by these evolutionary events continue to have an impact on modern populations even though the archaic hominin lineages are now extinct (Fig. 3b). Archaic hominins had considerably lower effective population sizes than AMHs, and thus they probably carried a larger fraction of weakly deleterious mutations than AMHs101. As a result, Neanderthal introgression is predicted to have substantially increased the genetic load of non-African AMHs130,131. Large-scale sequencing efforts, in combination with analysis of clinical biobanks and improved computational methods, have revealed the potential impacts of introgressed DNA on modern human genomes. Several recent studies link regions of archaic admixture in modern populations with a range of diseases, including immunological, neuropsychiatric and dermatological phenotypes102,132,133,134,135,136,137,138,139. This demonstrates the functional impact of introgressed sequence on disease risk in non-African humans today. However, some of these associations may be influenced by linked non-Neanderthal alleles140. For example, in addition to alleles of Neanderthal origin, introgression also reintroduced ancestral alleles that were lost in modern Eurasian populations prior to interbreeding (for example, in the out-of-Africa bottleneck)141. Some introgressed alleles may have initially lessened adverse effects from migration to northern climates, dietary changes and introduction to novel pathogens117,142,143. For example, Neanderthal alleles contribute to variation in innate immune response across populations125,132,134,144 and probably helped AMHs adapt to new viruses, in particular RNA viruses in Europe145. However, due to recent demographic and environmental changes, some previously adaptive Neanderthal alleles may no longer provide the same benefits146. For example, there is evidence that an introgressed Neanderthal haplotype increases risk for SARS-CoV-2 (ref.147).
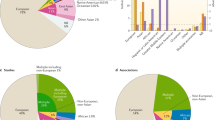
Physicians regularly rely on proxies for our more recent evolutionary history in the form of self-reported ancestry in their clinical practice; however, these measures fail to capture the complex evolutionary ancestry of each individual patient. For example, two individuals who identify as African Americans may both have 15% European ancestry, but this ancestry will be at different genomic loci and from different ancestral European and African populations (Fig. 3d). Thus, one may carry a disease-increasing European ancestry allele whereas the other does not. Mapping fine-scale genetic ancestry across patients’ genomes can improve our ability to summarize clinically relevant risk148, but such approaches require broad sampling across populations and awareness of human diversity (Box 4). The profound need to increase the sampling of diverse groups is demonstrated by the lack of diversity in genomic studies, and the potential for health disparities caused by the over-representation of European-ancestry populations149,150,151 (Fig. 4). In 2016, 81% of GWAS data were from studies conducted on European populations149. Although this is an improvement from 96% in 2009, most non-European populations still lack appropriate representation. The problem is more extreme for many phenotypes or traits of interest. For example, only 1.2% of the studies in a survey of 569 GWAS on neurological phenotypes included individuals of African ancestry150,152.

a | Interactions between the maternal killer cell inhibitory receptor (KIR) genotype and the fetal trophoblasts illustrate evolutionary trade-offs in pregnancy. Birthweight is under stabilizing selection in human populations. The interaction between maternal KIR genotypes (a diversity of which are maintained in the population) and the fetal trophoblasts influence birthweight. African (AFR) populations, relative to European (EUR) populations, maintain larger proportions of the KIR AA haplotype176, which is associated with improved maternal immune response to some viral challenges; however, it is also associated with low birthweight. Alternatively, the KIR BB haplotype is associated with higher birthweight but increased risk of pre-eclampsia. b | Current strategies for predicting genetic risk are confounded by a lack of inclusion of diverse human populations. Thus, they are more likely to fail in genetic risk prediction in populations that are under-represented in genetic databases. For example, polygenic risk score (PRS) models trained on European populations often perform poorly when applied to African populations. This poor performance stems from the fact that the genetic diversity of African populations, differences in effect sizes between populations and differential evolutionary pressures are not taken into account. The weights for each variant (blue circles) in the PRS derived from genome-wide association studies are signified by w1, w2 and w3. c | Population-specific adaptation and genetic hitch-hiking can produce different disease risk between populations. Haplotypes with protective effects against disease may rise to high frequency in specific populations through genetic hitch-hiking with nearby alleles under selection for a different trait. For example, selection for lighter skin pigmentation caused a haplotype that carried a variant associated with lighter skin (blue circle) to increase in frequency in European populations compared with African populations. This haplotype also carried a variant protective against prostate cancer (blue triangle).
Ancestry biases in genomic databases and GWAS propagate through other strategies that are designed to translate population genetic insights to the clinic, such as polygenic risk scores (PRSs)153,154 (Fig. 4b). PRSs hold the promise of predicting medical outcomes from genomic data alone. However, the evolutionary perspective suggests that the genetic architecture of diseases should differ between populations due to the effects of the demographic and environmental differences discussed above. Indeed, many PRSs generalize poorly across populations and are subject to biases155,156. Prioritization of Mendelian disease genes is also challenging in under-represented populations. Generally, African-ancestry individuals have significantly more variants, yet we know less about the pathogenicity of variants that are absent from or less frequent in European populations157. Patients of African and Asian ancestry are currently more likely than those of European ancestry to receive ambiguous genetic test results after exome sequencing or be told that they have variants of uncertain significance (VUS)158. Indeed, disease-causing variants of African origin are under-represented in common databases159. This under-representation covers a range of phenotypic traits and outcomes, including interpreting the effects of CYP2D6 variants on drug response160,161, risk identification and classification for breast cancer across populations162, and disparate effects of GWAS associations for traits including body mass index (BMI) and type 2 diabetes in non-European populations163. In a study on hypertrophic cardiomyopathy, benign variants in African Americans were incorrectly classified as pathogenic on the basis of GWAS results from a European ancestry cohort. Inclusion of individuals of African descent in the initial GWAS could have prevented these errors164.
Conclusions and future perspectives
All diseases have evolutionary histories, and the signatures of those histories are archived in our genomes. Recent advances in genomics are enabling us to read these histories with high accuracy, resolution and depth. Insights from evolutionary genomics reveal that there is not one answer to the question of why we get sick. Rather, diseases affect patchworks of ancient biological systems that evolved over millennia, and although the systems involved are ancient, the variation that is relevant for human disease is recent. Furthermore, evolutionary genomics approaches have the power to identify potential mechanisms, pathways and networks and to suggest clinical targets. In this context, we argue that an evolutionary perspective can aid the implementation of precision medicine in the era of genome sequencing and editing165 (Box 4).
