
Abstract
Hepatocellular carcinoma (HCC) is a prevalent disease with a progression that is modulated by the immune system. Systemic therapy is used in the advanced stage and until 2017 consisted only of antiangiogenic tyrosine kinase inhibitors (TKIs). Immunotherapy with checkpoint inhibitors has shown strong anti-tumour activity in a subset of patients and the combination of the anti-PDL1 antibody atezolizumab and the VEGF-neutralizing antibody bevacizumab has or will soon become the standard of care as a first-line therapy for HCC, whereas the anti-PD1 agents nivolumab and pembrolizumab are used after TKIs in several regions. Other immune strategies such as adoptive T-cell transfer, vaccination or virotherapy have not yet demonstrated consistent clinical activity. Major unmet challenges in HCC checkpoint immunotherapy are the discovery and validation of predictive biomarkers, advancing treatment to earlier stages of the disease, applying the treatment to patients with liver dysfunction and the discovery of more effective combinatorial or sequential approaches. Combinations with other systemic or local treatments are perceived as the most promising opportunities in HCC and some are already under evaluation in large-scale clinical trials. This Review provides up-to-date information on the best use of currently available immunotherapies in HCC and the therapeutic strategies under development.
Key points
-
Multiple immune mechanisms are important in the development and progression of hepatocellular carcinoma (HCC) and correlate with prognosis.
-
Checkpoint inhibitors targeting PD1 and PDL1 and CTLA4 are active, tolerable and clinically beneficial against advanced HCC.
-
At present, the best available first-line treatment for advanced HCC is a combination of PDL1 blockade with atezolizumab and VEGF blockade with bevacizumab.
-
There is as yet an almost complete lack of suitable biomarkers to guide the development of checkpoint inhibitors and their combinations in HCC.
-
Immunotherapy is likely to synergize with local and locoregional interventions in earlier stages of HCC.
-
Other promising forms of immunotherapy for HCC such as additional checkpoint inhibitors, adoptive cell transfer, vaccination and virotherapy are being actively pursued.
Similar content being viewed by others
Introduction
Liver cancer is one of the most common cancers worldwide and its incidence is rising in Western countries1. Worldwide, it has a mortality to incidence ratio of 0.91, occurs 2.3 times more frequently in men than in women, and 72% of new cases are diagnosed in Asia2. Hepatocellular carcinoma (HCC) is the most common primary liver cancer and most frequently develops on a background of chronic liver disease caused by infection with hepatitis B virus (HBV) or hepatitis C virus (HCV), alcohol abuse or the metabolic syndrome3. HCC is commonly multinodular at diagnosis due to synchronic carcinogenesis or early dissemination inside the liver, and it has a distinct affinity to grow inside the blood vessels, invading the portal or hepatic veins3. α-Fetoprotein (AFP) is the serum biomarker most widely used in HCC and it helps in refining prognosis and monitoring response to therapy. Treatment recommendations differ from region to region, indicating a relative lack of strong scientific evidence for several disease scenarios4,5,6,7. When tumours have not expanded outside the liver, locoregional treatments are applied. The choice between liver transplantation, resection, percutaneous ablation, transarterial chemoembolization (TACE) and radioembolization largely depends on tumour burden, location and comorbidities4. Due to a strong and broad resistance of HCC to cytotoxic chemotherapy, systemic therapy was for many years a deferred option in HCC. In 2008, the oral multi-tyrosine kinase inhibitor (TKI) sorafenib was shown to prolong the survival of patients with advanced stage HCC with preserved liver function (Child–Pugh class A)8. However, the efficacy of TKIs in the advanced stage was not replicated when combined with TACE in the intermediate stage9,10 or when given to prevent recurrence after resection or ablation11. Since 2017 three other multi-TKIs have been approved worldwide. Lenvatinib was found to be not inferior to sorafenib in the first-line setting12 whereas regorafenib13 and cabozantinib14 were found to prolong survival in the second-line setting compared with placebo. Ramucirumab, an inhibitor of the vascular endothelial growth factor (VEGF) receptor 2, also showed efficacy after sorafenib among patients with AFP levels >400 ng/ml15.
The immune system plays an important part in controlling cancer progression16. The innate and adaptive immune systems interact to enable effective anticancer immune surveillance. Dysfunctional tumour–immune system interactions lead to immune evasion through impaired antigen recognition or by generating an immunosuppressive tumour microenvironment (TME)17. Reduced recognition of tumour-associated antigens (TAAs) by immune cells can occur through epigenetic and post-transcriptional silencing or by alterations in the antigen-presenting or peptide-processing machinery18. The presence of an immunosuppressive TME can be due to: accumulation of cells with negative regulatory immune activity such as regulatory T cells (Treg), inhibitory B cells, myeloid-derived suppressor cells (MDSCs) or M2-polarized tumour-associated macrophages (TAMs); upregulation of co-inhibitory lymphocyte signals including immune checkpoint ligands and receptors; elevated levels of tolerogenic enzymes such as indoleamine 2,3-dioxygenase-1 (IDO) or arginase-1; reduced immunoglobulin-mediated opsonization; and the presence of a metabolically unfavourable milieu for immune cells19. Any attempt to overcome these barriers to effective tumour cell killing by the immune system represents a form of immunotherapy worthy of therapeutic evaluation.
Immune checkpoints include co-inhibitory molecules expressed by effector lymphocytes that prevent their overactivation. Liver tumours and other cancers exploit this physiological mechanism to evade anti-tumour immune responses by expressing the corresponding ligands in tumour and stromal cells20. Co-inhibitory receptors include cytotoxic T lymphocyte-associated antigen 4 (CTLA4), PD1, T cell immunoglobulin and mucin domain containing-3 (TIM3), lymphocyte-activation gene 3 (LAG3) and others21. CTLA4 is expressed by activated T cells and mostly by Treg cells. It prevents the activation of effector T cells and serves as an effector molecule for Treg cells22. PD1 is expressed by activated T cells, natural killer (NK) cells, Treg cells, MDSCs, monocytes and dendritic cells (DCs), whereas its ligand, PDL1, is expressed by a number of stromal and tumour cells, and myeloid cells including DCs. PD1 inhibits effector functions and leads to exhaustion or dysfunction of effector T cells. Immune checkpoint inhibitors (ICIs) are monoclonal antibodies that block the interaction of checkpoint proteins with their ligands, thereby preventing the inactivation of T cells. Across tumour types, ICIs have proven that an effective immune response is able to eliminate tumour cells23 in a way that has transformed cancer therapy. ICIs are the first, but hopefully not the last, immunotherapy agents to prove effective against HCC (Box 1).
In 2015, available evidence supporting the development of immune interventions and therapies against HCC was reviewed in this journal24. Some of the best predictions in that Review have since become reality, and immunotherapy is now the core of translational and clinical research in the field. In this update, we focus on the rationale, preclinical evidence and clinical use of immunotherapy in HCC, with a special focus on ICIs. After summarizing the latest findings related to the immune microenvironment, we discuss the available evidence on the clinical use of ICIs, provide the reader with important clues to the management of such therapies in HCC, and examine the mechanisms of response and resistance to ICIs. We finally provide a brief overview of the status of other immunotherapies such as cell therapies or therapeutic cancer vaccines, or the combination of ICIs and locoregional therapies.
The immune microenvironment of HCC
Antigenicity of HCC
The first step for the development of a tumour-specific T cell response is the expression of tumour antigens. A spontaneous immune response might be elicited during hepatocarcinogenesis through deregulated expression of oncofetal and cancer testis antigen genes25. Naturally occurring CD8+ T cells specific for AFP, glypican 3 (GPC3), melanoma-associated gene 1, and New York oesophageal squamous cell carcinoma 1 (NY-ESO1) can be detected in blood and tumour samples from patients with HCC25. These tumour-specific T-cell responses correlate with patient survival25. On the other hand, genomic mutations occurring during hepatocarcinogenesis might lead to amino acid changes that eventually create cancer neoantigens26. The new amino acids might enhance the HLA-binding capacity of the peptide, making the novel epitope noticeable to T cells, or it might result in new contact points with the T cell receptor (TCR), thereby enabling recognition by T cells not removed by immune tolerance mechanisms27. Some neoantigens arising from driver mutations are shared by different tumours and patients (for example, mutations in TP53 (ref.28)), but most are private neoepitopes resulting from seemingly passenger somatic mutations29.
Next-generation sequencing technologies have drafted the mutational landscape of many tumours30. Tumour mutational burden (TMB) is frequently used as a surrogate for the number of neoantigens, as the probability of identifying T lymphocytes specific for neoantigens correlates with TMB31. TMB is usually high in tumours with >20 somatic mutations per megabase such as melanoma, and only sporadic in tumours with less than one somatic mutation per megabase such as pancreatic cancer32. Compared with other tumours, HCC typically shows a low to moderate TMB, with an average of five somatic mutations per megabase, corresponding to approximately 60 non-synonymous substitutions33. In theory, the higher the TMB, the higher the chances of a tumour being antigenic. However, the frequency and relevance of neoantigens in HCC have not yet been described in detail.
The immune cell microenvironment of HCC
The liver has an anti-inflammatory immune environment to foster tolerance to foreign, harmless molecules such as food antigens34. In humans, non-parenchymal resident liver cells such as Kupffer cells, hepatic stellate cells (HSCs) and liver sinusoidal endothelial cells (LSECs) cooperate in the maintenance of this tolerogenic milieu. Kupffer cells are the liver-resident macrophages and together with LSECs and HSCs can act as antigen-presenting cells (APCs)35. Kupffer cells produce inhibitory molecules such as IL-10, prostaglandins and IDO36 and promote the activation of Treg cells37. LSECs express high levels of PDL1 (ref.38) and drive a TGFβ-dependent induction of Treg cells. HSCs release hepatocyte growth factor (HGF), which promotes MDSC39 and Treg cell accumulation40 inside the liver, and can also induce T cell apoptosis through PDL1 expression41.
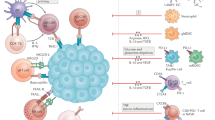
The TME of HCC is a complex and spatially structured mixture of hepatic non-parenchymal resident cells, tumour cells, immune cells and tumour-associated fibroblasts (Fig. 1). All these cellular populations dynamically interact through cell–cell contacts and the release or recognition of cytokines and other soluble factors. This complex cellular interplay has a substantial influence on tumour immune evasion. The adaptive immune response in patients with HCC is blunted, as shown by the enrichment of the TME with exhausted or dysfunctional tumour-infiltrating lymphocytes (TILs)25. The innate immune response is dampened too, and mechanisms implicated in NK cell dysfunction include expression of inhibitory receptors42,43, MDSC-mediated immune suppression44 and increased frequency of dysfunctional NK cells45.

