
Abstract
Protein kinases regulate nearly all aspects of cell life, and alterations in their expression, or mutations in their genes, cause cancer and other diseases. Here, we review the remarkable progress made over the past 20 years in improving the potency and specificity of small-molecule inhibitors of protein and lipid kinases, resulting in the approval of more than 70 new drugs since imatinib was approved in 2001. These compounds have had a significant impact on the way in which we now treat cancers and non-cancerous conditions. We discuss how the challenge of drug resistance to kinase inhibitors is being met and the future of kinase drug discovery.
Similar content being viewed by others
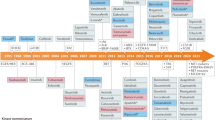

Introduction
In 2001, the first kinase inhibitor, imatinib, received FDA approval, providing the catalyst for an article with the provocative title ‘Protein kinases — the major drug targets of the twenty-first century?’1. Imatinib inhibits the Abelson (ABL) tyrosine kinase, which is expressed as a deregulated fusion protein, termed BCR–ABL, in nearly all cases of chronic myeloid leukaemia (CML)2 and is produced by a chromosome rearrangement that fuses the genes encoding the breakpoint cluster region protein (BCR) and ABL to form the Philadelphia chromosome. Although fasudil (an inhibitor of RHO-dependent protein kinases) and rapamycin (sirolimus, an inhibitor of the protein kinase TORC1) were approved earlier than 2001 (Supplementary Table 1), these compounds were developed and approved without knowledge of the identity of their target proteins. Imatinib was therefore the first drug that was developed by targeting a specific protein kinase to treat a disease to be approved.
At the time of writing the article highlighted above, small-molecule inhibitors of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) were on the cusp of approval and, as predicted, two such inhibitors — gefitinib (also known as Iressa) and erlotinib (also known as Tarceva) — were approved soon afterwards for the treatment of non-small-cell lung cancer (NSCLC) (Fig. 1). These drugs were originally designed to inhibit the wild-type version of the EGFR, which had been shown in the late 1980s to be overexpressed in many cancer types and to be associated with a poor prognosis3,4. However, it was the discovery that profound tumour sensitivity to these inhibitors was associated with particular activating EGFR mutations that are present in 10–15% of Western and 30–50% of Asian patients with NSCLC5,6,7,8 that ultimately led to the clinical use of these drugs in patients with EGFR mutations.

Events involving tyrosine kinases are in pink boxes, those involving serine/threonine-specific protein kinases are in blue boxes and those involving phosphatidylinositol 3-kinases (PI3Ks) are in grey boxes. BTK, Bruton’s tyrosine kinase; CML, chronic myeloid leukaemia; CLL, chronic lymphocytic leukaemia; EGFR, epidermal growth factor receptor; FGFR, fibroblast growth factor receptor; GIST, gastrointestinal tumour; NTRK, neurotrophic receptor tyrosine kinase; TKI, tyrosine kinase inhibitor.
The success of these pioneering compounds, especially the spectacular efficacy of imatinib as soon as it entered clinical trials in 1998, changed the perception of protein kinases as drug targets, which had previously received scepticism from many pharmaceutical companies. Since then, hundreds of protein kinase inhibitors have been developed and tested in humans and, at the time of writing, 76 have been approved for clinical use, mainly for the treatment of various cancers (Fig. 1; Supplementary Table 1). The clinical application of kinase inhibitors has also been facilitated by large-scale genomic efforts, which have identified subsets of cancers that can potentially be treated with a kinase inhibitor9,10.
In this review marking the 20th anniversary of the approval of imatinib, we discuss the progress that has been made in improving the development of potent and specific small-molecule tyrosine kinase inhibitors (TKIs) and in combatting the problem of resistance to these inhibitors in cancer therapy, the potential of combination therapies, the efficacy of kinase inhibitors in the clinic compared with other types of therapy and the exploitation of kinase inhibitors for the treatment of diseases other than cancer. Finally, we try to predict what the future of kinase drug discovery will be over the next 20 years.
Lessons from imatinib, gefitinib and erlotinib
When imatinib was first discovered, it was not initially given high priority for further development because of the low incidence of CML. Consequently, it only entered clinical trials 5 years later. So why did imatinib become one of the world’s most commercially successful drugs with peak sales of US$4.6 billion in 2012 before it went off patent in 2015? The first reason is that imatinib transformed CML from a rapidly fatal disease to a manageable condition, so that CML is no longer a rare leukaemia, with more than 100,000 patients now requiring imatinib daily to ensure their survival. It is indeed an irony that such a spectacular cancer therapy actually increases the incidence of cancer because it manages, but does not cure, the disease.
Second, imatinib was found to be equally effective in treating other rare cancers, such as gastrointestinal tumours (GIST), because it inhibits not only BCR–ABL but also two other protein kinases (the KIT and PDGF receptors), which are overexpressed or mutated in these and other cancers. The ‘polypharmacology’ of imatinib therefore turned out to be beneficial because it enabled the same drug to be exploited for the treatment of several types of cancer. Finally, because of its remarkable efficacy, the approval of imatinib was, at the time, the fastest in FDA history and relatively little marketing was needed compared with most drugs, as there were no other treatment options for patients with CML. Thus, the huge costs of drug development were minimized.
The remarkable efficacy of imatinib in CML can also be attributed to another factor. First, like other haematological cancers, CML is accessible to liquid biopsy, which improves the speed and efficiency of diagnosis, enabling treatment to begin at an early stage of disease progression (chronic phase). However, imatinib treatment in advanced disease (for example, blast crisis) is considerably less effective. The realization that early diagnosis and treatment can have a major impact on the efficacy of kinase inhibitors is now emerging as a crucial area for the improved treatment of solid tumours, as discussed later.
The significant benefit observed for BCR–ABL and EGFR inhibitors in specific molecularly characterized tumour settings contributed to an enduring paradigm shift towards precision medicine, which has influenced cancer treatment for the past 20 years and continues to do so. Since these initial observations, the clinical development of kinase inhibitors has moved away from the serendipitous discovery of exceptional responders within a broad population of patients, to the specific evaluation of new inhibitors in genomically defined subsets of cancer patients.
Trends in targeting tyrosine kinase activity
First-in-class versus next generation
The early selection of kinase targets for therapeutic intervention in oncology broadly followed two strategies. First, to focus on patients with mutations in the targets of approved kinase inhibitors. Second, to develop more effective next-generation versions of approved TKIs against these oncogenic kinase targets, which has become a successful and enduring theme.
Early examples of next-generation drugs include nilotinib11 and dasatinib12, which are more potent inhibitors of BCR–ABL than imatinib and give a more complete cytogenetic response13. Subsequently, bosutinib was approved as a second-generation inhibitor of BCR–ABL, and it was reported to achieve longer progression-free survival (PFS) and overall survival than either dasatinib or nilotinib, although its cytogenetic response was inferior14. Importantly, bosutinib, dasatinib and nilotinib also inhibit many imatinib-resistant mutants, and bosutinib and dasatinib inhibit imatinib-resistant mutants that nilotinib does not15. Bosutinib has been approved as a first-line therapy for CML because of its advantages over imatinib16. In addition, in contrast to imatinib, bosutinib does not inhibit the KIT receptor or PDGFR significantly. However, bosutinib and nilotinib are ineffective against the T315I mutant17, which led to the development and approval of ponatinib, the only ABL kinase inhibitor effective against this gatekeeper mutant18,19.
Lapatinib, another next-generation inhibitor, targets not only EGFR receptors but also the closely related HER2 (ref.20), which led to its approval for breast cancer (Fig. 1; Supplementary Table 1). Recently, more potent and specific inhibitors of HER2, neratinib and tucatinib, have been approved for breast cancer (Supplementary Table 1).
Crizotinib, which was initially developed as a MET inhibitor, was later discovered during its clinical development to also be an ALK and ROS1 inhibitor. Although its inhibition of ALK was sufficient to gain regulatory approval, the subsequent development of more selective and potent next-generation ALK inhibitors, including alectinib, brigatinib and lorlatinib, has led to crizotinib largely being replaced21,22,23,24,25,26,27. All three agents led to an improvement in PFS compared with crizotinib, while alectinib also leads to an improvement in overall survival compared with crizotinib, when used as the initial therapy in advanced ALK-positive NSCLC21,22,23,24,25,26,27.
This paradigm has continued with the development and approval of multiple drugs that target ROS1 (entrectinib and crizotinib), RET (selpercatinib and pralsetinib) and MET (capmatinib and tepotinib) for NSCLC, FGF receptors (erdafitinib) for bladder cancer, gene fusions of the neurotrophic receptor tyrosine kinase (NRTK) for thyroid, salivary and other solid tumours (larotrectinib and entrectinib) and Bruton’s tyrosine kinase for haematological cancers (ibrutinib, acalabrutinib and zanubrutinib) (Fig. 1; Supplementary Table 1).
Targeting angiogenesis
The development of TKIs that inhibit angiogenesis held promise because, at least in theory, such drugs should have clinical benefit for the treatment of many solid tumours by destroying blood vessels supplying these tumours28. Thus, many TKIs were developed with polypharmacology that included inhibition of VEGFRs and FLTRs, the receptor tyrosine kinases (RTKs) with crucial roles in angiogenesis. Several TKIs were approved for renal cell carcinoma (RCC) and other solid tumours. Early examples were sorafenib29 and sunitinib30, followed by the second-generation inhibitors pazopanib31,32 and cabozantinib33 (Supplementary Table 1). However, some of these drugs were later found to be multi-targeted, inhibiting so many protein kinases that it is unclear whether their clinical efficacy stems from inhibition of tyrosine kinases controlling angiogenesis or from the inhibition of other protein kinases. The most convincing evidence for the efficacy of VEGFR inhibitors comes from the success of the anti-VEGF antibody bevacizumab (Avastin), the world’s seventh best-selling drug in 2018, which is used to treat several solid tumours.
Polypharmacology versus selective inhibitors
It was the introduction of large panels of protein kinases to assess the specificities of protein kinase inhibitors34,35 that led to the realization that many inhibitors developed by targeting one particular protein kinase also inhibited others, resulting in some drugs being repurposed for the treatment of different cancers. Although the most selective kinase inhibitors are not entirely specific, potent activity is frequently restricted to just a few kinases that are closely related, which may enhance their efficacy for particular cancers. They include the targeting of both HER2 and EGFR by afatinib in lung cancer, and the targeting of both CDK4 and CDK6 by palbociclib in breast cancer. It also became clear that the clinical efficacy of some drugs was explained entirely by their off-target effects. For example, sorafenib (also known as BAY-43-9006) was originally developed as an inhibitor of the RAF family of serine/threonine kinases and approved for the treatment of RCC. However, the inhibition of RAF is unlikely to underlie its efficacy in RCC because, in contrast to many other cancers (see the following section), BRAF mutations are not observed in RCC. Similarly, the efficacy of ibrutinib in treating Waldenstrom’s macroglobulinaemia and the ABC subtype of diffuse large B cell lymphoma may be explained by its ability to inhibit haematopoietic cell kinase as well as Bruton’s tyrosine kinase, and not just the latter as believed initially36. A further example is crizotinib, which was originally designed and developed as a MET-targeted TKI, but was subsequently found to be a potent inhibitor of ALK and ROS1, enabling it to be approved for other indications37,38.
Protein kinase inhibitors that exhibit polypharmacology can have improved efficacy and the ability to treat several types of cancer, but this may also underlie their more serious adverse side effects. For example, dasatinib inhibits a number of other protein kinases in addition to BCR–ABL, which may underlie some of its more serious side effects compared with nilotinib39.
