
Abstract
Atherosclerosis, a dominant and growing cause of death and disability worldwide, involves inflammation from its inception to the emergence of complications. Targeting inflammatory pathways could therefore provide a promising new avenue to prevent and treat atherosclerosis. Indeed, clinical studies have now demonstrated unequivocally that modulation of inflammation can forestall the clinical complications of atherosclerosis. This progress pinpoints the need for preclinical investigations to refine strategies for combatting inflammation in the human disease. In this Review, we consider a gamut of attractive possibilities for modifying inflammation in atherosclerosis, including targeting pivotal inflammatory pathways such as the inflammasomes, inhibiting cytokines, manipulating adaptive immunity and promoting pro-resolution mechanisms. Along with lifestyle measures, pharmacological interventions to mute inflammation could complement traditional targets, such as lipids and hypertension, to make new inroads into the management of atherosclerotic risk.
Similar content being viewed by others

Introduction
Atherosclerosis now comprises the major contributor to a global epidemic of cardiovascular disease, which has overtaken communicable diseases to become the leading cause of death and disability worldwide1. The spread of obesity and attendant diabetes has aggravated the risk of atherosclerosis both in high-income regions and, alarmingly, in the developing world as well. Atherosclerosis is not only the main underlying cause in coronary heart disease but also causes many strokes and affects peripheral arteries. This lipid-driven disease arises from the accumulation of low-density lipoprotein (LDL) and remnant lipoprotein particles in focal areas of medium and large arteries. Predilection sites for lipid entry and retention in the subendothelial space localize in regions of disturbed, non-laminar flow at arterial branch points. Although lipids undoubtedly contribute causally to atherosclerosis, the ensuing inflammatory response orchestrates the progression and outcome of the disease.
Until recently, after lifestyle measures, non-invasive therapeutic intervention for atherosclerosis focused primarily on pharmacologically limiting risk factors such as arterial hypertension and hypercholesterolaemia. However, inflammation provides a set of pathways that link traditional risk factors to the altered behaviour of cells of the artery wall and the recruitment of leukocytes, mechanisms that promote the disease and its complications. The recent Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) moved targeting inflammation in atherosclerosis from conjecture to clinical reality2. This study targeted a specific pro-inflammatory cytokine, IL-1β, which has been implicated in atherogenesis through decades of experimental work. CANTOS showed that administration of an anti-IL-1β antibody to patients who were cardiovascularly stable after a myocardial infarction and were being treated according to current guidelines (including with statins) reduced recurrent major adverse cardiovascular events. Subsequent clinical trials with colchicine, another anti-inflammatory agent, have likewise demonstrated clinical benefit in patients with recent or temporally remote acute coronary syndromes (ACS)3,4.
This Review summarizes our current understanding of inflammatory processes and cellular participants in atherosclerosis. We then discuss how current clinical trials have started to target some of these processes and which experimental strategies could quell inflammation in the clinic. We also discuss means to promote inflammation resolution. However, both inhibiting inflammation and stimulating its resolution may cause unwanted effects. Hence, we elaborate on means to limit adverse effects, including chronopharmacology and targeted drug delivery with nanoparticles. Beyond pharmacological intervention, we also consider lifestyle changes and how their beneficial effects may arise, in part, from alleviating arterial inflammation. Taming inflammation in atherosclerosis could substantially benefit human health, adding to established targets such as LDL and hypertension, to stem the growing global burden of cardiovascular disease.
Key cells in arterial inflammation
Atherosclerotic lesions evolve from an orderly structured arterial wall to form a complex conglomerate of resident and newly recruited cells. Plaques, also known as atheromas, form in the inner layer of arteries (the tunica intima). Mature atheromas typically contain a lipid-rich central core underneath a fibrous cap (Fig. 1). A monolayer of endothelial cells rests on the surface of plaques, providing the crucial interface with the blood. In this section, we discuss three key cell types that contribute to arterial inflammation: smooth muscle cells (SMCs), macrophages and neutrophils. For other cell types, including endothelial cells and cells of the lymphoid lineage, we refer the reader to other excellent review articles5,6,7.

a | Overview of inflammatory processes. At the early stages of atherosclerosis, activated platelets secrete chemokines (such as C-C motif chemokine 5 (CCL5)) that promote adhesion of monocytes and neutrophils. Neutrophils themselves secrete chemotactic granule proteins (including cathelicidin, cathepsin G and CCL2), thus paving the way for arterial monocyte infiltration. The chemokine milieu is supplemented by chemokines secreted by activated smooth muscle cells (SMCs), such as CCL2 and CCL5. In progressing atherosclerotic lesions, medial SMCs migrate towards the developing fibrous cap where they undergo clonal expansion. SMC lipid loading triggers phenotype switching towards SMCs that express α-smooth muscle actin (αSMA+ SMCs), macrophage-like SMCs and smooth muscle foam cells. Heightened lipid loading of SMCs induces SMC apoptosis and — if not cleared quickly — necrosis. SMCs also undergo cell death after interaction with histone H4 presented in neutrophil extracellular traps (NETs). NET-associated cytotoxicity is observed during plaque erosion when NETs released at sites of disturbed flow induce endothelial cell desquamation. In systemic infections with Gram-negative organisms, which produce lipopolysaccharide (LPS), NET-associated histones promote the adhesion of monocytes, hence contributing to accelerated plaque growth under these conditions. Monocyte-derived macrophages ingest modified lipids and, in response, secrete inflammatory chemokines and cytokines. Excessive lipid uptake triggers macrophage proliferation or even cell death. b–d | Core inflammatory processes fuelled by SMCs (part b), macrophages (part c) and neutrophils (part d). b | Cholesterol uptake induces cell death in SMCs. SMC death, in turn, reduces the amount of extracellular matrix that is produced, which further fuels SMC death. Senescent SMCs release pro-inflammatory cytokines and matrix-degrading enzymes, including matrix metalloproteinases (MMPs). c | Priming and activation of the NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome. Priming by cholesterol crystals, modified lipids such as oxidized low-density lipoproteins (oxLDL) or impaired cholesterol efflux triggers the nuclear factor-κB (NF-κB) signalling pathway, promoting the transcription of NLRP3 and pro-IL-1β. Assembly of the NLRP3 inflammasome induces activation of caspase 1, which cleaves pro-IL-1β into mature IL-1β. d | Release of NETs is licensed by cholesterol crystals, LPS, modified lipids and chemokines such as CCL7. NETs exert cytotoxicity by means of NET-resident histones, prime the NLRP3 inflammasome in macrophages and induce coagulation by cleavage of factor XII and tissue factor pathway inhibitor (TFPI), and by direct platelet activation.
SMCs: more than just stability?
The majority of cardiovascular complications result from plaque rupture, an event whose likelihood correlates inversely with lesional SMC content. Hence, the prevailing view of SMCs in advanced atherosclerosis posits that these cells exert predominantly beneficial roles as they furnish the stabilizing fibrous cap and generate the extracellular matrix. However, the use of fate-mapping and lineage-tracing approaches, in combination with single-cell RNA sequencing, have unveiled a much more diverse picture of the SMC population throughout the various stages of atherosclerosis8 (Box 1). At early stages of atherosclerosis, lipids accumulate in the subendothelial region, where they become trapped as a consequence of the interaction between positively charged apolipoproteins and negatively charged proteoglycan side chains and components of the extracellular matrix produced by SMCs9. SMCs exposed to modified lipids or pro-inflammatory cytokines then release chemoattractants, including C-C motif chemokine 2 (CCL2) and CCL5 (refs10,11), which promote the recruitment of monocytes12 (Fig. 1a).
During lesion evolution, macrophages and SMCs die and contribute to the formation of the necrotic core (Fig. 1b). This highly inflammatory milieu evokes a healing response that initially fosters formation of the plaque’s protective fibrous cap. This structure contains abundant extracellular matrix and SMCs that contain α-smooth muscle actin (αSMA), which is a marker of contractile SMCs. In mouse models of atherosclerosis, fibrous cap SMCs arise from the tunica media13. These cells migrate towards the fibrous cap in a process that depends on the transcription factor OCT4 (also known as POU5F1)14, where they undergo clonal expansion15. Somatic mutations drive clonal expansion in non-malignant tissues during ageing, and clonal expansion in myeloid cells has been linked to an increased risk for cardiovascular events16. Similar mechanisms may occur in SMCs in atherosclerotic lesions. Indeed, early observations made in humans showed that lesional SMCs are monotypic17.
Once in the fibrous cap, SMCs are major producers of the extracellular matrix. Although definitive evidence is missing, data from mice lacking COL15A1, which encodes a collagen chain, specifically in SMCs18 and secretome proteomics of lipid-laden SMCs19 firmly suggest that SMCs contribute to fibrous cap formation. However, lipid uptake and changes in the interactions between SMCs and the extracellular matrix alter the SMC phenotype, increasing the expression of markers typically ascribed to macrophages20. In advanced atherosclerotic plaques in mice, 30–70% of cells with macrophage markers21,22 (also known as foam cells) are SMCs; similar observations have been made in human atherosclerotic plaques23,24. The detrimental consequences of this phenotypic switch towards macrophage-like SMCs derives support from a study in which the transcription factor Krueppel-like factor 4 (KLF4), a transcription factor earlier reported to control the phenotypic transition of SMCs during development25, was deleted specifically in SMCs. These mice show reduced SMC switching, a clear-cut increase in fibrous cap thickness and increased αSMA+ cell content of the fibrous cap21.
The SMC transition to macrophage-like cells is likely pro-inflammatory and contributes to plaque vulnerability, but SMCs can also transition to fibroblast cells, which have previously been thought to have the opposite effects. A recent study combining single-cell RNA sequencing and fate-mapping technologies revealed that lesional SMCs predominantly become fibroblast-like cells (sometimes called fibromyocytes)25. This transition depends on the transcription factor TCF21, deletion of which reduced the transition to fibromyocytes and, consequently, thinned fibrous caps. Single-cell RNA sequencing analyses of human atherosclerotic lesions and data sets from genome-wide association studies support a key role for TCF21: carriers of single-nucleotide polymorphisms that are associated with lower TCF21 expression have more coronary events26. These findings could accelerate SMC-targeted therapeutics, as both fibroblast-like and macrophage-like cells derived from SMCs could have roles in progressing lesions.
Macrophages: the central regulators
Macrophages are the most abundant leukocyte subset in atherosclerotic lesions. In healthy arteries, macrophages reside in the adventitia where they contribute to steady-state functions such as regulating the blood vessel diameter by interacting with SMCs27. In certain areas of the aortic tree, such as the lesser curvature of the aortic arch and at the branch points of large arteries, macrophages localize underneath the endothelium where they contribute to lipid retention28. Resident arterial macrophages in mice derive largely from embryogenic progenitors and can renew themselves29,30. In response to endothelial activation, namely as a consequence of hypertension, hyperglycaemia or hypercholesterolaemia, classical monocytes are recruited to the intima in a process involving integrins (such as α4β1; also known as VLA4) and chemokine receptors (including C-C chemokine receptor type 2 (CCR2) and CCR5)12,31 (Fig. 1a). Targeting monocyte integrins or chemokine receptors consistently reduced monocyte and macrophage accumulation and lesion burden in atherosclerotic mice, suggesting that recruitment is a main driver of lesional macrophage accumulation12,31. This is in agreement with a recent study that used a lineage-tracing approach to study lesional macrophage turnover29. Previous work has established that macrophages also proliferate locally32, predominantly in advanced atherosclerotic plaques; the mechanisms and relative importance of lesional macrophage proliferation remain unclear. Scavenger receptor A signalling32 and LXRα phosphorylation33, which control macrophage cholesterol content, seem to alter lesional macrophage proliferation, and administration of a statin decreased macrophage proliferation and lesion inflammation34. In addition to contributing to endothelial cell activation along the arterial tree, metabolic risk factors also activate blood cell production in the bone marrow in a myeloid cell-biased fashion, thereby boosting the monocyte and neutrophil supply for delivery to the developing atheroma (Box 2).
Activation of atheroma macrophages decisively determines the inflammatory environment in the plaque. Cholesterol crystals in the plaque co-activate the NACHT, LRR and PYD domains-containing protein 3 (NLRP3)-containing inflammasome, which has received considerable attention in the context of atherosclerosis. Inhibition of NLRP3 or its genetic deletion reduces atherosclerosis in mice35,36. The output of the NLRP3 inflammasome requires two hits — priming and activation — thus unleashing caspase 1, which cleaves IL-1β and IL-18 to their mature forms (Fig. 1c). Oxidized LDL can prime and activate the NLRP3 inflammasome in atherosclerotic mice. Priming by oxidized LDL depends on the lipid binding to a complex containing CD36, Toll-like receptor 4 (TLR4) and TLR6; activation of NLRP3 follows lysosomal damage after internalization of the oxidized LDL37. Inhibition of cholesterol efflux in myeloid cells increases inflammasome priming and activation, whereas induction of cholesterol efflux has the opposite effects38. NLRP3 inflammasome activation can also be induced by cholesterol crystals through lysosomal damage35. In summary, modified lipids activate the NLRP3 inflammasome in macrophages and cholesterol depletion from macrophages may mute the ensuing inflammatory response.
Macrophage function and phenotype can vary widely depending on several factors, including the microenvironment39, the macrophage origin and the stage of the disease. Such determinants define macrophages in their global appearance and functionality but also define their heterogeneity on a single-cell level, and different subsets can coexist within one tissue40. Although single-cell data from human plaques have become available41, we await detailed analysis of the macrophage compartment and alignment with data from mouse studies.