Hepatocellular carcinoma (HCC) tumour cells can escape immune attack from the host if they fail to effectively present antigens and remain unrecognized by the immune system, or if the tumour microenvironment is rich in cells and soluble molecules that deactivate or interfere with the action of tumour-killing cytotoxic T lymphocytes. A summary of this complex network of interactions is shown. Negative effects on the immune response are indicated by red arrows and enhancing effects are indicated by black arrows. Cells and molecules involved represent potential therapeutic targets through the blockade of negative signals or the stimulation of positive signals. Currently available therapeutic agents in orange boxes indicate their main mechanism of action. Effector T cells, natural killer (NK) cells and dendritic cells (DC) have an overall positive effect on immune tumour rejection, whereas regulatory T cells (Treg), myeloid-derived suppressor cells (MDSC), M2-polarized tumour-associated macrophages (TAM M2) and neutrophils have a negative effect. To be targeted by the immune system, HCC cells should express antigens through gene mutations leading to neoantigens (neoAgs) or gene deregulations leading to tumour-associated antigens (TAAs). Mutations in the β-catenin gene might impair the recruitment of conventional type 1 dendritic cells (cDC1) that are key in attracting immune effector cells, whereas the chemokine receptor 6 (CCR6) and chemokine ligand 20 (CCL20) axis attracts Treg cells. anti-CTLA4, CTLA4 inhibitor; anti-VEGF, VEGF inhibitors; anti-VEGFR, VEGFR inhibitors; CTLA4, cytotoxic T lymphocyte-associated antigen 4; GM-CSF, granulocyte–macrophage colony-stimulating factor; HGF, hepatocyte growth factor; IDO, indoleamine 2,3-dioxygenase-1; TGFβ, transforming growth factor-β; TKI, tyrosine kinase inhibitor; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
A number of immune or stromal cell types that are abundant in HCC tumours cooperate in the generation of an immunosuppressive TME and their presence generally correlates with a worse prognosis. The best known examples are Treg cells and TAMs; for example, the density of Treg cells in the liver correlates with a poor prognosis in HCC46,47. These immunosuppressive T lymphocytes are recruited by the chemokine receptor 6 (CCR6) and chemokine ligand 20 (CCL20) axis48, and are also induced to locally differentiate from CD4+ T cells under the influence of a specific subset of MDSCs49. An alternative mechanism by which MDSCs support tumour progression involves the production of VEGF, which promotes vascularization and angiogenesis of the malignant tissue50. TAMs tend to correlate with a worse prognosis in HCC, especially if skewed towards an M2-like polarization51. Less common immunosuppressive cell types in human HCC include a novel regulatory B cell population that expresses high levels of PD1 (ref.52), T helper 17 (TH17) cells, CD4+ T cells expressing CCR4 and CCR6 (ref.53), CD14+ DCs expressing high levels of CTLA4 and PD1 (ref.54), tumour-associated fibroblasts that inhibit NK cell function55 and neutrophils that recruit macrophages and Treg cells56. The peritumoural environment in the forefront of HCC is also important. Kupffer cells in the peritumoural boundary express higher levels of PDL1 than non-tumoural liver57 while activated HSCs in those boundaries contribute to a poor prognosis58.
HCC molecular features that model the immune microenvironment
In metastatic melanoma, loss-of-function mutations in the β-catenin pathway result in markedly less infiltration by TILs59,60. The mechanism has been traced to poor production of the CCL4 chemokine so that DCs are not chemoattracted to the TME. Indeed, the presence of a subpopulation of DCs named conventional type 1 DCs (DC1) seems to be the most critical factor in gathering a T cell and NK cell infiltrate in melanoma61,62. These DC1s are also commonly referred to as BATF3-dependent DCs and are critical for cross-presentation of tumour antigens to CD8+ T lymphocytes and for the action of ICIs63,64.
Echoing this line of research, a similar set of events has been shown in mouse models of genetic HCC carcinogenesis by hydrodynamic liver gene transfer. In such models, HCC tumours with activating mutations of β-catenin fail to produce the DC1-attracting CCL5 chemokine, resulting in a conspicuous paucity of DC1 in the mouse tumours65. Around a quarter of HCCs have mutations in β-catenin3. Preliminary clinical observations suggest that β-catenin-mutated tumours tend to be ‘cold’ in their lymphocyte infiltration and it has been proposed that they might fail to respond to PD1 blockade66, although this finding needs confirmation in clinical trials.
Across tumour types, the number and activation status of T cells has a clear influence on tumour progression and response to ICIs67. Research on the control of baseline T cell infiltration is very active68 and in patients and animal models genetic alterations of PTEN69, RAS and LBK1 (ref.70) have been shown to give rise to lymphocyte depletion and lymphocyte-exclusion phenotypes71. Emerging evidence also implicates the epigenetic status of several genetic loci in the outcome of PD1 and PDL1 checkpoint inhibitor therapy in lung cancer72. Several factors produced by tumour cells might help establish an immunosuppressive TME in HCC by accrual of MDSCs and Treg cells, two cell types with a negative effect on prognosis73. VEGF, IL-8, HGF and TGFβ are examples of these factors that can be therapeutically targeted74,75,76,77. The role of AFP as an immunomodulatory molecule in HCC is less clear although in vitro AFP produced by HCC shows a suppressive effect on NK cells and T cells78 and DCs exposed to AFP have a reduced effect in stimulating antigen-specific T cell activation and proliferation79. Other genetic traits that contribute to HCC progression include reduced expression of HLA molecules80, decreased expression of NKG2D ligands81, and secretion by tumour cells of soluble molecules such as TGFβ, IL-10, IDO and arginase, or increased expression of inhibitory checkpoint ligands82. In summary, baseline immune contexture in HCC is the result of genetic and epigenetic features that largely remain to be determined and in which the control of the presence and function of DC1s seems to be critical83.
Soluble molecules that favour immune evasion in HCC
The TME in HCC is enriched in soluble mediators that modulate the anti-tumour immune response. Adding a new layer of complexity, these molecules are pleiotropic and their effects can be different depending on the target immune cell population, or in acute versus chronic exposure84. TGFβ is abundant in the HCC TME, produced by tumour cells, macrophages or Treg cells, where it downregulates the anti-tumour response at different levels: it inhibits the activation of DCs85; promotes M2 polarization of TAMs86; impairs the effector functions of T cells87 and NK cells88; and promotes the generation of induced Treg cells89. High tissue expression of TGFβ is associated with a poor prognosis90 and high circulating levels determine the response to sorafenib91 and pembrolizumab92. Type I IFN favours the immune response but it also triggers negative signals such as the production of IL-10 (ref.93). IL-10 is upregulated in HCC, produced by DCs, TAMs and regulatory B and T cells. It down-modulates the T cell stimulatory capacity of APCs, promotes upregulation of PDL1 in monocytes94 and impairs the recruitment of tumour-specific human T cells95. High serum IL-10 levels are associated with increased numbers of MDSCs96 and reduced TIL activity in patients with HCC97.
VEGF is produced by tumour cells and the surrounding stroma98. Besides promoting tumour angiogenesis, VEGF inhibits the antigen-presenting functions and T cell stimulatory ability of DCs, and generates MDSCs and Treg cells99. The immunomodulatory effects of VEGF inhibitors might be partly responsible for their anti-tumour activity.
Besides cytokines, the HCC TME is enriched in metabolites with immunomodulatory properties. In HCC models, resistance to ICIs is associated with IDO upregulation promoted by IFNγ, and blockade of IDO enhances the efficacy of ICIs100. Adenosine is another immunosuppressive metabolite generated by the ectonucleotidases CD39 and CD73. Patients with HCC with high tumour levels of CD39 have increased recurrence rates after resection and poor survival101. CD73 is upregulated in human HCC cell lines, where its expression promotes tumour growth and metastasis102.
Finally, IFNγ is key to an effective anti-tumour immune response. In patients with HCC, lower serum levels of IFNγ are associated with more advanced tumour stage and worse prognosis103. Mechanisms of escape of HCC from the IFNγ pathway include loss of its receptor104, induction of immunosuppressive cytokines such as IL-10 or VEGF, checkpoint ligands such as PDL1 (ref.105) and enzymes such as IDO106. Altogether, these data provide a rationale for testing the efficacy of agents that counteract the immunosuppressive actions of TGFβ, VEGF or IDO in HCC. The presence of so many immunoregulatory targets in the HCC TME, both cell-associated and soluble, has prompted the investigation of immunotherapies already known to be effective in other malignancies.
Immune checkpoint inhibitors in HCC
Single agents
Only 4 years after a pilot clinical trial showed for the first time that CTLA4 blockade with tremelimumab can induce durable objective remission in patients with HCC and HCV infection107, the PD1 inhibitor nivolumab was granted accelerated approval in the USA for the treatment of patients with advanced HCC after sorafenib. Today, all phase III trials testing systemic therapy in treatment-naive patients with HCC involve ICIs. Given the relatively small fraction of patients who respond to ICIs108,109, this situation is mostly a reflection of the scarcity of effective systemic therapies for HCC.
PD1 and PDL1 inhibitors are the backbone of systemic therapies in clinical practice or under development for HCC. Nivolumab and pembrolizumab have shown unequivocal signs of activity in single-arm phase II trials in the second-line setting, after sorafenib failure or unacceptable toxicity108,109,110. As shown in Table 1, nivolumab and pembrolizumab produce a 15–20% rate of objective remissions (including 1–5% complete responses) that are durable and associated with prolonged survival. In the CheckMate 040 trial, the median duration of response to nivolumab among 48 patients in the dose-escalation cohort was 17 months (95% CI 6–24 months), and the 2-year survival rate among responders was over 80%108. Based on these results, nivolumab and pembrolizumab were approved by several regulatory agencies as second-line therapies after sorafenib. The consistency of results with the different monoclonal antibodies speaks in favour of a class effect. KEYNOTE-240, a phase III trial testing pembrolizumab versus placebo after sorafenib in 413 patients, showed a statistically significant prolongation of survival (HR 0.78; P = 0.023) that nevertheless did not meet the prespecified statistical threshold110. This statistically negative result was possibly influenced by the dual primary end point of overall survival and progression-free survival (PFS), and by the fact that effective therapies such as regorafenib and nivolumab became available during the study and probably improved survival after progression. PFS and overall survival curves indicated that some patients derived long-term benefit from pembrolizumab. Indeed, almost 20% of the patients receiving pembrolizumab remained free from progression for more than a year compared to less than 7% of controls110.
In the CheckMate 459 phase III trial comparing nivolumab versus sorafenib in 743 patients naive to systemic agents, the predefined threshold of statistical significance for overall survival was also not met, although patients survived longer after nivolumab than after sorafenib (median survival 16.4 versus 14.7 months, HR 0.85; P = 0.07)111. Survival curves overlapped completely for the first 6 months while 1-year and 2-year survival rates were 60% and 37% for nivolumab compared with 55% and 33% for sorafenib, respectively. Again, post-progression therapies probably had an effect on overall survival as at least 31% of patients treated with sorafenib later received an ICI or an investigational agent (often an ICI), while a more similar proportion of patients in both arms received a TKI (36% and 23%). Other end points in the CheckMate 459 trial also favoured nivolumab over sorafenib, with a more durable disease control (median 7.5 months versus 5.7 months), a better safety profile with fewer grade 3 or grade 4 treatment-related adverse events (TRAEs) (22% versus 49%), fewer events leading to treatment discontinuation and a better health-related quality of life. A more prolonged follow-up of CheckMate 459 has provided confirmation of the capacity of nivolumab versus sorafenib to increase the rate of long-term survivors (29% versus 21% at 33 months)112.
Other immune checkpoint inhibitors
There are other immune checkpoint molecules beyond PD1, PDL1 and CTLA4 that can be targeted to stimulate an anti-tumour immune response. In HCC, the abundance of PD1+ CD8+ T cells in the immune infiltrate105 and PDL1+ tumour cells113 is associated with a worse prognosis. TIM3 is expressed on CD4+ and CD8+ TILs114 and TAMs115 from human HCC tumours and it negatively regulates T cell effector function116, whereas its expression on Treg cells induces enhanced suppressor activity117. TIM3 is strongly expressed on, and associated with, less differentiated HCC tumours118. LAG3 binds MHC class II molecules with high affinity, is upregulated upon activation of T cells, and provides a negative signal to T cells119. Expression of LAG3 is substantially higher on tumour-specific CD4+ and CD8+ TILs than in other immune compartments in patients with HCC120. LAG3 has another functional soluble ligand named fibrinogen-like protein 1 which is synthetized by hepatocytes121. Together, these preclinical data provide support to the investigation of LAG3 and TIM3 inhibitors in HCC in combination with PD1 and PDL1 blockade.