Combining knowledge of evolutionary events along the human lineage with results from recent genomic studies provides an explanatory framework beyond descriptions of disease risk or association. For example, a recent analysis of the higher incidence of prostate cancer among men of African ancestry not only discovered a set of genetic variants associated with increased risk, but also used measures of selection to propose an evolutionary explanation of genetic hitch-hiking for the lower incidence in non-African populations166. Haplotypes with protective effects against prostate cancer may have risen to higher frequency in non-African populations because of selection on the nearby variants associated with skin pigmentation (Fig. 4c). Thus, evolutionary perspectives not only help answer the question of how we get sick but also why we get sick.
As the genetic information available from diverse populations increases, we can specifically map the genetics of traits in different populations and more precisely define disease risk on an individual basis167,168. However, we emphasize that environmental and social factors are major determinants of disease risk that often contribute more than genetics, and thus must be prioritized. Studying diverse human populations will provide additional power to discover trait-associated loci and understand genetic architecture across different environmental exposures and evolutionary histories150,169. For example, a GWAS with small sample size in a Greenlandic Inuit population found a variant in a fatty-acid enzyme that affects height in both this population and European populations170. Previous GWAS probably missed this variant due to its low frequency in European populations (0.017 compared with 0.98 in the Inuit); nevertheless, it has a much greater effect on height than other variants previously identified through GWAS170. Similarly, a recent study of height in 3,000 Peruvians identified another variant with an even greater influence on height171. The growth of large DNA biobanks in which hundreds of thousands of patients’ EHRs are linked to DNA samples represents a substantial untapped resource for evolutionary medicine5,86,87. These data enable testing of the functional effects of genetic variants on diverse traits at minimal additional cost. Shifting from single-ancestry GWAS to trans-ethnic or multi-ethnic GWAS will capitalize on the benefits of both a larger sample size and the inherent diversity of human populations for replication of established signals and discovery of new ones172,173,174,175.
Although evolutionary assumptions are tacit in medical practice, until recently self-reported family history remained the best representation of our evolutionary ancestry’s imprint on our disease risk. However, a family history cannot fully capture the complex evolutionary and demographic history of each individual. New technologies now enable the collection and interpretation of an individual’s family history in a much longer and complementary form — their genome. New data and methods are substantially increasing the resolution and depth with which these histories can be quantified, providing opportunities for evolution to inform medical practice.
References
Alföldi, J. & Lindblad-Toh, K. Comparative genomics as a tool to understand evolution and disease. Genome Res. 23, 1063–1068 (2013).
Meadows, J. R. S. & Lindblad-Toh, K. Dissecting evolution and disease using comparative vertebrate genomics. Nat. Rev. Genet. 18, 624–636 (2017).
Shendure, J. et al. DNA sequencing at 40: past, present and future. Nature 550, 345–353 (2017).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Dewey, F. E. et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 354, aaf6814 (2016).
McCarty, C. A. et al. The eMERGE Network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med. Genomics 4, 13 (2011).
Timpson, N. J., Greenwood, C. M. T., Soranzo, N., Lawson, D. J. & Richards, J. B. Genetic architecture: the shape of the genetic contribution to human traits and disease. Nat. Rev. Genet. 19, 110–124 (2018).
Orlando, L., Gilbert, M. T. P. & Willerslev, E. Reconstructing ancient genomes and epigenomes. Nat. Rev. Genet. 16, 395–408 (2015).
Skoglund, P. & Mathieson, I. Ancient genomics of modern humans: the first decade. Annu. Rev. Genomics Hum. Genet. 19, 381–404 (2018).
Ramamoorthy, A., Yee, S. W. & Karnes, J. Unveiling the genetic architecture of human disease for precision medicine. Clin. Transl. Sci. 12, 3–5 (2019).
Stearns, S. C. & Medzhitov, R. Evolutionary Medicine (Sinauer Associates, 2016). This foundational textbook provides an introduction to the field of evolutionary medicine.
Carroll, S. P. et al. Applying evolutionary biology to address global challenges. Science 346, 1245993 (2014).
Young, M. J. & Copeland, W. C. Human mitochondrial DNA replication machinery and disease. Curr. Opin. Genet. Dev. 38, 52–62 (2016).
Muñoz, S. & Méndez, J. DNA replication stress: from molecular mechanisms to human disease. Chromosoma 126, 1–15 (2017).
Ackermann, M., Chao, L., Bergstrom, C. T. & Doebeli, M. On the evolutionary origin of aging. Aging Cell 6, 235–244 (2007).
Flatt, T. & Partridge, L. Horizons in the evolution of aging. BMC Biol. 16, 93 (2018).
Rokas, A. The origins of multicellularity and the early history of the genetic toolkit for animal development. Annu. Rev. Genet. 42, 235–251 (2008).
Albuquerque, T. A. F., Drummond do Val, L., Doherty, A. & de Magalhães, J. P. From humans to hydra: patterns of cancer across the tree of life. Biol. Rev. 93, 1715–1734 (2018).
Crespi, B. & Summers, K. Evolutionary biology of cancer. Trends Ecol. Evol. 20, 545–552 (2005).
Kinzler, K. W. & Vogelstein, B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 386, 761 (1997).
Michor, F., Iwasa, Y. & Nowak, M. A. Dynamics of cancer progression. Nat. Rev. Cancer 4, 197–205 (2004).
Domazet-Lošo, T. & Tautz, D. Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa. BMC Biol. 8, 66 (2010).
Thomas, F. et al. Applying ecological and evolutionary theory to cancer: a long and winding road. Evol. Appl. 6, 1–10 (2013).
Vogelstein, B. et al. Cancer genome landscapes. Science 340, 1546–1558 (2013).
Aktipis, C. A. & Nesse, R. M. Evolutionary foundations for cancer biology. Evol. Appl. 6, 144–159 (2013).
Gerlinger, M. et al. Cancer: evolution within a lifetime. Annu. Rev. Genet. 48, 215–236 (2014).
Enriquez-Navas, P. M., Wojtkowiak, J. W. & Gatenby, R. A. Application of evolutionary principles to cancer therapy. Cancer Res. 75, 4675–4680 (2015).
