
Abstract
Currently, metformin is the first-line medication to treat type 2 diabetes mellitus (T2DM) in most guidelines and is used daily by >200 million patients. Surprisingly, the mechanisms underlying its therapeutic action are complex and are still not fully understood. Early evidence highlighted the liver as the major organ involved in the effect of metformin on reducing blood levels of glucose. However, increasing evidence points towards other sites of action that might also have an important role, including the gastrointestinal tract, the gut microbial communities and the tissue-resident immune cells. At the molecular level, it seems that the mechanisms of action vary depending on the dose of metformin used and duration of treatment. Initial studies have shown that metformin targets hepatic mitochondria; however, the identification of a novel target at low concentrations of metformin at the lysosome surface might reveal a new mechanism of action. Based on the efficacy and safety records in T2DM, attention has been given to the repurposing of metformin as part of adjunct therapy for the treatment of cancer, age-related diseases, inflammatory diseases and COVID-19. In this Review, we highlight the latest advances in our understanding of the mechanisms of action of metformin and discuss potential emerging novel therapeutic uses.
Key points
-
The liver and gut are the main target organs for metformin.
-
Mitochondria and lysosomes are the organelle targets in the glucose-lowering effect of metformin.
-
Host–gut microbiota interactions contribute to metformin’s therapeutic effects.
-
Metformin has anti-inflammatory and immunomodulatory properties in various immune-related diseases through AMPK-dependent and AMPK-independent mechanisms involving both the innate and adaptive immune systems.
-
Metformin therapy in patients with type 2 diabetes mellitus enhances the release of GDF15, which might facilitate weight loss but is not required for the effect in reducing blood levels of glucose.
Similar content being viewed by others

Introduction
For the past 60 years, metformin (1,1-dimethylbiguanide hydrochloride) has been the most commonly used glucose-lowering agent and has become the first-line medication for individuals newly diagnosed with type 2 diabetes mellitus (T2DM) in many clinical guidelines1,2. Metformin was first synthesized in 1922, and the first report of it being used to lower blood levels of glucose (in rabbits) was published in 1929 (refs. 3,4). In 1949, metformin (called flumamine at that time) was used for the treatment of an epidemic influenza outbreak in the Philippines and was noted to lower blood levels of glucose in some of the patients with influenza5. Interestingly, in the past few years, the use of metformin has been explored for the management of patients with T2DM and obesity who contract influenza6 or COVID-19 (refs. 7,8,9) (Box 1). In 1957, the French physician Jean Sterne introduced the clinical use of metformin for the treatment of adult-onset diabetes mellitus3,4. The benefits of metformin therapy in T2DM have been well documented; it has long-term safety and efficacy data, low risk of hypoglycaemia, cardiovascular benefits, mortality benefits, additive or synergistic effects in combination therapy, low cost and wide availability1,2,10,11,12. In addition, metformin moderately reduces body weight gain, possibly through upregulation of the anorectic cytokine growth differentiation factor 15 (refs. 13,14) (Box 2). Metformin is now used daily by >200 million patients with T2DM worldwide as monotherapy or in combination with sulfonylureas or dipeptidyl peptidase 4 inhibitors1,2. Administered orally as an immediate-release or extended-release formulation, the usual dose of metformin (0.5–2.5 g daily) is effective for long-term glycaemic control, with notable changes in HbA1c from baseline1,2.
It is important to note that metformin is increasingly being used during pregnancy for the management of gestational diabetes mellitus and in those with polycystic ovary syndrome or T2DM15. As metformin crosses the placenta and circulates in the developing fetus, the long-term effects of fetal exposure to metformin have raised concerns about the potential risk to growth and development of the fetus, and later, on offspring health16. While in utero exposure to metformin was considered safe, some studies have found higher occurrence of small for gestational age birthweight and increased risk of childhood obesity17. However, reassuring data were provided in a long-term follow-up study showing similar anthropometrics in children exposed or not to metformin in utero18.
Metformin is a synthetic biguanide (two coupled molecules of guanidine) that is mainly absorbed in the upper small intestine and exhibits flip–flop pharmacokinetics with limited oral bioavailability19,20. Metformin is a basic hydrophilic drug with a pKa value of 11.5, and exists as an organic cation at physiological pH. The biodistribution and pharmacodynamics of the drug are dependent on the expression of transporters for cationic compounds, such as organic cation transporters (OCTs), plasma membrane monoamine transporter and multidrug and toxin extrusion proteins21,22. The half-life of metformin in plasma is short, at 2–6 h, and leads to a steady-state plasma concentration of metformin in patients with T2DM of ~4–15 μM (~0.5–2.0 μg/ml)23. Concerns have been expressed regarding the therapeutically appropriate concentrations to establish the cellular actions of metformin in preclinical models24,25,26,27. It should be noted that metformin accumulates in multiple tissues, including tumours, to concentrations higher than those in plasma, which highlights uncertainty about the relevant metformin concentrations for in vitro studies19,24,27,28. Kinetic and biodistribution studies in humans using 11C-metformin PET–CT showed that the highest metformin uptake is in the gastrointestinal tract, liver and kidneys29. However, 11C-metformin PET–CT lacks the resolution to determine the subcellular distribution of metformin and cannot be used to establish whether metformin accumulates within specific organelles.
In 2019, we extensively reviewed the glucoregulatory mechanisms of metformin action in T2DM in Nature Reviews Endocrinology26. As such, the objective of this Review is to provide an update of our current understanding of the molecular mechanisms of metformin action. Early studies highlighted the liver as the major site of metformin action for the control of hepatic glucose production, through both AMP-activated protein kinase (AMPK)-dependent and AMPK-independent mechanisms. However, there is increasing evidence that other sites of action might also be important, including the gastrointestinal tract, the gut microbiota and the tissue-resident immune cells. In addition, owing to its pleiotropic modes of action, with multiple sites of action and signalling pathways, the repurposing of metformin is being expanded to include various pathophysiological conditions9,30,31. In this Review, we mostly focus on selected papers published since our previous Review and discuss the interaction of metformin with novel factors, most of which were revealed by studies in preclinical models. We also examine current knowledge on the immune-modulating and anti-inflammatory effects of metformin that underlie its therapeutic benefits as a potential anti-inflammatory and anti-ageing drug. Finally, we discuss the benefits of repurposing metformin in the treatment of local and systemic inflammation, cancer, age-related diseases and coronavirus disease 2019 (COVID-19).
Paradigm changes in glucoregulatory actions
Metformin target organs
Gastrointestinal system
The antihyperglycaemic effects of metformin are traditionally thought to primarily arise through effects in the liver; however, emerging evidence supports the role of extrahepatic mechanisms26. In the past few years, clinical studies conducted in individuals with recent-onset T2DM (duration of T2DM less than 50 months) and in non-diabetic control individuals demonstrated a metformin-associated increase in endogenous glucose production, which indicates that the glucose-lowering action of metformin is not entirely governed by a reduction in hepatic gluconeogenesis32,33. Over the past few years, the gastrointestinal tract has been the focus of attention as an additional or even alternative site of metformin action in the management of T2DM; multiple mechanisms, not necessarily mutually exclusive, have been proposed, including direct actions of the drug on intestinal cells or alterations in the composition and metabolic profile of the gut microbiota26. Specifically, the role of intestinal AMPK has been highlighted in the glucose-lowering and metabolic actions of metformin in rodents34,35,36,37. High accumulation of metformin in the gut has been reported with concentrations up to 30–300-fold times greater than in plasma and other tissues, suggesting that the gut acts as an important metformin reservoir in both humans and animal models19,37,38,39. The ‘sponge’ hypothesis has been proposed to explain the slow and dose-dependent absorption of metformin along the gastrointestinal tract40. In humans, metformin enters enterocytes via saturable apical transport but its release through the basolateral membrane is inefficient due to the absence of efflux intestinal transporters, which sequester and concentrate the drug within enterocytes. Hence, metformin in the intestinal lumen might undergo a predominantly saturable paracellular transport across the human intestine to access circulating blood40,41.
Metformin inhibits the intestinal absorption of dietary glucose in rodents and minipigs42,43,44 and patients with T2DM45. This finding was confirmed by PET–CT imaging showing accumulation of 18F-labelled fluorodeoxyglucose (FDG), a non-metabolizable glucose analogue, in the intestinal lumen of Goto–Kakizaki diabetic rats treated with a single oral dose of metformin46. The inhibition of intestinal glucose absorption resulted from the transient decrease in the abundance of sodium–glucose transporter 1 (SGLT1) at the apical membrane of enterocytes in the jejunum. The reduction in postprandial glucose response mediated by a single administration of metformin was abrogated in mice lacking SGLT1, but not in those lacking glucose transporter 2 (GLUT2)46. Interestingly, the glucose-lowering efficacy after a single administration of metformin into the jejunum of minipigs was combined with a reduction in intestinal glucose absorption and an increase in glucagon-like peptide 1 (GLP1) release, suggesting that delayed intestinal glucose absorption and the exposure of glucose to more distal regions of the intestine is sufficient to stimulate GLP1 secretion following oral intake of glucose46. In line with this observation, a single administration of metformin into the proximal and distal small intestine markedly attenuated the glycaemic response to oral glucose in patients with T2DM, concomitantly with enhanced GLP1 secretion47.
Based on observations in patients with T2DM receiving metformin, PET–CT imaging after intravenous administration of 18F-FDG, which enters enterocytes via basolateral GLUT2, showed that increased glucose uptake from the circulation into the gastrointestinal system contributes to the glucose-lowering effect of the drug and the improvement in glycaemic control37,48,49. Similarly, 18F-FDG PET–CT imaging in mice fed a high-fat diet (HFD) confirmed that metformin-induced basolateral intestinal glucose uptake is accompanied by an improvement in glucose tolerance, which occurs in a dose-dependent manner37. Taking advantage of newly developed 18F-FDG PET–MRI techniques, a dose-dependent metformin-induced accumulation of 18F-FDG was demonstrated in both the intestinal wall and the luminal space of the ileum and the colon of participants with T2DM receiving metformin50,51, which suggests that metformin also promotes the release of glucose from enterocytes into the intraluminal space. In addition, after acute oral administration of 18F-FDG and metformin in HFD-fed mice, 18F-FDG PET showed reduced transepithelial glucose transport from the proximal small intestine lumen into the circulation52.
Altogether, these studies suggest that intestinal glucose uptake along the gastrointestinal tract from both the bloodstream and the intestinal lumen is critical to the glucose-lowering capacity of metformin. Therefore, the gut might predominantly act as a glucose sink through the uptake of glucose by enterocytes in response to metformin action. In this context, the contribution of GLUT2 expression at the basolateral surface and at the apical surface of enterocytes that is triggered by metformin53, possibly via an AMPK-dependent mechanism37, is consistent with the modified glycaemic response to metformin of patients expressing a particular GLUT2 variant54. Similarly, metformin-induced GLUT1 expression in the colon and ileum, secondary to increased expression of activating transcription factor 4, might also contribute to the metformin-mediated basolateral intestinal glucose uptake in HFD-fed mice37. After uptake into enterocytes, glucose anaerobic metabolism resulted in lactate and acetate accumulation in the wall of the small intestine and release into the circulation; this finding is supported by preclinical37,55,56 (note that ref. 56 is a preprint and has not yet been peer-reviewed) and clinical studies57. Intestinal lactate and acetate production establishes a gut–liver crosstalk to blunt hepatic glucose production, possibly through reduction in the activity of the hepatic pyruvate carboxylase by reducing pH in the portal vein (a consequence of increased lactate) and of the hepatic mitochondrial pyruvate carriers 1 and 2 by acetylation (a consequence of increased acetate)37. Intestinal lactate production might also participate in an intestinal–liver futile cycle that results in increased energy expenditure during long-term treatment with metformin, which has been reported in HFD-fed mice58.
Brown adipose tissue
Brown adipose tissue (BAT) is a highly metabolically active organ and is well-recognized for its thermogenic role by dissipating energy to heat. However, a growing number of studies have shown that BAT contributes to the regulation of whole-body glucose homeostasis59,60 and it has been suggested that BAT could be a therapeutic target in the treatment and prevention of T2DM. Using 11C-metformin PET imaging, uptake of metformin was demonstrated in the interscapular BAT depot of mice61, which supports the suggestion that BAT could be a metformin target (Box 3).