Research in the past 10 years indicates that macrophage programming in response to numerous stimuli can persist even after the trigger has subsided. In fact, macrophages can acquire characteristics of immunological memory that were initially identified after brief exposure to microorganisms42,43. This phenomenon, denoted ‘trained immunity’, involves a heightened cytokine response to a second stimulus, a process that reflects long-term cellular reprogramming. Although the initial concept of trained immunity focused on pathogens as the first stimuli, we now recognize that non-microbial endogenous stimuli, such as lipoproteins, can also initiate trained immunity. In mice, for example, a cholesterol-rich diet leads to a profound inflammatory transcriptional and epigenetic rewiring of monocytes in circulation and their bone marrow progenitors44. Feeding-induced hypercholesterolaemia in these mice elicited a heightened inflammatory response to subsequent inflammatory stimuli, a response maintained even weeks after switching them to a normal chow diet44.
Support for the clinical importance of such studies stems from observations made in patients with familial hypercholesterolaemia. Monocytes from these patients have a higher capacity to produce cytokines and have epigenetic marks — namely enrichment of histone H3 trimethylated at lysine 4 (H3K4me3), along with a decrease of H3K9me3 — in the promoter region of at least one of those cytokines, tumour necrosis factor (TNF)45. Although in vitro studies suggested that statins can reverse such training46, the in vivo response remained heightened even after statin treatment45. As IL-1β occupies a central role in inflammatory signalling, it is not surprising that this cytokine also contributes to innate immune training. IL-1β drives epigenetic reprogramming in monocytes reminiscent of the programme acquired by monocytes trained by β-glucan, the Bacillus Calmette–Guérin (BCG) vaccine or oxidized LDL47,48. In addition, IL-1β exposure can elicit the metabolic changes seen during trained immunity in monocytes49. Indeed, pharmacological inhibition of IL-1 using an anti-IL-1 receptor antibody, anakinra, annulled the metabolic switch, myeloid skewing and cell cycle modulation that occurred in haematopoietic stem cells following treatment with β-glucan47.
Granulocytes: unusual suspects
Much attention has focused on macrophages as important regulators of atherosclerosis. Neutrophils, the most abundant circulating white blood cells in humans, have, until recently, received far less notice. Epidemiological evidence, however, indicates that counts of circulating neutrophils predict future cardiovascular events in humans, suggesting an association between neutrophils and atherosclerosis50. Blood neutrophil counts in hypercholesterolaemic mice correlate with lesion sizes51. Depletion of neutrophils during atherogenesis in hyperlipidaemic mice reduces lesional macrophage accumulation, indicating that neutrophils, in part, control macrophage migration. Indeed, neutrophils secrete several proteins with chemotactic activity for monocytes including CCL2, cathelicidin, cathepsin G and α-defensins52,53,54,55 (Fig. 1a). The latter forms heterodimers with platelet-derived CCL5, and therapeutic disruption of this complex reduces monocyte recruitment in mouse models of vascular inflammation53.
Neutrophil secretory products not only attract but also activate macrophages. Neutrophil extracellular traps (NETs) prime macrophages in atherosclerotic mice to produce IL-1β by stimulating the NLRP3 inflammasome56 (Fig. 1d). In addition, macrophages may sense NETs through absent in melanoma 2 (AIM2), a cytosolic DNA-recognizing component of the inflammasome57. NRLP3-containing and AIM2-containing inflammasomes have similar outcomes, through the activation of caspase 1, but differ in their upstream signals. Activation of AIM2 results in production of IL-1β and IL-18 and plaques with signs of instability, and its genetic deletion or therapeutic inhibition improved indices of plaque stability57. However, not all cells can sense and respond to NETs in an orderly fashion. A recent study has revealed that NETs are rich in cytotoxic histone H4, which can puncture SMCs and cause their death; when this occurs repeatedly, the process can thin the fibrous cap58. Neutralizing antibodies to H4 or the use of peptides that shield the cationic charge of the H4 amino terminus, the motif that mediates cytotoxicity, alleviated plaque destabilization58.
Although human atheromas grow slowly, lesions can undergo ‘crises’ that episodically accelerate their evolution. For example, the risk of a cardiovascular event is several times higher after the onset of respiratory infection and the risk of a cardiovascular event is proportional to the severity of the infection59 (see next section for details on infection as a risk factor). Administration of bacterial lipopolysaccharide, which mimics a systemic Gram-negative infection, to rabbits or mice augments inflammation in pre-existing atheromas and provokes neutrophils to contribute to plaque characteristics implicated in destabilization. Such treatment can also elicit epigenetic changes that mediate trained immunity44,47 and increase the production of leukotriene B4 (LTB4), a lipid with strong chemoattractant activity for neutrophils, in lesions60, and induces the release of NETs along the arterial lumen61. The heightened infiltration of neutrophils into advanced lesions during endotoxaemia can induce collagen degradation and necrosis in the lesions60. Exposure of histone H2a within NETs at the intimal surface facilitates adhesion of monocytes61. Such hyper-acute monocyte recruitment accelerates lesion growth and may contribute to thrombotic events associated with acute infections. Neutralization of H2a with blocking antibodies or peptides that target the N terminus prevent accelerated monocyte recruitment in this setting61.
Although fibrous cap rupture causes the majority of cases of ACS, superficial erosion, which appears to be on the rise in the era of better LDL control, can also cause acute coronary artery thrombosis62. Similar to ruptured plaques, neutrophils localize at sites of plaque erosion63. In human specimens, these co-localize with patches of TLR2 expression. The number of adherent neutrophils correlates negatively with endothelial continuity, a process regulated by TLR2-dependent activation of endothelial cells64. Depletion of neutrophils or inhibition of neutrophil adhesion prevented endothelial erosion, thus consolidating the role of neutrophils in this pathophysiology. More recent work provided genetic evidence for the involvement of NETs during a process resembling endothelial erosion in mice. Therapeutically, inhibition of NETosis or treatment with DNase I reduced endothelial discontinuity and endothelial cell apoptosis65.
Risk factors in arterial inflammation
Plaque characteristics
The most dreaded and dramatic consequences of atherosclerosis involve thrombotic complications. A great deal of interest over many decades has focused on the rupture of a plaque’s protective fibrous cap as a thrombosis trigger. Indeed, the term ‘vulnerable plaque’ has entered common parlance and refers to a lipid-rich lesion with a thin (<60 µm) fibrous cap overlying a necrotic core packed with macrophage foam cells. When the weak fibrous cap bursts open, coagulation proteins in the blood gain access to the thrombogenic mediator tissue factor in the lipid core, thus triggering thrombus formation.
A second type of plaque disruption involves erosion of the superficial layer of the intima without a fracture of the fibrous cap. Superficial erosion can occur in plaques that are extracellular matrix-rich and relatively lipid-poor. Numerous observational studies have shown that, coincident with the advent of effective lipid lowering and other preventive therapies, there has been a shift from ST-elevation myocardial infarction (STEMI) to more cases of non-ST-elevation myocardial infarction (NSTEMI). Contemporary data suggest that plaque erosion — rather than accounting for a fifth of ACS, as described previously — causes approximately a third of ACS cases66,67. The application of optical coherence tomography, an intravascular imaging technique, to patients with ACS has shown that plaque erosion associates with NSTEMI whereas plaque rupture more commonly causes STEMI68. As plaque rupture becomes less common, in part because of better control of classical risk factors, the proportion of ACS caused by superficial erosion and, thus, NSTEMI may be on the rise. LDL elevation may be permissive to atheroma formation, and likely contributes to all causes of plaque disruption. The decrease in the lipid content and the relative increase in fibrous tissue in the plaques of patients who have undergone intensive LDL lowering, which renders these lesions relatively lipid-poor, raises the possibility that erosion might strongly contribute to the residual burden of ACS events in these individuals, despite highly effective lipid control62,67.
Attack of the clones
Cells in many tissues acquire somatic mutations over time. Most such somatic mutations will never manifest as disease. However, acquired mutations in a small subset of known driver genes for leukaemia can give rise to clones of myeloid cells that circulate in peripheral blood16. Exome sequencing of blood has shown that up to 10% of septuagenarians have such clones of mutant leukocytes69,70. The transition to a haematological malignancy requires the serial mutation of three or more leukaemia driver genes. Individuals with a single acquired mutation, which can lead to clonal haematopoiesis of indeterminate potential (CHIP), transition to acute leukaemia at a rate of 1% per year or lower.
However, individuals with CHIP have a strikingly increased risk for cardiovascular events that far outstrips their tendency to develop leukaemia16. Fully adjusted for all traditional risk factors, having clonal haematopoiesis confers an almost twofold increase in the risk of acute myocardial infarction or stroke16,67. Individuals with CHIP also have worse outcomes when affected by heart failure or following percutaneous aortic valve replacement. The two most commonly mutated genes in CHIP regulate the methylation of DNA. The augmented cardiovascular risk may thus derive from epigenetic changes in gene expression. Interestingly, the cause and consequence may also be inverted in some cases; traits of atherosclerosis can cause CHIP through continuous stimulation of stem cell proliferation71. In agreement with previous observations72, a combination of risk factors of atherosclerosis accelerated stem cell cycling and, hence, increased the expansion of clones bearing the somatic mutations that cause CHIP.
Convergent data from several sources implicate altered inflammatory signalling as a key consequence of clonal haematopoiesis due to mutations in DNMT3A, TET2 and JAK2 (refs16,73,74). Recent work identified increased macrophage proliferation and prominent necrotic core formation in atherosclerotic lesions of mice expressing JAK2V617F (ref.75), a gain-of-function mutation that increases JAK–STAT signalling and associates with a substantial risk of premature coronary heart disease16. Deletion of the essential inflammasome components caspase 1 and caspase 11, the pyroptosis executioner gasdermin D or the double-stranded DNA-sensing inflammasome AIM2 (but not NLRP3) reversed these adverse changes75. CHIP — a potent, common, age-related, independent and newly recognized risk factor for adverse cardiovascular outcomes — could be a novel link between inflammation and cardiovascular disease that was completely unsuspected just a few years ago. These recent observations provide a fertile field for further exploration of the roles of inflammation in cardiovascular disease. Curiously, CHIP associates inconsistently with increases in the widely used biomarker of inflammation, C-reactive protein (CRP)69. This apparent paradox implies that there are aspects of cardiovascular inflammation beyond those captured by elevations in CRP.
Acute infections
Excess mortality from cardiovascular disease during influenza epidemics was initially recognized early in the twentieth century, and recent reports indicate that COVID-19 may be associated with a similar increase76,77. In a recent study, the risk for an acute coronary event within 1 week after laboratory-confirmed infection with respiratory syncytial virus or influenza virus was four or six times higher, respectively, than the risk during the previous year78. Several-fold increases in ACS risk were also found for acute bacterial pneumonia, urinary tract infection and bacteraemia with one of several bacterial strains79,80. Of note, the increased risk peaks within the first couple of weeks after infection and relates to the severity of the infection. Thus, acute viral or bacterial infection can augment a pre-existing cardiovascular risk. A connection between infection and ACS pertains to a variety of pathogens, and as severity of infection correlates with ACS risk, it is likely that the host response to the infection is a major determinant. Despite this overwhelming epidemiological evidence, there is surprisingly little experimental understanding of how an acute infection accelerates atherosclerosis. As one obvious mechanism, circulating cytokines, a consequence of acute infection, may activate lesion-resident inflammatory cells81,82. This mechanism doubtlessly contributes to advanced COVID-19, which is characterized by excessive cytokine production and multi-organ system failure83. Polymicrobial sepsis in mice aggravated atherosclerosis in as few as 24 h, and atherosclerosis increased with time84. Mechanistically, abdominal sepsis induced the expansion of the lesional macrophage population, likely as a consequence of increased recruitment. In addition, increases in lesional TNF, IL-6 and CCL2 levels indicate induction of the local arterial cytokine and chemokine network. At the intimal surface, increases in platelet activity associate with myocardial infarction in patients with pneumonia85. This observation may relate to the direct prothrombotic state evoked by activated platelets, the ability of platelets to induce NETs86 or their ability to team up with neutrophils to promote arterial monocyte recruitment53,55. Treating the inciting infection and providing haemodynamic and respiratory support are part of the established tools to fight the complications of sepsis. A suite of clinical trials currently underway will evaluate the efficacy of various anti-thrombotic and anticoagulant regimens in severe COVID-19 infections.
Clinical studies
Thousands of publications describe experimental studies of inflammation in atherosclerosis, and numerous biomarker studies have probed the relationship between inflammation and cardiovascular events. Yet, until recently, we lacked direct evidence from properly powered and well-controlled clinical trials that an anti-inflammatory intervention could improve cardiovascular outcomes. We have now entered an era when clinical investigations have begun to close this gap between experiment and observation, and clinical practice (Table 1).
Targeting innate immunity
CANTOS was the first trial to validate the concept that targeting inflammation could be therapeutically relevant in human atherosclerosis2. CANTOS enrolled more than 10,000 individuals who had sustained an acute myocardial infarction but were in the stable phase at least 1 month after the qualifying event2. The enrollees all received standard care: guideline-directed medical therapy including statin administration. Indeed, the baseline LDL concentration of approximately 2 mM (81 mg/dl) established excellent treatment of this classical risk factor. CANTOS selected individuals who had CRP levels, measured with a high sensitivity CRP assay (hsCRP), greater than 2 mg/l; this cut-off value is approximately the median for a general population. Thus, CANTOS preselected individuals deemed likely to benefit from anti-inflammatory intervention by showing evidence of residual inflammatory burden despite standard therapy. CANTOS met its primary end point and showed a 15% reduction in major adverse cardiovascular events, including myocardial infarction, stroke or cardiovascular death, in individuals treated with the anti-IL-1β antibody canakinumab (150 mg four times per year). In addition, in an exploratory analysis, individuals treated with canakinumab had a reduction in incident cancer and a striking decrease in mortality from lung cancer2.
A secondary analysis of CANTOS stratified outcomes based on the achieved hsCRP levels 3 months after initiation of therapy. Participants who responded to the anti-inflammatory drug by a greater than median reduction in hsCRP levels showed a 26% decrease in the prespecified primary end point of myocardial infarction, stroke or cardiovascular death, and a reduction in total mortality as well. Gout attacks decreased by more than half87, and patients treated with canakinumab were hospitalized or died from heart failure less often88 and had less incident anaemia89 than those treated with placebo.