Gerstung, M. et al. The evolutionary history of 2,658 cancers. Nature 578, 122–128 (2020). This study analyses the evolution of somatic mutational processes and reconstructs the life history of 38 different types of cancer based on whole-genome sequences from 2,658 tumours.
Hibino, T. et al. The immune gene repertoire encoded in the purple sea urchin genome. Dev. Biol. 300, 349–365 (2006).
Jones, J. D. G. & Dangl, J. L. The plant immune system. Nature 444, 323–329 (2006).
Flajnik, M. F. & Kasahara, M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat. Rev. Genet. 11, 47–59 (2010).
Chuong, E. B., Elde, N. C. & Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science. 351, 1083–1087 (2016). This study provides an example of the co-option of ancient genomic elements (endogenous retroviruses) to create new regulatory elements in mammalian immune systems.
Dunne, D. W. & Cooke, A. A worm’s eye view of the immune system: consequences for evolution of human autoimmune disease. Nat. Rev. Immunol. 5, 420–426 (2005).
Shubin, N., Tabin, C. & Carroll, S. Deep homology and the origins of evolutionary novelty. Nature 457, 818–823 (2009).
Shubin, N. Your Inner Fish: A Journey into the 3.5-billion-year History of the Human Body (Knopf Doubleday, 2008).
Guernsey, M. W., Chuong, E. B., Cornelis, G., Renfree, M. B. & Baker, J. C. Molecular conservation of marsupial and eutherian placentation and lactation. eLife 6, e27450 (2017).
Abbot, P. & Capra, J. A. What is a placental mammal anyway?: many developmental functions in marsupials and eutherian mammals are accomplished by different tissues, but similar genes. eLife 6, e30994 (2017).
Katz, P. S. ‘Model organisms’ in the light of evolution. Curr. Biol. 26, R649–R650 (2016).
Bolker, J. A. Selection of models: evolution and the choice of species for translational research. Brain Behav. Evol. 93, 82–91 (2019). This review provides guidelines for considering evolutionary context in the selection of model organisms for translational research.
Bart van der Worp, H. et al. Can animal models of disease reliably inform human studies? PLoS Med. 7, 1–8 (2010).
Mak, I. W. Y., Evaniew, N. & Ghert, M. Lost in translation: animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 6, 114–118 (2014).
Caulin, A. F. & Maley, C. C. Peto’s paradox: evolution’s prescription for cancer prevention. Trends Ecol. Evol. 26, 175–182 (2011).
Tollis, M. et al. Return to the sea, get huge, beat cancer: an analysis of cetacean genomes including an assembly for the humpback whale (Megaptera novaeangliae). Mol. Biol. Evol. 36, 1746–1763 (2019).
Sulak, M. et al. TP53 copy number expansion is associated with the evolution of increased body size and an enhanced DNA damage response in elephants. eLife 5, e11994 (2016).
Vazquez, J. M., Sulak, M., Chigurupati, S. & Lynch, V. J. A zombie LIF gene in elephants is upregulated by TP53 to induce apoptosis in response to DNA damage. Cell Rep. 24, 1765–1776 (2018). This study finds a molecular explanation for Peto’s paradox — why do elephants not have high cancer rates given their large number of cells? — in the refunctionalization of LIF6, a pseudogene with pro-apoptotic functions.
O’Bleness, M., Searles, V. B., Varki, A., Gagneux, P. & Sikela, J. M. Evolution of genetic and genomic features unique to the human lineage. Nat. Rev. Genet. 13, 853–866 (2012).
Patterson, N., Richter, D. J., Gnerre, S., Lander, E. S. & Reich, D. Genetic evidence for complex speciation of humans and chimpanzees. Nature 441, 1103–1108 (2006).
Crespi, B. J. & Go, M. C. Diametrical diseases reflect evolutionary-genetic tradeoffs: evidence from psychiatry, neurology, rheumatology, oncology and immunology. Evol. Med. Public Health 2015, 216–253 (2015). This article describes the broad imprint of genetic trade-offs on diverse human diseases.
Stearns, S. C. Trade-offs in life-history evolution. Funct. Ecol. 3, 259 (1989).
Ricklefs, R. E. & Wikelski, M. The physiology/life-history nexus. Trends Ecol. Evol. 17, 462–468 (2002).
Zera, A. J. & Harshman, L. G. The physiology of life history trade-offs in animals. Annu. Rev. Ecol. Syst. 32, 95–126 (2001).
Brady, S. P. et al. Understanding maladaptation by uniting ecological and evolutionary perspectives. Am. Nat. 194, 495–515 (2019).
Ellison, P. T. Evolutionary tradeoffs. Evol. Med. Public Health 2014, 93 (2014).
Al-Nbaheen, M. et al. Human stromal (mesenchymal) stem cells from bone marrow, adipose tissue and skin exhibit differences in molecular phenotype and differentiation potential. Stem Cell Rev. Rep. 9, 32–43 (2013).
Jacqueline, C. et al. Cancer: a disease at the crossroads of trade-offs. Evol. Appl. 10, 215–225 (2017).
Marques-Bonet, T. & Eichler, E. E. The evolution of human segmental duplications and the core duplicon hypothesis. Cold Spring Harb. Symposia Quant. Biol. 74, 355–362 (2009).
Dennis, M. Y. & Eichler, E. E. Human adaptation and evolution by segmental duplication. Curr. Opin. Genet. Dev. 41, 44–52 (2016).
Fiddes, I. T. et al. Human-specific NOTCH2NL genes affect notch signaling and cortical neurogenesis. Cell 173, 1356–1369.e22 (2018).
Suzuki, I. K. et al. Human-specific NOTCH2NL genes expand cortical neurogenesis through Delta/Notch regulation. Cell 173, 1370–1384.e16 (2018).
Guerrier, S. et al. The F-BAR domain of srGAP2 induces membrane protrusions required for neuronal migration and morphogenesis. Cell 138, 990–1004 (2009).
Dennis, M. Y. et al. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell 149, 912–922 (2012).
Florio, M. et al. Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 347, 1465–1470 (2015).
Antonacci, F. et al. Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolutionary instability. Nat. Genet. 46, 1293–1302 (2014).
Sikela, J. M. & Searles Quick, V. B. Genomic trade-offs: are autism and schizophrenia the steep price of the human brain? Hum. Genet. 137, 1–13 (2018).
