
Abstract
Oestrogens and their receptors contribute broadly to physiology and diseases. In premenopausal women, endogenous oestrogens protect against cardiovascular, metabolic and neurological diseases and are involved in hormone-sensitive cancers such as breast cancer. Oestrogens and oestrogen mimetics mediate their effects via the cytosolic and nuclear receptors oestrogen receptor-α (ERα) and oestrogen receptor-β (ERβ) and membrane subpopulations as well as the 7-transmembrane G protein-coupled oestrogen receptor (GPER). GPER, which dates back more than 450 million years in evolution, mediates both rapid signalling and transcriptional regulation. Oestrogen mimetics (such as phytooestrogens and xenooestrogens including endocrine disruptors) and licensed drugs such as selective oestrogen receptor modulators (SERMs) and downregulators (SERDs) also modulate oestrogen receptor activity in both health and disease. Following up on our previous Review of 2011, we herein summarize the progress made in the field of GPER research over the past decade. We will review molecular, cellular and pharmacological aspects of GPER signalling and function, its contribution to physiology, health and disease, and the potential of GPER to serve as a therapeutic target and prognostic indicator of numerous diseases. We also discuss the first clinical trial evaluating a GPER-selective drug and the opportunity of repurposing licensed drugs for the targeting of GPER in clinical medicine.
Key points
-
Oestrogens exert multiple activities in physiology, including reproduction, immunity, cardiovascular and endocrine functions, and ageing, as well as in diseases such as hormone-sensitive cancers, arterial hypertension, atherosclerosis and osteoporosis.
-
Oestrogen signalling mediates both acute (non-genomic) and chronic (transcriptional) effects through cytosolic or nuclear oestrogen receptors ERα and ERβ and membrane subpopulations and the G protein‐coupled oestrogen receptor (GPER), which is a 7-transmembrane protein.
-
Molecules that activate oestrogen receptors include natural oestrogens, phytooestrogens, mycooestrogens and synthetic compounds, such as selective oestrogen receptor modulators and downregulators and xenooestrogens (also known as endocrine disruptors), activate oestrogen receptors and/or GPER.
-
Research using Gper-deficient animals, GPER‐selective agonists and antagonists, and non-selective compounds has revealed multiple roles of GPER in physiology and disease, including as a constitutive activator of the reactive oxygen species-producing enzyme NOX1.
-
GPER holds potential to become a diagnostic, prognostic and therapeutic target in clinical medicine, including the repurposing of licensed drugs targeting GPER and the ongoing first-in-human clinical trial of the GPER-selective agonist G-1.
Similar content being viewed by others


Introduction
Although actions of sex steroid hormones were described more than 2,000 years ago1, the concept of a ‘hormone’ was first introduced in 1910 by Starling2. It has been a hundred years since the chemical structures of oestrogens (and other steroids) were determined3,4 (Box 1). Identification and characterization of oestrogen receptors began in the 1950s by Jensen, Szego and others5,6,7, leading to the cloning of oestrogen receptor-α (ERα) by Chambon and associates in 1985 (ref. 8) (Box 1). In 1996, Kuiper et al.9 and Mosselman et al.10 cloned and identified oestrogen receptor-β (ERβ) contemporaneously with several reports describing the cloning of the orphan G protein-coupled receptor GPR30 (reviewed in refs. 7,11) (Box 1). GPR30 is a protein that predates the evolutionary divergence of fish and tetrapods more than 450 million years ago12. The discoveries that oestrogen binds to and activates cell signalling via GPR30 (refs. 13,14,15), establishing it as a transmembrane oestrogen receptor, resulted in its designation as the G protein-coupled oestrogen receptor (GPER) by the International Union of Basic and Clinical Pharmacology in 2008 (refs. 11,16). Following up on our previous article in Nature Reviews Endocrinology11, we now provide an update on the field of GPER research over the past decade. We will discuss advances made in cell signalling, molecular biology, pharmacology and genetics related to GPER. Special emphasis is given to the roles of GPER in pathophysiology and human disease and as a potential diagnostic, prognostic and therapeutic target in numerous and diverse areas of clinical medicine.
Molecular signalling mediated by GPER
G protein-coupled receptors (GPCRs) are 7-transmembrane spanning proteins that conventionally reside at the plasma membrane and signal to heterotrimeric G proteins, among other proteins, upon binding of ligands to their extracellular surface or within their transmembrane helices. GPER is predominantly expressed on intracellular membranes (the endoplasmic reticulum and Golgi apparatus), with little detected at the plasma membrane in many cell types14. While most investigations support this localization, limited expression in the plasma membrane in certain cell types (for example, uterine and renal epithelium), with constitutive internalization, has been reported17. Nuclear localization of GPER has also been observed and was suggested to be required for the GPER-mediated induction of transcription and cell migration18.
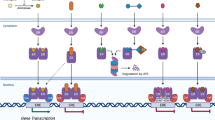
GPER signals through multiple G proteins, including Gαs15,19 and Gαi14,20 proteins, as well as via Gβγ signalling13, and possibly Gαq/11 protein21 (Fig. 1). GPER signalling involves, or possibly requires, epidermal growth factor (EGF) receptor transactivation13, a mechanism that, at the time this study was published in 2000, had only recently been discovered22. In addition to adenylyl cyclase19 and ERK1/2, GPER also activates PI3K–Akt signalling, which has been implicated in tumour cell survival23, activation of endothelial nitric oxide synthase (NOS3, also known as eNOS), nitric oxide (NO) formation and, thus, in cGMP-dependent vasodilation24,25 (Fig. 1). GPER also regulates ion channels, including those for calcium26, sodium27 and potassium28, and has been implicated in mTOR signalling and autophagy29.

Non-genomic and genomic signalling pathways are activated by oestrogen and oestrogenic ligands (in yellow) through binding to the three known oestrogen receptors, oestrogen receptor-α (ERα), oestrogen receptor-β (ERβ) and the G protein-coupled oestrogen receptor (GPER). 17β-Oestradiol (E2), selective agonists such as G-1, or selective oestrogen receptor modulators (SERMs) and selective oestrogen receptor downregulators and/or degraders (SERDs) activate GPER (1), which is localized predominantly intracellularly at the endoplasmic reticulum. GPER activates several heterotrimeric G proteins (2), leading to multiple downstream cascades, including cAMP production (3) and activation of PKA (3) and CREB (3). G protein activation also leads to calcium (Ca2+) mobilization from intracellular stores, which activates PKC and leads to activation of plasma membrane calcium channels. GPER activation can also lead to regulation of gene expression via activation of the YAP–TAZ transcription factors via Rho–ROCK signalling (4). Activation of SRC via G proteins can also lead to activation of matrix metalloproteinases (MMPs) (5) that cleave pro-heparin-binding epidermal growth factor (HB-EGF) (5), releasing free HB-EGF. HB-EGF then transactivates the EGF receptor (5), which in turn activates MAPK (ERK1/2), Akt and other pathways. These induce additional, rapid (non-genomic) effects such as activation of the l-arginine–endothelial nitric oxide synthase (NOS3)–NO–cGMP pathway (in combination with mobilization of calcium stores). Akt causes phosphorylation of endothelial NOS3 (6), which releases nitric oxide (NO) and leads to juxtacrine signalling from endothelial to vascular smooth muscle cells (7), and activation of PKG. Activation of MAPK and Akt signalling also causes genomic effects regulating gene transcription such as FOXO3 phosphorylation and degradation (8). In the classic, genomic oestrogen receptor pathway, 17β-oestradiol binds cytosolic and nuclear oestrogen receptors (9), inducing receptor dimerization and binding to the promoters of target genes. Alternatively, activated oestrogen receptors modulate the function of other classes of transcription factors (TF) through protein–protein interactions (10). Subpopulations of membrane-bound oestrogen receptors (mER) are present at the plasma membrane (11). Once activated, these oestrogen receptors interact with adaptor proteins (adaptor) and signalling molecules, such as SRC, which mediate rapid signalling events (for example, PI3K–Akt and MAPK signalling) (11). Oestrogen receptor ERα, potentially following transactivation of EGFR by GPER, is regulated by phosphorylation through kinases (such as MAPK and Akt), resulting in the regulation of gene expression (12). HIF1α, following GPER activation, induces γ-secretase-dependent activation of NOTCH (13) and VEGF signalling (13). Basal expression and/or activity of GPER constitutively induces expression of the NADPH oxidase NOX1 (14).
Transcriptional regulation is often a consequence of rapid signalling, yielding sustained genomic effects (Fig. 1). Rapid signalling pathways initiated by GPER that lead to transcriptional regulation include adenylyl cyclase-generated cAMP-dependent phosphorylation of CREB30 and MITF31 by PKA. GPER inactivates the FOXO3 transcription factor via Akt, promoting cell survival23. GPER-mediated ERK1 and ERK2 activation leads to Elk1-mediated transcription, which upregulates FOS and subsequently CTGF, FGF2 and CYP1B1 production32,33. GPER can either activate or inhibit NF-κB transcriptional activity, depending on the cellular context34,35; GPER also γ-secretase-dependent activation of Notch, resulting in expression of HES1 and SNAIL36. GPER stimulation can activate YAP and TAZ, two homologous transcription coactivators and key effectors of the Hippo tumour suppressor pathway, via Gαq/11, PLCβ–PKC, ERK1/2 and the Rho–ROCK signalling pathways37 (Fig. 1). GPER expression, and therefore function, is also regulated by multiple microRNAs38. Finally, basal expression and activity of GPER constitutively regulate expression and activity of the NADPH oxidase NOX1 (ref. 39) (Fig. 1), a reactive oxygen species (ROS)-producing enzyme implicated in many non-communicable diseases40.
Natural and synthetic ligands of GPER
Oestrogen receptors are activated by a wide range of chemical entities derived from diverse sources, including endogenous oestrogens, phytooestrogens (plant-derived oestrogens), mycooestrogens (fungus-derived oestrogens) and xenooestrogens (synthetic molecules also known as ‘endocrine disruptors’) (Fig. 2). The identification and characterization of oestrogen receptors facilitated the development of targeted drugs, including selective oestrogen receptor modulators (SERMs) and selective oestrogen receptor downregulators (or degraders) (SERDs), some of which were, in fact, already available in the 1960s41 (Box 1). In the following section, we will discuss GPER-targeting steroidal ligands, xenooestrogens, plant-derived and fungus-derived molecules, and synthetic receptor-selective ligands and their activities with respect to GPER (Fig. 2).

Shown are examples of natural steroids, phytooestrogens, xenooestrogens/endocrine disrupting chemicals (EDCs), therapeutic agents and experimental compounds that display varying activities towards oestrogen receptor-α (ERα), oestrogen receptor-β (ERβ) and the G protein-coupled oestrogen receptor (GPER) but are generally non-selective. Also shown are synthetic experimental compounds that exhibit selectivity for ERα and/or ERβ, such as propylpyrazoletriol (PPT), diarylpropionitrile (DPN) and AB-1, or for GPER, such as G-1, G15, G36 and CIMBA. p,p′-DDT, p,p′-dichlorodiphenyltrichloroethane.
Steroid hormones
GPER, at the time still known as the orphan receptor GPR30, was first linked to oestrogen-mediated signalling, in 2000, through the activation of ERK via transactivation of the EGF receptor13 (Box 1). High-affinity, competitive binding of 17β-oestradiol to GPER was first demonstrated in 2005 (refs. 14,15). In contrast to 17β-oestradiol, oestrogens, such as oestrone and oestriol, exhibit poor binding to GPER15. GPER shows no binding to other steroids, such as testosterone, progesterone, aldosterone and cortisol15,42,43,44, although aldosterone has been shown to be involved in crosstalk between the mineralocorticoid receptor and GPER and between the EGF receptor and GPER43. The catecholoestrogen 2-methoxy-oestradiol45 and the glucuronic acid metabolite 17β-oestradiol-17-d-glucuronide46 act as GPER agonists, whereas another catecholoestrogen, 2-hydroxy-oestradiol, is reported to act as an antagonist47. Dehydroepiandrosterone shows agonistic behaviour towards GPER48,49, whereas its metabolite 7β-hydroxy-epiandrosterone antagonizes GPER-mediated oestrogenic responses50. Most recently, 27-hydroxycholesterol, a cholesterol metabolite implicated in oestrogen receptor-negative breast cancer, was reported to bind and activate GPER, although with relatively low affinity compared with its most important physiological ligand, 17β-oestradiol51.
Xenooestrogens and natural oestrogenic molecules
Xenooestrogens are a large family of chemically stable synthetic molecules with oestrogenic activities often referred to as environmental oestrogens or endocrine-disrupting chemicals (EDCs). They are found in a wide range of consumer products and plastics, and most of them are toxic52. Endocrine-disrupting chemicals can be found in detergents, surfactants, resins, lubricants, plasticizers, fire retardants and pesticides52. Xenooestrogens that bind and/or regulate the activity of GPER (typically acting as agonists) include bisphenol A (BPA), polychlorinated biphenyls (PCBs), diethylstilbestrol (DES), nonylphenol, dichlorodiphenyltrichloroethane (DDT) and dichlorodiphenyltrichloroethylene isomers, kepone, methoxychlor and atrazine (Fig. 2).
Many molecules present in soy or green tea plants also target oestrogen receptors. Such naturally occurring phytooestrogens include flavonoids, isoflavonoids, chalcones, coumestans, stilbenes, lignans, ginsenosides and tetrahydrofurandiols53. Phytooestrogens that bind and/or activate GPER include genistein54, daidzein55, equol56, quercetin57, resveratrol58, oleuropein59, icariin60 and the green tea polyphenol (-)-epicatechin61. The mycooestrogen zearalenone also shows agonism towards GPER54,62.
Discovery of GPER-selective ligands
Owing to the highly conserved nature of the binding sites in ERα and ERβ, the typical affinity difference for oestrogen receptor subtype-specific compounds ranges from ~30-fold to 300-fold63. Oestrogen receptor subtype-biased ligands, such as propylpyrazoletriol (PPT, an ERα-selective agonist) and diarylpropionitrile (DPN, an ERβ-selective agonist) (Fig. 2), have been developed and are widely used64,65. PPT, however, also acts as a GPER agonist66, complicating the interpretation of its use.