Metformin target organelles
Since the early 2000s, mitochondria have been considered the classic target organelles for the glucose-lowering actions of metformin, based on matrix enrichment and the specific, mild and reversible inhibition of the mitochondrial respiratory chain complex I26. Remarkably, a study published in 2023 combining cryo-electron microscopy and enzyme kinetics identified three possible independent binding or interaction sites for biguanides on various complex I protein subunits62. The major inhibitory site is located in the amphipathic region of the quinone-binding channel (Q-channel), adjacent to a mobile structural element in the NDUFS7 subunit; when a biguanide binds to this site, reactivation of the enzymatic deactivated state is prevented62, as previously reported63. These structural data obtained using a synthetic biguanide-like molecule supports a specific effect on complex I. Of note, it is worth mentioning that inhibition of mitochondrial glycerol-3-phosphate dehydrogenase (mGPDH) and mitochondrial respiratory chain complex IV have also been suggested as alternative mechanisms of metformin action, according to studies performed in rodents64,65. However, the arguments supporting a role for these other putative mitochondrial sites of action of metformin remain highly debated66,67,68,69. Finally, the identification of metformin-binding proteins associated with isolated lysosomes in human and mouse cells highlights lysosomes as an alternative or additional functional target of metformin36.
AMPK-dependent and AMPK-independent mechanisms
Initially, it was thought that metformin acts principally through the activation of the LKB1–AMPK signalling pathway70,71. Metformin-induced AMPK activation can occur through AMP-dependent and AMP-independent AMPK-activation pathways that are correlated with the concentrations of metformin used and the target organelles (mitochondria or lysosome)36,72,73. However, AMPK-independent mechanisms of metformin action have also been documented extensively26,74,75. Of note, the acute glucose-lowering effect of metformin is preserved in liver-specific, intestine-specific and skeletal muscle-specific AMPK-knockout mouse models, which indicates that at least some direct effects of metformin are AMPK-independent56,76,77. By contrast, intestinal AMPK is required for the therapeutic effects of chronic metformin administration in HFD-fed mice35,36. Even though metformin-induced AMPK activation does not directly inhibit hepatic gluconeogenesis36,76, the effect of chronic metformin administration might indirectly improve the ability of insulin to lower hepatic glucose production as a result of an AMPK-dependent reduction in hepatic lipid-induced insulin resistance. Supporting this suggestion, impairments in the glucose-lowering and lipid-lowering effects of chronic metformin administration in mice have been associated with compromised AMPK signalling36,78. Lastly, to determine the phosphorylation events induced by metformin independently of the LKB1–AMPK signalling pathway, a quantitative proteomic approach based on selective enrichment of the phospho-scaffolding protein 14-3-3ζ interactors was conducted in liver-specific LKB1-knockout and AMPK-knockout mice in response to acute metformin administration79. Roughly half of the phosphorylation events were independent of LKB1–AMPK signalling, indicating that other stress kinases acutely activated by metformin might participate in the pleiotropic action of the drug79.
New glucose-lowering mechanisms of metformin
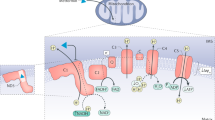
The antidiabetic effect of metformin is mainly mediated through inhibition of hepatic gluconeogenesis80; however, emerging evidence suggests that the gastrointestinal tract also has a role in the glucose-lowering action of metformin. It is widely accepted that metformin targets hepatic mitochondria and inhibits mitochondrial respiratory chain complex I; the inhibition is reversible and weak26,81. This inhibition leads to a moderate decrease in ATP synthesis by mitochondrial oxidative phosphorylation (OXPHOS) and an increase in the AMP to ATP ratio in hepatocytes. Consequently, because gluconeogenesis is a costly ATP-dependent metabolic pathway, the reduction in cellular energy charge could be sufficient to account for the decrease in hepatic gluconeogenic flux76,82. Furthermore, the metformin-induced mild increase in intracellular levels of AMP leads to inhibition of AMP-regulated enzymes involved in hepatic gluconeogenesis (such as fructose-1,6-bisphosphatase and adenylate cyclase), which contributes to a decrease in hepatic glucose production and activation of the cellular energy sensor AMPK; however, AMPK activation has no direct effect on glucose production26,74,75,76 (Fig. 1). The physiological relevance of these mechanisms has been questioned due to the use of suprapharmacological (millimolar) concentrations of metformin in the studies25,80. Indeed, studies showed that clinically relevant (micromolar) metformin concentrations suppress glucose production in primary mouse hepatocytes via mechanisms independent of apparent changes in adenine nucleotide levels36,64,83,84.

Left: Complex I inhibition-dependent mechanisms. In the liver, metformin induces a mild inhibition of mitochondrial respiratory chain complex I, leading to a moderate decrease in ATP synthesis and a concomitant increase in cellular levels of AMP. The metformin-induced decrease in hepatic gluconeogenic flux, an ATP-dependent metabolic process, could result from this reduction in ATP levels. In addition, increased AMP levels lead to inhibition of the activity of enzymes that are regulated by AMP and are involved in gluconeogenesis, such as adenylate cyclase and fructose-1-6-bisphosphatase (FBP1), which contributes to decreased glucose output. Of note, the metformin-induced increase in the AMP to ATP ratio also activates AMP-activated protein kinase (AMPK), but this has no direct effect on the regulation of glucose production. The inhibition of complex I by metformin is also accompanied by an increase in cellular redox potential (NADH:NAD+). Middle: Mitochondrial glycerol-3-phosphate dehydrogenase (mGPDH)-dependent and complex IV inhibition-dependent mechanisms. Metformin directly inhibits mGPDH, resulting in an increased cytosolic redox state (NADH:NAD+), reduced gluconeogenesis from lactate and reduced activity of the glycerol–phosphate shuttle (which transfers NADH from the cytosol to mitochondria). In addition, metformin raises the hepatic redox state through an increase in the glutathione to oxidized glutathione ratio (GSH:GSSG), leading to inhibition of genes encoding enzymes involved in gluconeogenesis through a let-7–TET3–HNF-4α pathway. Finally, metformin inhibits mitochondrial respiratory chain complex IV, which can also result in an indirect inhibition of mGPDH activity. Right: AMPK activation-dependent mechanisms in lysosomes. Metformin at low concentrations binds presenilin enhancer 2 (PEN2), which is recruited to ATPase H+ transporting accessory protein 1 (ATP6AP1) independent of changes in AMP levels, leading to inhibition of v-ATPase and phosphorylation and/or activation of AMPK in lysosomes through the formation of a supercomplex containing the v-ATPase, Ragulator, AXIN, liver kinase B1 (LKB1) and AMPK. Thereafter, metformin-activated AMPK from lysosomes reduces lipid accumulation in the liver via acetyl-CoA carboxylase (ACC) inhibition and increases glucagon-like peptide 1 (GLP1) secretion in the gut, inducing reductions in blood levels of glucose. cGPDH, cytosolic glycerol-3-phosphate dehydrogenase; HNF-4α, hepatocyte nuclear factor 4α; LDH, lactate dehydrogenase; OCT1, organic transporter 1; TET3, Tet methylcytosine dioxygenase 3.
Lysosomal PEN2–ATP6AP1 axis
It has been reported that, in primary mouse hepatocytes, low concentrations of metformin activate AMPK in lysosomes through an AMP-independent mechanism involving the recruitment of a complex composed of AXIN and the upstream kinase LKB1 to the surface of lysosomes via docking onto the vacuolar H+-ATPase (v-ATPase)–Ragulator complex72,85. In this model, low-dose metformin inhibited v-ATPase in lysosomes, which acts as a sensor of low metformin concentrations. In a study published in 2022, the membrane protein presenilin enhancer 2 (PEN2; a subunit of γ-secretase complex) was identified as a partner of metformin36. Notably, metformin at concentrations as low as 5 µM could trigger robust activation of AMPK in both primary mouse and human hepatocytes after only 2 h of treatment, which contrasts with other reports that concentrations <40 µM failed to induce AMPK phosphorylation after a 24-h incubation period84 (Foretz, M., unpublished work). In that 2022 study36, primary hepatocytes were treated with concentrations of metformin found in the bloodstream (5–10 µM) rather than those measured in the liver (100–200 µM)19,64, calling into question whether a PEN2-dependent mechanism of action is relevant in in vivo settings.
At the molecular level, the phenylalanine-35 (F35), glutamate-40 (E40) and tyrosine-47 (Y47) residues of PEN2 are critical for binding to metformin, as these residues interact with the biguanide group of the molecule. Metformin-bound PEN2 is then recruited to ATP6AP1, an accessory protein of v-ATPase, leading to the inhibition of v-ATPase and the activation of AMPK at the lysosome surface without altering cellular levels of AMP36 (Fig. 1). Of note, high-dose metformin (>100 μM) bypassed the requirement of PEN2–ATP6AP1 signalling for AMPK activation at the lysosome surface, because high doses of metformin increase the intracellular levels of AMP36,76. In terms of the physiological response, intestine-specific deletion of PEN2 in mice impaired improvements in glucose tolerance associated with GLP1 secretion in response to metformin36. However, it remains unclear whether these effects are mediated by PEN2–AMPK signalling in enterocytes that are exposed to metformin concentrations much higher than 5 µM19,36. Furthermore, PEN2 ablation in the liver abolished the metformin-induced reduction in hepatic lipid content in HFD-fed mice, supporting the suggestion that metformin might indirectly suppress gluconeogenesis in the long term through an AMPK-dependent reduction in lipid-induced insulin resistance36,76,78. However, the relevance of this mechanism in humans is questionable as metformin has no substantial efficacy in the treatment of fatty liver in patients with non-alcoholic fatty liver disease86,87.
Interestingly, in response to low concentrations of metformin, the PEN2–ATP6AP1 axis leads to activation of the AMPK pool in lysosomes in primary hepatocytes without affecting other AMPK pools, such as those of the endoplasmic reticulum (ER) and mitochondria36. Of note, it is unclear how AMPK anchored at the lysosome surface can phosphorylate its lipogenic target acetyl-CoA carboxylase and inhibit lipogenesis, which takes place in the cytoplasm and ER, to ultimately reduce hepatic lipid content. Lastly, knockdown of PEN2 or ATP6AP1 in Caenorhabditis elegans abrogated the metformin-induced extension of lifespan36. However, these experiments were conducted using extremely high concentrations of metformin (50 mM) that are 10,000 times higher than those used in vitro (5 µM)36. Additionally, the PEN2 residues involved in metformin binding in mice and humans (F35, E40 and Y47) are not conserved in C. elegans (F35, D40 and N47)88, which questions the real contribution of PEN2 in this metformin effect. The lysosomal metformin–PEN2 model raises other essential questions about the mechanism of action of metformin. Notably, this model suggests that the glucose-lowering effects of metformin occur through AMPK in the intestine but does not explain how low concentrations of metformin directly inhibit acute hepatic glucose production, which is clearly AMPK-independent64,76,89,90.
Mitochondrial glycerol-3-phosphate dehydrogenase
Other mechanisms involving changes in hepatic redox state, but independent of changes in adenine nucleotide levels and AMPK activation, have been proposed to explain the inhibition of hepatic glucose production in response to low concentrations of metformin. The cytosolic reducing equivalents (NADH) produced by intermediate metabolism are transferred from the cytosol to mitochondria through NADH shuttle systems to be oxidized by the mitochondrial electron transport chain (ETC) to generate ATP. The malate–aspartate shuttle and the glycerol–phosphate shuttle are the two major redox shuttle systems maintaining the redox balance between cytosolic and mitochondrial compartments. Clinically relevant concentrations of metformin increase the hepatic cytosolic NADH to NAD+ ratio (lactate to pyruvate ratio) independently of changes in intracellular levels of ATP, which results in inhibition of glucose production from reduced gluconeogenic substrates (lactate and glycerol) but not from oxidized substrates (alanine and pyruvate)64,91. The increase in cytosolic redox state induced by metformin was postulated to be mediated by direct inhibition of mGPDH activity (Fig. 1). mGPDH is located on the outer side of the inner membrane and, with its cytosolic partner cGPDH, constitutes a glycerol–phosphate shuttle.