Combinations
Based on the activity of single-agent ICIs and a better understanding of the TME, a number of combination strategies can be considered and many of them have already entered clinical development (Fig. 2). Table 1 and Table 2 summarize the available experience, which comes mostly from patients treated in the second-line setting after sorafenib. Altogether, combinations result in a consistent twofold increase in response rate, with ~5% complete remissions and encouragingly long survivals in excess of 18 months. In parallel, additive toxicities from the combinations increase the number of high-grade TRAEs, serious adverse events and adverse events leading to treatment discontinuation, as shown in Table 3. There are no obvious signs of synergistic toxicities for overlapping adverse events such as skin toxicities, diarrhoea and hepatitis.

Combination strategies are shown for immune checkpoint inhibitors (ICIs) with other therapeutic tools, established or in development, that are being explored in preclinical or clinical studies based on their additive or potentially synergistic mechanisms of action. HCC, hepatocellular carcinoma; mAb, monoclonal antibody.
The increased response rates of around 30% observed with the dual blockade of CTLA4 and PD1/PDL1 confirm initial reports of HCC as a CTLA4-responsive tumour107,122. Contrary to PD1 and PDL1 inhibitors, dosing and timing seem important for CTLA4 inhibitors. From preliminary findings with the combination of ipilimumab and nivolumab, the best median overall survival (22.8 months) was obtained with the highest dose (3 mg/kg once every 6 weeks) of the former and the lower dose of the latter (1 mg/kg once every 2 weeks)123. Such encouraging results have led to accelerated approval of this combination by the FDA to treat patients with HCC after sorafenib. A similar effect was observed with the combination of a single 300 mg dose of tremelimumab combined with a continuous dose of the PDL1 inhibitor durvalumab124. Interestingly, this single, high priming dose of tremelimumab resulted in an early burst of proliferating CD8+ T cells in peripheral blood124. These findings are in line with observations in melanoma indicating that the activity of CTLA4 inhibitors is dose-dependent125 and that the first doses of CTLA4 inhibitors cause a proliferative burst of CD4+ and CD8+ T cells126, probably related to the increased efficacy of the combination. In HCC, as in other tumour types, combination regimens increase the rate of TRAEs that are nevertheless tolerable, as shown in Table 3. The combination with lenvatinib is associated with double the response rate compared with the response rate observed with single-agent pembrolizumab, and leads to a promising 20 months median overall survival, but at the cost of increased toxicity127. The combination of cabozantinib and nivolumab is also associated with double the response rate compared with the response rate observed with single-agent nivolumab, and leads to prolonged survival, but at the cost of more frequent and serious toxicity128.
Based on such encouraging observations, a number of ICI combinations have entered into phase III clinical trials comparing them with standard of care in patients naive to systemic therapy (Box 1; Fig. 3). Two such combinations have already proved superior to sorafenib after more than a decade of failing trials. In the IMbrave150 clinical trial, 501 patients were randomly assigned at a ratio of 2:1 to receive the standard dose of atezolizumab (1,200 mg) plus a high dose (15 mg/kg) of the VEGF inhibitor bevacizumab every 3 weeks, or sorafenib129. The trial had the dual primary end point of PFS and overall survival and was stopped at the first interim analysis after a median follow-up of only 8.6 months, when improved overall survival (HR 0.58, 95% CI 0.42–0.79; P = 0.0006) and improved confirmed PFS (HR 0.59, 95% CI 0.47–0.76; P < 0.0001) were observed. The target population was comparable with those in other trials, with the main exception of a lower proportion of patients with distant metastases (Table 2). With a more prolonged follow-up (median 15.6 months), median overall survival was 19.2 months in the atezolizumab plus bevacizumab arm and 13.4 months in the sorafenib arm, with 52% and 40% of patients surviving at 18 months, respectively130. Median PFS and response rate by central review using RECIST 1.1 criteria were also better with the combination than with sorafenib. Importantly, health-related quality of life was also significantly preserved in the combination treatment arm. Median time to deterioration in patient-reported quality of life was longer with the combination (11.2 versus 3.6 months; HR 0.63, 95% CI 0.46–0.85), and much longer than median PFS in the combination arm but not in the sorafenib arm129. The trial lacked an arm with single-agent atezolizumab or bevacizumab; however, the contribution of both agents was probably driving the clinical benefit of the combination. On the one hand, PFS and response rate were better with the combination than with single-agent atezolizumab in a randomized cohort of 119 patients in a phase Ib trial131. On the other hand, bevacizumab showed response rates of 13–14% by mRECIST in single-agent phase II studies in HCC132,133. In late 2020, the combination of the PDL1 inhibitor sintilimab with a bevacizumab biosimilar also proved superior to sorafenib among 571 patients with HCC naive to systemic therapy, who were enrolled in the ORIENT-32 trial134. After a median follow-up of 10 months, median overall survival was not reached in the combination arm and was 10.4 months in the sorafenib arm (HR 0.57, 95% CI 0.43–0.75; P < 0.0001).

Combination immune checkpoint inhibitor (ICI) strategies reported or in ongoing phase III clinical trials are presented. CTLA4, cytotoxic T lymphocyte-associated antigen 4; VEGF, vascular endothelial growth factor.
The FDA, EMA and other regulatory agencies worldwide have approved the atezolizumab plus bevacizumab combination for first-line therapy in HCC. This combination will therefore set a new standard of care for treatment-naive patients. Yet, the story of immunotherapy in the first-line therapy setting of HCC is not finished. The beneficial interaction of VEGF or VEGFR blockade and PD1 and PDL1 inhibitors acts at different levels. Reduction and normalization of the tumour vasculature is probably only part of the effect and various lines of research concur to indicate that functional control by VEGF of innate immune populations including MDSCs is key135. In this respect, bevacizumab has demonstrated a capacity to reduce MDSCs in patients with other tumour types such as lung or colorectal cancer136,137. Moreover, data from preclinical mouse models of HCC have shown that the superior therapeutic activity of a combination of anti-PD1 and anti-VEGFR2 antibodies is associated with enhanced M1 and decreased M2 TAM levels, as well as with an increased level of infiltrating CD8+ T cells138. A key question is what kind of agent would make the most of the ICI–anti-angiogenic agent combination. Bevacizumab neutralizes VEGFA while VEGFR inhibitors curtail proximal signal transduction from VEGF receptors. The only possible definitive answer to this question would come from clinical trials, which will probably not involve direct comparisons. In this regard, it must be considered that the spectra of tyrosine kinases inhibited to some extent by the commonly used agents139 reaches those that are able to directly or indirectly regulate anti-tumour immune responses. The development of combinations of anti-angiogenic agents for advanced HCC promises to be as interesting as the situation in metastatic renal cell carcinoma in which physicians have to choose from two or three combination regimens in any clinical scenario.
If one or more of the phase III trials testing dual blockade of CTLA4–PDL1 or combinations of ICIs with TKIs (Table 4) also show positive results, the choice of first-line treatment will depend substantially on patient characteristics, tolerability and toxicity profile. With this scenario, is there any role for single-agent ICI in the first-line therapy of HCC? Based on the results of the CheckMate 459 trial112, we believe that nivolumab is a valuable option for patients with strong contraindications to the use of anti-angiogenics, usually related to their cardiovascular risk, and for those in whom the risk of variceal bleeding cannot be evaluated or managed adequately in a timely manner (particularly relevant in the current times of the coronavirus disease 2019 pandemic).
Management of ICI-based therapies in HCC
Because of their key role in maintaining immune homeostasis, the blockade of inhibitory checkpoint molecules often produces a wide range of immune-mediated adverse events (IMAEs) that result from impaired self-tolerance. IMAEs can involve almost every organ resulting in, for example, hepatitis, colitis or pneumonitis, to mention some of the most common IMAEs, but they are usually manageable although they can also be life-threatening140. Among patients with HCC, the safety profile and overall incidence of IMAEs are comparable to those of other tumours141. However, the coexistence of chronic liver disease, most usually at the cirrhotic stage, has two consequences: liver toxicities are more frequent and more severe in patients with HCC than in those without chronic liver disease, and the diagnosis of other IMAEs might be challenging because of the confounding effect of organ dysfunctions associated with such a chronic condition. The latter has been reviewed and specific recommendations would help in daily practice141.
Most IMAEs can be successfully treated with corticosteroids alone or in combination with azathioprine or mycophenolate mofetil (Box 2). Drug discontinuation due to TRAEs occurs in 10–20% of patients with HCC, almost double that with single-agent ICIs142. With the dual blockade of CTLA4 and PD1, hepatic TRAEs developed after 4–5 weeks of therapy and around 90% resolved after 6–8 weeks142. No significant differences in their safety profiles in terms of IMAEs and adverse events causing death or treatment discontinuation have been reported for anti-PD1 agents versus anti-PDL1 agents143. Use and dose levels of anti-CTLA4 agents are more related to serious IMAEs. In the combination cohort of the CheckMate 040 trial, corticosteroids were used in 50% of patients treated with the higher dose of ipilimumab compared to 24% with the lower dose142. Steroid usage in other tumours such as melanoma or lung cancer seems not to curtail efficacy if given to treat IMAEs144. Clinically significant hepatitis flares have not been reported among patients with HCC chronically infected with HCV or HBV and treated with ICIs. Nevertheless, patients with hepatitis B have been excluded from all trials if they were not under effective treatment with direct antiviral agents. The compromised liver safety profile of TNF-blocking agents such as infliximab discourages its use for hepatitis IMAEs and in patients with HCC145, although no direct evidence for this contraindication has been reported yet146.
The combination of ICIs and TKIs also causes more severe toxicity than single ICIs127. More important than the increased frequency is probably the more diverse spectrum of toxicities that result from non-overlapping adverse events, and the increased severity of overlapping adverse events such as skin toxicities, diarrhoea and hepatitis. Highlighting this additive effect of ICI and TKI combinations, some lethal adverse events were reported with the combination of pembrolizumab and lenvatinib in patients with HCC127. The effect on quality of life will be critical in the event that these combinations prove to be useful as first-line agents.
The combination of atezolizumab and bevacizumab seems to be better tolerated than the combination of ICIs and TKIs. Hypertension was the only adverse event occurring in >10% of patients (15.2%)129. Bevacizumab causes a fourfold increase in the risk of gastrointestinal haemorrhage or perforation in patients with advanced cancer147. As patients with cirrhosis are at risk of gastrointestinal bleeding due to gastroesophageal varices, hypertensive gastropathy or gastrointestinal angiodysplasia, concerns were raised early about the safety of bevacizumab in patients with HCC. On the other hand, VEGF is key in the pathophysiology of portal hypertension. It might aggravate portal hypertension by increasing splanchnic vasodilation and enhancing angiogenesis in the splanchnic circulation148. VEGF inhibition, therefore, reduces portal pressure in animal models149. However, when the hepatic venous pressure gradient was measured in 13 patients with HCC before and after chemoembolization alone or combined with bevacizumab (5 mg/kg), no changes were observed between groups of patients after acute and chronic exposure to bevacizumab150. This circumstantial evidence suggests that at least bevacizumab would be unlikely to aggravate portal pressure. In prior studies in HCC132,133, bevacizumab was associated with a 7–10% rate of severe haemorrhagic events, including but not restricted to gastrointestinal and variceal bleeding. In the IMbrave150 trial, patients were excluded if they had untreated or incompletely treated oesophageal or gastric varices with bleeding, or if they were at high risk of bleeding as assessed by upper gastrointestinal endoscopy129. Among these well-selected patients, the incidence of upper gastrointestinal bleeding observed with the combination of atezolizumab and bevacizumab was 7% compared with 4.5% after sorafenib129. Thus, it seems that the risk of bevacizumab-induced gastrointestinal bleeding is not much higher in patients with cirrhosis than it is in other patients with cancer.