The discovery and development of highly GPER-selective ligands were essential to facilitating research into the physiology and pathophysiology related to this receptor. In 2006, compound library screening led to the identification of G-1 (1-(4-(6-bromobenzo[1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)-ethanone), a small molecule that acts as a selective agonist of GPER67 (Fig. 2). The discovery of GPER-selective antagonists G15 and G36 complemented the use of G-1 as an agonist in understanding the roles of GPER in cell biology and physiology68,69. Some reports suggest that the activity of these compounds can vary depending on the system employed70,71. Other reported GPER-selective ligands include the agonists GPERL1 and GPERL2 (ref. 72), a series of indole-thiazole derivates that act as GPER agonists73, the antagonist CIMBA (an acyclic analogue of G36)74, as well as the pan-oestrogen receptor and GPER antagonist MIBE75 (Fig. 2). Proteolysis-targeting chimaeras (PROTACs), which are molecules that induce degradation of specific proteins (via selective recruitment of E3 ubiquitin ligases and target ubiquitination followed by degradation in proteosomes), were developed to target ERα as early as 2005 (ref. 76), with a pair of 17β-oestradiol–proteolysis-targeting chimaeras shown to degrade GPER in addition to ERα in a study published in 2021 (ref. 44). The 2019 discovery of AB-1, an agonist of ERα and ERβ that lacks affinity for GPER, should allow further dissection of the functions of ERα and/or ERβ compared with GPER in cells that express multiple oestrogen receptors77. Of these GPER-targeting ligands, only G-1 has so far entered clinical trials, specifically for use in combination therapy with immune checkpoint inhibitors (ICIs) in cancer. G-1 exhibits a favourable safety profile in these trials, either alone or in combination with pembrolizumab, with encouraging initial antitumour activity observed to date (NCT04130516)78,79,80.
Roles of GPER in physiology and disease
In the following sections, we will review advances in understanding the functions of GPER in cardiovascular and kidney disease, endocrinology and metabolism, gastrointestinal and liver diseases, immunity and immunology, neurology, and the physiological ageing process. Findings are frequently based on effects due to phenotypes of Gper-deficient mice or the effects of GPER-selective ligands (Fig. 3). Reported phenotypes of multiple differently derived Gper-deficient mice are not entirely consistent, probably due to differences in genetic background and other factors, including age81. The available evidence points to multiple roles of GPER in oestrogen-dependent and oestrogen-independent functions and pathologies, allowing the development of possible diagnostic and therapeutic approaches with regard to GPER.

The G protein-coupled oestrogen receptor (GPER) regulates many physiological functions (white background) and is involved in multiple pathologies and diseases (pink background) CKD, chronic kidney disease; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; PAH, pulmonary arterial hypertension; VSMC, vascular smooth muscle cell.
Clinical genetics
Sex chromosomes, sex steroids and sex steroid receptors contribute to and determine disease risk and efficacy of pharmacological therapy82,83. In humans, the GPER gene maps to chromosome 7p22.3, a region associated with arterial hypertension in genetic linkage studies84. The GPER single-nucleotide polymorphism rs11544331, which results in a Pro16Leu alteration in the receptor (amino acid substitution of proline 16 to leucine), produces a hypofunctional variant of GPER. The Leu variant is associated with slightly higher blood pressure than the Pro variant in women but not in men, and its allele frequency is two-fold higher in women with hypertension compared with age-matched men85. The inhibitory effect of GPER on pro-inflammatory gene expression in induced pluripotent stem cell-derived endothelial cells is reduced in the GPER Leu variant compared with the Pro variant86. Moreover, GPER activation induces LDL receptor expression, in part by downregulating proprotein convertase subtilisin–kexin type 9 (PCSK9) resulting in increased plasma levels of LDL cholesterol in Pro16Leu variant carriers87. Finally, expression of the Pro16Leu variant of GPER in cancer-associated fibroblasts increases secretion of paracrine factors promoting migration of breast cancer cells88. Together, these genetic observations support potentially important roles for GPER for human diseases.
Cardiovascular and kidney diseases
Endogenous oestrogens in premenopausal women protect against cardiovascular diseases in general, and particularly against arterial hypertension, coronary heart disease (including myocardial infarction) and heart failure11,89,90. GPER is widely expressed in the cardiovascular system in mammals, including the arterial wall and the heart11. In the cardiovascular system, physiological functions of GPER include the regulation of arterial blood pressure, angiogenesis, myocardial contractility and suppression of inflammation11. Activation of GPER results in acute vasodilatation of human, pig, rat and mouse arteries91,92,93. The underlying mechanisms include direct effects on vascular smooth muscle91,92,94 and activation of the endothelial l-arginine–NOS3–NO–cGMP pathway24,95,96 (Fig. 1). GPER-mediated vasodilatation also involves cAMP-dependent97 and Rho kinase-dependent mechanisms98 as well as inhibiting contractile factors such as endothelial vasoconstrictor prostanoids99 and endothelin-1 (refs. 92,100). GPER-dependent vasodilation is augmented during pregnancy101 and is reduced by ageing39,102,103. Systemic deletion of Gper prevents age-dependent, endothelium-dependent dysfunction, probably due to a reduction in NOX1 abundance39,103; the main effects are summarized in Fig. 3.
Blood pressure and arterial hypertension
Endothelium-derived contracting factors, such as cyclooxygenase-derived vasoconstrictor prostanoids and endothelin 1, are involved in the pathogenesis of arterial hypertension89; their activity is suppressed by constitutive GPER activity and augmented by systemic deletion of Gper99. Similarly, acute (seconds to minutes)91 and chronic treatment (hours to days) with the GPER agonist G-1, via its nitric oxide (NO)-liberating and antioxidant effects24,95, induces vasodilation and lowers blood pressure. Interestingly, deletion of Gper prevents angiotensin II-induced elevations of blood pressure, which are also markedly lowered by the GPER antagonist and NOX1 downregulator G36 (refs. 39,40). These data suggest that either agonist-dependent activation (through increased NO bioactivity) or chronic antagonism of GPER (via NOX1 downregulation) could be suitable for the treatment of different forms of arterial hypertension and related diseases such as atherosclerosis, stroke and chronic kidney disease (CKD).
The GPER agonist G-1 prevents hypertension in intrauterine growth-restricted female rat offspring later in life, suggesting a potential role in embryonic priming of adult hypertension104. Arterial blood pressure in Gper-deficient animals is normal91,105 or slightly reduced compared with animals expressing GPER106. Crosstalk between GPER and endothelin receptors has been described, resulting in natriuretic effects107. Aldosterone, which also has natriuretic effects, has been implicated in the actions of GPER, yet there is no evidence of aldosterone binding to GPER42,43,44,108,109. Consistent with this, deletion of Gper has no effect on the hypertensive effects induced by aldosterone110; however, GPER does regulate autocrine aldosterone synthesis in the renal medulla111. In addition, crosstalk between the mineralocorticoid receptor and GPER has been reported43. Correspondingly, mineralocorticoid receptor antagonists downregulate the expression of GPER112. Moreover, aldosterone triggers both direct interactions between the mineralocorticoid receptor and GPER involving the EGF receptor, which is abrogated by GPER gene silencing in endothelial and SkBr3 breast cancer cells in vitro43. Such interactions between the mineralocorticoid receptor and GPER might also contribute to aldosterone-mediated regulation of the sodium–chloride cotransporter, which is reduced in male mice lacking Gper113.
Atherosclerosis and coronary artery disease
Atherosclerosis is a chronic systemic inflammatory vascular disease89 and the underlying cause of coronary artery disease (CAD), peripheral artery disease and stroke. The main complications of CAD are myocardial infarction, fatal ventricular arrhythmias following reperfusion injury after infarction, and heart failure89. Natural or surgical menopause accelerates CAD progression and can be alleviated by oestrogen therapy, which activates all three oestrogen receptors89. In mice of both sexes fed either a regular diet or a high-calorie diet rich in fat and sugars, deletion of Gper results in moderate dyslipidaemia114,115. In endothelial cells, oestrogen-mediated activation of GPER attenuates transcytosis of LDL cholesterol into endothelial cells, compatible with an indirect vasculoprotective effect116. G-1 also reduces cardiac lipid accumulation and PPARα expression in surgically postmenopausal rats with type 2 diabetes mellitus (T2DM)117. In human monocytes, which contribute to the earliest stages of atherogenesis118, the anti-inflammatory effects of oestrogen might involve both direct effects via GPER119 as well as crosstalk between ERα and GPER120.
In the arteries of patients with coronary artery disease, GPER expression is sensitive to 17β-oestradiol regulation121. Activation of GPER by G-1 or green tea polyphenols inhibits the growth of coronary vascular smooth muscle cells61,91,118,122,123,124, a crucial step during atherogenesis. Deletion of Gper increases both perivascular adipose tissue growth and the production of cyclooxygenase-dependent adipose-derived contracting factor (ADCF), suggesting that endogenous GPER activity negatively regulates these processes125. In ovariectomized, that is, surgically postmenopausal, ApoE-deficient mice or in surgically postmenopausal C57BL/6J mice fed a cholate-containing atherogenic diet, G-1 reduces inflammation and atherosclerosis126. G-1 also reduces steady-state mRNA levels of the angiotensin AT1 receptor in ApoE-deficient mice123, a receptor protein that mediates angiotensin II-dependent vasoconstriction, vascular cell growth, inflammation and oxidative stress.
Myocardial disease and heart failure
GPER activation attenuates reperfusion injury following myocardial infarction through pathways involving GSK3β, mitophagy and mechanisms regulating mitochondrial permeability127,128,129. Arterial hypertension, T2DM and the resulting coronary artery disease and loss of myocardial tissue from myocardial infarction are the most frequent causes of heart failure. While heart failure with reduced ejection fraction (HFrEF) is often due to the loss of contractile tissue following myocardial infarction, heart failure with preserved ejection fraction (HFpEF) is a consequence of diabetes mellitus, arterial hypertension and ageing, all resulting in myocardial fibrosis and stiffening89,90. Patients with HFpEF are primarily perimenopausal or early postmenopausal women, suggesting that the cessation of endogenous oestrogen production contributes to the pathogenesis of HFpEF.
In experimental models of HFrEF, oestrogen therapy can reverse heart failure-induced myocardial fibrosis130. ERα and ERβ, as well as GPER, are all involved in the inhibitory effects of oestrogen on cardiomyocyte proliferation131,132. Interestingly, SERMs and SERDs, which are also GPER agonists, also inhibit cardiomyocyte proliferation133. Hypoxia and/or hypoxaemia, which occur during myocardial ischaemia and heart failure, upregulate GPER134. GPER controls myocardial contractility involving crosstalk between GPER and β1 adrenoceptors135. In a model of ageing-associated HFpEF, systemic deletion of Gper in male mice prevents the development of heart failure and myocardial fibrosis, an effect that is related to downregulation of NOX1 protein expression and associated reduction of NOX1 function39. In vitro studies using Nox1-knock-in experiments in aortic vascular smooth muscle cells from Gper-deficient mice further underscored that constitutive NOX1 expression and activity require GPER expression, which, probably through ligand-independent or basal activity, enables ROS formation, inflammation and myocardial fibrosis39. By contrast, in young female mice, cardiomyocyte-specific deletion of Gper worsens cardiomyocyte function compared with wild-type mice both in vitro and in vivo, which can be partly rescued by inhibiting cardiac NLRP3 inflammatory pathways136. G-1 reduces diastolic dysfunction in experimental HFpEF137 and in rats with hypertensive cardiomyopathy137,138; G-1 treatment also improves cardiac function and reduces cardiac fibrosis in surgically postmenopausal rats139. Taken together, either reducing constitutive NOX1-dependent production of ROS by blocking GPER or increasing NO bioactivity by activating GPER, holds potential for pharmacological intervention in heart failure, possibly in a sex-dependent manner.
Renal physiology and disease
Loss of functional kidney tissue, particularly due to CKD, facilitates the development of arterial hypertension and cardiovascular disease. Similar to cardiovascular diseases, CKD displays sex differences with premenopausal women being largely protected from CKD development compared with age-matched men, implicating a role for oestrogens and oestrogen receptors140. GPER regulates renal artery and intrarenal vascular tone103,141, and its activation increases Ca2+ flux and H+-ATPase activity in renal tubular cells142; GPER also regulates natriuresis107 via crosstalk with endothelin ETA and ETB receptors143. Deletion of Gper counteracts the development of focal segmental glomerulosclerosis (FSGS) and the resulting proteinuria144 and tubulo-interstitial injury caused by inflammation and oxidative stress, by reducing NOX1 upregulation145. Activation of GPER also reduces glomerular mesangial cell proliferation induced by hyperglycaemia in vitro (which is associated with oxidative stress)60, and Gper silencing in these cells markedly reduces NOX1 abundance144. The GPER antagonist and NOX1 downregulator G36 reduces mRNA expression of podocyte injury markers NPHS1 (coding for nephrin), COL4A1 (collagen IV) and WT1 (Wilms-tumour 1) in human podocytes in vitro144. Protective effects of GPER signalling on podocytes have also been demonstrated for treatment with GPER agonists, probably via activation of the l-arginine–NOS–nitric oxide pathway146. In a model of hypertensive nephropathy, GPER activation reduces proteinuria as well as tubular injury but not glomerular injury via pressure-independent mechanisms147,148. Possibly, the stimulating effect of G-1 on tubular epithelial cell proliferation could contribute to this effect149. Protective effects of GPER signalling have also been reported for methotrexate-induced human renal epithelial cell injury in vitro150 and for acute renal endothelial cell injury following renal ischaemia in female mice151.
Pulmonary diseases
Pulmonary arterial hypertension (PAH) is a chronic fibroproliferative disorder of the pulmonary vasculature, ultimately leading to right-heart failure. Four out of five patients are women, suggesting a role for sex chromosomes, sex steroids, or sex steroid receptors. In experimental rat models of PAH, ovariectomy increases mortality152, while 17β-oestradiol (a non-selective oestrogen receptor and GPER agonist)153 or the GPER agonists G-1 (ref. 154) or 2-ME152 partially reduce or even reverse established cardiopulmonary injury. G-1 also improves skeletal muscle function and exercise intolerance in rats with PAH, possibly through normalization of SERCA2a and phospholamban expression154,155. Finally, in experimental hypoxia-induced PAH in rats, blocking GPER using G36 improves cardiac function by lowering right ventricular pressure, probably involving the downregulation of NOX1 (refs. 156,157). Thus, both agonists and antagonists of GPER might aid in the treatment of PAH.
Endocrinology and metabolism
Metabolic homeostasis is differentially regulated in men and women, with the metabolic actions of oestrogens mediated through both ERα158,159,160 and GPER; discussed later in this section. Premenopausal women exhibit lower incidences of obesity and T2DM compared with age-matched men; these protective effects are lost following menopause, with similar effects seen in rodents. Oestrogen therapy can alleviate weight gain and its associated adverse metabolic effects present in postmenopausal women and in surgically postmenopausal mice161,162,163.
