
Abstract
One hundred years have passed since Warburg discovered alterations in cancer metabolism, more than 70 years since Sidney Farber introduced anti-folates that transformed the treatment of childhood leukaemia, and 20 years since metabolism was linked to oncogenes. However, progress in targeting cancer metabolism therapeutically in the past decade has been limited. Only a few metabolism-based drugs for cancer have been successfully developed, some of which are in — or en route to — clinical trials. Strategies for targeting the intrinsic metabolism of cancer cells often did not account for the metabolism of non-cancer stromal and immune cells, which have pivotal roles in tumour progression and maintenance. By considering immune cell metabolism and the clinical manifestations of inborn errors of metabolism, it may be possible to isolate undesirable off-tumour, on-target effects of metabolic drugs during their development. Hence, the conceptual framework for drug design must consider the metabolic vulnerabilities of non-cancer cells in the tumour immune microenvironment, as well as those of cancer cells. In this Review, we cover the recent developments, notable milestones and setbacks in targeting cancer metabolism, and discuss the way forward for the field.
Similar content being viewed by others
Introduction
Before Sidney Farber published his seminal paper in 1948 in The New England Journal of Medicine describing anti-folate-induced remissions1, childhood acute lymphocytic leukaemia (ALL) was universally fatal. Thirty years before this article was published, Otto Warburg’s determination to conquer cancer led him to discover that many tumours used aerobic glycolysis (also known as the Warburg effect) to convert almost all glucose to lactate even in the presence of oxygen2. Aerobic glycolysis, however, has never been successfully exploited clinically, particularly as the use of 2-deoxyglucose, which inhibits glycolysis, was proved to have undesirable side effects and limited efficacy in humans3. Farber noted the clinical ‘accelerated phenomenon’ among 11 children treated with an active form of folate, pteroyltriglutamic acid, which was thought to have broad anticancer activity, and thus sought folate antagonists as therapeutic agents. Working with chemist Yellapragada Subbarow, the anti-folate aminopterin was synthesized, and Farber was able to induce remissions in children with ALL, thus providing the foundation for cancer chemotherapy4. Today, another folate antagonist, methotrexate, is still used in the multi-chemotherapy treatment of childhood ALL and, used together with l-asparaginase, induces a remarkable 90% cure rate. Indeed, many anti-metabolite drugs, particularly those targeting nucleotide metabolism, have been approved and used in the clinic (Table 1). The recent approval of drugs that target mutant isocitrate dehydrogenases in acute myeloid leukaemias (AMLs) (Table 1) provides a milestone in targeting cancer metabolism with precision and establishes that metabolic therapy can be highly effective.
Warburg’s and Farber’s ideas were conceptually displaced in the 1980s as oncogenes and tumour suppressors became recognized as targets in human cancer treatments. Molecular biology took centre stage, and the drive to target oncogenes began with vigour, resulting in many effective anticancer kinase drugs that displaced interest in targeting metabolism. However, the links between oncogenes, tumour suppressors and metabolism began to emerge in the 1990s, ushering in a resurgence of interest in cancer metabolism5,6,7. More recently, the success of immunotherapy underscores the importance of non-cancer-cell autonomous components of the tumour immune microenvironment (TIME)8,9,10, which is a neoplastic hub of metabolically active cells comprising tumour cells, immune cells, stromal cells and blood and lymph vessel cells, all of which are involved in tumour growth. In this regard, targeting cancer metabolism must be based on a thorough understanding of how inhibiting specific metabolic pathways affects the TIME cells, which can either dampen or promote tumour progression. In this Review, we cover the fundamentals of cancer metabolism and focus on recent small-molecule drug discovery efforts to target cancer.
Metabolism: concepts and vulnerabilities
Glucose, amino acid and fatty acid metabolism
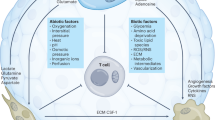
Glucose provides the major source of energy and carbon skeleton for biosynthesis (Figs 1,2). Hence, insulin and glucagon have evolved to regulate glucose levels during feeding and fasting states. To maintain cell homeostasis, glucose is metabolized to pyruvate via glycolysis and can then be imported into the mitochondrion via mitochondrial pyruvate carriers11. Mitochondrial pyruvate carriers are thought to be tumour suppressive, as documented in several models12,13, emphasizing the role of glycolysis in tumorigenesis and suggesting that mitochondrial pyruvate carriers might not be ideal cancer targets for developing inhibitors. Mitochondrial pyruvate dehydrogenase converts pyruvate to acetyl-Coenzyme A (acetyl-CoA), which is further oxidized through the tricarboxylic acid (TCA) cycle, culminating in oxaloacetate, which joins the newly synthesized acetyl-CoA to produce citrate, which replenishes the metabolic cycle. Carbon skeletons and ATP are generated by glycolysis and mitochondrial oxidative metabolism, providing energy and building blocks to maintain cellular integrity (Figs 1,2).

The deregulated tyrosine kinase receptors activate RAS or PI3K, MYC and/or mTOR. mTORC1 increases protein synthesis from MYC-induced mRNAs to enhance influx of amino acids (AAs, including glutamine), glucose and fatty acids to drive fatty acid, nucleotide and protein synthesis. Hypoxia-inducible factors HIF1α and HIF2α are induced by mTORC1 or hypoxia, resulting in activation of the glycolytic pathway with the conversion of glucose to lactate, which is exported extracellularly. MYC transactivates genes involved in glycolysis and shunting of 3-phosphoglycerate to produce serine, which is converted to glycine and contributes to one-carbon (1C) metabolism. MYC also induces glutamine metabolism, enhancing glutamine and glucose uptake and their catabolism in the tricarboxylic acid (TCA) cycle, providing scaffolds for fatty acids and nucleotide synthesis. Oxygen is shown to assist the mitochondrial electron transport chain (ETC, with complexes I–V shown) or inhibit HIF1α and HIF2α by mediating its proteasomal degradation. aKG, α-ketoglutarate; ARNT, aryl hydrocarbon receptor nuclear translocator; MAX, MYC-associated factor X; OAA, oxaloacetate; Suc-CoA, succinyl-Coenzyme A.

Glucose is taken up into the cell by glucose transporters (GLUT) and phosphorylated by hexokinases HK1 and HK2. Glucose 6-phosphate (P) and its downstream intermediates can either be converted to pyruvate or fuel biosynthesis through the hexosamine biosynthesis pathway, the pentose phosphate pathway (PPP), via glycerol 3-P production or via serine biosynthesis pathways. Hexosamine has a key role in glycosylation. The PPP provides ribose 5-P for nucleotide synthesis and NADPH. Glycerol 3-P provides the backbone for lipid synthesis. Serine biosynthesis has a key part in amino acid metabolism and nucleotide metabolism through control of one-carbon (1C) metabolism (see Fig. 5). Pyruvate can either enter the tricarboxylic acid (TCA) cycle as acetyl-CoA through the mitochondrial pyruvate carrier (MPC) and pyruvate dehydrogenase, be converted to lactate by lactate dehydrogenase (LDH) and exported through the monocarboxylate transporter (MCT) or be converted to alanine via glutamic–pyruvic transaminase (GPT). Inhibitors (blue) and activators (purple) of key enzymes (red) or transporters (white) are shown. In addition to producing energy by contributing reducing equivalents to the electron transport chain, the TCA cycle is an important source of aspartate for nucleotide metabolism (see Fig. 5). The conversion of pyruvate to lactate has a key role in maintaining cellular NAD+ to NADH ratios. aKG, α-ketoglutarate; OAA, oxaloacetate; PDH, pyruvate dehydrogenase; PHGDH, phosphoglycerate dehydrogenase; PKM2, pyruvate kinase muscle isozyme 2; SHMT, serine hydroxymethyl transferase.
Similarly, amino acids and fatty acids are taken up by normal cells to maintain structure and function (Fig. 1). Of the amino acids, histidine, lysine, methionine, phenylalanine, threonine, tryptophan and branched-chain amino acids (isoleucine, leucine and valine) are essential and cannot be synthesized de novo in humans. These dietary amino acids and non-essential amino acids, such as glutamine (Fig. 3), serine and glycine, are imported into cells by various transporters and used for one-carbon (1C) metabolism, nucleic acid and protein synthesis. Branched-chain amino acids can be metabolized as fuel sources via the mitochondrial branched-chain α-ketoacid dehydrogenase complex, which converts these amino acids into ketoacids, generating succinyl-CoA and acetyl-CoA for oxidation by the TCA cycle14. The TCA cycle intermediate oxaloacetate can be transaminated to produce the pivotal amino acid aspartate, which also accumulates as a result of loss of arginosuccinate synthetase (ASS1), which consumes aspartate to produce arginosuccinate from citrulline15. Aspartate accumulation supports cell proliferation through nucleotide and asparagine synthesis16,17,18,19.

Glutamine acts as a key carbon and nitrogen donor for biosynthesis. Glutamine is taken up by transporters, including alanine–serine–cysteine transporter 2 (ASCT2, also known as SLC1A5) and can be exported or imported through large neutral amino acid transporter 1 (LAT1, also known as SLC7A5) in exchange for branched-chain amino acids (BCAAs). Following its production from glutamine by glutaminase enzymes (GLS1 and GLS2), glutamate can be converted to α-ketoglutarate (aKG), be exported in exchange for cystine by the xCT transporter, or fuel proline biosynthesis. Glutamate can be converted to aKG by either glutamate dehydrogenase (GDH) or aminotransferases. The glutamic-oxaloacetic transaminase (GOT) produces aspartate, which has a key part in nucleotide synthesis. Glutamine can act as a nitrogen donor for the synthesis of asparagine, hexosamine, purine and pyrimidine. Aminotransferases use glutamate as a nitrogen donor for the synthesis of alanine, serine and aspartate. Glutamate, cystine-derived cysteine and serine-derived glycine contribute to glutathione biosynthesis, which has a key role in modulating oxidative stress. Inhibitors of key enzymes or transporters are shown (blue). ASNS, asparagine synthetase; CAD, carbamoyl-phosphate synthetase 2, aspartate transcarbamylase and dihydroorotase; CTPS, CTP synthase; GFAT, fructose 6-phosphate transaminase (GFPT1, GFPT2); GMPS, guanine monophosphate synthase; GPT, glutamic–pyruvic transaminase; OAA, oxaloacetate; PFAS, phosphoribosylformylglycinamidine synthase; PPAT, phosphoribosyl pyrophosphate amidotransferase; PSAT1, phosphoserine aminotransferase 1.
Dietary fatty acids and cholesterol are packaged by the liver and delivered to peripheral tissues for uptake, storage or metabolism. In particular, fatty acids are imported into cells and bound to fatty acid-binding proteins and stored in droplets or delivered to the mitochondrion or peroxisomes for oxidation (Figs 1,4). Fatty acid oxidation provides a major source of energy because complete oxidation of one 16-carbon fatty acid could generate 129 ATP molecules compared with the 38 generated from one molecule of glucose. Fatty acyl-CoAs are converted to fatty acyl-carnitines by mitochondrial membrane-bound carnitine palmitoyltransferase 1 (CPT1) and transported into the mitochondrion by the carnitine–acylcarnitine translocase. Fatty acyl-carnitine is then reconverted to fatty acyl-CoA by CPT2 and then oxidized into acetyl-CoA, which enters the TCA cycle for further catabolism (Fig. 4).

Fatty acid synthesis can be used along with exogenous fatty acids for lipid production, which can be used to produce membranes and lipid droplets and to modulate signalling pathways. Mitochondrial acetyl-CoA derived from fatty acid oxidation (which can be an important tricarboxylic acid (TCA) cycle carbon source in some cancers), glucose or other sources can condense with oxaloacetate (OAA) to form citrate, which can then be exported from the mitochondrion. Citrate and acetate — via ATP citrate lyase (ACLY) and synthetase short-chain family member 2 (ACSS2), respectively — are two major sources of cytoplasmic acetyl-CoA, from which acetyl-CoA carboxylase (ACC1 and ACC2) makes malonyl-CoA, which is then cyclically extended by the addition of carbons from acetyl-CoA by fatty acid synthase (FASN) to make saturated fatty acids. Fatty acids are desaturated by stearoyl-CoA desaturase 1 (SCD1). Membrane desaturation has a key role in maintaining membrane fluidity. Fatty acid catabolism is initiated with the formation of fatty acyl-CoA by an acyl-CoA ligase. Fatty acyl-CoA is converted by carnitine palmitoyltransferase 1 (CPT1) to an acylcarnitine, which is then imported into the mitochondrion via carnitine–acylcarnitine translocase (CAT, SLC25A20). Inside the mitochondrion, CPT2 converts the acyl-carnitine back to a fatty acyl-CoA, which can then be cyclically oxidized to two carbon acetyl-CoA molecules. Inhibitors of key enzymes or transporters are shown (blue). aKG, α-ketoglutarate; CoA, Coenzyme A; CTP, citrate transporter protein (SLC25A1); PDH, pyruvate dehydrogenase.
Nucleotide synthesis
Cancer cell growth and proliferation depends on de novo nucleotide synthesis from intermediates in the TCA cycle, glucose-derived ribose sugars from the pentose phosphate pathway (PPP), and amino acids that generate purines and pyrimidine nucleotides. In particular, 1C metabolism, which was targeted by Sidney Farber, is pivotal for nucleic acid synthesis. Folate as tetrahydrofolate (THF) is essential and serves as the 1C carrier, with three carbon oxidation states20. Methylene-THF (5,10-CH2-THF) participates in the production of dTMP from dUMP by thymidylate synthase (TYMS), which is inhibited by fluorodeoxyuridine monophosphate (FdUMP), a product the chemotherapeutic agent 5-fluorouracil. De novo pyrimidine synthesis consists of six steps (Fig. 5) with the notable involvement of mitochondrial dihydroorotate dehydrogenase (DHODH), which requires a functional electron transport chain (ETC) for ubiquinone-mediated oxidation of dihydroorotate to orotate. Formyl-THF (10-CHO-THF) provides 1C units and participates in the 11-step de novo synthesis of purines. In de novo purine synthesis, carbon and nitrogen atoms from glycine, glutamine, aspartate and formyl groups from the folate carrier are sequentially added onto ribose derived from glucose (Fig. 5). De novo syntheses of pyrimidines and purines are essential for mRNA synthesis and DNA replication of growing and proliferating cancer cells (Fig. 5). Therefore, these pathways contain multiple potential therapeutic targets. As normal proliferative tissues in humans also depend on these pathways, bone marrow suppression, gastrointestinal mucositis and hair loss are common clinical manifestations of chemotherapies that target nucleotide synthesis.