The introduction and exploitation of kinobead technology40 to assess the specificities of protein kinase inhibitors led to the realization that they can also inhibit proteins that are not protein kinases, such as G protein-coupled receptors41 and bromodomain (BRD)-containing proteins42. The polypharmacology of protein kinase inhibitors can therefore extend beyond the inhibition of protein kinases alone. Kinase inhibitors that are undergoing clinical trials (the polo-like kinase 1 (PLK1) inhibitor BI2536) or are approved (the JAK2 inhibitor fedratinib) target BRD4. Since BRD inhibitors profoundly affect the transcription of many genes, it is likely that such polypharmacology contributes to the efficacy and/or adverse side effects of these compounds. The inhibition of BRD4, and not PLK1, explains why BI2536 blocks interferon-β gene transcription43 and may therefore greatly increase susceptibility to infection by viruses, including coronaviruses. Indeed, 10% of life-threatening COVID-19 infections are caused by the presence of neutralizing autoantibodies to type I interferons in patients44, and a further 3.5% have genetic deficiencies that prevent type I interferon production45.
Targeting the classical MAP kinase cascade by combination therapy
The activation of RTKs triggers the conversion of the small guanine nucleotide-binding protein RAS from its inactive GDP-bound state into the active GTP-bound state. GTP–RAS then switches on the classical mitogen-activated protein (MAP) kinase pathway, a ‘cascade’ of protein serine/threonine kinases that activate one another sequentially. GTP–RAS activates the isoforms of RAF, which then activate MEK1 and MEK2. MEK1/MEK2 activate ERK1 and ERK2, which, in turn, activate several other kinases (Fig. 2). Together, these protein kinases phosphorylate a myriad of intracellular targets to drive the proliferation or differentiation of cells46,47,48.

The classical mitogen-activated protein (MAP) kinase cascade is frequently hyperactivated in lung and other cancers owing to the overexpression or mutation of receptor tyrosine kinases, such as the epidermal growth factor receptor (EGFR), ALK and MET, and more downstream effectors that are most commonly RAS and BRAF. Examples of drugs that target the EGFR (gefitinib, erlotinib and osimertinib), ALK (crizotinib, brigatinib and alectinib) and MET (capmatinib and tepotinib) are highlighted in red. The three approved inhibitors of BRAF and the four approved inhibitors of MEK1 and MEK2 are also shown.
An early success that stemmed from sequencing the entire human genome was the discovery that the gene encoding the BRAF isoform is mutated to deregulated and highly active forms in many cancers. One particularly frequent mutation in which valine 600 is mutated to glutamic acid is found in 50% of patients with malignant melanoma49, the most aggressive type of skin cancer. Vemurafenib inhibits the BRAF-V600E mutant about fivefold more potently than wild-type BRAF and about 100-fold more potently than CRAF50, and was the first RAF inhibitor to be approved for clinical use. The initial effects of vemurafenib were impressive, with overt signs of melanoma disappearing within weeks in many patients. However, the remission was not long-lasting owing to the presence and subsequent proliferation of residual melanoma cells that were resistant to vemurafenib, causing the melanoma to return within weeks to months. Similar observations were made with dabrafenib, another RAF inhibitor, and with trametinib, an inhibitor of MEK1 and MEK2, the protein kinases activated by RAF (Fig. 2).
Interestingly, the combination of dabrafenib and trametinib delays the reappearance of metastatic melanoma significantly, with survival rates of 37% and 34% after 4 and 5 years, respectively, and with complete responses seen in 19% of these patients51. The combination of dabrafenib and trametinib improves relapse-free survival in patients with NSCLC expressing BRAF-V600E when given after surgery52,53,54,55. The combination of vemurafenib and the MEK inhibitor cobimetinib, as well as the combination of the BRAF-V600E inhibitor encorafenib and the MEK inhibitor binimetinib, have also been approved for treatment of melanoma (Fig. 2).
The enhanced efficacy of combining BRAF inhibitors with MEK inhibitors may be explained not only by targeting two kinases that act sequentially in the same signalling pathway, but also because RAF inhibitors when used alone paradoxically activate cells expressing wild-type RAF. This is caused, at least in part, by drug-induced heterodimerization and activation of different RAF isoforms56,57,58. MEK inhibitors can therefore suppress bypass caused by the activation of other RAF isoforms and other RAF–MEK complexes and so ameliorate serious safety issues such as the development of squamous cell carcinomas, which have been observed in patients treated with BRAF-V600E inhibitors alone59.
RAF and MEK interact with KSR1, an inactive pseudokinase that is structurally related to RAF. Interestingly, trametinib causes MEK to engage KSR1 more effectively, which may facilitate the ability of particular combinations of RAF and MEK inhibitors to target RAF–KSR–MEK complexes60. The first dual RAF–MEK inhibitor VS6766 (also known as CH5126766) has entered phase I clinical trials and has shown some efficacy in solid tumours and in multiple myeloma harbouring various RAS–RAF mutations61.
Other early-stage clinical trials are ongoing in which MEK inhibitors are being combined with other kinase inhibitors, for example, with one of the HER/EGFR inhibitors lapatinib or dacomitinib62 for KRAS-mutation-positive colorectal, non-small cell lung and pancreatic cancers.
Combining kinase inhibitors with checkpoint inhibitors
The proteins programmed cell death 1 ligand 1 (PDL1) and its receptor programmed cell death 1 (PD1) restrict the activation of immune cells, and antibodies targeting these proteins, termed immuno-oncology checkpoint inhibitors, enhance the ability of immune cells to destroy tumours. This can have significant benefit in many patients with advanced metastatic tumours in multiple areas of oncology. However, there is still a need to improve overall response rates and survival63. One strategy that is emerging is to combine immuno-oncology checkpoint inhibitors with small-molecule kinase inhibitors. A rationale for using these combinations is that angiogenesis is important in establishing an immunosuppressive microenvironment64. Second, some tumours, such as renal tumours, are associated with high sensitivity to VEGFR inhibitors owing to alterations in the Von Hippel–Landau gene that lead to upregulation of pro-angiogenesis genes65. Consequently, multiple studies are now investigating whether the combination of VEGFR inhibitors with antibodies that target PD1 or PDL1 (for example, axitinib and pembrolizumab) improves the treatment of RCC and other cancers.
There is also evidence that some kinase inhibitors can potentially prime and stimulate immune responses against tumours by modulating the tumour microenvironment, for example, by preventing the increased expression of PDL1 or by enhancing T cell infiltration, or because their cytotoxic activity is associated with an ‘immunogenic cell death’ phenotype66,67. Therefore, combining immuno-oncology checkpoint inhibitors with appropriate kinase inhibitors may confer a synergistic response in oncogenic addiction settings. This has led to the evaluation of other drug combinations, such as the PD1 inhibitor spartalizumab with dabrafenib and trametinib in BRAF-V600E melanoma68, although this combination recently failed to reach its primary PFS end point in a phase III trial69. The combination of protein kinase inhibitors and immuno-oncology checkpoint inhibitors may also cause enhanced toxicity that could limit clinical utility, for example, as seen when osimertinib was combined with the PDL1 inhibitor durvalumab70 or crizotinib with nivolumab71. Therefore, the effectiveness of this strategy requires further evaluation.
There is also evidence that some signalling pathways driven by RTKs can modulate the tumour microenvironment independently of cancer cells. For example, MET has been implicated in regulating tumour-associated neutrophil recruitment72, which has led to the initiation of studies to explore whether the combination of MET inhibitors with immuno-oncology checkpoint inhibitors has clinical benefit in MET-driven and broader disease populations, such as the combination of capmatinib and pembrolizumab in NSCLC73.
Targeting the PI3K pathway
The activation of RTKs triggers activation not only of the RAS–RAF–MEK–ERK pathway, but also of type 1 PI3Ks. These plasma membrane-associated lipid kinases catalyse the conversion of phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-trisphosphate (PIP3), which mediates some of the intracellular actions of RTKs by activating the protein serine/threonine kinases PDK1 and AKT. The actions of PI3Ks are antagonized by phosphatases that reconvert PIP3 into PIP2, which include phosphatase and tensin homologue deleted on chromosome 10 (PTEN)74.
Activating hotspot mutations in PI3Kα such as the E545K, H1047 and E542K mutations, or deletion of the gene encoding PTEN, frequently lead to oncogenic transformation, which has made PI3Ks an intensive focus of drug discovery. However, so far, there has been limited success in developing approved drugs against PI3Ks. Idelalisib75, a specific inhibitor of PI3Kδ, which is expressed in B and T cells, has been approved for relapsed follicular B cell non-Hodgkin lymphoma (FL), relapsed chronic lymphocytic leukaemia (CLL) and relapsed small lymphocytic lymphoma (SLL), and was followed by the next-generation inhibitors copanlisib (for relapsed FL) and duvelisib (for CLL, SLL and FL)76. Only recently has a PI3Kα selective inhibitor, alpelisib, been approved for the treatment of hormone receptor-positive and HER2/neu-negative breast cancer harbouring mutations in the gene encoding PI3Kα77. In combination with fulvestrant, an oestrogen receptor antagonist, alpelisib doubles PFS in these patients compared with fulvestrant alone. The paucity of drugs that target PI3Ks may reflect the toxicity associated with inhibition of this pathway due to its importance for normal homeostatic functions, perhaps compounded by the difficulty in developing inhibitors with the required level of selectivity.
Inhibitors of AKT (also known as PKB) have yet to be approved, but the AKT inhibitor capivasertib was found to be well tolerated and to show promising anticancer activity in a phase I trial, when used alone in patients with advanced oestrogen receptor-positive metastatic breast cancer expressing the AKT-E17K mutant78. This same mutation has been identified in several cancers but is found most frequently in breast cancer where it is present in more than 6% of patients79. A separate phase II trial reported that capivasertib more than doubled PFS when given in combination with fulvestrant80, and more advanced clinical trials are ongoing.
The clinical impact of kinase inhibitors
The development and approval of kinase inhibitors has transformed the clinical care of multiple malignancies including CML, GIST, melanoma and NSCLC. In CML, the use of allogeneic bone marrow transplantation as initial therapy for patients with chronic phase disease has been replaced by the use of inhibitors of BCR–ABL, and is now reserved for multidrug-resistant disease or more advanced phase disease. The clinical use of systemic chemotherapy for GIST or BRAF mutant melanoma has virtually disappeared or is reserved for last-line treatment following exhaustion of all other targeted treatment modalities. In NSCLC, the use of genotype-directed therapies including kinase inhibitors has supplanted the use of systemic chemotherapy as initial therapy for patients with advanced NSCLC. Multiple clinical trials have also demonstrated that the use of genotype-directed therapy (for EGFR mutation and ALK-rearranged NSCLC) leads to improved response rates and PFS, as well as reduced adverse side effects compared with chemotherapy23,81,82. Although previously felt not to be beneficial, there is now also a resurgence of interest in evaluating the efficacy of systemic chemotherapy and a kinase inhibitor administered concurrently. This is based on two recent randomized studies demonstrating an improvement in PFS and overall survival when patients with EGFR mutation were treated with carboplatin, pemetrexed and gefitinib compared with gefitinib alone, as initial systemic therapy for advanced NSCLC83,84. Whether this same approach will be applicable in other genotype-directed settings in NSCLC remains to be determined.
Another feature of kinase inhibitors that has transformed clinical care is their ability to penetrate the blood–brain barrier (BBB) to treat brain metastases and/or leptomeningeal disease. As systemic therapies have become more effective, and patients with advanced cancer are living longer with their disease, the incidence of central nervous system (CNS) metastases has increased. Few chemotherapeutic agents cross the BBB, and the long-term cognitive side effects of radiation therapy can be devastating. In addition, the use of intrathecal chemotherapy is largely ineffective in the treatment of leptomeningeal disease from solid tumours. Several next-generation kinase inhibitors including osimertinib, alectinib, brigatinib, lorlatinib, selpercatinib and capmatinib have demonstrated clinical CNS activity in patients with NSCLC85,86,87,88,89. Patients presenting with asymptomatic CNS metastases can often be treated with systemic therapy alone, allowing radiation therapy and/or the use of focal radiation therapy to be delayed or avoided for incompletely treated lesions. Osimertinib and alectinib have also been used successfully to treat patients with leptomeningeal disease90,91. Despite these successes, there continues to be a need to develop additional kinase inhibitors that can penetrate the CNS, as most current inhibitors do not effectively achieve this. Furthermore, there is little knowledge about how resistance develops in the CNS following successful treatment with a kinase inhibitor, which is clearly an important area of future research.