These benefits of canakinumab therapy in this population came at a price. There was a small but statistically significant increase in infections, including fatal infections, in those receiving canakinumab. These infections were from common viral and bacterial agents; there was no increase in infections with opportunistic pathogens or tuberculosis2. These findings can inform the design of future trials and guide the safer deployment of anti-inflammatory therapies in the future, and indicate the need for further anti-inflammatory therapies to optimize the benefit–risk ratio.
Smaller trials blocked both IL-1α and IL-1β with the receptor antagonist anakinra. The MRC-ILA study enrolled patients with NSTEMI and showed a decrease in the area under the curve of the inflammatory biomarker CRP90. Likewise, Virginia Commonwealth University Anakinra Remodeling Trial 3 (VCUART3) showed a decrease in the area under the CRP curve in patients with STEMI treated with anakinra91.
A second anti-inflammatory trial addressed the use of low-dose weekly methotrexate in individuals who had had or were at high risk for coronary events92. Methotrexate, a disease-modifying antirheumatic drug, has transformed the practice of rheumatology and has also benefited patients with psoriasis. Observations of cardiovascular events in individuals enrolled in the studies of low-dose methotrexate for rheumatological or dermatological indications showed strong evidence for a cardiovascular benefit. The Cardiovascular Inflammation Reduction Trial (CIRT) tested this hypothesis prospectively in a randomized controlled study. However, CIRT halted prematurely on the advice of the data safety monitoring committee because of futility93. The enrolled population in CIRT, which was not selected a priori on the basis of elevated inflammation as indicated by hsCRP, had a much lower inflammatory burden than individuals enrolled in CANTOS. The mean baseline CRP in CANTOS was 4.2 mg/l, compared with 1.5 mg/l in CIRT. Moreover, although canakinumab handily decrease CRP levels in participants of CANTOS, biomarkers of inflammation, including hsCRP, IL-1β and IL-6, did not fall in individuals receiving methotrexate. In contrast with CANTOS, which showed a reduction in cancer, CIRT showed a significant increase in cutaneous cancer, raising another potential limitation of anti-inflammatory therapy: interference with immune surveillance for malignancy.
In sum, the results of CIRT may indicate that, despite the observational data, methotrexate does not reduce cardiovascular events. Alternatively, the enrolled population in CIRT may have already exhibited tamed inflammation, as indicated by the normal baseline hsCRP levels, and stood to benefit little from additional anti-inflammatory therapy. The negative result from CIRT underscores the vital importance of conducting rigorous randomized controlled clinical trials in this arena as the results of CIRT differed dramatically from the observational analyses, which were not designed to evaluate and did not duly adjudicate cardiovascular events.
Colchicine, a natural product, has been used for millennia for inflammatory diseases. In the cardiovascular field, colchicine has become the mainstay for management of pericarditis, particularly recurrent pericarditis. The low-dose colchicine (LoDoCo) study enrolled 532 individuals in an open-label, non-placebo-controlled study with 3 years of follow up. There was a remarkable reduction of more than two-thirds in the primary end point of major adverse cardiovascular events94. Despite the small size of this study, which only had 55 primary end point events, the LoDoCo results spawned two large clinical trials, the Colchicine Cardiovascular Outcomes Trial (COLCOT) and LoDoCo2. COLCOT enrolled individuals within 30 days following an ACS event3. Nearly 5,000 participants received 0.5 mg of colchicine daily or a placebo. The participants treated with colchicine, after almost 2 years of follow up, showed a statistically significant reduction in a primary end point that combined “hard” events such as myocardial infarction and stroke with other end points, notably hospitalization for angina leading to coronary revascularization. The clinical benefit was a 23% reduction in the primary end point, driven primarily by a reduction in urgent hospitalizations for angina requiring revascularization. LoDoCo2, which was of similar size to COLCOT, focused on individuals more than 6 months after their last coronary event or revascularization4. After a 30-day open-label run-in with 0.5 mg of colchicine daily, those who tolerated the therapy were randomly allocated to either continue on colchicine or receive placebo. This second large study met its primary, event-driven end point, which included cardiovascular death, non-fatal ACS or non-fatal stroke.
COLCOT and LoDoCo2 neatly bracket two important populations: those with recent ACS, and those in the stable phase of coronary artery disease. The information from these two trials could well change practice and make anti-inflammatory therapy a mainstay of care for individuals post ACS. In COLCOT, colchicine showed unexpectedly little gastrointestinal intolerance. As in CANTOS, colchicine treatment was associated with a small but significant increase in infections, notably pneumonia.
In the wake of COLCOT and LoDoCo2, a raft of trials employing colchicine in various scenarios, ranging from acute myocardial infarction to post-percutaneous intervention, have been performed, are underway or are planned. The colchicine in patients with STEMI/synergy stent registry (NCT03048825) is evaluating, using a factorial design, the combination of colchicine and spironolactone on end points including heart failure. Thus, the upcoming results from a number of studies will inform us regarding the clinical utility of colchicine in various forms of cardiovascular disease.
Beyond IL-1β and colchicine, a number of other targets appear worthy of investigation in the realm of innate immunity. Like methotrexate, agents that antagonize TNF have proven highly effective in diseases such as rheumatoid arthritis and inflammatory bowel disease. TNF antagonism, however, yielded a safety concern in one of the trials that used this strategy in patients with heart failure95. Herein lies another important message: despite robust experimental evidence in animals and smaller pilot studies in humans that showed a benefit, the large-scale randomized trial not only showed no benefit but suggested possible harm at a higher dose. For these reasons, anti-TNF therapy has not engendered enthusiasm in patients at risk for heart failure, including those with coronary artery disease after ACS.
IL-6 is strongly induced by IL-1 (refs96,97), and likely participates causally in human cardiovascular events as shown by concordant Mendelian randomization studies98,99. Thus, agents that neutralize this cytokine also merit evaluation for their capacity to reduce cardiovascular events. However, tocilizumab, a readily available therapeutic that targets the IL-6 receptor (IL-6R), consistently causes an increase in triglycerides. This finding has diminished enthusiasm for long-term studies with this agent in individuals with coronary artery disease. Abundant human genetic evidence points to triglyceride-rich lipoproteins as causal factors in human cardiovascular events. Nonetheless, studies on the short-term administration of an IL-6R antagonist during acute myocardial infarction have yielded encouraging signals for benefit based on cardiac magnetic resonance imaging100. A larger clinical trial is now underway. A phase II study in patients with chronic kidney disease treated with the monoclonal antibody ziltivekimab, which neutralizes IL-6, will be completed in 2021 and could serve as a prelude to a large phase III trial.
Targeting other cytokines may also be valuable. IL-17 isoforms have a controversial role in experimental atherosclerosis101. Although antibodies that neutralize IL-17A benefit patients with certain inflammatory diseases, no rigorous trial has tested their use in patients with atherosclerosis. IL-23, another potential adaptive immune target, has been considered in atherosclerosis but in some studies may produce cardiovascular harm, limiting the enthusiasm for anti-atherosclerotic therapeutics that target IL-23 (ref.102).
Targeting adaptive immunity
The adaptive immune system has also received attention as a potential target in atherosclerotic cardiovascular disease. Seemingly paradoxically, immunization with oxidized forms of LDL can mitigate experimental atherosclerosis103,104. These observations led to a number of attempts to modulate atherosclerosis using vaccination. Although preclinical data were promising, despite considerable effort, none of these vaccination strategies have yielded clinical data that substantiate cardiovascular event reduction in patients104.
Strong preclinical evidence supports the operation of regulatory T cells (Treg cells) in experimental atherosclerosis. Mitigation of inflammation by transforming growth factor-β (TGFβ) and other mediators released by Treg cells can mute experimental atherogenesis105. This observation has led to an innovative clinical trial using low-dose IL-2 to skew the T cell balance towards Treg cells106. Biomarker studies in humans are substantiating the importance of Treg cells107. We look forward to a large-scale outcomes-based trial that would substantiate the approach of modulating adaptive immunity in patients with atherosclerosis.
The importance of biomarkers
As shown by contrasting the results of CANTOS and those of CIRT, the use of biomarkers to select individuals who are particularly likely to benefit from a therapy provides a potential pathway to success. This kind of patient stratification has been used for therapies that alter LDL levels and blood pressure for generations. In the oncology field, the use of genetic markers to target therapies has become routine. Today in the cardiovascular arena, save perhaps for transthyretin mutations, we have no therapies targeted on the basis of genetic testing.
Harnessing the emerging field of genetic markers of cardiovascular risk might provide a path to precisely target particular anti-inflammatory agents to certain patients. For example, the proposal that individuals with CHIP due to loss-of-function mutations in DNMT3A or TET2 might benefit from an anti-IL-6 strategy warrants further investigation. Likewise, those with clonal haematopoiesis due to mutations in JAK2 might benefit from administration of a JAK1/2 inhibitor such as ruxolitinib74,108. The use of biomarkers, including genetic variants, as a strategy for precision targeting of anti-inflammatory therapies could permit smaller, quicker and more economical end point studies that are ultimately needed to establish benefit and receive approval from regulatory authorities.
Preclinical strategies
Limiting arterial inflammation
With the success of CANTOS, several strategies have emerged to interfere with IL-1β or its production, including inhibition of inflammasome activation. In addition, there are currently several promising avenues of research that have not yet entered the clinical trial phase but are in active development. In this section, we focus on selected aspects of controlling arterial inflammation and direct the reader to recent overview articles for broader information109,110.
Closing the gates for myeloid cells
A group of small chemotactic cytokines known as chemokines orchestrate immune cell trafficking. Upon binding to G-protein-coupled receptors, chemokines regulate immune cell movement in steady state as well as during inflammation. Given the importance of intimal leukocyte accumulation during atheroprogression, antagonizing chemokine–receptor interactions may be a promising therapeutic avenue.
Activated endothelial cells can release C-X-C motif chemokine 1 (CXCL1), which interacts with C-X-C chemokine receptor type 2 (CXCR2) on myeloid cells, thereby promoting mobilization from the bone marrow and recruitment to sites of inflammation, including the atherosclerotic lesion. Thus, genetic deletion of CXCR2 from bone marrow cells or antibody-mediated neutralization of CXCL1 reduces the atherosclerotic burden and lesional macrophage accumulation in mice12. Mobilization of classical monocytes from the bone marrow is under control of the CCR2–CCL2 axis111 and genetic deletion of Ccr2 greatly reduces atherosclerotic lesion sizes, likely due to monocytopenia in this model12,112. In myocardial infarction in mice, small interfering RNA (siRNA)-mediated silencing of CCR2 or delivery of a non-agonistic CCL2-competing mutant protein that exhibits strong proteoglycan binding lowered monocyte recruitment, ventricular remodelling and ischaemia–reperfusion injury113,114 (Fig. 2a). In humans, higher plasma CCL2 levels are associated with a higher risk of cardiovascular events and higher lesional CCL2 associates with features of plaque destabilization115,116. Blocking CCR2 with an antibody that blocks CCL2 binding lowers CRP levels in patients at risk of cardiovascular disease117. Upon ligation of the chemokine receptor, a signalling cascade ensues that, in many cases, leads to integrin activation and, consequently, to cell adhesion. In atherosclerosis, platelets are a prominent source of CCL5, which, when immobilized on arterial endothelium, promotes monocyte adhesion and recruitment118. Consequently, lack of CCR5, a receptor of CCL5, or inhibition of CCR5 with maraviroc, a FDA-approved inhibitor of HIV entry, lowers the degree of atherosclerosis and lesional macrophage content in hypercholesterolaemic mice119,120. In a clinical setting, treatment of patients with HIV with maraviroc reduces atheroprogression121.

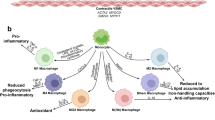
a | Reducing monocyte recruitment. Silencing of C-C chemokine receptor 2 (CCR2) or timed inhibition of CCR2 signalling reduces monocyte adhesion. Overriding chemokine receptor signalling, for example with the small molecule Ac2-26, reduces integrin activation and monocyte arrest. Heterodimers of C-C motif chemokine 5 (CCL5) and C-X-C motif chemokine 4 (CXCL4) as well as of CCL5 and neutrophil defensin 1 (DEFA1) promote monocyte adhesion. Small peptides that disrupt these interactions reduce monocyte adhesion during cardiovascular inflammation. b | Inhibiting neutrophil extracellular traps (NETs). Inhibition of protein-arginine deiminase type 4 (PAD4) halts NET release. DNase I cleaves DNA strands in NETs. Neutralization of gasdermin D prevents NET discharge. Neutralization of NET-resident proteins by antibodies or small-molecule inhibitors reduces NET-driven inflammation. c | Examples of strategies to inhibit immune checkpoints. Neutralization of CD80/86 can reduce T cell and dendritic cell responses. The CD40–CD40 ligand (CD40L) interaction activates macrophages via intracellular TNF receptor-associated factor 6 (TRAF6) signalling, a cascade that can be inhibited with small molecules. d | Increasing inflammation resolution. Putrescine improves the ability of macrophages to engulf dead cells. Resolving N-formyl peptide receptor 2 (FPR2) agonists (Ac2-26, lipoxin A4, resolvin D1) and chemokine-like receptor 1 (CMKLR1) agonists (chemerin C15, resolvin E1) lower the production of inflammatory cytokines and improve the efferocytosis capacity. NF-κB, nuclear factor-κB.
An intriguing aspect of chemokines is their ability to form oligomers either with themselves or with another partner. In this context, CCL5 exhibits a high degree of promiscuity by forming complexes with chemokines CXCL4 or CCL17, the neutrophil-derived peptide neutrophil defensin 1 (DEFA1; also known as HNP1) as well as galectin 3 (refs53,122,123,124). Functionally, CCL5–CXCL4 complexes as well as CCL5–DEFA1 complexes increase inflammatory monocyte recruitment, and selective disruption with cyclical peptides decreases atherosclerosis formation or improves recovery after myocardial information, respectively (Fig. 2a).