a | One-carbon (1C) metabolism. Nucleotide synthesis uses ribose 5-P produced by the pentose phosphate pathway (PPP) and aspartate produced from oxaloacetate, as well as glycine and tetrahydrofolate methyl donors — methylene-THF (5,10-CH-THF) and formyl-THF (10-CHO-THF) — arising from 1C metabolism linked to the conversion of serine to glycine. 1C metabolism occurs in both the cytoplasm and mitochondria. b | Purine synthesis is a multistep, multienzyme pathway that uses ribose 5-P, glutamine (Gln), glycine (Gly), aspartate (Asp) and 10-CHO-THF to make inosine monophosphate (IMP). IMP is converted to ADP and GDP, which can then be converted to dADP and dGDP. c | Pyrimidine synthesis is a multistep process that uses phosphoribosyl pyrophosphate (PRPP) as a scaffold to produce UDP from glutamine, carbonate and aspartate. Ribonucleotide reductase (RNR) converts UDP to dUDP and CDP to dCDP. Thymidylate synthase (TYMS), which can be inhibited by the clinical agents pemetrexed or 5-fluorouracil (5-FU), converts dUMP to dTMP. CoQH2, reduced Coenzyme Q; DHF, dihydrofolate; DHODH, dihydroorotate dehydrogenase; ETC, electron transport chain; GCS, glycine cleavage system; HCO3, bicarbonate; IMPDH, inosine monophosphate dehydrogenase; MTHFD1, methylenetetrahydrofolate dehydrogenase 1; MTHFD1L, methylenetetrahydrofolate dehydrogenase 1-like; MTHFD2, methylenetetrahydrofolate dehydrogenase 2; MTX, methotrexate; PHGDH, phosphoglycerate dehydrogenase; PPAT, phosphoribosyl pyrophosphate amidotransferase; RNR, ribonucleotide reductase; SHMT1, serine hydroxymethyl transferase 1.
Driven by cellular receptors that trigger the RAS–RAF–MEK–ERK and PI3K–AKT–mTOR signalling transduction pathways (Fig. 1), proliferating cells import nutrients and amino acids that stimulate the mammalian target of rapamycin complex 1 (mTORC1) and activate transcriptional reprogramming by inducing MYC and other transcription factors21,22. MYC accelerates the expression of many ‘housekeeping’ genes, which are largely metabolic, mitochondrial and ribosomal genes, permitting rapid transcriptional amplification of growth signalling. New transcripts, particularly those encoding nutrient transporters, are translated to increase the nutrient importation required for cell growth and proliferation. Imported amino acids further stimulate mTORC1, which promotes protein synthesis and ribosomal biogenesis, providing translational amplification of growth signalling (Fig. 1). Conceptually, normal proliferating cells can be distinguished from neoplastic cells by the ability of normal cells to sense nutrient deficiency and stop proliferating, whereas neoplastic cells with deregulated growth driven by oncogenes are constitutively dependent on nutrients. In this regard, deprivation of nutrients could trigger cell death23,24 in neoplastic cells, whereas normal cells could retreat into the G0/G1 cell cycle state.
Cancer cell metabolic vulnerabilities
Although oncogenic activation of tumour metabolism renders cancer cells potentially vulnerable to inhibition of different metabolic pathways, these pathways are also used by immune cells25, particularly upon receptor stimulation, such as T cell receptor (TCR) engagement together with CD28 activation. In this regard, precise inhibition of specific nutrient transporters or enzymes may derail the accurate targeting of tumour cells versus antitumour cells in the TIME. In fact, metabolic inhibition of antitumour immune cells such as cytotoxic T lymphocytes (CTL) and natural killer (NK) cells could counteract any desirable inhibition of cancer cell survival. Hence, it is of paramount importance that targeting metabolism for cancer therapy should be studied in the context of a normal immune system, particularly for solid tumours, such as pancreatic cancer tumours that can contain more non-cancer cells than cancer cells. By contrast, haematological tumours, particularly acute leukaemic cells that depend on metabolism for rapid proliferation, could be more vulnerable to metabolic inhibition, as seen with induction leukaemic therapy using l-asparaginase and methotrexate.
Cancer cell metabolic vulnerabilities have been uncovered using a candidate approach, including targeting of glycolysis, aspartate, glutamine or fatty acid metabolism, or conducting genetic synthetic lethality screens using small interfering RNAs (siRNAS), short hairpin RNAs (shRNAs) or CRISPR–Cas9. Not surprisingly, there are substantial cell type-specific vulnerabilities uncovered by CRISPR–Cas9 screening depending on the cell of origin26. One study integrating metabolomics and CRISPR–Cas9 screening uncovered asparagine synthetase (ASNS) as a vulnerability in solid tumour cell lines, such as gastric and liver cancer cell lines, that have lower levels of ASNS, suggesting that the therapeutic use of l-asparaginase could be expanded beyond its use in ALL to solid tumours26. Specific oncogenic mutations may also confer selective metabolic vulnerability. In lung adenocarcinoma, mutations in Kelch-like ECH-associated protein 1 (KEAP1), which constitutively activates the transcription factor nuclear factor erythroid 2-related factor 2 (NFE2L2, also known as NRF2), induce the expression of the cystine/glutamate antiporter xCT (encoded by SLC7A11), which exports glutamate in exchange for cysteine, which is required for glutathione synthesis. KEAP1-mutant cells also depend on glutamine, which is used to generate glutathione, further rendering them vulnerable to glutaminase inhibition27. Loss of von Hippel–Lindau (pVHL) function in renal cell carcinoma (RCC) activates the hypoxia inducible factor 1 (HIF1α), making these cells vulnerable to inhibition of glycolysis28. Further, KRAS activation in a mouse model of lung cancer result in different metabolic dependencies on branched-chain amino acids than in pancreatic cancer29. Moreover, MYC-driven transgenic murine lung cancer cells express glutamine synthetase, whereas MYC-driven murine liver cancer cells do not30, underscoring the importance of cell type in determining the specific metabolic dependencies of different cancers. It is also notable that dietary influence on tumorigenicity has been documented in different mouse tumour models31,32,33,34, highlighting the role of diet, which affects nutrient availability (serine and glycine, for example), in the sensitivity of cancers to metabolic therapy.
Metabolism-based drug development
A key consideration for targeting metabolism is drug specificity. Specifically, medicinal chemistry, coupled with structural biology, can be used to generate highly specific drugs whose interactions with their targets are defined by crystallography. There are several highly specific metabolic inhibitors in this category, including those acting on catalytic and allosteric sites. A key challenge to targeting active sites is the prevalence of hydrophobic pockets in metabolic enzymes, and, as such, allosteric inhibitors provide additional opportunities with perhaps better specificity. One example is the development of active site inhibitors of lactate dehydrogenase (LDH), with the latest compounds having nanomolar potency and in vivo efficacy in tumour models35. Another notable example is the development of allosteric glutaminase (GLS) inhibitors from the tool compound BPTES to the clinically tested CB-839, which has been co-crystallized with GLS1 (refs36,37). The CB-839 compound is specific for GLS1 and spares GLS2, which is encoded by a different gene. In contrast to GLS1-specific inhibition, the glutamine mimetic drug 6-diazo-5-oxo-l-norleucine (DON) binds covalently to multiple enzymes that use glutamine38. A prodrug of DON, although lacking single-enzyme precision, has remarkable antitumour activity owing to its ability to inactivate multiple metabolic targets and therefore blunt the ability of cancer cells to rewire metabolic pathways39. This ability of cancer cells to rewire metabolic pathways may pose a challenge to precision therapies owing to development of resistance. Thus, combination therapies or therapies that block multiple pathways may have advantages over a single-agent therapy40.
Although generating metabolic inhibitors with half-maximal inhibitory concentrations (IC50 values) in the nanomolar range is paramount for drug development, the intracellular concentrations of enzymes could pose a problem for effective dosage. For example, lactate dehydrogenase A (LDHA) has an estimated intracellular concentration of 4 μM compared with 0.27 μM for hexokinase 2 (HK2) or 0.07 μM for GLS1 in HeLa cells41. High cellular concentration will require sufficiently high drug concentrations to ensure nearly complete target neutralization, posing additional pharmacokinetics issues. As such, targeted protein degradation through proteolysis targeting chimaera (PROTAC) technology42 — which tethers small-molecule binders to warheads that target proteins for degradation via the ubiquitin-proteasome system — could mitigate this problem. However, there are currently few examples of proof-of-concept PROTAC metabolic drugs. Notably, degraders have been designed to target the solute carrier transmembrane (SLC) transporters, opening the opportunity to study the functions of these transporters43. A DHODH PROTAC has been reported44, but it was incapable of degrading its target protein.
As cancer metabolism has been a burgeoning field over the past 20 years, many metabolic inhibitors have been touted in the literature, despite a lack of evidence for specificity or proper pharmacokinetic or pharmacodynamic markers of target engagement. Notwithstanding the use of tool compounds for proof-of-concept studies in targeting metabolism, rigorous development of drugs will require a multidisciplinary development paradigm. Therefore, it is critically important that the advancement to the clinic of specific inhibitors of enzymes or transporters should be accompanied by robust medicinal chemistry, structural biology and proper pharmacokinetics and pharmacodynamics studies. Imaging technologies based on metabolic tracers can be highly useful for monitoring drug activity in vivo. For example, the on-target effect of an LDH inhibitor was documented by in vivo hyperpolarized 13C-pyruvate tumour imaging45, the in vivo effects of the glutaminase inhibitor CB-839 were established by labelled 18F-glutamine positron emission tomography (PET)46, and the effect of loss of acetyl-CoA synthetase short-chain family member 2 (ACSS2), which generates acetyl-CoA from acetate, was confirmed by 11C-acetate PET47.
Lessons from inborn errors of metabolism
Knowledge of inherited human disorders of metabolism could provide valuable information regarding therapeutic windows and the potential side effects of targeting specific metabolic pathways with drugs (Box 1). For example, glucose 6-phosphate dehydrogenase deficiency is globally prevalent and is associated with having a normal life but can be complicated by drug-induced haemolysis. The phenotypes of genetically modified mouse strains may also reveal potential side effects of targeting specific metabolic enzymes.
The dependency of some tumours on aerobic glycolysis suggests that glucose transporters and glycolytic enzymes could be therapeutic targets, particularly as oncogenic transformation by RAS, SRC kinase48 or MYC enhances glucose uptake49. As such, specific hereditary deficiencies resulting from mutations in components of the glycolytic pathway could phenocopy the effect of drugs that potentially inhibit glycolysis. Mutations in glycolysis and its clinical manifestations have been documented (Box 1) with haemolytic anaemia being the dominant clinical presentation50. Intriguingly, a specific inhibitor of LDH triggers haemolysis in mice, phenocopying acute LDHA genetic knockout51, whereas human mutations in LDHA or LDHB are associated with exertional muscle fatigue without haemolysis52,53. However, a mouse mutation in Ldha is associated with chronic anaemia54. Hence, species-specific differences may complicate assessment of the side effects of drugs targeting glycolysis in humans from preclinical studies in mice.
Glucose-derived pyruvate can be further converted to lactate by LDH, or to alanine by glutamic–pyruvic transaminase (GPT), or can enter the mitochondrion for further catabolism by the TCA cycle (Fig. 2). Mutations in TCA-cycle components, particularly the pyruvate dehydrogenase (PDH) complex, are associated with congenital lactic acidosis (see the Online Mendelian Inheritance in Man (OMIM) database in Related links), indicating that pyruvate diverted from mitochondria is shunted towards lactic acid production. Other mutations of TCA-cycle enzymes are associated with brain developmental disorders or, in the case of succinate dehydrogenase, with familial paraganglioma or leiomyoma (see the OMIM).
Similar to glucose deprivation, glutamine deprivation in MYC-transformed cells triggers apoptosis24, indicating that oncogenic transformation could confer vulnerabilities to the inhibition of glutamine metabolism pathways (Fig. 3). Indeed, inhibitors of glutamine transporters or glutaminases are documented to have in vivo activity in mouse models of cancers (see Glutamine metabolism), and side effects of these drugs seem to be minimal. Glutamine is taken up into cells through seven transporters that are expressed in a tissue-specific pattern with alanine–serine–cysteine transporter 2 (ASCT2, encoded by SLC1A5) and large neutral amino acids transporter small subunit 1 (LAT1) being most highly implicated in tumorigenesis. Mutations in sodium-dependent neutral amino acid transporter (B(0)AT1) are associated with Hartnup disorder, which comprises pellagra and progressive neurological disorders(see the OMIM). Mutations in other enzymes involved in glutamine metabolism have been documented (Box 1), many of which are associated with developmental abnormalities affecting the central nervous system, in which glutamate–glutamine cycling is essential for neurotransmission (see the OMIM). Thus, glutamine metabolic drugs that penetrate the central nervous system may have neurological effects.
In addition to fatty acids and cholesterol influx from the circulating blood, de novo fatty acid synthesis (Fig. 4) seems to be essential for cancer cell proliferation, because inhibition of components of the fatty acid synthesis pathway can curb tumorigenesis in mouse models of cancer (see Fatty acid synthesis). The effects of inborn errors in the fatty acid synthesis pathway (ATP citrate lyase (ACLY), acetyl-CoA carboxylases (ACC1 and ACC2, encoded by ACACA and ACACB, respectively), fatty acid synthase (FASN) or stearoyl-CoA desaturase (SCD1) have not been documented; however, we note that loss of murine acetyl-CoA carboxylase 1 (Acaca) is embryonic lethal, whereas loss of ACC2 (Acacb) is tolerated55. Fasn heterozygosity causes death at various developmental stages; by contrast, Fasn-null animals die before implantation56. Genetic ablation of Scd1 in mice is associated with atrophy of sebaceous and Meibomian glands leading to skin and eyelid abnormalities57. Hence, targeting fatty acid metabolism in humans could result in similar sebaceous gland disorders and/or dry eyes from dysfunctional Meibomian glands, and possibly other toxicities.