Srinivasan, S. et al. Genetic markers of human evolution are enriched in schizophrenia. Biol. Psychiatry 80, 284–292 (2016).
Polimanti, R. & Gelernter, J. Widespread signatures of positive selection in common risk alleles associated to autism spectrum disorder. PLoS Genet. 13, 1–14 (2017).
Xu, K., Schadt, E. E., Pollard, K. S., Roussos, P. & Dudley, J. T. Genomic and network patterns of schizophrenia genetic variation in human evolutionary accelerated regions. Mol. Biol. Evol. 32, 1148–1160 (2015).
Doan, R. N. et al. Mutations in human accelerated regions disrupt cognition and social behavior. Cell 167, 341–354.e12 (2016).
Sharp, P. M. & Hahn, B. H. The evolution of HIV-1 and the origin of AIDS. Phil. Trans. R. Soc. B 365, 2487–2494 (2010).
Nguyen, D. H., Hurtado-Ziola, N., Gagneux, P. & Varki, A. Loss of Siglec expression on T lymphocytes during human evolution. Proc. Natl Acad. Sci. USA 103, 7765–7770 (2006).
Soto, P. C., Karris, M. Y., Spina, C. A., Richman, D. D. & Varki, A. Cell-intrinsic mechanism involving Siglec-5 associated with divergent outcomes of HIV-1 infection in human and chimpanzee CD4 T cells. J. Mol. Med. 91, 261–270 (2013).
Arora, G., Polavarapu, N. & McDonald, J. F. Did natural selection for increased cognitive ability in humans lead to an elevated risk of cancer? Med. Hypotheses 73, 453–456 (2009).
Varki, N. M. & Varki, A. On the apparent rarity of epithelial cancers in captive chimpanzees. Phil. Trans. R. Soc. B 370, 20140225 (2015).
Varki, A. Loss of N-glycolylneuraminic acid in humans: mechanisms, consequences, and implications for hominid evolution. Yearb. Phys. Anthropol. 44, 54–69 (2001).
Chou, H. H. et al. A mutation in human CMP-sialic acid hydroxylase occurred after the Homo–Pan divergence. Proc. Natl Acad. Sci. USA 95, 11751–11756 (1998).
Irie, A., Koyamat, S., Kozutsumi, Y., Kawasaki, T. & Suzuki, A. The molecular basis for the absence of N-glycolylneuraminic acid in humans. J. Biol. Chem. 273, 15866–15871 (1998).
Martin, M. J., Rayner, J. C., Gagneux, P., Barnwell, J. W. & Varki, A. Evolution of human–chimpanzee differences in malaria susceptibility: relationship to human genetic loss of N-glycolylneuraminic acid. Proc. Natl Acad. Sci. USA 102, 12819–12824 (2005).
Varki, A. & Gagneux, P. Human-specific evolution of sialic acid targets: explaining the malignant malaria mystery? Proc. Natl Acad. Sci. USA 106, 14739–14740 (2009).
Deng, L. et al. Host adaptation of a bacterial toxin from the human pathogen Salmonella typhi. Cell 159, 1290–1299 (2014).
Varki, A. Uniquely human evolution of sialic acid genetics and biology. Proc. Natl Acad. Sci. USA 107, 8939–8946 (2010). This article demonstrates that genes involved in sialic acid biology are a ‘hot spot’ of genetic and physiological changes during recent evolution with implications for human-specific disease.
Quach, H. & Quintana-Murci, L. Living in an adaptive world: genomic dissection of the genus Homo and its immune response. J. Exp. Med. 214, 877–894 (2017).
Quintana-Murci, L. Human immunology through the lens of evolutionary genetics. Cell 177, 184–199 (2019).
Seluanov, A., Gladyshev, V. N., Vijg, J. & Gorbunova, V. Mechanisms of cancer resistance in long-lived mammals. Nat. Rev. Cancer 18, 433–441 (2018).
He, S. & Sharpless, N. E. Senescence in health and disease. Cell 169, 1000–1011 (2017).
Coppola, L. et al. Biobanking in health care: evolution and future directions. J. Transl. Med. 17, 172 (2019).
Bowton, E. et al. Biobanks and electronic medical records: enabling cost-effective research. Sci. Transl. Med. 6, 234cm3 (2014).
Bush, W. S., Oetjens, M. T. & Crawford, D. C. Unravelling the human genome–phenome relationship using phenome-wide association studies. Nat. Rev. Genet. 17, 129–145 (2016).
van Manen, D. et al. Genome-wide association scan in HIV-1-infected individuals identifying variants influencing disease course. PLoS ONE 6, e22208 (2011).
Sáez-Cirión, A. & Pancino, G. HIV controllers: a genetically determined or inducible phenotype? Immunol. Rev. 254, 281–294 (2013).
McLaren, P. J. et al. Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc. Natl Acad. Sci. USA 112, 14658–14663 (2015).
Parham, P. & Ohta, T. Population biology of antigen presentation by MHC class I molecules. Science 272, 67–74 (1996).
Klepiela, P. et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432, 769–774 (2004).
Martin, M. P. et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat. Genet. 39, 733–740 (2007).
De Groot, N. G. et al. AIDS-protective HLA-B*27/B*57 and chimpanzee MHC class I molecules target analogous conserved areas of HIV-1/SIVcpz. Proc. Natl Acad. Sci. USA 107, 15175–15180 (2010).
Fan, S., Hansen, M. E. B., Lo, Y. & Tishkoff, S. A. Going global by adapting local: a review of recent human adaptation. Science 354, 54–59 (2016).
Marciniak, S. & Perry, G. H. Harnessing ancient genomes to study the history of human adaptation. Nat. Rev. Genet. 18, 659–674 (2017).
Nielsen, R. et al. Tracing the peopling of the world through genomics. Nature 541, 302–310 (2017).
Azevedo, L., Serrano, C., Amorim, A. & Cooper, D. N. Trans-species polymorphism in humans and the great apes is generally maintained by balancing selection that modulates the host immune response. Hum. Genomics 9, 21 (2015).
Gravel, S. When is selection effective? Genetics 203, 451–462 (2016).
Prüfer, K. et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49 (2014).
Vernot, B. & Akey, J. M. Resurrecting surviving Neandertal linages from modern human genomes. Science 343, 1017–1021 (2014).