Multiple ways to target NETs
The recognition of the importance of NETs in arterial thrombosis, vascular inflammation and vascular injury during the past decade demonstrates the potential of NET release pathways, the NET structure and components of NETs as therapeutic targets. Numerous experimental studies have utilized an inhibitor of protein-arginine deiminase type 4 (PAD4), chloramidine, to inhibit histone citrullination and NET release58,65,125 (Fig. 2b). PAD4 is an enzyme enriched in the cytosol of haematopoietic cells. Upon translocation to the nucleus, PAD4 citrullinates transcription factors, thereby participating in epigenetic regulation of gene expression and stem cell differentiation126,127. The citrullination of argine residues in histones is an important step in chromatin decondensation during NET release128. Although it is useful as a preclinical agent, chloramidine inhibits several isoforms of PAD, and hence lacks the desired specificity of a clinical therapeutic. A therapeutic anti-citrullinated protein antibody (tAPCA) can inhibit NET release and improves signs of inflammation in a mouse model of arthritis129. Although the mechanism through which this antibody inhibits NET formation is unknown, these observations made in mice are encouraging. Beyond limiting NET release, dissolving NETs provides an alternative therapeutic approach. DNase I, for example, breaks up double-stranded DNA and, hence, dissolves NETs. DNase is a FDA-approved drug used in patients with cystic fibrosis to clear bronchial mucous, which is rich in neutrophil products. DNase administration has proven useful in murine atherosclerosis58, and there is also early clinical evidence that it reduces myocardial infarction following ischaemia–reperfusion injury130,131. Gasdermin D is also critically involved in NET release132. Gasdermin D itself is cleaved by neutrophil elastase during NETosis and, in turn, activates neutrophil elastase, furnishing a feedforward loop132. Given the importance of gasdermin D during NET formation, inhibitors of gasdermin D, such as necrosulfonamide, may harbour therapeutic potential during cardiovascular inflammation133.
Targeting proteins embedded within the NETs could also be therapeutically useful. For example, inhibitors of myeloperoxidase might quell one of the NET-associated generators of reactive oxygen species. Neutralization of NET-associated neutrophil elastase and cathepsin G may prevent NET-driven coagulation134. In addition, NET-bound H4 can trigger death of intimal SMCs and accelerate plaque destabilization in mice58. Therapeutic interference using either antibodies or a cyclical histone interference peptide that neutralizes the positive charge at the N terminus of H4 reduced SMC death and conferred plaque stability. A similar approach was chosen to target H2a during endotoxaemia. In this experiment, neutralization of H2a with an antibody or a small peptide lowered myeloid cell adhesion to the carotid artery and the consequent lesion expansion promoted by NETs61.
Titrating the immune response
Immune checkpoint regulators comprise central control elements in inflammation and can accelerate or halt leukocyte activation upon interaction with antigen-presenting cells. Upon T cell receptor ligation, co-stimulatory molecules induce T cell activation. Co-inhibitory receptor–ligand interactions, in turn, dampen T cell responses and thereby contribute to tolerance towards self-antigens. The potential for therapeutic interference in immune checkpoint modulation has been demonstrated in the field of oncology, in which agents targeting PD1 or CTLA4 are now used routinely135. By contrast, this knowledge is just now being applied to vascular inflammation. Mice lacking functional PD1 and PD2 exhibit augmented atherosclerosis, supporting the notion that checkpoint inhibition operates in this disease136.
The interaction between CD40 and CD40 ligand (CD40L) promotes T cell activation but also stimulates macrophages and dendritic cells. Deletion or antibody-assisted inhibition of either CD40 or CD40L lowers plaque burden and induces plaque stability in mouse models of atherosclerosis137,138,139,140. Clinical observations indicate that the CD40–CD40L axis is also important in human cardiovascular disease. Levels of soluble CD40L, for example, associate with primary risk factors such as hypercholesterolaemia and diabetes and can predict future cardiovascular disease141. Expression of CD40 and CD40L in human plaque specimens associates with features of instability. CD40 signals through TNF receptor-associated factors (TRAFs), and activation of TRAF6 but not TRAF2, TRAF3 or TRAF5 augmented atherosclerosis in mice139. This finding stimulated the generation of small-molecule inhibitors that selectively target the interaction between CD40 and TRAF6. These so-called TRAF-STOPs can reduce atherogenesis and prevent atheroprogression in hypercholesterolaemic mice142 (Fig. 2c). Because these molecules do not interfere with TRAF2, TRAF3 or TRAF5, they should impair host defences less than inhibiting CD40–CD40L interactions does. Nanodelivery techniques might further alleviate such unwanted effects. Indeed, encapsulation of TRAF-STOPs into recombinant high-density lipoprotein (rHDL) nanoparticles (rHDL-6877002), delivered into mice with pre-established lesions, reduced macrophage accumulation142,143. rHDL-6877002 was safe, and had a favourable biodistribution in non-human primates143.
The interactions between the immune checkpoint proteins CD80/86 and CD28 or CTLA4 have received considerable attention in cardiovascular inflammation. In human lesions, the expression of CD80/86 correlates with lesion vulnerability144. In turn, mice lacking CD80/86 develop less atherosclerosis with lower effector T cell activity, suggesting a connection between CD80/86 and plaque development145. Overexpression of CTLA4 in mice likewise reduced T cell activation and plaque development146. Pharmacological inhibition of the interaction between CD28 and CD80/86, using a CTLA4 fusion protein, lowered atherosclerosis development in hypercholesterolaemic mice147, supporting the overall viability of therapeutically targeting CD80/86 co-stimulatory signalling in cardiovascular inflammation.
Harnessing trained immunity
As described above, trained immunity involves immune cell reprogramming upon an initial inflammatory stimulus that elicits a hyper-responsive state to a secondary stimulus; this phenomenon’s underlying molecular mechanisms provide an innovative targeting framework (reviewed elsewhere148). Although only at its infancy, therapeutic targeting of trained immunity could have relevance to cardiovascular inflammation. In the context of blockade of cholesterol synthesis, fluvastatin blunts trained immunity induction by β-glucan in vitro46, and hence delivery of statins to macrophages, in the form of HDL nanoparticles149, may reduce vascular inflammation, in part, by reducing immune training. IL-1β and granulocyte–macrophage colony-stimulating factor (GM-CSF) are key regulators of trained immunity in bone marrow progenitors47, so antibody-assisted neutralization of these two factors may also inhibit trained immunity. Antibodies to IL-1β tamed cardiovascular inflammation as shown in CANTOS, and antibodies to GM-CSF are at an advanced phase of clinical development for various inflammatory conditions including asthma and rheumatoid arthritis. Finally, trained immunity involves epigenetic regulation, so inhibitors of histone or DNA methylation can modulate this process. A recent article provides details and a broader overview of targeting of trained immunity148.
Inflammasomes and their output
In the wake of CANTOS, several approaches have emerged that therapeutically interfere with inflammasome priming and activation as well as the main product of inflammasome activation, mature IL-1β. Inhibition of nuclear factor-κB (NF-κB) signalling has been the primary goal for targeting inflammasome priming, as NF-κB transcriptionally regulates NLRP3, and several small-molecule drugs targeting the NF-κB pathway have been successfully employed in preclinical models of cardiovascular inflammation. However, inhibition of NF-κB signalling will have broad effects beyond the expression of components of the inflammasome complex — non-specific immunosuppression and impaired host defence may result.
The most thoroughly characterized inhibitor of the NLRP3 inflammasome is MCC950 (also known as CP-456,773). This compound potently inhibits NLRP3 activation, preventing ATP-triggered, NLRP3-driven IL-1β release150. In mice, MCC950 reduced the lesion burden and macrophage accumulation in hypercholesterolaemia and hyperglycaemia-induced atherosclerosis36,151. MCC950 was also tested in phase II clinical trials for rheumatoid arthritis. However, the compound was not tested further owing to liver toxicity. Key reports on the importance of NLRP3 in atherosclerosis35 and other manifestations of chronic inflammation have engendered intense interest in the development of specific antagonists of the NLRP3-containing inflammasome152. Recent work indicates that the AIM2 inflammasome is also important hypercholesterolaemia-driven57,153 and CHIP-accelerated atherosclerosis75, and consequently AIM2 may be an alternative target in ageing-associated atherosclerosis.
Downstream of inflammasome activation, inhibiting caspase 1 could reduce the release of active IL-1β. The small-molecule inhibitors VX-740 and VX-765 are caspase 1 inhibitor prodrugs that are intracellularly activated by esterases. VX-765 can mitigate atherosclerosis in mice154. However, as in the case of MCC950, clinical studies did not proceed beyond phase II clinical trials due to concerns regarding hepatotoxicity. We have provided a few illustrative examples of inflammasome targeting in this section, and refer the reader to comprehensive reviews of this topic152,155.
Stimulating inflammation resolution
A typical acute inflammatory response is self-limiting: a resolution phase follows the acute inflammatory reaction to an injury, limiting inflammation and evoking reparative processes. Such organized responses promote the return to tissue integrity and function. Pro-resolving pathways and mediators have been mapped156. Whereas agents neutralizing pro‐inflammatory mediators have long been investigated as potential drugs, transferring the concepts of inflammation resolution from bench to bedside requires a paradigm shift, from antagonism towards agonism. The main advantage of immunoresolvents is that they would limit several core processes of atherosclerosis simultaneously: reducing leukocyte infiltration and the production of pro‐inflammatory mediators, and increasing the containment and phagocytosis of cellular debris and apoptotic cells. Therapies that actively promote resolution may also enhance innate immune responses to bacterial infections157, whereas established anti‐inflammatory therapies, including TNF and IL-1β neutralizing strategies, may compromise host defence. For example, in CANTOS, IL-1β neutralization led to a small yet statistically significant increase in lethal infections2. Inflammation resolution is controlled by various classes of mediators including peptides, lipids and gases (including NO and H2S). Numerous cell types, such as Treg cells and myeloid-derived suppressor cells, regulate resolution as well as vagal innervation158,159,160. In this section, we focus on peptides and lipids, as these have been extensively studied in the context of atherosclerosis. Inflammation resolution has been reviewed elsewhere161.
Inflammatory and resolving mediators converge at FPR2 and CMKLR1
N-Formyl peptide receptor 2 (FPR2) is a G-protein-coupled receptor expressed on the cell membrane of many cell types relevant to atherosclerosis, including monocytes, macrophages and neutrophils as well as SMCs. The FPR gene family has a complex evolutionary history and, as a consequence, these proteins sense not just formylated peptides of prokaryotic and eukaryotic origin but also an array of endogenous pro-inflammatory and pro-resolving peptides and lipids. In addition to formylated peptides, serum amyloid A and cathelicidin trigger inflammatory responses via FPR2. In the context of atherosclerosis, cathelicidin serves as a chemoattractant for monocytes and, hence, promotes the early stages of atherosclerosis54. With regard to resolving ligands, lipoxin A4, aspirin-triggered lipoxin, resolvin D1, resolvin D3 and annexin A1 all act via FPR2. Functionally, these ligands limit leukocyte recruitment, promote efferocytosis and stimulate leukocyte egress from sites of inflammation161. With such a heterogeneous array of ligands and functional consequences, it is not surprising that genetic deletion of Fpr2 in hyperlipidaemic mice has yielded variable outcomes in the context of atherosclerosis. In intermediate and advanced stages of atherosclerosis generated in Apoe–/– or Ldlr–/– mice, lack of FPR2 yielded either protective effects162,163 or no effects on lesion size164. By contrast, mice lacking FPR2 during the early stages of lesion development generated larger lesions with a higher macrophage and neutrophil content165. A possible explanation for such apparent stage-dependent differences may be the imbalance between pro-inflammatory and pro-resolving FPR2 agonists that are present at different stages of lesion formation. As an example, resolvin D1 levels decrease substantially during the advanced stages of atherosclerosis166.
Chemokine-like receptor 1 (CMKLR1; also known as ChemR23), a class A G-protein-coupled receptor, has structural similarity to the FPRs. It is widely expressed with highest expression in dendritic cells, monocytes and macrophages. Endothelial cells, vascular SMCs and adipocytes also have high CMKLR1 expression. The CMKLR1 ligand, chemerin, is a chemotactic factor that is secreted as a proprotein. Successive proteolytic cleavages can produce chemerin variants with pro-inflammatory or anti-inflammatory activity, so the effects of chemerin depend on the class of proteases that dominate the microenvironment. Of note, the C15 isoform of chemerin can repolarize macrophages and dampen inflammation167,168,169. Resolvin E1, an anti-inflammatory lipid mediator derived from the ω-3 fatty acid eicosapentaenoic acid (EPA), resolves inflammation by limiting neutrophil recruitment, producing inflammatory cytokines in macrophages and increasing phagocytosis169,170,171,172 through CMKLR1. Two recent studies aimed to understand the role of CMKLR1 in atherosclerosis. Both studies were performed in Apoe–/– mice and animals received a diet with a similar fat content for comparable periods of time. However, the study investigating plaque development at an earlier stage reported accelerated atherosclerosis in mice lacking CMKLR1 (ref.172), a finding that contrasts with those in mice developing advanced lesions173. As for FPR2, such dichotomous reports on the role of CMKLR1 may result from the relative abundance of pro-inflammatory versus anti-inflammatory CMKLR1 ligands.