Targeting nucleotide synthesis (Fig. 5), which is required for RNA and DNA replication, is primarily associated with side effects in highly proliferative tissues, including bone marrow suppression and gastrointestinal mucosal disruption. Human mutations in folate, pyrimidine and purine synthesis pathways are largely associated with neurodevelopmental defects, perhaps reflecting rapid growth during fetal development in which nucleotide synthesis is essential, and hence the need for adequate prenatal folate supplementation.
The mitochondrion is perhaps the most challenging of targets. Its pivotal role as a metabolic and biosynthetic organelle makes in an attractive anticancer target, but this approach thus far has been limited by toxicities predicted by inborn errors of mitochondria. Specifically, the targeting of the ETC is likely to be fraught with severe side effects, particularly as some of the ETC inhibitors are lethally toxic, including cyanide, which inhibits cytochrome a3 and mitochondrial respiration. The biguanide metformin, which is clinically used for insulin-resistant diabetes, is thought to inhibit mitochondrial complex I. A unique feature of metformin is that is imported into the mitochondrion in a membrane potential-dependent manner58. As such, after import and inhibition of complex I, the mitochondrial membrane potential diminishes, leading to decreased metformin importation, thereby creating a rheostat that prevents toxicity while gaining therapeutic benefits. Based on this property of metformin, additional complex I inhibitors have been sought in cancer treatment. In particular, the compound IACS-010759 has been advanced to clinical trials59, but is associated with neuropathy and visual changes. In this regard, it is notable that many inherited mitochondrial DNA mutations are associated with hyperlactaemia and diseases afflicting tissues that are highly dependent on mitochondrial function, resulting in encephalopathy, myopathy and optic atrophy.
Metabolic targets
In this section we will focus on small-molecule inhibitors that have been substantially characterized and reported to target cancer metabolism, highlighting on-target versus off-target effects and in vivo efficacy as a stepping stone towards the clinic (Table 1).
Aerobic glycolysis
Inhibition of glucose uptake is a therapeutic opportunity and has been investigated in several studies (Fig. 2). STF-31, a small-molecule inhibitor60 of glucose transporter 1 (GLUT1), had features of a glucose transporter inhibitor and exhibited efficacy against RCC tumour xenografts in vivo; however, this compound has off-target effects. Specifically, STF-31 also inhibits nicotinamide phosphoribosyltransferase (NAMPT)61, and adding nicotinic acid or expressing a drug-resistant NAMPT mutant conferred resistance to STF-31, suggesting that GLUT1 inhibition is not the only mechanism by which STF-31 inhibits tumour growth. Another GLUT inhibitor, Glutor, was identified through screening for inhibitors of 2-deoxyglucose uptake62. Glutor targets GLUT1, GLUT2 and GLUT3 to inhibit glycolytic flux, and overexpression of these glucose transporters led to higher IC50 values. BAY-876 (ref.63) was identified by screening for compounds that reduced ATP production in GLUT1+ DLD1 cells from colorectal adenocarcinoma versus GLUT1− DLD1 cells. Structure–activity relationship studies were used in developing BAY-876, which has nanomolar values of IC50 for GLUT1 but is 100-fold less active against GLUT2, GLUT3 and GLUT4. Although no in vivo efficacy was provided for Glutor or BAY-876 in these studies62,63, a related compound BAY-897 was reported to impair tumour growth in a triple-negative breast cancer patient-derived xenograft model64. Whether these compounds have the desired pharmacokinetic properties to advance to the clinic remains to be established.
Hexokinase is the first step in glycolysis, which produces and ionically traps negatively charged glucose 6-phosphate intracellularly. HK2, in particular, shows increased expression in cancer, as it is induced by HIF1 and MYC, both independently and cooperatively65. Loss of HK2 diminishes tumorigenesis in vivo66, underscoring its attractiveness as a therapeutic target. 3-Bromopyruvate, a pyruvate analogue and highly reactive compound, was proposed as a HK2 inhibitor, but unbiased proteomic studies using radiolabelled 3-bromopyruvate revealed that GAPDH is the most pyruvylated protein and is probably the primary target of 3-bromopyruvate in SK-Hep1 cells67. HK2 inhibitors have been developed, including a series of glucosamine derivatives with nanomolar values of IC50 against purified HK2 (ref.68), which also inhibit hexokinase 1 (HK1). Several members of this series are more selective for HK2, with co-crystal structures showing inhibitors bound to the catalytic pocket (Fig. 6). No in vivo efficacy data were provided. Another putative HK2 selective inhibitor with sub-micromolar potency, benitrobenrazide69, was developed from a lead compound identified through virtual ligand screening of six million compounds using the crystal structure of HK2. Oral administration of benitrobenrazide demonstrated efficacy in two tumour xenograft models in immunocompromised mice70.

a | Tetramer of glutaminase 1 (GLS1) bound to the CB-839 allosteric inhibitor (arrow) at the tetramerization interface. b | NCGC00420737-09 inhibitor (arrow) bound to tetramer of lactate dehydrogenase A (LDHA) with NAD (dashed arrow). c | GSK compound 27, a 2-amido-6-benzenesulfonamide glucosamine inhibitor (arrow), bound to hexokinase 2 (HK2) with different domains colour highlighted. d | AZ3965 inhibitor (arrow) bound to monocarboxylate transporter 1 (MCT1) (red transmembrane helices) and basigin (yellow sheet linked to a single transmembrane red helix on the right) as determined by cryogenic electron microscopy. The Protein Database IDs are: GLS1 (5HL1); LDHA (6Q13); HK2 (5HFU) and MCT1 (6LLY). Structures are displayed by iCn3D at the NCBI structure website (https://www.ncbi.nlm.nih.gov/Structure/icn3d/full.html).
Vulnerabilities of tumours to highly specific enzyme inhibitors are heightened when related isozymes, such as HK1, are silenced, rendering HK2+ tumour cells sensitive to hexokinase inhibition71. In this regard, passenger deletions, particularly of enolase 1 (ENO1) in gliomas associated with deletion of the 1p36 tumour-suppressor locus, provide vulnerability to inhibitors of enolase72. Based on this concept, POMHEX, a potent prodrug inhibitor of ENO2, was developed by conducting structure–activity relationship studies of SF2312 (refs73,74). The active inhibitor HEX, released from the POMHEX ester, binds the active site of ENO2, as documented by crystallography, and is potent against ENO1-deleted cells75. Further, POMHEX prolongs survival in a murine intracranial glioma model75. Interestingly, this study also uncovers the exquisite sensitivity of melanoma and neuroblastoma cell lines to POMHEX. Importantly, POMHEX has favourable pharmacokinetic and safety profiles in primates75.
Pyruvate kinase is another key glycolytic enzyme with two alternatively spliced muscle forms, PKM1 and PKM2, and a liver and red blood cell isoform (PKLR). Low activity of PKM2 seems to slow glycolytic flux from phosphoenolpyruvate to pyruvate so that upstream intermediates can be shunted towards biosynthetic pathways to produce lipids and nucleotides. Thought to alter biosynthesis by decreasing flux from glucose to lactate, a PKM2 inhibitor had in vivo antitumour effects against H1299 xenografts from non-small-cell lung cancer (NSCLC)76. It is notable that PKM2 is not required for tumorigenesis in several models, and in fact PKM2 loss accelerated tumorigenesis in a Brca1 mutant murine breast cancer model77,78,79. Hence, whether inhibitors or activators (TEPP-46) of PKM2 should be further investigated for cancer therapy is unclear. Nonetheless, allosteric activators of pyruvate kinase, stemming from the work on PKM2 activators, have been further developed to treat haemolytic anaemias associated with hereditary red blood cell PKLR mutations80.
Lactate dehydrogenase, which exists as homotetramers and heterotetramers of LDHA and LDHB, is essential for the Warburg effect. LDHA has been explored as a therapeutic target, because knockout of Ldha resulted in tumour inhibition in a mouse model of lung cancer51, and a tool compound with nonspecific LDHA inhibition reduced xenograft growth81. LDHA, which is induced by MYC or hypoxia, catalyses the reduction of pyruvate to lactate by NADH with regeneration of NAD+, which is used upstream by GAPDH. LDHB can mediate oxidation of lactate to pyruvate in vitro82, and commensal production of lactate by tumour or stromal cells that could be converted to pyruvate for oxidation by tumour cells has been reported83. As such, inhibition of both LDHA and LDHB could be therapeutically useful. Most attempts have targeted LDHA, and although several potent LDHA inhibitors have been developed, its selective inhibition by small molecules has only limited success84,85,86,87. GSK2837808A has LDHA inhibitory potency at 2 nM with more than 10-fold selectivity over LDHB87, but no efficacy studies were provided or performed because of the low in vivo pharmacokinetic exposure. In vitro pretreatment of patient-derived melanoma cells with an LDH inhibitor rendered them more susceptible to tumour-infiltrating lymphocyte killing8. Further, in vivo, adoptive T cell therapy in a B16 melanoma immunocompetent model was enhanced by co-treatment with GSK2837808A8, which alone had virtually no efficacy. Another orally available potent LDH inhibitor, GNE-140 (ref.88), was developed with 3 nM potency against purified enzyme and nanomolar levels of activity against the MIA PaCa-2 cell line. Co-crystal structures reveal drug occupancy of the pyruvate pocket of LDHA, and pharmacokinetics studies reveal high protein binding with extended exposure following high-dose oral administration. Treatment with GNE-140 in vitro revealed rewiring of metabolism with heightened oxidative phosphorylation driven by activation of the AMPK–mTORC1 pathway, which could be dampened by co-treatment with mitochondrial or mTORC1 inhibitors88,89. However, neither study provided in vivo efficacy of the compound.
Among the many LDH catalytic site inhibitors reported, the novel pyrazole-based compound series (such as NCI-006) has been the most rigorously optimized, having over 900 molecules with structure–activity relationships, resulting in nanomolar values of IC50 for lead compounds, co-crystal structures (Fig. 6), desirable pharmacokinetics and documented in vivo target engagement and efficacy with oral availability35,90. Specifically, in vivo pharmacodynamics studies using hyperpolarized 13C-pyruvate tumour imaging showed that one of the compounds inhibited the conversion of pyruvate to lactate in vivo, accompanied by inhibition of MIA PaCa-2 and HT29 tumour xenograft growth. Further, oxidative rewiring due to LDH inhibition was exploited to show the synergistic therapeutic effect with the mitochondrial complex I inhibitor IACS-10759. This class of LDH inhibitors also impairs Ewing sarcoma tumour growth in xenograft mouse models91, with haemolysis as the major toxicity, as would be expected given the dependency of erythrocytes on glycolysis50.
An enzymatic screen of a library of 3.2 million compounds uncovered phthalimide and dibenzofuran derivatives as highly selective LDHA inhibitors92 that did not interfere with LDHB activity, suggesting a non-catalytic pocket mode of action when compared to other inhibitors. Consistent with this observation, these compounds are allosteric inhibitors, according to X-ray crystallography that illustrates the rearrangement of the drug-bound LDHA tetramer, and have nanomolar values of IC50 against recombinant LDHA activity, inhibiting cellular lactate production at low (micromolar) concentrations. Although the cell growth inhibitory activities of the compounds were not provided in vitro or in vivo against tumour cell lines or xenografts, these allosteric inhibitors represent the first highly selective LDHA inhibitors, illustrating that targeting unique allosteric pockets as opposed to conserved catalytic domains can be highly specific, particularly for dehydrogenases that use NAD+ or NADH as cofactors. Whether LDHA specific inhibitors will prove to be generally effective remains to be established, particularly because the B16 melanoma cell line93 (rendered deficient for LDHA, LDHB or both LDHA and LDHB by CRISPR–Cas9 genome editing) showed blunted in vivo tumorigenesis only when both isozymes were deleted. It is thus not surprising that the two isozymes could function redundantly in tumours and hence inhibitors targeting both LDHA and LDHB could be more advantageous.
Although many tumour cells undergo aerobic glycolysis to produce and efflux lactate, lactate derived from labelled glucose in vivo is taken up by tumour cells94,95 as a source for TCA cycle metabolism, underscoring the importance of the lactate-proton symporters from the SLC16A family. Despite controversy regarding lactate as a tumour fuel96, the blocking of the SLC16A family members monocarboxylate transporter 1 (MCT1 encoded by SLC16A1) or MCT4 (encoded by SLC16A3) causes intracellular lactate accumulation that in turn decreases recycling of NADH to NAD+ and inhibits glycolysis97. MCT1, which is transactivated by MYC, is thought to be involved in lactate influx, whereas MCT4, a target of HIF1α, is involved in lactate efflux. However, in a MYC-driven tumour model, MCT1 was involved in lactate efflux resulting from MYC-induced glycolysis98. Hence, inhibiting MCT1 or MCT4 could have antitumour therapeutic effects. One potent MCT1 inhibitor, a precursor to AZD3965, was generated as a potential immunosuppressive agent99,100. AZD3965 (ref.101) has an inhibitory constant Ki = 1.9 nM and a desirable pharmacokinetics profile, with anti-lymphoma activity in tumour xenograft models but little in vivo activity in the 4T1 mouse syngeneic breast cancer model102. A cryogenic electron microscopy structure of micellar MCT1 bound to AZD3965 (ref.103) revealed that the inhibitor binds to the central channel in the outward (towards the extracellular space) open conformation of MCT1 (Fig. 6). Further, non-conserved amino acids between MCT1 and MCT4 in the central channel contact the inhibitor and account for the specificity of AZD3965 for MCT1. Importantly, AZD3965 is undergoing phase I studies in patients with advanced-stage cancers with the finding that urinary lactate level is elevated after oral treatment, suggesting that renal resorption of lactate may be inhibited by the drug. Concerningly, patients treated with AZD3965 (ref.104) had retinal disturbance at all but the lowest dose. Consistent with this observation, this inhibitor is documented to cause reversible reduced visual acuity in rats105, probably related to MCT function in the retina106. The clinical efficacy of AZD3965 awaits additional clinical trials.
Glutamine metabolism
Glutamine is an abundant circulating non-essential amino acid with plasma levels of 0.5 mM in contrast to a 10-fold higher level of glucose. Glutamine is actively transported into cells through ASCT2, and is then converted to glutamate via deamination by mitochondrial glutaminases GLS1 and GLS2. Subsequent conversion of glutamate to α-ketoglutarate can be accomplished by glutamate dehydrogenase, or aminotransferases including glutamic-oxaloacetic transaminase (GOT) or GPT (Fig. 3). α-Ketoglutarate enters the TCA cycle and undergoes forward cycling to succinate or is converted to isocitrate via reductive carboxylation, particularly under hypoxia. Transgenic mouse models of cancer illustrate tissue-selective dependency on glutamine and, therefore, potential differences in vulnerabilities to targeting glutamine metabolism107.