The successful implementation of precision therapy for patients with advanced cancers requires the timely identification of an appropriate genomic alteration predicted to lead to drug sensitivity. This is exemplified most clearly in NSCLC where there are seven different genomic alterations all with unique drug approvals. The large number of genetic tests required to deliver effective genotype-directed therapies is leading to replacement of the ineffective and slow strategy of sequential single gene testing in favour of more comprehensive and targeted next-generation sequencing (NGS)92. Other technological advances have also led to the development of NGS from tumour-derived cell-free DNA found in plasma93. This has greatly expanded the ability to genotype tumours, as not all lung cancers, for example, can be sampled successfully by biopsy for tumour-based sequencing.
Despite the medical advances made by the introduction of kinase inhibitors for the treatment of cancers, access to new therapies is not universal. Barriers, both regulatory and financial, still limit the ability of new treatments to reach patients equally worldwide. Progress is therefore needed to ensure that all patients are able to access the most effective treatments for their cancer.
Resistance to protein kinase inhibitors
Despite the huge benefits that approved kinase inhibitors have undoubtedly brought to many cancer patients, these drugs are not curative. Most only delay tumour progression because advanced tumours find escape routes to circumvent target inhibition, leading to drug resistance (Fig. 3).

Drug resistance occurs primarily through four main mechanisms. Acquired drug resistance mutations most commonly affect the binding of the drug to its target. Acquired oncogenic amplifications or rearrangements can activate downstream signalling to bypass inhibition of the drug target. Mutations in downstream effectors can activate signalling pathways despite effective inhibition of an upstream kinase target. State transformation can lead to kinase inhibitor insensitivity.
Resistance to kinase inhibitors can broadly be categorized as either innate or primary resistance, or acquired resistance. Even when tumours harbour an oncogenic driver mutation associated with sensitization to a specific kinase inhibitor, not all tumour cells respond and others exhibit only transient duration of benefit. This can be due to factors extrinsic to the tumour, such as the drug producing insufficient inhibition of the target kinase or not remaining associated with the kinase for long enough.
Primary or innate resistance
Intrinsic resistance can be caused by tumours that harbour a population of cancer cells that are refractory to target inhibition from the outset owing to, for example, co-existing genetic aberrations in multiple oncogenic pathways. As discussed above, advances in NGS technology92 and its application to circulating tumour DNA fragments (ctDNA) in plasma93 have enabled a paradigm shift in our understanding of how the genomic heterogeneity of advanced tumours adversely impacts treatment outcomes. For example, despite the oncogenic addiction within EGFR-mutant NSCLC, a subset of patients do not achieve tumour shrinkage when treated with early generation EGFR inhibitors owing to the existence of de novo MET amplification94 or the EGFRT790M mutation95, which mediate primary resistance to these drugs. Similar observations are emerging in other treatment settings, including other types of cancer in which kinase inhibitors are deployed96,97. Increasing access to NGS data derived from tumour tissues and plasma will continue to be key and lead to a deeper understanding of the molecular heterogeneity of tumours. In turn, this will guide the strategies needed to tackle primary resistance, such as combination therapies.
Acquired resistance
This type of resistance has been observed since the first kinase-inhibiting drugs were introduced, and dealing effectively with this problem has become a major challenge, because acquired resistance ultimately develops in most patients with advanced metastatic disease. As discussed below, acquired drug resistance is complex and the mechanisms are diverse, but they all result from cancer cells finding a route to maintain continued proliferative and survival signalling, despite the continued presence of the original kinase inhibitor (Fig. 3). Acquired resistance mechanisms can be caused by on-target secondary mutations, by the acquisition of ‘bypass’ signalling pathways or by histological transformation98. Alternatively, they can be tumour cell extrinsic and caused by an alteration in the biology of the tumour microenvironment, or result from metastatic tumour cells evading the inhibitor by finding a sanctuary that the inhibitor cannot gain access to, such as the CNS. One general mechanism by which tumour cells can become resistant to drugs is through increased expression and activity of drug transporters, such as multidrug resistance proteins or brain cancer resistance proteins, that actively export drugs from cells. Although more commonly associated with the loss of clinical efficacy to chemotherapeutic drugs, this mechanism may also prevent kinase-inhibiting drugs from reaching clinically effective concentrations within the CNS, in which high levels of these drug transporters are expressed99.
Nearly all tyrosine kinases, as well as a number of protein serine/threonine kinases, including RAF56, p38α MAP kinase100 and SIK101, possess amino acids with small side chains at the gatekeeper site, most frequently threonine. This creates a small hydrophobic pocket near the ATP-binding site, facilitating the development of potent and relatively specific inhibitors of these kinases that target both the hydrophobic pocket and the ATP-binding site itself. However, it is precisely the feature that makes these kinases so druggable that also makes them so susceptible to drug resistance, since all that is required to cause resistance is mutation of the gatekeeper threonine to an amino acid with a larger side chain100,101.
Early research showed that resistance to imatinib was associated with a single gatekeeper amino acid alteration, in which Thr315 of BCR–ABL is frequently changed to isoleucine, thereby disrupting drug binding102. A strikingly similar acquired resistance mechanism to the EGFR inhibitors gefitinib and erlotinib is seen in NSCLC where the similarly located gatekeeper residue Thr790 is mutated to methionine, accounting for about 50% of the cases of drug resistance103. Intriguingly, and in contrast to BCR–ABL, the EGFRT790M gatekeeper mutation also enhances affinity for ATP, making it even harder for gefitinib and erlotinib to out-compete ATP104 (Figs 4,5).

Epidermal growth factor receptor (EGFR) kinase inhibitors are a key example of the theme of next-generation agent development to continuously improve drug properties. First-generation inhibitors, gefitinib and erlotinib, have an anilinoquinazoline core backbone structure and bind reversibly in competition with ATP to potently inhibit EGFR catalytic activity. Second-generation inhibitors, afatinib and dacomitinib, share the quinazoline core structure, but contain a covalent warhead that enables irreversible binding and which increases potency but also toxicity. Afatinib and dacomitinib also did not overcome the common acquired resistance mutation EGFRT790M, which led to the development of the third-generation inhibitors osimertinib and olmutinib. To overcome the high affinity of EGFR-T790M for ATP, these inhibitors contain a covalent warhead. They also contain a pyrimidine core backbone that is structurally distinct from the quinazoline-based structures to optimize selective binding within the catalytic domain that harbours the EGFR-T790M alteration.

X-ray crystal structures highlighting the binding modes for first-generation, second-generation and third-generation EGFR inhibitors, showing key residues in the kinase domain. Highlighted is the gatekeeper Thr790, which when mutated to Met is a frequent cause of drug resistance to first-generation and second-generation inhibitors, and Leu858, which when mutated to Arg is a frequent cause of EGFR activation. Exon 19, deletion of which also causes activation, is included for orientation. a | Crystal structure of the wild-type kinase domain of the EGFR in complex with gefitinib (green) (Protein Data Bank (PDB) identifier 2ITY). b | Crystal structure of the wild-type kinase domain of the EGFR in complex with afatinib (green). Afatinib forms a covalent bond with Cys797 of the EGFR (PDB identifier 4G5J). c | Crystal structure of the EGFR-T790M mutant in complex with osimertinib (green), which also forms a covalent bond with Cys797. The Thr790Met mutation changes the conformation of the ATP-binding pocket, which increases affinity for ATP (PDB identifier 6JX4).
Mutations in the kinase domain that cause drug resistance, including gatekeeper, solvent-front mutations and DFG-loop mutations, have been identified in an increasing number of protein kinases, and become a common clinical theme. For example, the ALK G1202R, ROS1G2032R, TRKAG595R, TRKCG623R and RET G810X mutations have led to the development of specific inhibitors capable of overcoming these classes of mutation105,106,107. The increasing molecular and clinical knowledge of how different kinase inhibitors are able to trigger these mutational events, is making it increasingly possible to predict likely mechanisms of resistance, which in turn enables more rapid and effective drug discovery efforts to be made to optimize clinical treatment108.
The second major mechanism of acquired resistance involves the cancer cell bypassing the targeted kinase. This can occur by the overexpression or amplification of another kinase (or kinases) that then mimics the actions of the drugged kinase. A common theme is for cancer cells to switch their dependence to other non-targeted RTKs. For example, in NSCLC, tumour cells frequently respond to EGFR inhibitors by amplifying the gene encoding MET and, less frequently, the genes encoding HER2 and FGF. These RTKs then switch on the same downstream signalling pathways that would normally be activated by the mutant EGFR if it was not inhibited by the drug (that is, the RAF–MEK–ERK and PI3K–PDK1–AKT pathways)109. This phenomenon has also been observed in other NSCLC settings where the cells respond to inhibitors of mutant ALK by overexpressing the EGFR or KIT receptor, while MET amplification has been identified as an emerging resistance mechanism to RET inhibitors110. In breast cancer, resistance to agents targeting HER2 overexpression have similarly been associated with switching to alternative RTKs including MET, HER3 and IGF1R111,112. A variation on this theme occurs in oestrogen receptor-positive breast cancer, whereby resistance to the CDK4/CDK6 inhibitors ribociclib and palbociclib (Supplementary Table 1) occurs via the acquired upregulation of CDK6 (ref.113) and its activator cyclin D1 (ref.114), resulting in less effective inhibition of CDK6 by these drugs. The upregulation of CDK2/cyclin E activity is another cause of resistance to palbociclib115, which can be suppressed by the combined use of CDK2 and CDK4/CDK6 inhibitors116.
In addition to compensation by overexpression of the same or another kinase, bypass of the drugged kinase can also occur via alterations in downstream signalling components. For example, RTK inhibition can be overcome through acquired mutations affecting KRAS117,118, RAF119 or loss of neurofibromin (NF1)120, causing constitutive activation of the RAF–MEK–ERK pathway. Similarly, compensation caused by the loss of PTEN or mutations in the gene encoding PI3K121,122,123,124 can lead to constitutive activation of the PI3K pathway in various treatment regimens and across various disease settings. Such mechanistic heterogeneity and plasticity in how cancer cells can trigger bypass mechanisms to restore downstream signalling, coupled with an inability to predict which resistance mechanisms will be adopted by tumours, continues to provide significant challenges to tackling such resistance effectively.