Attempts have emerged to increase levels of pro-resolving mediators by exogenous administration (Fig. 2d). Intraperitoneal administration of resolvin D1 restores lesional resolvin D1 levels, improves lesion stability and inhibits progression of atherosclerosis166. Delivery of lipoxin A4 had similar effects162. Non-lipid FPR2 activators, such as the N-terminal annexin A1 fragment Ac2-26, can also reduce early atherosclerosis by inhibiting chemokine-mediated leukocyte activation165. However, such restoration strategies have limitations, including the unfavourable pharmacokinetics of these agonists as well as their potential side effects when delivered systemically. Ac2-26 encapsulated in collagen-targeted nanoparticles can revert advanced atherosclerosis, and these particles accumulate specifically in the atherosclerotic lesion164. To stimulate CMKLR1-driven inflammation resolution, resolvin E1 has been applied topically in the oral cavity of hypercholesterolaemic rabbits or via oral gavage in hypercholesterolaemic mice. Both treatments limited experimental atherosclerosis development174,175. Finally, treatment of mice with maresin 1 and resolvin D2 reduced atheroprogression, halting the expansion of the necrotic core and increasing the thickness of the fibrous cap176. These encouraging effects in animal studies raise the question of whether supplementation with ω-3 fatty acid, a precursor of resolving lipid mediators, could potentially increase the generation of resolving lipid mediators and hence counterbalance their decline in advanced lesions. The REDUCE-IT trial showed that a high dose of purified pharmaceutical-grade EPA improved cardiovascular outcomes in patients with high cardiovascular risk and high levels of triglycerides (150–499 mg/dl) treated with statins. Studies with lower doses of EPA or mixtures of ω-3 fatty acids have not yielded positive results177,178,179,180.
Efferocytosis: greenkeeping the lesion
The clearance of dying cells can limit inflammatory responses and maintain tissue homeostasis. Accordingly, efferocytosis — the clearance of dead cells by macrophages — comprises a critical cellular arm of the inflammation resolution machinery. In the context of atherosclerosis, cells that die during the early stages of the disease are swiftly cleared, but the efferocytosis capacity diminishes as lesion development progresses. Macrophages recognize apoptotic cells via receptors and their ligation triggers engulfment. In the context of atherosclerosis, MER proto-oncogene tyrosine kinase (MERTK) has received much attention. In advanced atherosclerosis in humans and mice, the macrophage surface expression of MERTK declines, and the expression of disintegrin and metalloproteinase domain-containing protein 17 (ADAM17), a protease that cleaves MERTK, increases, thus providing one mechanism of impaired efferocytosis in advanced atherosclerosis181. Further support for this notion stems from mice expressing cleavage-resistant MERTK, which have lesions with smaller necrotic cores and macrophages with higher efferocytosis capacity182. The enzyme calcium/calmodulin-dependent protein kinase IIγ (CaMKIIγ) regulates MERTK levels via a pathway that includes LXRα183. Mice lacking CaMKIIγ in myeloid cells show increased MERTK expression in lesional macrophages, increased efferocytosis and smaller necrotic cores. Efferocytosis, however, is not just impaired because of changes in the efferocyte — lesional apoptotic cells also acquire strategies to escape clearance. As an example, expression of the ‘do not eat me’ signal CD47 is inappropriately increased on lesional apoptotic cells, rendering them resistant to efferocytosis184. Antibody-assisted neutralization of CD47 is one possible way to revert this misdirected inflammatory response184.
Numerous nanoparticle-based strategies have emerged to alter the capacity for efferocytosis (details in section Targeted delivery). Alternative strategies may derive from an improved understanding of the metabolic regulation of continued efferocytosis. For example, the metabolism of apoptotic cell-derived arginine and ornithine to putrescine by macrophage arginase 1 and ornithine decarboxylase promotes continued effercytosis. Consequently, treatment with putrescine promotes atherosclerosis resolution in mice185.
Optimizing therapy in time and space
Targeting inflammation systemically may come at the cost of impairing host defences. Optimizing drug delivery in time and space furnishes two options to reduce such unwanted actions.
Chronopharmacology
In circadian medicine, two general strategies may be applicable: targeting the molecular clock or exploiting its rhythmic outputs. Sleep control, exercise and feeding can modulate the molecular clock to influence cardiovascular health. Exploiting circadian outputs for atherosclerosis treatment dates back to the recognition that a critical enzyme that regulates cholesterol synthesis oscillates within a 24-h period. Two decades later, when simvastatin, a short-acting statin, was approved for the treatment of hyperlipidaemia, it was prescribed to be taken “one time per day in the evenings”, thus marking the first clinical application of chronopharmacological treatment.
In the past 10 years we have learnt that levels of certain drug targets oscillate over the day and that such oscillations are phase-shifted between organs. Indeed, a recent comprehensive analysis of a high-resolution time series in 64 tissues revealed that 82% of all protein-coding genes oscillate in primates186, thus offering possibilities for time-optimized treatment strategies. Indeed, a recent study using the cyclic ordering by periodic structure (CYCLOPS) algorithm revealed that half of the protein-coding genes cycled in at least one of 13 analysed tissues in humans187. One thousand of these oscillating genes encode proteins that are drug targets themselves or are involved in drug metabolism187 (Fig. 3a). Of note, oscillating protein-coding genes include several chemokines, cytokines and adhesion molecules with reported relevance in cardiovascular biology, such as vascular cell adhesion protein 1 (VCAM1), IL-1β, IL-6 and CCL2 (ref.187). With regard to CCL2, a recent study has shown that discrete diurnal invasion of the arterial wall driven by the CCL2–CCR2 axis could sustain atherogenic growth52. Indeed, recruitment of neutrophils and monocytes into atherosclerotic lesions oscillates with a peak during the transition from the active to the resting phase, whereas recruitment to microvascular beds peaks 12 h later. Thus, time-optimized pharmacological CCR2 neutralization limited macrovascular myeloid cell recruitment and atheroprogression without disturbing microvascular recruitment. Beyond chemokines, arterial endothelial cell adhesion molecules were also found to oscillate in mice: intercellular adhesion molecule 1 (ICAM1) and VCAM1 peak during the transition from the active to the resting phase. This rhythmic expression depends on oscillatory sympathetic innervation throughout the day; local denervation or systemic administration of a β2-adrenergic receptor antagonist depleted the morning peak of myeloid cell influx into arteries188. Thus, two preclinical studies centred on arterial myeloid cell recruitment illustrate the potential of chronopharmacological intervention to alleviate the atherosclerosis burden.

a | Organ-specific oscillation of genes regulating expression of drug targets that are relevant to cardiovascular inflammation in humans. Group of selected oscillating gene targets in human tissues reported in CircaDB. Logarithmic peak of expression level from RNA sequencing data is shown in different colours over 24-hour clocks. The time of gene expression peaks vary between tissues; the night is presented as grey shadow. b | Examples of nanodelivery methods that can direct functionality to lesional cells. Vascular endothelial growth factor C (VEGF-C) conjugated to an antibody recognizing the extra domain A of fibronectin is presented to smooth muscle cells (SMCs) in the fibrous cap (example 1). These cells respond by improving cholesterol efflux and lowering cell death. Ac2-26 incorporated in collagen IV-targeting nanoparticles reprogrammes macrophages towards a resolving phenotype with improved ability to clear dead cells, lower release of matrix-degrading proteases and increase the secretion of anti-inflammatory cytokines (example 2). Nanoparticles that mimic high-density lipoprotein (HDL), loaded with small-molecule inhibitors of TNF receptor-associated factor 6 (TRAF6), lower inflammatory macrophage responses in atherosclerotic lesions (example 3). Single-walled carbon nanotubes loaded with a chemical inhibitor of the antiphagocytic CD47–SIRPα axis target lesional macrophages and improve their ability to clear dead cells (example 4). CCL2, C-C motif chemokine 2; FPR2, N-formyl peptide receptor 2; ICAM1, intercellular adhesion molecule 1; IL-1R1, IL-1 receptor type 1; IRAK1, IL-1 receptor-associated kinase 1; LDLR, low-density lipoprotein receptor; MALT1, mucosa-associated lymphoid tissue lymphoma translocation protein 1; MMPs, matrix metalloproteinases; NF-κB, nuclear factor-κB; PDE4D, cAMP-specific 3′,5′-cyclic phosphodiesterase 4D; PTGS2, prostaglandin G/H synthase 2.
Apart from the oscillation of the target, the pharmacokinetics of the drug can be modulated with chronopharmacology189. A recent meta-analysis of 106 clinical trials evaluating time-of-day administration of drugs revealed a time-of-day impact in efficacy and/or toxicity in 75% of the studies189. Because of pharmacokinetics, only drugs with short half-lives (typically less than 6 h) should show time-of-day dependency. Yet, in the above-mentioned meta-analysis, a time-of-day dependency was also found for most drugs with plasma half-lives between 8 and 15 h, thus including most drugs used in cardiovascular medicine. These observations demonstrate the enormous potential for timed intervention in cardiovascular diseases.
Targeted delivery
In addition to time-optimized delivery, the delivery of a therapeutic to specific sites may limit potential systemic side effects. Nanoparticle formulations are typically pursued to improve the pharmacokinetics and pharmacodynamics of medicines, for example to increase the plasma half-lives and improve the delivery of drugs with low water solubility. Although nanomedicines are still scarce in the cardiovascular field, several studies have examined nanomedicines in clinical trials, including a second-generation lipid nanoparticle that was used to deliver siRNA that targeted proprotein convertase subtilisin/kexin type 9 (PCSK9)190,191 and prednisolone-loaded liposomes192. However, nanomedicine’s importance goes beyond improving the pharmacology. Its full potential resides in the potential to couple systemic delivery with homing to specific sites. In the context of atherosclerosis, preclinical strategies have emerged to direct nanoparticles to the fibrous cap, to endothelial cells and to macrophages. As an example, in a study centred on resolving chronic vascular inflammation, nanoparticles smaller than 100 nm were generated to access the inflamed vascular wall by enhanced permeability and retention. The surface of these nanoparticles was decorated with a collagen IV-binding heptapeptide, which was identified from a phage display library193. Particles generated in this way accumulated in the lesion shoulder, and using them to deliver Ac2-26 stabilized atherosclerotic lesions164 (Fig. 3b). Similar effects were found when these copolymers were loaded with IL-10 (ref.194). Very recently, the same targeting strategy served to successfully deliver PAD4 inhibitors to eroded arteries in mice, thus inhibiting NET release and reducing vascular damage195. Another recent study used a much simpler approach, employing an immunocytokine to deliver vascular endothelial growth factor C (VEGF-C) to the fibrous cap. In this strategy, VEGF-C was fused to an antibody that recognizes the alternatively spliced extra domain A of fibronectin, a molecular motif expressed in the basement membrane of inflamed vessels196 (Fig. 3b). VEGF-C deposited at the fibrous cap reduced cholesterol overload-induced cell death of SMCs, and hence stabilized atherosclerotic plaques.
Beyond targeting the fibrous cap, strategies have emerged that enable targeting of lesional macrophages. To this end, a combinatorial library of nanoparticles with distinct physiochemical properties and immune cell specificities has been generated and screened in vivo197. This nanoparticle library was based on endogenous HDL particles that incorporate the LXR agonist GW3965, and sought to permit lesional macrophage targeting whilst minimizing known hepatotoxic side effects. In a more recent study, PEGylated single-walled carbon nanotubes were delivered to atherosclerotic mice and uptake of these nanotubes was reported to occur specifically in lesional macrophages198. Loading these nanotubes with a chemical inhibitor of the antiphagocytic CD47–SIRPα signalling axis reduced plaque burden without the toxic side effects that are typically associated with off-target cell clearance, such as anaemia. Incorporating stabilin 2-targeting peptides in nanoparticles could also target lesional macrophages199. Loading such particles with siRNA to CaMKIIγ improved lesional efferocytosis and limited the size of necrotic cores199. Alternatively, HDL-like particles reconstituted from human apolipoprotein A-I and phospholipids can target lesional macrophages. These 22 nm nanoparticles accumulate in lesional macrophages. Loading these cells with inhibitors of CD40 signalling reduced macrophage-driven inflammatory cascades in advanced atherosclerotic lesions142,143. For targeting endothelial cells, αvβ3 integrin targeted nanoparticles loaded with fumagilin were designed to specifically home to neovessels, thus reducing plaque neoangiogenesis200. These studies are some of the examples of how nanotechnology can be used to alleviate atherosclerosis. For an overview, we would like to redirect the reader to recent reviews201,202.
Lifestyle interventions
Although it is generally accepted that a healthy lifestyle reduces cardiovascular risk, its therapeutic application is not as easy as taking a tablet. Large cohort studies have consistently shown that adhering to a healthy lifestyle associates with reduced cardiovascular risk. As an example, two prospective observational studies revealed an almost 80% reduction in the incidence of myocardial infarction when adhering to a healthy lifestyle203,204. In both studies, a healthy lifestyle was defined as limited alcohol consumption, no smoking, a healthy diet and moderate physical activity. Although the results from these studies are striking, implementation may be difficult on an individual level and underlying mechanisms are not well defined. Moreover, lifestyle interventions can modify genetically determined cardiovascular risk205. Of note, many of the lifestyle modifications that are known to alter cardiovascular risk target inflammatory processes (Box 3).
Conclusion and outlook
Only a few years ago, many viewed atherosclerosis as a lipid storage disease, because cholesterol accumulates in plaques. Indeed, a popular formulation of the pathophysiology of atherosclerosis in the 1970s showed bland proliferative lesions containing SMCs and extracellular matrix but not immune cells. The ensuing decades have shone a bright light on inflammation as a common mediator of many risk factors, including levels of LDL and triglyceride-rich lipoproteins, and the altered behaviour of artery wall cells, which beckon leukocytes. These immune cells accumulate within complicated lesions, propagate the disease and produce its complications. The enormous advances in dissecting the cellular and molecular mechanisms of innate and adaptive immunity in atherosclerosis have moved from theory and promise to clinical practice, as evidenced by the success of recent trials in which anti-inflammatory intervention reduced recurrent events, even in patients with excellent control of LDL.