An ASCT2 antagonist, V-9302, was developed108 from the glutamylanilide scaffold that mimics glutamine. It has in vivo antitumour activity and was able to diminish [18F]-fluoroglutamine tumour influx, as determined by PET109. V-9302 is also documented to have in vivo efficacy in the immunocompetent E0771 mouse model of breast cancer by enhancing T cell activation, presumably owing to diminished ‘glutamine steal’ by tumour cells110. Further, V-9302 in combination with the glutaminase inhibitor CB-839 reduced human liver tumour xenograft growth111. However, V-9302 does not target ASCT2 selectively112, given that ASCT2-null human osteosarcoma 143B cells remained sensitive to V-9302, but it blocks glutamine influx through the transporters SNAT1 and LAT1, as determined by glutamine uptake when expressed ectopically in Xenopus oocytes112. As such, the off-target effect of V-9302 complicates the interpretation of the in vivo efficacy tumour studies. Nonetheless, the fact that V-9302 inhibits other glutamine transporters indicates that inhibition of glutamine influx (among other amino acids) might be the key therapeutic effect of V-9302 in vivo. Consistent with the antitumour activity of V-9302 and its inhibition of LAT1-dependent neutral amino acid transport, LAT1 was found to be essential for tumorigenesis in a KRAS-mutant model of colorectal cancer (CRC)113. Further, the LAT1 inhibitor JPH203 (also known as KYT-0353), showed efficacy against the HT-29 CRC xenografts in vivo114,115. Although JPH203 blocks the influx of many neutral amino acids, including glutamine, it is now in phase I and advancing to phase II clinical studies116.
DON is a reactive diazo glutamine analogue isolated from Streptomyces that demonstrated activity as an antitumour drug in animal tumour models38. As a glutamine analogue, DON targets multiple glutamine-utilizing enzymes (phosphoribosyl pyrophosphate amidotransferase (PPAT), phosphoribosylformylglycinamidine synthase (PFAS), guanine monophosphate synthase (GMPS), carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (CAD), CTP synthase (CTPS), glutamine-dependent NAD+ synthetase (NADSYN), GLS1, GLS2, asparagine synthetase (ASNS), glutamine synthetase (GLUL), and fructose 6-phosphate transaminase (GFAT, encoded by GFPT1 and GFPT2)38. However, to date, no biochemical experiments have demonstrated the degree of engagement with these specific putative targets in different cancer or non-transformed cell lines. Although DON displayed evidence of activity against Hodgkin lymphoma and other cancers in small clinical studies, they were insufficiently powered to be conclusive. DON is active owing to its covalent binding to targets, but its intolerable gastrointestinal toxicity (nausea and vomiting) in humans halted further clinical development.
To overcome GI toxicities, DON prodrugs have been generated117 with the strategy of incorporating pro-moieties that are preferentially cleaved by enzymes enriched in tumours (such as histone deacetylases (HDAC) or cathepsins)118, thereby diminishing the release of DON in non-tumour sites. Specifically, the tool compound prodrug JHU-083 (ethyl 2-(2-amino-4-methylpentanamido)-DON, was engineered with a leucyl amide and an ethyl ester117 on DON’s amine and carboxylate groups, respectively117. JHU-083 was found to be highly active in vivo in a medulloblastoma mouse model119 and importantly has therapeutic effects on the immune system39,120. Specifically, JHU-083, as a single agent, was able to reduce tumour growth more effectively than the combination of anti-PD1 and anti-CTLA4 in the 4T1 model120, accompanied by markedly diminished infiltration of myeloid-derived suppressor cells. When used as a single agent in the MC38 mouse model of CRC39, JHU-083 induced tumour regression and prolonged survival in a CD8+ T cell-dependent manner. Moreover, JHU-083 blockade of glutamine metabolism induced a shift in CD8+ tumour-infiltrating lymphocytes towards a more active long-lived phenotype. These observations have catapulted the DON prodrug, DRP-104, into the clinic with an active trial (NCT04471415) for patients with advanced-stage solid tumours in combination with atezolizumab (anti-PD1). Whereas DON prodrugs have shown striking antitumour activity with antitumour immunity effects, biomarkers for sensitivity to DON prodrugs will be important to determine the tumour type selectivity of the prodrugs and target engagement. Although the multi-hit nature of DON prodrugs may seem nonspecific, the preclinical responses in mouse models are remarkable and may be instructive in multi-hit drug therapy, as opposed to single-enzyme-hit therapy, which could be associated with metabolic rewiring and protective stress response.
The kidney-type glutaminase GLS1, which is activated by MYC121, and the liver-type glutaminase GLS2, which is induced by p53 (ref.122), are encoded by different genes. Both enzymes convert glutamine to glutamate. GLS2 is expressed in normal hepatocytes and suppressed in human hepatocellular carcinoma and in a MYC-induced mouse model of liver cancer123, and this suppression is associated with the induction of GLS1 (ref.123). It is notable that GLS2 is tumorigenic in breast cancer124, and its expression could compensate for loss of Gls in MYC-induced mouse liver tumours40. This suggests that GLS2 could confer resistance to GLS1-specific inhibitors, such as the allosteric inhibitor BPTES or CB-839, and this resistance could be hypothetically avoided by the use of a dual GLS1/GLS2 inhibitor125,126.
BPTES was discovered via screening of compounds that inhibit glutaminase and showed in vivo activity in a MYC-induced mouse model of liver cancer and in a lymphoma xenograft model127. A derivative of BPTES, CB-839, was produced as a potent orally available inhibitor128 with in vivo antitumour activity. Both BPTES and CB-839 occupy the interfaces of the GLS1 tetramer (Fig. 6), allosterically locking a phosphate-dependent activation peptide loop in the off-state36,37. Intriguingly, GLS1 inhibition seems to skew T cells towards the TH1 fate — in which IL-2 and IFNγ stimulate antitumour CTLs — and away from the inflammatory TH17 phenotype that is associated with autoimmunity129. As such, it is important to note that CB-839 has been assessed in two completed clinical trials and is now under evaluation in trials in combination with azacitidine for myelodysplastic syndrome (NCT03047993), with sapanisertib for NSCLC (NCT04250545), with nivolumab for melanoma, clear-cell (conventional) RCC and NSCLC (NCT02771626), and radiation therapy and temozolomide for isocitrate dehydrogenase (IDH)-mediated diffuse astrocytoma (NCT03528642). Disappointingly, CB-839 did not prove efficacious in RCC when treated in combination with carbozantinib130 and results from other trials are still pending. Owing to its properties, CB-839 is dosed at relatively high levels, 600–800 mg twice a day. Hence, another compound was generated from the BPTES co-crystal structure. Compared with CB-839, IPN60090, developed from a different scaffold, has a superior pharmacokinetic profile and in vivo efficacy at lower doses in a NSCLC patient-derived xenograft model in combination with an mTORC1/2 inhibitor131. It is undergoing open-label phase I studies (NCT03894540).
Glutamate generated from glutamine provides a carbon and nitrogen source for de novo proline synthesis through its conversion by pyrroline-5-carboxylate synthase (P5CS) to pyrroline-5-carboxylate (P5C) and ultimately to proline by pyrroline-5-carboxylate reductase (PYCR). Enzymes for de novo proline synthesis are activated by MYC132, whereas the first degradation step by proline dehydrogenase (PRODH) is suppressed by MYC and activated by p53 (ref.133). As such, targeting PYCR has been attempted134,135 with limited preclinical efficacy data. A suicide inhibitor for PRODH has been identified136 and found to be synthetically lethal with the p53-activated state and in combination with glutaminase inhibition. These inhibitors await further development and suitable preclinical cancer models on their path towards clinical application.
Fatty acid synthesis
Although certain tumours rely on fatty acid oxidation137,138, there is a lack of highly specific fatty acid oxidation inhibitors, and therefore here we focus on fatty acid synthesis (Fig. 4) and on inhibitors that have been developed to advanced stages. The use of etomoxir to inhibit fatty acid oxidation has off-target effects139, but etomoxir does have activity against a transgenic mouse model of MYC-inducible breast cancer137. It is notable that ST1326, which inhibits carnitine O-palmitoyltransferase 1 (CPT1A, which has a key role in fatty acid oxidation through the production of fatty acyl-carnitines), has in vivo activity against a MYC-inducible mouse lymphoma model140. However, there is no thorough understanding of whether ST1326 has off-target effects. Additional studies are needed to determine tumour dependency on fatty oxidation versus fatty acid synthesis.
Tumour cells rely on de novo fatty acid synthesis for growth and proliferation, and as such are expected to be vulnerable to inhibition of fatty acid synthetic enzymes141,142. Carbons from acetyl-CoA, which is generated from citrate that is exported from the mitochondria into the cytosol for conversion to acetyl-CoA by ACLY, are the key source for fatty acid chain elongation, which also occurs in the cytosol. ACC1 and ACC2 produce malonyl-CoA as a scaffold for 2-carbon chain elongation by FASN, culminating in an 18-chain stearate that is monounsaturated by SCD1, an oxygen-dependent and iron-containing enzyme, to produce oleate (Fig. 4). ACLY is necessary for tumorigenesis in mouse models of cancer, and tool compound inhibitors of ACLY with high IC50 values have been reported to have antitumour efficacy in xenograft models of lung and prostate cancer143. It is notable that ACLY is a desirable target for the treatment of hyperlipidaemia, and the ACLY inhibitor bempedoic acid144,145,146, which was developed from a long-chain hydrocarbon skeleton, was found to inhibit lipid synthesis and reduce non-high-density lipoprotein (HDL)-cholesterol in rats. Bempedoic acid has been advanced to clinical trials147 and was approved by the FDA in 2020 as a lipid-lowering drug. Further, a series of allosteric ACLY inhibitors with low (nanomolar) competitive inhibitory activity was discovered and characterized by cryogenic electron microscope structures of homotetramer ACLY bound to one of the inhibitors (NDI-091143)148. The efficacy of highly active ACLY inhibitors has not been reported for in vivo tumour models. Such studies will be highly instructive, particularly regarding how members of the ACSS family might impart resistance to ACLY antitumour therapy.
As ACSS2, which produces acetyl-CoA from acetate, supports acetate-dependent tumours47,149 and could bypass ACLY inhibition150, it has been a target for drug development. In two human hepatocellular carcinoma models driven by MYC activation and loss of PTEN, mice lacking ACSS2 had a diminished tumour burden47. It is encouraging that Acss2-knockout mice do not display any phenotypic deficits, which suggests the potential safety of ACSS2 inhibitors47,151. Interestingly, Acss2-knockout mice are resistant to steatosis when placed on a high-fat diet151. In vitro, ACSS2 (ref.152) is also induced by hypoxia and low serum cell culture media and maintains cancer cell growth under stress. Inhibition of ACSS2 by inducible shRNAs152 or CRISPR knockout153,154 suppressed in vivo tumorigenesis. These studies collectively indicate that ACSS2 inhibition could have beneficial antitumour effects. As such, ACSS2 inhibitors are being developed155 (patents WO/2019/097515, WO/2015/175845 and WO/2020/252407) and await testing in tumour models. A predicted transition-state mimetic inhibitor, VY-3-135, was recently documented to have nanomolar biological activity in vitro and has efficacy in xenograft mouse models with high ACSS2 expression but not in those with low expression153. Further, VY-3-135 inhibits the labelling of palmitate by D3-acetate, providing evidence for in vivo target engagement. A previously described ACSS2 inhibitor (N-(2,3-di-2-thienyl-6-quinoxalinyl)-N′-(2-methoxyethyl)urea47 inhibited myeloma growth both in vitro and in a diet-induced obese mouse model of myeloma154. Notably, ACSS2 amplification is found in breast cancer152, but, among cancer types, is highest in CRC (see CBioPortal)156, raising the question of whether intestinal microbial acetate sources (30–90 mM concentration in the colon) could contribute to the evolution of these tumours, which could therefore be uniquely sensitive to ACCS2 inhibition157.
Acetyl-CoA generation is the first step in fatty acid synthesis, followed by the production of malonyl-CoA by ACC1 or ACC2, which is required for tumorigenesis158. Treatment of MYC-induced kidney tumours with a low-potency ACC1 inhibitor, TOFA, caused tumour regression142. Consistent with the observation that ACC1 knockout blocks tumour cell line proliferation, the nanomolar inhibitor of ACC1, ND-646, was shown to curb both tumour fatty acid synthesis and tumour growth of A549 xenograft and KRAS-driven lung cancer in vivo158,159. Further, ND-646 reduces hepatic steatosis in rats159 and another orally administered liver-targeted ACC1 and ACC2 inhibitor, PF-05221304, is currently undergoing clinical studies (NCT03248882) in nonalcoholic fatty liver disease with fibrosis. Given its preliminary safety profile in phase I studies160, which indicated mild thrombocytopenia at high doses, assessing the activity of PF-05221304 in preclinical models, particularly of hepatocellular carcinoma, would be highly instructive.
Malonyl-CoA produced by an ACC is further elongated by FASN. FASN has been a target of intense interest for several decades161, starting with its discovery as a circulating prognostic marker162 — originally called OA-519 before its identification as FASN — in breast cancer. Since those discoveries, the proton pump inhibitor omeprazole has been studied as a weak, nonspecific FASN inhibitor, and more potent FASN inhibitors, including C75 (ref.163), were developed from cerulenin, an irreversible natural product FASN inhibitor and anti-fungal antibiotic produced by Cephalosporium caerulens164. C75 has been shown in preclinical studies to have in vivo antitumour effects165,166, but it has not advanced into the clinic because of undesirable side effects and lack of potency. FASN inhibitors, AZ22 and AZ65, were reported to inhibit the growth of 2D and 3D breast and prostate cancer cell cultures, as well as an in vivo breast tumour xenograft152. TVB-3664 (ref.167) is an orally available FASN inhibitor that has modest in vivo antitumour activity against CRC patient-derived xenografts, associated with large changes in lipid composition. Further, TVB-3664 or its related inhibitor TVB-3166 have efficacy in combination with taxane in three NSCLC xenograft models, including one patient-derived xenograft168, as well as in xenografts from ovarian (OVCAR8) and pancreatic (PANC1) cancer cell lines. The TVB-2640 FASN inhibitor is now in clinical studies of multiple solid tumours, including NSCLC (NCT03808558), CRC (NCT02980029), HER2+ advanced-stage breast cancer (NCT03179904) and high-grade astrocytoma (NCT03032484) as well as steatohepatitis (NCT03938246). The field is anxiously awaiting the results of these studies, decades after FASN was identified as a potential cancer therapeutic target.