Obesity and diabetes mellitus
Since the first reports demonstrating roles of endogenous GPER in the regulation of body weight, adipose tissue growth, obesity and insulin function in 2009 (refs. 91,164), studies in mice lacking Gper have found that these mice develop dyslipidaemia and show reduced energy expenditure compared with wild-type mice. These effects are probably responsible for the observed increases in visceral and subcutaneous adipose tissue depots, given that food intake and locomotor activity remain unaffected in Gper-deficient mice114,115,165. Compared with males, female ovary-intact Gper-deficient mice exhibit a lower sensitivity to acute leptin-stimulated food intake and short-term cholecystokinin-stimulated satiety signals165. The expression of thermogenic genes, such as those encoding uncoupling protein 1 (Ucp1) and the β3-adrenergic receptor, is reduced in brown adipose tissue of Gper-deficient mice consistent with the decreased energy expenditure.
17β-Oestradiol treatment protects β-cells from apoptosis and prevents diabetes mellitus in mice166. The severity of diabetes mellitus in mice lacking both ERα and ERβ worsens following surgical menopause167. 17β-Oestradiol supplementation improves glucose homeostasis in these mice, suggesting alternative mechanisms of oestrogen action other than signalling through ERα or ERβ, for example, through GPER167. Indeed, in mice lacking Gper, plasma levels of glucose are increased and these animals exhibit glucose intolerance, defective glucose-stimulated and oestrogen-stimulated insulin secretion, and insulin resistance114,164,165. Insulin secretion in response to both 17β-oestradiol and G-1 in healthy islets is reduced by pharmacological GPER inhibition and is absent in mouse islets lacking Gper26. In a mouse model of streptozotocin-induced diabetes mellitus, deletion of Gper results in greater loss of pancreatic β-cells, reduced pancreatic insulin content and, consequently, abnormally increased plasma levels of glucose compared with wild-type mice167.
GPER as a therapeutic target in obesity and diabetes mellitus
Therapeutic targeting of GPER in glucose homeostasis and lipid metabolism has been studied in models of Western diet-induced obesity in male mice and in models of surgical menopause in female mice, both of which result in obesity and metabolic dysfunction. G-1 treatment over a period of 6–8 weeks reduced overall body weight, adiposity and circulating levels of lipids compared with vehicle-treated mice, without affecting lean mass or bone density, via increased basal energy expenditure168. No changes in either daily food consumption or locomotion were observed in this study although, in surgically postmenopausal obese rats, G-1 treatment acutely and transiently decreased food intake169. G-1 treatment in surgically postmenopausal mice increased the expression of genes involved in mitochondrial biogenesis and fatty acid oxidation in brown and white adipose tissue and in skeletal muscle, while reducing the expression of genes involved in inflammation, hypoxia and angiogenesis168.
In line with previous results69,126, G-1 treatment of surgically postmenopausal obese mice was devoid of the feminizing effects of 17β-oestradiol as indicated by the absence of uterine imbibition168. In addition to weight loss and improved lipid profiles, G-1 also improved glucose homeostasis at the level of glucose and insulin tolerance tests, and reduced fasting blood levels of glucose and insulin168. In postmenopausal rats with streptozotocin-induced diabetes G-1 treatment reduced disease-induced weight loss to a comparable degree as did 17β-oestradiol treatment, and similarly improved glucose homeostasis and lipid profiles compared with vehicle-treated diabetic rats170. While surgically postmenopausal obese mice show improved glucose homeostasis in response to acute or chronic 17β-oestradiol treatment, deletion of Gper abrogates this response, indicating a key role of GPER in 17β-oestradiol-mediated glucose homeostasis in vivo164,165. Moreover, G-1 amplifies glucose-stimulated insulin secretion ex vivo in pancreatic islets obtained from patients with T2DM, while also suppressing glucagon and somatostatin secretion171,172. Thus, selective GPER agonists hold potential for the treatment of obesity and associated diseases such as diabetes mellitus.
Gastrointestinal and liver diseases
Oestrogens modulate multiple gastrointestinal and hepatic functions via their receptors173, including via GPER173. GPER is a cell-specific marker of gastric epithelium chief cells174 and also controls lower oesophageal sphincter tone175, colonic motility and severity of visceral pain176,177. In human Crohn’s disease178, ulcerative colitis179 and irritable bowel syndrome (IBS)180,181,182, the majority of studies found intestinal GPER expression to be increased compared with healthy individuals. GPER activation reduces inflammation, tissue injury and mortality in a mouse model of Crohn’s disease178 and G-1 reduces colonic crypt cell injury related to reperfusion injury following intestinal ischaemia183. Finally, intestinal inflammation in a mouse model of acute colitis induced by dextran sulfate sodium is reduced by GPER activation, improving intestinal mucosal barrier function184.
GPER regulates liver in zebrafish185 and contributes to oestrogen-dependent proliferation and lipid metabolism in human hepatocytes185,186. In addition, both GPER or ERα protect hepatocytes from fatty degeneration, a predisposing factor propagating non-alcoholic fatty liver disease and steatohepatitis187.
Obesity in premenopausal women is associated with an increased risk of developing gallstones, which are formed via GPER-dependent mechanisms188. Oestrogen-dependent cholesterol crystallization pathways differ markedly between those involving ERα or GPER189, yet deletion of Gper190 or its pharmacological inhibition74 completely prevents gallstone formation in female mice.
Cancer biology and oncology
GPER is expressed in tumours and tumour cells of cancer patients, including the mammary gland191,192,193,194,195, endometrium66,196, ovaries197, prostate198, pancreas199, thyroid200, colon201 and lung202. Increased GPER expression correlates with a worse outcome in breast191,192,193, endometrial196 and ovarian197 cancer. Although pharmacological activation of GPER can increase proliferation and associated signalling in breast203, endometrial204, thyroid200 and ovarian205 cancer cells, inhibition of proliferation due to GPER signalling has also been reported in breast206, pancreatic199 and melanoma207 cancer cells. With these — sometimes — opposing results in different cell lines, the role of GPER in cancer in vivo appears to be more complex than anticipated. Indeed, in certain forms of cancer, endogenous GPER activity might be protective, possibly through anti-inflammatory effects208.
Breast cancer
Much has been published regarding GPER and breast cancer due to obvious questions arising from the well-documented importance of presence or absence of ER for the efficacy of anti-oestrogen therapies in cancer treatment209. The fact that SERMs, such as tamoxifen14 and raloxifene66, as well as SERDs, such as fulvestrant13, act as GPER agonists to activate growth and survival pathways has led to the suggestion that GPER expression and/or activity could contribute to breast cancer recurrence194. This complex pharmacology has also led to a search for ERα-selective compounds that do not cross-react with GPER77.
Supporting roles for GPER in breast cancer recurrence and metastasis, GPER expression is elevated in metastases of patients with breast cancer compared with matched primary tumours210,211. However, this elevated GPER expression, where assessed, is only observed in women originally treated with tamoxifen211. Aromatase inhibitors are more effective than tamoxifen at inhibiting tumour growth in primary breast tumours that are both ERα-positive and GPER-positive, with this difference in treatment efficacy being absent in primary ERα-positive and GPER-negative breast tumours192. Moreover, aromatase inhibition resulted in better disease-free progression for patients with breast cancer compared with a tamoxifen-based therapy, consistent with a role for GPER in recurrence and metastasis193. Using a genetic mouse model of mammary gland tumorigenesis, systemic Gper deficiency resulted in reduced tumour size and metastasis compared with wild-type mice, consistent with a pro-tumorigenic role for GPER in vivo212.
In vitro, tamoxifen induces proliferation of tamoxifen-resistant MCF-7 cells through a GPER-dependent pathway210,213. This proliferation can be blocked by GPER knockdown or co-treatment with the GPER-selective antagonist G15 (refs. 69,210) as tamoxifen binds to and cross-activates GPER15,66,214. Breast cancer cell survival in the presence of tamoxifen might be mediated by Akt-induced inactivation of the pro-apoptotic transcription factor FOXO3, suggesting a mechanism to enhance eventual tamoxifen resistance23. Tamoxifen-mediated cross-activation of GPER also induces breast cancer cell migration215, potentially via the YAP–TAZ pathway37 (Fig. 1), and increases aromatase expression in tamoxifen-resistant (ERα-positive) cells216. In vivo, GPER also contributes to tamoxifen resistance in MCF-7 cells, with tamoxifen-resistant xenografts derived from MCF-7 cells regaining sensitivity to tamoxifen in female mice upon treatment with a combination of tamoxifen and G15, where neither alone had an effect210. GPER downregulation and G15 treatment also sensitize breast cancer cells to doxorubicin by inhibiting epithelial-to-mesenchymal transition217. Lastly, G-1 (as well as tamoxifen and fulvestrant) increases natural killer cell-mediated growth inhibition of both ERα-negative and ERα-positive breast cancer cells, suggesting a novel role for GPER in cancer therapy218.
Cancer-associated fibroblasts (CAFs) express GPER, with most studies to date employing breast CAFs, which have previously described roles supporting breast tumour progression18,219,220. In breast CAFs, GPER mediates expression of HIF1α and VEGF195 and has been implicated in promoting tumour progression by increasing migration and invasion221,222,223. Tamoxifen and G-1 induce increased aromatase expression in breast CAFs, resulting in increased oestrogen production219, potentially leading to tamoxifen resistance216.
The tumour microenvironment also contains adipocytes, particularly in adipose-rich tissues such as the breast. Obesity has been clinically established as an important contributor to multiple cancers224. Adipocytes not only express aromatase, resulting in intracrine oestrogen synthesis, but also adipokines and other (pro-inflammatory) cytokines and hormones that can promote tumorigenesis. The actions of GPER in reducing obesity and mitigating metabolic dysfunction168, inflammation194 and chemotherapy-associated cardiotoxicity225 could, in part, reduce the incidence of and improve outcomes in breast cancer and other cancers.
Malignant melanoma
Female patients with malignant melanoma have a better clinical outcome than male patients226, although ICIs, an effective treatment for melanoma, show better therapeutic efficacy in men than in women227. A role for GPER activity in melanoma was first suggested by the observation that GPER (but not ERα) mediates oestrogen-induced melanogenesis (melanocyte differentiation and melanin production)31,228. Treatment of mouse melanoma cells with G-1 or tamoxifen, interestingly, inhibits proliferation in vitro229. Combining ICIs (specifically an anti-PD1 antibody) with G-1 not only reduces tumour growth but also improves survival of melanoma-bearing female mice, far more than either anti-PD1 antibodies or G-1 treatment alone. Combination therapy utilizing immune checkpoint inhibition and G-1 can result in long-term clearance of tumours, indicating immunological memory207, with similar results in pancreatic cancer mouse xenograft models199. This effect is potentially mediated through lowering Myc levels, which results in decreased expression of PDL1 and increased expression of HLA class I in melanoma tumour cells, which together could lead to improved immune recognition of melanoma tumour cells207. In 2019, these results led to the initiation of the first Phase 1 clinical trial of G-1 for the treatment of malignant melanoma (NCT04130516)78.
Other forms of cancer
The type of cancer might determine whether GPER activity promotes or inhibits carcinogenesis and/or metastasis. Pharmacological activation of GPER reduces liver tumorigenesis, at least in part, through inhibiting inflammation and fibrosis208. In mouse models of non-small-cell lung cancer (urethane-induced adenocarcinoma), tumour burden increases following treatment with 17β-oestradiol or G-1, and decreases upon treatment with G15 (ref. 202), possibly with the involvement of NOTCH-dependent pathways230. GPER expression is increased in castration-resistant prostate cancer231, and its activation is associated with sustained cytotoxic ERK activation198. In a prostate cancer mouse xenograft model, chronic treatment with G-1 for several weeks inhibits cancer progression but only following cancer recurrence after castration231, suggesting the potential for GPER-targeted therapies in castration-resistant prostate cancer.
GPER expression and function have also been implicated in gastric epithelial metaplasia and gastric cancer173,174,232,233 as well as in colon cancer173,234. In mouse syngeneic pancreatic cancer xenograft models, G-1, alone or in combination with ICIs improves survival compared with vehicle only or ICIs alone, respectively, resulting in a substantial cure rate199. In line with the beneficial effects of G-1 on pancreatic cancer, tamoxifen, also acting as a GPER agonist, inhibits the recruitment and polarization of tumour-associated macrophages and interferes with myofibroblastic differentiation of pancreatic stellate cells in the tumour microenvironment235. This reduces the cells’ ability to remodel the extracellular matrix and to promote cancer cell invasion235. GPER is highly overexpressed in Waldenström macroglobulinaemia, yet G-1 treatment, both in vitro and in vivo, induces apoptosis of tumour cells, even in the protective bone marrow milieu236. In this study, G-1 treatment improved survival in a murine xenograft model but had no effect on B cells transplanted from healthy donors236.
Immune system and immunology
Regulation of fish granulocyte functions by oestrogens through GPER predates the evolutionary divergence of fish and tetrapods more than 450 million years ago, which indicates that oestrogens are modulators of the immune response and that GPER have played a pivotal role in immunity throughout evolution12. Sex plays an important role in immune responses with oestrogens frequently exerting anti-inflammatory effects, traditionally through ERα and, to a lesser extent, through ERβ237. However, 17β-oestradiol also mediates part of its anti-inflammatory effects through GPER, which is widely expressed in white blood cells, (including neutrophils, eosinophils, monocytes and lymphocytes) as well as in macrophages238.
Regulation of immune cells by GPER
GPER regulates apoptosis in eosinophils239, suggesting a role for GPER in allergic immune responses. Indeed, in a model of allergic pulmonary inflammation, G-1 attenuates airway hyper-responsiveness, reducing bronchoalveolar levels of inflammatory cells and the T helper 2 (TH2) cell cytokines IL-5 and IL-13, while increasing the frequency of splenic regulatory T cells (which produce the anti-inflammatory cytokine IL-10), thus establishing crosstalk between GPER and IL-10 (ref. 240). Moreover, G-1 treatment also promotes the formation of IL-10 in pro-inflammatory TH17 cells241,242. In macrophages, G-1 inhibits the production of lipopolysaccharide-induced cytokines, such as TNF and IL-6 (ref. 119), through the inhibition of NF-κB120, while also downregulating TLR4 expression243. Neutrophils show complex responses to G-1 in vitro, with G-1 treatment causing activation of human neutrophils244 and increased cell death-associated neutrophil extracellular trap formation245. In fish granulocytes, G-1 has multiple effects245, including suppression of ROS production12.