Having more active agents and combinations available implies that decisions to switch patients from one treatment to another will be needed. These decisions are usually made on the basis of tumour progression and treatment toxicity. With ICIs, such decisions might not be easy. On the one hand, tumour progression does not always mean lack of benefit. As in other tumour types, delayed responses or ‘pseudo-progressions’ occur in HCC151. Furthermore, the pattern of progression could also be relevant, and it has been shown with nivolumab that some patients might obtain a treatment benefit from prolonged therapy. In a sub-analysis published in abstract form, patients with progression in target lesions followed by a formal response, progression with new lesions followed by decreases in target lesions of ≥10%, or progression of target lesions or new lesions followed by stabilization had a median overall survival of 18.8 months compared with 8.4 months in the remaining progressors152. On the other hand, toxicities might mean an increased chance of benefit, as occurs with skin toxicity and sorafenib153. This possibility was suggested in a single-centre cohort of 114 patients with HCC, in which the presence of IMAEs in 68.4% of patients was independently associated with improved median PFS (HR 0.52) and overall survival (HR 0.38)154. This observation needs confirmation in larger series where the effect of a more prolonged time on treatment can be taken into consideration.
How to individualize therapies
It is clear that immunotherapy does not provide benefit in all patients. Identifying those with intrinsic resistance to ICIs would enable other therapies to be attempted and would save a substantial amount of money and health resources. Unfortunately, we ignore much more than we know. From a patient perspective, no strong clues were obtained from post-hoc subgroup analysis of randomized clinical trials although some signals deserve attention. Overall survival was slightly better among Asian patients and those with AFP levels of >200 ng/ml in the KEYNOTE-240 trial (pembrolizumab versus placebo)110 and the CheckMate 459 trial (nivolumab versus sorafenib)111. However, objective remissions occurred irrespective of AFP levels, region or aetiology after nivolumab or pembrolizumab monotherapies, or the combination of ipilimumab and nivolumab142. In the CheckMate 459 trial, better overall survival was also observed among patients with vascular invasion or extrahepatic disease while in the IMbrave150 trial, overall survival and PFS were better in all groups except in those with Barcelona Clinic Liver Cancer stage B, non-viral aetiology, high AFP levels, and absence of macroscopic vascular invasion and extrahepatic spread129.
Patient characteristics should not inform treatment decisions based on increased expected efficacy but rather on avoidable toxicities. Severe cardiovascular disease and thrombotic or bleeding events are contraindications to the use of TKIs and other anti-angiogenics including bevacizumab155. Contraindications with the former include chronic conditions such as uncontrolled hypertension, moderate to severe congestive heart failure or diabetic and ischaemic ulcers, and with the latter include unstable angina or myocardial infarction, transient ischaemic attack or cerebrovascular accident, pulmonary embolism, and bleeding events. Altogether, anti-angiogenics are contraindicated in at least 10–15% of patients with HCC156. Besides these formal contraindications, patient preferences based on expected toxicities should be discussed as the meaning of individual quality of life goes beyond overall group assessments.
Mechanisms of response and resistance
Establishing pretreatment baseline levels of T cell infiltration and activity are paramount to determining response to checkpoint inhibition in various types of malignancies67. However, the effect of CD4+ and CD8+ T cell infiltration on survival following second-line treatment of advanced HCC with PD1 inhibitors has been shown in the form of weak correlations and trends indicating that the effect is not as important as originally predicted157. Around 20% of advanced HCC tumours show expression of PDL1 in tumour cells by immunohistochemistry108,158. In the CheckMate 040 trial, tumour responses were observed regardless of PDL1 expression, although response rate was increased among patients with at least 1% of tumour cells expressing PDL1 (ref.157). PDL1 expression in either tumour cells or stromal immune cells was increased in patients with objective remission following treatment with pembrolizumab, but responses also occurred in the absence of expression in both cell types110. Median overall survival was 16.1 months and 8.6 months (HR 0.80) following nivolumab and sorafenib treatment, respectively, in naive patients with PDL1-positive HCC, and 16.7 months and 15.2 months (HR 0.84), respectively, in patients with PDL1-negative HCC111. With nivolumab monotherapy there was also a trend towards better survival in patients with higher tumour infiltration by CD3+ or CD8+ cells and several inflammatory gene signatures correlated with increased response rate and improved overall survival157. These gene signatures were related to cytolytic genes, inflammatory activity, IFNγ-associated genes, antigen presentation, exhaustion markers and NK cell markers, all of which are indicative of a superior presence and activity of T cells and NK cells that use IFNγ and cytolysis as main anti-tumour effector mechanisms. Interestingly, the most complex transcriptomic classification including a large number of genes159 was not identified as predictive of response in this analysis. However, the number of patients in whom RNA sequencing data were available was rather limited in this study159 and larger series might offer more conclusive positive or negative results. Linking transcriptomics and genomics correlates with the benefit of immunotherapy will need multifaceted integrative analyses and, importantly, biopsy sample collection before and during treatment160,161.
Among patients with paired HCC biopsy samples before and after two doses of tremelimumab, those with objective remissions had higher CD3+ and CD8+ infiltration than non-responders122. As for the combinations, objective HCC remissions occurred irrespective of expression of PDL1 in tumour cells treated with ipilimumab plus nivolumab123,142. It is unlikely that a single biomarker would be sensitive enough to inform clinical decisions in a timely fashion. However, extensive transcriptomic, mutational and immunohistological analyses must be undertaken as it is possible that integrative multifactorial indices might identify subsets of patients who would benefit from ICI therapy162. The importance of paired pretreatment and on-treatment biopsies cannot be overstated, and unfortunately diagnostic biopsy samples for HCC are seldom collected.
When considering the increased efficacy of combination ICI therapies, the question arises as to whether we are facing synergistic activity or just an additive effect. We still lack correlative studies from the main clinical trials. However, a subgroup analysis published in abstract form has suggested an association between response to tremelimumab, durvalumab or their combinations and an increase in proliferating Ki67+ CD8+ T cells among blood mononuclear cells on day 15 after treatment initiation124. The increase in this population of peripheral effector T cells was maximal for responders to a high priming dose of tremelimumab plus durvalumab, the combination that achieved the best overall survival, compared with tremelimumab or durvalumab monotherapies, or the combination of a repeated lower dose of tremelimumab with the same dose of durvalumab. The analysis of this sort of easily accessible biomarkers might be useful to inform the design of new combinations and to interpret data between combinations. In animal models in which VEGFR blockade increases the efficacy of PD1 inhibitors, the TME shows meaningful changes. For example, dual blockade of VEGFR and PD1 in mice reduces Treg cells and M2-polarized macrophages, increases PDL1 expression in TAMs and HCC cells, and promotes normalized vessel formation mediated by CD4+ cells138. Interestingly, efficacy was also seen with lower doses (vascular normalizing rather than anti-vascular) of anti-angiogenics, opening an interesting field of research into avoiding the combined toxicities.
One important point regarding ICIs is their efficacy and safety compared to one another. In a meta-analysis of studies of PD1 and PDL1 inhibitors including 5,744 patients with advanced non-small-cell lung carcinoma (NSCLC)163, overall response rates were similar at 19% and 18.6%. However, in a meta-analysis assessing differences in overall survival following treatment with PD1 and PDL1 inhibitors across different cancer types in a set of 19 randomized trials involving almost 12,000 patients, anti-PD1 agents led to superior overall survival (HR 0.75; P < 0.001) and PFS (HR 0.73; P = 0.02) compared with anti-PDL1 agents143. PDL2 present in tumour cells can also bind PD1 and inhibit T cell activation164. The lack of inhibition of PDL2 signalling with anti-PDL1 agents might perhaps partly explain this difference. However, currently, it is unclear whether PD1 and PDL1 blockade result in different immunobiology. The functional importance of PDL1 interaction with CD80 (ref.165) remains a matter of speculation as does the potential roles of PDL2. Direct comparisons of the best-performing PDL1 and PD1 agents either as monotherapy or in combinations are not being pursued in clinical trials and, regarding CTLA4, it remains to be seen if intratumour Treg cell depletion mediates a major contribution to the effect of ipilimumab or tremelimumab.
Another potential cause of tumour resistance is the development of anti-drug antibodies (ADAs) that are able to alter the clearance of these agents or neutralize their activity. Across tumour types, the reported incidence of ADAs during monotherapies with anti-PD1 (nivolumab, pembrolizumab and cemiplimab), anti-CTLA4 (ipilimumab) and anti-PDL1 (avelumab and durvalumab) agents is low at 0–12.7%166, and a relevant effect of ADAs on efficacy was not observed for nivolumab, pembrolizumab or ipilimumab in large clinical trials167. However, ADAs were detected in up to 36% of patients with NSCLC treated with atezolizumab, with a negative effect on systemic exposure to the drug and a deleterious effect on anti-tumour efficacy168. Regarding HCC, in a subanalysis of the IMbrave150 trial, efficacy (effect on overall survival) was lower in the 20% of patients receiving atezolizumab and bevacizumab who were ADA-positive by week 6 (ref.168).
Other HCC immunotherapies
Adoptive cell therapy
Adoptive cell therapy (ACT) with effector cells is a form of passive therapy in which lymphocytes are sensitized and/or expanded ex vivo and then reinfused into the patient169. Cells used include lymphokine-activated killer (LAK) cells, cytokine-induced killer (CIK) cells, NK cells, TILs and redirected peripheral blood T cells. For the peripheral blood cells, immune T cells are genetically recoded to render them able to target tumour cells. The two main approaches are based either on transgenic tumour antigen-specific TCRs or chimeric antigen receptors (CARs)169,170. TIL, LAK and CIK therapies are independent of target tumour genes. TIL and TCR-redirected cells have MHC-restricted anti-tumour function, whereas LAK, CIK and CAR T cells do not169,171. In general, patients treated with ACT receive a preconditioning regimen with cyclophosphamide and fludarabine to induce lymphodepletion and thereby support in vivo expansion of adoptively transferred cells. Patients receiving LAK, CIK and TIL therapy are also frequently treated with IL-2 to sustain in vivo survival of transferred cells172.
Initial attempts to develop ACT in HCC have failed to reach clinical stage due to a lack of efficacy and the relative complexity of the technology. To illustrate such complexity, LAK cells expand insufficiently ex vivo and exhibit low cytolytic effects in vivo in patients with melanoma173. Adjuvant use of LAK cells after resection delayed time to recurrence of HCC but did not prolong survival174. CIK cells usually show a higher cytotoxic effect and proliferative response than LAK cells. They are a heterogeneous population where NKT cells are primarily responsible for the anti-tumour activity175. A large number of single arm or controlled trials with different methodological shortcomings suggested activity of CIK cells against HCC. A multicentre randomized phase III trial in 2015 including 226 patients proved that adjuvant immunotherapy with CIK cells improved both PFS and overall survival of patients with HCC after curative surgical resection or percutaneous ablation compared with no adjuvant therapy176. ACT was delivered over a maximum of 60 weeks and resulted in a median PFS of 44 months compared with 30 months in the control group. Despite these positive results, ACT is not used in most centres as an adjuvant therapy, probably due to the limitations of in-house cell therapy facilities.