However, this model of metformin-induced mGPDH inhibition raises some issues. The malate–aspartate shuttle is the main NADH shuttle in the liver and inhibition of the glycerol–phosphate shuttle might be insufficient to reduce gluconeogenesis92. Indeed, mice lacking the glycerol–phosphate shuttle display normal fasting blood levels of glucose, whereas mice lacking the malate–aspartate shuttle have reduced blood levels of glucose and an increased cytosolic NADH to NAD+ ratio93. Although many studies have demonstrated that low doses of metformin cause an increase in the cytosolic NADH to NAD+ ratio, several studies did not find a decrease in glucose production from lactate or a direct inhibition of mGPDH activity in response to metformin66,83,94. As such, a body of evidence questions whether mGPDH is a direct molecular target for metformin68,81,95.
Mitochondrial respiratory chain complex IV
In an attempt to address the controversies described in the previous sections, an alternative interpretation was proposed in 2022, in which clinically relevant concentrations of metformin inhibit complex IV activity, resulting in inhibition of mGPDH activity, an increased cytosolic redox state and a reduction in hepatic gluconeogenesis65. In this newly proposed mechanism, it has been postulated that the inhibition of complex IV by metformin blocks the ETC, leading to indirect inhibition of mGPDH activity through a decrease in the ubiquinone pool, which is the electron acceptor of mGPDH (Fig. 1). The interaction between metformin and complex IV could be driven by the ability of biguanides to bind metal ions, such as iron and copper96, which are both present in complex IV. However, all complexes of the ETC contain iron and/or copper ions, which are essential for the transfer of electrons, therefore making it unlikely that metformin specifically targets complex IV in this manner. Altogether, the fundamental concern with the hypothesis of metformin-induced complex IV inhibition is that it postulates an increase in the redox potential without impaired cellular energy charge, which goes against the OXPHOS bioenergetic process. Indeed, disruption of the ETC through the inhibition of complex IV by metformin would presumably, like inhibition of complex I, impact redox potential-dependent ATP synthesis, leading to a decrease in cellular levels of ATP. Of note, early studies that suggested that metformin induces complex I inhibition simultaneously showed an increase in the AMP to ATP and the NADH to NAD+ ratios82,97.
It is worth noting that the concept of linking complex I inhibition by metformin to suppression of energy-demanding gluconeogenic flux has been discredited because low concentrations of the drug repress glucose production in hepatocytes independently of any detectable changes in the AMP to ATP ratio64,83,84. However, the measurement of small and transient physiological changes in adenine nucleotide levels might sometimes be impeded by technical limitations. Notably, the use of AMPK as a sensitive probe for assessing subtle changes in the AMP to ATP ratio revealed metformin-dependent changes in cellular energy charge that are not detectable with other methods98. Thus, a reduction in energy charge dependent on complex I inhibition cannot be ruled out as a cause of the inhibition of hepatic gluconeogenesis in response to low concentrations of metformin.
Let-7 microRNA
In line with the redox effects of metformin, a study suggested that clinically relevant concentrations of metformin could inhibit hepatic gluconeogenesis through redox-dependent transcriptional regulation99. Mechanistically, metformin induces let-7 microRNA expression in a redox-dependent manner through an increase in the reduced glutathione to oxidized glutathione (GSH to GSSG) ratio. In turn, let-7 downregulates TET3 and changes in the ratio of HNF-4α isoforms, leading to inhibition of the expression of genes encoding enzymes involved in gluconeogenesis (Fig. 1). However, this mechanism cannot explain the acute reduction in gluconeogenic flux induced by metformin, in which the drug can inhibit hepatic glucose production in the absence of transcriptional changes in the gluconeogenic programme76.
Metformin and microbiota interactions
Modulation of microbial communities
In the past few years, the relationship between metformin and the gut microbiota has attracted much attention26. Metagenomics studies indicate that metformin induces alterations in the overall structure and functions of gut microbial communities, leading to amelioration of dysbiosis associated with T2DM and subsequently of host metabolism100,101,102,103,104,105,106,107. Of note, the effect of metformin on the gut microbiota in people with T2DM could differ with ethnicity and shows high variability102. Nevertheless, a signature of metformin treatment across different ethnicities has been established, with the enrichment of two operational taxonomic units from Bacteroides and reduced abundance of one operational taxonomic unit from Faecalibacterium108. In healthy people, metformin also affects the composition of the gut microbiota, validating the concept that the interaction between metformin and the gut microbiota is independent of the dysbiosis induced by T2DM or prevailing blood levels of glucose109,110. The gut microbial signature following metformin treatment is associated with increased levels of Escherichia spp. and decreased levels of Intestinibacter spp. in both healthy normoglycaemic people and those with T2DM104,106,110. In addition to its interactions with intestinal microbial communities, metformin treatment in patients with T2DM and periodontitis was associated with changes in oral microbiota composition111. However, it remains unclear whether the oral microbiota is directly targeted by metformin or by the influence of systemic glycaemic changes on the oral environment.
Similarly, alterations in the gut microbiota related to metformin treatment were reported in rats and mice fed a regular diet112 or a HFD113,114,115, as well as in various animal models of obesity and diabetes mellitus, in parallel to improvements in glucose tolerance107. Of interest, sex-related differences were reported for metformin-induced changes in gut microbiota composition and function in HFD-fed mice, indicating the relevance of interactions between sex hormones and the microbiota116. Importantly, metformin-induced changes in the diversity of the gut microbiota are linked to oral, but not intraperitoneal, metformin delivery in mice117. Metformin-induced changes in gut microbiota diversity of HFD-fed mice were associated with reduced microbiota encroachment into the mucus layer, which is a driver of low-grade inflammation118. Improvements in the metabolic parameters in HFD-fed mice recipients of faecal microbiota transplants from metformin-treated donors have given support to the overall beneficial metabolic effect of metformin-induced changes in gut microbiota104,114. In addition, depletion of the gut microbiota by antibiotic treatment abrogated the glucose-lowering action of metformin in HFD-fed mice113. However, some studies demonstrated the ability of metformin to reduce glucose levels in HFD-fed mice under antibiotic treatment118, making the contribution of gut microbiota to the glucose-lowering action of metformin still a matter of debate119.
Most of the clinical evidence concerning the action of metformin on the microbiota was established from the observed changes in the faecal microbiota that relate mainly to the large intestine, which is where the highest concentrations of metformin have been reported19,37,38. However, metformin can also accumulate and reach high concentrations in the small intestine19,39, triggering specific changes in the local microbiota of the duodenum, jejunum and ileum in rodents114,120. Alteration of the microbiota composition in the small intestine is accompanied by modifications in the expression of genes related to intestinal glucose and fatty acid uptake that contribute to the metabolic effects of metformin114,120,121. The distinctive role of the different microbiotas along the gastrointestinal tract was demonstrated by metformin-treated microbiota transplants to the upper small intestine that resulted in the restoration of glucose-sensing mechanisms114. In addition, the glucose-lowering efficacy of a delayed-release formulation of metformin that was delivered to the distal small intestine also favours compartmentalized gut-based mechanisms of metformin action in humans122.
The influence of metformin on gut microbiota composition can result from direct effects on bacterial growth and changes in the intestinal environment. Metformin treatment alters gut microbiota composition, which is dependent on AMPK in the intestine and probably results from the modulation of expression of antimicrobial peptides, including regenerating islet-derived protein 3γ35. In addition, accumulating evidence shows that metformin enhances the protection of the intestinal mucosal barrier function by increasing the relative abundance of Akkermansia muciniphila and the number of goblet cells, leading to thickening of the mucus layer113,123,124,125. Furthermore, metformin also promotes the expression of the tight-junction protein occludin113,124,125,126, which reduces the translocation of lipopolysaccharide, colonic inflammation and dysfunctional gut permeability. Using in vitro gut microbiota culture models, metformin was shown to directly alter gut microbiota growth curves and compositional profiles104,127,128. By combining data from metatranscriptomics and metaproteomics analyses, metformin–microbiota interactions were found to be associated with changes in microbiota functional activities linked to the modification of multiple metabolic pathways104,105,106,127,128, including the production of short-chain fatty acids (SCFAs) and glucose metabolism (glycolysis, aerobic oxidation and pentose phosphate pathways). Metformin use was linked to the enrichment of bacterial genes with products involved in carbohydrate, lipid, amino acid and nitrogen metabolism, drug resistance and lipopolysaccharide biosynthesis104,105,127. By selectively altering the metabolic profile of the human gut microbiota, metformin provides a competitive advantage and promotes specific bacterial populations, including Escherichia coli and A. muciniphila102,103,104,105,127,129.
Metformin–microbiota relationship
Previous studies have shown that metformin impairs folate and methionine production in the microbiota of the nematode C. elegans (using worms fed with live E. coli), leading to altered host methionine metabolism and healthspan extension130. In subsequent studies, a high-throughput host–microbiota–drug–nutrient screen approach identified a bacterial signalling pathway that integrates metformin and nutrient signals to alter metabolite production by the microbiota131. In support of metformin–microbiota interactions, an in silico human microbiota metabolic modelling study showed a predicted increase in agmatine production capacity by E. coli in metformin-treated patients with T2DM131. In addition, using systems biology methodologies, a competing behaviour of prevalent gut microbiota from metformin-treated patients with T2DM was linked to their capacity to utilize and produce SCFAs and amino acids129. Similarly, using global metabolomic approaches, the metabolism of branched-chain amino acids was correlated to changes in the gut microbiota and the hypoglycaemic effect associated with metformin administration in healthy individuals132.
The crosstalk between metformin and gut microbiota metabolism was further emphasized by the causal relationship reported between the long-term durability of the glycaemic response to metformin monotherapy in T2DM and different gut microbiota compositions that were characterized by unique microbial metabolic pathways133. For instance, pathways involved in thiamine biosynthesis could be involved in ensuring the sustained durability of the glycaemic response to metformin monotherapy133. Another well-documented metformin–gut microbiota interaction is the shift towards SCFA-producing bacteria and increased faecal concentrations of both butyrate and propionate in patients with T2DM and obesity102,103,104,106. SCFAs exhibit beneficial effects on glucose metabolism via multiple pathways, including stimulation of the release of the incretin hormone GLP1 and peptide YY from enteroendocrine L cells and modulation of intestinal gluconeogenesis, which enhances glucose management134,135. Metformin use is also associated with modulation of the bile acid pool by decreasing the abundance of Bacteroides fragilis and its bile salt hydrolase activity121. This effect is accompanied by increased levels of the bile acid glycoursodeoxycholic acid, which improves glucose metabolism homeostasis through inhibition of intestinal farnesoid X receptor signalling via an AMPK-independent mechanism that is associated with increased GLP1 levels121,136. These findings are consistent with the therapeutic effect of metformin-induced selective alterations in intestinal microbial composition and metabolism on host targets, which improve host metabolism102,104,134,137.
Dysbiosis is associated with pathologies (including cancer and cardiovascular, metabolic and inflammatory bowel diseases). Thus, therapeutic strategies could be developed that are based on metformin-induced modulation of the microbiome and/or metabolite production independently of the antidiabetic effect of metformin. Metformin reduces circulating levels of trimethylamine N-oxide, a microbiota–host co-metabolite synthesized from choline that is associated with an increased risk of cardiovascular disease in mice fed a high-choline diet138,139. In addition, metformin treatment is correlated with the remodelling of the gut microbiota, with increased relative abundance of Bifidobacterium and Akkermansia and decreased abundance of cutC (which encodes a protein involved in choline metabolism in bacteria)138. In addition, metformin has shown potential as an intervention for preventing cognitive decline related to dysbiosis caused by obesity and ageing in mice125,140,141.