Regulation of inflammation by GPER
Deletion of Gper in mice increases circulating levels of pro-inflammatory cytokines, with a concomitant decrease in adiponectin levels compared with the wild type114,165. In a mouse model of diethylnitrosamine-induced liver cancer, deletion of Gper increases inflammation, fibrosis and tumorigenesis208. Consistent with this, GPER activation reduces expression of fibrosis markers in hepatic stellate cells in vitro, suggesting a possible role for GPER in counteracting liver inflammation and liver cancer208. In a mouse model of atherosclerosis, G-1 treatment reduces the increased number of CD68+ macrophages but not of CD3+ T cells, whereas deletion of Gper has the opposite effect126.
Modulation of GPER activity in immunity, inflammation and infection
In surgically postmenopausal mice with diet-induced obesity, chronic treatment with G-1 reduces levels of TNF, MCP1 and IL-6 as well as the expression of inflammatory genes in multiple metabolic tissues168. GPER may also play a role in inflammatory bowel diseases; in a model of Crohn’s disease, G-1 treatment reduces mortality, improves macroscopic and microscopic injury scores, and lowers C-reactive protein levels173,178. In a mouse model of Staphylococcus aureus skin and soft tissue infection, G-1 reduces dermonecrosis and increases bacterial clearance, indicating a role of GPER for the innate immune system246,247. These effects are more pronounced in females, suggesting a sex-specific response, and are absent in Gper-deficient mice, confirming the selectivity of G-1 for its target GPER247.
Clinical data suggest a sex bias in COVID-19 severity following SARS-CoV-2 infection, with men exhibiting increased hospitalization and mortality compared with women. A role for GPER in this sex bias is suggested based on experimental models of both overexpression of GPER and treatment with G-1, each of which (similar to 17β-oestradiol treatment) leads to reduced SARS-CoV-2 viral load in infected bronchial cells in vitro compared with uninfected cells. These reductions in viral load caused by 17β-oestradiol and G-1 treatment are reversed by treatment with G15 (ref. 248). GPER activation also results in anti-inflammatory immune responses in numerous neurological diseases249,250,251. Lastly, in a genome-wide CRISPR–Cas9 screen, GPER was identified as a downregulator of type I interferon252. GPER expression during pregnancy is both necessary and sufficient to suppress IFNγ signalling, which is elevated in reproductive and fetal tissues in influenza A virus-infected female mice. During virus-induced maternal inflammation, blocking GPER with G15 delays fetal development and promotes fetal demise compared with vehicle-treated mice252. Thus, GPER expression and activity are required to protect the fetus during maternal infection. Taken together, pharmacological activation of GPER holds promise for the treatment of diseases and conditions that are associated with activation of inflammation (due to infectious pathogens such as bacteria or viruses) and of conditions associated with an abnormal immune response.
Ageing and neurological diseases
Cardiovascular and renal ageing
Physiological ageing is an unmodifiable risk factor for arterial hypertension, myocardial disease and atherosclerotic vascular disease. In addition, vascular ageing is further accelerated by modifiable risk factors, including obesity (which is often associated with hypertension and diabetes) and smoking89. Endogenous Gper expression is associated with suppresion of the age-dependent increases in endothelin ETB receptors, and endothelin-converting enzyme-2 in the heart253. Moreover, Gper deficiency abrogates age-dependent impairment of vasodilatation by interfering with NOX1-dependent ROS formation, specifically by reducing NOX1 expression, which is induced by GPER39,103. Accordingly, Gper deficiency prevents ageing-induced myocardial fibrosis and the associated development of diastolic heart failure (HFpEF) and for the most part prevents angiotensin-induced hypertension39. In addition, Gper deficiency is associated with a supression of development of age-dependent CKD due to FSGS144. The effect of Gper deficiency could be partly recapitulated pharmacologically by reducing NOX1 abundance and the associated production of ROS with G36, the first NOX1 downregulator39. Thus, blocking the GPER–NOX1 axis holds therapeutic opportunities for ageing-associated non-communicable diseases, including arterial hypertension.
Neurological diseases
In premenopausal women, endogenous oestrogens protect against stroke and dementia254. GPER, like ERα and ERβ, regulates arterial tone of the cerebral vasculature255. Antisense oligonucleotide knockdown of Gper in vivo largely abrogates the protective effects of oestrogen on cerebral ischaemia256, whereas activation of GPER with G-1 reduces reperfusion injury following cerebral ischaemia in both male and female mice257,258. This involves inhibition of both apoptosis259 and inflammatory pathways, such as TLR4 (ref. 258), with concomitant activation of anti-inflammatory pathways260. GPER-dependent protective effects have been demonstrated in rodent models of ischaemic261 and haemorrhagic stroke262. G-1-dependent protection from ischaemic stroke is completely abrogated by systemic deletion of Gper, while only partial protection was observed in animals with astrocyte- or neuronal cell-specific Gper deletion261. Activation of GPER by G-1 also attenuates blood–brain barrier injury263 and improves immunoprotection following stroke264. GPER also might play a role in psychiatric disorders such as anxiety265, depression266 and addiction267. Systemic deletion of Gper increases anxiety in rats265; accordingly, activation of GPER by G-1 has anxiolytic and also antidepressant effects in rodents69,266. Finally, deletion of Gper or GPER antagonism enhances morphine analgesia and reduces pain involving µ-type opioid receptors, suggesting the potential of GPER blockade for the treatment of pain, substance addiction, and opioid tolerance268.
Ageing is the main risk factor for Parkinson disease and Alzheimer disease as well as for vascular dementia. Studies in neurotoxic mouse models of Parkinson disease have shown that 17β-oestradiol-dependent, ERα-mediated protective effects on dopaminergic neurons require crosstalk with GPER and that GPER also has independent protective effects against Parkinson disease267. In a mouse model of Parkinson disease, G-1 treatment reduces the release of pro-inflammatory cytokines251 and also mediates part of the neuroprotective effects of IGF1 on dopaminergic neuronal injury269. G-1 treatment also reduces microglial activation and decreases pro-inflammatory cytokine production251. GPER is important for maintaining long-term memory, and G-1 enhances object recognition and long-term memory in male mice270. Accordingly, in a mouse model of Alzheimer disease and after traumatic brain injury in rats, improvements in neuropsychological functions are observed upon G-1 treatment271,272,273. GPER also mediates the anti-inflammatory effects of genistein in microglia250.
Elevated levels of 17β-oestradiol present in pregnant women are associated with reduced severity of multiple sclerosis274, and 17β-oestradiol supplementation reduces symptom severity and immune infiltration in a mouse model of MS (experimental autoimmune encephalomyelitis) in mice of both sexes275. In this model, female Gper-deficient mice exhibit reduced 17β-oestradiol-mediated protection against multiple sclerosis disease severity and reduced protective effects of 17β-oestradiol on white matter damage compared with wild-type mice119,249,276. Conversely, GPER activation by G-1 reduces multiple sclerosis severity, an effect absent in female Gper-deficient mice. Mechanistically, G-1 reduced inflammatory cytokine production in macrophages and upregulated PD1 to enhance the activity of T regulatory cells249.
Conclusions
Progress made in the past decade in the field of GPER has broadened our understanding of the multiple functions of this receptor at the cell, tissue and organismal level, including in humans. Widely expressed, GPER mediates both rapid and genomic effects in all main organs, being involved in multiple aspects of health and disease (Fig. 3). In addition to oestrogens, many natural and synthetic molecules target GPER, either as selective or combined oestrogen receptor agonists or antagonists. Importantly, clinically approved ERα antagonists, such as the SERMS tamoxifen and raloxifene or the SERD fulvestrant, licensed for the treatment of breast cancer209, show agonistic activity towards GPER13,14,66. Diverse molecules present in plants (such as genistein, daidzein and green tea polyphenols) and EDCs also activate GPER; further study is required to determine how their effects on health or disease involve GPER. Utilizing GPER expression as a diagnostic marker in tissues or in circulating cells provides new opportunities to further characterize pathological conditions at different stages during disease progression or even before diseases develop. Targeting GPER pharmacologically could provide new opportunities to treat diseases for which no or only a few effective therapies exist (such as malignant melanoma and other cancers), including inhibition of the constitutive inducing effect of GPER on NOX1 activity. Clinical studies that should also consider sex, genetics and hormonal status are needed to determine whether utilizing or targeting GPER could improve diagnosis, prognosis, therapy and the clinical course of human diseases and thus overall health82.
References
Aristotélēs. Historia Animalium Books 1–10 (transl. and ed. Peck, A. L., Balme, D. M. & Gotthelf, A.) 1–624 (Harvard Univ. Press, 1965).
Starling, E. H. Croonian lecture: on the chemical correlation of the functions of the body. Lancet 2, 579–583 (1905).
Allen, E. & Doisy, E. A. An ovarian hormone: preliminary report on its localization, extraction and partial purification, and action in test animals. JAMA 81, 819–821 (1923).
Butenandt, A. Über “Progynon”, ein krystallisiertes weibliches Sexualhormon. Die Naturwissenschaften 17, 879 (1929).
Jensen, E. A conversation with Elwood Jensen. Interview by David D. Moore. Annu. Rev. Physiol. 74, 1–11 (2012).
Soloff, M. S. & Szego, C. M. Purification of estradiol receptor from rat uterus and blockade of its estrogen-binding function by specific antibody. Biochem. Biophys. Res. Commun. 34, 141–147 (1969).
Barton, M. et al. Twenty years of the G protein-coupled estrogen receptor GPER: historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 176, 4–15 (2018).
Walter, P. et al. Cloning of the human estrogen receptor cDNA. Proc. Natl Acad. Sci. USA 82, 7889–7893 (1985). The cloning of ERα was a landmark accomplishment in the oestrogen and nuclear hormone receptor fields.
Kuiper, G. G., Enmark, E., Pelto-Huikko, M., Nilsson, S. & Gustafsson, J. A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl Acad. Sci. USA 93, 5925–5930 (1996). One of two papers reporting the cloning of ERβ, revealing the existence of a second oestrogen receptor.
Mosselman, S., Polman, J. & Dijkema, R. ERβ: identification and characterization of a novel human estrogen receptor. FEBS Lett. 392, 49–53 (1996). One of two papers reporting the cloning of ERβ, revealing the existence of a second oestrogen receptor.
Prossnitz, E. R. & Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 7, 715–726 (2011). A comprehensive review summarizing knowledge in the GPER field in the first 10 years after GPER was identified as an oestrogen receptor.
Cabas, I., Chaves-Pozo, E., Mulero, V. & Garcia-Ayala, A. Role of estrogens in fish immunity with special emphasis on GPER1. Dev. Comp. Immunol. 89, 102–110 (2018).
Filardo, E. J., Quinn, J. A., Bland, K. I. & Frackelton, A. R. Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 14, 1649–1660 (2000). A landmark paper reporting the discovery of GPR30/GPER activation by oestrogen.
Revankar, C. M., Cimino, D. F., Sklar, L. A., Arterburn, J. B. & Prossnitz, E. R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307, 1625–1630 (2005). This is one of two key studies that first reported oestrogen binding to and signalling via GPER.
Thomas, P., Pang, Y., Filardo, E. J. & Dong, J. Identity of an oestrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146, 624–632 (2005). This is one of two key studies that first reported oestrogen binding to and signalling via GPER.
Alexander, S. P. H., Mathie, A. & Peters, J. A. Guide to Receptors and Channels (GRAC), 3rd edn. Br. J. Pharmacol. 153 (Suppl. 2), 1–209 (2008).
Gaudet, H. M., Cheng, S. B., Christensen, E. M. & Filardo, E. J. The G-protein coupled estrogen receptor, GPER: the inside and inside-out story. Mol. Cell Endocrinol. 418, 207–219 (2015).
Madeo, A. & Maggiolini, M. Nuclear alternate estrogen receptor GPR30 mediates 17β-estradiol-induced gene expression and migration in breast cancer-associated fibroblasts. Cancer Res. 70, 6036–6046 (2010).
Filardo, E. J., Quinn, J. A., Frackelton, A. R. Jr. & Bland, K. I. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 16, 70–84 (2002).
Lucas, T. F., Royer, C., Siu, E. R., Lazari, M. F. & Porto, C. S. Expression and signaling of G protein-coupled estrogen receptor 1 (GPER) in rat Sertoli cells. Biol. Reprod. 83, 307–317 (2010).
Deng, Q. et al. GPER/Hippo-YAP signal is involved in bisphenol S induced migration of triple negative breast cancer (TNBC) cells. J. Hazard. Mater. 355, 1–9 (2018).
Daub, H., Weiss, F. U., Wallasch, C. & Ullrich, A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379, 557–560 (1996).
Zekas, E. & Prossnitz, E. R. Estrogen-mediated inactivation of FOXO3a by the G protein-coupled estrogen receptor GPER. BMC Cancer 15, 702 (2015).
Fredette, N. C., Meyer, M. R. & Prossnitz, E. R. Role of GPER in estrogen-dependent nitric oxide formation and vasodilation. J. Steroid Biochem. Mol. Biol. 176, 65–72 (2018).
Peixoto, P., Aires, R. D., Lemos, V. S., Bissoli, N. S. & Santos, R. L. D. GPER agonist dilates mesenteric arteries via PI3K-Akt-eNOS and potassium channels in both sexes. Life Sci. 183, 21–27 (2017).
Sharma, G. & Prossnitz, E. R. Mechanisms of estradiol-induced insulin secretion by the G protein-coupled estrogen receptor GPR30/GPER in pancreatic β-cells. Endocrinology 152, 3030–3039 (2011).
Greenlee, M. M. et al. Estradiol activates epithelial sodium channels in rat alveolar cells through the G protein-coupled estrogen receptor. Am. J. Physiol. Lung Cell Mol. Physiol. 305, L878–889 (2013).
Evanson, K. W., Goldsmith, J. A., Ghosh, P. & Delp, M. D. The G protein-coupled estrogen receptor agonist, G-1, attenuates BK channel activation in cerebral arterial smooth muscle cells. Pharmacol. Res. Perspect. 6, e00409 (2018).
Yue, J. et al. Activation of G-protein-coupled receptor 30 protects neurons against excitotoxicity through inhibiting excessive autophagy induced by glutamate. ACS Chem. Neurosci. 10, 4227–4236 (2019).
Zhang, H. et al. Mechanisms of estradiol-induced EGF-like factor expression and oocyte maturation via G protein-coupled estrogen receptor. Endocrinology 161, bqaa190 (2020).
Sun, M. et al. G protein-coupled estrogen receptor enhances melanogenesis via cAMP-protein kinase (PKA) by upregulating microphthalmia-related transcription factor-tyrosinase in melanoma. J. Steroid Biochem. Mol. Biol. 165, 236–246 (2017).