Approaches to overcoming resistance
Over the past 20 years, considerable progress has been made in understanding the mechanisms of acquired resistance, and in developing strategies to prolong the duration of treatment benefit to patients. A dominating approach has been to develop next-generation drugs that not only have enhanced selectivity and reduced adverse effects, but are also capable of tackling on-target resistance caused by mutations at the gatekeeper and other sites. This approach has led to the sequential approval of different drugs against the same target in multiple disease settings, for example, bosutinib, ponatinib and radotinib have all been approved for imatinib-resistant BCR–ABL (Fig. 1; Supplementary Table 1). Another important example that demonstrates the clinical impact of this strategy, and how the approach to kinase drug discovery has evolved, is the EGFR inhibitors developed to tackle on-target resistance to gefitinib and erlotinib (Figs 4,5) caused by acquisition of the T790M mutation at the gatekeeper site of the EGFR kinase domain. Initially, the second-generation inhibitors afatinib and dacomitinib were designed to enhance potency and try to overcome the resistance limitations of first-generation inhibitors. Although sharing the same chemical template, afatinib and dacomitinib were differentiated by the addition of a reactive acrylamide warhead that enabled them to bind covalently and irreversibly to cysteine 797 of the EGFR125,126 (Figs 4,5). However, although these compounds were approved for clinical use, a key drawback was their increased activity against wild-type EGFR, which exacerbated commonly observed side effects such as skin rash and diarrhoea. As these side effects were dose limiting in most patients, it was impossible to use these compounds at the concentrations needed to deal effectively with the T790M mutation in a clinical setting. This challenge led several companies to embark on innovative strategies to develop ‘third-generation’ inhibitors that were specifically designed to target the EGFR-T790M mutant, while maintaining activity against the initial activating mutations and retaining sufficient selectivity over wild-type EGFR to address the limitations of afatinib and dacomitinib. These efforts led to the development and approval of the first third-generation EGFR inhibitor osimertinib127 (Figs 4,5), with other agents now emerging, for example, olmutinib128. Osimertinib has not only improved PFS compared with chemotherapy in the EGFR-T790M population129 but also in first-line treatment for EGFR mutations compared with the first-generation inhibitors gefitinib and erlotinib130,131. Consequently, osimertinib has become the standard of care and is now being used as the initial first-line treatment for EGFR-mutant NSCLC, enabling all patients to benefit from improved survival, regardless of whether they harbour the EGFRT790M mutation. However, despite its effectiveness, cancer cells still find additional routes of acquired resistance to escape from inhibition by osimertinib, including the EGFRC797S mutation, which prevents osimertinib from binding covalently to the EGFR, or bypass mechanisms such as the amplification of MET132,133.
Another good example of the enhanced clinical impact of next-generation inhibitors is provided by ALK. The first-in-class ALK inhibitor crizotinib induced resistance in NSCLC through multiple on-target mutations, including not only the gatekeeper L1196M mutation, but also the L1152R, C1156Y and G1202R mutations134. This prompted the development of the second-generation inhibitors ceritinib135, alectinib136 and brigatinib137, each of which inhibited one or more of the mutant ALKs that are resistant to crizotinib. However, although these agents had improved potency and targeted key resistance mutations, they did not target all the ALK mutants resistant to crizotinib. The third-generation inhibitor lorlatinib, which is a modified version of crizotinib with increased potency against the G1202R and other early-generation resistance mutations138, was therefore developed. Lorlatinib has been approved for the treatment of patients with cancer that has progressed after treatment with both first-generation and second-generation ALK inhibitors. However, compound ALK mutations have now been observed in patients treated with lorlatinib, in which dual mutations can reside on the same allele, such as the C1156Y and L1198 mutations139. This suggests that additional strategies to inhibit ALK are needed140.
The development of next-generation inhibitors has become entrenched in current kinase drug discovery, underpinned by its successes with drug-resistant mutants of BCR–ABL, EGFR and ALK. However, as exemplified by osimertinib and lorlatinib, acquired resistance to third-generation agents unfortunately still occurs, and so despite the advances, these agents are not curative in advanced metastatic disease settings. New treatment approaches will therefore continue to be needed. One promising approach is the use of combination therapy that combines inhibitors of the original kinase mutation with inhibitors of a second kinase to block the predominant cause of resistance. Currently, the only examples of this strategy are the combination of RAF and MEK inhibitors to delay resistance in malignant melanoma and the combination of MEK inhibitors with inhibitors of RTKs (see above). Another approach is the continual development of further next-generation agents that directly target the drug-resistant kinase mutant.
Advances in kinase inhibitor development
Over the past 20 years the design of kinase inhibitors has been improved in multiple ways, principally aimed at improving selectivity and overcoming the challenges of resistance. These medicinal chemistry efforts have been facilitated by several key technical advances. First, following the sequencing of the human genome, the comprehensive classification and annotation of the 500 plus protein kinases (the human kinome) and its subdivision into a number of subfamilies, has led to a holistic understanding of kinome relationships141 and facilitated a step change in the way that kinase drug discovery can be tackled. Second, the introduction of kinase ‘profiling’ panels of gradually increasing size34,35, which today encompass most of the kinome, have become less expensive owing to miniaturization, robotics and the development of novels ways to measure the activities of kinases and their interaction with small chemical entities. These developments have enabled drug developers to profile inhibitors against off-target kinases routinely, as well as the primary target, enabling promiscuous lead compounds to be discarded at early stages, and later to help guide improved selectivity within key series of compounds. Extensive kinase inhibitor profiling has also enabled new clinical opportunities to be identified and undesired side effects to be explained40. Third, increasing access to high-resolution structures of kinase catalytic domains, facilitated by the Structural Genomics Consortium142, has enabled routine integration of structural and computational chemistry into drug discovery, including fragment-based drug design approaches143,144. This is providing improved information about structure–activity relationships, allowing better drug design by enabling chemical groups to be positioned more precisely. Fourth, mechanisms of drug resistance that have been observed in patients have helped both to identify potential resistance mechanisms that are relevant clinically and to inform the opportunities and characteristics of the next-generation inhibitors needed to overcome the effects of these mutations in patients. Therefore, the ongoing iterative cycle between bench and bedside will continue to be key to ongoing developments and their success.
The increased molecular and structural understanding of human kinases, coupled with the increasing experience of chemists in the protein kinase arena, has enabled the pharmaceutical industry to move away from high-throughput screening to the use of smaller more focused chemical libraries enriched for kinase scaffolds, originally pioneered by the biotechnology company Biofocus. This shift away from screening vast numbers of diverse compound collections that required the expression and purification of huge amounts of the target kinase, has improved the feasibility of undertaking extensive kinase profiling on more limited numbers of compounds, and increased the success rate in transforming initial hits into promising optimized leads. It has also enabled pharmaceutical companies to explore alternative approaches and to start to move away from ATP-competitive agents, whose disadvantages are discussed in the section Resistance to protein kinase inhibitors.
Although there had been a reluctance to pursue covalent inhibition strategies owing to the potential risk of idiosyncratic toxicities caused, for example, by triggering immune responses through covalent attachment of such inhibitors to proteins, the development of inhibitors that bind covalently to protein kinases has received renewed interest in recent years. Covalent inhibitors are typically developed by structure-guided incorporation of a reactive warhead designed to target the nucleophilic thiol groups of cysteine residues within kinase catalytic domains145. Many kinases have cysteine residues in and around the ATP-binding site, enabling the design of inhibitors that can form an irreversible covalent bond with the kinase, thereby permanently disabling enzymatic activity146. Kinase function can only be restored by the expression of new kinase molecules. These covalent approaches can drive both potency and high selectivity towards the primary target. Examples of approved kinase inhibitors that bind covalently to their targets include afatinib, ibrutinib, neratinib, dacomitinib and osimertinib.
RET inhibitor development nicely exemplifies the shift to develop more potent and specific inhibitors. RET is a proto-oncogenic RTK in which activating mutations and rearrangements lead to medullary thyroid carcinoma and other cancers147. RET inhibitors were initially identified by profiling the specificities of compounds that had already been developed for other reasons and led them to be repurposed for cancers driven by RET alterations. These compounds, which included cabozantinib, sorafenib and vandetanib, had sufficient clinical benefit to achieve approval for the treatment of RET-driven thyroid cancer, but their overall benefit was reduced by dose-limiting toxicities, probably caused by the inhibition of other protein kinases, such as VEGFRs. In RET-rearranged NSCLC, these agents also had limited activity and substantial toxicity148. This problem led to medicinal chemistry efforts to develop compounds that were more RET-selective, resulting in much higher clinical activity and the approval of selpercatinib149 and pralsetinib150 for thyroid and lung cancers.
There are important classes of protein kinase for which it has proved impossible, so far, to develop clinically useful inhibitors, owing to an inability to achieve a sufficient therapeutic index between the effect on tumours and the effect on primary cells. Protein kinases that drive the cell division cycle, such as PLKs, aurora kinases and some cyclin-dependent kinases, are prominent examples151,152. Continued and sustained medicinal chemistry efforts may be needed to develop sufficiently selective inhibitors with improved tolerability. This has already happened in the case of CDK4/CDK6 inhibitors, as discussed earlier153. Another innovative approach currently under clinical investigation is to encapsulate an inhibitor of the aurora B kinase within a polymeric nanoparticle to improve its therapeutic index through selective delivery to the tumour154. It will be interesting to see whether this approach also improves the clinical benefit of approved kinase inhibitors.
Finally, as discussed above, another important issue that is now driving kinase inhibitor development is the generation of drugs that cross the BBB, because metastasis to the brain is a common clinical issue now that patients with advanced cancer are surviving longer, and historically the brain is a sanctuary site for cancer cells evading drug targeting. It is now clear that kinase inhibitors require specific physiochemical properties to enable them to penetrate the BBB effectively and achieve adequate brain exposure155. In recent years, a deeper understanding of the properties of a drug needed to cross the BBB and avoid efflux transporters has enabled medicinal chemists to optimize the pharmacokinetic parameters needed, which include minimal molecular size, lipophilicity, partition coefficients, surface polarity and numbers of hydrogen bond donors156. This is resulting in the development and exploitation of increasing numbers of CNS-penetrant kinase inhibitors. Lorlatinib and AZD3759 are good examples of how CNS penetration properties can now be optimized as an integrated discovery programme157,158. However, balancing and maintaining all the other desired properties of a drug, such as potency and selectivity, alongside optimal BBB permeability, remains a significant challenge.
Kinase inhibitors in diseases other than cancer
Smooth muscle relaxation and immunosuppression
Despite the impact that protein kinase inhibitors have had on the treatment of cancer, relatively few protein kinase inhibitors have been approved so far for the treatment of other diseases. Fasudil (previously known as AT877 and HA107) shows efficacy for the treatment of cerebral vasospasm159 and was approved for this purpose in Japan in 1995, and later in China, but has not yet been approved in either the USA or Europe. Fasudil reduced pulmonary vascular resistance in a clinical trial and was efficacious in the treatment of pulmonary hypertension160, and so may be approved for other indications in the future. Fasudil was developed from a class of molecules originally identified as calmodulin antagonists and was later found to inhibit several protein kinases of the AGC subfamily. Only after its approval for cerebral vasospasm was it shown to inhibit Rho-dependent kinases, which leads to reduced phosphorylation of the myosin P light chain, increased smooth muscle relaxation and hence the dilation of blood vessels161.
Rapamycin, a naturally occurring compound produced by the soil bacterium Streptomyces hygroscopicus, was identified about 50 years ago. Originally developed as an antifungal agent, it was initially discarded because of its undesirable immunosuppressive side effects. Many years later, the target for rapamycin was identified as FK506-binding protein (FKBP), and the FKBP–rapamycin complex was shown to be a potent and specific inhibitor of the TORC1 protein kinase complex162. Renewed interest in exploiting the immunosuppressant properties of rapamycin led to its approval in 1999 as an immunosuppressant for recipients of renal transplant, and later for the treatment of lymphangioleiomyomatosis, a rare lung disease. Interestingly, rapamycin would never have been identified using the conventional approach of screening of libraries of small molecules against the catalytic subunit of TORC1, as it only inhibits the intact TORC1 complex.
Inflammatory and autoimmune diseases
Inflammatory and autoimmune diseases, including arthritis, asthma, colitis, fibrosis, systemic lupus erythematosus (SLE), psoriasis and sepsis, are frequently caused by deregulation of the immune system, leading to the overproduction of inflammatory mediators. One strategy for the treatment of these diseases is to target protein kinases that either mediate the intracellular actions of cytokines or are required for their production. However, as the normal function of cytokines is to combat microbial pathogens, one challenge is to develop effective drugs that do not carry an unacceptable risk of infection163,164.