The perception that inflammation replaces traditional risk factors poses a false dichotomy206. Rather than displacing classic risk factors such as LDL, the concept of inflammation as a driver of atherosclerosis provides a rich set of pathways by which the traditional drivers can give rise to the disease and its clinical expression. The ample preclinical implications of inflammation in atherosclerosis have opened the door to many new therapeutic targets. The recent demonstration that anti-inflammatory interventions can forestall atherosclerotic complications merely scratches the surface of the potential for developing novel therapeutics. The success of targeting IL-1β highlights the inflammasome pathway as a promising pathway for further therapeutic interventions. For example, the NLRP3 inflammasome as well as the downstream cytokines IL-1β, IL-18 and IL-6 are attractive candidate targets for intervention. Selective neutralization of certain chemokines also merits consideration in the context. The challenge remains to optimize net benefit, as interference with inflammatory pathways can impair host defences.
Promoting resolution without impairing defences furnishes one possible avenue to optimize benefit over risk in novel therapeutics to combat inflammation. Another opportunity lies in targeting the most appropriate therapeutic agent to particular patients. Achieving the goal of personalized intervention or precision therapeutics will require aligning biomarkers to identify susceptible individuals for particular interventions. It is likely that the advent of multi-omic technologies, machine learning and artificial intelligence will help solve the multidimensional problem presented by the myriad mediators and plentiful pathways that are susceptible to therapeutic manipulation. Thus, although we have achieved much, much more remains to be done in harnessing the concept of inflammation in atherosclerosis to ameliorate the disease and forestall its clinical complications. Mining this exciting area promises to unearth further fundamental discoveries, and hasten the translation of research to the clinic to confront the rising worldwide tide of cardiovascular disease.
References
Roth, G. A. et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J. Am. Coll. Cardiol. 76, 2982–3021 (2020).
Ridker, P. M. et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131 (2017). This paper presented the first clinical study to show that anti-inflammatory treatment can reduce cardiovascular complications.
Tardif, J. C. et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 381, 2497–2505 (2019).
Nidorf, S. M. et al. Colchicine in patients with chronic coronary disease. N. Engl. J. Med. 383, 1838–1847 (2020).
Gimbrone, M. A. Jr. & García-Cardeña, G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 118, 620–636 (2016).
Sage, A. P., Tsiantoulas, D., Binder, C. J. & Mallat, Z. The role of B cells in atherosclerosis. Nat. Rev. Cardiol. 16, 180–196 (2019).
Gisterå, A. & Hansson, G. K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 13, 368–380 (2017).
Basatemur, G. L., Jørgensen, H. F., Clarke, M. C. H., Bennett, M. R. & Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 16, 727–744 (2019).
Skålén, K. et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 417, 750–754 (2002).
Cushing, S. D. et al. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl Acad. Sci. USA 87, 5134–5138 (1990).
Quinn, M. T., Parthasarathy, S., Fong, L. G. & Steinberg, D. Oxidatively modified low density lipoproteins: a potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc. Natl Acad. Sci. USA 84, 2995–2998 (1987).
Soehnlein, O. et al. Distinct functions of chemokine receptor axes in the atherogenic mobilization and recruitment of classical monocytes. EMBO Mol. Med. 5, 471–481 (2013).
Jacobsen, K. et al. Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight 2, e95890 (2017).
Cherepanova, O. A. et al. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat. Med. 22, 657–665 (2016).
Misra, A. et al. Integrin β3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat. Commun. 9, 2073 (2018).
Jaiswal, S. et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121 (2017). This study established clonal haematopoiesis as an important non-classical risk factor of atherosclerotic cardiovascular disease.
Murry, C. E., Gipaya, C. T., Bartosek, T., Benditt, E. P. & Schwartz, S. M. Monoclonality of smooth muscle cells in human atherosclerosis. Am. J. Pathol. 151, 697–705 (1997).
Durgin, B. G. et al. Smooth muscle cell-specific deletion of Col15a1 unexpectedly leads to impaired development of advanced atherosclerotic lesions. Am. J. Physiol. Heart Circ. Physiol. 312, H943–H958 (2017).
Langley, S. R. et al. Extracellular matrix proteomics identifies molecular signature of symptomatic carotid plaques. J. Clin. Invest. 127, 1546–1560 (2017).
Vengrenyuk, Y. et al. Cholesterol loading reprograms the microRNA-143/145–myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler. Thromb. Vasc. Biol. 35, 535–546 (2015).
Shankman, L. S. et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 21, 628–637 (2015).
Chappell, J. et al. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ. Res. 119, 1313–1323 (2016).
Wang, Y. et al. Smooth muscle cells contribute the majority of foam cells in ApoE (apolipoprotein E)-deficient mouse atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 39, 876–887 (2019).
Allahverdian, S., Chehroudi, A. C., McManus, B. M., Abraham, T. & Francis, G. A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 129, 1551–1559 (2014).
Cordes, K. R. et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460, 705–710 (2009).
Wirka, R. C. et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 25, 1280–1289 (2019).
Lim, H. Y. et al. Hyaluronan receptor LYVE-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity 49, 1191 (2018).
Paulson, K. E. et al. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ. Res. 106, 383–390 (2010).
Williams, J. W. et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat. Immunol. 21, 1194–1204 (2020).
Ensan, S. et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1+ precursors and circulating monocytes immediately after birth. Nat. Immunol. 17, 159–168 (2016).
Combadière, C. et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6Chi and Ly6Clo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 117, 1649–1657 (2008).
Robbins, C. S. et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 19, 1166–1172 (2013).
Gage, M. C. et al. Disrupting LXRα phosphorylation promotes FoxM1 expression and modulates atherosclerosis by inducing macrophage proliferation. Proc. Natl Acad. Sci. USA 115, E6556–E6565 (2018).
Tang, J. et al. Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci. Adv. 1, e1400223 (2015).
Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010). This study established the importance of the NLRP3 inflammasome in the development of atherosclerosis.
van der Heijden, T. et al. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein e-deficient mice-brief report. Arterioscler. Thromb. Vasc. Biol. 37, 1457–1461 (2017).
Sheedy, F. J. et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 14, 812–820 (2013).
Westerterp, M. et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation 138, 898–912 (2018).
Lavin, Y. et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326 (2014).
Zernecke, A. et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ. Res. 127, 402–426 (2020).
Fernandez, D. M. et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 25, 1576–1588 (2019).
Saeed, S. et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345, 1251086 (2014).
Cheng, S. C. et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345, 1250684 (2014).
Christ, A. et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell 172, 162–175 (2018). This study establishes an important link between nutrition and long-term inflammatory responses.
Bekkering, S. et al. Treatment with statins does not revert trained immunity in patients with familial hypercholesterolemia. Cell Metab. 30, 1–2 (2019).
Bekkering, S. et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell 172, 135–146 (2018).
Mitroulis, I. et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 172, 147–161 (2018). This study introduced the bone marrow, specifically myelopoiesis, as an important regulator of trained immunity.
Moorlag, S. J. C. F. M., Röring, R. J., Joosten, L. A. B. & Netea, M. G. The role of the interleukin-1 family in trained immunity. Immunol. Rev. 281, 28–39 (2018).
Arts, R. J. W. et al. BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe 23, 89–100 (2018).
Friedman, G. D., Klatsky, A. L. & Siegelaub, A. B. The leukocyte count as a predictor of myocardial infarction. N. Engl. J. Med. 290, 1275–1278 (1974).
Drechsler, M., Megens, R. T., van Zandvoort, M., Weber, C. & Soehnlein, O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 122, 1837–1845 (2010).
Winter, C. et al. Chrono-pharmacological targeting of the CCL2–CCR2 axis ameliorates atherosclerosis. Cell Metab. 28, 175–182 (2018). This study revealed that myeloid cell recruitment in macrocirculation and microcirculation peaks at different times of day, hence establishing a window of opportunity for time-optimized treatments.
Alard, J. E. et al. Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci. Transl Med. 7, 317ra196 (2015).
Döring, Y. et al. Lack of neutrophil-derived CRAMP reduces atherosclerosis in mice. Circ. Res. 110, 1052–1056 (2012).
Ortega-Gomez, A. et al. Cathepsin G controls arterial but not venular myeloid cell recruitment. Circulation 134, 1176–1188 (2016).
Warnatsch, A., Ioannou, M., Wang, Q. & Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 349, 316–320 (2015).
Paulin, N. et al. Double-strand DNA sensing Aim2 inflammasome regulates atherosclerotic plaque vulnerability. Circulation 138, 321–323 (2018).
Silvestre-Roig, C. et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 569, 236–240 (2019). This study showed an intriguing NET-centred mechanism of neutrophil-driven plaque destabilization.
Musher, D. M., Abers, M. S. & Corrales-Medina, V. F. Acute infection and myocardial infarction. N. Engl. J. Med. 380, 171–176 (2019).
Mawhin, M. A. et al. Neutrophils recruited by leukotriene B4 induce features of plaque destabilization during endotoxaemia. Cardiovasc. Res. 114, 1656–1666 (2018).
Schumski, A. et al. Endotoxinemia accelerates atherosclerosis via electrostatic charge-mediated monocyte adhesion. Circulation 143, 254–266 (2021).
Libby, P. & Pasterkamp, G. Requiem for the ‘vulnerable plaque’. Eur. Heart J. 36, 2984–2987 (2015).
Quillard, T. et al. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur. Heart J. 36, 1394–1404 (2015).
Franck, G. et al. Flow perturbation mediates neutrophil recruitment and potentiates endothelial injury via TLR2 in mice: implications for superficial erosion. Circ. Res. 121, 31–42 (2017).
Franck, G. et al. Roles of PAD4 and NETosis in experimental atherosclerosis and arterial injury: implications for superficial erosion. Circ. Res. 123, 33–42 (2018). Together with Franck et al. (2017), this article shed light on the importance of neutrophils and NETs during endothelial erosion.
Partida, R. A., Libby, P., Crea, F. & Jang, I. K. Plaque erosion: a new in vivo diagnosis and a potential major shift in the management of patients with acute coronary syndromes. Eur. Heart J. 39, 2070–2076 (2018).
Pasterkamp, G., den Ruijter, H. M. & Libby, P. Temporal shifts in clinical presentation and underlying mechanisms of atherosclerotic disease. Nat. Rev. Cardiol. 14, 21–29 (2017).
Kolte, D., Libby, P. & Jang, I. K. New insights into plaque erosion as a mechanism of acute coronary syndromes. JAMA 325, 1043–1044 (2021).
Jaiswal, S. & Libby, P. Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat. Rev. Cardiol. 17, 137–144 (2020).
Libby, P. & Ebert, B. L. CHIP (clonal hematopoiesis of indeterminate potential): potent and newly recognized contributor to cardiovascular risk. Circulation 138, 666–668 (2018).
Heyde, A. et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell 184, 1348–1361 (2021).
Yvan-Charvet, L. et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328, 1689–1693 (2010).
Fuster, J. J. et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 (2017).
Wang, W. et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in Jak2V617F mice. Circ. Res. 123, e35–e47 (2018).
Fidler, T. P. et al. The Aim2 inflammasome and Gasdermin D promote atherosclerotic plaque necrosis in Jak2V617F clonal hematopoiesis. Nature 592, 296–301 (2021). This recent work generated a connection between ageing-associated clonal haematopoiesis and AIM2 inflammasome activation in atheroprogression.
Collins, S. D. Excess mortality from causes other than influenza and pneumonia during influenza epidemics. Public Health Rep. 47, 2159–2179 (1932).
Guo, T. et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 5, 1–8 (2020).
Kwong, J. C. et al. Acute myocardial infarction after laboratory-confirmed influenza infection. N. Engl. J. Med. 378, 345–353 (2018).
Corrales-Medina, V. F. et al. Association between hospitalization for pneumonia and subsequent risk of cardiovascular disease. JAMA 313, 264–274 (2015).
Dalager-Pedersen, M., Søgaard, M., Schønheyder, H. C., Nielsen, H. & Thomsen, R. W. Risk for myocardial infarction and stroke after community-acquired bacteremia: a 20-year population-based cohort study. Circulation 129, 1387–1396 (2014).
Mauriello, A. et al. Diffuse and active inflammation occurs in both vulnerable and stable plaques of the entire coronary tree: a histopathologic study of patients dying of acute myocardial infarction. J. Am. Coll. Cardiol. 45, 1585–1593 (2005).
Madjid, M. et al. Influenza epidemics and acute respiratory disease activity are associated with a surge in autopsy-confirmed coronary heart disease death: results from 8 years of autopsies in 34,892 subjects. Eur. Heart J. 28, 1205–1210 (2007).
Libby, P. & Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 41, 3038–3044 (2020).
Kaynar, A. M. et al. Effects of intra-abdominal sepsis on atherosclerosis in mice. Crit. Care 18, 469 (2014).
Cangemi, R. et al. Platelet activation is associated with myocardial infarction in patients with pneumonia. J. Am. Coll. Cardiol. 64, 1917–1925 (2014).
Clark, S. R. et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 13, 463–469 (2007).
Solomon, D. H. et al. Relationship of interleukin-1β blockade with incident gout and serum uric acid levels: exploratory analysis of a randomized controlled trial. Ann. Intern. Med. 169, 535–542 (2018).
Everett, B. M. et al. Anti-Inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 139, 1289–1299 (2019).
Vallurupalli, M. et al. Effects of interleukin-1β inhibition on incident anemia: exploratory analyses from a randomized trial. Ann. Intern. Med. 172, 523–532 (2020).
Morton, A. C. et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: the MRC-ILA Heart Study. Eur. Heart J. 36, 377–384 (2015).
Abbate, A. et al. Interleukin-1 blockade inhibits the acute inflammatory response in patients with ST-segment-elevation myocardial infarction. J. Am. Heart Assoc. 9, e014941 (2020).
Everett, B. M. et al. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am. Heart J. 166, 199–207 (2013).