Pandey, D. P. et al. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 28, 523–532 (2009).
Cirillo, F. et al. GPER is involved in the regulation of the estrogen-metabolizing cyp1b1 enzyme in breast cancer. Oncotarget 8, 106608–106624 (2017).
Zhu, P. et al. GPER/ERK&AKT/NF-κB pathway is involved in cadmium-induced proliferation, invasion and migration of GPER-positive thyroid cancer cells. Mol. Cell Endocrinol. 442, 68–80 (2017).
Chen, Z. J. et al. Activation of GPER suppresses epithelial mesenchymal transition of triple negative breast cancer cells via NF-κB signals. Mol. Oncol. 10, 775–788 (2016).
De Francesco, E. M., Maggiolini, M. & Musti, A. M. Crosstalk between Notch, HIF-1α and GPER in breast cancer EMT. Int. J. Mol. Sci. 19, 2011 (2018).
Zhou, X. et al. Estrogen regulates Hippo signaling via GPER in breast cancer. J. Clin. Invest. 125, 2123–2135 (2015).
Zhu, L. et al. MicroRNA-2861 and microRNA-5115 regulates myocardial ischemia-reperfusion injury through the GPR30/mTOR signaling pathway by binding to GPR30. J. Cell Physiol. 235, 7791–7802 (2020).
Meyer, M. R. et al. Obligatory role for GPER in cardiovascular aging and disease. Sci. Signal. 9, ra105 (2016). This study reports regulation of the NADPH oxidase NOX1 by GPER expression and the discovery of GPER antagonists as NOX1 downregulators.
Barton, M., Meyer, M. R. & Prossnitz, E. R. Nox1 downregulators: a new class of therapeutics. Steroids 152, 108494 (2019).
Jensen, E. V. & Jordan, V. C. The estrogen receptor: a model for molecular medicine. Clin. Cancer Res. 9, 1980–1989 (2003).
Cheng, S. B. et al. Anatomical location and redistribution of G protein-coupled estrogen receptor-1 during the estrus cycle in mouse kidney and specific binding to estrogens but not aldosterone. Mol. Cell Endocrinol. 382, 950–959 (2014).
Rigiracciolo, D. C. et al. GPER is involved in the stimulatory effects of aldosterone in breast cancer cells and breast tumor-derived endothelial cells. Oncotarget 7, 94–111 (2016).
Lu, A. S., Rouhimoghadam, M., Arnatt, C. K., Filardo, E. J. & Salem, A. K. Proteolytic targeting chimeras with specificity for plasma membrane and intracellular estrogen receptors. Mol. Pharm. 18, 1455–1469 (2021).
Koganti, S., Snyder, R., Gumaste, U., Karamyan, V. T. & Thekkumkara, T. 2-Methoxyestradiol binding of GPR30 down-regulates angiotensin AT(1) receptor. Eur. J. Pharmacol. 723, 131–140 (2014).
Zucchetti, A. E. et al. G-protein-coupled receptor 30/adenylyl cyclase/protein kinase A pathway is involved in estradiol 17ss-D-glucuronide-induced cholestasis. Hepatology 59, 1016–1029 (2014).
Chourasia, T. K., Pang, Y. & Thomas, P. The catecholestrogen, 2-hydroxyestradiol-17beta, acts as a G protein-coupled estrogen receptor 1 (GPER/GPR30) antagonist to promote the resumption of meiosis in zebrafish oocytes. Biol. Reprod. 92(69), 1–13 (2015).
Teng, Y. et al. Dehydroepiandrosterone activation of G-protein-coupled estrogen receptor rapidly stimulates MicroRNA-21 transcription in human hepatocellular carcinoma cells. J. Biol. Chem. 290, 15799–15811 (2015).
Cao, J., Lu, M., Yan, W., Li, L. & Ma, H. Dehydroepiandrosterone alleviates intestinal inflammatory damage via GPR30-mediated nrf2 activation and NLRP3 inflammasome inhibition in colitis mice. Free Radic. Biol. Med. 172, 386–402 (2021).
Sandra, N., Ester, P., Marie-Agnes, P., Robert, M. & Olivier, H. The DHEA metabolite 7β-hydroxy-epiandrosterone exerts anti-estrogenic effects on breast cancer cell lines. Steroids 77, 542–551 (2012).
Avena, P. et al. 27-Hydroxycholesterol binds GPER and induces progression of estrogen receptor-negative breast cancer. Cancers 14, 1521 (2022).
Diamanti-Kandarakis, E. et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr. Rev. 30, 293–342 (2009).
Lorand, T., Vigh, E. & Garai, J. Hormonal action of plant derived and anthropogenic non-steroidal estrogenic compounds: phytoestrogens and xenoestrogens. Curr. Med. Chem. 17, 3542–3574 (2010).
Thomas, P. & Dong, J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: a potential novel mechanism of endocrine disruption. J. Steroid Biochem. Mol. Biol. 102, 175–179 (2006).
Zhou, C., Li, P., Han, M. & Gao, X. Daidzein stimulates fatty acid-induced fat deposition in C2C12 myoblast cells via the G protein-coupled receptor 30 pathway. Anim. Biotechnol. 33, 851–863 (2020).
Moriyama, M. et al. S-equol, a major isoflavone from soybean, inhibits nitric oxide production in lipopolysaccharide-stimulated rat astrocytes partially via the GPR30-mediated pathway. Int. J. Inflam. 2018, 8496973 (2018).
Maggiolini, M. et al. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17β-estradiol and phytoestrogens in breast cancer cells. J. Biol. Chem. 279, 27008–27016 (2004).
Dong, W. H., Chen, J. C., He, Y. L., Xu, J. J. & Mei, Y. A. Resveratrol inhibits K(v)2.2 currents through the estrogen receptor GPR30-mediated PKC pathway. Am. J. Physiol. Cell Physiol. 305, C547–557 (2013).
Chimento, A. et al. Oleuropein and hydroxytyrosol activate GPER/ GPR30-dependent pathways leading to apoptosis of ER-negative SKBR3 breast cancer cells. Mol. Nutr. Food Res. 58, 478–489 (2013).
Li, Y. C., Ding, X. S., Li, H. M. & Zhang, C. Icariin attenuates high glucose-induced type IV collagen and fibronectin accumulation in glomerular mesangial cells by inhibiting transforming growth factor-β production and signalling through G protein-coupled oestrogen receptor 1. Clin. Exp. Pharmacol. Physiol. 40, 635–643 (2013).
Moreno-Ulloa, A. et al. (-)-Epicatechin stimulates mitochondrial biogenesis and cell growth in C2C12 myotubes via the G-protein coupled estrogen receptor. Eur. J. Pharmacol. 822, 95–107 (2018).
Lo, E. K., Lee, J. C., Turner, P. C. & El-Nezami, H. Low dose of zearalenone elevated colon cancer cell growth through G protein-coupled estrogenic receptor. Sci. Rep. 11, 7403 (2021).
Paterni, I., Granchi, C., Katzenellenbogen, J. A. & Minutolo, F. Estrogen receptors alpha (ERα) and beta (ERβ): subtype-selective ligands and clinical potential. Steroids 90, 13–29 (2014).
Stauffer, S. R. et al. Pyrazole ligands: structure-affinity/activity relationships and estrogen receptor-α-selective agonists. J. Med. Chem. 43, 4934–4947 (2000).
Meyers, M. J. et al. Estrogen receptor-β potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J. Med. Chem. 44, 4230–4251 (2001).
Petrie, W. K. et al. G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obstet. Gynecol. Int. 2013, 472720 (2013).
Bologa, C. G. et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2, 207–212 (2006). This study reports the first GPER-selective ligand G-1.
Dennis, M. K. et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J. Steroid Biochem. Mol. Biol. 127, 358–366 (2011).
Dennis, M. K. et al. In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 5, 421–427 (2009). This study reports the discovery of the first GPER-selective antagonist.
Urban, N., Leonhardt, M. & Schaefer, M. Multiplex GPCR screen reveals reliably acting agonists and a Gq-phospholipase C coupling mode of GPR30/GPER1. Mol. Pharmacol. 103, 48–62 (2023).
Tutzauer, J. et al. Ligand-independent G protein-coupled estrogen receptor/G protein-coupled receptor 30 activity: lack of receptor-dependent effects of G-1 and 17β-estradiol. Mol. Pharmacol. 100, 271–282 (2021).
Lappano, R. et al. Two novel GPER agonists induce gene expression changes and growth effects in cancer cells. Curr. Cancer Drug Targets 12, 531–542 (2012).
O’Dea, A., Sondergard, C., Sweeney, P. & Arnatt, C. K. A series of indole-thiazole derivatives act as GPER agonists and inhibit breast cancer cell growth. ACS Med. Chem. Lett. 9, 901–906 (2018).
DeLeon, C. et al. A novel GPER antagonist protects against the formation of estrogen-induced cholesterol gallstones in female mice. J. Lipid Res. 61, 767–777 (2020).
Lappano, R. et al. MIBE acts as antagonist ligand of both estrogen receptor α and GPER in breast cancer cells. Breast Cancer Res. 14, R12 (2012).
Bargagna-Mohan, P., Baek, S. H., Lee, H., Kim, K. & Mohan, R. Use of PROTACS as molecular probes of angiogenesis. Bioorg. Med. Chem. Lett. 15, 2724–2727 (2005).
Revankar, C. M. et al. A selective ligand for estrogen receptor proteins discriminates rapid and genomic signaling. Cell. Chem. Biol. 26, 1692–1702 (2019). This study reports the first oestrogen receptor-selective ligand (agonist) that does not bind GPER.
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04130516 (2019).
Muller, C. et al. Phase 1 trial of a novel, first-in-class G protein-coupled estrogen receptor (GPER) agonist, LNS8801 in patients with advanced or recurrent treatment-refractory solid malignancies. J. Clin. Oncol. 39, 3084 (2021).
Muller, C. et al. Phase 1b study of the novel first-in-class G protein-coupled estrogen receptor (GPER) agonist, LNS8801 in combination with pembrolizumab in patients with immune checkpoint inhibitor (ICI)-relapsed and refractory solid malignancies and dose escalation update. J. Clin. Oncol. 40, 2574 (2022).
Prossnitz, E. R. & Hathaway, H. J. What have we learned about GPER function in physiology and disease from knockout mice? J. Steroid Biochem. Mol. Biol. 153, 114–126 (2015). A review of GPER physiology based on studies with different Gper-knockout mouse strains.
Mielke, M. M. & Miller, V. M. Improving clinical outcomes through attention to sex and hormones in research. Nat. Rev. Endocrinol. 17, 625–635 (2021).
Mauvais-Jarvis, F. et al. Sex and gender: modifiers of health, disease, and medicine. Lancet 396, 565–582 (2020). An excellent overview of the roles of sex and sex steroids and their receptors in human physiology and disease.
Lafferty, A. R. et al. A novel genetic locus for low renin hypertension: familial hyperaldosteronism type II maps to chromosome 7 (7p22). J. Med. Genet. 37, 831–835 (2000).
Feldman, R. D. et al. A common hypofunctional genetic variant of GPER is associated with increased blood pressure in women. Br. J. Clin. Pharmacol. 78, 1441–1452 (2014). The first clinical genetics study to suggest a role of GPER in the regulation of arterial blood pressure and possibly in the pathogenesis of arterial hypertension in humans.
Fredette, N. C., Malik, E., Mukhtar, M. L., Prossnitz, E. R. & Terada, N. A hypertension patient derived induced pluripotent stem cell model demonstrates a role for GPER in hypertension risk and development. Am. J. Physiol. Cell. Physiol. 319, C825–C838 (2020).
Hussain, Y. et al. G-protein estrogen receptor as a regulator of low-density lipoprotein cholesterol metabolism: cellular and population genetic studies. Arterioscler. Thromb. Vasc. Biol. 35, 213–221 (2015).
Pupo, M. et al. A genetic polymorphism repurposes the G-protein coupled and membrane-associated estrogen receptor GPER to a transcription factor-like molecule promoting paracrine signaling between stroma and breast carcinoma cells. Oncotarget 8, 46728–46744 (2017).
Meyer, M. R. & Barton, M. Estrogens and coronary artery disease: new clinical perspectives. Adv. Pharmacol. 77, 307–360 (2016).
Barton, M. & Meyer, M. R. Heart failure with preserved ejection fraction in women: new clues to causes and treatment. JACC Basic Transl. Sci. 5, 296–299 (2020).
Haas, E. et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ. Res. 104, 288–291 (2009). The first study reporting acute changes in vascular tone and arterial blood pressure following activation of GPER and a regulatory role for endogenous GPER in obesity.
Meyer, M. R., Baretella, O., Prossnitz, E. R. & Barton, M. Dilation of epicardial coronary arteries by the G protein-coupled estrogen receptor agonists G-1 and ICI 182,780. Pharmacology 86, 58–64 (2010).
Arefin, S. et al. Vasodilatory effects of the selective GPER agonist G-1 is maximal in arteries of postmenopausal women. Maturitas 78, 123–130 (2014).
Yu, X. et al. Activation of G protein-coupled estrogen receptor induces endothelium-independent relaxation of coronary artery smooth muscle. Am. J. Physiol. Endocrinol. Metab. 301, E882–888 (2011).
Lindsey, S. H., Liu, L. & Chappell, M. C. Vasodilation by GPER in mesenteric arteries involves both endothelial nitric oxide and smooth muscle cAMP signaling. Steroids 81, 99–102 (2014).
Pang, Y. & Thomas, P. Additive effects of low concentrations of estradiol-17β and progesterone on nitric oxide production by human vascular endothelial cells through shared signaling pathways. J. Steroid Biochem. Mol. Biol. 165, 258–267 (2017).
Yu, X., Stallone, J. N., Heaps, C. L. & Han, G. The activation of G protein-coupled estrogen receptor induces relaxation via cAMP as well as potentiates contraction via EGFR transactivation in porcine coronary arteries. PLoS ONE 13, e0191418 (2018).
Yu, X. et al. Activation of G protein-coupled estrogen receptor 1 induces coronary artery relaxation via Epac/Rap1-mediated inhibition of RhoA/Rho kinase pathway in parallel with PKA. PLoS ONE 12, e0173085 (2017).
Meyer, M. R., Fredette, N. C., Barton, M. & Prossnitz, E. R. G protein-coupled estrogen receptor inhibits vascular prostanoid production and activity. J. Endocrinol. 227, 61–69 (2015).