TILs are generated from a fresh tumour sample and tumour-reactive, expanding cells are selected based on their recognition of autologous tumour cells, and then further expanded to obtain several billion active cells169. The feasibility of adjuvant TIL therapy was shown in a phase I trial in patients with HCC177. TILs could be expanded in 15 out of 17 patients, and patients received doses up to 3 × 109 cells with minimal adverse effects. Increasing the content of T cells specific for tumour neoepitopes and scaling up the process are the main challenges for clinical application.
NK cells are characterized by a high receptor diversity that enables them to recognize tumour cells without prior sensitization or acquired receptor rearrangement. Their poor expansion capacity in vitro might be overcome for clinical use178. Ongoing phase II clinical trials are evaluating the use of allogeneic NK cells for the treatment of patients with HCC at high risk of recurrence after resection (NCT02008929) and patients after TACE (NCT02854839).
CAR T cell therapy is a promising strategy that has shown great success in treating haematological malignancies, while their application in solid tumours is still in development. CAR T cells typically have an extracellular antigen-recognition domain, a transmembrane domain and an intracellular signalling domain170. The extracellular antigen-recognition domain usually consists of a single-chain variable fragment derived from the variable heavy and light chains of a monoclonal antibody specific for an appropriate tumour cell target that can be a tumour antigen. GPC3 has become the most specific and attractive target in HCC. Efficacy has been shown in animal models with orthotopic xenografts179 and in patient-derived xenografts180. One of the main problems of CAR T cells is the off-target toxicity, in which the target molecule is expressed in non-tumour tissues. One clinical trial is underway to assess the safety and efficacy of GPC3-directed CAR T cells given as an intravenous injection (NCT04121273).
AFP is commonly overexpressed in HCC but is expressed intracellularly and secreted, and is therefore more appropriate for designing TCR-based than CAR-based therapies. Four HLA-A2-restricted AFP epitopes have been described in patients with HCC181. Some epitope-specific TCRs from human T cells show low affinity and anti-tumour effect due to central and peripheral tolerance182. Mutations in the complementarity-determining region might enhance the affinity183, and objective remissions have been reported in an ongoing clinical trial in HCC with such AFP-specific TCR T cells (NCT03132792). Other potential targets for TCR-engineered T cells include TP53 hotspot mutations frequent in HCC184, and HBV antigens (NCT03899415).
Therapeutic vaccines
The main rationale for the use of vaccines against cancer is the generation of tumour-specific responses with increased potency. This effect can be achieved by: de novo priming of T cells against antigens expressed by tumour cells that are unable to spontaneously trigger a response; enhancing already existing responses; or widening the repertoire and breadth of tumour-specific responses185. Although vaccines were traditionally considered as a stand-alone therapy, it is now clear that they should be part of combinatorial strategies with ICIs or ACT. The immunosuppressive TME impairs T cell activity, suggesting that combinations of ICIs might block these factors and facilitate the effector function of anti-tumour lymphocytes186.
In situ vaccines rely on the activation of tumour-infiltrating APCs that will engulf and present endogenous TAAs. Conversely, classic tumour vaccines involve the exogenous administration of antigens or antigen-pulsed DCs187. Tumour antigens should provide sufficient immunogenicity to break the tolerance imposed by many self-molecules expressed by tumour cells, while at the same time conferring specificity for the tumour cells and avoiding unwanted recognition of non-tumour cells188. In tumour lysates, antigen identity is unknown, and they mainly contain self-molecules that cannot guarantee the correct presentation of relevant TAA. In HCC, many clinical trials based on tumour lysates have failed to produce consistent results24.
HCC peptide vaccines based on defined antigens have typically targeted TAAs such as telomerase, GPC-3 and AFP186. Spontaneous T cell responses against these antigens have been observed in patients with HCC25, indicating that they are somehow immunogenic. However, T cell affinity for their cognate antigen is unknown in most cases. Only a few strategies targeting telomerase and GPC-3 have reached the clinical stage189,190 and none of them has provided clinically meaningful results leading to pharmaceutical development.
More immunogenic vaccines should probably rely on the identification of true tumour-specific antigens. A first approach is the use of peptides identified through techniques of HLA peptidomics. The HLA peptidome is the group of HLA class I-bound peptides expressed by a particular cell and represents a distinctive immunological signature that can be selectively recognized by CTLs191. In HCC, a vaccination clinical trial based on this approach has finished recruitment and its results will be reported soon192.
A second strategy is based on the use of neoantigens. Their identification relies on a complex pipeline193 that includes the analysis of mutations in tumour cells versus wild-type cells, the expression of the mutated gene and analysis of immune-related parameters such as epitope processability and binding to HLA molecules. The ability to predict highly immunogenic neoantigens with anti-tumour activity as vaccines using this approach has been shown in other tumours such as melanoma194 and glioblastoma195. In HCC we still lack evidence from clinical studies, and there are few data associating the presence of mutations with specific immune responses. Ongoing studies by our group show that mutations found in patients with HCC might originate peptides with a higher HLA-binding capacity than non-mutated wild-type sequences. These peptides are immunogenic in HLA transgenic mice and induce T cells that selectively recognize the mutated, but not the wild-type, sequence, suggesting that this strategy might also be suitable as a vaccine in HCC196.
Enhancing with locoregional therapies
The immune system has evolved to respond to the presence of microbial pathogens and stressful tissue damage. Both circumstances are detected by intertwined innate receptors whose function dictates whether an adaptive immune response is mounted and progresses or fails to do so197. Conceivably, these functions also determine whether immunity, tolerance or ignorance occurs against tumour antigens. Hence, over the years, many attempts have been pursued to make cancer tissue look like infected or stressed tissue that would favour the adaptive activities of cytotoxic immunity. In this regard, there are two important concepts to consider: immunogenic cell death198 and the use of pathogen-associated molecular patterns199.
Immunogenic cell death is a form of death in which cells release or produce substances collectively known as alarmins, which alert and activate APCs such as DCs200. Immunogenic cell death involves reticular stress, necrosis or necroptosis, release of mitochondrial and nuclear components and the triggering of the type I interferon system198. Along these lines, a number of chemotherapy agents, radiotherapies and other physical forces cause cell and tissue destruction and the resulting improvement in immunity against cancer contributes to the therapeutic benefit198,201.
The immune system reacts to substances that are exclusive of viruses or prokaryotes. This reactivity provides a means to ignite local immunity through the local delivery of agents that include Toll-like receptor agonists202, cGAS–STING agonists203, RIGI or MDA5 agonists204 and inflammasome agonists205. These agents are often derived from microbial products or synthetic analogues and they are usually quite toxic when given systemically, causing sepsis-like cytokine release syndromes and systemic inflammation. When delivered intratumourally they might not only be more potent in eliciting an immune response, but also be less toxic. Microorganisms themselves and not their immune-stimulating molecules can be used to enhance anti-tumour immunity and this is the basis for the use of Bacillus Calmette–Guérin vaccine to locally treat superficial bladder cancer206. Oncolytic virotherapy is a cancer treatment strategy that uses viruses that replicate selectively in cancer cells to destroy them, with potential efficacy in HCC that is under study207. Yet, it is increasingly clear that most of the beneficial effects of virotherapy are due to the elicitation or activation of an immune response that targets tumour antigens rather than the cytopathic effects of the viruses208.
Image-guided interventions are common practice in HCC therapy, including percutaneous ablation or intraarterial therapies4. Such accessibility to tumour sites makes HCC ideal for local interventions that can cause immunogenic cell death or local delivery of pathogen-associated molecular patterns. Importantly, to obtain a strong immune stimulation, different locoregional therapies can be combined sequentially or simultaneously, and also combined with systemic immunotherapy209. Activation of immune responses and T cell infiltration is observed in HCC after radiofrequency ablation210. Partial tumour ablation with radiofrequency ablation or TACE in patients with advanced HCC receiving tremelimumab resulted in a response rate of 26% and a disease control rate of 89%, with 45% of the stabilizations lasting longer than 6 months, and an overall survival of 12.3 months122. These encouraging data have fuelled clinical trials in which ICIs alone or in combination with other ICIs or bevacizumab are given in combination with chemoembolization or radioembolization, or after complete surgical or percutaneous ablation (Table 4).
Future directions
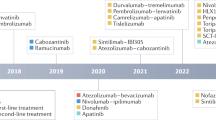
Progress in HCC immunotherapy has advanced even though the introduction of ICIs in this disease has lagged behind that in other tumours (Fig. 4). Looking to the past, the 2015 Review on immunotherapy in HCC in this journal is now outdated mainly because of clinical trial advances24. In the near future (Table 4), checkpoint-based immunotherapy might be found to increase the efficacy of locoregional and radical treatments for HCC, and neoadjuvant therapy in patients with resectable or non-resectable disease will probably pave the way to an unprecedented drop in mortality rates in this deadly disease. Ideally, the road ahead should include the development of new immunotherapy agents such as agonist immunostimulatory monoclonal antibodies, bispecific antibodies, engineered cytokines, antibody–drug conjugates, adoptive T cell therapy and neoantigen vaccination (Fig. 2). Importantly, it should also include the identification of the molecular mechanisms of sensitivity and resistance to individual agents or combinations so that useful biomarkers are made available that might help advance personalized therapy. To achieve this goal, efforts should be made to embed correlative research studies in every new clinical trial or prospective study, and information from clinical trials should ideally be made available to researchers while protecting the rights and interests of patients and companies.

Single agents and combinations approved or under study in randomized trials across tumour stages are shown. Those in bold type are already approved in at least one country. HCC, hepatocellular carcinoma; VEGF, vascular endothelial growth factor.
Conclusions
Within a decade, research into immunotherapy for HCC has grown dramatically and changed the treatment paradigm. ICIs are now well-established as active agents in the advanced stage. A better understanding of the local factors that give rise to, or abort, an anti-tumour immune response in the TME is shaping the design of new agents and combinatorial therapies. The development of such therapies in synergistic combinations will probably be the key task in the future, with the hope that a new and effective immunotherapeutic breakthrough is made. Along the way, the quest for accurate, user-friendly biomarkers will probably also be successful in opening the door to personalized immunotherapies.
References
Ferlay, J. et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 144, 1941–1953 (2019).
WHO. Liver. http://gco.iarc.fr/today/data/factsheets/cancers/11-Liver-fact-sheet.pdf (2018).
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2, 16018 (2016).
European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 69, 182–236 (2018). A thorough and updated guidance on the management of HCC.
Heimbach, J. K. et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 67, 358–380 (2018).
Vogel, A. et al. Hepatocellular carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 29, iv238–iv255 (2018).
Omata, M. et al. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: a 2017 update. Hepatol. Int. 11, 317–370 (2017).
Llovet, J. M. et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, 378–390 (2008).
Lencioni, R. et al. Sorafenib or placebo plus TACE with doxorubicin-eluting beads for intermediate stage HCC: the SPACE trial. J. Hepatol. 64, 1090–1098 (2016).
Kudo, M. et al. Brivanib as adjuvant therapy to transarterial chemoembolization in patients with hepatocellular carcinoma: a randomized phase III trial. Hepatology 60, 1697–1707 (2014).
Bruix, J. et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 16, 1344–1354 (2015).
Kudo, M. et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 391, 1163–1173 (2018).
Bruix, J. et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 389, 56–66 (2017).
Abou-Alfa, G. K. et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 379, 54–63 (2018).
Zhu, A. X. et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Oncol. 20, 282–296 (2019).
Schreiber, R. D., Old, L. J. & Smyth, M. J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570 (2011).