Metformin-induced alterations in the gut microbiota also contribute to the antitumour effects of metformin. Orally ingested metformin, but not intraperitoneally injected metformin, suppresses tumour growth in HFD-fed mice117. Furthermore, faecal transplants of the microbiota from metformin-treated donor mice were sufficient to recapitulate the metformin-induced reduction in tumour growth and were associated with increased abundance of SCFA-producing bacteria and reduced expression of genes with protein products critical for regulating cholesterol synthesis in the tumour117. In addition, in a mouse model of colorectal cancer induced by Fusobacterium nucleatum, which is enriched in colorectal carcinogenesis, metformin decreases tumour size and the abundance of F. nucleatum in tumour tissue142. Additionally, the use of metformin in the treatment of inflammatory bowel disease demonstrated positive effects through its action on inflammatory pathways, intestinal barrier integrity and gut microbiota143. Metformin increased the relative abundance of Lactobacillus and Akkermansia species and alleviated gut dysbiosis, colonic inflammation and mucus barrier disruption induced by experimental colitis123,144.
Available data suggest that metformin and the gut microbiota work synergistically to produce beneficial therapeutic effects. Furthermore, adding gut microbiota modulators, such as probiotics or prebiotics, to metformin therapy results in improved T2DM outcomes in animal and human studies145. However, the metabolites produced by the gut microbiota can also influence the efficacy of metformin treatment and contribute to the interindividual variability in response to the drug. For example, metabolomic analysis of plasma from people treated with metformin who had high blood levels of glucose and from those with low blood levels of glucose identified a microbial metabolite, imidazole propionate, as a negative regulator of metformin action in humans146. Imidazole propionate reduced the acute glucose-lowering effect of metformin in mice fed a western diet by inducing inhibition of AMPK signalling in the liver through a p38γ-dependent mechanism146. Interestingly, inhibition of p38γ prevented the inhibitory effect of imidazole propionate on acute metformin action, highlighting potential therapeutic opportunities in patients with T2DM who are not fully responsive to metformin146.
Metformin intolerance
The gut microbiota is increasingly recognized as a potential mediator of the gastrointestinal adverse effects of metformin147. Prevalent gastrointestinal adverse effects after metformin intake have been attributed to gases (such as CO2 and H2S) produced by the gut microbiota, seemingly through metformin-induced metabolic modelling of Escherichia spp. and A. muciniphila102,110,129. A study comparing the gut microbiota profile in metformin-tolerant and metformin-intolerant patients with T2DM established that a shift in the composition of the gut microbiota on the introduction of metformin is responsible for the drug intolerance148. The use of microbiota modulators, including probiotic, prebiotic and symbiotic supplementation, might be a viable approach to achieving improved tolerability of metformin145. Altogether, metformin-induced changes in individual gut microbiotas and a subsequent shift in bacterial metabolite production could contribute to its therapeutic efficacy, but also to its adverse gastrointestinal effects in the host102,104,109,128,131,149.
Metformin and the immune system
Dampening of (meta)inflammation
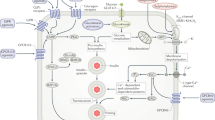
The immune system has a central role in the initiation and progression of many pathologies. For cardiometabolic diseases, the contribution of chronic low-grade inflammation to metabolic dysfunction and the development of atherosclerosis is now well documented150,151. Obesity induces various degrees of inflammation in adipose tissues, pancreatic islets and the liver, which contributes to hepatic steatosis, systemic insulin resistance and progression towards T2DM, non-alcoholic steatohepatitis (NASH) and cardiovascular diseases152,153,154. This so-called metaflammation also predisposes the individual to co-morbidities, such as Mycobacterium tuberculosis infection155 or severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (Box 1). Although incompletely understood, the crosstalk between parenchymal and tissue-resident immune cells within local niches in metabolic organs is central in these inflammatory processes153,156. Remarkably, metformin had some anti-inflammatory effects through both AMPK-dependent and AMPK-independent mechanisms in cross-sectional studies in patients with T2DM, various interventional studies in rodent models of obesity and T2DM, and in in vitro and ex vivo experiments in several immune cell types26,157 (Fig. 2a).

a, Following putative transporter-mediated internalization in various immune cell subsets, metformin inhibits the mitochondrial respiratory chain complex I and can modulate cell-specific inflammatory processes by both AMP-activated protein kinase (AMPK)-independent and AMPK-dependent mechanisms. b, Metformin can modulate the immune system, which could have beneficial effects in various pathological conditions (such as certain cancers, infections and hyperinflammatory diseases). These effects have been reported to involve various innate and adaptive immune cells, leading to modulation of several cell–cell interactions in local niches, as well as in immunometabolic and cellular processes. ATF3, activating transcription factor 3; CXCL1, chemokine (C-X-C motif) ligand 1; FASN, fatty acid synthase; FOXO3, forkhead box O3; IFNγ, interferon-γ; ISGs, interferon-stimulated genes; mTOR, mammalian target of rapamycin; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; NF-κB, nuclear factor-κB; NGAL, neutrophil gelatinase-associated lipocalin; PD1, programmed cell death protein 1; Pi, inorganic phosphate; PRF1, perforin 1; ROS, reactive oxygen species; STAT1 and STAT3, signal transducer and activator of transcription 1 and 3; TGFβ, transforming growth factor-β; TNF, tumour necrosis factor.
Metformin treatment also improves mitochondrial functions in peripheral blood mononuclear cells (PBMCs) from patients with T2DM, which is associated with increased AMPK phosphorylation and mitophagy and reduced levels of reactive oxygen species (ROS) and serum levels of the pro-inflammatory cytokines TNF and IL-6 (refs. 158,159). In addition, in a placebo-controlled trial in patients with prediabetes, treatment with metformin reduced the concentrations of the neutrophil-derived extracellular trap (NET) components elastase, proteinase 3, histones and double-strand DNA, independently of its effect on normalizing glucose levels160. This finding suggests that metformin can dampen NETosis in activated neutrophils, which is important for host defence against pathogens and is involved in inflammatory-mediated tissue damage and thrombosis. Metformin also induces a dose-dependent inhibition of NETosis induced by phorbol 12-myristate 13-acetate and ionomycin in vitro at concentrations within the therapeutic range160. This effect was associated with reduced PKC-βII membrane translocation, which is consistent with a direct effect of the drug on the NETosis machinery161. However, further studies are required to decipher the exact mechanism by which neutrophil functions are regulated, directly or indirectly, by metformin.
Changes in intrinsic metabolism of tissue-resident and newly recruited macrophages resulting from alterations in the local microenvironment within metabolic organs are thought to shape their functions and be among the main drivers of obesity-associated metaflammation162. Interestingly, metformin reduces pro-inflammatory activation of macrophages in vitro by interfering with the cellular metabolic reprogramming that underpins inflammation163. At the molecular level, although a contribution of AMPK activation could not be excluded, metformin reduced fatty acid synthesis mediated by fatty acid synthase (FASN) and suppressed FASN-dependent palmitoylation of AKT in lipopolysaccharide-stimulated mouse bone marrow-derived macrophages, leading to reduced NF-κB activation and reduced expression of pro-inflammatory cytokines163 (Fig. 2a). A novel pathway by which metformin could dampen inflammation independently of the AMPK–NF-κB axis has also been uncovered in mice. This pathway involves direct inhibition of the mitochondrial respiratory chain complex I and downstream inhibition of inflammasomes in alveolar macrophages164. Metformin inhibited lipopolysaccharide-induced mitochondrial ATP synthesis and subsequent generation and cytosolic release of mtDNA, a potent NLRP3 ligand, ultimately leading to reduction in inflammasome activation and IL-1β production in mouse bone marrow-derived macrophages164 (Fig. 2a).
Remarkably, metformin treatment in mouse models of acute respiratory distress syndrome blunts lipopolysaccharide-induced and SARS-CoV-2-induced pulmonary inflammation, an effect mimicked by myeloid-specific ablation of cytidine monophosphate kinase 2, a rate-limiting enzyme in mtDNA synthesis164. By contrast, the inhibitory action of metformin on mitochondrial complex I was also shown to prevent pro-inflammatory activation of alveolar macrophages by urban particulate matter air pollution, notably through reducing levels of complex III-derived ROS and the subsequent decrease in calcium-mediated IL-6 release165 (Fig. 2a). Of note, a considerable number of in vitro and ex vivo mechanistic studies used concentrations of metformin exceeding the therapeutic range and should therefore be considered with caution.
Prevention of inflammaging
Like cardiometabolic diseases, ageing and age-related disorders are often associated with chronic low-grade inflammation (also called inflammaging) and remodelling of the immune system166,167. Metformin has been considered as a treatment for attenuating detrimental consequences of ageing, notably for its possible beneficial effects on immune senescence168. Immune senescence is characterized by inflammatory activation of immune cells, a decline in their functions and reduced immune response efficacy, leading to increased vulnerability to infectious diseases, diminished responses to vaccination and increased susceptibility to metaflammation and age-related inflammatory diseases167. Interestingly, metformin treatment reduced the frequencies of pro-inflammatory B cell subsets in blood from older patients (>70 years old) with T2DM and increased influenza vaccine-specific antibody responses after vaccination169. These results were associated with reduced expression of senescence-associated secretory phenotype markers and reduced secretion of pathogenic autoimmune IgG antibodies by B cells isolated from these patients, supporting an anti-ageing effect of metformin on humoral immunity169.
Ovarian fibrosis is associated with ageing and an increased risk of developing ovarian cancer, notably in people with T2DM170. In a study using single-cell RNA sequencing, metformin treatment modulated the composition of ovarian fibroblastic and immune cells and prevented age-associated ovarian fibrosis in female mice171. The underlying mechanism remains unclear and its translation to humans is still questionable. However, this study identifies a unique macrophage subset induced by metformin in the ovary that might contribute to the clearance of senescence-associated secretory phenotype-producing fibroblasts and parallel expansion of myofibroblasts involved in extracellular matrix remodelling, resulting in maintenance of tissue homeostasis during ageing171 (Fig. 2b). Considering the use of metformin as a potential anti-inflammaging therapeutic strategy was also strengthened with the demonstration that metformin can alleviate ageing-associated T cell inflammation by preventing the age-induced appearance of a T helper 17 inflammaging profile through maintenance of CD4+ T cell autophagy and mitochondrial OXPHOS172.
Action on immunomodulation
Tuberculosis
A variety of other immunomodulatory properties of metformin have been reported, including enhanced immunosuppressive capacity in several autoimmune hyperinflammatory diseases173,174,175,176, improved immune response to infection177 and potentiation of antitumour immunity178. Metformin can reduce M. tuberculosis infection, lower progression to active tuberculosis and decrease mortality in both mice and humans30,179. Two studies have shown that part of this beneficial effect can be mediated by enhanced host antimycobacterial immune responses180,181. Indeed, although performed using supratherapeutic concentrations, ex vivo treatment of PBMCs from healthy donors with metformin induces mTORC1 inhibition and immune cell metabolic reprogramming, leading to decreased pro-inflammatory cytokine production and increased phagocytosis activity in response to stimulation with M. tuberculosis lysate180 (Fig. 2a). Furthermore, PBMCs from metformin-treated healthy donors display increased AMPK activation, downregulation of genes with protein products involved in the type 1 interferon response and altered cellular composition, with a shift in myeloid cells from classic to non-classic monocytes180. On the adaptive side of the immune response, metformin treatment expanded a population of memory-like CD8+CXCR3+ T cells in spleen and lungs, and enhanced immunogenicity and protective efficacy against M. tuberculosis challenge in mice vaccinated with Bacillus Calmette–Guérin181. Also observed in healthy individuals and patients with T2DM treated with metformin, this metformin-induced CD8+ T cell subset has features of metabolic reprogramming, such as increased mitochondrial OXPHOS and fatty acid oxidation, survival capacity and antimycobacterial properties181 (Fig. 2b). Collectively, these results might support the use of metformin as a new therapeutic option for patients with tuberculosis and/or as an adjunct drug to the tuberculosis vaccine.
Cancer
Metformin has also been reported to promote anticancer immunity through modulation of the tumour immune microenvironment, notably by increasing levels of tumour-infiltrating cytotoxic CD8+ T cells and decreasing levels of tumour-promoting CD163+ macrophages in human oesophageal squamous cell carcinoma182. At the molecular level, metformin triggers AMPK activation and STAT3 inactivation, leading to altered production of the effector cytokines TNF (increase) and IL-10 (decrease) by immune cells182. Metformin treatment also increased natural killer cell infiltration and cytotoxicity in head and neck squamous cell carcinoma in patients by promoting perforin release secondary to an AMPK-independent and mTORC1–STAT1-mediated downregulation of CXCL1 (ref. 183) (Fig. 2b).