Meyer, M. R., Field, A. S., Kanagy, N. L., Barton, M. & Prossnitz, E. R. GPER regulates endothelin-dependent vascular tone and intracellular calcium. Life Sci. 91, 623–627 (2012).
Tropea, T. et al. Pregnancy augments G protein estrogen receptor (GPER) induced vasodilation in rat uterine arteries via the nitric oxide–cGMP signaling pathway. PLoS ONE 10, e0141997 (2015).
Gurrala, R. et al. Alterations in the estrogen receptor profile of cardiovascular tissues during aging. Geroscience 43, 433–442 (2021).
Meyer, M. R., Rosemann, T., Barton, M. & Prossnitz, E. R. GPER mediates functional endothelial aging in renal arteries. Pharmacology 100, 188–193 (2017).
Davis, G. K., Newsome, A. D., Cole, A. B., Ojeda, N. B. & Alexander, B. T. Chronic estrogen supplementation prevents the increase in blood pressure in female intrauterine growth-restricted offspring at 12 months of age. Hypertension 73, 1128–1136 (2019).
Luo, P. et al. Stress-related arterial hypertension in Gper-deficient rats. Acta Physiol. Sin. 69, 532–540 (2017).
Waghulde, H. et al. Attenuation of microbiotal dysbiosis and hypertension in a CRISPR/Cas9 gene ablation rat model of GPER1. Hypertension 72, 1125–1132 (2018).
Gohar, E. Y. et al. Evidence for G-protein-coupled estrogen receptor as a pronatriuretic factor. J. Am. Heart Assoc. 9, e015110 (2020). This study reports the identification of GPER as a modulator of natriuresis.
Barton, M. & Meyer, M. R. Nicolaus Copernicus and the rapid vascular responses to aldosterone. Trends Endocrinol. Metab. 26, 396–398 (2015).
Wehling, M. Rapid actions of aldosterone revisited: receptors in the limelight. J. Steroid Biochem. Mol. Biol. 176, 94–98 (2018).
Dinh, Q. N. et al. Aldosterone-induced hypertension is sex-dependent, mediated by T cells and sensitive to GPER activation. Cardiovasc. Res. 117, 960–970 (2021). An important study identifying T cell-mediated mechanisms involving GPER as sex-dependent modifiers of aldosterone-dependent arterial hypertension.
Caroccia, B. et al. Aldosterone stimulates its biosynthesis via a novel GPER-mediated mechanism. J. Clin. Endocrinol. Metab. 104, 6316–6324 (2019). This study reports the regulation of adrenal aldosterone biosynthesis involving GPER-dependent mechanisms.
Wang, D., Wang, M., Sun, P. & Gao, Q. Eplerenone inhibits oxidized low-density lipoprotein-induced proliferation and migration of vascular smooth muscle cells by downregulating GPER expression. Adv. Clin. Exp. Med. 30, 405–412 (2021).
Cheng, L. et al. Rapid aldosterone-mediated signaling in the DCT increases activity of the thiazide-sensitive NaCl cotransporter. J. Am. Soc. Nephrol. 30, 1454–1470 (2019).
Sharma, G. et al. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology 154, 4136–4145 (2013).
Meoli, L. et al. Sex- and age-dependent effects of Gpr30 genetic deletion on the metabolic and cardiovascular profiles of diet-induced obese mice. Gene 540, 210–216 (2014).
Ghaffari, S., Naderi Nabi, F., Sugiyama, M. G. & Lee, W. L. Estrogen inhibits LDL (low-density lipoprotein) transcytosis by human coronary artery endothelial cells via GPER (G-protein-coupled estrogen receptor) and SR-BI (scavenger receptor class B type 1). Arterioscler. Thromb. Vasc. Biol. 38, 2283–2294 (2018). This study demonstrated that oestrogen-dependent inhibition of LDL cholesterol transcytosis, which is involved in early atherogenesis, is mediated by GPER and scavenger receptor B1.
Jafarynezhad, F. et al. The G-protein-coupled estrogen receptor agonist prevents cardiac lipid accumulation by stimulating cardiac peroxisome proliferator-activated receptor alpha: a preclinical study in ovariectomized-diabetic rat model. Int. J. Endocrinol. Metab. 20, e123560 (2022).
Locher, R., Emmanuele, L., Suter, P. M., Vetter, W. & Barton, M. Green tea polyphenols inhibit human vascular smooth muscle cell proliferation stimulated by native low-density lipoprotein. Eur. J. Pharmacol. 434, 1–7 (2002).
Blasko, E. et al. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J. Neuroimmunol. 214, 67–77 (2009). One of two studies to first suggest a role for GPER in experimental multiple sclerosis.
Pelekanou, V. et al. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERα36 and GPR30/GPER1. J. Leukoc. Biol. 99, 333–347 (2016).
Haas, E. et al. Differential effects of 17β-estradiol on function and expression of estrogen receptor alpha, estrogen receptor beta, and GPR30 in arteries and veins of patients with atherosclerosis. Hypertension 49, 1358–1363 (2007).
Li, F. et al. Activation of GPER induces differentiation and inhibition of coronary artery smooth muscle cell proliferation. PLoS ONE 8, e64771 (2013).
Jehle, J. et al. G protein-coupled estrogen receptor GPR30 exerts vasoprotective effects in apolipoprotein E-deficient mice. Arch. Med. Sci. https://doi.org/10.5114/aoms/127200 (2021).
Moreno-Ulloa, A. et al. The effects of (-)-epicatechin on endothelial cells involve the G protein-coupled estrogen receptor (GPER). Pharmacol. Res. 100, 309–320 (2015).
Meyer, M. R., Fredette, N. C., Barton, M. & Prossnitz, E. R. Regulation of vascular smooth muscle tone by adipose-derived contracting factor. PLoS ONE 8, e79245 (2013).
Meyer, M. R. et al. G protein-coupled estrogen receptor protects from atherosclerosis. Sci. Rep. 4, 7564 (2014).
Bopassa, J. C., Eghbali, M., Toro, L. & Stefani, E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 298, H16–23 (2010). First study reporting a protective role of GPER in myocardial reperfusion injury from myocardial ischaemia following coronary occlusion.
Kabir, M. E. et al. G protein-coupled estrogen receptor 1 mediates acute estrogen-induced cardioprotection via MEK/ERK/GSK-3β pathway after ischemia/reperfusion. PLoS ONE 10, e0135988 (2015).
Feng, Y., Madungwe, N. B., da Cruz Junho, C. V. & Bopassa, J. C. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br. J. Pharmacol. 174, 4329–4344 (2017).
Iorga, A. et al. Rescue of pressure overload-induced heart failure by estrogen therapy. J. Am. Heart Assoc. 5, e002482 (2016).
Goncalves, G. K. et al. Neonatal cardiomyocyte hypertrophy induced by endothelin-1 is blocked by estradiol acting on GPER. Am. J. Physiol. Cell Physiol. 314, C310–C322 (2018).
Watanabe, T. et al. 17β-Estradiol inhibits cardiac fibroblast growth through both subtypes of estrogen receptor. Biochem. Biophys. Res. Commun. 311, 454–459 (2003).
Mercier, I., Mader, S. & Calderone, A. Tamoxifen and ICI 182,780 negatively influenced cardiac cell growth via an estrogen receptor-independent mechanism. Cardiovasc. Res. 59, 883–892 (2003).
Recchia, A. G. et al. The G protein-coupled receptor 30 is up-regulated by hypoxia-inducible factor-1α (HIF-1α) in breast cancer cells and cardiomyocytes. J. Biol. Chem. 286, 10773–10782 (2011).
Whitcomb, V. et al. Regulation of beta adrenoceptor-mediated myocardial contraction and calcium dynamics by the G protein-coupled estrogen receptor 1. Biochem. Pharmacol. 171, 113727 (2020).
Wang, H., Sun, X., Hodge, H. S., Ferrario, C. M. & Groban, L. NLRP3 inhibition improves heart function in GPER knockout mice. Biochem. Biophys. Res. Commun. 514, 998–1003 (2019). This study reports that inhibition of the NLRP3 inflammasome ameliorates impaired systolic and diastolic myocardial function in mice lacking GPER.
Wang, H. et al. Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc. Res. 94, 96–104 (2012).
Jessup, J. A., Lindsey, S. H., Wang, H., Chappell, M. C. & Groban, L. Attenuation of salt-induced cardiac remodeling and diastolic dysfunction by the GPER agonist G-1 in female mRen2.Lewis rats. PLoS ONE 5, e15433 (2010). The first report to suggest that pharmacological activation of GPER reduces functional and structural cardiac injury in salt-sensitive hypertension.
Alencar, A. K. et al. Effect of age, estrogen status, and late-life GPER activation on cardiac structure and function in the Fischer344xBrown Norway female rat. J. Gerontol. A Biol. Sci. Med. Sci. 72, 152–162 (2017).
Bairey Merz, C. N. et al. Sex and the kidneys: current understanding and research opportunities. Nat. Rev. Nephrol. 15, 776–783 (2019).
Chang, Y. et al. G protein-coupled estrogen receptor activation improves contractile and diastolic functions in rat renal interlobular artery to protect against renal ischemia reperfusion injury. Biomed. Pharmacother. 112, 108666 (2019).
Hofmeister, M. V. et al. 17β-Estradiol induces nongenomic effects in renal intercalated cells through G protein-coupled estrogen receptor 1. Am. J. Physiol. Ren. Physiol. 302, F358–368 (2012).
Gohar, E. Y. & Pollock, D. M. Functional interaction of endothelin receptors in mediating natriuresis evoked by G protein-coupled estrogen receptor 1. J. Pharmacol. Exp. Ther. 376, 98–105 (2021).
Meyer, M. R. et al. GPER is required for age-dependent albuminuria and glomerulosclerosis: evidence for its role in podocyte injury and mesangial Nox1 regulation. Hypertension 72, AP261 (2018).
Meyer, M. R. et al. Deletion of GPER protects from age-related renovascular dysfunction and tubulo-interstitial injury. Hypertension 66, AP607 (2015).
Qiao, C., Ye, W., Li, S., Wang, H. & Ding, X. Icariin modulates mitochondrial function and apoptosis in high glucose-induced glomerular podocytes through G protein-coupled estrogen receptors. Mol. Cell. Endocrinol. 473, 146–155 (2018).
Lindsey, S. H., Yamaleyeva, L. M., Brosnihan, K. B., Gallagher, P. E. & Chappell, M. C. Estrogen receptor GPR30 reduces oxidative stress and proteinuria in the salt-sensitive female mRen2.Lewis rat. Hypertension 58, 665–671 (2011). The first study to report the antiproteinuric effects of pharmacological activation of GPER.
Gohar, E. Y. et al. Activation of G protein-coupled estrogen receptor 1 ameliorates proximal tubular injury and proteinuria in Dahl salt-sensitive female rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 320, R297–R306 (2021).
Sanchez, D. S. et al. Estradiol stimulates cell proliferation via classic estrogen receptor-alpha and G protein-coupled estrogen receptor-1 in human renal tubular epithelial cell primary cultures. Biochem. Biophys. Res. Commun. 512, 170–175 (2019).
Kurt, A. H., Bozkus, F., Uremis, N. & Uremis, M. M. The protective role of G protein-coupled estrogen receptor 1 (GPER-1) on methotrexate-induced nephrotoxicity in human renal epithelium cells. Ren. Fail. 38, 686–692 (2016).
Hutchens, M. P., Fujiyoshi, T., Komers, R., Herson, P. S. & Anderson, S. Estrogen protects renal endothelial barrier function from ischemia-reperfusion in vitro and in vivo. Am. J. Physiol. Ren. Physiol. 303, F377–385 (2012).
Tofovic, S. P., Zhang, X., Jackson, E. K., Zhu, H. & Petrusevska, G. 2-Methoxyestradiol attenuates bleomycin-induced pulmonary hypertension and fibrosis in estrogen-deficient rats. Vasc. Pharmacol. 51, 190–197 (2009).
Umar, S. et al. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am. J. Respir. Crit. Care Med. 184, 715–723 (2011).
Alencar, A. K. N. et al. Cardioprotection induced by activation of GPER in ovariectomized rats with pulmonary hypertension. J. Gerontol. A Biol. Sci. Med. Sci. 73, 1158–1166 (2018).
Alencar, A. K. et al. Activation of GPER ameliorates experimental pulmonary hypertension in male rats. Eur. J. Pharm. Sci. 97, 208–217 (2017). This is the first study to report therapeutic efficacy and protection of the right ventricle of the heart by pharmacological activation of GPER in experimental pulmonary arterial hypertension.
Ahmadian, R. et al. GPER contributes to the development of pulmonary hypertension in female rats. FASEB J. 34, 1 (2020).
Meyer, M. R. & Barton, M. GPER blockers as Nox downregulators: a new drug class to target chronic non-communicable diseases. J. Steroid Biochem. Mol. Biol. 176, 82–87 (2018).
Mauvais-Jarvis, F., Clegg, D. J. & Hevener, A. L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 34, 309–338 (2013). A comprehensive review of oestrogen receptor-dependent and GPER-dependent regulation glucose homeostasis and energy balance.
Meyer, M. R., Clegg, D. J., Prossnitz, E. R. & Barton, M. Obesity, insulin resistance and diabetes: sex differences and role of oestrogen receptors. Acta Physiol. 203, 259–269 (2011).
Madak-Erdogan, Z. et al. Design of pathway preferential estrogens that provide beneficial metabolic and vascular effects without stimulating reproductive tissues. Sci. Signal. 9, ra53 (2016). This study reports the identification of ‘pathway-preferential’ oestrogens with beneficial vascular and metabolic effects without stimulating reproductive tissues.
Gurney, E. P., Nachtigall, M. J., Nachtigall, L. E. & Naftolin, F. The Women’s Health Initiative Trial and related studies: 10 years later: a clinician’s view. J. Steroid Biochem. Mol. Biol. 142, 4–11 (2014).
Stubbins, R. E., Holcomb, V. B., Hong, J. & Nunez, N. P. Estrogen modulates abdominal adiposity and protects female mice from obesity and impaired glucose tolerance. Eur. J. Nutr. 51, 861–870 (2012).
Bonds, D. E. et al. The effect of conjugated equine oestrogen on diabetes incidence: the women’s health initiative randomised trial. Diabetologia 49, 459–468 (2006).
Mårtensson, U. E. et al. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology 150, 687–698 (2009).
Davis, K. E. et al. Sexually dimorphic role of G protein-coupled estrogen receptor (GPER) in modulating energy homeostasis. Horm. Behav. 66, 196–207 (2014).
Le May, C. et al. Estrogens protect pancreatic β-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc. Natl Acad. Sci. USA 103, 9232–9237 (2006).