The huge success of the anti-TNF antibody adalimumab (Humira; the world’s top selling drug in 2018) and the TNF-binding protein etanercept (Enbrel; the world’s fifth best-selling drug in 2018) in treating rheumatoid arthritis, Crohn’s disease and ankylosing spondylitis, may explain why interest in protein kinase inhibitors for the treatment of immune diseases waned for a number of years. However, up to one-third of patients with rheumatoid arthritis do not respond to Humira or Enbrel, and renewed interest in the potential of kinase inhibitors led to the approval of the Janus kinase (JAK) inhibitor tofacitinib for the treatment of moderate to severe rhuematoid arthritis in 2012, the first drug to be developed outside the field of cancer by targeting a specific protein kinase, and the first orally active drug to treat rheumatoid arthritis for more than 50 years (Fig. 1). Tofacitinib has subsequently been approved for psoriatic arthritis, ulcerative colitis and juvenile idiopathic arthritis.
Tofacitinib targets the four members of the JAK family of protein tyrosine kinases, JAK1, JAK2, JAK3 and TYK2, which mediate the actions of about 25 cytokines and interferons. The JAK family members control the transcription of many genes, principally by phosphorylating and activating members of the STAT family of transcription factors165. The success of tofacitinib stimulated the development of additional JAK inhibitors (collectively termed JAKitinibs). One of these, baracitinib, has been approved for the treatment of moderate to severe rheumatoid arthritis in patients with an inadequate response to one or more TNF antagonists, and is on the cusp of approval in Europe for atopic dermatitis. About a dozen other JAKitinibs are undergoing clinical trials166,167.
The involvement of JAKs in mediating the actions of so many cytokines can account for the efficacy of tofacitinib and baricitinib. However, it is also likely to underlie concerns about their adverse side effects, which delayed the approval of tofacitinib for rheumatoid arthritis in Europe for 4 years. Treatment with tofacitinib carries an increased risk of microbial infection, especially of the upper respiratory tract, and more recently, an increased risk of venous thromboembolism in 1% of treated patients has been recognized166,167. It is also worth noting that JAKitinibs would be expected to suppress the beneficial effects of the anti-inflammatory cytokine IL-10, which are also mediated by JAK family members.
The various JAKitinibs undergoing clinical trials inhibit all JAK family members but to differing degrees, for example, tofacitinib inhibits JAK1 and JAK3 more potently than JAK2 and TYK2. It is still too early to tell whether the adverse side effects of JAK inhibitors differ significantly between different JAKitinibs and, if so, whether they correlate with inhibition of a particular JAK isoform or heterodimer. In summary, the development of JAKitinibs is currently a very active area of drug development, and more JAKitinibs are likely to be approved for the treatment of inflammatory and autoimmune diseases in the future.
A notable feature of the JAKs is that they each contain two kinase domains, one encoding a catalytically inactive pseudokinase and the other an active kinase. Interestingly, it is the Val617Phe mutation in the pseudokinase domain of JAK2 that is a principal cause of human myeloproliferative diseases168,169, including primary polycythaemia, thrombocythaemia and myelofibrosis. This mutation generates a form of JAK2 that is catalytically active even in cells not stimulated with cytokines. These observations suggest that one physiological role of the pseudokinase domain of JAK2 is to prevent activation of the other kinase domain until it is required.
Ruxolitinib, a JAKitinib that inhibits the active kinase domain of JAK2 (and other JAKs), was approved for the treatment of myeloproliferative disorders in 2011. However, many patients treated with ruxolitinib lose or have a suboptimal response to this drug, or develop cytopenia during treatment, resulting in ruxolitinib being discontinued within months170,171. Indeed, in a couple of phase III trials, discontinuation rates for ruxolitinib at 3 and 5 years were 50% and 70%, respectively. In 2019, fedratinib was approved for the treatment of adult patients with intermediate-2 or high-risk primary or secondary myelofibrosis or as a second-line therapy for patients previously treated with ruxolitinib. Although fedratinib inhibits JAK2, it also inhibits BRD-containing proteins, such as BRD4, which may contribute to its clinical efficacy. In summary, fedratinib may be a promising new therapy for patients with advanced myelofibrosis, but more extensive clinical experience with this compound is needed to determine how good it is relative to ruxolitinib and whether biomarkers will be able to identify a subset of patients most likely to respond to fedratinib. Perhaps drugs that target the pseudokinase domain of JAK2, and hence the primary cause of these diseases, will prove to be even more effective if they are ever developed in the future.
There is increasing interest in targeting protein kinases that suppress the production of inflammatory cytokines. One interesting candidate is IRAK4 (interleukin 1 receptor-associated kinase 4), which is required for the production of pro-inflammatory cytokines and chemokines induced by the activation of Toll-like receptors (TLRs)172,173. Mice expressing kinase-inactive mutants of IRAK4 are protected in multiple models of inflammatory and autoimmune disease174 and an orally active IRAK4 inhibitor prevents both the autoimmune and autoinflammatory aspects of SLE in a mouse model of relevance to human SLE173. At least four compounds that inhibit IRAK4 specifically or are dual inhibitors of IRAK4 and IRAK1 (PF-06650833, BAY1834845, BAY1830839 and CA-4948) are currently undergoing phase I/II clinical trials for inflammatory and autoimmune diseases.
The potential concern about IRAK4 inhibitors causing increased susceptibility to infection by microbial pathogens has been addressed by a remarkable 30+ year study of about 50 individuals from more than 30 families, who lack IRAK4 expression and whose cells produce little IL-6 or IL-8 in response to ligands that activate TLRs175. Young children with IRAK4 deficiency are extremely susceptible to infection by pyogenic bacteria, half dying before the age of 8, even when broad-spectrum antibiotics are administered daily. Fascinatingly, these individuals no longer die when they reach the age of 8, and invasive bacterial infections gradually decline and are rarely seen after they reach the age of 20. They also have normal resistance to common fungi, parasites, viruses and many other types of bacteria. Therefore, the essential roles of IRAK4 in humans appear to be confined to the protection of young children against infection by a few bacterial species175,176 and suggest that IRAK4 inhibitors could be administered safely to adults. In adults, other innate immune signalling pathways may assume greater importance as the first line of defence. Consistent with this notion, the IRAK4 inhibitor PF-06650833 was reported to be well tolerated with no dose-limiting adverse events observed in two phase I clinical trials177. In a phase II trial, only 6% of the 187 participants stopped treatment owing to an adverse effect occurring during treatment178. Further information about the efficacy of these and other IRAK4 inhibitors in chronic inflammatory diseases is awaited with interest.
The inhibition of members of the SIK subfamily of protein kinases enhances the production of IL-10 by dephosphorylating and activating CRTC3, a co-activator of the transcription factor CREB, transforming macrophages from an inflammatory M1 to an anti-inflammatory M2b state that promotes the resolution of inflammation and the repair of damaged tissue after infection101,179.
This suggests that SIK-inhibiting drugs may have therapeutic potential for the treatment of inflammatory diseases. SIK inhibitors also suppress the TLR-dependent production of pro-inflammatory cytokines in macrophages by an unknown IL-10-independent mechanism101,179. SIK inhibitors (for example, GLPG3970; Galapagos Plc) are now entering advanced clinical trials for rheumatoid arthritis, ulcerative colitis, psoriasis and other diseases. SIK inhibitors have additionally shown preclinical activity in models of fibrotic disease180.
SIK inhibitors, via their ability to enhance activation of CREB, also stimulate the synthesis of melanin and hence the tanning of skin181. Consequently, SIK inhibitors are being developed to protect skin against the damaging effects of UV radiation. SIK inhibition, also by activating CREB, stimulates the proliferation of osteoblasts and bone matrix mineralization, suggesting a potential use in treating osteoporosis182,183. Although these studies are at a preliminary stage, SIK-inhibiting drugs may turn out to have multiple uses in treating various human diseases and conditions.
Alzheimer disease
Many years ago, a class of anti-inflammatory drugs called cytokine synthesis anti-inflammatory drugs (CSAIDs), were found to inhibit a protein kinase now called p38α MAP kinase184,185,186, sparking the development of a plethora of potent and specific inhibitors of this protein kinase. However, although these compounds displayed impressive efficacy in mouse models of inflammatory disease and many passed phase I/II clinical trials, none progressed to phase III trials for chronic inflammatory diseases owing to lack of efficacy187. More recently, interest in brain microglia dysfunction and neuroinflammation as potential causes of neurodegenerative diseases, led one BBB-penetrant p38α MAP kinase inhibitor neflamapimod (previously VX745) to be repurposed for Alzheimer disease. This compound reversed amyloid-β (Aβ)-induced synaptic dysfunction and loss and reduced Aβ production in a mouse model of Alzheimer disease, and improved performance in the Morris water maze (MWM) test in rats with cognitive impairment188. In phase IIb human clinical trials, patients with the highest plasma concentrations of neflamapimod showed improvement in the Hopkins Verbal Learning Test, Revised and the Wechsler Memory Scale immediate and delayed recall test189. Statistically significant reductions in cerebrospinal fluid markers of Alzheimer disease were also observed. For these reasons, further clinical trials at higher doses of neflamapimod are ongoing. Several TKIs that cross the BBB and have passed phase I/II trials are also being repurposed for Alzheimer disease, including saracitinib (previously AZD0530) originally developed as an inhibitor of Src family members, such as Fyn, and the BCR–ABL inhibitor nilotinib already approved for CML190.
Glycogen synthase kinase 3 (GSK3) is one of the protein kinases that phosphorylates Tau; hyperphosphorylation of tau leads to its aggregation and the formation of brain deposits called ‘tangles’, which are a hallmark of Alzheimer disease and other tauopathies191. The GSK3 inhibitor tideglusib192 was found to reduce tau hyperphosphorylation, and other markers of Alzheimer disease, such as amyloid deposition, neuron loss and gliosis in mouse brain, and it also reversed spatial memory deficit in a mouse model of Alzheimer disease193. Moreover, tideglusib reduced Aβ production and ameliorated the Alzheimer disease-like neuropathology and behavioural deficits in the water maze test in another mouse model of Alzheimer disease190. Tideglusib was well tolerated in phase I and phase II clinical trials. A pilot, double-blind, randomized phase II trial (NCT00948259) of tideglusib in 30 patients with mild to moderate Alzheimer disease reported positive trends in several cognitive benefit tests. However, a larger phase II trial involving 306 patients with Alzheimer disease missed its primary cognitive end point. Further studies are ongoing190.
Parkinson disease
Between 1% and 2% of Parkinson’s disease worldwide is caused by mutations in the protein kinase LRRK2 gene, although such mutations are more prevalent in particular populations194. The protein kinase activity of LRRK2 is elevated not only as a result of the most prevalent mutation in the kinase catalytic domain (LRRK2-Gly2019Ser), but also from the other seven major pathogenic mutations that have been identified, which are located outside the kinase catalytic domain195. Moreover, independent of mutation, LRRK2 activity was reported to be increased in the dopaminoceptive neurons of individuals with idiopathic Parkinson disease, as judged by increased phosphorylation of its substrate RAB10 (ref.196). It was suggested that LRRK2 may be activated by oxidative mechanisms involving α-synuclein and mitochondrial impairment, leading to endolysosomal dysfunction, the accumulation of phosphorylated α-synuclein and parkinsonism196. Increased LRRK2 activity may therefore contribute to pathogenesis in a significant percentage of patients with Parkinson disease. A small number of LRRK2 inhibitors, including DNL201 (Denali) have advanced to clinical trials.
Other diseases caused by activating mutations in protein kinases
Activating mutations in protein kinase Cγ (PKCγ), the neuron-specific isoform of PKC, may underlie some cases of spinocerebellar ataxia type 14 (ref.197), and increased expression of the protein kinases WNK1 or WNK4, caused by their mutation and other mechanisms, underlies Gordon syndrome, an inherited hypertension resulting from defective excretion of potassium ions. Hypertension is mediated by enhanced WNK-catalysed phosphorylation and activation of the protein kinases SPAK and OSR1, which control the activities of several ion cotransporters198.