Chen, L., Wolf, A. B., Fu, W., Li, L. & Akey, J. M. Identifying and interpreting apparent Neanderthal ancestry in African individuals. Cell 180, 677–687.e16 (2020).
Hammer, M. F., Woerner, A. E., Mendez, F. L., Watkins, J. C. & Wall, J. D. Genetic evidence for archaic admixture in Africa. Proc. Natl Acad. Sci. USA 108, 15123–15128 (2011).
Hsieh, P. H. et al. Model-based analyses of whole-genome data reveal a complex evolutionary history involving archaic introgression in Central African Pygmies. Genome Res. 26, 291–300 (2016).
Lachance, J. et al. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell 150, 457–469 (2012).
Keinan, A. & Clark, A. G. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science 336, 740–743 (2012).
Fu, W. & Akey, J. M. Selection and adaptation in the human genome. Annu. Rev. Genom. Hum. Genet. 14, 467–489 (2013).
Uricchio, L. H., Zaitlen, N. A., Ye, C. J., Witte, J. S. & Hernandez, R. D. Selection and explosive growth alter genetic architecture and hamper the detection of causal rare variants. Genome Res. 26, 863–873 (2016). This study demonstrates how the recent demographic and selective history of human populations poses challenges for the identification and interpretation of genetic variants that cause disease.
Fu, Q. et al. A revised timescale for human evolution based on ancient mitochondrial genomes. Curr. Biol. 23, 553–559 (2013).
Do, R. et al. No evidence that selection has been less effective at removing deleterious mutations in Europeans than in Africans. Nat. Genet. 47, 126–131 (2015).
Simons, Y. B., Turchin, M. C., Pritchard, J. K. & Sella, G. The deleterious mutation load is insensitive to recent population history. Nat. Genet. 46, 220–224 (2014).
Simons, Y. B. & Sella, G. The impact of recent population history on the deleterious mutation load in humans and close evolutionary relatives. Curr. Opin. Genet. Dev. 41, 150–158 (2016).
Sella, G. & Barton, N. H. Thinking about the evolution of complex traits in the era of genome-wide association studies. Annu. Rev. Genomics Hum. Genet. 20, 461–493 (2019).
Kim, M. S., Patel, K. P., Teng, A. K., Berens, A. J. & Lachance, J. Genetic disease risks can be misestimated across global populations. Genome Biol. 19, 179 (2018).
Rees, J. S., Castellano, S. & Andrés, A. M. The genomics of human local adaptation. Trends Genet. 36, 415–428 (2020).
Huerta-Sanchez, E. et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512, 194–197 (2014).
Mathieson, I. et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 528, 499–503 (2015).
Di Rienzo, A. & Hudson, R. R. An evolutionary framework for common diseases: the ancestral-susceptibility model. Trends Genet. 21, 596–601 (2005).
Manus, M. B. Evolutionary mismatch. Evol. Med. Public Health 2018, 190–191 (2018).
Gibson, G. Decanalization and the origin of complex disease. Nat. Rev. Genet. 10, 134–140 (2009).
Lachance, J. Disease-associated alleles in genome-wide association studies are enriched for derived low frequency alleles relative to HapMap and neutral expectations. BMC Med. Genomics 3, 57 (2010).
Kwiatkowski, D. P. How malaria has affected the human genome and what human genetics can teach us about malaria. Am. J. Hum. Genet. 77, 171–192 (2005).
Minster, R. L. et al. A thrifty variant in CREBRF strongly influences body mass index in Samoans. Nat. Genet. 48, 1049–1054 (2016). This paper argues that bottlenecks and extreme past environments created a ‘thrifty’ gene variant in Samoans that is metabolically beneficial in times of starvation, but may contribute to obesity risk in modern environments.
Nédélec, Y. et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell 167, 657–669.e21 (2016). This work shows that the human inflammatory response differs based on the different evolutionary histories (including local adaptations and archaic introgression) of human populations.
Raj, T. et al. Common risk alleles for inflammatory diseases are targets of recent positive selection. Am. J. Hum. Genet. 92, 517–529 (2013).
Barber, M. F., Lee, E. M., Griffin, H. & Elde, N. C. Rapid evolution of primate type 2 immune response factors linked to asthma susceptibility. Genome Biol. Evol. 9, 1757–1765 (2017).
Smallwood, T. B. et al. Helminth immunomodulation in autoimmune disease. Front. Immunol. 8, 453 (2017).
Sobotková, K. et al. Helminth therapy—from the parasite perspective. Trends Parasitol. 35, 501–515 (2019).
Harris, K. & Nielsen, R. The genetic cost of neanderthal introgression. Genetics 203, 881–891 (2016).
Juric, I., Aeschbacher, S. & Coop, G. The strength of selection against Neanderthal introgression. PLoS Genet. 12, 1–25 (2016).
Dannemann, M., Andrés, A. M. & Kelso, J. Introgression of Neandertal- and Denisovan-like haplotypes contributes to adaptive variation in human Toll-like receptors. Am. J. Hum. Genet. 98, 22–33 (2016).
Simonti, C. N. The phenotype legacy of admixture between modern humans and Neandertals. Science 351, 737–742 (2016). This paper uses a large biobank cohort to demonstrate that admixture between Neanderthals and the ancestors of modern Eurasians influences disease risk.
Quach, H. et al. Genetic adaptation and neandertal admixture shaped the immune system of human populations. Cell 167, 643–656.e17 (2016).
Dannemann, M. & Kelso, J. The contribution of neanderthals to phenotypic variation in modern humans. Am. J. Hum. Genet. 101, 578–589 (2017).
Sankararaman, S. et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507, 354–357 (2014).
Sankararaman, S., Mallick, S., Patterson, N. & Reich, D. The combined landscape of Denisovan and Neanderthal ancestry in present-day humans. Curr. Biol. 26, 1241–1247 (2016).
McCoy, R. C., Wakefield, J. & Akey, J. M. Impacts of Neanderthal-introgressed sequences on the landscape of human gene expression. Cell 168, 916–927.e12 (2017).
McArthur, E., Rinker, D. C. & Capra, J. A. Quantifying the contribution of Neanderthal introgression to the heritability of complex traits. Preprint at bioRxiv https://doi.org/10.1101/2020.06.08.140087 (2020).