The de novo production of long-chain saturated fatty acids by FASN in growing cells must be balanced with the production of unsaturated fatty acids via SCD1 to maintain proper membrane fluidity, because stiff membranes from excessive saturated fatty acids cause detrimental stress response. In particular, excessive palmitate is toxic to cells because it causes lipid bilayer stress and triggers the unfolded protein response through serine/threonine-protein kinase and endoribonuclease IRE1169. As such, inhibition of SCD1 would induce stress and cell growth arrest or death, making it a potential cancer therapeutic target170. The crystal structures of SCD1 bound to substrate have been solved for the human protein171, and for the mouse protein without bound drug172. Notably, early interest in targeting SCD1 (ref.173) was focused on treating insulin-resistant diabetes and dyslipidaemia174,175, which is improved in mice lacking SCD1. In particular, SCD1-deficient mice are resistant to obesity or diabetes when fed with a high-fat diet176. Many SCD1 inhibitors have been generated by more than ten pharmaceutical companies, mostly for obesity, dyslipidaemia and diabetes173. Many of these compounds have nanomolar values of IC50. The inhibitor GSK1940029 has been in a phase I clinical trial as a topical treatment for acne and found to be well tolerated177. Intriguingly, SCD1 inhibition can alleviate α-synuclein cytotoxicity associated with Parkinson disease178, and clinical trials are being implemented. Hence, it seems that inhibitors of SCD1 will probably be useful for non-oncology indications. However, these studies will provide the safety profiles of these drugs and may set the stage for their application for cancer therapy.
Nucleotide metabolism
Many approved metabolic drugs in clinical use target nucleotide metabolism (Fig. 5), particularly interrupting DNA synthesis (Table 1). Given the early success of Sidney Farber, with anti-folates as the first mechanism-based metabolic cancer therapy, it is understandable that an interest in 1C metabolism has resurged recently20,179. Methotrexate targets dihydrofolate reductase (DHFR) and has played a key part in the development of cancer chemotherapy. Pemetrexed emerged later and targets thymidylate synthetase (TYMS) and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) transformylase, adding to the therapy regimens against NSCLC. Serine and glycine are also critical for nucleotide synthesis by mediating 1C metabolism. Serine can be synthesized from 3-phosphoglycerate through phosphoglycerate dehydrogenase (PHGDH) and phosphoserine aminotransferase 1 (PSAT1), which accepts a nitrogen from glutamate. De novo-synthesized serine is then converted to glycine by the serine hydroxymethyl transferases, SHMT1 and SHMT2, which produce glycine and donate a methyl group to THF. Glycine, in turn, can donate a methyl group via the glycine cleavage system to THF, which donates 1C towards thymidine and purine synthesis. It is notable that a screen with a cDNA library for genes that could rescue the slowed growth of Myc-null rat fibroblasts revealed that mitochondrial Shmt2, a MYC target, can partially increase Myc-null cell proliferation180, illustrating the importance of 1C metabolism downstream of MYC-driven proliferation.
PHGDH catalyses the first rate-limiting step of de novo serine synthesis to provide 1C units for purine and deoxythymidine synthesis, as well as supplying a critical substrate for protein synthesis. PHGDH is amplified in 4% of cases of melanoma and pancreatic cancer, and PHGDH inhibitors have been developed to interrupt 1C and nucleotide metabolism in cancer156. In particular, numerous tumours have been reported to be sensitive to PHGDH loss or inhibition, particularly in the context of serine/glycine deprivation, which suggests that dietary manipulation may affect tumorigenesis32,181,182. Several efforts have generated PHGDH inhibitors183 — including ketothioamides184, BI-4924 (ref.185), indole amides186, piperazine-1-thiourea-based inhibitors187, and an allosteric inhibitor188 — that have in vivo efficacy. The use of NCT-503, which was developed from a screen of 400,000 compounds, revealed sensitivities of PHGDH-dependent cancer cells in vitro189, and in orthotopic breast tumour xenografts. NCT-503 caused decreased conversion of glucose carbons to serine, but also unexpectedly decreased incorporation of exogenous serine into AMP and dTMP. These changes were accompanied by SHMT1-mediated production of serine from glycine and 1C units, which diminishes the substrate availability for nucleotide synthesis. Another PHGDH inhibitor, PH-755 (WO2016040449A1), was able to diminish brain metastasis from MDA-MB-231-derived xenografts and prolonged survival181. None of the PHGDH inhibitors have yet entered human clinical studies.
As cytosolic SHMT1 and mitochondrial SHMT2 are essential for the use of 1C units from serine and glycine for nucleotide synthesis, inhibitors of these enzymes have been developed190,191,192,193. Intriguingly, antifolate inhibitors also have activity against SHMTs194 — with the co-crystal structure of SHMT2-lometrexol195 showing docking of the drug in the active site — and can reduce SHMT1 and SHMT2 activities by more than 50%, albeit at a high (100 μM) drug concentration. By contrast, the pyrrolo[3,2-d]pyrimidine AGF347 targets SHMT1 and SHMT2 with Ki = 2 μM and has efficacy against MIA PaCa-2 xenografts191. The inhibitor SHIN1 has a 10 nM IC50 against SHMT1 and SHMT2 (ref.193). SHIN2, related to SHIN1, has antitumour activity in vivo in a Notch1-induced ALL immunocompetent mouse model196 and is associated with prolonged survival. Target engagement was demonstrated in vivo through the use of 13C-serine tracing in tumours. Synergy between SHIN2 and methotrexate has been documented. SHMT2 is also essential for mitochondrial translation197, indicating that the antitumour effects of SHMTs inhibitors could be manifold. Lead candidates from these molecules need further development to improve drug metabolism and pharmacokinetic properties in order to advance towards the clinic. Owing to the unfavourable pharmacokinetics of the currently available SHMT inhibitors, the antidepressant sertraline, previously described to inhibit serotonin reuptake, has been repurposed as an SHMT1 and SHMT2 inhibitor and shown to have in vivo effect on breast tumour MDA-MB468 xenografts in combination with the anti-malarial and putative mitochondrial inhibitor, artemether198.
How dietary sources and the availability of serine/glycine in the tumour microenvironment affect inhibition of 1C metabolism remains to be further established. Intriguingly, serine catabolism through methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) provides a major source of NADH for survival of cells that have slowed respiration after treatment with mitochondrial inhibitor metformin199. Dietary serine starvation enhanced oxidative phosphorylation and prolonged the survival of Eu-Myc lymphoma mice via a cooperative antitumour effect with phenformin, a mitochondrial inhibitor that is more potent than metformin32. Similarly, serine starvation and PHGDH inhibition cooperatively curb in vivo tumour growth in various xenograft mouse models182.
In addition to the nucleoside antimetabolites used in the clinic (Table 1), recent efforts have been made to develop additional drugs to target purine or pyrimidine metabolic enzymes. One of those, inosine monophosphate dehydrogenase (IMPDH), which is involved in guanine synthesis, is inhibited by mycophenolate mofetil, a clinically approved immunosuppressant used to treat autoimmune diseases and suppress allograft rejections200. Mycophenolic acid can suppress growth of liver cancer organoids and decreased tumour recurrence in a small clinical study in patients with human hepatocellular carcinoma201, and other inhibitors of IMPDH2 have been developed as potential anticancer drugs202. Another target is the key pyrimidine synthesis enzyme DHODH, which resides in the mitochondrion. The DHODH inhibitor, leflunomide, used clinically for the treatment of rheumatoid arthritis, has antitumour activity203 in a KrasG12D/Lkb-null immunocompetent mouse model of lung adenocarcinoma, despite its immunosuppressive activity. It has also shown in vivo activity in a PTEN-deficient prostate cancer mouse model. In a phase I study in refractory myeloma204, leflunomide showed manageable side effects, with 9 out of 11 patients achieving stable disease. On the basis of these results, more potent inhibitors of DHODH are being developed for treatment of rheumatoid arthritis and SARS-CoV-2 (ref.205) as well as cancer206,207,208. Another DHODH inhibitor, AG-636 (ref.208), with a 17 nM IC50, has activity against tumour growth in xenograft models of diffuse large B cell lymphoma (OCILY19), and mantle-cell lymphoma (Z138), but limited activity in models of lung (A549) and colon (HCT-116) cancer208. Notably, DHODH function is intimately coupled to the mitochondrial ETC for ubiquinone-mediated oxidation of dihydroorotate to orotate209.
The mitochondrion
The mitochondrion is a respiratory and metabolic organelle that is essential for tumorigenesis and is hence an attractive target206 (Fig. 1). However, direct targeting of its respiratory activity is a major challenge210. Metformin and phenformin, which was withdrawn because of lactic acidosis, are biguanides that target mitochondrial complex I and have been used clinically for the treatment of diabetes (Table 1). Many studies have demonstrated the antitumour efficacy of metformin and phenformin206, particularly in combination with other drugs, such as LDH inhibitors45. Further, a more potent biguanide, IM156, has efficacy and enhanced survival of mice bearing Myc-induced mouse lymphoma with ectopic expression of the miR-17-92 microRNA cluster and reduced LKB1 expression211. IM156 has been studied in a phase I clinical trial for solid tumours and lymphoma (NCT03272256) and phase II studies are pending. Efforts to generate a more potent, non-biguanide, mitochondrial inhibitor were achieved with IACS-010759 (ref.212), which has striking in vivo preclinical activity and demonstrated a pharmacodynamic response of reversing consumptive hypoxia in vivo213,214. IACS-010759 has advanced into the clinic for advanced-stage solid tumours and refractory AML with early reports of clinical response, but has neurological side effects and some patients have reported hyperlactaemia59.
The nonspecific mitochondrial inhibitor CPI-613 has progressed into clinical trials, and has a structure analogous to lipoate, a fatty acid derivative used by several mitochondrial enzymes (PDH and OGDH). CPI-613 inhibits mitochondrial respiratory activity in vitro and has demonstrated preclinical antitumour effects210,215. Particularly remarkable are the clinical responses documented in metastatic pancreatic cancer with complete remissions216, increased remissions of relapse in elderly patients with AML217, and responses in advanced-stage haematological malignancies218. With positive clinical signals in phase I studies, definitive clinical trials of CPI-613 combination therapy in pancreatic cancer219 and AML220 are ongoing.
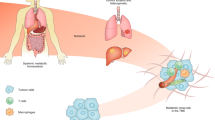
Targeting TIME-associated metabolism
The success of the blossoming field of cancer metabolism is marked by the milestone of the FDA approval of inhibitors of mutant IDH2 and IDH1 for AML (Table 1), based on their ability to target mutant enzymes, reversing the production of 2-hydroxyglutarate and promoting AML differentiation with clinical benefit. Other notable milestones include registration trials with CPI-613 in pancreatic cancer and AML, clinical trials with the glutaminase inhibitor CB-839, which are still awaiting beneficial outcomes for approval, and the impending launch of DRP-104 DON prodrug in the clinic (NCT04471415). It is notable, however, that the deployment of these drugs does not account for the possible beneficial or untoward effect on the tumour microenvironment, except for DRP-104, which is a class of compound that seems to increase the antitumour immune response while damaging cancer cell metabolism. Although drug targeting of metabolism in immune cells (Table 1) is not covered here, it is an emerging area that holds additional hope for new cancer metabolic drugs. As such, fundamental research to provide a deeper understanding of how metabolic drugs affect the TIME, particularly immune cells, is necessary to enable precision oncology221,222,223.
The TIME224 is complex and varies with tumour composition of cancer, stromal, vascular, lymphatic and immune cells that are antagonistic to or supportive of cancer cell survival. Metabolic interactions in the TIME are still poorly understood, but a richer understanding is accumulating9,225,226,227. Notably, cancer cells can produce various immunosuppressive metabolites, such as adenosine, 2-hydroxyglutarate, kynurenine, lactate and methylthioadenosine, particularly in methylthioadenosine phosphorylase-deficient tumours228,229,230, which are barriers to cancer immunotherapy. Different cancer types vary in the composition of TIME — from desmoplastic pancreatic cancers, densely occupied by stromal cells and fibrosis, to leukaemias, which consist of largely circulating tumour cells surviving without a significant TIME, apart from leukaemic stem cells in the bone marrow. Importantly, stromal cells can provide nutrients, such as alanine in pancreatic cancer231,232, and growth factors for cancer cells, whereas immune cells such as M2 macrophages and myeloid-derived suppressor cells can dampen antitumour immunity mediated by CD8+ CTLs and NK cells. Further, intratumour dendritic cells that present processed tumour antigen in situ have been identified, opening up an additional layer of complexity along with non-NK innate lymphoid cells. Given that a solid tumour is a neoplastic organ, targeting metabolism therapeutically is expected to cause inhibition of the same enzyme in cancer cells and antitumour immune cells. In this regard, it is critically important that metabolic cancer drug development advances from assessment of cellular activity to assessment of efficacy in immunocompromised mice and then ultimately in immunocompetent mice or humanized mouse models.
The exemplary study of broadly active DON prodrugs illustrates a beneficial effect on the antitumour immune arm with diminished myeloid-derived suppressor cells and heightened CD8+ T cell antitumour activity39. As TCR activation is associated with increased glycolysis and mitochondrial metabolism and respiration233,234, how targeting various metabolic enzymes affect T cell function is critically important to know. The glutaminase inhibitor CB-839 skews T cells towards TH1 CTL fate110, and the glutamine transporter inhibitor V-9302 enhances CD8+ T cell effector function110. HK2 loss in T cells does not seem to affect their activity235, suggesting that it would be a desirable target. A previously described LDH inhibitor (NCI-737)35 enhances T memory stem cells (TSCM) when combined with IL-21 treatment, enabling improved ex vivo preparation of tumour specific T cells for adoptive therapy236. One study suggested that LDH inhibition could diminish the immunosuppressive effect of tumour-derived lactate8. Conversely, MCT1 inhibitors were among the most immunosuppressive compounds identified in a high-throughput screen using a β-galactosidase reporter as a readout of nuclear factor of activated T cells (NFAT)-driven transcription in T cell receptor-activated Jurkat cells97,99,100. These studies illustrate the importance of understanding how manipulation of metabolism affects immune cells. As such, metabolic enhancement of antitumour CTLs could enhance metabolic cancer therapy, such as the use of nicotinamide riboside to boost mitochondrial clearance237 and enhance T cell activity in combination with anti-PD1 treatment.