We anticipate that drugs that target hyperactivated, disease-causing forms of protein kinases, such as PKCγ and WNK1/WNK4, will be developed in the future and their therapeutic potential examined in human clinical trials.
The next 20 years of kinase drug discovery
We predict that oncology will continue to dominate the field of kinase drug discovery over the next 20 years. Although only 50 of the 500 protein kinases encoded by the human genome have so far been targeted for the treatment of cancer (Supplementary Table 1), we predict that there will be only a modest increase in the number of new kinases targeted for cancer drug development in the future, as new mutations in kinases that are drivers of particular cancers have not been identified in recent years. Inhibitors of some other protein kinases, such as CK2 (refs199,200), are being developed where there is evidence that the viability of cancer cells is affected more profoundly by their inhibition compared with primary cells, but they have yet to enter clinical trials. We therefore think that the focus of research on kinase inhibitors in oncology will mostly be elsewhere, with major efforts being directed towards tackling the war against acquired resistance mechanisms. We envisage an accelerating trend towards the development of next-generation kinase inhibitors with improved selectivity, the ability to combat resistance mechanisms and the capability of CNS penetration (see above). Additionally, we envisage a continued focus on increasing the range of novel rationally designed combinations of kinase inhibitors, as well as the combination of kinase inhibitors with other therapeutic modalities to overcome and prevent resistance. Among other creative and innovative approaches needed to tackle resistance, one strategy gaining increasing attention is to build our understanding of drug-tolerant ‘persister’ cells (DTPs), which are thought to be a reservoir of long-lived cells that may eventually acquire genetic alterations leading to drug resistance. More effective targeting of DTP populations may be a new strategy to delay or prevent acquired resistance from occurring201. Early intervention may also be facilitated by monitoring molecular tumour progression routinely and non-invasively using ctDNA from patient plasma samples, enabling earlier and more informed changes in treatment management to be deployed on the basis of molecular signals before clinical progression202,203. Opportunities in this area will expand significantly, as non-invasive technologies for detecting biomarkers become more sensitive and more widely available.
The role of the tumour stromal microenvironment and immune biology in the protection of cancer cells will be another area of intensive research focus to identify and develop small-molecule inhibitors that can target kinases such as the TAM kinases (TYRO3, AXL and MER)204 and colony-stimulating factor 1 receptor (CSF1R)205 that have key roles in immune signalling, and which may complement or replace immuno-oncology approaches using the biologic drugs highlighted above. We predict that this will lead to the approval of compounds that target kinases with alternative modes of action. Many such early-stage clinical studies are currently ongoing.
Although future innovations will undoubtedly increase patient benefit in advanced metastatic settings, the challenge of treating such heterogeneous and biologically plastic late-stage tumours will remain. Experience in treating CML has revealed that the earlier imatinib is given during disease progression, the less likely it is that acquired resistance will occur206, and post-surgical imatinib treatment has also been shown to increase survival in high-risk patients with GIST207. Similarly, osimertinib given after post-surgical resection for the treatment of early-stage NSCLC caused by EGFR mutations, has shown an improvement in disease-free patient survival compared with patients given a placebo208 and has just received FDA approval for this purpose. We therefore predict that much earlier interventions with kinase inhibitors before the onset of more advanced metastatic disease will be a significant step forward in improving the overall survival efficacy of kinase-inhibiting drugs in the treatment of many cancers. However, the use of kinase inhibitors in early-stage disease settings will also require financial and toxicity barriers to be overcome, as well as the demonstration of a sufficiently significant improvement in clinical efficacy. In the future, the ability to identify high-risk patients with the greatest potential to benefit from the use of kinase inhibitors (for example, identifying patients with minimal residual disease)209 would be an important advance.
So far, the only rationally designed drugs to treat diseases other than cancer that have been developed and approved by targeting a specific protein kinase subfamily are the JAKitinibs (discussed above). Although inhibitors of other kinases are in clinical trials for the treatment of inflammatory and neurodegenerative diseases (discussed above), we think that the number of new protein kinases that will be targeted to treat diseases other than cancer will also increase only gradually as fundamental advances in our understanding of different protein kinases and their roles in disease accumulate. However, we do foresee a considerable increase in the repurposing of kinase inhibitors that were initially developed for other indications where there is unmet medical need, especially kinase inhibitors that have passed phase I/II clinical trials for other indications but were not approved. This is not only because many protein kinases are multi-targeted but also because they have multiple functions in cells. The repurposing of neflamapimod from rheumatoid arthritis to Alzheimer disease and saracitinib and nilotinib from cancer to Alzheimer disease (discussed above) are early signs of a trend that we think is likely to expand.
The development and application of GSK3 inhibitors is a striking example of how far drug repurposing can extend. Although the GSK3 inhibitor tideglusib is undergoing clinical trials for Alzheimer disease (see above), GSK3 also has a crucial role in suppressing activation of the WNT signalling pathway, which is required in many physiological processes, such as the repair of tissue damage210. For example, WNT signalling is activated when teeth are damaged211. In a remarkable development, low doses of tideglusib were applied to biodegradable collagen sponges and inserted into tooth cavities. The sponges gradually degraded over time and were replaced by new dentine, the main supporting structure of the tooth. This finding has the potential to transform the way that teeth cavities are treated in the future, potentially making man-made fillings obsolete212.
GSK3 was originally identified as a protein kinase that regulates the synthesis of glycogen, and the insulin-induced inhibition of GSK3, mediated by AKT, has an important role in regulating the insulin-dependent conversion of blood glucose into tissue glycogen213. For this reason, small-molecule inhibitors of GSK3 were initially developed as potential treatments for type 2 diabetes mellitus191. Subsequently, interest in GSK3 inhibitors for this purpose waned because of concerns that chronic GSK3 inhibition would cause aberrant WNT–β-catenin signalling, a hallmark of many cancers, especially colon cancer. Now that we know that GSK3 inhibitors are well tolerated in humans, these concerns may be unfounded and perhaps the potential of GSK3 inhibitors to lower blood glucose levels in type 2 diabetes should be revisited.
An increase in the development of protein kinase inhibitors that are not small chemical entities is another area that we predict will gain increased traction in the years to come. Antibodies that target RTKs or their activating ligands have been approved for a number of years, such as the anti-HER2 antibody trastuzumab (herceptin) for patients with HER2-positive breast cancer214 and the anti-VEGF antibody bevacizumab (Avastin) for the treatment of solid tumours215. We predict that next-generation antibody approaches will lead to more drug approvals, which will include bi-specific antibodies that block more than one kinase. An example that is already showing encouraging data in clinical development is amivantamab (JNJ-61186372), a dual MET/EGFR antibody216. Antibody–drug conjugates (ADCs), in which a specific antibody is linked to a cytotoxic payload, is another exciting therapeutic platform of the future. Proof of clinical concept has already been achieved with approvals of anti-HER2 ADCs, such as trastuzumab–emtansine (T-DM1) and trastuzumab–deruxtecan (T-DXd) for breast cancer217,218, in which the anti-HER2 is linked to a cytotoxic microtubule inhibitor and a topoisomerase 1 inhibitor, respectively. Similarly, patritumab, a HER3 antibody linked to DXd (HER3–DXd) is in early clinical development for the treatment of solid tumours such as colorectal cancer219.
We predict that breakthroughs in the development of small-molecule inhibitors will continue to take place. For example, the development of drugs that induce the degradation of specific proteins has been making significant progress in recent years. PROTAC technology (proteolysis-targeting chimeras) exploits chemical entities to direct the ubiquitylation-mediated degradation of specific proteins220. Although still in preclinical development, there are multiple examples of the application of this technology to reduce the expression in cancer cells of protein kinases such as AKT, SGK3 and ABL221,222,223, which illustrates the potential of this approach if the challenge of translating it into clinical drugs can be overcome. It is important to emphasize that the specific proteolytic destruction of a protein kinase is not equivalent to the inhibition of its catalytic activity because many kinases are multi-domain proteins. Every domain is eliminated when the expression of a protein kinase is ablated, which could improve efficacy, but also cause more serious adverse side effects. As discussed above, we predict a continued focus on the development of inhibitors that bind covalently to protein kinases146.
The approved small-molecule inhibitors of MEK1/MEK2 (trametinib, selumetinib) and the development of MK2206, an AKT inhibitor that advanced to a phase II trial for recurrent, platinum-resistant ovarian and fallopian tube cancers, has highlighted the possibility of developing allosteric drugs that block kinase activity by binding to regions outside the catalytic domain224. A potential advantage of such allosteric inhibitors is that competition with intracellular ATP for binding to the catalytic site is avoided, increasing the potential to develop more potent, as well as more specific inhibitors. A further example is ascinimib, which is an allosteric ABL inhibitor that binds to the myristoyl-binding site of BCR–ABL. In preclinical and clinical studies this compound has been shown to be effective against resistance mechanisms, including the ABLT315I gatekeeper mutation225,226. In addition, allosteric inhibitors can potentially co-bind with ATP-competitive inhibitors leading to more effective and durable target inhibition or increased effectiveness against compound drug resistance mutations, as has been demonstrated in preclinical models for EGFR-mutant NLSLC227 and CML228.
In summary, the potential for developing novel types of kinase inhibitor is huge, and we confidently predict that this will continue to be a major growth area over the next 20 years.
References
Cohen, P. Protein kinases — the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 1, 309–315 (2002). A review of the history of the development of protein kinase inhibitors up to the time that imatinib was approved for clinical use, and which provides the background to this article.
Witte, O. N., Dasgupta, A. & Baltimore, D. Abelson murine leukaemia virus protein is phosphorylated in vitro to form phosphotyrosine. Nature 283, 826–831 (1980).
Veale, D., Ashcroft, T., Marsh, C., Gibson, G. J. & Harris, A. L. Epidermal growth factor receptors in non-small cell lung cancer. Br. J. Cancer 55, 513–516 (1987).
Haeder, M. et al. Epidermal growth factor receptor expression in human lung cancer cell lines. Cancer Res. 48, 1132–1136 (1988).
Lynch, T. J. et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 350, 2129–2139 (2004).
Paez, J. G. et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500 (2004).
Pao, W. et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl Acad. Sci. USA 101, 13306–13311 (2004).
Shigematsu, H. et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl Cancer Inst. 97, 339–346 (2005).
Berger, A. H. et al. High-throughput phenotyping of lung cancer somatic mutations. Cancer Cell 30, 214–228 (2016).
Campbell, J. D. et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 48, 607–616 (2016).
Breccia, M. & Alimena, G. Nilotinib: a second-generation tyrosine kinase inhibitor for chronic myeloid leukemia. Leuk. Res. 34, 129–134 (2010).
Kantarjian, H., Jabbour, E., Grimley, J. & Kirkpatrick, P. Dasatinib. Nat. Rev. Drug Discov. 5, 717–718 (2006).
Mealing, S. et al. The relative efficacy of imatinib, dasatinib and nilotinib for newly diagnosed chronic myeloid leukemia: a systematic review and network meta-analysis. Exp. Hematol. Oncol. 2, 5 (2013).
Cortes, J. E. et al. Matching-adjusted indirect comparison of bosutinib, dasatinib and nilotinib effect on survival and major cytogenetic response in treatment of second-line chronic phase chronic myeloid leukemia. Curr. Med. Res. Opin. 35, 1615–1622 (2019).
Redaelli, S. et al. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J. Clin. Oncol. 27, 469–471 (2009).
Cortes, J. E. et al. Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: results from the randomized BFORE trial. J. Clin. Oncol. 36, 231–237 (2018).
Noronha, G. et al. Inhibitors of ABL and the ABL-T315I mutation. Curr. Top. Med. Chem. 8, 905–921 (2008).