Finally, metformin treatment rescues NASH-associated impairment in CD8+ T cell response to immune PD1–PDL1 checkpoint inhibitor therapy for advanced hepatocellular carcinoma184. Mechanistically, metformin induces metabolic reprogramming of hepatic CD8+ T cells, leading to increased motility of these tumour-infiltrating cells and improved efficacy of anti-PD1 antibody therapy against liver tumours in mouse models of NASH and hepatocellular carcinoma184 (Fig. 2b). Boosting anti-PD1 therapy with metformin-loaded macrophage-derived microparticles targeting M2-like tumour-associated macrophages has been suggested for resetting the polarization state of these cells towards an antitumoural M1-like phenotype185. Interestingly, this macrophage-targeted approach results in remodelling of the tumour immune microenvironment. This remodelling increases recruitment of CD8+ T cells, decreases infiltration of both immunosuppressive myeloid-derived suppressor cells and regulatory T cells and enhances tumour penetration and anticancer activity of the anti-PD1 antibody185.
Repurposing metformin
Metformin has a long history as the first-choice oral antihyperglycaemic drug for treating T2DM, with a high safety profile. In the past decade, repurposing metformin for additional applications has received increased attention. Indeed, metformin has shown promising effects beyond T2DM that are now being investigated in several planned and active clinical trials.
An anticancer agent
A retrospective observational study published in 2005 showed reduced risk of a cancer diagnosis in people taking metformin186. Subsequently, the effect of metformin treatment in preventing the development and progression of various types of cancer has been extensively investigated11,31,187. Repurposing an inexpensive licensed drug for the treatment of cancer was also encouraged by results from cellular and preclinical studies demonstrating antineoplastic effects and inhibition of tumour growth by metformin via inhibition of mitochondrial OXPHOS associated with AMPK-dependent and AMPK-independent mechanisms188. However, the high concentrations of metformin used in in vitro studies might not be readily translated to effects in preclinical and clinical studies24,189, which raises questions as to what the appropriate drug exposure to obtain a direct anticancer effect might be. Importantly, supportive evidence for a protective effect of metformin on cancer risk was not always consistent190,191. Thus, validation of the advantage of metformin treatment as a single agent or as an adjuvant in cancer therapy, notably for its emerging role in regulating cancer immunity, awaits further clarification.
Inflammatory and immune-mediated diseases
Studies on the anti-inflammatory properties of metformin and studies highlighting its possible immunomodulatory functions in various pathophysiological conditions suggest that it could be used in the context of several infectious, autoimmune and hyperinflammatory diseases. However, although the initiation of an increasing number of clinical trials supported by these new findings are being undertaken, further work is needed to clarify whether modulation of the immune response results from a direct action of metformin on various immune cell subsets and/or is mostly the consequence of indirect effects of the drug on their microenvironment (for example, nutrients, oxygen tension and non-immune cells present in the local niche).
Of note, based on the immune cell transcriptional profiles available from the ImmGen Consortium192 and the Tabula Sapiens Consortium193, the expression levels of genes encoding the main metformin transporters OCT1–3 (SLC22A1–3), plasma membrane monoamine transporter (SLC29A4) and multidrug and toxin extrusion protein 1 (SLC47A1) are all very low to undetectable in both mouse splenocytes and human PBMCs. Therefore, it would be important to investigate whether metformin can be taken up and accumulate in specific innate or adaptive immune cell subsets, in both healthy and disease states. In addition, whether clinically relevant concentrations of the drug, which are much lower than the ones generally used in most of the in vitro and ex vivo studies, can directly trigger immunomodulation, notably through immune cell metabolic reprogramming, needs to be determined. In this context, it should be noted that oral metformin did not enter the thymus to activate AMPK and did not reduce the occurrence of T cell acute lymphoblastic leukaemia in mice, presumably due to a lack of appropriate transporters194. By contrast, the cell-permeable biguanide phenformin did enter the thymus and activate AMPK, causing a delayed onset of T cell acute lymphoblastic leukaemia194. Thus, using phenformin when metformin is not taken up by cancer or immune cells could provide better efficacy than metformin in current clinical trials195.
Ageing
Metformin has been proposed as an anti-ageing drug as it increased median lifespan and maximum lifespan in studies performed in several species, including C. elegans, Drosophila melanogaster, rodents and humans168. The interplay between the microbiota and the host organism seemed to be crucial to extend the maximum lifespan in both C. elegans and D. melanogaster131, which suggests there is evolutionary conservation of microorganism-derived metabolites in response to metformin. Metformin use has been associated with increased overall survival (for example, in relation to the incidence of cancer and cardiovascular disease in patients with T2DM compared with that in matched control individuals without T2DM196); however, the ability of metformin to promote health or increase lifespan in healthy populations remains to be clearly determined. In addition, further studies are also needed to directly link metformin-induced increased longevity with alterations in the profile of the human gut microbiota. Interestingly, the benefits of metformin on longevity are restricted to mice treated with metformin from young adulthood, and are not seen when treatment is initiated in older animals197. This finding suggests that delaying the occurrence of (immune) senescence and related inflammaging might be one of the important underlying mechanisms. Interestingly, in old C. elegans, metformin causes a severe metabolic failure that leads to ATP exhaustion and cell death198. More insights into the benefits of metformin on healthspan and lifespan in humans will hopefully be provided by the MILES (Metformin in Longevity Study) and TAME (Targeting Aging with Metformin) clinical trials199.
Conclusions
Metformin is one of the most common medications used worldwide and has been used for >60 years. However, the mechanisms underlying its therapeutic effects remain incompletely understood. The latest advances regarding metformin’s glucoregulatory mechanisms of action have identified new potential factors, adding some layers of complexity and confusion that might fuel ongoing controversies. Whether cellular energy charge is altered or not in response to low metformin concentrations in the liver remains to be properly addressed, specifically using sensitive adenine nucleotide quantification approaches. In addition, whether these bioenergetic effects are the unique consequence of metformin’s inhibitory action on complex I or on other mitochondrial targets needs to be clarified. Furthermore, considering the lysosome as an additional organelle target provides insights for novel mechanisms of metformin action through the AMPK signalling pathway. Moreover, although it has long been assumed that the antihyperglycaemic effects of metformin are due to its exclusive action in the liver, there is now strong evidence that extrahepatic sites of action, notably the gut and its microbiota, are involved in its various clinical benefits. In addition, metformin has immunomodulatory properties in various pathological contexts (such as cancer, hyperinflammatory diseases and infectious diseases) involving direct or indirect regulation of the host innate and adaptive immune response. Some of these new insights from preclinical studies are currently being explored in clinical trials that are repurposing metformin for a range of diseases beyond T2DM. Lastly, it is worth mentioning that if the therapeutic prescription of metformin is expanded to other indications in the future, resulting in more widespread use, contamination of global waters with the drug might increase, which could raise environmental concerns (Box 4).
References
Schernthaner, G. & Schernthaner, G. H. The right place for metformin today. Diabetes Res. Clin. Pract. 159, 107946 (2020).
Ahmad, E. et al. Where does metformin stand in modern day management of type 2 diabetes? Pharmaceuticals 13, 427 (2020).
Triggle, C. R. et al. Metformin: Is it a drug for all reasons and diseases? Metabolism 133, 155223 (2022).
Bailey, C. J. Metformin: historical overview. Diabetologia 60, 1566–1576 (2017).
Garcia, E. Y. Flumamine, a new synthetic analgesic and anti-flu drug. J. Philipp. Med. Assoc. 26, 287–293 (1950).
Cummings, T. H., Magagnoli, J., Hardin, J. W. & Sutton, S. S. Patients with obesity and a history of metformin treatment have lower influenza mortality: a retrospective cohort study. Pathogens 11, 270 (2022).
Khunti, K. et al. Prescription of glucose-lowering therapies and risk of COVID-19 mortality in people with type 2 diabetes: a nationwide observational study in England. Lancet Diabetes Endocrinol. 9, 293–303 (2021).
Bramante, C. T. et al. Metformin and risk of mortality in patients hospitalised with COVID-19: a retrospective cohort analysis. Lancet Healthy Longev. 2, e34–e41 (2021).
Lalau, J. D. et al. Metformin use is associated with a reduced risk of mortality in patients with diabetes hospitalised for COVID-19. Diabetes Metab. 47, 101216 (2021).
Flory, J. & Lipska, K. Metformin in 2019. JAMA 321, 1926–1927 (2019).
Foretz, M., Guigas, B., Bertrand, L., Pollak, M. & Viollet, B. Metformin: from mechanisms of action to therapies. Cell Metab. 20, 953–966 (2014).
Matthews, D. R. et al. Glycaemic durability of an early combination therapy with vildagliptin and metformin versus sequential metformin monotherapy in newly diagnosed type 2 diabetes (VERIFY): a 5-year, multicentre, randomised, double-blind trial. Lancet 394, 1519–1529 (2019).
Day, E. A. et al. Metformin-induced increases in GDF15 are important for suppressing appetite and promoting weight loss. Nat. Metab. 1, 1202–1208 (2019).
Coll, A. P. et al. GDF15 mediates the effects of metformin on body weight and energy balance. Nature 578, 444–448 (2020).
Newman, C. & Dunne, F. P. Metformin for pregnancy and beyond: the pros and cons. Diabet. Med. 39, e14700 (2022).
Verma, V. & Mehendale, A. M. A review on the use of metformin in pregnancy and its associated fetal outcomes. Cureus 14, e30039 (2022).
Nguyen, L., Chan, S. Y. & Teo, A. K. K. Metformin from mother to unborn child – are there unwarranted effects? EBioMedicine 35, 394–404 (2018).
Feig, D. S. et al. Outcomes in children of women with type 2 diabetes exposed to metformin versus placebo during pregnancy (MiTy Kids): a 24-month follow-up of the MiTy randomised controlled trial. Lancet Diabetes Endocrinol. 11, 191–202 (2023).
Wilcock, C. & Bailey, C. J. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 24, 49–57 (1994).
Pentikäinen, P. J., Neuvonen, P. J. & Penttilä, A. Pharmacokinetics of metformin after intravenous and oral administration to man. Eur. J. Clin. Pharmacol. 16, 195–202 (1979).
Jensen, J. B. et al. [11C]-Labeled metformin distribution in the liver and small intestine using dynamic positron emission tomography in mice demonstrates tissue-specific transporter dependency. Diabetes 65, 1724–1730 (2016).
Chan, P., Shao, L., Tomlinson, B., Zhang, Y. & Liu, Z. M. Metformin transporter pharmacogenomics: insights into drug disposition – where are we now? Expert. Opin. Drug. Metab. Toxicol. 14, 1149–1159 (2018).
Graham, G. G. et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 50, 81–98 (2011).
Chandel, N. S. et al. Are metformin doses used in murine cancer models clinically relevant? Cell Metab. 23, 569–570 (2016).
He, L. & Wondisford, F. E. Metformin action: concentrations matter. Cell Metab. 21, 159–162 (2015).
Foretz, M., Guigas, B. & Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 15, 569–589 (2019).
Dowling, R. J. et al. Metformin pharmacokinetics in mouse tumors: implications for human therapy. Cell Metab. 23, 567–568 (2016).
Quinn, B. J. et al. Inhibition of lung tumorigenesis by metformin is associated with decreased plasma IGF-I and diminished receptor tyrosine kinase signaling. Cancer Prev. Res. 6, 801–810 (2013).
Sundelin, E., Jensen, J. B., Jakobsen, S., Gormsen, L. C. & Jessen, N. Metformin biodistribution: a key to mechanisms of action? J. Clin. Endocrinol. Metab. 105, dgaa332 (2020).
Singhal, A. et al. Metformin as adjunct antituberculosis therapy. Sci. Transl. Med. 6, 263ra159 (2014).
Thakkar, B., Aronis, K. N., Vamvini, M. T., Shields, K. & Mantzoros, C. S. Metformin and sulfonylureas in relation to cancer risk in type II diabetes patients: a meta-analysis using primary data of published studies. Metabolism 62, 922–934 (2013).