Future directions
Many metabolic enzymes have been studied as targets for cancer therapy, but vulnerabilities of specific tumour types to specific inhibitors either as single agents or in combination with chemotherapy, radiation, targeted therapy (such as kinase inhibitors) and/or immunotherapy remain to be decoded. It is notable that metabolic plasticity is a challenge when targeting specific metabolic enzymes, but passenger deletions or amplification of metabolic genes, such as ENO1 (ref.75), PHGDH238 or ACSS2 (ref.152) could provide a roadmap for the clinical stratification of vulnerable tumours. Given the broad opportunities in targeting metabolism for cancer therapy, a key question is whether we have exhausted nearly all metabolic hubs as targets. An avenue towards the identification of metabolic targets of interest could be gleaned from unbiased CRISPR–Cas9 synthetic lethality screening of metabolic genes that favour antitumour responses, particularly in vivo. An in vitro genome-scale sgRNA library revealed the key role for the PBAF complex — which participates in the regulation of HIF1 metabolic targets — in resisting T cell-mediated killing of B16F10 melanoma cells239. NAMPT was also found as a target that was not further pursued in this study. Notably, NAMPT inhibitors have been developed240 but are not covered in this Review because previous clinical studies of potent NAMPT inhibitor were halted by on-target clinically significant thrombocytopenia. In vivo studies with sgRNA libraries targeting pancreatic ductal KRASG12D tumour cells241,242 identified haem synthesis as a vulnerability, independent of host immunity, and autophagy as being central to tumour immune evasion in vivo, which was also documented for in vivo B16 melanoma tumours screened with sgRNAs against 19,000 protein-coding genes243. Whichever future avenues are pursued to target metabolism for cancer therapy, the desirable outcome would be drugs that simultaneously disable cancer cells while synergizing with targeted therapies and favouring antitumour immunity.
References
Farber, S. & Diamond, L. K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 238, 787–793 (1948). This landmark paper was the first to describe the use of folate metabolism inhibition to treat cancer.
Warburg, O. On respiratory impairment in cancer cells. Science 124, 269–270 (1956).
Landau, B. R., Laszlo, J., Stengle, J. & Burk, D. Certain metabolic and pharmacologic effects in cancer patients given infusions of 2-deoxy-d-glucose. J. Natl Cancer Inst. 21, 485–494 (1958).
Miller, D. R. A tribute to Sidney Farber — the father of modern chemotherapy. Br. J. Haematol. 134, 20–26 (2006).
Dang, C. V. & Semenza, G. L. Oncogenic alterations of metabolism. Trends Biochem. Sci. 24, 68–72 (1999).
DeBerardinis, R. J. & Chandel, N. S. Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200 (2016).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Cascone, T. et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 27, 977–987 (2018).
Elia, I. & Haigis, M. C. Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat. Metab. https://doi.org/10.1038/s42255-020-00317-z (2021).
Li, X., Sun, X. & Carmeliet, P. Hallmarks of endothelial cell metabolism in health and disease. Cell Metab. 30, 414–433 (2019).
Olson, K. A., Schell, J. C. & Rutter, J. Pyruvate and metabolic flexibility: illuminating a path toward selective cancer therapies. Trends Biochem. Sci. 41, 219–230 (2016).
Bensard, C. L. et al. Regulation of tumor initiation by the mitochondrial pyruvate carrier. Cell Metab. 31, 284–300 (2020).
Schell, J. C. et al. A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol. Cell 56, 400–413 (2014).
Neinast, M. D. et al. Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell Metab. 29, 417–429 (2019).
Sullivan, L. B., Gui, D. Y. & Vander Heiden, M. G. Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat. Rev. Cancer 16, 680–693 (2016).
Garcia-Bermudez, J. et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 20, 775–781 (2018).
Sullivan, L. B. et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 20, 782–788 (2018).
Pavlova, N. N. et al. As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab. 27, 428–438 (2018).
Birsoy, K. et al. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 (2015). This paper was the first report to establish a key link between the TCA cycle and nucleotide metabolism.
Ducker, G. S. & Rabinowitz, J. D. One-carbon metabolism in health and disease. Cell Metab. 25, 27–42 (2017).
Liu, G. Y. & Sabatini, D. M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 21, 183–203 (2020).
Stine, Z. E., Walton, Z. E., Altman, B. J., Hsieh, A. L. & Dang, C. V. MYC, metabolism, and cancer. Cancer Discov. 5, 1024–1039 (2015).
Shim, H., Chun, Y. S., Lewis, B. C. & Dang, C. V. A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc. Natl Acad. Sci. USA 95, 1511–1516 (1998). This article provided a link between an oncogene and glucose dependency.
Yuneva, M., Zamboni, N., Oefner, P., Sachidanandam, R. & Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 178, 93–105 (2007). This article provided a link between an oncogene and glutamine dependency.
Andrejeva, G. & Rathmell, J. C. Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metab. 26, 49–70 (2017).
Li, H. et al. The landscape of cancer cell line metabolism. Nat. Med. 25, 850–860 (2019).
Romero, R. et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 23, 1362–1368 (2017).
Srinivasan, R., Ricketts, C. J., Sourbier, C. & Linehan, W. M. New strategies in renal cell carcinoma: targeting the genetic and metabolic basis of disease. Clin. Cancer Res. 21, 10–17 (2015).
Mayers, J. R. et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 353, 1161–1165 (2016). This article shows the complexity of the tissue of origin-specific link between RAS and metabolism.
Yuneva, M. O. et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 15, 157–170 (2012). This article shows the complexity of the tissue of origin-specific link between MYC and metabolism.
Maddocks, O. D. et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546 (2013). This article illustrates the dietary effects on p53-dependent tumorigenesis.
Maddocks, O. D. K. et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376 (2017).
Goncalves, M. D. et al. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 363, 1345–1349 (2019).
Vernieri, C. et al. Targeting cancer metabolism: dietary and pharmacologic interventions. Cancer Discov. 6, 1315–1333 (2016).
Rai, G. et al. Pyrazole-based lactate dehydrogenase inhibitors with optimized cell activity and pharmacokinetic properties. J. Med. Chem. 63, 10984–11011 (2020).
Ramachandran, S. et al. Structural basis for exploring the allosteric inhibition of human kidney type glutaminase. Oncotarget 7, 57943–57954 (2016).
Thangavelu, K. et al. Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc. Natl Acad. Sci. USA 109, 7705–7710 (2012).
Lemberg, K. M., Vornov, J. J., Rais, R. & Slusher, B. S. We’re not “DON” yet: optimal dosing and prodrug delivery of 6-diazo-5-oxo-L-norleucine. Mol. Cancer Ther. 17, 1824–1832 (2018).
Leone, R. D. et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021 (2019). This paper describes the anti-tumour and pro-immune effects of DON prodrugs targeting glutamine metabolism.
Mendez-Lucas, A. et al. Identifying strategies to target the metabolic flexibility of tumours. Nat. Metab. 2, 335–350 (2020).
Hein, M. Y. et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163, 712–723 (2015).
Nalawansha, D. A. & Crews, C. M. PROTACs: an emerging therapeutic modality in precision medicine. Cell Chem. Biol. 27, 998–1014 (2020).
Bensimon, A. et al. Targeted degradation of SLC transporters reveals amenability of multi-pass transmembrane proteins to ligand-induced proteolysis. Cell Chem. Biol. 27, 728–739 (2020).
Madak, J. T., Cuthbertson, C. R., Chen, W., Showalter, H. D. & Neamati, N. Design, synthesis, and characterization of brequinar conjugates as probes to study DHODH inhibition. Chemistry 23, 13875–13878 (2017).
Oshima, N. et al. Dynamic imaging of LDH inhibition in tumors reveals rapid in vivo metabolic rewiring and vulnerability to combination therapy. Cell Rep. 30, 1798–1810 (2020). This article describes the efficacy and imaging pharmacodynamics of targeting the Warburg effect with a potent LDH inhibitor in vivo.
Zhou, R. et al. [18F](2 S,4 R)4-fluoroglutamine PET detects glutamine pool size changes in triple-negative breast cancer in response to glutaminase inhibition. Cancer Res. 77, 1476–1484 (2017).
Comerford, S. A. et al. Acetate dependence of tumors. Cell 159, 1591–1602 (2014). This article established acetate as an important source of acetyl-CoA in tumours.
Flier, J. S., Mueckler, M. M., Usher, P. & Lodish, H. F. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science 235, 1492–1495 (1987).
Shim, H. et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc. Natl Acad. Sci. USA 94, 6658–6663 (1997).
van Wijk, R. & van Solinge, W. W. The energy-less red blood cell is lost: erythrocyte enzyme abnormalities of glycolysis. Blood 106, 4034–4042 (2005).
Xie, H. et al. Targeting lactate dehydrogenase-a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 19, 795–809 (2014).
Maekawa, M., Sudo, K., Nagura, K., Li, S. S. & Kanno, T. Population screening of lactate dehydrogenase deficiencies in Fukuoka prefecture in Japan and molecular characterization of three independent mutations in the lactate dehydrogenase-B(H) gene. Hum. Genet. 93, 74–76 (1994).
Kanno, T. et al. Lactate dehydrogenase M-subunit deficiency: a new type of hereditary exertional myopathy. Clin. Chim. Acta 173, 89–98 (1988).
Kremer, J. P., Datta, T., Pretsch, W., Charles, D. J. & Dormer, P. Mechanisms of compensation of hemolytic anemia in a lactate dehydrogenase mouse mutant. Exp. Hematol. 15, 664–670 (1987).
Abu-Elheiga, L. et al. Mutant mice lacking acetyl-CoA carboxylase 1 are embryonically lethal. Proc. Natl Acad. Sci. USA 102, 12011–12016 (2005).
Chirala, S. S. et al. Fatty acid synthesis is essential in embryonic development: fatty acid synthase null mutants and most of the heterozygotes die in utero. Proc. Natl Acad. Sci. USA 100, 6358–6363 (2003).
Miyazaki, M., Man, W. C. & Ntambi, J. M. Targeted disruption of stearoyl-CoA desaturase1 gene in mice causes atrophy of sebaceous and meibomian glands and depletion of wax esters in the eyelid. J. Nutr. 131, 2260–2268 (2001).
Bridges, H. R., Jones, A. J., Pollak, M. N. & Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 462, 475–487 (2014).
Yap, T. A. et al. Phase I trial of IACS-010759 (IACS), a potent, selective inhibitor of complex I of the mitochondrial electron transport chain, in patients (pts) with advanced solid tumors. J. Clin. Oncol. 37, 3014–3014 (2019).
Chan, D. A. et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl Med. 3, 94ra70 (2011).
Adams, D. J. et al. NAMPT is the cellular target of STF-31-like small-molecule probes. ACS Chem. Biol. 9, 2247–2254 (2014).
Reckzeh, E. S. et al. Inhibition of glucose transporters and glutaminase synergistically impairs tumor cell growth. Cell Chem. Biol. 26, 1214–1228 (2019).
Siebeneicher, H. et al. Identification and optimization of the first highly selective GLUT1 inhibitor BAY-876. ChemMedChem 11, 2261–2271 (2016).
Wu, Q. et al. GLUT1 inhibition blocks growth of RB1-positive triple negative breast cancer. Nat. Commun. 11, 4205 (2020).
Kim, J. W., Gao, P., Liu, Y. C., Semenza, G. L. & Dang, C. V. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell Biol. 27, 7381–7393 (2007).
Patra, K. C. et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 24, 213–228 (2013).
Ganapathy-Kanniappan, S. et al. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is pyruvylated during 3-bromopyruvate mediated cancer cell death. Anticancer. Res. 29, 4909–4918 (2009).
Lin, H. et al. Discovery of a novel 2,6-disubstituted glucosamine series of potent and selective hexokinase 2 inhibitors. ACS Med. Chem. Lett. 7, 217–222 (2016).
Liu, Y. et al. Structure based discovery of novel hexokinase 2 inhibitors. Bioorg. Chem. 96, 103609 (2020).
Zheng, M. et al. Novel selective hexokinase 2 inhibitor Benitrobenrazide blocks cancer cells growth by targeting glycolysis. Pharmacol. Res. https://doi.org/10.1016/j.phrs.2020.105367 (2020).
Xu, S. et al. An HK2 antisense oligonucleotide induces synthetic lethality in HK1-HK2+ multiple myeloma. Cancer Res. 79, 2748–2760 (2019).
Muller, F. L. et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 488, 337–342 (2012).
Leonard, P. G. et al. SF2312 is a natural phosphonate inhibitor of enolase. Nat. Chem. Biol. 12, 1053–1058 (2016).
Anderson, V. E., Weiss, P. M. & Cleland, W. W. Reaction intermediate analogues for enolase. Biochemistry 23, 2779–2786 (1984).
Lin, Y. H. et al. An enolase inhibitor for the targeted treatment of ENO1-deleted cancers. Nat. Metab. 2, 1413–1426 (2020).
Suzuki, A. et al. Subcellular compartmentalization of PKM2 identifies anti-PKM2 therapy response in vitro and in vivo mouse model of human non-small-cell lung cancer. PLoS ONE 14, e0217131 (2019).
Lau, A. N. et al. PKM2 is not required for colon cancer initiated by APC loss. Cancer Metab. 5, 10 (2017).
Hillis, A. L. et al. PKM2 is not required for pancreatic ductal adenocarcinoma. Cancer Metab. 6, 17 (2018).
Israelsen, W. J. et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155, 397–409 (2013).
Grace, R. F. et al. Safety and efficacy of mitapivat in pyruvate kinase deficiency. N. Engl. J. Med. 381, 933–944 (2019).
Le, A. et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl Acad. Sci. USA 107, 2037–2042 (2010).
Kaplan, N. O. & Everse, J. Regulatory characteristics of lactate dehydrogenases. Adv. Enzym. Regul. 10, 323–336 (1972).