Rabinovich, G. A., Gabrilovich, D. & Sotomayor, E. M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 25, 267–296 (2007).
Cai, L. et al. Defective HLA class I antigen processing machinery in cancer. Cancer Immunol. Immunother. 67, 999–1009 (2018).
Velcheti, V. & Schalper, K. Basic overview of current immunotherapy approaches in cancer. Am. Soc. Clin. Oncol. Educ. Book 35, 298–308 (2016).
Chen, L. & Flies, D. B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 13, 227–242 (2013).
He, X. & Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 30, 660–669 (2020).
Jain, N., Nguyen, H., Chambers, C. & Kang, J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc. Natl Acad. Sci. USA 107, 1524–1528 (2010).
Ribas, A. & Wolchok, J. D. Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018).
Prieto, J., Melero, I. & Sangro, B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 12, 681–700 (2015).
Flecken, T. et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology 59, 1415–1426 (2014).
Yarchoan, M., Johnson, B. A., Lutz, E. R., Laheru, D. A. & Jaffee, E. M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 17, 209–222 (2017).
Luksza, M. et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 551, 517–520 (2017).
Malekzadeh, P. et al. Antigen experienced T cells from peripheral blood recognize p53 neoantigens. Clin. Cancer Res. 26, 1267–1276 (2020).
Efremova, M., Finotello, F., Rieder, D. & Trajanoski, Z. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front. Immunol. 8, 1679 (2017).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Chan, T. A. et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann. Oncol. 30, 44–56 (2019).
Schumacher, T. N. & Schreiber, R. D. Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015).
Fujimoto, A. et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 48, 500–509 (2016).
Crispe, I. N. The liver as a lymphoid organ. Annu. Rev. Immunol. 27, 147–163 (2009).
Ebrahimkhani, M. R., Mohar, I. & Crispe, I. N. Cross-presentation of antigen by diverse subsets of murine liver cells. Hepatology 54, 1379–1387 (2011).
Krenkel, O. & Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 17, 306–321 (2017).
Ormandy, L. et al. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 65, 2457–2464 (2005).
Shetty, S., Lalor, P. F. & Adams, D. H. Liver sinusoidal endothelial cells – gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 15, 555–567 (2018).
Höchst, B. et al. Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion. J. Hepatol. 59, 528–535 (2013).
Dunham, R. M. et al. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J. Immunol. 190, 2009–2016 (2013).
Yu, M. C. et al. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology 40, 1312–1321 (2004).
Cariani, E. et al. HLA and killer immunoglobulin-like receptor genes as outcome predictors of hepatitis C virus-related hepatocellular carcinoma. Clin. Cancer Res. 19, 5465–5473 (2013).
Cariani, E. et al. Natural killer cells phenotypic characterization as an outcome predictor of HCV-linked HCC after curative treatments. Oncoimmunology 5, e1154249 (2016).
Hoechst, B. et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 50, 799–807 (2009).
Zhang, Q. F. et al. Liver-infiltrating CD11b-CD27- NK subsets account for NK-cell dysfunction in patients with hepatocellular carcinoma and are associated with tumor progression. Cell. Mol. Immunol. 14, 819–829 (2017).
Fu, J. et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 132, 2328–2339 (2007).
Gao, Q. et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J. Clin. Oncol. 25, 2586–2593 (2007).
Chen, K. J. et al. Selective recruitment of regulatory T cell through CCR6-CCL20 in hepatocellular carcinoma fosters tumor progression and predicts poor prognosis. PLoS ONE 6, e24671 (2011).
Hoechst, B. et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 135, 234–243 (2008).
Veglia, F., Perego, M. & Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nature Immunology 19, 108–119 (2018).
Yeung, O. W. H. et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 62, 607–616 (2015).
Xiao, X. et al. PD-1hi identifies a novel regulatory B-cell population in human hepatoma that promotes disease progression. Cancer Discov. 6, 546–559 (2016).
Zhao, F. et al. Human CCR4+ CCR6+ Th17 cells suppress autologous CD8+ T cell responses. J. Immunol. 188, 6055–6062 (2012).
Han, Y. et al. Human CD14+CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology 59, 567–579 (2014).
Affo, S., Yu, L.-X. & Schwabe, R. F. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu. Rev. Pathol. Mech. Dis. 12, 153–186 (2017).
Zhou, S. L. et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology 150, 1646–1658.e17 (2016).
Wu, K., Kryczek, I., Chen, L., Zou, W. & Welling, T. H. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 69, 8067–8075 (2009).
Ji, J. et al. Hepatic stellate cell and monocyte interaction contributes to poor prognosis in hepatocellular carcinoma. Hepatology 62, 481–495 (2015).
Spranger, S., Bao, R. & Gajewski, T. F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015).
Luke, J. J., Bao, R., Sweis, R. F., Spranger, S. & Gajewski, T. F. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin. Cancer Res. 25, 3074–3083 (2019).
Spranger, S. et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc. Natl Acad. Sci. USA 113, E7759–E7768 (2016).
Barry, K. C. et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 24, 1178–1191 (2018).
Bray, S. M., Vujanovic, L. & Butterfield, L. H. Dendritic cell-based vaccines positively impact natural killer and regulatory T cells in hepatocellular carcinoma patients. Clin. Dev. Immunol. 2011, 249281 (2011).
Sánchez-Paulete, A. R. et al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov. 6, 71–79 (2016).
de Galarreta, M. R. et al. β-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 9, 1124–1141 (2019). A link between a frequent genetic alteration in HCC and resistance to ICIs in animal models.
Harding, J. J. et al. Prospective genotyping of hepatocellular carcinoma: clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin. Cancer Res. 25, 2116–2126 (2019).
Cristescu, R. et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362, eaar3593 (2018).
Fridman, W. H., Zitvogel, L., Sautès-Fridman, C. & Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 14, 717–734 (2017).
Peng, W. et al. Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. 6, 202–216 (2016).
Skoulidis, F. et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 8, 822–835 (2018).
Spranger, S. & Gajewski, T. F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 18, 139–147 (2018).
Duruisseaux, M. et al. Epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: a multicentre, retrospective analysis. Lancet Respir. Med. 6, 771–781 (2018).
Zhou, J. et al. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int. J. Cancer 125, 1640–1648 (2009).
Horwitz, E. et al. Human and mouse VEGFA-amplified hepatocellular carcinomas are highly sensitive to sorafenib treatment. Cancer Discov. 4, 730–743 (2014).
Glodde, N. et al. Reactive neutrophil responses dependent on the receptor tyrosine kinase c-MET limit cancer immunotherapy. Immunity 47, 789–802.e9 (2017).
Sanmamed, M. F. et al. Serum interleukin-8 reflects tumor burden and treatment response across malignancies of multiple tissue origins. Clin. Cancer Res. 20, 5697–5707 (2014).
Tsai, J. H. F. et al. Elevated urinary transforming growth factor-β1 level as a tumour marker and predictor of poor survival in cirrhotic hepatocellular carcinoma. Br. J. Cancer 76, 244–250 (1997).
Ritter, M. et al. Immunoregulation of dendritic and T cells by alpha-fetoprotein in patients with hepatocellular carcinoma. J. Hepatol. 41, 999–1007 (2004).
Pardee, A. D., Shi, J. & Butterfield, L. H. Tumor-derived α-fetoprotein impairs the differentiation and T cell stimulatory activity of human dendritic cells. J. Immunol. 193, 5723–5732 (2014).
Dong, L. Q. et al. Heterogeneous immunogenomic features and distinct escape mechanisms in multifocal hepatocellular carcinoma. J. Hepatol. 72, 896–908 (2020).
Kamimura, H. et al. Reduced NKG2D ligand expression in hepatocellular carcinoma correlates with early recurrence. J. Hepatol. 56, 381–388 (2012).
Makarova-Rusher, O. V., Medina-Echeverz, J., Duffy, A. G. & Greten, T. F. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 62, 1420–1429 (2015).
Berraondo, P., Ochoa, M. C., Olivera, I. & Melero, I. Immune desertic landscapes in hepatocellular carcinoma shaped by β-catenin activation. Cancer Discov. 9, 1003–1005 (2019).
Montfort, A. et al. The TNF paradox in cancer progression and immunotherapy. Front. Immunol. 10, 1818 (2019).
Zhong, M. et al. Induction of tolerogenic dendritic cells by activated TGF-β/Akt/Smad2 signaling in RIG-I-deficient stemness-high human liver cancer cells. BMC Cancer 19, 439 (2019).
Zhang, F. et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 7, 52294–52306 (2016).
Ahmadzadeh, M. & Rosenberg, S. A. TGF-β1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J. Immunol. 174, 5215–5223 (2005).
Okumoto, K. et al. Possible contribution of circulating transforming growth factor-beta1 to immunity and prognosis in unresectable hepatocellular carcinoma. Liver Int. 24, 21–28 (2004).
Yamagiwa, S., Gray, J. D., Hashimoto, S. & Horwitz, D. A. A role for TGF-β in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J. Immunol. 166, 7282–7289 (2001).
Peng, L. et al. High TGF-β1 expression predicts poor disease prognosis in hepatocellular carcinoma patients. Oncotarget 8, 34387–34397 (2017).
Lin, T. H. et al. High serum transforming growth factor-β1 levels predict outcome in hepatocellular carcinoma patients treated with sorafenib. Clin. Cancer Res. 21, 3678–3684 (2015).
Feun, L. G. et al. Phase 2 study of pembrolizumab and circulating biomarkers to predict anticancer response in advanced, unresectable hepatocellular carcinoma. Cancer 125, 3603–3614 (2019).
Stewart, C. A. et al. Interferon-dependent IL-10 production by Tregs limits tumor Th17 inflammation. J. Clin. Invest. 123, 4859–4874 (2013).
Kuang, D. M. et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 206, 1327–1337 (2009).
Yi, Y. et al. The functional impairment of HCC-infiltrating γδ T cells, partially mediated by regulatory T cells in a TGFβ- and IL-10-dependent manner. J. Hepatol. 58, 977–983 (2013).
Arihara, F. et al. Increase in CD14+HLA-DR-/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol. Immunother. 62, 1421–1430 (2013).
Shi, Y., Song, Q., Hu, D., Zhuang, X. & Yu, S. Tumor-infiltrating lymphocyte activity is enhanced in tumors with low IL-10 production in HBV-induced hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 461, 109–114 (2015).
Apte, R. S., Chen, D. S. & Ferrara, N. VEGF in signaling and disease: beyond discovery and development. Cell 176, 1248–1264 (2019).
Courau, T. et al. TGF-β and VEGF cooperatively control the immunotolerant tumor environment and the efficacy of cancer immunotherapies. JCI Insight 1, e85974 (2016).
Brown, Z. J. et al. Indoleamine 2,3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunol. Immunother. 67, 1305–1315 (2018).
Cai, X. Y. et al. Overexpression of CD39 in hepatocellular carcinoma is an independent indicator of poor outcome after radical resection. Medicine 95, e4989 (2016).
Shali, S. et al. Ecto-5′-nucleotidase (CD73) is a potential target of hepatocellular carcinoma. J. Cell. Physiol. 234, 10248–10259 (2019).
Lee, I. C. et al. Serum interferon gamma level predicts recurrence in hepatocellular carcinoma patients after curative treatments. Int. J. Cancer 133, 2895–2902 (2013).
Nagao, M. et al. The impact of interferon gamma receptor expression on the mechanism of escape from host immune surveillance in hepatocellular carcinoma. Hepatology 32, 491–500 (2000).
Shi, F. et al. PD-1 and PD-L1 upregulation promotes CD8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer 128, 887–896 (2011).