Skov, L. et al. The nature of Neanderthal introgression revealed by 27,566 Icelandic genomes. Nature 582, 78–83 (2020).
Rinker, D. C. et al. Neanderthal introgression reintroduced functional ancestral alleles lost in Eurasian populations. Nat. Ecol. Evol. 4, 1332–1341 (2020).
Racimo, F., Marnetto, D. & Huerta-Sánchez, E. Signatures of archaic adaptive introgression in present-day human populations. Mol. Biol. Evol. 34, 296–317 (2017).
Bigham, A. W. & Lee, F. S. Human high-altitude adaptation: forward genetics meets the HIF pathway. Genes Dev. 28, 2189–2204 (2014).
Deschamps, M. et al. Genomic signatures of selective pressures and introgression from Archaic hominins at human innate immunity genes. Am. J. Hum. Genet. 98, 5–21 (2016).
Enard, D. & Petrov, D. A. Evidence that RNA viruses drove adaptive introgression between Neanderthals and modern humans. Cell 175, 360–371.e13 (2018).
Gittelman, R. M. et al. Archaic hominin admixture facilitated adaptation to out-of-Africa environments. Curr. Biol. 26, 3375–3382 (2016).
Zeberg, H. & Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 587, 610–612 (2020).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018). This study develops PRSs for five common diseases and argues that these scores could stratify patients into clinically meaningful risk groups.
Popejoy, A. B. & Fullerton, S. M. Genomics is failing on diversity. Nature 538, 161–164 (2016).
Kelly, D. E., Hansen, M. E. B. & Tishkoff, S. A. Global variation in gene expression and the value of diverse sampling. Curr. Opin. Syst. Biol. 1, 102–108 (2017).
Hindorff, L. A. et al. Prioritizing diversity in human genomics research. Nat. Rev. Genet. 19, 175–185 (2018).
Quansah, E. & McGregor, N. W. Towards diversity in genomics: the emergence of neurogenomics in Africa? Genomics 110, 1–9 (2018).
Mostafavi, H. et al. Variable prediction accuracy of polygenic scores within an ancestry group. eLife 9, e48376 (2020).
Torkamani, A., Wineinger, N. E. & Topol, E. J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 19, 581–590 (2018).
Martin, A. R. et al. Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet. 100, 635–649 (2017). This study demonstrates that PRSs for eight phenotypes using existing GWAS summary statistics are inconsistent for populations that are genetically distant from the original GWAS population.
Martin, A. R. et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 51, 584–591 (2019).
Sirugo, G., Williams, S. M. & Tishkoff, S. A. The missing diversity in human genetic studies. Cell 177, 26–31 (2019).
Petrovski, S. & Goldstein, D. B. Unequal representation of genetic variation across ancestry groups creates healthcare inequality in the application of precision medicine. Genome Biol. 17, 157 (2016).
Kessler, M. D. et al. Challenges and disparities in the application of personalized genomic medicine to populations with African ancestry. Nat. Commun. 7, 12521 (2016).
Maisano Delser, P. & Fuselli, S. Human loci involved in drug biotransformation: worldwide genetic variation, population structure, and pharmacogenetic implications. Hum. Genet. 132, 563–577 (2013).
Madian, A. G., Wheeler, H. E., Jones, R. B. & Dolan, M. E. Relating human genetic variation to variation in drug responses. Trends Genet. 28, 487–495 (2012).
Huo, D. et al. Comparison of breast cancer molecular features and survival by African and European ancestry in The Cancer Genome Atlas. JAMA Oncol. 3, 1654–1662 (2017).
Carlson, C. S. et al. Generalization and dilution of association results from European GWAS in populations of non-European ancestry: the PAGE study. PLoS Biol. 11, e1001661 (2013).
Manrai, A. K. et al. Genetic misdiagnoses and the potential for health disparities. N. Engl. J. Med. 375, 655–665 (2016). This article demonstrates the misclassification of ‘causal’ hypertrophic cardiomyopathy variants in diverse individuals based on the lack of diversity in existing genetic databases.
Lynch, M. Mutation and human exceptionalism: our future genetic load. Genetics 202, 869–875 (2016). This article explores the implications of the recent removal of many selective pressures from the natural environment on the human genome, risk for disease and precision medicine.
Lachance, J. et al. Genetic hitchhiking and population bottlenecks contribute to prostate cancer disparities in men of African descent. Cancer Res. 78, 2432–2443 (2018). This analysis shows that haplotypes with protective effects against prostate cancer may have risen to higher frequency in non-African populations because of selection on the nearby variants associated with skin pigmentation.
Márquez-Luna, C. et al. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet. Epidemiol. 41, 811–823 (2017).
Shi, H. et al. Localizing components of shared transethnic genetic architecture of complex traits from GWAS summary data. Am. J. Hum. Genet. 106, 805–817 (2020).
Crawford, N. G. et al. Loci associated with skin pigmentation identified in African populations. Science. 358, eaan8433 (2017).
Fumagalli, M. et al. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science 349, 1343–1347 (2015).
Asgari, S. et al. A positively selected FBN1 missense variant reduces height in Peruvian individuals. Nature 582, 234–239 (2020).
Li, Y. R. & Keating, B. J. Trans-ethnic genome-wide association studies: advantages and challenges of mapping in diverse populations. Genome Med. 6, 1–14 (2014).
Cook, J. P. & Morris, A. P. Multi-ethnic genome-wide association study identifies novel locus for type 2 diabetes susceptibility. Eur. J. Hum. Genet. 24, 1175–1180 (2016).
Mägi, R. et al. Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution. Hum. Mol. Genet. 26, 3639–3650 (2017).
Fernández-Rhodes, L. et al. Trans-ethnic fine-mapping of genetic loci for body mass index in the diverse ancestral populations of the Population Architecture using Genomics and Epidemiology (PAGE) study reveals evidence for multiple signals at established loci. Hum. Genet. 136, 771–800 (2017).
Hiby, S. E. et al. Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J. Exp. Med. 200, 957–965 (2004).
Yang, J. et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 47, 1114–1120 (2015).
Corbett, S., Courtiol, A., Lummaa, V., Moorad, J. & Stearns, S. The transition to modernity and chronic disease: mismatch and natural selection. Nat. Rev. Genet. 19, 419–430 (2018).