O’Hare, T., Zabriskie, M. S., Eiring, A. M. & Deininger, M. W. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat. Rev. Cancer 12, 513–526 (2012).
O’Hare, T. et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 16, 401–412 (2009).
Moy, B., Kirkpatrick, P., Kar, S. & Goss, P. Lapatinib. Nat. Rev. Drug Discov. 6, 431–432 (2007).
Christensen, J. G. et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther. 6, 3314–3322 (2007). An interesting preclinical example of the polypharmacology of a kinase inhibitor.
Kwak, E. L. et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 363, 1693–1703 (2010).
Solomon, B. J. et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 371, 2167–2177 (2014).
Mok, T. et al. Updated overall survival and final progression-free survival data for patients with treatment-naive advanced ALK-positive non-small-cell lung cancer in the ALEX study. Ann. Oncol. 31, 1056–1064 (2020).
Peters, S. et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N. Engl. J. Med. 377, 829–838 (2017).
Camidge, D. R. et al. Brigatinib versus crizotinib in ALK-positive non-small-cell lung cancer. N. Engl. J. Med. 379, 2027–2039 (2018).
Shaw, A. T. et al. First-line lorlatinib or crizotinib in advanced ALK-positive lung cancer. N. Engl. J. Med. 383, 2018–2029 (2020).
Rajabi, M. & Mousa, S. A. The role of angiogenesis in cancer treatment. Biomedicines 5, 34 (2017).
Llovet, J. M. et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, 378–390 (2008).
Faivre, S., Demetri, G., Sargent, W. & Raymond, E. Molecular basis for sunitinib efficacy and future clinical development. Nat. Rev. Drug Discov. 6, 734–745 (2007).
Bukowski, R. M., Yasothan, U. & Kirkpatrick, P. Pazopanib. Nat. Rev. Drug Discov. 9, 17–18 (2010).
Keating, G. M. Axitinib: a review in advanced renal cell carcinoma. Drugs 75, 1903–1913 (2015).
Rathi, N., Maughan, B. L., Agarwal, N. & Swami, U. Mini-review: cabozantinib in the treatment of advanced renal cell carcinoma and hepatocellular carcinoma. Cancer Manag. Res. 12, 3741–3749 (2020).
Davies, S. P., Reddy, H., Caivano, M. & Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105 (2000).
Bain, J. et al. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315 (2007). This paper and Davies et al. (2000) introduce and popularize the use of kinase profiling panels to assess the specificities of kinase inhibitors.
Yang, G. et al. HCK is a survival determinant transactivated by mutated MYD88, and a direct target of ibrutinib. Blood 127, 3237–3252 (2016).
Shaw, A. T. et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 371, 1963–1971 (2014).
Rodig, S. J. & Shapiro, G. I. Crizotinib, a small-molecule dual inhibitor of the c-Met and ALK receptor tyrosine kinases. Curr. Opin. Investig. Drugs 11, 1477–1490 (2010).
Suh, K. J. et al. Analysis of adverse events associated with dasatinib and nilotinib treatments in chronic-phase chronic myeloid leukemia patients outside clinical trials. Int. J. Hematol. 106, 229–239 (2017).
Klaeger, S. et al. The target landscape of clinical kinase drugs. Science 358, eaan4368 (2017).
Elkins, J. M. et al. Comprehensive characterization of the published kinase inhibitor set. Nat. Biotechnol. 34, 95–103 (2016).
Ciceri, P. et al. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat. Chem. Biol. 10, 305–312 (2014). The paper demonstrating that protein kinases inhibitors frequently bind to BRD-containing proteins, and that dual kinase–BRD inhibitors can be designed rationally.
Malik, N. et al. Suppression of interferon β gene transcription by inhibitors of bromodomain and extra-terminal (BET) family members. Biochem. J. 468, 363–372 (2015).
Bastard, P. et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370, eabd4585 (2020).
Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370, eabd4570 (2020).
Kolch, W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 351, 289–305 (2000).
Roy, F., Laberge, G., Douziech, M., Ferland-McCollough, D. & Therrien, M. KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev. 16, 427–438 (2002).
Terrell, E. M. & Morrison, D. K. Ras-mediated activation of the Raf family kinases. Cold Spring Harb. Perspect. Med. 9, a033746 (2019).
Davies, H. et al. Mutations of the BRAF gene in human cancer. Nature 417, 949–954 (2002). This study exploits the sequence of the human genome to identify protein kinases mutated in human cancers. The analysis reveals the prevalence of the BRAF V600E mutation in malignant melanoma and other cancers.
Peng, S. B. et al. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell 28, 384–398 (2015).
Robert, C. et al. Five-year outcomes with dabrafenib plus trametinib in metastaticmelanoma. N. Engl. J. Med. 381, 626–636 (2019). Review of the clinical studies that revealed the remarkable improvement in the treatment of metastatic melanoma produced by combining inhibitors of BRAF and MEK, compared with either kinase inhibitor used alone.
Long, G. V. et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N. Engl. J. Med. 377, 1813–1823 (2017).
Dummer, R. et al. Five-year analysis of adjuvant dabrafenib plus trametinib in stage III melanoma. N. Engl. J. Med. 383, 1139–1148 (2020).
Planchard, D. et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol. 17, 984–993 (2016).
Planchard, D. et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 18, 1307–1316 (2017).
Hall-Jackson, C. A. et al. Paradoxical activation of Raf by a novel Raf inhibitor. Chem. Biol. 6, 559–568 (1999).
Poulikakos, P. I., Zhang, C., Bollag, G., Shokat, K. M. & Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430 (2010).
Karoulia, Z., Gavathiotis, E. & Poulikakos, P. I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 17, 676–691 (2017).
Su, F. et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 366, 207–215 (2012).
Khan, Z. M. et al. Structural basis for the action of the drug trametinib at KSR-bound MEK. Nature 588, 509–514 (2020).
Poh, A. Dual RAF-MEK inhibitor assessed. Cancer Discov. 11, 5–6 (2020).
van Geel, R. et al. Phase 1 study of the pan-HER inhibitor dacomitinib plus the MEK1/2 inhibitor PD-0325901 in patients with KRAS-mutation-positive colorectal, non-small-cell lung and pancreatic cancer. Br. J. Cancer 122, 1166–1174 (2020).
de Miguel, M. & Calvo, E. Clinical challenges of immune checkpoint inhibitors. Cancer Cell 38, 326–333 (2020).
Giraldo, N. A. et al. The clinical role of the TME in solid cancer. Br. J. Cancer 120, 45–53 (2019).
Baldewijns, M. M. et al. VHL and HIF signalling in renal cell carcinogenesis. J. Pathol. 221, 125–138 (2010).
Liu, P. et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat. Commun. 10, 1486 (2019). The first evidence that a protein TKI is capable of inducing immunogenic cell death.
Frederick, D. T. et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 19, 1225–1231 (2013).
Dummer, R. et al. Combined PD-1, BRAF and MEK inhibition in advanced BRAF-mutant melanoma: safety run-in and biomarker cohorts of COMBI-i. Nat. Med. 26, 1557–1563 (2020).
P. Nathan, R. D. et al. LBA43 - Spartalizumab plus dabrafenib and trametinib (Sparta-DabTram) in patients (pts) with previously untreated BRAF V600–mutant unresectable or metastatic melanoma: Results from the randomized part 3 of the phase III COMBI-i trial. Ann. Oncol. 31, S1142–S1215 (2020).
Yang, J. C. et al. Osimertinib plus durvalumab versus osimertinib monotherapy in EGFR T790M-positive NSCLC following previous EGFR TKI therapy: CAURAL Brief Report. J. Thorac. Oncol. 14, 933–939 (2019).
Spigel, D. R. et al. Phase 1/2 study of the safety and tolerability of nivolumab plus crizotinib for the first-line treatment of anaplastic lymphoma kinase translocation - positive advanced non-small cell lung cancer (CheckMate 370). J. Thorac. Oncol. 13, 682–688 (2018).
Glodde, N. et al. Reactive neutrophil responses dependent on the receptor tyrosine kinase c-MET limit cancer immunotherapy. Immunity 47, 789–802.e789 (2017).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04139317 (2021).
Hemmings, B. A. & Restuccia, D. F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 4, a011189 (2012).
Flinn, I. W. et al. Idelalisib, a selective inhibitor of phosphatidylinositol 3-kinase-delta, as therapy for previously treated indolent non-Hodgkin lymphoma. Blood 123, 3406–3413 (2014).
Sapon-Cousineau, V., Sapon-Cousineau, S. & Assouline, S. PI3K inhibitors and their role as novel agents for targeted therapy in lymphoma. Curr. Treat. Options Oncol. 21, 51 (2020).
Andre, F. et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 380, 1929–1940 (2019).
Smyth, L. M. et al. Capivasertib, an AKT kinase inhibitor, as monotherapy or in combination with fulvestrant in patients with AKT1 E17K-mutant, ER-positive metastatic breast cancer. Clin. Cancer Res. 26, 3947–3957 (2020).
Rudolph, M. et al. AKT1 (E17K) mutation profiling in breast cancer: prevalence, concurrent oncogenic alterations, and blood-based detection. BMC Cancer 16, 622 (2016).
Jones, R. H. et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 21, 345–357 (2020).
Maemondo, M. et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 362, 2380–2388 (2010).
Oizumi, S. et al. Quality of life with gefitinib in patients with EGFR-mutated non-small cell lung cancer: quality of life analysis of North East Japan Study Group 002 Trial. Oncologist 17, 863–870 (2012).
Hosomi, Y. et al. Gefitinib alone versus gefitinib plus chemotherapy for non-small-cell lung cancer with mutated epidermal growth factor receptor: NEJ009 study. J. Clin. Oncol. 38, 115–123 (2020).
Noronha, V. et al. Gefitinib versus gefitinib plus pemetrexed and carboplatin chemotherapy in EGFR-mutated lung cancer. J. Clin. Oncol. 38, 124–136 (2020).
Reungwetwattana, T. et al. CNS response to osimertinib versus standard epidermal growth factor receptor tyrosine kinase inhibitors in patients with untreated EGFR-mutated advanced non-small-cell lung cancer. J. Clin. Oncol. 36, 3290–3297 (2018).
Camidge, D. R. et al. Exploratory analysis of brigatinib activity in patients with anaplastic lymphoma kinase-positive non-small-cell lung cancer and brain metastases in two clinical trials. J. Clin. Oncol. 36, 2693–2701 (2018).
Solomon, B. J. et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol. 19, 1654–1667 (2018).
Subbiah, V. et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann. Oncol. 29, 1869–1876 (2018).
Lin, J. J. et al. Efficacy of alectinib in patients with ALK-positive NSCLC and symptomatic or large CNS metastases. J. Thorac. Oncol. 14, 683–690 (2019).
Yang, J. C. H. et al. Osimertinib in patients with epidermal growth factor receptor mutation-positive non-small-cell lung cancer and leptomeningeal metastases: the BLOOM study. J. Clin. Oncol. 38, 538–547 (2020).
Ou, S. H., Sommers, K. R., Azada, M. C. & Garon, E. B. Alectinib induces a durable (>15 months) complete response in an ALK-positive non-small cell lung cancer patient who progressed on crizotinib with diffuse leptomeningeal carcinomatosis. Oncologist 20, 224–226 (2015).
Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351 (2016).
Cescon, D. W., Bratman, S. V., Chan, S. M. & Siu, L. L. Circulating tumor DNA and liquid biopsy in oncology. Nat. Cancer 1, 276–290 (2020). This review discusses the current landscape of ctDNA liquid-biopsy applications across cancer and highlights opportunities for clinical investigation.
Li, J. W., Cao, S. H., Xu, J. L. & Zhong, H. De novo MET amplification promotes intrinsic resistance to first-generation EGFR tyrosine kinase inhibitors. Cancer Biol. Ther. 20, 1183–1186 (2019).