Ridker, P. M. et al. Low-dose methotrexate for the prevention of atherosclerotic events. N. Engl. J. Med. 380, 752–762 (2019).
Nidorf, S. M., Eikelboom, J. W., Budgeon, C. A. & Thompson, P. L. Low-dose colchicine for secondary prevention of cardiovascular disease. J. Am. Coll. Cardiol. 61, 404–410 (2013).
Mann, D. L. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ. Res. 116, 1254–1268 (2015).
Loppnow, H. & Libby, P. Adult human vascular endothelial cells express the IL6 gene differentially in response to LPS or IL1. Cell Immunol. 122, 493–503 (1989).
Loppnow, H. & Libby, P. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin-6. J. Clin. Invest. 85, 731–738 (1990).
Swerdlow, D. I. et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet 379, 1214–1224 (2012).
Sarwar, N. et al. IL6R genetics consortium emerging risk factors collaboration. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet 379, 1205–1213 (2012).
Kleveland, O. et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: a double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 37, 2406–2413 (2016).
Taleb, S., Tedgui, A. & Mallat, Z. Interleukin-17: friend or foe in atherosclerosis? Curr. Opin. Lipidol. 21, 404–408 (2010).
Ait-Oufella, H., Libby, P. & Tedgui, A. Anticytokine immune therapy and atherothrombotic cardiovascular risk. Arterioscler. Thromb. Vasc. Biol. 39, 1510–1519 (2019).
Binder, C. J., Hartvigsen, K. & Witztum, J. L. Promise of immune modulation to inhibit atherogenesis. J. Am. Coll. Cardiol. 50, 547–550 (2007).
Nilsson, J. & Hansson, G. K. Vaccination strategies and immune modulation of atherosclerosis. Circ. Res. 126, 1281–1296 (2020).
Ketelhuth, D. F., Gistera, A., Johansson, D. K. & Hansson, G. K. T cell-based therapies for atherosclerosis. Curr. Pharm. Des. 19, 5850–5858 (2013).
Zhao, T. X. et al. Low-dose interleukin-2 in patients with stable ischaemic heart disease and acute coronary syndromes (LILACS): protocol and study rationale for a randomised, double-blind, placebo-controlled, phase I/II clinical trial. BMJ Open 8, e022452 (2018).
Han, S. F. et al. The opposite-direction modulation of CD4+CD25+ Tregs and T helper 1 cells in acute coronary syndromes. Clin. Immunol. 124, 90–97 (2007).
Wolach, O. et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl Med. 10, eaan8292 (2018).
Zhao, T. X. & Mallat, Z. Targeting the immune system in atherosclerosis: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 73, 1691–1706 (2019).
Lutgens, E. et al. Immunotherapy for cardiovascular disease. Eur. Heart J. 40, 3937–3946 (2019).
Serbina, N. V. & Pamer, E. G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7, 311–317 (2006).
Boring, L., Gosling, J., Cleary, M. & Charo, I. F. Decreased lesion formation in CCR2–/– mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 394, 894–897 (1998).
Majmudar, M. D. et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 127, 2038–2046 (2013).
Liehn, E. A. et al. A new monocyte chemotactic protein-1/chemokine CC motif ligand-2 competitor limiting neointima formation and myocardial ischemia/reperfusion injury in mice. J. Am. Coll. Cardiol. 56, 1847–1857 (2010).
Georgakis, M. K. et al. Genetically determined levels of circulating cytokines and risk of stroke. Circulation 139, 256–268 (2019).
Georgakis, M. K. et al. Monocyte-chemoattractant protein-1 levels in human atherosclerotic lesions associate with plaque vulnerability. Preprint at medRxiv https://doi.org/10.1101/2020.09.04.20187955 (2020).
Gilbert, J. et al. Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am. J. Cardiol. 107, 906–911 (2011).
von Hundelshausen, P. et al. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation 103, 1772–1777 (2001).
Veillard, N. R. et al. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ. Res. 94, 253–261 (2004).
Cipriani, S. et al. Efficacy of the CCR5 antagonist maraviroc in reducing early, ritonavir-induced atherogenesis and advanced plaque progression in mice. Circulation 127, 2114–2124 (2013).
Maggi, P. et al. Effects of therapy with maraviroc on the carotid intima media thickness in HIV-1/HCV co-infected patients. Vivo 31, 125–131 (2017).
von Hundelshausen, P. et al. Chemokine interactome mapping enables tailored intervention in acute and chronic inflammation. Sci. Transl Med. 9, eaah6650 (2017).
Koenen, R. R. et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat. Med. 15, 97–103 (2009).
Eckardt, V. et al. Chemokines and galectins form heterodimers to modulate inflammation. EMBO Rep. 21, e47852 (2020).
Knight, J. S. et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 114, 947–956 (2014).
Christophorou, M. A. et al. Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 507, 104–108 (2014).
Wang, Y. et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 306, 279–283 (2004).
Wang, Y. et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 184, 205–213 (2009).
Chirivi, R. G. S. et al. Therapeutic ACPA inhibits NET formation: a potential therapy for neutrophil-mediated inflammatory diseases. Cell Mol. Immunol. https://doi.org/10.1038/s41423-020-0381-3 (2020).
Vogel, B., Shinagawa, H., Hofmann, U., Ertl, G. & Frantz, S. Acute DNase1 treatment improves left ventricular remodeling after myocardial infarction by disruption of free chromatin. Basic. Res. Cardiol. 110, 15 (2015).
Ge, L. et al. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am. J. Physiol. Heart Circ. Physiol. 308, H500–H509 (2015).
Sollberger, G. et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 3, eaar6689 (2018).
Rathkey, J. K. et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 3, eaat2738 (2018).
Massberg, S. et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 16, 887–896 (2010).
Gotwals, P. et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 17, 286–301 (2017).
Gotsman, I. et al. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J. Clin. Invest. 117, 2974–2982 (2007).
Mach, F., Schönbeck, U., Sukhova, G. K., Atkinson, E. & Libby, P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature 394, 200–203 (1998).
Lutgens, E. et al. Both early and delayed anti-CD40L antibody treatment induces a stable plaque phenotype. Proc. Natl Acad. Sci. USA 97, 7464–7469 (2000).
Lutgens, E. et al. Deficient CD40–TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J. Exp. Med. 207, 391–404 (2010).
Lutgens, E. et al. Requirement for CD154 in the progression of atherosclerosis. Nat. Med. 5, 1313–1316 (1999).
Garlichs, C. D. et al. Upregulation of CD40 and CD40 ligand (CD154) in patients with moderate hypercholesterolemia. Circulation 104, 2395–2400 (2001).
Seijkens, T. T. P. et al. Targeting CD40-induced TRAF6 signaling in macrophages reduces atherosclerosis. J. Am. Coll. Cardiol. 71, 527–542 (2018).
Lameijer, M. et al. Efficacy and safety assessment of a TRAF6-targeted nanoimmunotherapy in atherosclerotic mice and non-human primates. Nat. Biomed. Eng. 2, 279–292 (2018). Together with Seijkens et al. (2018) this study shed light on the possibility of using nanoimmunotherapy to treat atherosclerosis by targeting the CD40–TRAF6 axis.
de Boer, O. J. et al. Costimulatory molecules in human atherosclerotic plaques: an indication of antigen specific T lymphocyte activation. Atherosclerosis 133, 227–234 (1997).
Buono, C. et al. B7-1/B7-2 costimulation regulates plaque antigen-specific T-cell responses and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation 109, 2009–2015 (2004).
Matsumoto, T. et al. Overexpression of cytotoxic T-lymphocyte-associated antigen-4 prevents atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 36, 1141–1151 (2016).
Ma, K. et al. CTLA4-IgG ameliorates homocysteine-accelerated atherosclerosis by inhibiting T-cell overactivation in apoE–/– mice. Cardiovasc. Res. 97, 349–359 (2013).
Mulder, W. J. M., Ochando, J., Joosten, L. A. B., Fayad, Z. A. & Netea, M. G. Therapeutic targeting of trained immunity. Nat. Rev. Drug Discov. 18, 553–566 (2019).
Duivenvoorden, R. et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat. Commun. 5, 3065 (2014).
Coll, R. C. et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255 (2015).
Sharma, A. et al. Specific NLRP3 inhibition protects against diabetes-associated atherosclerosis. Diabetes 70, 772–787 (2021).
Mangan, M. S. J. et al. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 17, 688 (2018).
Lüsebrink, E. et al. AIM2 stimulation impairs reendothelialization and promotes the development of atherosclerosis in mice. Front. Cardiovasc. Med. 7, 582482 (2020).
Li, Y. et al. VX-765 attenuates atherosclerosis in ApoE deficient mice by modulating VSMCs pyroptosis. Exp. Cell Res. 389, 111847 (2020).
Abbate, A. et al. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ. Res. 126, 1260–1280 (2020).
Serhan, C. N. et al. The Atlas of Inflammation Resolution (AIR). Mol. Asp. Med. 74, 100894 (2020).
Chiang, N. et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 484, 524–528 (2012).
Mirakaj, V., Dalli, J., Granja, T., Rosenberger, P. & Serhan, C. N. Vagus nerve controls resolution and pro-resolving mediators of inflammation. J. Exp. Med. 211, 1037–1048 (2014).
Proto, J. D. et al. Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity 49, 666–677 (2018).
Ortega-Gómez, A., Perretti, M. & Soehnlein, O. Resolution of inflammation: an integrated view. EMBO Mol. Med. 5, 661–674 (2013).
Bäck, M., Yurdagul, A. Jr., Tabas, I., Öörni, K. & Kovanen, P. T. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat. Rev. Cardiol. 16, 389–406 (2019).
Petri, M. H. et al. Aspirin-triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E–/– mice. Br. J. Pharmacol. 174, 4043–4054 (2017).
Petri, M. H. et al. The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc. Res. 105, 65–74 (2015).
Fredman, G. et al. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci. Transl Med. 7, 275ra20 (2015).
Drechsler, M. et al. Annexin A1 counteracts chemokine-induced arterial myeloid cell recruitment. Circ. Res. 116, 827–835 (2015).
Fredman, G. et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 7, 12859 (2016).
Cash, J. L. et al. Resolution mediator chemerin15 reprograms the wound microenvironment to promote repair and reduce scarring. Curr. Biol. 24, 1406–1414 (2014).
Cash, J. L. et al. Chemerin15 inhibits neutrophil-mediated vascular inflammation and myocardial ischemia-reperfusion injury through ChemR23. EMBO Rep. 14, 999–1007 (2013).
Chang, C. et al. Chemerin15-ameliorated cardiac ischemia–reperfusion injury is associated with the induction of alternatively activated macrophages. Mediators Inflamm. 2015, 563951 (2015).
El Kebir, D., Gjorstrup, P. & Filep, J. G. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc. Natl Acad. Sci. USA 109, 14983–14988 (2012).
López-Vicario, C. et al. Association of a variant in the gene encoding for ERV1/ChemR23 with reduced inflammation in visceral adipose tissue from morbidly obese individuals. Sci. Rep. 7, 15724 (2017).
Laguna-Fernandez, A. et al. ERV1/ChemR23 signaling protects against atherosclerosis by modifying oxidized low-density lipoprotein uptake and phagocytosis in macrophages. Circulation 138, 1693–1705 (2018).
van der Vorst, E. P. C. et al. Hematopoietic ChemR23 (chemerin receptor 23) fuels atherosclerosis by sustaining an M1 macrophage-phenotype and guidance of plasmacytoid dendritic cells to murine lesions—brief report. Arterioscler. Thromb. Vasc. Biol. 39, 685–693 (2019).
Hasturk, H. et al. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler. Thromb. Vasc. Biol. 35, 1123–1133 (2015).
Salic, K. et al. Resolvin E1 attenuates atherosclerosis in absence of cholesterol-lowering effects and on top of atorvastatin. Atherosclerosis 250, 158–165 (2016).
Viola, J. R. et al. Resolving lipid mediators Maresin 1 and Resolvin D2 prevent atheroprogression in mice. Circ. Res. 119, 1030–1038 (2016).
ASCEND Study Collaborative Group, Bowman, L. et al. Effects of n-3 fatty acid supplements in diabetes mellitus. N. Engl. J. Med. 379, 1540–1550 (2018).
Aung, T. et al. Associations of ω-3 fatty acid supplement use with cardiovascular disease risks: meta-analysis of 10 trials involving 77917 individuals. Omega-3 Treatment Trialists’ Collaboration. JAMA Cardiol. 3, 225–234 (2018).
Manson, J. E. et al. Marine n-3 fatty acids and prevention of cardiovascular disease and cancer. N. Engl. J. Med. 380, 23–32 (2019).
Nicholls, S. J. et al. Effect of high-dose ω-3 fatty acids vs corn oil on major adverse cardiovascular events in patients at high cardiovascular risk: the STRENGTH randomized clinical trial. JAMA 324, 2268–2280 (2020).
Thorp, E. et al. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase C, and p38 mitogen-activated protein kinase (MAPK). J. Biol. Chem. 286, 33335–33344 (2011).
Cai, B. et al. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J. Clin. Invest. 127, 564–568 (2017).
Doran, A. C. et al. CAMKIIγ suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis. J. Clin. Invest. 127, 4075–4089 (2017).
Kojima, Y. et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536, 86–90 (2016).
Yurdagul, A. Jr. et al. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. 31, 518–533 (2020).
Mure, L. S. et al. Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 359, eaao0318 (2018). This important study generated an atlas of diurnal transcription of 64 tissues harvested in 2-h intervals from baboons.
Ruben, M. D. et al. A database of tissue-specific rhythmically expressed human genes has potential applications in circadian medicine. Sci. Transl Med. 10, eaat8806 (2018).
de Juan, A. et al. Artery-associated sympathetic innervation drives rhythmic vascular inflammation of arteries and veins. Circulation 140, 1100–1114 (2019).
Ruben, M. D., Smith, D. F., FitzGerald, G. A. & Hogenesch, J. B. Dosing time matters. Science 365, 547–549 (2019).