Rodríguez, J. A. et al. Antagonistic pleiotropy and mutation accumulation influence human senescence and disease. Nat. Ecol. Evol. 1, 1–5 (2017).
Byars, S. G. et al. Genetic loci associated with coronary artery disease harbor evidence of selection and antagonistic pleiotropy. PLoS Genet. 13, e1006328 (2017).
Gluckman, P. D. & Hanson, M. A. Changing times: the evolution of puberty. Mol. Cell. Endocrinol. 254–255, 26–31 (2006).
Arnold, A. J. & Fristrup, K. The theory of evolution by natural selection: a hierarchical expansion. Paleobiology 8, 113–129 (1982).
Futuyma, D. J. Evolutionary constraint and ecological consequences. Evolution 64, 1865–1884 (2010).
Gluckman, P. & Hanson, M. Developmental Origins of Health and Disease (Cambridge Univ. Press, 2006).
Barreiro, L. B. & Quintana-Murci, L. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat. Rev. Genet. 11, 17–30 (2010).
Fay, J. C. Disease consequences of human adaptation. Appl. Transl. Genomics 2, 42–47 (2013).
Frank, S. A. & Crespi, B. J. Pathology from evolutionary conflict, with a theory of X chromosome versus autosome conflict over sexually antagonistic traits. Proc. Natl Acad. Sci. USA 108, 10886–10893 (2011).
Abbot, P. & Rokas, A. Mammalian pregnancy. Curr. Biol. 27, R127–R128 (2017).
Aungst, H. et al. in Power, M. & Schulkin J. in Integrating Evolutionary Biology into Medical Education: For Maternal and Child Healthcare Students, Clinicians, and Scientists Ch. 5 (eds Schulkin, J. & and Power, M.) 91–118 (Oxford Univ. Press, 2019).
Redman, C. W. G. & Sargent, I. L. Immunology of pre-eclampsia. Am. J. Reprod. Immunol. 63, 534–543 (2010).
Than, N. G. et al. Integrated systems biology approach identifies novel maternal and placental pathways of preeclampsia. Front. Immunol. 9, 1661 (2018).
Bergmann, A. et al. Reduction of circulating soluble Flt-1 alleviates preeclampsia-like symptoms in a mouse model. J. Cell. Mol. Med. 14, 1857–1867 (2010).
Turanov, A. A. et al. RNAi modulation of placental sFLT1 for the treatment of preeclampsia. Nat. Biotechnol. 36, 1164–1173 (2018).
Robertson, S. A. Preventing preeclampsia by silencing soluble Flt-1? N. Engl. J. Med. 380, 1080–1082 (2019).
Moorjani, P., Amorim, C. E. G., Arndt, P. F. & Przeworski, M. Variation in the molecular clock of primates. Proc. Natl Acad. Sci. USA 113, 10607–10612 (2016).
Marinić, M. & Lynch, V. J. Relaxed constraint and functional divergence of the progesterone receptor (PGR) in the human stem-lineage. PLoS Genet. 16, e1008666 (2020).
Zeberg, H., Kelso, J. & Pääbo, S. The Neandertal progesterone receptor. Mol. Biol. Evol. 37, 2655–2660 (2020).
Clark, A. G. et al. Positive selection in the human genome inferred from human–chimp–mouse orthologous gene alignments. Cold Spring Harb. Symp. Quant. Biol. 68, 479–486 (2003).
Chen, C. et al. The human progesterone receptor shows evidence of adaptive evolution associated with its ability to act as a transcription factor. Mol. Phylogenet. Evol. 47, 637–649 (2008).
LaBella, A. L. et al. Accounting for diverse evolutionary forces reveals mosaic patterns of selection on human preterm birth loci. Nat. Commun. 11, 3731 (2020). This article demonstrates that a mosaic of diverse selective pressures has shaped the genetic loci that influence risk for preterm birth, a common complex disease.
Schmidt, A., Morales-Prieto, D. M., Pastuschek, J., Fröhlich, K. & Markert, U. R. Only humans have human placentas: molecular differences between mice and humans. J. Reprod. Immunol. 108, 65–71 (2015).
Hammer, A. Immunological regulation of trophoblast invasion. J. Reprod. Immunol. 90, 21–28 (2011).
Erlebacher, A. Immunology of the maternal–fetal interface. Annu. Rev. Immunol. 31, 387–411 (2013).
Robinson, D. P. & Klein, S. L. Pregnancy and pregnancy-associated hormones alter immune responses and disease pathogenesis. Hormones Behav. 62, 263–271 (2012).
Kourtis, A. P., Read, J. S. & Jamieson, D. J. Pregnancy and infection. N. Engl. J. Med. 370, 2211–2218 (2014).
Hiby, S. E. et al. Maternal activating KIRs protect against human reproductive failure mediated by fetal HLA-C2. J. Clin. Invest. 120, 4102–4110 (2010).
Moffett, A., Chazara, O., Colucci, F. & Johnson, M. H. Variation of maternal KIR and fetal HLA-C genes in reproductive failure: too early for clinical intervention. Reprod. Biomed. Online 33, 763–769 (2016).
Moon, J. M., Capra, J. A., Abbot, P. & Rokas, A. Immune regulation in eutherian pregnancy: live birth coevolved with novel immune genes and gene regulation. BioEssays 41, 1900072 (2019).
Muehlenbachs, A., Fried, M., Lachowitzer, J., Mutabingwa, T. K. & Duffy, P. E. Natural selection of FLT1 alleles and their association with malaria resistance in utero. Proc. Natl Acad. Sci. USA 105, 14488–14491 (2008).
Andersson, D. I. et al. Antibiotic resistance: turning evolutionary principles into clinical reality. FEMS Microbiol. Rev. 44, 171–188 (2020).
Enriquez-Navas, P. M. et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci. Transl. Med. 8, 327ra24 (2016).
Gatenby, R. A. & Brown, J. S. Integrating evolutionary dynamics into cancer therapy. Nat. Rev. Clin. Oncol. 17, 675–686 (2020).
Fairlamb, A. H., Gow, N. A. R., Matthews, K. R. & Waters, A. P. Drug resistance in eukaryotic microorganisms. Nat. Microbiol. 1, 16092 (2016).
Li, R., Chen, Y., Ritchie, M. D. & Moore, J. H. Electronic health records and polygenic risk scores for predicting disease risk. Nat. Rev. Genet. 21, 493–502 (2020).