Zabala, M. et al. Induction of immunosuppressive molecules and regulatory T cells counteracts the antitumor effect of interleukin-12-based gene therapy in a transgenic mouse model of liver cancer. J. Hepatol. 47, 807–815 (2007).
Sangro, B. et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 59, 81–88 (2013). The first clinical trial of immunotherapy in HCC, showing activity of CTLA4 blockade.
El-Khoueiry, A. B. et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389, 2492–2502 (2017). The first clinical trial to show consistent antitumor activity of PD1 blockade in HCC.
Zhu, A. X. et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 19, 940–952 (2018).
Finn, R. S. et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase III trial. J. Clin. Oncol. 38, 193–202 (2020).
Yau, T. et al. CheckMate 459: a randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC) [abstract LBA38_PR]. Ann. Oncol. 30 (Suppl. 5), v874–v875 (2019).
Sangro, B. et al. CheckMate 459: long-term (minimum follow-up 33.6 months) survival outcomes with nivolumab versus sorafenib as first-line treatment in patients with advanced hepatocellular carcinoma [abstract LBA-3]. Ann. Oncol. 31 (Suppl. 3), S241–S242 (2020).
Gao, Q. et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin. Cancer Res. 15, 971–979 (2009).
Li, H. et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 56, 1342–1351 (2012).
Yan, W. et al. Tim-3 fosters HCC development by enhancing TGF-β-mediated alternative activation of macrophages. Gut 64, 1593–1604 (2015).
Du, W. et al. Tim-3 as a target for cancer immunotherapy and mechanisms of action. Int. J. Mol. Sci. 18, 645 (2017).
Gautron, A. S., Dominguez-Villar, M., de Marcken, M. & Hafler, D. A. Enhanced suppressor function of TIM-3+FoxP3+ regulatory T cells. Eur. J. Immunol. 44, 2703–2711 (2014).
Li, Z. et al. Immune checkpoint proteins PD-1 and TIM-3 are both highly expressed in liver tissues and correlate with their gene polymorphisms in patients with HBV-related hepatocellular carcinoma. Medicine 95, e5749 (2016).
Andrews, L. P., Marciscano, A. E., Drake, C. G. & Vignali, D. A. A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 276, 80–96 (2017).
Zhou, G. et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas. Gastroenterology 153, 1107–1119.e10 (2017).
Wang, J. et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 176, 334–347.e12 (2019).
Duffy, A. G. et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 66, 545–551 (2017).
Yau, T. et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): results from CheckMate 040 [abstract]. J. Clin. Oncol. 37 (Suppl. 15), 4012 (2019).
Kelley, R. K. et al. Efficacy, tolerability, and biologic activity of a novel regimen of tremelimumab (T) in combination with durvalumab (D) for patients (pts) with advanced hepatocellular carcinoma (aHCC) [abstract]. J. Clin. Oncol. 38 (Suppl. 15), 4508 (2020).
Ascierto, P. A. et al. Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 18, 611–622 (2017).
Callahan, M. K. et al. Peripheral and tumor immune correlates in patients with advanced melanoma treated with combination nivolumab (anti-PD-1, BMS-936558, ONO-4538) and ipilimumab [abstract]. J. Clin. Oncol. 31 (Suppl. 15), 3003 (2013).
Zhu, A. X. et al. A phase Ib study of lenvatinib (LEN) plus pembrolizumab (PEMBRO) in unresectable hepatocellular carcinoma (uHCC) [abstract]. J. Clin. Oncol. 38 (Suppl. 15), 4519 (2020).
Yau, T. et al. Nivolumab (NIVO) + ipilimumab (IPI) + cabozantinib (CABO) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): results from CheckMate 040 [abstract]. J. Clin. Oncol. 38 (Suppl. 4), 478 (2020).
Finn, R. S. et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 382, 1894–1905 (2020). The pivotal clinical trial that has established a combination of an anti-PD1 and an anti -VEGF as the standard of care for first-line systemic therapy in HCC.
Finn, R. S. et al. IMbrave150: updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC) [abstract]. J. Clin. Oncol. 39 (Suppl. 3), 267 (2021).
Lee, M. et al. Randomised efficacy and safety results for atezolizumab (atezo) + bevacizumab (bev) in patients (pts) with previously untreated, unresectable hepatocellular carcinoma (HCC) [abstract LBA39]. Ann. Oncol. 30 (Suppl. 5), v875 (2019).
Siegel, A. B. et al. Phase II trial evaluating the clinical and biologic effects of bevacizumab in unresectable hepatocellular carcinoma. J. Clin. Oncol. 26, 2992–2998 (2008).
Boige, V. et al. Efficacy, safety, and biomarkers of single-agent bevacizumab therapy in patients with advanced hepatocellular carcinoma. Oncologist 17, 1063–1072 (2012).
Ren, Z. et al. Sintilimab plus bevacizumab biosimilar vs sorafenib as first-line treatment for advanced hepatocellular carcinoma (ORIENT-32)2 [abstract LBA2]. Ann. Oncol. 31 (Suppl. 6), S1287 (2020).
Rahma, O. E. & Hodi, F. S. The intersection between tumor angiogenesis and immune suppression. Clin. Cancer Res. 25, 5449–5457 (2019).
Feng, P.-H. et al. Bevacizumab reduces S100A9-positive MDSCs linked to intracranial control in patients with EGFR-mutant lung adenocarcinoma. J. Thorac. Oncol. 13, 958–967 (2018).
Limagne, E. et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res. 76, 5241–5252 (2016).
Shigeta, K. et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology 71, 1247–1261 (2020).
Fabian, M. A. et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 23, 329–336 (2005).
Postow, M. A., Sidlow, R. & Hellmann, M. D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 378, 158–168 (2018).
Sangro, B. et al. Diagnosis and management of toxicities of immune checkpoint inhibitors in hepatocellular carcinoma. J. Hepatol. 72, 320–341 (2020). A thorough guidance on how to manage the toxicity of ICIs in HCC.
Yau, T. et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the CheckMate 040 randomized clinical trial. JAMA Oncol. 6, e204564 (2020). The first clinical trial to show improved activity with the dual blockade of CTLA4 and PD1.
Duan, J. et al. Use of immunotherapy with programmed cell death 1 vs programmed cell death ligand 1 inhibitors in patients with cancer: a systematic review and meta-analysis. JAMA Oncol. 6, 375–384 (2020).
Spain, L., Diem, S. & Larkin, J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat. Rev. 44, 51–60 (2016).
Vollmer, O. et al. Characterization of auto-immune hepatitis associated with the use of anti-TNFα agents: an analysis of 389 cases in VigiBase. Autoimmun. Rev. 19, 102460 (2020).
Peeraphatdit, T. B. et al. Hepatotoxicity from immune checkpoint inhibitors: a systematic review and management recommendation. Hepatology 72, 315–329 (2020).
Geiger-Gritsch, S. et al. Safety of bevacizumab in patients with advanced cancer: a meta-analysis of randomized controlled trials. Oncologist 15, 1179–1191 (2010).
Brusilovskaya, K., Königshofer, P., Schwabl, P. & Reiberger, T. Vascular targets for the treatment of portal hypertension. Semin. Liver Dis. 39, 483–501 (2019).
Reiberger, T. et al. Sorafenib attenuates the portal hypertensive syndrome in partial portal vein ligated rats. J. Hepatol. 51, 865–873 (2009).
Scheiner, B. et al. Short- and long-term effects of transarterial chemoembolization on portal hypertension in patients with hepatocellular carcinoma. United Eur. Gastroenterol. J. 7, 850–858 (2019).
Rimola, J. et al. Radiological response to nivolumab in patients with hepatocellular carcinoma: a multicenter analysis of real-life practice. Eur. J. Radiol. 135, 109484 (2021).
El-Khoueiry, A. B. et al. Impact of antitumor activity on survival outcomes, and nonconventional benefit, with nivolumab (NIVO) in patients with advanced hepatocellular carcinoma (aHCC): subanalyses of CheckMate-040 [abstract]. J. Clin. Oncol. 36 (Suppl. 4), 475 (2018).
Reig, M. et al. Early dermatologic adverse events predict better outcome in HCC patients treated with sorafenib. J. Hepatol. 61, 318–324 (2014).
Wong, L. et al. Association between immune-related adverse events and efficacy of immune checkpoint inhibitors in patients with advanced hepatocellular carcinoma [abstract 325P]. Ann. Oncol. 30 (Suppl. 9), ix110 (2019).
Gordan, J. D. et al. Systemic therapy for advanced hepatocellular carcinoma: ASCO guideline. J. Clin. Oncol. 38, 4317–4345 (2020).
Pelizzaro, F. et al. Capecitabine in advanced hepatocellular carcinoma: a multicenter experience. Dig. Liver Dis. 51, 1713–1719 (2019).
Sangro, B. et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 73, 1460–1469 (2020).
Calderaro, J. et al. Programmed death ligand 1 expression in hepatocellular carcinoma: relationship with clinical and pathological features. Hepatology 64, 2038–2046 (2016). A comprehensive study of the clinical and pathological correlations of PDL1 expression in HCC.
Sia, D. et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology 153, 812–826 (2017).
Liu, X. S. & Mardis, E. R. Applications of immunogenomics to cancer. Cell 168, 600–612 (2017).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24, 1550–1558 (2018).
Chen, D. S. & Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 (2017).
Pillai, R. N. et al. Comparison of the toxicity profile of PD-1 versus PD-L1 inhibitors in non-small cell lung cancer: a systematic analysis of the literature. Cancer 124, 271–277 (2018).
Latchman, Y. et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2, 261–268 (2001).
Sugiura, D. et al. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 364, 558–566 (2019).
Davda, J. et al. Immunogenicity of immunomodulatory, antibody-based, oncology therapeutics. J. Immunother. Cancer 7, 105 (2019).
van Brummelen, E. M. J., Ros, W., Wolbink, G., Beijnen, J. H. & Schellens, J. H. M. Antidrug antibody formation in oncology: clinical relevance and challenges. Oncologist 21, 1260–1268 (2016).
FDA. Highlights of Prescribing Information: TECENTRIQ. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761034s025lbl.pdf (2020).
Rosenberg, S. A. & Restifo, N. P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015).
June, C. H. & Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 379, 64–73 (2018).
Cappuzzello, E., Sommaggio, R., Zanovello, P. & Rosato, A. Cytokines for the induction of antitumor effectors: the paradigm of cytokine-induced killer (CIK) cells. Cytokine Growth Factor Rev. 36, 99–105 (2017).
Rosenberg, S. A. et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N. Engl. J. Med. 313, 1485–1492 (1985).
Rosenberg, S. A., Spiess, P. & Lafreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 233, 1318–1321 (1986).
Takayama, T. et al. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: a randomised trial. Lancet 356, 802–807 (2000).
Lu, P. H. & Negrin, R. S. A novel population of expanded human CD3+CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency. J. Immunol. 153, 1687–1696 (1994).
Lee, J. H. et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 148, 1383–1391.e6 (2015).
Jiang, S. S. et al. A phase I clinical trial utilizing autologous tumor-infiltrating lymphocytes in patients with primary hepatocellular carcinoma. Oncotarget 6, 41339–41349 (2015).
Lee, H. J., Kim, S. K., Cho, D. & Lee, J. J. Cellular immunotherapy as a beacon of hope for hematological malignancies. Blood Res. 50, 126–128 (2015).
Gao, H. et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin. Cancer Res. 20, 6418–6428 (2014).
Jiang, Z. et al. Anti-GPC3-CAR T cells suppress the growth of tumor cells in patient-derived xenografts of hepatocellular carcinoma. Front. Immunol. 7, 690 (2017).