Liu, S. et al. Importance of extranuclear estrogen receptor-α and membrane G protein-coupled estrogen receptor in pancreatic islet survival. Diabetes 58, 2292–2302 (2009).This study is the first to report a role of GPER in the regulation of pancreatic islet survival.
Sharma, G. et al. Preclinical efficacy of the GPER-selective agonist G-1 in mouse models of obesity and diabetes. Sci. Transl Med. 12, eaau5956 (2020). The study reports efficacy of the GPER agonist G-1 for the treatment of obesity, and the associated insulin resistance and diabetes mellitus.
Butler, M. J., Hildebrandt, R. P. & Eckel, L. A. Selective activation of estrogen receptors, ERα and GPER-1, rapidly decreases food intake in female rats. Horm. Behav. 103, 54–61 (2018).
Azizian, H., Khaksari, M., Asadi Karam, G., Esmailidehaj, M. & Farhadi, Z. Cardioprotective and anti-inflammatory effects of G-protein coupled receptor 30 (GPR30) on postmenopausal type 2 diabetic rats. Biomed. Pharmacother. 108, 153–164 (2018).
Balhuizen, A., Kumar, R., Amisten, S., Lundquist, I. & Salehi, A. Activation of G protein-coupled receptor 30 modulates hormone secretion and counteracts cytokine-induced apoptosis in pancreatic islets of female mice. Mol. Cell. Endocrinol. 320, 16–24 (2010).
Kumar, R., Balhuizen, A., Amisten, S., Lundquist, I. & Salehi, A. Insulinotropic and antidiabetic effects of 17β-estradiol and the GPR30 agonist G-1 on human pancreatic islets. Endocrinology 152, 2568–2579 (2011).
Jacenik, D., Beswick, E. J., Krajewska, W. M. & Prossnitz, E. R. G protein-coupled estrogen receptor in colon function, immune regulation and carcinogenesis. World J. Gastroenterol. 25, 4092–4104 (2019).
Hata, M. et al. GPR30-expressing gastric chief cells do not dedifferentiate but are eliminated via PDK-dependent cell competition during development of metaplasia. Gastroenterology 158, 1650–1666.e15 (2020).
Tsai, C. C. et al. Estradiol mediates relaxation of porcine lower esophageal sphincter. Steroids 136, 56–62 (2018).
Zielinska, M. et al. G protein-coupled estrogen receptor and estrogen receptor ligands regulate colonic motility and visceral pain. Neurogastroenterol. Motil. 29, 13025 (2017).
Li, Y. et al. G protein-coupled estrogen receptor is involved in modulating colonic motor function via nitric oxide release in C57BL/6 female mice. Neurogastroenterol. Motil. 28, 432–442 (2016).
Jacenik, D. et al. G protein-coupled estrogen receptor mediates anti-inflammatory action in Crohn’s disease. Sci. Rep. 9, 6749 (2019). This study reports that pharmacological activation of GPER reduces inflammatory activation and mortality in experimental Crohn’s disease.
Wlodarczyk, M. et al. G protein-coupled receptor 30 (GPR30) expression pattern in inflammatory bowel disease patients suggests its key role in the inflammatory process. A preliminary study. J. Gastrointestin. Liver. Dis. 26, 29–35 (2017).
Qin, B. et al. Expression of G protein-coupled estrogen receptor in irritable bowel syndrome and its clinical significance. Int. J. Clin. Exp. Pathol. 7, 2238–2246 (2014).
Shao, X., Li, J., Xu, F., Chen, D. & Liu, K. Mir-155-mediated deregulation of GPER1 plays an important role in the gender differences related to inflammatory bowel disease. Can. J. Infect. Dis. Med. Microbiol. 2020, 8811477 (2020).
Jacenik, D. et al. Estrogen signaling deregulation related with local immune response modulation in irritable bowel syndrome. Mol. Cell. Endocrinol. 471, 89–96 (2018).
Chai, S. et al. Activation of G protein-coupled estrogen receptor protects intestine from ischemia/reperfusion injury in mice by protecting the crypt cell proliferation. Clin. Sci. 133, 449–464 (2019).
Wang, Q. et al. Activation of the G protein-coupled estrogen receptor prevented the development of acute colitis by protecting the crypt cell. J. Pharmacol. Exp. Ther. 376, 281–293 (2021).
Chaturantabut, S. et al. Estrogen activation of G-protein-coupled estrogen receptor 1 regulates phosphoinositide 3-kinase and mTOR signaling to promote liver growth in zebrafish and proliferation of human hepatocytes. Gastroenterology 156, 1788–1804.e13 (2019). An important paper reporting that pharmacological activation of GPER stimulates hepatocyte proliferation and thus liver regeneration.
Tian, L. et al. The developmental wnt signaling pathway effector β-catenin/TCF mediates hepatic functions of the sex hormone estradiol in regulating lipid metabolism. PLoS Biol. 17, e3000444 (2019).
Farruggio, S. et al. Genistein improves viability, proliferation and mitochondrial function of cardiomyoblasts cultured in physiologic and peroxidative conditions. Int. J. Mol. Med. 44, 2298–2310 (2019).
Wang, H. H., Liu, M., Clegg, D. J., Portincasa, P. & Wang, D. Q. New insights into the molecular mechanisms underlying effects of estrogen on cholesterol gallstone formation. Biochim. Biophys. Acta 1791, 1037–1047 (2009).
de Bari, O., Wang, T. Y., Liu, M., Portincasa, P. & Wang, D. Q. Estrogen induces two distinct cholesterol crystallization pathways by activating ERα and GPR30 in female mice. J. Lipid Res. 56, 1691–1700 (2015).
Wang, H. H. et al. Activation of a novel estrogen receptor GPR30 enhances cholesterol cholelithogenesis in female mice. Hepatology 72, 2077–2089 (2020). This study causally links GPER to oestrogen-dependent gallstone formation in female mice.
Filardo, E. J. et al. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res. 12, 6359–6366 (2006). The first report showing an association between GPER expression and clinical outcome in breast cancer.
Ignatov, T., Treeck, O., Kalinski, T., Ortmann, O. & Ignatov, A. GPER-1 expression is associated with a decreased response rate to primary tamoxifen therapy of breast cancer patients. Arch. Gynecol. Obstet. 301, 565–571 (2020).
Ignatov, T. et al. G-protein-coupled estrogen receptor GPER-1 expression in hormone receptor-positive breast cancer is associated with poor benefit of tamoxifen. Breast Cancer Res. Treat. 174, 121–127 (2019).
Pepermans, R. A., Sharma, G. & Prossnitz, E. R. G protein-coupled estrogen receptor in cancer and stromal cells: functions and novel therapeutic perspectives. Cells 10, 672 (2021).
De Francesco, E. M. et al. GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer Res. 74, 4053–4064 (2014).
Smith, H. O. et al. GPR30: a novel indicator of poor survival for endometrial carcinoma. Am. J. Obstet. Gynecol. 196 (386), e381–e389 (2007). This study was the first to suggest an association between GPER expression and clinical outcome and survival in patients with endometrial cancer.
Smith, H. O. et al. GPR30 predicts poor survival for ovarian cancer. Gynecol. Oncol. 114, 465–471 (2009). This study was the first to suggest an association between GPER expression and clinical outcome and survival in patients with ovarian cancer.
Chan, Q. K. et al. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G(2) cell-cycle arrest. Cell. Death Differ. 17, 1511–1523 (2010).
Natale, C. A. et al. Pharmacologic activation of the G protein-coupled estrogen receptor inhibits pancreatic ductal adenocarcinoma. Cell. Mol. Gastroenterol. Hepatol. 10, 868–880 (2020). This study reports that pharmacological activation of GPER enhances the efficacy of immune checkpoint inhibitors (ICIs) and prolongs survival in a model of pancreatic cancer.
Vivacqua, A. et al. 17β-Estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the G protein-coupled receptor GPR30. Mol. Pharmacol. 70, 1414–1423 (2006).
Gilligan, L. C. et al. Estrogen activation by steroid sulfatase increases colorectal cancer proliferation via GPER. J. Clin. Endocrinol. Metab. 102, 4435–4447 (2017).
Liu, C. et al. G-protein-coupled estrogen receptor antagonist G15 decreases estrogen-induced development of non-small cell lung cancer. Oncol. Res. 27, 283–292 (2019).
Scaling, A. L., Prossnitz, E. R. & Hathaway, H. J. GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast. Horm. Cancer 5, 146–160 (2014).
Vivacqua, A. et al. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17β-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol. Endocrinol. 20, 631–646 (2006).
Albanito, L. et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17β-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 67, 1859–1866 (2007).
Ariazi, E. A. et al. The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res. 70, 1184–1194 (2010).
Natale, C. A. et al. Activation of G protein-coupled estrogen receptor signaling inhibits melanoma and improves response to immune checkpoint blockade. eLife 7, e31770 (2018). This study reports that pharmacological activation of GPER enhances the efficacy of ICI and prolongs survival in a mouse model of malignant melanoma.
Wei, T. et al. G protein-coupled estrogen receptor deficiency accelerates liver tumorigenesis by enhancing inflammation and fibrosis. Cancer Lett. 382, 195–202 (2016).
McDonnell, D. P., Wardell, S. E., Chang, C. Y. & Norris, J. D. Next-generation endocrine therapies for breast cancer. J. Clin. Oncol. 39, 1383–1388 (2021).
Mo, Z. et al. GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Res. 15, R114 (2013).
Ignatov, A. et al. G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res. Treat. 128, 457–466 (2011).
Marjon, N. A., Hu, C., Hathaway, H. J. & Prossnitz, E. R. G protein-coupled estrogen receptor regulates mammary tumorigenesis and metastasis. Mol. Cancer Res. 12, 1644–1654 (2014).
Ignatov, A., Ignatov, T., Roessner, A., Costa, S. D. & Kalinski, T. Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res. Treat. 123, 87–96 (2010).
Prossnitz, E. R. & Arterburn, J. B. International Union of Basic and Clinical Pharmacology. XCVII. G protein-coupled estrogen receptor and its pharmacologic modulators. Pharmacol. Rev. 67, 505–540 (2015).
Li, Y. et al. 4-Hydroxytamoxifen-stimulated processing of cyclin E is mediated via G protein-coupled receptor 30 (GPR30) and accompanied by enhanced migration in MCF-7 breast cancer cells. Toxicology 309, 61–65 (2013).
Catalano, S. et al. Tamoxifen through GPER upregulates aromatase expression: a novel mechanism sustaining tamoxifen-resistant breast cancer cell growth. Breast Cancer Res. Treat. 146, 273–285 (2014).
Liu, Y. et al. G15 sensitizes epithelial breast cancer cells to doxorubicin by preventing epithelial-mesenchymal transition through inhibition of GPR30. Am. J. Transl Res. 7, 967–975 (2015).
Wolfson, B., Padget, M. R., Schlom, J. & Hodge, J. W. Exploiting off-target effects of estrogen deprivation to sensitize estrogen receptor negative breast cancer to immune killing. J. Immunother. Cancer 9, e002258 (2021).
Luo, H. et al. GPER-mediated proliferation and estradiol production in breast cancer-associated fibroblasts. Endocr. Relat. Cancer 21, 355–369 (2014).
Yang, K. & Yao, Y. Mechanism of GPER promoting proliferation, migration and invasion of triple-negative breast cancer cells through CAF. Am. J. Transl Res. 11, 5858–5868 (2019).
Ren, J. et al. GPER in CAFs regulates hypoxia-driven breast cancer invasion in a CTGF-dependent manner. Oncol. Rep. 33, 1929–1937 (2015).
Santolla, M. F. et al. GPER mediates a feedforward FGF2/FGFR1 paracrine activation coupling CAFs to cancer cells toward breast tumor progression. Cells 8, 223 (2019).
De Marco, P. et al. GPER signalling in both cancer-associated fibroblasts and breast cancer cells mediates a feedforward IL1β/IL1R1 response. Sci. Rep. 6, 24354 (2016). This study provides evidence for a role of GPER in inflammatory signalling in breast cancer in cancer-associated fibroblasts.
Divella, R., De Luca, R., Abbate, I., Naglieri, E. & Daniele, A. Obesity and cancer: the role of adipose tissue and adipo-cytokines-induced chronic inflammation. J. Cancer 7, 2346–2359 (2016).
De Francesco, E. M. et al. Protective role of GPER agonist G-1 on cardiotoxicity induced by doxorubicin. J. Cell Physiol. 232, 1640–1649 (2017).
Smalley, K. S. Why do women with melanoma do better than men? eLife 7, e33511 (2018).
Chakraborty, B. et al. Inhibition of estrogen signaling in myeloid cells increases tumor immunity in melanoma. J. Clin. Invest. 131, e151347 (2021).
Natale, C. A. et al. Sex steroids regulate skin pigmentation through nonclassical membrane-bound receptors. eLife 5, e15104 (2016).
Ribeiro, M. P. C., Santos, A. E. & Custodio, J. B. A. The activation of the G protein-coupled estrogen receptor (GPER) inhibits the proliferation of mouse melanoma K1735-M2 cells. Chem. Biol. Interact. 277, 176–184 (2017).
Shen, Y., Li, C., Zhou, L. & Huang, J. A. G protein-coupled oestrogen receptor promotes cell growth of non-small cell lung cancer cells via YAP1/QKI/circNOTCH1/m6A methylated NOTCH1 signalling. J. Cell. Mol. Med. 25, 284–296 (2021).
Lam, H. M. et al. Targeting GPR30 with G-1: a new therapeutic target for castration-resistant prostate cancer. Endocr. Relat. Cancer 21, 903–914 (2014).
Lee, S. J. et al. G protein-coupled estrogen receptor-1 agonist induces chemotherapeutic effect via ER stress signaling in gastric cancer. BMB Rep. 52, 647–652 (2019).
Zheng, S. et al. Screening and survival analysis of hub genes in gastric cancer based on bioinformatics. J. Comput. Biol. 26, 1316–1325 (2019).
Bustos, V. et al. GPER mediates differential effects of estrogen on colon cancer cell proliferation and migration under normoxic and hypoxic conditions. Oncotarget 8, 84258–84275 (2017).
Cortes, E. et al. GPER is a mechanoregulator of pancreatic stellate cells and the tumor microenvironment. EMBO Rep. 20, e46556 (2019).
Morelli, E. et al. Therapeutic activation of G protein-coupled estrogen receptor 1 in Waldenstrom macroglobulinemia. Exp. Hematol. Oncol. 11, 54 (2022).
Klein, S. L. & Flanagan, K. L. Sex differences in immune responses. Nat. Rev. Immunol. 16, 626–638 (2016).