Sonveaux, P. et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 118, 3930–3942 (2008).
Granchi, C., Bertini, S., Macchia, M. & Minutolo, F. Inhibitors of lactate dehydrogenase isoforms and their therapeutic potentials. Curr. Med. Chem. 17, 672–697 (2010).
Rajeshkumar, N. V. et al. Therapeutic targeting of the Warburg effect in pancreatic cancer relies on an absence of p53 function. Cancer Res. 75, 3355–3364 (2015).
Zhang, S. L., He, Y. & Tam, K. Y. Targeting cancer metabolism to develop human lactate dehydrogenase (hLDH)5 inhibitors. Drug Discov. Today 23, 1407–1415 (2018).
Billiard, J. et al. Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 1, 19 (2013).
Boudreau, A. et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Chem. Biol. 12, 779–786 (2016).
Pusapati, R. V. et al. mTORC1-dependent metabolic reprogramming underlies escape from glycolysis addiction in cancer cells. Cancer Cell 29, 548–562 (2016).
Rai, G. et al. Discovery and optimization of potent, cell-active pyrazole-based inhibitors of lactate dehydrogenase (LDH). J. Med. Chem. 60, 9184–9204 (2017).
Yeung, C. et al. Targeting glycolysis through inhibition of lactate dehydrogenase impairs tumor growth in preclinical models of Ewing sarcoma. Cancer Res. 79, 5060–5073 (2019).
Friberg, A. et al. Structural evidence for isoform-selective allosteric inhibition of lactate dehydrogenase A. ACS Omega 5, 13034–13041 (2020).
Zdralevic, M. et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J. Biol. Chem. 293, 15947–15961 (2018).
Hui, S. et al. Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118 (2017).
Faubert, B. et al. Lactate metabolism in human lung tumors. Cell 171, 358–371 (2017).
Liu, S., Dai, Z., Cooper, D. E., Kirsch, D. G. & Locasale, J. W. Quantitative analysis of the physiological contributions of glucose to the TCA cycle. Cell Metab. 32, 619–628 (2020).
Puri, S. & Juvale, K. Monocarboxylate transporter 1 and 4 inhibitors as potential therapeutics for treating solid tumours: a review with structure–activity relationship insights. Eur. J. Med. Chem. 199, 112393 (2020).
Doherty, J. R. et al. Blocking lactate export by inhibiting the Myc target MCT1 disables glycolysis and glutathione synthesis. Cancer Res. 74, 908–920 (2014).
Michne, W. F. et al. Novel inhibitors of the nuclear factor of activated T cells (NFAT)-mediated transcription of beta-galactosidase: potential immunosuppressive and antiinflammatory agents. J. Med. Chem. 38, 2557–2569 (1995).
Murray, C. M. et al. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat. Chem. Biol. 1, 371–376 (2005).
Curtis, N. J. et al. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 8, 69219–69236 (2017).
Guan, X. & Morris, M. E. In vitro and in vivo efficacy of AZD3965 and alpha-cyano-4-hydroxycinnamic acid in the murine 4T1 breast tumor model. AAPS J. 22, 84 (2020).
Wang, N. et al. Structural basis of human monocarboxylate transporter 1 inhibition by anti-cancer drug candidates. Cell https://doi.org/10.1016/j.cell.2020.11.043 (2020). This article provides structural insight into the mechanism of a clinical stage MCT1 inhibitor.
Halford, S. E. R. et al. A first-in-human first-in-class (FIC) trial of the monocarboxylate transporter 1 (MCT1) inhibitor AZD3965 in patients with advanced solid tumours. J. Clin. Oncol. 35, 2516–2516 (2017).
Allen, A. E. et al. Effects of a monocarboxylate transport 1 inhibitor, AZD3965, on retinal and visual function in the rat. Br. J. Pharmacol. 177, 4734–4749 (2020).
Han, J. Y. S. et al. Role of monocarboxylate transporters in regulating metabolic homeostasis in the outer retina: insight gained from cell-specific Bsg deletion. FASEB J. 34, 5401–5419 (2020).
Altman, B. J., Stine, Z. E. & Dang, C. V. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16, 773 (2016).
Schulte, M. L., Khodadadi, A. B., Cuthbertson, M. L., Smith, J. A. & Manning, H. C. 2-Amino-4-bis(aryloxybenzyl)aminobutanoic acids: a novel scaffold for inhibition of ASCT2-mediated glutamine transport. Bioorg. Med. Chem. Lett. 26, 1044–1047 (2016).
Schulte, M. L. et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 24, 194–202 (2018).
Edwards, D. N. et al. Selective glutamine metabolism inhibition in tumor cells improves anti-tumor T lymphocyte activity in triple-negative breast cancer. J. Clin. Invest. https://doi.org/10.1172/JCI140100 (2020).
Jin, H. et al. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. eLife https://doi.org/10.7554/eLife.56749 (2020).
Broer, A., Fairweather, S. & Broer, S. Disruption of amino acid homeostasis by novel ASCT2 inhibitors involves multiple targets. Front. Pharmacol. 9, 785 (2018).
Najumudeen, A. K. et al. The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat. Genet. 53, 16–26 (2021).
Oda, K. et al. L-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci. 101, 173–179 (2010).
Okunushi, K. et al. JPH203, a newly developed anti-cancer drug, shows a preincubation inhibitory effect on L-type amino acid transporter 1 function. J. Pharmacol. Sci. 144, 16–22 (2020).
Enomoto, K. et al. A novel therapeutic approach for anaplastic thyroid cancer through inhibition of LAT1. Sci. Rep. 9, 14616 (2019).
Rais, R. et al. Discovery of 6-diazo-5-oxo-l-norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: a potential treatment for glioblastoma. J. Med. Chem. 59, 8621–8633 (2016).
Tenora, L. et al. Tumor-targeted delivery of 6-diazo-5-oxo-l-norleucine (DON) using substituted acetylated lysine prodrugs. J. Med. Chem. 62, 3524–3538 (2019).
Hanaford, A. R. et al. Orally bioavailable glutamine antagonist prodrug JHU-083 penetrates mouse brain and suppresses the growth of MYC-driven medulloblastoma. Transl Oncol. 12, 1314–1322 (2019).
Oh, M. H. et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Invest. 130, 3865–3884 (2020).
Gao, P. et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 (2009).
Hu, W. et al. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl Acad. Sci. USA 107, 7455–7460 (2010).
Xiang, Y. et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Invest. 125, 2293–2306 (2015).
Dias, M. M. et al. GLS2 is protumorigenic in breast cancers. Oncogene 39, 690–702 (2020).
Lukey, M. J. et al. Liver-type glutaminase GLS2 is a druggable metabolic node in luminal-subtype breast cancer. Cell Rep. 29, 76–88.e77 (2019).
Wang, J. B. et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18, 207–219 (2010).
Le, A. et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121 (2012).
Gross, M. I. et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 13, 890–901 (2014). This article describes the development of the clinical stage glutaminase inhibitor CB-839.
Johnson, M. O. et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell 175, 1780–1795 (2018).
Tannir, N. M. et al. CANTATA: Primary analysis of a global, randomized, placebo (Pbo)-controlled, double-blind trial of telaglenastat (CB-839) + cabozantinib versus Pbo + cabozantinib in advanced/metastatic renal cell carcinoma (mRCC) patients (pts) who progressed on immune checkpoint inhibitor (ICI) or anti-angiogenic therapies. J. Clin. Oncol. 39, 4501–4501 (2021). This paper reports the clinical outcome of targeting glutaminase in renal cell carcinoma and failure to meet the primary end point.
Soth, M. J. et al. Discovery of IPN60090, a clinical stage selective glutaminase-1 (GLS-1) inhibitor with excellent pharmacokinetic and physicochemical properties. J. Med. Chem. 63, 12957–12977 (2020). This article describes the development of a glutaminase inhibitor at the clinical stage.
Liu, W. et al. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl Acad. Sci. USA 109, 8983–8988 (2012).
Nagano, T. et al. Proline dehydrogenase promotes senescence through the generation of reactive oxygen species. J. Cell Sci. 130, 1413–1420 (2017).
Christensen, E. M. et al. In crystallo screening for proline analog inhibitors of the proline cycle enzyme PYCR1. J. Biol. Chem. 295, 18316–18327 (2020).
Milne, K. et al. A fragment-like approach to PYCR1 inhibition. Bioorg. Med. Chem. Lett. 29, 2626–2631 (2019).
Scott, G. K. et al. Targeting mitochondrial proline dehydrogenase with a suicide inhibitor to exploit synthetic lethal interactions with p53 upregulation and glutaminase inhibition. Mol. Cancer Ther. 18, 1374–1385 (2019).
Camarda, R. et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 22, 427–432 (2016).
Duman, C. et al. Acyl-CoA-binding protein drives glioblastoma tumorigenesis by sustaining fatty acid oxidation. Cell Metab. 30, 274–289 (2019).
Yao, C. H. et al. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of beta-oxidation. PLoS Biol. 16, e2003782 (2018).
Pacilli, A. et al. Carnitine-acyltransferase system inhibition, cancer cell death, and prevention of myc-induced lymphomagenesis. J. Natl Cancer Inst. 105, 489–498 (2013).
Montesdeoca, N., Lopez, M., Ariza, X., Herrero, L. & Makowski, K. Inhibitors of lipogenic enzymes as a potential therapy against cancer. FASEB J. 34, 11355–11381 (2020).
Gouw, A. M. et al. The MYC oncogene cooperates with sterol-regulated element-binding protein to regulate lipogenesis essential for neoplastic growth. Cell Metab. 30, 556–572 (2019).
Granchi, C. ATP citrate lyase (ACLY) inhibitors: an anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur. J. Med. Chem. 157, 1276–1291 (2018).
Cramer, C. T. et al. Effects of a novel dual lipid synthesis inhibitor and its potential utility in treating dyslipidemia and metabolic syndrome. J. Lipid Res. 45, 1289–1301 (2004).
Pinkosky, S. L. et al. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J. Lipid Res. 54, 134–151 (2013).
Pinkosky, S. L. et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat. Commun. 7, 13457 (2016).
Ray, K. K. et al. Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N. Engl. J. Med. 380, 1022–1032 (2019). This article reports the clinical trial results for a non-cancer indication for an ACLY inhibitor.
Wei, J. et al. An allosteric mechanism for potent inhibition of human ATP-citrate lyase. Nature 568, 566–570 (2019).
Schug, Z. T., Vande Voorde, J. & Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 16, 708–717 (2016).
Zhao, S. et al. ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep. 17, 1037–1052 (2016).
Huang, Z. et al. ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc. Natl Acad. Sci. USA 115, E9499–E9506 (2018).
Schug, Z. T. et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71 (2015).
Miller, K. D. et al. Targeting ACSS2 with a transition state mimetic inhibits triple-negative breast cancer growth. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-20-1847 (2021).
Li, Z. et al. Acetyl-CoA synthetase 2: a critical linkage in obesity-induced tumorigenesis in myeloma. Cell Metab. 33, 78–93 (2021).
Kargbo, R. B. Inhibition of ACSS2 for treatment of cancer and neuropsychiatric diseases. ACS Med. Chem. Lett. 10, 1100–1101 (2019).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Louis, P., Hold, G. L. & Flint, H. J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 12, 661–672 (2014). This Review discusses the importance of the gut microbiota metabolites in colon cancer.
Svensson, R. U. et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 22, 1108–1119 (2016).
Harriman, G. et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl Acad. Sci. USA 113, E1796–E1805 (2016).
Bergman, A. et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of a liver-targeting acetyl-CoA carboxylase inhibitor (PF-05221304): a three-part randomized phase 1 study. Clin. Pharmacol. Drug Dev. 9, 514–526 (2020).
Kuhajda, F. P. et al. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc. Natl Acad. Sci. USA 91, 6379–6383 (1994).
Kuhajda, F. P., Piantadosi, S. & Pasternack, G. R. Haptoglobin-related protein (Hpr) epitopes in breast cancer as a predictor of recurrence of the disease. N. Engl. J. Med. 321, 636–641 (1989).
Kuhajda, F. P. et al. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc. Natl Acad. Sci. USA 97, 3450–3454 (2000).
Bills, G. F., Platas, G. & Gams, W. Conspecificity of the cerulenin and helvolic acid producing ‘Cephalosporium caerulens’, and the hypocrealean fungus Sarocladium oryzae. Mycol. Res. 108, 1291–1300 (2004).
Alli, P. M., Pinn, M. L., Jaffee, E. M., McFadden, J. M. & Kuhajda, F. P. Fatty acid synthase inhibitors are chemopreventive for mammary cancer in neu-N transgenic mice. Oncogene 24, 39–46 (2005).
Chen, H. W., Chang, Y. F., Chuang, H. Y., Tai, W. T. & Hwang, J. J. Targeted therapy with fatty acid synthase inhibitors in a human prostate carcinoma LNCaP/tk-luc-bearing animal model. Prostate Cancer Prostatic Dis. 15, 260–264 (2012).
Zaytseva, Y. Y. et al. Preclinical evaluation of novel fatty acid synthase inhibitors in primary colorectal cancer cells and a patient-derived xenograft model of colorectal cancer. Oncotarget 9, 24787–24800 (2018). This paper reports the preclinical assessment of a FASN inhibitor that is a precursor of a compound that has already reached clinical trials.
Heuer, T. S. et al. FASN inhibition and taxane treatment combine to enhance anti-tumor efficacy in diverse xenograft tumor models through disruption of tubulin palmitoylation and microtubule organization and FASN inhibition-mediated effects on oncogenic signaling and gene expression. EBioMedicine 16, 51–62 (2017).
Halbleib, K. et al. Activation of the unfolded protein response by lipid bilayer stress. Mol. Cell 67, 673–684 (2017).
Peck, B. et al. Inhibition of fatty acid desaturation is detrimental to cancer cell survival in metabolically compromised environments. Cancer Metab. 4, 6 (2016).
Wang, H. et al. Crystal structure of human stearoyl-coenzyme A desaturase in complex with substrate. Nat. Struct. Mol. Biol. 22, 581–585 (2015).
Bai, Y. et al. X-ray structure of a mammalian stearoyl-CoA desaturase. Nature 524, 252–256 (2015).