Duncan, L. et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun. 10, 3328 (2019).
Acknowledgements
The authors thank L. Muglia and members of the Capra, Rokas and Abbot laboratories for helpful discussions. They also thank the National Institutes of Health (NIH) (T32LM012412 to M.L.B. and R35GM127087 to J.A.C.), the Burroughs Wellcome Fund Preterm Birth Initiative (A.R. and J.A.C.), the March of Dimes Prematurity Research Center Ohio Collaborative (P.A., A.R. and J.A.C.) and the American Heart Association (A.A.) for support.
Author information
Authors and Affiliations
Contributions
The authors contributed to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- Genetic architecture
-
All aspects of the genetic variants that influence variation of a trait in a population. Commonly studied attributes of genetic architecture include the number of genetic loci that influence a trait, the frequency of these variants, the magnitudes of their effects and how they interact with one another and the environment. The genetic architectures of traits vary along these axes; for example, some traits are influenced by many common variants of small effect, whereas others are driven by a few rare variants of large effect.
- Reaction norms
-
Representations of how the expressed phenotype for a genotype varies in response to a range of environments. Reaction norms can be used to illustrate many of the concepts described in this Review, including evolutionary mismatches and antagonistic pleiotropy.
- Evolutionary trade-off
-
Adaptations that are advantageous for one phenotype have costs for others. Evolutionary trade-offs often result when genes influence multiple phenotypes (pleiotropy) or when there is a limited resource that must be apportioned to different functions. Because of trade-offs, there is not an optimal genotype across all environments.
- Pleiotropic
-
Pertaining to pleiotropy, which is when a genetic locus (for example, a gene or regulatory element) has effects on multiple unrelated phenotypes. Antagonistic pleiotropy results when a locus has a beneficial effect on one trait and a detrimental effect on another.
- Homology
-
Similarity in traits, bodily structures or genomic sequences due to shared ancestry between two species. Homology is considered when selecting model systems to study a particular phenotype; however, it does not guarantee underlying functional or mechanistic similarity.
- Diametric diseases
-
Linked diseases for which decreasing the risk for one increases the risk for the other, such as protection from infectious disease increasing risk for autoimmune disease. Diametric disorders result from evolutionary trade-offs.
- Viviparous
-
An animal that gives birth to live young, rather than laying eggs.
- Human accelerated regions
-
(HARs). Genomic loci conserved across mammalian species that experienced an increase in substitution rate specific to the human lineage. Genetic changes in HARs are responsible for some attributes of human-specific biology.
- Bottlenecks
-
Rapid decreases in the size of populations that lead to a decrease in genetic diversity. Genetic bottlenecks can be caused by environmental factors (such as famine or disease) or demographic factors (such as migration). The ancestors of most modern non-African populations experienced a bottleneck as they left Africa, which is often referred to as the out-of-Africa bottleneck.
- Introgression
-
The flow of genetic material between two species through interbreeding followed by backcrossing. Analyses of ancient DNA have revealed that introgression was common in human history over the past several hundred thousand years.
- Anatomically modern humans
-
(AMHs). Individuals, both modern and ancient, with the physical characteristics of humans (Homo sapiens) living today.
- Founder effects
-
Reduced genetic diversity as a result of a small number of individuals establishing a new population from a larger original population. Founder effects can lead to genetic conditions that were rare in the original population becoming common in the new population. Serial founder effects occurred as anatomically modern humans spread out of Africa and colonized the world.
- Mutation load
-
The component of the genetic load contributed by recent deleterious variants; other factors that contribute to the overall genetic load include the amount of heterozygote advantage and inbreeding.
- Admixture
-
The creation of novel genotypes from interbreeding between two genetically differentiated populations.
- Archaic hominin
-
Ancient individuals on the human lineage, such as Neanderthals and Denisovans, that diverged before the origin of anatomically modern humans. Use of this terminology is established in human evolutionary genetics, but is not consistent across fields due to historical differences in the use of taxonomic terms and the fluidity of the species concept in the presence of substantial introgression.
- Ancestral susceptibility model
-
A model proposing that ancestral alleles adapted to ancient environments can increase disease risk in modern environments due to evolutionary mismatches. Many human populations are likely to be subject to such mismatches due to rapidly changing environments.
- Hygiene hypothesis
-
A hypothesis proposing that immune systems adapted for environments with a high pathogen load are now mismatched to current environments with low pathogen load. This mismatch is further hypothesized to contribute to the higher incidence of autoimmune and inflammatory diseases.
- Genetic load
-
The decrease in population fitness caused by the presence of non-optimal alleles compared with the most fit genotype: (Wmax – Wmean) / Wmax, where Wmax is the maximum possible fitness and Wmean is the average fitness over all observed genotypes.
- Polygenic risk scores
-
(PRSs). Results of a mathematical model to estimate the genetic risk of a disease for an individual based on the sum of the effects of all their genetic variants as estimated in a genome-wide association study. The clinical utility of PRSs is a topic of current debate (Box 4).
- Mendelian disease
-
A disease caused by a variant in a single gene, such as sickle cell anaemia, cystic fibrosis and phenylketonuria. Mendelian (also known as monogenic) disorders are usually rare and follow simple dominant or recessive inheritance patterns.
Rights and permissions
About this article

Cite this article
Benton, M.L., Abraham, A., LaBella, A.L. et al. The influence of evolutionary history on human health and disease. Nat Rev Genet 22, 269–283 (2021). https://doi.org/10.1038/s41576-020-00305-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41576-020-00305-9
This article is cited by
-
Differentiated genomic footprints suggest isolation and long-distance migration of Hmong-Mien populations
BMC Biology (2024)
-
Evolutionary origin of germline pathogenic variants in human DNA mismatch repair genes
Human Genomics (2024)
-
PDDGCN: A Parasitic Disease–Drug Association Predictor Based on Multi-view Fusion Graph Convolutional Network
Interdisciplinary Sciences: Computational Life Sciences (2024)
-
Strategies for Dissecting the Genetic Driving of Conserved Noncoding-Elements for Evolutionary Development of the Corpus Callosum
Neuroscience Bulletin (2024)
-
Elevated genetic risk for multiple sclerosis emerged in steppe pastoralist populations
Nature (2024)