Chakraborty, B. et al. Estrogen receptor signaling in the immune system. Endocr. Rev. 44, 117–141 (2023).
Tamaki, M. et al. Expression and functional roles of G-protein-coupled estrogen receptor (GPER) in human eosinophils. Immunol. Lett. 160, 72–78 (2014).
Itoga, M. et al. G-protein-coupled estrogen receptor agonist suppresses airway inflammation in a mouse model of asthma through IL-10. PLoS ONE 10, e0123210 (2015).
Brunsing, R. L., Owens, K. S. & Prossnitz, E. R. The G protein-coupled estrogen receptor (GPER) agonist G-1 expands the regulatory T-cell population under TH17-polarizing conditions. J. Immunother. 36, 190–196 (2013).
Brunsing, R. L. & Prossnitz, E. R. Induction of interleukin-10 in the T helper type 17 effector population by the G protein coupled estrogen receptor (GPER) agonist G-1. Immunology 134, 93–106 (2011).
Rettew, J. A., McCall, S. H. & Marriott, I. GPR30/GPER-1 mediates rapid decreases in TLR4 expression on murine macrophages. Mol. Cell. Endocrinol. 328, 87–92 (2010).
Rodenas, M. C. et al. G protein-coupled estrogen receptor 1 regulates human neutrophil functions. Biomed. Hub. 2, 1–13 (2017).
Yasuda, H. et al. 17-β-Estradiol enhances neutrophil extracellular trap formation by interaction with estrogen membrane receptor. Arch. Biochem. Biophys. 663, 64–70 (2019).
Castleman, M. J. et al. Innate sex bias of Staphylococcus aureus skin infection is driven by α-hemolysin. J. Immunol. 200, 657–668 (2018).
Triplett, K. D. et al. GPER activation protects against epithelial barrier disruption by Staphylococcus aureus α-toxin. Sci. Rep. 9, 1343 (2019).
Costa, A. J. et al. Overexpression of estrogen receptor GPER1 and G1 treatment reduces SARS-CoV-2 infection in BEAS-2B bronchial cells. Mol. Cell. Endocrinol. 558, 111775 (2022).
Wang, C. et al. Membrane estrogen receptor regulates experimental autoimmune encephalomyelitis through up-regulation of programmed death 1. J. Immunol. 182, 3294–3303 (2009). One of two studies to first suggest a role for GPER in multiple sclerosis.
Du, Z. R. et al. G protein-coupled estrogen receptor is involved in the anti-inflammatory effects of genistein in microglia. Phytomedicine 43, 11–20 (2018).
Guan, J., Yang, B., Fan, Y. & Zhang, J. GPER agonist G1 attenuates neuroinflammation and dopaminergic neurodegeneration in parkinson disease. Neuroimmunomodulation 24, 60–66 (2017).
Harding, A. T., Goff, M. A., Froggatt, H. M., Lim, J. K. & Heaton, N. S. GPER1 is required to protect fetal health from maternal inflammation. Science 371, 271–276 (2021). This study reveals a crucial immune regulatory role for GPER in protecting the fetus from maternal inflammation and infection.
Meyer, M. R., Fredette, N. C., Sharma, G., Barton, M. & Prossnitz, E. R. GPER is required for the age-dependent upregulation of the myocardial endothelin system. Life Sci. 159, 61–65 (2016).
Robison, L. S., Gannon, O. J., Salinero, A. E. & Zuloaga, K. L. Contributions of sex to cerebrovascular function and pathology. Brain Res. 1710, 43–60 (2019).
Patkar, S., Farr, T. D., Cooper, E., Dowell, F. J. & Carswell, H. V. Differential vasoactive effects of oestrogen, oestrogen receptor agonists and selective oestrogen receptor modulators in rat middle cerebral artery. Neurosci. Res. 71, 78–84 (2011).
Tang, H. et al. GPR30 mediates estrogen rapid signaling and neuroprotection. Mol. Cell. Endocrinol. 387, 52–58 (2014).
Murata, T., Dietrich, H. H., Xiang, C. & Dacey, R. G. Jr. G protein-coupled estrogen receptor agonist improves cerebral microvascular function after hypoxia/reoxygenation injury in male and female rats. Stroke 44, 779–785 (2013).
Zhang, Z. et al. The novel estrogenic receptor GPR30 alleviates ischemic injury by inhibiting TLR4-mediated microglial inflammation. J. Neuroinflammation 15, 206 (2018).
Han, Z. W. et al. GPER agonist G1 suppresses neuronal apoptosis mediated by endoplasmic reticulum stress after cerebral ischemia/reperfusion injury. Neural Regen. Res. 14, 1221–1229 (2019).
Bai, N. et al. G-protein-coupled estrogen receptor activation upregulates interleukin-1 receptor antagonist in the hippocampus after global cerebral ischemia: implications for neuronal self-defense. J. Neuroinflammation 17, 45 (2020).
Wang, X. S. et al. Activation of G protein-coupled receptor 30 protects neurons by regulating autophagy in astrocytes. Glia 68, 27–43 (2020).
Peng, J. et al. Activation of GPR30 with G1 attenuates neuronal apoptosis via src/EGFR/stat3 signaling pathway after subarachnoid hemorrhage in male rats. Exp. Neurol. 320, 113008 (2019).
Lu, D. et al. Activation of G protein-coupled estrogen receptor 1 (GPER-1) ameliorates blood-brain barrier permeability after global cerebral ischemia in ovariectomized rats. Biochem. Biophys. Res. Commun. 477, 209–214 (2016).
Zhang, B. et al. Estradiol and G1 reduce infarct size and improve immunosuppression after experimental stroke. J. Immunol. 184, 4087–4094 (2010). The first study to suggest protective effects of GPER activation in a preclinical model of ischaemic stroke.
Zheng, Y. et al. GPER-deficient rats exhibit lower serum corticosterone level and increased anxiety-like behavior. Neural Plast. 2020, 8866187 (2020).
Wang, J. et al. The antidepressant and anxiolytic effect of GPER on translocator protein (TSPO) via protein kinase a (PKA) signaling in menopausal female rats. J. Steroid Biochem. Mol. Biol. 207, 105807 (2021).
Bourque, M., Morissette, M. & Di Paolo, T. Neuroprotection in Parkinsonian-treated mice via estrogen receptor alpha activation requires G protein-coupled estrogen receptor 1. Neuropharmacology 95, 343–352 (2015).
Jiao, Y. et al. Molecular identification of bulbospinal on neurons by GPER which drives pain and morphine tolerance. J. Clin. Invest. 133, e154588 (2023).
Yuan, L. J. et al. G protein-coupled estrogen receptor is involved in the neuroprotective effect of IGF-1 against MPTP/MPP+-induced dopaminergic neuronal injury. J. Steroid Biochem. Mol. Biol. 192, 105384 (2019).
de Souza, L. O. et al. The G protein-coupled estrogen receptor (GPER) regulates recognition and aversively-motivated memory in male rats. Neurobiol. Learn. Mem. 184, 107499 (2021).
Kubota, T., Matsumoto, H. & Kirino, Y. Ameliorative effect of membrane-associated estrogen receptor G protein coupled receptor 30 activation on object recognition memory in mouse models of Alzheimer’s disease. J. Pharmacol. Sci. 131, 219–222 (2016).
Wang, Z. F., Pan, Z. Y., Xu, C. S. & Li, Z. Q. Activation of G-protein coupled estrogen receptor 1 improves early-onset cognitive impairment via PI3K/Akt pathway in rats with traumatic brain injury. Biochem. Biophys. Res. Commun. 482, 948–953 (2017).
Amirkhosravi, L. et al. E2-BSA and G1 exert neuroprotective effects and improve behavioral abnormalities following traumatic brain injury: the role of classic and non-classic estrogen receptors. Brain Res. 1750, 147168 (2021).
Confavreux, C., Hutchinson, M., Hours, M. M., Cortinovis-Tourniaire, P. & Moreau, T. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in multiple sclerosis group. N. Engl. J. Med. 339, 285–291 (1998).
Seifert, H. A. et al. Estrogen protects both sexes against EAE by promoting common regulatory cell subtypes independent of endogenous estrogen. Metab. Brain Dis. 32, 1747–1754 (2017).
Subramanian, S., Miller, L. M., Grafe, M. R., Vandenbark, A. A. & Offner, H. Contribution of GPR30 for 1,25 dihydroxyvitamin D(3) protection in EAE. Metab. Brain Dis. 27, 29–35 (2012).
Markee, J. E. Rhythmic uterine vascular changes. Am. J. Physiol. 100, 32–39 (1932).
Carloni, E. L’azione degli estratti ovarici sull’atteggiamento dei capillari e sulla loro pressione, nelle varie fasi della rivoluzione funziozionale utero-ovarica. Arch. Ostet. Ginecol. 17, 327i (1930).
Collin, J. B., Browne, J. S. & Thomson, D. L. The chemical nature of emmenin. Endocrinology 18, 71–74 (1934).
Dawson, R. F. & Robson, J. M. The pharmacological actions of diethylstilboestrol and other oestrogenic and non-oestrogenic substances. J. Physiol. 95, 420–430 (1939).
Reynolds, S. R. & Foster, F. I. Peripheral vascular action of estrogen in the human male. J. Clin. Invest. 18, 649–655 (1939).
Vance, D. A. Premarin: the intriguing history of a controversial drug. Int. J. Pharm. Compd. 1, 282–286 (2007).
Veurink, M., Koster, M. & Berg, L. T. The history of DES, lessons to be learned. Pharm. World Sci. 27, 139–43 (2005).
Pincus, G., Rock, J., Chang, M. C. & Garcia, C. R. Effects of certain 19-nor steroids on reproductive processes and fertility. Fed. Proc. 18, 1051–1056 (1959).
Lerner, L. J., Holthaus, F. J. & Thompson, C. R. A non-steroidal estrogen antagonist 1-(p-2-diethylaminoethoxyphenyl)-1-phenyl-2-p-methoxyphenyl ethanol. Endocrinology 63, 295–318 (1958).
Jordan, V. C. Tamoxifen: a most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2, 205–13 (2003).
Toft, D. & Gorski, J. A receptor molecule for estrogens: isolation from the rat uterus and preliminary characterization. Proc. Natl Acad. Sci. USA 55, 1574–1581 (1966).
Jensen, E. V. et al. A two-step mechanism for the interaction of estradiol with rat uterus. Proc. Natl Acad. Sci. USA 59, 632–638 (1968).
Szego, C. M. & Davis, J. S. Adenosine 3’,5’-monophosphate in rat uterus: acute elevation by estrogen. Proc. Natl Acad. Sci. USA 58, 1711–1718 (1967).
Pietras, R. J. & Szego, C. M. Endometrial cell calcium and oestrogen action. Nature 253, 357–359 (1975).
Jordan, V. C. & Dowse, L. J. Tamoxifen as an anti-tumour agent: effect on oestrogen binding. J. Endocrinol. 68, 297–303 (1976).
Pietras, R. J. & Szego, C. M. Estrogen receptors in uterine plasma membrane. J. Steroid Biochem. 11, 1471–1483 (1979).
Owman, C., Blay, P., Nilsson, C. & Lolait, S. J. Cloning of human cDNA encoding a novel heptahelix receptor expressed in Burkitt’s lymphoma and widely distributed in brain and peripheral tissues. Biochem. Biophys. Res. Commun. 228, 285–292 (1996).
Carmeci, C., Thompson, D. A., Ring, H. Z., Francke, U. & Weigel, R. J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 45, 607–617 (1997).
Feng, Y. & Gregor, P. Cloning of a novel member of the G protein-coupled receptor family related to peptide receptors. Biochem. Biophys. Res. Commun. 231, 651–654 (1997).
Takada, Y., Kato, C., Kondo, S., Korenaga, R. & Ando, J. Cloning of cDNAs encoding G protein-coupled receptor expressed in human endothelial cells exposed to fluid shear stress. Biochem. Biophys. Res. Commun. 240, 737–741 (1997).
Kvingedal, A. M. & Smeland, E. B. A novel putative G-protein-coupled receptor expressed in lung, heart and lymphoid tissue. FEBS Lett. 407, 59–62 (1997).
O’Dowd, B. F. et al. Discovery of three novel G-protein-coupled receptor genes. Genomics 47, 310–313 (1998).
Bonini, J. A., Anderson, S. M. & Steiner, D. F. Molecular cloning and tissue expression of a novel orphan G protein-coupled receptor from rat lung. Biochem. Biophys. Res. Commun. 234, 190–193 (1997).
Acknowledgements
E.R.P. is supported by grants from the US National Institutes of Health (R01 CA163890 and R01 CA194496), from Dialysis Clinic, Inc., and by the UNM Comprehensive Cancer Center (NIH P30 CA118100) and the Autophagy, Inflammation and Metabolism (AIM) Center of Biomedical Research Excellence (CoBRE, NIH P20 GM121176). M.B. is supported by grants 108 258 and 122 504 from the Swiss National Science Foundation (SNSF).
Author information
Authors and Affiliations
Contributions
Both authors contributed equally to all aspects of this manuscript.
Corresponding authors
Ethics declarations
Competing interests
M.B. and E.R.P. are inventors on U.S. patent Nos. 10,251,870, 10,682,341 and 10,980,785, and E.R.P. is an inventor on U.S. Patent Nos. 10,471,047 and 10,561,648, all for the therapeutic use of compounds targeting GPER (“Method for treating obesity, diabetes, cardiovascular and kidney diseases by regulating GPR30/GPER”). E.R.P. is an inventor on U.S. Patent Nos. 7,875,721 and 8,487,100 for GPER-selective ligands and imaging agents (“Compounds for binding to ERα/β and GPR30, methods of treating disease states and conditions mediated through these receptors and identification thereof”). M.B. has served or serves as a consultant to Abbott, Inc., Abbvie, Inc., Travere, Inc. and Pharmazz, Inc.
Peer review
Peer review information
Nature Reviews Endocrinology thanks Guichun Han and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Review criteria
Abstracts of all articles published on GPR30 or GPER, published between January 1996 and February 2023, were retrieved from the U.S. National Library of Medicine (PubMed.gov). Articles were assessed for relevance, importance and scientific rigour, with a focus on publication in the past 10 years. The authors apologize to their colleagues whose work could not be included due to space and reference restrictions.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Prossnitz, E.R., Barton, M. The G protein-coupled oestrogen receptor GPER in health and disease: an update. Nat Rev Endocrinol 19, 407–424 (2023). https://doi.org/10.1038/s41574-023-00822-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41574-023-00822-7