Zhang, Z., Dales, N. A. & Winther, M. D. Opportunities and challenges in developing stearoyl-coenzyme A desaturase-1 inhibitors as novel therapeutics for human disease. J. Med. Chem. 57, 5039–5056 (2014).
Flowers, J. B. et al. Loss of stearoyl-CoA desaturase-1 improves insulin sensitivity in lean mice but worsens diabetes in leptin-deficient obese mice. Diabetes 56, 1228–1239 (2007).
Liu, G. Recent advances in stearoyl-CoA desaturase 1 inhibitors for dyslipidemia and obesity. Curr. Top. Med. Chem. 10, 419–433 (2010).
Ntambi, J. M. et al. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl Acad. Sci. USA 99, 11482–11486 (2002).
Brigandi, R. A., Zhu, J., Murnane, A. A., Reedy, B. A. & Shakib, S. A phase 1 randomized, placebo-controlled trial with a topical inhibitor of stearoyl-coenzyme A desaturase 1 under occluded and nonoccluded conditions. Clin. Pharmacol. Drug Dev. 8, 270–280 (2019).
Vincent, B. M. et al. Inhibiting stearoyl-CoA desaturase ameliorates alpha-synuclein cytotoxicity. Cell Rep. 25, 2742–2754.e2731 (2018).
Locasale, J. W. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer 13, 572–583 (2013).
Nikiforov, M. A. et al. A functional screen for Myc-responsive genes reveals serine hydroxymethyltransferase, a major source of the one-carbon unit for cell metabolism. Mol. Cell Biol. 22, 5793–5800 (2002).
Ngo, B. et al. Limited environmental serine and glycine confer brain metastasis sensitivity to PHGDH inhibition. Cancer Discov. 10, 1352–1373 (2020).
Tajan, M. et al. Serine synthesis pathway inhibition cooperates with dietary serine and glycine limitation for cancer therapy. Nat. Commun. 12, 366 (2021).
Zhao, X., Fu, J., Du, J. & Xu, W. The role of D-3-phosphoglycerate dehydrogenase in cancer. Int. J. Biol. Sci. 16, 1495–1506 (2020).
Ravez, S. et al. Alpha-ketothioamide derivatives: a promising tool to interrogate phosphoglycerate dehydrogenase (PHGDH). J. Med. Chem. 60, 1591–1597 (2017).
Weinstabl, H. et al. Intracellular trapping of the selective phosphoglycerate dehydrogenase (PHGDH) inhibitor BI-4924 disrupts serine biosynthesis. J. Med. Chem. 62, 7976–7997 (2019).
Mullarky, E. et al. Inhibition of 3-phosphoglycerate dehydrogenase (PHGDH) by indole amides abrogates de novo serine synthesis in cancer cells. Bioorg. Med. Chem. Lett. 29, 2503–2510 (2019).
Rohde, J. M. et al. Discovery and optimization of piperazine-1-thiourea-based human phosphoglycerate dehydrogenase inhibitors. Bioorg. Med. Chem. 26, 1727–1739 (2018).
Wang, Q. et al. Rational design of selective allosteric inhibitors of PHGDH and serine synthesis with anti-tumor activity. Cell Chem. Biol. 24, 55–65 (2017).
Pacold, M. E. et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 12, 452–458 (2016).
Dekhne, A. S. et al. Cellular pharmacodynamics of a novel pyrrolo[3,2-d]pyrimidine inhibitor targeting mitochondrial and cytosolic one-carbon metabolism. Mol. Pharmacol. 97, 9–22 (2020).
Dekhne, A. S. et al. Novel pyrrolo[3,2-d]pyrimidine compounds target mitochondrial and cytosolic one-carbon metabolism with broad-spectrum antitumor efficacy. Mol. Cancer Ther. 18, 1787–1799 (2019).
Schwertz, G. et al. Conformational aspects in the design of inhibitors for serine hydroxymethyltransferase (SHMT): biphenyl, aryl sulfonamide, and aryl sulfone motifs. Chemistry 23, 14345–14357 (2017).
Ducker, G. S. et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc. Natl Acad. Sci. USA 114, 11404–11409 (2017).
Paiardini, A. et al. Screening and in vitro testing of antifolate inhibitors of human cytosolic serine hydroxymethyltransferase. ChemMedChem 10, 490–497 (2015).
Scaletti, E., Jemth, A. S., Helleday, T. & Stenmark, P. Structural basis of inhibition of the human serine hydroxymethyltransferase SHMT2 by antifolate drugs. FEBS Lett. 593, 1863–1873 (2019).
Garcia-Canaveras, J. C. et al. SHMT inhibition is effective and synergizes with methotrexate in T-cell acute lymphoblastic leukemia. Leukemia https://doi.org/10.1038/s41375-020-0845-6 (2020).
Minton, D. R. et al. Serine catabolism by SHMT2 Is required for proper mitochondrial translation initiation and maintenance of formylmethionyl-tRNAs. Mol. Cell 69, 610–621.e615 (2018).
Geeraerts, S. L. et al. Repurposing the antidepressant sertraline as SHMT inhibitor to suppress serine/glycine synthesis-addicted breast tumor growth. Mol. Cancer Ther. 20, 50–63 (2021).
Yang, L. et al. Serine catabolism feeds NADH when respiration is impaired. Cell Metab. 31, 809–821 (2020).
Johnson, M. C. & Kollman, J. M. Cryo-EM structures demonstrate human IMPDH2 filament assembly tunes allosteric regulation. eLife https://doi.org/10.7554/eLife.53243 (2020).
Chen, K. et al. Suppression of hepatocellular carcinoma by mycophenolic acid in experimental models and in patients. Transplantation 103, 929–937 (2019).
Shah, C. P. & Kharkar, P. S. Discovery of novel human inosine 5′-monophosphate dehydrogenase 2 (hIMPDH2) inhibitors as potential anticancer agents. Eur. J. Med. Chem. 158, 286–301 (2018).
Jin, R. et al. Leflunomide suppresses the growth of LKB1-inactivated tumors in the immunocompetent host and attenuates distant cancer metastasis. Mol. Cancer Ther. https://doi.org/10.1158/1535-7163.MCT-20-0567 (2020).
Rosenzweig, M. et al. Repurposing leflunomide for relapsed/refractory multiple myeloma: a phase 1 study. Leuk. Lymphoma 61, 1669–1677 (2020).
Hahn, F. et al. IMU-838, a developmental DHODH inhibitor in phase II for autoimmune disease, shows anti-SARS-CoV-2 and broad-spectrum antiviral efficacy in vitro. Viruses https://doi.org/10.3390/v12121394 (2020).
Vasan, K., Werner, M. & Chandel, N. S. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 32, 341–352 (2020).
Petrovic, M. M. et al. Potent human dihydroorotate dehydrogenase inhibitory activity of new quinoline-4-carboxylic acids derived from phenolic aldehydes: synthesis, cytotoxicity, lipophilicity and molecular docking studies. Bioorg. Chem. 105, 104373 (2020).
McDonald, G. et al. Selective vulnerability to pyrimidine starvation in hematologic malignancies revealed by AG-636, a novel clinical-stage inhibitor of dihydroorotate dehydrogenase. Mol. Cancer Ther. 19, 2502–2515 (2020).
Martinez-Reyes, I. et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292 (2020).
Anderson, R. G., Ghiraldeli, L. P. & Pardee, T. S. Mitochondria in cancer metabolism, an organelle whose time has come? Biochim. Biophys. Acta Rev. Cancer 1870, 96–102 (2018).
Izreig, S. et al. Repression of LKB1 by miR-17~92 sensitizes MYC-dependent lymphoma to biguanide treatment. Cell Rep. Med. 1, 100014 (2020).
Molina, J. R. et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 24, 1036–1046 (2018). This paper describes the preclinical activity of a complex I inhibitor that advanced to phase I clinical trials.
Gammon, S. T. et al. Mechanism-specific pharmacodynamics of a novel complex-I inhibitor quantified by imaging reversal of consumptive hypoxia with [18F]FAZA PET in vivo. Cells https://doi.org/10.3390/cells8121487 (2019).
Vashisht Gopal, Y. N. et al. A novel mitochondrial inhibitor blocks MAPK pathway and overcomes MAPK inhibitor resistance in melanoma. Clin. Cancer Res. 25, 6429–6442 (2019).
Egawa, Y., Saigo, C., Kito, Y., Moriki, T. & Takeuchi, T. Therapeutic potential of CPI-613 for targeting tumorous mitochondrial energy metabolism and inhibiting autophagy in clear cell sarcoma. PLoS ONE 13, e0198940 (2018).
Alistar, A. et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 18, 770–778 (2017). This paper reports the phase I clinical trials results of CPI-613 in pancreatic cancer.
Pardee, T. S. et al. A phase I study of CPI-613 in combination with high-dose cytarabine and mitoxantrone for relapsed or refractory acute myeloid leukemia. Clin. Cancer Res. 24, 2060–2073 (2018).
Pardee, T. S. et al. A phase I study of the first-in-class antimitochondrial metabolism agent, CPI-613, in patients with advanced hematologic malignancies. Clin. Cancer Res. 20, 5255–5264 (2014).
Philip, P. A. et al. A phase III open-label trial to evaluate efficacy and safety of CPI-613 plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas. Future Oncol. 15, 3189–3196 (2019).
Pardee, T. S., Luther, S., Buyse, M., Powell, B. L. & Cortes, J. Devimistat in combination with high dose cytarabine and mitoxantrone compared with high dose cytarabine and mitoxantrone in older patients with relapsed/refractory acute myeloid leukemia: ARMADA 2000 Phase III study. Future Oncol. 15, 3197–3208 (2019).
Wolpaw, A. J. & Dang, C. V. Exploiting metabolic vulnerabilities of cancer with precision and accuracy. Trends Cell Biol. 28, 201–212 (2018).
Augustin, R. C., Delgoffe, G. M. & Najjar, Y. G. Characteristics of the tumor microenvironment that influence immune cell functions: hypoxia, oxidative stress, metabolic alterations. Cancers https://doi.org/10.3390/cancers12123802 (2020).
Varanasi, S. K., Kumar, S. V. & Rouse, B. T. Determinants of tissue-specific metabolic adaptation of T cells. Cell Metab. 32, 908–919 (2020).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550 (2018).
Reina-Campos, M., Moscat, J. & Diaz-Meco, M. Metabolism shapes the tumor microenvironment. Curr. Opin. Cell Biol. 48, 47–53 (2017).
Bader, J. E., Voss, K. & Rathmell, J. C. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol. Cell 78, 1019–1033 (2020). This Review discusses the importance of metabolic complexity of the tumour immune microenvironment.
Lyssiotis, C. A. & Kimmelman, A. C. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 27, 863–875 (2017).
Henrich, F. C. et al. Suppressive effects of tumor cell-derived 5′-deoxy-5′-methylthioadenosine on human T cells. Oncoimmunology 5, e1184802 (2016).
DePeaux, K. & Delgoffe, G. M. Metabolic barriers to cancer immunotherapy. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-021-00541-y (2021).
Galluzzi, L. & Kroemer, G. Potent immunosuppressive effects of the oncometabolite R-2-hydroxyglutarate. Oncoimmunology 7, e1528815 (2018).
Parker, S. J. et al. Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discov. 10, 1018–1037 (2020).
Sousa, C. M. et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483 (2016).
Teijeira, A. et al. Metabolic consequences of T-cell costimulation in anticancer immunity. Cancer Immunol. Res. 7, 1564–1569 (2019).
Menk, A. V. et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. 22, 1509–1521 (2018).
Mehta, M. M. et al. Hexokinase 2 is dispensable for T cell-dependent immunity. Cancer Metab. 6, 10 (2018).
Hermans, D. et al. Lactate dehydrogenase inhibition synergizes with IL-21 to promote CD8+ T cell stemness and antitumor immunity. Proc. Natl Acad. Sci. USA 117, 6047–6055 (2020).
Yu, Y. R. et al. Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat. Immunol. 21, 1540–1551 (2020).
Mullarky, E. et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc. Natl Acad. Sci. USA 113, 1778–1783 (2016).
Pan, D. et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775 (2018).
Galli, U. et al. Recent advances in NAMPT inhibitors: a novel immunotherapic strategy. Front. Pharmacol. 11, 656 (2020).
Zhu, X. G. et al. Functional genomics in vivo reveal metabolic dependencies of pancreatic cancer cells. Cell Metab. https://doi.org/10.1016/j.cmet.2020.10.017 (2020).
Biancur, D. E. et al. Functional genomics identifies metabolic vulnerabilities in pancreatic cancer. Cell Metab. 33, 199–210 (2021).
Lawson, K. A. et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 586, 120–126 (2020).
Acknowledgements
Owing to space limitation, we regret omitting citations of papers that have contributed to the field. This work was partially supported by the Ludwig Institute for Cancer Research, NIH/NCI grants CA057341 (to C.V.D.), CA051497 (to C.V.D.), CA252225 (to C.V.D.), CA249950 (to Z.T.S.), CA114046 (to Z.T.S.) and P30CA010815 (to Wistar Institute). We thank B. J. Altman, B. Keith and B. Slusher for comments.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
C.V.D. is a consultant of the Barer Institute, Inc. and member of the board of directors of Rafael Pharmaceuticals, Inc. J.S. receives sponsored research support from the Barer Institute, Inc. Z.E.S. is an employee of the Barer Institute. Z.T.S. declares no competing interests.
Additional information
Peer review information
Nature Reviews Drug Discovery thanks Sarah-Maria Fendt and the other, anonymous, reviewers for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
cBioPortal: https://www.cbioportal.org
OMIM: https://omim.org
Glossary
- One-carbon (1C) metabolism
-
Refers to a series of interlinking folate and methionine metabolic pathways, acting as a methyl (1C) donor for pathways that include epigenetic modifications and the synthesis of DNA and amino acids.
- Tool compound
-
A reagent compound that requires further development before clinical use, but that can be used to investigate the drugging of a target.
- Lactic acidosis
-
A build-up of lactic acid in the blood due to overproduction or under-utilization.
- Pro-moieties
-
Modifications of a pharmacologically active compound to improve its drug-like properties.
- Thrombocytopenia
-
A condition of low platelet counts that can result in bleeding.
Rights and permissions
About this article

Cite this article
Stine, Z.E., Schug, Z.T., Salvino, J.M. et al. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov 21, 141–162 (2022). https://doi.org/10.1038/s41573-021-00339-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-021-00339-6