Gormsen, L. C. et al. Metformin increases endogenous glucose production in non-diabetic individuals and individuals with recent-onset type 2 diabetes. Diabetologia 62, 1251–1256 (2019).
McCreight, L. J. et al. Metformin increases fasting glucose clearance and endogenous glucose production in non-diabetic individuals. Diabetologia 63, 444–447 (2020).
Duca, F. A. et al. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med. 21, 506–511 (2015).
Zhang, E. et al. Intestinal AMPK modulation of microbiota mediates crosstalk with brown fat to control thermogenesis. Nat. Commun. 13, 1135 (2022).
Ma, T. et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature 603, 159–165 (2022).
Tobar, N. et al. Metformin acts in the gut and induces gut–liver crosstalk. Proc. Natl Acad. Sci. USA 120, e2211933120 (2023).
Gormsen, L. C. et al. In vivo imaging of human 11C-metformin in peripheral organs: dosimetry, biodistribution, and kinetic analyses. J. Nucl. Med. 57, 1920–1926 (2016).
Bailey, C. J., Wilcock, C. & Scarpello, J. H. Metformin and the intestine. Diabetologia 51, 1552–1553 (2008).
Proctor, W. R. et al. Why does the intestine lack basolateral efflux transporters for cationic compounds? A provocative hypothesis. J. Pharm. Sci. 105, 484–496 (2016).
Shirasaka, Y. et al. Multiple transport mechanisms involved in the intestinal absorption of metformin: impact on the nonlinear absorption kinetics. J. Pharm. Sci. 111, 1531–1541 (2022).
Wilcock, C. & Bailey, C. J. Reconsideration of inhibitory effect of metformin on intestinal glucose absorption. J. Pharm. Pharmacol. 43, 120–121 (1991).
Ikeda, T., Iwata, K. & Murakami, H. Inhibitory effect of metformin on intestinal glucose absorption in the perfused rat intestine. Biochem. Pharmacol. 59, 887–890 (2000).
Wu, T. et al. Metformin reduces the rate of small intestinal glucose absorption in type 2 diabetes. Diabetes Obes. Metab. 19, 290–293 (2017).
Bailey, C. J. Metformin and intestinal glucose handling. Diabetes Metab. Rev. 11, S23–S32 (1995).
Zubiaga, L. et al. Oral metformin transiently lowers post-prandial glucose response by reducing the apical expression of sodium-glucose co-transporter 1 in enterocytes. iScience 26, 106057 (2023).
Borg, M. J. et al. Comparative effects of proximal and distal small intestinal administration of metformin on plasma glucose and glucagon-like peptide-1, and gastric emptying after oral glucose, in type 2 diabetes. Diabetes Obes. Metab. 21, 640–647 (2019).
Koffert, J. P. et al. Metformin treatment significantly enhances intestinal glucose uptake in patients with type 2 diabetes: results from a randomized clinical trial. Diabetes Res. Clin. Pract. 131, 208–216 (2017).
Chang, H. S., Kim, S. J. & Kim, Y. H. Association between colonic 18F-FDG uptake and glycemic control in patients with diabetes mellitus. Nucl. Med. Mol. Imaging 54, 168–174 (2020).
Ito, J. et al. Dose-dependent accumulation of glucose in the intestinal wall and lumen induced by metformin as revealed by 18F-labelled fluorodeoxyglucose positron emission tomography-MRI. Diabetes Obes. Metab. 23, 692–699 (2021).
Morita, Y. et al. Enhanced release of glucose into the intraluminal space of the intestine associated with metformin treatment as revealed by [18F]fluorodeoxyglucose PET-MRI. Diabetes Care 43, 1796–1802 (2020).
Horakova, O. et al. Metformin acutely lowers blood glucose levels by inhibition of intestinal glucose transport. Sci. Rep. 9, 6156 (2019).
Ait-Omar, A. et al. GLUT2 accumulation in enterocyte apical and intracellular membranes: a study in morbidly obese human subjects and ob/ob and high fat-fed mice. Diabetes 60, 2598–2607 (2011).
Rathmann, W. et al. A variant of the glucose transporter gene SLC2A2 modifies the glycaemic response to metformin therapy in recently diagnosed type 2 diabetes. Diabetologia 62, 286–291 (2019).
McCreight, L. J., Bailey, C. J. & Pearson, E. R. Metformin and the gastrointestinal tract. Diabetologia 59, 426–435 (2016).
Kjøbsted, R. et al. Metformin improves glycemia independently of skeletal muscle AMPK via enhanced intestinal glucose clearance. Preprint at bioRxiv https://www.biorxiv.org/content/10.1101/2022.05.22.492936v1 (2022).
Rittig, N. et al. Metformin stimulates intestinal glycolysis and lactate release: a single-dose study of metformin in patients with intrahepatic portosystemic stent. Clin. Pharmacol. Ther. 110, 1329–1336 (2021).
Schommers, P. et al. Metformin causes a futile intestinal-hepatic cycle which increases energy expenditure and slows down development of a type 2 diabetes-like state. Mol. Metab. 6, 737–747 (2017).
Chondronikola, M. et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes 63, 4089–4099 (2014).
Sponton, C. H. et al. The regulation of glucose and lipid homeostasis via PLTP as a mediator of BAT–liver communication. EMBO Rep. 21, e49828 (2020).
Breining, P. et al. Metformin targets brown adipose tissue in vivo and reduces oxygen consumption in vitro. Diabetes Obes. Metab. 20, 2264–2273 (2018).
Bridges, H. R. et al. Structural basis of mammalian respiratory complex I inhibition by medicinal biguanides. Science 379, 351–357 (2023).
Bridges, H. R., Jones, A. J., Pollak, M. N. & Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 462, 475–487 (2014).
Madiraju, A. K. et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510, 542–546 (2014).
LaMoia, T. E. et al. Metformin, phenformin, and galegine inhibit complex IV activity and reduce glycerol-derived gluconeogenesis. Proc. Natl Acad. Sci. USA 119, e2122287119 (2022).
MacDonald, M. J., Ansari, I. H., Longacre, M. J. & Stoker, S. W. Metformin’s therapeutic efficacy in the treatment of diabetes does not involve inhibition of mitochondrial glycerol phosphate dehydrogenase. Diabetes 70, 1575–1580 (2021).
Fontaine, E. Metformin-induced mitochondrial complex I inhibition: facts, uncertainties, and consequences. Front. Endocrinol. 9, 753 (2018).
Pecinová, A., Brázdová, A., Drahota, Z., Houštěk, J. & Mráček, T. Mitochondrial targets of metformin – are they physiologically relevant? Biofactors 45, 703–711 (2019).
Vial, G., Detaille, D. & Guigas, B. Role of mitochondria in the mechanism(s) of action of metformin. Front. Endocrinol. 10, 294 (2019).
Zhou, G. et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174 (2001).
Shaw, R. J. et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 (2005).
Zhang, C. S. et al. Metformin activates AMPK through the lysosomal pathway. Cell Metab. 24, 521–522 (2016).
Howell, J. J. et al. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 25, 463–471 (2017).
Hunter, R. W. et al. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat. Med. 24, 1395–1406 (2018).
Miller, R. A. et al. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 494, 256–260 (2013).
Foretz, M. et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 120, 2355–2369 (2010).
Olivier, S. et al. Deletion of intestinal epithelial AMP-activated protein kinase alters distal colon permeability but not glucose homeostasis. Mol. Metab. 47, 101183 (2021).
Fullerton, M. D. et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 19, 1649–1654 (2013).
Stein, B. D. et al. Quantitative in vivo proteomics of metformin response in liver reveals AMPK-dependent and -independent signaling networks. Cell Rep. 29, 3331–3348.e7 (2019).
LaMoia, T. E. & Shulman, G. I. Cellular and molecular mechanisms of metformin action. Endocr. Rev. 42, 77–96 (2021).
Yoval-Sánchez, B., Ansari, F., Lange, D. & Galkin, A. Effect of metformin on intact mitochondria from liver and brain: concept revisited. Eur. J. Pharmacol. 931, 175177 (2022).
Owen, M. R., Doran, E. & Halestrap, A. P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 348, 607–614 (2000).
Alshawi, A. & Agius, L. Low metformin causes a more oxidized mitochondrial NADH/NAD redox state in hepatocytes and inhibits gluconeogenesis by a redox-independent mechanism. J. Biol. Chem. 294, 2839–2853 (2019).
Cao, J. et al. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem. 289, 20435–20446 (2014).
Zhang, C. S. et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 20, 526–540 (2014).
Blazina, I. & Selph, S. Diabetes drugs for nonalcoholic fatty liver disease: a systematic review. Syst. Rev. 8, 295 (2019).
Li, Y., Liu, L., Wang, B., Wang, J. & Chen, D. Metformin in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Biomed. Rep. 1, 57–64 (2013).
Holmes, O., Paturi, S., Selkoe, D. J. & Wolfe, M. S. Pen-2 is essential for γ-secretase complex stability and trafficking but partially dispensable for endoproteolysis. Biochemistry 53, 4393–4406 (2014).
Hasenour, C. M. et al. 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) effect on glucose production, but not energy metabolism, is independent of hepatic AMPK in vivo. J. Biol. Chem. 289, 5950–5959 (2014).
Cokorinos, E. C. et al. Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab. 25, 1147–1159.e10 (2017).
Madiraju, A. K. et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 24, 1384–1394 (2018).
Baur, J. A. & Birnbaum, M. J. Control of gluconeogenesis by metformin: does redox trump energy charge? Cell Metab. 20, 197–199 (2014).
Saheki, T. et al. Citrin/mitochondrial glycerol-3-phosphate dehydrogenase double knock-out mice recapitulate features of human citrin deficiency. J. Biol. Chem. 282, 25041–25052 (2007).
Calza, G. et al. Lactate-induced glucose output is unchanged by metformin at a therapeutic concentration – a mass spectrometry imaging study of the perfused rat liver. Front. Pharmacol. 9, 141 (2018).
Glossmann, H. H. & Lutz, O. M. D. Commentary: lactate-induced glucose output is unchanged by metformin at a therapeutic concentration – a mass spectrometry imaging study of the perfused rat liver. Front. Pharmacol. 10, 90 (2019).
Logie, L. et al. Cellular responses to the metal-binding properties of metformin. Diabetes 61, 1423–1433 (2012).
El-Mir, M. Y. et al. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 275, 223–228 (2000).
Hawley, S. A. et al. Use of cells expressing γ subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 11, 554–565 (2010).
Xie, D. et al. Let-7 underlies metformin-induced inhibition of hepatic glucose production. Proc. Natl Acad. Sci. USA 119, e2122217119 (2022).
Karlsson, F. H. et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498, 99–103 (2013).
Napolitano, A. et al. Novel gut-based pharmacology of metformin in patients with type 2 diabetes mellitus. PLoS ONE 9, e100778 (2014).
Forslund, K. et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528, 262–266 (2015).
de la Cuesta-Zuluaga, J. et al. Metformin is associated with higher relative abundance of mucin-degrading Akkermansia muciniphila and several short-chain fatty acid-producing microbiota in the gut. Diabetes Care 40, 54–62 (2017).
Wu, H. et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 23, 850–858 (2017).
Vich Vila, A. et al. Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat. Commun. 11, 362 (2020).
Mueller, N. T. et al. Metformin affects gut microbiome composition and function and circulating short-chain fatty acids: a randomized trial. Diabetes Care 44, 1462–1471 (2021).
Zhang, Q. & Hu, N. Effects of metformin on the gut microbiota in obesity and type 2 diabetes mellitus. Diabetes Metab. Syndr. Obes. 13, 5003–5014 (2020).
Alvarez-Silva, C. et al. Trans-ethnic gut microbiota signatures of type 2 diabetes in Denmark and India. Genome Med. 13, 37 (2021).
Elbere, I. et al. Association of metformin administration with gut microbiome dysbiosis in healthy volunteers. PLoS ONE 13, e0204317 (2018).
Bryrup, T. et al. Metformin-induced changes of the gut microbiota in healthy young men: results of a non-blinded, one-armed intervention study. Diabetologia 62, 1024–1035 (2019).