Fitzgerald, K. et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N. Engl. J. Med. 376, 41–51 (2017).
Fitzgerald, K. et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet 383, 60–68 (2014).
van der Valk, F. M. et al. Prednisolone-containing liposomes accumulate in human atherosclerotic macrophages upon intravenous administration. Nanomedicine 11, 1039–1046 (2015).
Chan, J. M. et al. Spatiotemporal controlled delivery of nanoparticles to injured vasculature. Proc. Natl Acad. Sci. USA 107, 2213–2218 (2010).
Kamaly, N. et al. Targeted interleukin-10 nanotherapeutics developed with a microfluidic chip enhance resolution of inflammation in advanced atherosclerosis. ACS Nano 10, 5280–5292 (2016).
Molinaro, R. et al. Targeted delivery of protein arginine deiminase-4 inhibitors to limit arterial intimal NETosis and preserve endothelial integrity. Cardiovasc Res. https://doi.org/10.1093/cvr/cvab074 (2021).
Silvestre-Roig et al. Arterial delivery of VEGF-C stabilizes atherosclerotic lesions. Circ. Res. 128, 284–286 (2021).
Tang, J. et al. Immune cell screening of a nanoparticle library improves atherosclerosis therapy. Proc. Natl Acad. Sci. USA 113, E6731–E6740 (2016).
Flores, A. M. et al. Pro-efferocytic nanoparticles are specifically taken up by lesional macrophages and prevent atherosclerosis. Nat. Nanotechnol. 15, 154–161 (2020). This recent study utilized ‘trojan horse’ single-walled carbon nanotubes to specifically interfere with the functionality of lesional macrophages.
Tao, W. et al. siRNA nanoparticles targeting CaMKIIγ in lesional macrophages improve atherosclerotic plaque stability in mice. Sci. Transl Med. 12, eaay1063 (2020).
Winter, P. M. et al. Endothelial αvβ3 integrin-targeted fumagillin nanoparticles inhibit angiogenesis in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 26, 2103–2109 (2006).
Flores, A. M. et al. Nanoparticle therapy for vascular diseases. Arterioscler. Thromb. Vasc. Biol. 39, 635–646 (2019).
Duivenvoorden, R. et al. Nanoimmunotherapy to treat ischaemic heart disease. Nat. Rev. Cardiol. 16, 21–32 (2019).
Chomistek, A. K. et al. Healthy lifestyle in the primordial prevention of cardiovascular disease among young women. J. Am. Coll. Cardiol. 65, 43–51 (2015).
Akesson, A., Larsson, S. C., Discacciatim, A. & Wolk, A. Low-risk diet and lifestyle habits in the primary prevention of myocardial infarction in men: a population-based prospective cohort study. J. Am. Coll. Cardiol. 64, 1299–1306 (2014).
Khera, A. V. et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N. Engl. J. Med. 375, 2349–2358 (2016).
Strandberg, T. E., Libby, P. & Kovanen, P. T. A tale of two therapies lipid-lowering vs anti-inflammatory therapy — a false dichotomy? Eur. Heart J. Cardiovasc. Pharmacother. https://doi.org/10.1093/ehjcvp/pvaa131 (2020).
O’Donoghue, M. L. et al. Effect of losmapimod on cardiovascular outcomes in patients hospitalized with acute myocardial infarction: a randomized clinical trial. JAMA 315, 1591–1599 (2016).
Mease, P. J. et al. Secukinumab inhibition of interleukin-17A in patients with psoriatic arthritis. N. Engl. J. Med. 373, 1329–1339 (2015).
Winkels, H. et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Circ. Res. 122, 1675–1688 (2018).
Xie, X. et al. Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat. Immunol. 21, 1119–1133 (2020).
Ballesteros, I. et al. Co-option of neutrophil fates by tissue environments. Cell 183, 1282–1297 (2020).
Pan, H. et al. Single-cell genomics reveals a novel cell state during smooth muscle cell phenotypic switching and potential therapeutic targets for atherosclerosis in mouse and human. Circulation 142, 2060–2075 (2020).
Ma, W. F. et al. Single-cell RNA-seq analysis of human coronary arteries using an enhanced workflow reveals SMC transitions and candidate drug targets. Preprint at bioRxiv https://doi.org/10.1101/2020.10.27.357715 (2020).
Keren, L. et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell 174, 1373–1387 (2018).
Goltsev, Y. et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174, 968–981 (2018).
Vickovic, S. et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 16, 987–990 (2019).
Eng, C. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239 (2019).
Liu, Y. et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell 183, 1665–1681 (2020).
Westerterp, M. et al. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell 11, 195–206 (2012).
Gu, Q. et al. AIBP-mediated cholesterol efflux instructs hematopoietic stem and progenitor cell fate. Science 363, 1085–1088 (2019).
Takubo, K. et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12, 49–61 (2013).
Nagareddy, P. R. et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 17, 695–708 (2013).
McAlpine, C. S. et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature 566, 383–387 (2019).
Heidt, T. et al. Chronic variable stress activates hematopoietic stem cells. Nat. Med. 20, 754–758 (2014).
Frodermann, V. et al. Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells. Nat. Med. 25, 1761–1771 (2019). Together with McAlpine et al. (2019) and Heidt et al. (2014), this paper established the bone marrow as an important link between cardiovascular risk factors and lesional inflammation.
Pietras, E. M. et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 18, 607–618 (2016).
Sager, H. B. et al. Targeting interleukin-1β reduces leukocyte production after acute myocardial infarction. Circulation 132, 1880–1890 (2015).
Méndez-Ferrer, S., Lucas, D., Battista, M. & Frenette, P. S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452, 442–447 (2008).
Dutta, P. et al. Myocardial infarction accelerates atherosclerosis. Nature 487, 325–329 (2012).
Krohn-Grimberghe, M. et al. Nanoparticle-encapsulated siRNAs for gene silencing in the haematopoietic stem-cell niche. Nat. Biomed. Eng. 4, 1076–1089 (2020).
Lee, D. C. et al. Leisure-time running reduces all-cause and cardiovascular mortality risk. J. Am. Coll. Cardiol. 64, 472–481 (2014).
Noz, M. P. et al. Sixteen-week physical activity intervention in subjects with increased cardiometabolic risk shifts innate immune function towards a less proinflammatory state. J. Am. Heart Assoc. 8, e013764 (2019).
Berg, K. E. et al. Elevated CD14++CD16– monocytes predict cardiovascular events. Circ. Cardiovasc. Genet. 5, 122–131 (2012).
Horne, B. D. et al. Which white blood cell subtypes predict increased cardiovascular risk? J. Am. Coll. Cardiol. 45, 1638–1643 (2005).
Martínez-González, M. A., Gea, A. & Ruiz-Canela, M. The Mediterranean diet and cardiovascular health. Circ. Res. 124, 779–798 (2019).
Estruch, R. et al. Primary prevention of cardiovascular disease with a mediterranean diet supplemented with extra-virgin olive oil or nuts. N. Engl. J. Med. 378, e34 (2018).
Neeland, I. J. et al. Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: a position statement. Lancet Diabetes Endocrinol. 7, 715–725 (2019).
Koeth, R. A. et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 19, 576–585 (2013).
Wang, Z. et al. Impact of chronic dietary red meat, white meat, or non-meat protein on trimethylamine N-oxide metabolism and renal excretion in healthy men and women. Eur. Heart J. 40, 583–594 (2019).
Haghikia, A. et al. Gut microbiota-dependent trimethylamine N-oxide predicts risk of cardiovascular events in patients with stroke and is related to proinflammatory monocytes. Arterioscler. Thromb. Vasc. Biol. 38, 2225–2235 (2018).
Malinowski, B. et al. Intermittent fasting in cardiovascular disorders — an overview. Nutrients 11, 673 (2019).
Teng, N. I. et al. Improvement of metabolic parameters in healthy older adult men following a fasting calorie restriction intervention. Aging Male 16, 177–183 (2013).
Bhutani, S., Klempel, M. C., Kroeger, C. M., Trepanowski, J. F. & Varady, K. A. Alternate day fasting and endurance exercise combine to reduce body weight and favorably alter plasma lipids in obese humans. Obesity 21, 1370–1379 (2013).
Erdem, Y. et al. The effect of intermittent fasting on blood pressure variability in patients with newly diagnosed hypertension or prehypertension. J. Am. Soc. Hypertens. 12, 42–49 (2018).
Sutton, E. F. et al. Early time-restricted feeding improves insulin sensitivity, blood pressure, and oxidative stress even without weight loss in men with prediabetes. Cell Metab. 27, 1212–1221 (2018).
Wilkinson, M. J. et al. Ten-hour time-restricted eating reduces weight, blood pressure, and atherogenic lipids in patients with metabolic syndrome. Cell Metab. 31, 92–104 (2020).
Trepanowski, J. F. et al. Effect of alternate-day fasting on weight loss, weight maintenance, and cardioprotection among metabolically healthy obese adults: a randomized clinical trial. JAMA Intern. Med. 177, 930–938 (2017).
Harvie, M. et al. The effect of intermittent energy and carbohydrate restriction v. daily energy restriction on weight loss and metabolic disease risk markers in overweight women. Br. J. Nutr. 110, 1534–1547 (2013).
Moro, T. et al. Effects of eight weeks of time-restricted feeding (16/8) on basal metabolism, maximal strength, body composition, inflammation, and cardiovascular risk factors in resistance-trained males. J. Transl Med. 14, 290 (2016).
Jordan, S. et al. Dietary intake regulates the circulating inflammatory monocyte pool. Cell 178, 1102–1114 (2019).
van der Valk, F. M. et al. Increased haematopoietic activity in patients with atherosclerosis. Eur. Heart J. 38, 425–432 (2017).
Seijkens, T. et al. Hypercholesterolemia-induced priming of hematopoietic stem and progenitor cells aggravates atherosclerosis. FASEB J. 28, 2202–2213 (2014).
van Kampen, E., Jaminon, A., van Berkel, T. J. & Van Eck, M. Diet-induced (epigenetic) changes in bone marrow augment atherosclerosis. J. Leukoc. Biol. 96, 833–841 (2014).
Chevre, R., Silvestre-Roig, C. & Soehnlein, O. Nutritional modulation of innate immunity: the fat–bile–gut connection. Trends Endocrinol. Metab. 29, 686–698 (2018).
Acknowledgements
The authors thank K. Szeler (Karolinska Institute, Stockholm) for analysing data displayed in Fig. 3a. The authors receive funding from the Deutsche Forschungsgemeinschaft (SFB914 TP B8, SFB1123 TP A6, TP B5), the Vetenskapsrådet (2017-01762), the Else-Kröner-Fresenius Stiftung, the Swedish Heart–Lung Foundation and the Leducq foundation. P.L. receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892), the American Heart Association (18CSA34080399), the RRM Charitable Fund and the Simard Fund.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
O.S. holds two patents on targeting histones in inflammation and one on disrupting CCL5-HNP1 heteromers, and also receives funding from Novo Nordisk to study chronopharmacological treatment strategies in cardiovascular disease. P.L. is a member of the scientific advisory board for Amgen, Corvidia Therapeutics, DalCor Pharmaceuticals, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis and XBiotech, Inc. P.L.’s laboratory has received research funding in the past 2 years from Novartis. P.L. is on the Board of Directors of XBiotech, Inc. P.L. has a financial interest in Xbiotech, a company developing therapeutic human antibodies. P.L.’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies.
Additional information
Peer review information
Nature Reviews Drug Discovery thanks Martin Lefkowitz, Willem Mulder and the other, anonymous, reviewer for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Glossary
- Myocardial infarction
-
A disruption of blood flow to heart tissue. Commonly known as a heart attack.
- Statins
-
A class of drugs, also known as 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors, that reduce levels of low-density lipoprotein by impairing cholesterol biosynthetic pathways.
- Acute coronary syndromes
-
(ACS). A set of symptoms indicative of reduced blood flow to the heart. Myocardial infarction is a subtype of ACS.
- Tunica media
-
The thickest layer of the arterial wall, composed of smooth muscle cells, fibroblasts and extracellular matrix.
- Adventitia
-
The outermost layer of the wall of a blood vessel.
- β-Glucan
-
One of the main components of bacterial cell walls.
- Neutrophil extracellular traps
-
(NETs). Networks of extracellular macromolecules secreted by neutrophils. Bacteria bind to these traps as part of the host defence system.
- Endotoxaemia
-
The presence of bacterially produced endotoxins in the blood. Endotoxaemia can progress to septic shock.
- NETosis
-
Neutrophil extracellular trap activation and release.
- ST-elevation myocardial infarction
-
(STEMI). A subtype of myocardial infarction, along with non-ST-elevation myocardial infarction, defined by changes in electrocardiogram recordings.
- Ischaemia–reperfusion injury
-
Tissue damage caused when blood supply is restored.
- Enhanced permeability and retention
-
The proposed mechanism through which small molecules tend to accumulate in highly vascularized tissues, such as tumours.
Rights and permissions
About this article

Cite this article
Soehnlein, O., Libby, P. Targeting inflammation in atherosclerosis — from experimental insights to the clinic. Nat Rev Drug Discov 20, 589–610 (2021). https://doi.org/10.1038/s41573-021-00198-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-021-00198-1
This article is cited by
-
Effects of lifestyle factors on leukocytes in cardiovascular health and disease
Nature Reviews Cardiology (2024)
-
Cardiovascular disease and cancer: shared risk factors and mechanisms
Nature Reviews Cardiology (2024)
-
Dysregulated cellular metabolism in atherosclerosis: mediators and therapeutic opportunities
Nature Metabolism (2024)
-
A lipidome landscape of aging in mice
Nature Aging (2024)
-
Specialized pro-resolving mediators in vascular inflammation and atherosclerotic cardiovascular disease
Nature Reviews Cardiology (2024)