Butterfield, L. H. et al. A phase I/II trial testing immunization of hepatocellular carcinoma patients with dendritic cells pulsed with four alpha-fetoprotein peptides. Clin. Cancer Res. 12, 2817–2825 (2006).
Sun, L. et al. Engineered cytotoxic T lymphocytes with AFP-specific TCR gene for adoptive immunotherapy in hepatocellular carcinoma. Tumor Biol. 37, 799–806 (2016).
Gerry, A. B. et al. Improved affinity AFP-specific T cell receptor for hepatocellular carcinoma. J. Immunother. Cancer 1, P10 (2013).
Malekzadeh, P. et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Invest. 129, 1109–1114 (2019).
Hu, Z., Ott, P. A. & Wu, C. J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 18, 168–182 (2018). A review on the field of tumour neoantigen-based vaccines, discussing developments and future directions for this treatment strategy.
Tagliamonte, M. et al. Potentiating cancer vaccine efficacy in liver cancer. Oncoimmunology 7, e1488564 (2018).
Hammerich, L. et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 25, 814–824 (2019).
Coulie, P. G., Van Den Eynde, B. J., Van Der Bruggen, P. & Boon, T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat. Rev. Cancer 14, 135–146 (2014).
Sawada, Y. et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin. Cancer Res. 18, 3686–3696 (2012).
Greten, T. F. et al. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer 10, 209 (2010).
Rammensee, H.-G. & Singh-Jasuja, H. HLA ligandome tumor antigen discovery for personalized vaccine approach. Expert Rev. Vaccin. 12, 1211–1217 (2013).
Buonaguro, L. & HEPAVAC Consortium. Developments in cancer vaccines for hepatocellular carcinoma. Cancer Immunol. Immunother. 65, 93–99 (2016).
Chen, F. et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J. Clin. Invest. 129, 2056–2070 (2019).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017).
Keskin, D. B. et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239 (2019).
Vercher, E. et al. Identification of neoantigen-reactive T cells in hepatocellular carcinoma: implication in adoptive T cell therapy. J. Hepatol. 73, S39–S40 (2020).
Matzinger, P. The danger model: a renewed sense of self. Science 296, 301–305 (2002).
Galluzzi, L., Buqué, A., Kepp, O., Zitvogel, L. & Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2017).
Vanpouille-Box, C., Hoffmann, J. A. & Galluzzi, L. Pharmacological modulation of nucleic acid sensors – therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 18, 845–867 (2019).
Yang, D., Han, Z. & Oppenheim, J. J. Alarmins and immunity. Immunol. Rev. 280, 41–56 (2017).
Rodriguez-Ruiz, M. E., Vitale, I., Harrington, K. J., Melero, I. & Galluzzi, L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat. Immunol. 21, 120–134 (2020).
Smith, M. et al. Trial Watch: Toll-like receptor agonists in cancer immunotherapy. OncoImmunology 7, e1526250 (2018).
Flood, B. A., Higgs, E. F., Li, S., Luke, J. J. & Gajewski, T. F. STING pathway agonism as a cancer therapeutic. Immunol. Rev. 290, 24–38 (2019).
Márquez-Rodas, I. et al. Intratumoral nanoplexed poly I:C BO-112 in combination with systemic anti-PD-1 for patients with anti-PD-1-refractory tumors. Sci. Transl. Med. 12, eabb0391 (2020).
Bhatia, S. et al. Intratumoral G100, a TLR4 agonist, induces antitumor immune responses and tumor regression in patients with Merkel cell carcinoma. Clin. Cancer Res. 25, 1185–1195 (2019).
Aznar, M. A. et al. Intratumoral delivery of immunotherapy–act locally, think globally. J. Immunol. 198, 31–39 (2017).
Tenneti, P., Borad, M. J. & Babiker, H. M. Exploring the role of oncolytic viruses in hepatobiliary cancers. Immunotherapy 10, 971–986 (2018).
Melero, I. et al. Repurposing infectious disease vaccines for intratumoral immunotherapy. J. Immunother. Cancer 8, e000443 (2020).
Rodríguez-Ruiz, M. E. et al. Combined immunotherapy encompassing intratumoral poly-ICLC, dendritic-cell vaccination and radiotherapy in advanced cancer patients. Ann. Oncol. 29, 1312–1319 (2018).
Mizukoshi, E. et al. Enhancement of tumor-associated antigen-specific T cell responses by radiofrequency ablation of hepatocellular carcinoma. Hepatology 57, 1448–1457 (2013).
Sangro, B. et al. Nivolumab in sorafenib-naive and -experienced patients with advanced hepatocellular carcinoma (HCC): survival, hepatic safety, and biomarker assessments in CheckMate-040 [abstract 141]. Hepatology 66 (Suppl. 1), 82A (2017).
Qin, S. et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: a multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 21, 571–580 (2020).
Acknowledgements
S.H.-S. receives funding from ISCIII/FEDER, UE (PI18/00556), and Gobierno de Navarra (Departamento de Salud (045-2017)) co-financed (50%) with FEDER funds (UE, FEDER 2014-2020 “Una manera de hacer Europa”). I.M. receives funding from MINECO SAF2014-52361-R and SAF 2017-83267-C2-1R; Worldwide Cancer Research Grant (15-1146); the Asociacion Española Contra el Cancer (AECC) Foundation under grant GCB15152947MELE; and the European Union’s Horizon 2020 Program (grant agreement no. 635122 PROCROP). B.S. receives funding from ISCIII/EU TRANSCAN-2 (AC16/00065) and ISCIII FIS (PI19/00742). P.S. receives funding from Instituto de Salud Carlos III co-financed by European FEDER funds (PI17/00249 and PI20/00260), Fundación Bancaria La Caixa “Hepacare” project, and “Murchante contra el Cáncer” initiative.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
I.M. reports advisory roles with Roche-Genentech, Bristol-Myers Squibb, CYTOMX, Incyte, MedImmune, Tusk, F-Star, Genmab, Molecular Partners, Alligator, Bioncotech, MSD, Merck Serono, Boehringer Ingelheim, Astra Zeneca, Numab, Catalym and Bayer, and research funding from Roche, BMS, Alligator and Bioncotech. B.S. reports consultancy fees from Adaptimmune, Astra Zeneca, Bayer, BMS, BTG, Eli Lilly, Ipsen, Novartis, Merck, Roche, Sirtex Medical and Terumo and speaker fees from Astra Zeneca, Bayer, BMS, BTG, Eli Lilly, Ipsen, Novartis, Merck, Roche, Sirtex Medical, Terumo BMS and Sirtex Medical. P.S. and S.H.-S. report no competing interests.
Additional information
Peer review information
Nature Reviews Gastroenterology & Hepatology thanks A.-L. Cheng, P. Galle, A. Zhu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ClinicalTrial.gov: https://www.clinicaltrials.gov/
Glossary
- Biomarker
-
Biological parameter assessed in the laboratory that enables predicting or following the effects of a given treatment; identification of predictive biomarkers is important in selecting patients who would benefit from treatment.
- Vascular endothelial growth factor
-
(VEGF). A family of soluble proteins that regulate angiogenesis of blood and lymphatic vessels acting on receptors encompassing intracellular tyrosine kinase domains; VEGFA is considered the main pro-angiogenic factor in human malignancies including HCC.
- Tumour microenvironment
-
Cellular and molecular components of the malignant tissue in which tumour and stromal cells exist in a mutual relationship in which each cell type shapes the functions of the other.
- Myeloid-derived suppressor cells
-
Immature haematopoietic cells of myeloid lineage resembling either macrophages or neutrophils that are abundant in patients with advanced cancer and are able to inhibit T cell responses.
- Immune checkpoint
-
Surface receptor expressed on immune system cells that downregulates the intensity of immune responses or prevents their onset; neutralization with monoclonal antibodies of checkpoint receptors or their ligands known as checkpoint inhibitors restores immune responses to cancer.
- Cytotoxic T lymphocyte-associated antigen 4
-
(CTLA4). Co-inhibitory surface glycoprotein also known as CD152 that is expressed on activated T cells and constitutively on regulatory T cells and that shows homology and shares ligands with CD28; it is known as an immune checkpoint inhibitor that upon ligation downregulates T cell-mediated immune responses to avoid autoimmunity.
- PD1
-
Checkpoint inhibitor molecule also known as CD279 expressed on the membrane of activated and exhausted T cells that, upon ligation by its cognate ligand PDL1, downregulates immune responses bringing tyrosine phosphatases, mainly SHP-2, to activating immune synapses.
- Therapeutic cancer vaccines
-
Formulation of cancer-expressed antigens in an immunogenic fashion, given to patients to elicit specific immune responses against malignant cells; adjuvant components enhance immunogenicity by promoting focal inflammation at the site of administration.
- Cancer neoantigens
-
Exclusive protein sequence expressed by cancer cells as a result of a non-synonymous mutation that is presented by self-MHC antigen-presenting molecules and is able to elicit cancer-specific immune responses.
- T cell receptor
-
(TCR). A clonally distributed antigen-specific receptor expressed on the surface of T cells that specifically recognizes peptides presented by MHC antigen-presenting molecules and initiates intracellular signalling via the CD3 complex.
- Tumour mutational burden
-
(TMB). Number of mutations per megabase of exonic DNA in tumour cells as compared with non-transformed cells of the same individual; it is usually expressed as number of mutations per megabase and correlates with the probability of T cell recognizable neoantigens in a given tumour.
- Ectonucleotidases CD39 and CD73
-
Surface enzymes expressed by tumour and non-tumour cells in the tumour microenvironment that synthesize the immunosuppressive metabolite adenosine from ATP.
- Immune-mediated adverse events
-
(IMAEs). Series of adverse effects following treatment with checkpoint inhibitors that resemble organ-specific autoimmune conditions.
- Chimeric antigen receptors
-
(CARs). Fusion proteins composed of a single-chain antibody that recognizes a surface tumour protein linked to a transmembrane domain; these constructs are usually transfected to T cells with retroviral vectors to artificially confer the ability to recognize and destroy cancer cells.
- Immunogenic cell death
-
A form of cell death that provokes immunity against the cell’s antigenic components and that usually accompanies stressful or non-programmed cell death; it is mediated by alarmins that are recognized by innate receptors on immune cells, especially dendritic cells.
- Toll-like receptor
-
A member of the family of surface and endosomal proteins that have evolved to detect pathogen-associated molecular patterns that denote the presence of moieties exclusively present in prokaryotic cells.
Rights and permissions
About this article

Cite this article
Sangro, B., Sarobe, P., Hervás-Stubbs, S. et al. Advances in immunotherapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 18, 525–543 (2021). https://doi.org/10.1038/s41575-021-00438-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41575-021-00438-0
This article is cited by
-
Expression of PD-L1 clones (22C3 and 28-8) in hepatocellular carcinoma: a tertiary cancer care hospital experience
Egyptian Liver Journal (2024)
-
Construction of a prognostic model for hepatocellular carcinoma patients receiving transarterial chemoembolization treatment based on the Tumor Burden Score
BMC Cancer (2024)
-
Nano-modified viruses prime the tumor microenvironment and promote the photodynamic virotherapy in liver cancer
Journal of Biomedical Science (2024)
-
High LGALS3 expression induced by HCP5/hsa-miR-27b-3p correlates with poor prognosis and tumor immune infiltration in hepatocellular carcinoma
Cancer Cell International (2024)
-
Efficacy and safety of the combination of camrelizumab and apatinib in the treatment of liver cancer: a systematic review and single-arm meta-analysis
BMC Gastroenterology (2024)