Yang, Y. et al. Changes of saliva microbiota in the onset and after the treatment of diabetes in patients with periodontitis. Aging 12, 13090–13114 (2020).
Lee, H. & Ko, G. Effect of metformin on metabolic improvement and gut microbiota. Appl. Env. Microbiol. 80, 5935–5943 (2014).
Shin, N. R. et al. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 63, 727–735 (2014).
Bauer, P. V. et al. Metformin alters upper small intestinal microbiota that impact a glucose-SGLT1-sensing glucoregulatory pathway. Cell Metab. 27, 101–117 (2018).
Zhang, X. et al. Modulation of gut microbiota by berberine and metformin during the treatment of high-fat diet-induced obesity in rats. Sci. Rep. 5, 14405 (2015).
Silamiķele, L. et al. Metformin strongly affects gut microbiome composition in high-fat diet-induced type 2 diabetes mouse model of both sexes. Front. Endocrinol. 12, 626359 (2021).
Broadfield, L. A. et al. Metformin-induced reductions in tumor growth involves modulation of the gut microbiome. Mol. Metab. 61, 101498 (2022).
Adeshirlarijaney, A., Zou, J., Tran, H. Q., Chassaing, B. & Gewirtz, A. T. Amelioration of metabolic syndrome by metformin associates with reduced indices of low-grade inflammation independently of the gut microbiota. Am. J. Physiol. Endocrinol. Metab. 317, E1121–E1130 (2019).
Adeshirlarijaney, A. & Gewirtz, A. T. Considering gut microbiota in treatment of type 2 diabetes mellitus. Gut Microbes 11, 253–264 (2020).
Bravard, A. et al. Metformin treatment for 8 days impacts multiple intestinal parameters in high-fat high-sucrose fed mice. Sci. Rep. 11, 16684 (2021).
Sun, L. et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 24, 1919–1929 (2018).
DeFronzo, R. A. et al. Once-daily delayed-release metformin lowers plasma glucose and enhances fasting and postprandial GLP-1 and PYY: results from two randomised trials. Diabetologia 59, 1645–1654 (2016).
Ke, H. et al. Metformin exerts anti-inflammatory and mucus barrier protective effects by enriching Akkermansia muciniphila in mice with ulcerative colitis. Front. Pharmacol. 12, 726707 (2021).
Zhou, Z. Y. et al. Metformin exerts glucose-lowering action in high-fat fed mice via attenuating endotoxemia and enhancing insulin signaling. Acta Pharmacol. Sin. 37, 1063–1075 (2016).
Ahmadi, S. et al. Metformin reduces aging-related leaky gut and improves cognitive function by beneficially modulating gut microbiome/goblet cell/mucin axis. J. Gerontol. A Biol. Sci. Med. Sci. 75, e9–e21 (2020).
Brandt, A. et al. Metformin attenuates the onset of non-alcoholic fatty liver disease and affects intestinal microbiota and barrier in small intestine. Sci. Rep. 9, 6668 (2019).
Li, L. et al. An in vitro model maintaining taxon-specific functional activities of the gut microbiome. Nat. Commun. 10, 4146 (2019).
Hao, Z. et al. Metaproteomics reveals growth phase-dependent responses of an in vitro gut microbiota to metformin. J. Am. Soc. Mass. Spectrom. 31, 1448–1458 (2020).
Rosario, D. et al. Understanding the representative gut microbiota dysbiosis in metformin-treated type 2 diabetes patients using genome-scale metabolic modeling. Front. Physiol. 9, 775 (2018).
Cabreiro, F. et al. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 153, 228–239 (2013).
Pryor, R. et al. Host-microbe-drug-nutrient screen identifies bacterial effectors of metformin therapy. Cell 178, 1299–1312.e29 (2019).
Lee, Y. et al. Changes in the gut microbiome influence the hypoglycemic effect of metformin through the altered metabolism of branched-chain and nonessential amino acids. Diabetes Res. Clin. Pract. 178, 108985 (2021).
Hung, W. W. et al. Gut microbiota compositions and metabolic functions in type 2 diabetes differ with glycemic durability to metformin monotherapy. Diabetes Res. Clin. Pract. 174, 108731 (2021).
De Vadder, F. et al. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 156, 84–96 (2014).
Holst, J. J., Gasbjerg, L. S. & Rosenkilde, M. M. The role of incretins on insulin function and glucose homeostasis. Endocrinology 162, bqab065 (2021).
Sansome, D. J. et al. Mechanism of glucose-lowering by metformin in type 2 diabetes: role of bile acids. Diabetes Obes. Metab. 22, 141–148 (2020).
Zhernakova, A. et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–569 (2016).
Su, C. et al. Metformin alleviates choline diet-induced TMAO elevation in C57BL/6J mice by influencing gut-microbiota composition and functionality. Nutr. Diabetes 11, 27 (2021).
Kuka, J. et al. Metformin decreases bacterial trimethylamine production and trimethylamine N-oxide levels in db/db mice. Sci. Rep. 10, 14555 (2020).
Ji, S., Wang, L. & Li, L. Effect of metformin on short-term high-fat diet-induced weight gain and anxiety-like behavior and the gut microbiota. Front. Endocrinol. 10, 704 (2019).
Deng, W. et al. Metformin alleviates autistic-like behaviors elicited by high-fat diet consumption and modulates the crosstalk between serotonin and gut microbiota in mice. Behav. Neurol. 2022, 6711160 (2022).
Huang, X. et al. Metformin elicits antitumour effect by modulation of the gut microbiota and rescues Fusobacterium nucleatum-induced colorectal tumourigenesis. EBioMedicine 61, 103037 (2020).
Wanchaitanawong, W., Thinrungroj, N., Chattipakorn, S. C., Chattipakorn, N. & Shinlapawittayatorn, K. Repurposing metformin as a potential treatment for inflammatory bowel disease: evidence from cell to the clinic. Int. Immunopharmacol. 112, 109230 (2022).
Liu, Z. et al. Metformin affects gut microbiota composition and diversity associated with amelioration of dextran sulfate sodium-induced colitis in mice. Front. Pharmacol. 12, 640347 (2021).
Seicaru, E. M., Popa Ilie, I. R., Cătinean, A., Crăciun, A. M. & Ghervan, C. Enhancing metformin effects by adding gut microbiota modulators to ameliorate the metabolic status of obese, insulin-resistant hosts. J. Gastrointestin Liver Dis. 31, 344–354 (2022).
Koh, A. et al. Microbial imidazole propionate affects responses to metformin through p38γ-dependent inhibitory AMPK phosphorylation. Cell Metab. 32, 643–653 (2020).
Bonnet, F. & Scheen, A. Understanding and overcoming metformin gastrointestinal intolerance. Diabetes Obes. Metab. 19, 473–481 (2017).
Díaz-Perdigones, C. M., Muñoz-Garach, A., Álvarez-Bermúdez, M. D., Moreno-Indias, I. & Tinahones, F. J. Gut microbiota of patients with type 2 diabetes and gastrointestinal intolerance to metformin differs in composition and functionality from tolerant patients. Biomed. Pharmacother. 145, 112448 (2022).
Nakajima, H. et al. The effects of metformin on the gut microbiota of patients with type 2 diabetes: a two-center, quasi-experimental study. Life 10, 195 (2020).
Hotamisligil, G. S. Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185 (2017).
Lee, Y. S., Wollam, J. & Olefsky, J. M. An integrated view of immunometabolism. Cell 172, 22–40 (2018).
Rohm, T. V., Meier, D. T., Olefsky, J. M. & Donath, M. Y. Inflammation in obesity, diabetes, and related disorders. Immunity 55, 31–55 (2022).
Hoogerland, J. A., Staels, B. & Dombrowicz, D. Immune-metabolic interactions in homeostasis and the progression to NASH. Trends Endocrinol. Metab. 33, 690–709 (2022).
Roy, P., Orecchioni, M. & Ley, K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat. Rev. Immunol. 22, 251–265 (2022).
Eckold, C. et al. Impact of intermediate hyperglycemia and diabetes on immune dysfunction in tuberculosis. Clin. Infect. Dis. 72, 69–78 (2021).
Caputa, G., Castoldi, A. & Pearce, E. J. Metabolic adaptations of tissue-resident immune cells. Nat. Immunol. 20, 793–801 (2019).
Kristófi, R. & Eriksson, J. W. Metformin as an anti-inflammatory agent: a short review. J. Endocrinol. 251, R11–R22 (2021).
Bhansali, S., Bhansali, A. & Dhawan, V. Metformin promotes mitophagy in mononuclear cells: a potential in vitro model for unraveling metformin’s mechanism of action. Ann. N. Y. Acad. Sci. 1463, 23–36 (2020).
de Marañón, A. M. et al. Metformin modulates mitochondrial function and mitophagy in peripheral blood mononuclear cells from type 2 diabetic patients. Redox Biol. 53, 102342 (2022).
Menegazzo, L. et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetol. 55, 593–601 (2018).
Wang, H., Li, T., Chen, S., Gu, Y. & Ye, S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. 67, 3190–3200 (2015).
Wculek, S. K., Dunphy, G., Heras-Murillo, I., Mastrangelo, A. & Sancho, D. Metabolism of tissue macrophages in homeostasis and pathology. Cell Mol. Immunol. 19, 384–408 (2022).
Xiong, W. et al. Metformin alleviates inflammation through suppressing FASN-dependent palmitoylation of Akt. Cell Death Dis. 12, 934 (2021).
Xian, H. et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity 54, 1463–1477 (2021).
Soberanes, S. et al. Metformin targets mitochondrial electron transport to reduce air-pollution-induced thrombosis. Cell Metab. 29, 335–347 (2019).
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C. & Santoro, A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14, 576–590 (2018).
Aiello, A. et al. Immunosenescence and its hallmarks: how to oppose aging strategically? A review of potential options for therapeutic intervention. Front. Immunol. 10, 2247 (2019).
Kulkarni, A. S., Gubbi, S. & Barzilai, N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. 32, 15–30 (2020).
Frasca, D., Diaz, A., Romero, M. & Blomberg, B. B. Metformin enhances B cell function and antibody responses of elderly individuals with type-2 diabetes mellitus. Front. Aging 2, 715981 (2021).
Lee, J. Y. et al. Diabetes mellitus and ovarian cancer risk: a systematic review and meta-analysis of observational studies. Int. J. Gynecol. Cancer 23, 402–412 (2013).
Landry, D. A. et al. Metformin prevents age-associated ovarian fibrosis by modulating the immune landscape in female mice. Sci. Adv. 8, eabq1475 (2022).
Bharath, L. P. et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 32, 44–55 (2020).
Ursini, F. et al. Metformin and autoimmunity: a “new deal” of an old drug. Front. Immunol. 9, 1236 (2018).
Titov, A. A., Baker, H. V., Brusko, T. M., Sobel, E. S. & Morel, L. Metformin inhibits the type 1 IFN response in human CD4+ T cells. J. Immunol. 203, 338–348 (2019).
Duan, W. et al. Metformin mitigates autoimmune insulitis by inhibiting Th1 and Th17 responses while promoting Treg production. Am. J. Transl. Res. 11, 2393–2402 (2019).
Sun, F. et al. Safety and efficacy of metformin in systemic lupus erythematosus: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Rheumatol. 2, e210–e216 (2020).
Malik, F. et al. Is metformin poised for a second career as an antimicrobial? Diabetes Metab. Res. Rev. 34, e2975 (2018).
Kim, K., Yang, W. H., Jung, Y. S. & Cha, J. H. A new aspect of an old friend: the beneficial effect of metformin on anti-tumor immunity. BMB Rep. 53, 512–520 (2020).
Leow, M. K. et al. Latent tuberculosis in patients with diabetes mellitus: prevalence, progression and public health implications. Exp. Clin. Endocrinol. Diabetes 122, 528–532 (2014).
Lachmandas, E. et al. Metformin alters human host responses to Mycobacterium tuberculosis in healthy subjects. J. Infect. Dis. 220, 139–150 (2019).
Böhme, J. et al. Metformin enhances anti-mycobacterial responses by educating CD8+ T-cell immunometabolic circuits. Nat. Commun. 11, 5225 (2020).
Wang, S. et al. Low-dose metformin reprograms the tumor immune microenvironment in human esophageal cancer: results of a phase II clinical trial. Clin. Cancer Res. 26, 4921–4932 (2020).
Crist, M. et al. Metformin increases natural killer cell functions in head and neck squamous cell carcinoma through CXCL1 inhibition. J. Immunother. Cancer 10, e005632 (2022).
Wabitsch, S. et al. Metformin treatment rescues CD8+ T-cell response to immune checkpoint inhibitor therapy in mice with NAFLD. J. Hepatol. 77, 748–760 (2022).
Wei, Z. et al. Boosting anti-PD-1 therapy with metformin-loaded macrophage-derived microparticles. Nat. Commun. 12, 440 (2021).
Evans, J. M., Donnelly, L. A., Emslie-Smith, A. M., Alessi, D. R. & Morris, A. D. Metformin and reduced risk of cancer in diabetic patients. BMJ 330, 1304–1305 (2005).
Heckman-Stoddard, B. M., DeCensi, A., Sahasrabuddhe, V. V. & Ford, L. G. Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia 60, 1639–1647 (2017).
Misirkic Marjanovic, M. S., Vucicevic, L. M., Despotovic, A. R., Stamenkovic, M. M. & Janjetovic, K. D. Dual anticancer role of metformin: an old drug regulating AMPK dependent/independent pathways in metabolic, oncogenic/tumorsuppresing and immunity context. Am. J. Cancer Res. 11, 5625–5643 (2021).
Badrick, E. & Renehan, A. G. Diabetes and cancer: 5 years into the recent controversy. Eur. J. Cancer 50, 2119–2125 (2014).
Goodwin, P. J. et al. Effect of metformin vs placebo on invasive disease-free survival in patients with breast cancer: the MA.32 randomized clinical trial. JAMA 327, 1963–1973 (2022).
Skuli, S. J. et al. Metformin and cancer, an ambiguanidous relationship. Pharmaceuticals 15, 626 (2022).
Heng, T. S. & Painter, M. W. The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol. 9, 1091–1094 (2008).
Jones, R. C. et al. The Tabula Sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896 (2022).
Vara-Ciruelos, D. et al. Phenformin, but not metformin, delays development of T cell acute lymphoblastic leukemia/lymphoma via cell-autonomous AMPK activation. Cell Rep. 27, 690–698.e4 (2019).
Zhao, H., Swanson, K. D. & Zheng, B. Therapeutic repurposing of biguanides in cancer. Trends Cancer 7, 714–730 (2021).
Valencia, W. M., Palacio, A., Tamariz, L. & Florez, H. Metformin and ageing: improving ageing outcomes beyond glycaemic control. Diabetologia 60, 1630–1638 (2017).
Anisimov, V. N. et al. If started early in life, metformin treatment increases life span and postpones tumors in female SHR mice. Aging 3, 148–157 (2011).
Espada, L. et al. Loss of metabolic plasticity underlies metformin toxicity in aged Caenorhabditis elegans. Nat. Metab. 2, 1316–1331 (2020).
Mohammed, I., Hollenberg, M. D., Ding, H. & Triggle, C. R. A critical review of the evidence that metformin is a putative anti-aging drug that enhances healthspan and extends lifespan. Front. Endocrinol. 12, 718942 (2021).
Simonnet, A. et al. High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obesity 28, 1195–1199 (2020).
Petrilli, C. M. et al. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York city: prospective cohort study. BMJ 369, m1966 (2020).
Cai, Q. et al. Obesity and COVID-19 severity in a designated hospital in Shenzhen, China. Diabetes Care 43, 1392–1398 (2020).
Zhu, L. et al. Association of blood glucose control and outcomes in patients with COVID-19 and pre-existing type 2 diabetes. Cell Metab. 31, 1068–1077 (2020).
Stefan, N., Birkenfeld, A. L. & Schulze, M. B. Global pandemics interconnected – obesity, impaired metabolic health and COVID-19. Nat. Rev. Endocrinol. 17, 135–149 (2021).
Cheng, X. et al. Metformin is associated with higher incidence of acidosis, but not mortality, in individuals with COVID-19 and pre-existing type 2 diabetes. Cell Metab. 32, 537–547.e3 (2020).
Gordon, D. E. et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020).
Schaller, M. A. et al. Ex vivo SARS-CoV-2 infection of human lung reveals heterogeneous host defense and therapeutic responses. JCI Insight 6, e148003 (2021).
Cory, T. J., Emmons, R. S., Yarbro, J. R., Davis, K. L. & Pence, B. D. Metformin suppresses monocyte immunometabolic activation by SARS-CoV-2 spike protein subunit 1. Front. Immunol. 12, 733921 (2021).
Reis, G. et al. Effect of early treatment with metformin on risk of emergency care and hospitalization among patients with COVID-19: the TOGETHER randomized platform clinical trial. Lancet Reg. Health Am. 6, 100142 (2022).
Bramante, C. T. et al. Randomized trial of metformin, ivermectin, and fluvoxamine for Covid-19. N. Engl. J. Med. 387, 599–610 (2022).
Bramante, C. T. et al. Outpatient treatment of Covid-19 with metformin, ivermectin, and fluvoxamine and the development of Long Covid over 10-month follow-up. Preprint at medRxiv https://www.medrxiv.org/content/10.1101/2022.12.21.22283753v1 (2022).
Gerstein, H. C. et al. Growth differentiation factor 15 as a novel biomarker for metformin. Diabetes Care 40, 280–283 (2017).
Natali, A. et al. Metformin is the key factor in elevated plasma growth differentiation factor-15 levels in type 2 diabetes: a nested, case-control study. Diabetes Obes. Metab. 21, 412–416 (2019).
Klein, A. B. et al. The GDF15-GFRAL pathway is dispensable for the effects of metformin on energy balance. Cell Rep. 40, 111258 (2022).
Aguilar-Recarte, D. et al. A positive feedback loop between AMPK and GDF15 promotes metformin antidiabetic effects. Pharmacol. Res. 187, 106578 (2022).
Klein, A. B., Kleinert, M., Richter, E. A. & Clemmensen, C. GDF15 in appetite and exercise: essential player or coincidental bystander? Endocrinology 163, bqab242 (2022).
Yang, L. et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 23, 1158–1166 (2017).
Mullican, S. E. et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 23, 1150–1157 (2017).
Diabetes Prevention Program Research Group. Long-term safety, tolerability, and weight loss associated with metformin in the Diabetes Prevention Program Outcomes Study. Diabetes Care 35, 731–737 (2012).
Ning, H. H. et al. The effects of metformin on simple obesity: a meta-analysis. Endocrine 62, 528–534 (2018).
Gao, F. et al. Growth differentiation factor 15 is not associated with glycemic control in patients with type 2 diabetes mellitus treated with metformin: a post-hoc analysis of AIM study. BMC Endocr. Disord. 22, 256 (2022).
Al-Kuraishy, H. M. et al. Metformin and growth differentiation factor 15 (GDF15) in type 2 diabetes mellitus: a hidden treasure. J. Diabetes 14, 806–814 (2022).
Kincaid, J. W. R. & Coll, A. P. Metformin and GDF15: where are we now? Nat. Rev. Endocrinol. 19, 6–7 (2022).
Karise, I., Bargut, T. C., Del Sol, M., Aguila, M. B. & Mandarim-de-Lacerda, C. A. Metformin enhances mitochondrial biogenesis and thermogenesis in brown adipocytes of mice. Biomed. Pharmacother. 111, 1156–1165 (2019).
Yang, Q. et al. AMPK/α-ketoglutarate axis dynamically mediates DNA demethylation in the Prdm16 promoter and brown adipogenesis. Cell Metab. 24, 542–554 (2016).
Geerling, J. J. et al. Metformin lowers plasma triglycerides by promoting VLDL-triglyceride clearance by brown adipose tissue in mice. Diabetes 63, 880–891 (2014).
Tokubuchi, I. et al. Beneficial effects of metformin on energy metabolism and visceral fat volume through a possible mechanism of fatty acid oxidation in human subjects and rats. PLoS ONE 12, e0171293 (2017).
English, P. J. et al. Metformin prolongs the postprandial fall in plasma ghrelin concentrations in type 2 diabetes. Diabetes Metab. Res. Rev. 23, 299–303 (2007).
Rouru, J., Isaksson, K., Santti, E., Huupponen, R. & Koulu, M. Metformin and brown adipose tissue thermogenetic activity in genetically obese Zucker rats. Eur. J. Pharmacol. 246, 67–71 (1993).
Pescador, N. et al. Metformin reduces macrophage HIF1α-dependent proinflammatory signaling to restore brown adipocyte function in vitro. Redox Biol. 48, 102171 (2021).
Yang, D. et al. A nationwide wastewater-based assessment of metformin consumption across Australia. Env. Int. 165, 107282 (2022).
He, Y., Zhang, Y. & Ju, F. Metformin contamination in global waters: biotic and abiotic transformation, byproduct generation and toxicity, and evaluation as a pharmaceutical indicator. Env. Sci. Technol. 56, 13528–13545 (2022).
Elizalde-Velázquez, G. A. & Gómez-Oliván, L. M. Occurrence, toxic effects and removal of metformin in the aquatic environments in the world: recent trends and perspectives. Sci. Total. Environ. 702, 134924 (2020).
Balakrishnan, A., Sillanpää, M., Jacob, M. M. & Vo, D. N. Metformin as an emerging concern in wastewater: occurrence, analysis and treatment methods. Environ. Res. 213, 113613 (2022).
Ambrosio-Albuquerque, E. P. et al. Metformin environmental exposure: a systematic review. Environ. Toxicol. Pharmacol. 83, 103588 (2021).
Wilkinson, J. L. et al. Pharmaceutical pollution of the world’s rivers. Proc. Natl Acad. Sci. USA 119, e2113947119 (2022).
Niemuth, N. J. & Klaper, R. D. Low-dose metformin exposure causes changes in expression of endocrine disruption-associated genes. Aquat. Toxicol. 195, 33–40 (2018).
Barros, S. et al. Metformin disrupts Danio rerio metabolism at environmentally relevant concentrations: a full life-cycle study. Sci. Total Environ. 846, 157361 (2022).
Phillips, J. et al. Developmental phenotypic and transcriptomic effects of exposure to nanomolar levels of metformin in zebrafish. Environ. Toxicol. Pharmacol. 87, 103716 (2021).
Barros, S. et al. Are fish populations at risk? Metformin disrupts zebrafish development and reproductive processes at chronic environmentally relevant concentrations. Env. Sci. Technol. 57, 1049–1059 (2023).
Li, T., Xu, Z. J. & Zhou, N. Y. Aerobic degradation of the antidiabetic drug metformin by Aminobacter sp. strain NyZ550. Environ. Sci. Technol. 57, 1510–1519 (2023).
Acknowledgements
The authors acknowledge the support of grants from Inserm, CNRS, Université Paris Cité, Agence Nationale de la Recherche (ANR), Société Francophone du Diabète (SFD), Fondation pour la Recherche Médicale (FRM), the Dutch Organization for Scientific Research (ZonMW) and DiabetesFonds.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Endocrinology thanks Michael Pollak and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ImmGen Consortium: https://www.immgen.org/
Tabula Sapiens Consortium: https://tabula-sapiens-portal.ds.czbiohub.org/
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Foretz, M., Guigas, B. & Viollet, B. Metformin: update on mechanisms of action and repurposing potential. Nat Rev Endocrinol 19, 460–476 (2023). https://doi.org/10.1038/s41574-023-00833-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41574-023-00833-4
This article is cited by
-
Association of glucose-lowering drug target and risk of gastrointestinal cancer: a mendelian randomization study
Cell & Bioscience (2024)
-
Metformin acts through appetite-suppressing metabolite: Lac-Phe
Nature Reviews Endocrinology (2024)
-
Metformin protects ovarian granulosa cells in chemotherapy-induced premature ovarian failure mice through AMPK/PPAR-γ/SIRT1 pathway
Scientific Reports (2024)
-
Glycolysis is reduced in dengue virus 2 infected liver cells
Scientific Reports (2024)
-
Metformin and feeding increase levels of the appetite-suppressing metabolite Lac-Phe in humans
Nature Metabolism (2024)