Yu, H. A. et al. Poor response to erlotinib in patients with tumors containing baseline EGFR T790M mutations found by routine clinical molecular testing. Ann. Oncol. 25, 423–428 (2014).
Wander, S. A. et al. The genomic landscape of intrinsic and acquired resistance to cyclin-dependent kinase 4/6 inhibitors in patients with hormone receptor-positive metastatic breast cancer. Cancer Discov. 10, 1174–1193 (2020).
Guo, R. et al. MET inhibitor resistance in patients with MET exon 14-altered lung cancers. J. Clin. Oncol. 37, 9006–9006 (2019).
Lovly, C. M. & Shaw, A. T. Molecular pathways: resistance to kinase inhibitors and implications for therapeutic strategies. Clin. Cancer Res. 20, 2249–2256 (2014).
Robey, R. W. et al. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 18, 452–464 (2018).
Eyers, P. A., Craxton, M., Morrice, N., Cohen, P. & Goedert, M. Conversion of SB 203580-insensitive MAP kinase family members to drug-sensitive forms by a single amino-acid substitution. Chem. Biol. 5, 321–328 (1998).
Clark, K. et al. Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc. Natl Acad. Sci. USA 109, 16986–16991 (2012).
Gorre, M. E. et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293, 876–880 (2001). One of the first examples of clinical drug resistance to a kinase inhibitor mediated by a gatekeeper mutation.
Nguyen, K. S., Kobayashi, S. & Costa, D. B. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin. Lung Cancer 10, 281–289 (2009).
Yun, C. H. et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl Acad. Sci. USA 105, 2070–2075 (2008).
Drilon, A. et al. Repotrectinib (TPX-0005) Is a next-generation ROS1/TRK/ALK inhibitor that potently inhibits ROS1/TRK/ALK solvent-front mutations. Cancer Discov. 8, 1227–1236 (2018).
Solomon, B. J. et al. RET solvent front mutations mediate acquired resistance to selective RET inhibition in RET-driven malignancies. J. Thorac. Oncol. 15, 541–549 (2020).
Cocco, E. et al. TRK xDFG mutations trigger a sensitivity switch from type I to II kinase inhibitors. Cancer Discov. 11, 126–141 (2021).
Volm, M. & Efferth, T. Prediction of cancer drug resistance and implications for personalized medicine. Front. Oncol. 5, 282 (2015).
Husain, H. et al. Strategies to overcome bypass mechanisms mediating clinical resistance to EGFR tyrosine kinase inhibition in lung cancer. Mol. Cancer Ther. 16, 265–272 (2017).
Lin, J. J. et al. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann. Oncol. 31, 1725–1733 (2020).
Lavaud, P. & Andre, F. Strategies to overcome trastuzumab resistance in HER2-overexpressing breast cancers: focus on new data from clinical trials. BMC Med. 12, 132 (2014).
D’Amato, V. et al. Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat. Rev. 41, 877–883 (2015).
Yang, C. et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 36, 2255–2264 (2017).
Herrera-Abreu, M. T. et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 76, 2301–2313 (2016).
Min, A. et al. Cyclin E overexpression confers resistance to the CDK4/6 specific inhibitor palbociclib in gastric cancer cells. Cancer Lett. 430, 123–132 (2018).
Pandey, K. et al. Combined CDK2 and CDK4/6 inhibition overcomes palbociclib resistance in breast cancer by enhancing senescence. Cancers 12, 3566 (2020).
Bordi, P. et al. Detection of ALK and KRAS mutations in circulating tumor DNA of patients with advanced ALK-positive NSCLC with disease progression during crizotinib treatment. Clin. Lung Cancer 18, 692–697 (2017).
Suzawa, K. et al. Activation of KRAS mediates resistance to targeted therapy in MET exon 14-mutant non-small cell lung cancer. Clin. Cancer Res. 25, 1248–1260 (2019).
Ohashi, K. et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc. Natl Acad. Sci. USA 109, E2127–E2133 (2012).
de Bruin, E. C. et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 4, 606–619 (2014).
Jamme, P. et al. Alterations in the PI3K pathway drive resistance to MET inhibitors in NSCLC harboring MET exon 14 skipping mutations. J. Thorac. Oncol. 15, 741–751 (2020).
Costa, C. et al. PTEN loss mediates clinical cross-resistance to CDK4/6 and PI3Kα inhibitors in breast cancer. Cancer Discov. 10, 72–85 (2020).
Juric, D. et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 518, 240–244 (2015).
Sos, M. L. et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 69, 3256–3261 (2009).
Solca, F. et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 343, 342–350 (2012).
Engelman, J. A. et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 67, 11924–11932 (2007).
Cross, D. A. E. et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR Inhibitors in lung cancer. Cancer Discov. 4, 1046–1061 (2014).
Kim, E. S. Olmutinib: first global approval. Drugs 76, 1153–1157 (2016).
Mok, T. S. et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung Ccancer. N. Engl. J. Med. 376, 629–640 (2017).
Soria, J. C. et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 378, 113–125 (2018).
Ramalingam, S. S. et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N. Engl. J. Med. 382, 41–50 (2019).
Leonetti, A. et al. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 121, 725–737 (2019).
Thress, K. S. et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat. Med. 21, 560–562 (2015).
Katayama, R. et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl Med. 4, 120ra117 (2012).
Crinò, L. et al. Multicenter phase II study of whole-body and intracranial activity with ceritinib in patients with ALK-rearranged non-small-cell lung cancer previously treated with chemotherapy and crizotinib: results from ASCEND-2. J. Clin. Oncol. 34, 2866–2873 (2016).
Shaw, A. T. et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: a single-group, multicentre, phase 2 trial. Lancet Oncol. 17, 234–242 (2016).
Gettinger, S. N. et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: a single-arm, open-label, phase 1/2 trial. Lancet Oncol. 17, 1683–1696 (2016).
Gainor, J. F. et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 6, 1118–1133 (2016).
Yoda, S. et al. Sequential ALK inhibitors can select for lorlatinib-resistant compound ALK mutations in ALK-positive lung cancer. Cancer Discov. 8, 714–729 (2018).
Recondo, G. et al. Diverse resistance mechanisms to the third-generation ALK inhibitor lorlatinib in ALK-rearranged lung cancer. Clin. Cancer Res. 26, 242–255 (2020).
Manning, G., Whyte, D. B., Martinez, R., Hunter, T. & Sudarsanam, S. The protein kinase complement of the human genome. Science 298, 1912–1934 (2002). The comprehensive classification and annotation of the 500 plus protein kinases (the human kinome) and its subdivision into subfamilies, which has greatly facilitated the understanding of kinome relationships.
Morgan Jones, M. et al. The Structural Genomics Consortium: A Knowledge Platform for Drug Discovery (RAND Corporation, 2014).
Murray, C. W. & Blundell, T. L. Structural biology in fragment-based drug design. Curr. Opin. Struct. Biol. 20, 497–507 (2010).
Thomas, S. E. et al. Structure-guided fragment-based drug discovery at the synchrotron: screening binding sites and correlations with hotspot mapping. Phil. Trans. R. Soc. A 377, 20180422 (2019).
Abdeldayem, A., Raouf, Y. S., Constantinescu, S. N., Moriggl, R. & Gunning, P. T. Advances in covalent kinase inhibitors. Chem. Soc. Rev. 49, 2617–2687 (2020).
Liu, Q. et al. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 20, 146–159 (2013). This article and Abdeldayem et al. (2020) review advances in developing kinase inhibitors that bind covalently to conserved cysteine residues located near the catalytic site, and contributed to the increasing numbers of these inhibitors that are being developed and approved for clinical use.
Arighi, E., Borrello, M. G. & Sariola, H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor. Rev. 16, 441–467 (2005).
Gautschi, O. et al. Targeting RET in patients with RET-rearranged lung cancers: results from the global, multicenter RET registry. J. Clin. Oncol. 35, 1403–1410 (2017).
Drilon, A. et al. Efficacy of selpercatinib in RET fusion-positive non-small-cell lung cancer. N. Engl. J. Med. 383, 813–824 (2020).
Subbiah, V. et al. Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discov. 8, 836–849 (2018).
Kollareddy, M. et al. Aurora kinase inhibitors: progress towards the clinic. Invest. New Drugs 30, 2411–2432 (2012).
Komlodi-Pasztor, E., Sackett, D. L. & Fojo, A. T. Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin. Cancer Res. 18, 51–63 (2012).
Sherr, C. J. & Bartek, J. Cell cycle–targeted cancer therapies. Annu. Rev. Cancer Biol. 1, 41–57 (2017).
Ashton, S. et al. Aurora kinase inhibitor nanoparticles target tumors with favorable therapeutic index in vivo. Sci. Transl Med. 8, 325ra317 (2016).
Heffron, T. P. Challenges of developing small-molecule kinase inhibitors for brain tumors and the need for emphasis on free drug levels. Neuro-Oncol. 20, 307–312 (2017).
Skerratt, S. E. & Storer, R. I. in Kinase Drug Discovery: Modern Approaches (eds Ward, R. A. & Goldberg, F. W.) 128–180 (Royal Society of Chemistry, 2019). This article and Heffron (2017) discuss the unmet need for kinase inhibitors to achieve free brain penetration, and the chemistry design approaches and challenges to achieve inhibitors with appropriate properties.
Johnson, T. W. et al. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J. Med. Chem. 57, 4720–4744 (2014).
Zeng, Q. et al. Discovery and evaluation of clinical candidate AZD3759, a potent, oral active, central nervous system-penetrant, epidermal growth factor receptor tyrosine kinase inhibitor. J. Med. Chem. 58, 8200–8215 (2015).
Shibuya, M. & Suzuki, Y. Treatment of cerebral vasospasm by a protein kinase inhibitor AT 877 [Japanese]. No Shinkei 45, 819–824 (1993).
Doggrell, S. A. Rho-kinase inhibitors show promise in pulmonary hypertension. Expert Opin. Investig. Drugs 14, 1157–1159 (2005).
Nagumo, H. et al. Rho kinase inhibitor HA-1077 prevents Rho-mediated myosin phosphatase inhibition in smooth muscle cells. Am. J. Physiol. Cell Physiol. 278, C57–65 (2000).
Heitman, J., Movva, N. R. & Hall, M. N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253, 905–909 (1991).
Cohen, P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell Biol. 21, 317–324 (2009).
Zarrin, A. A., Bao, K., Lupardus, P. & Vucic, D. Kinase inhibition in autoimmunity and inflammation. Nat. Rev. Drug Discov. 20, 39–63 (2021).
Shuai, K. & Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 3, 900–911 (2003).
Fragoulis, G. E., McInnes, I. B. & Siebert, S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology 58, i43–i54 (2019). Recent review of the increasing number of JAK inhibitors being developed for the treatment of rheumatoid arthritis and other immune diseases.
A, T. V., Haikarainen, T., Raivola, J. & Silvennoinen, O. Selective JAKinibs: prospects in inflammatory and autoimmune diseases. BioDrugs 33, 15–32 (2019).
Baxter, E. J. et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365, 1054–1061 (2005).
Kralovics, R. et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 352, 1779–1790 (2005).
Kuykendall, A. T. et al. Between a rux and a hard place: evaluating salvage treatment and outcomes in myelofibrosis after ruxolitinib discontinuation. Ann. Hematol. 97, 435–441 (2018).
Talpaz, M., Erickson-Viitanen, S., Hou, K., Hamburg, S. & Baer, M. R. Evaluation of an alternative ruxolitinib dosing regimen in patients with myelofibrosis: an open-label phase 2 study. J. Hematol. Oncol. 11, 101 (2018).
Kim, T. W. et al. A critical role for IRAK4 kinase activity in Toll-like receptor-mediated innate immunity. J. Exp. Med. 204, 1025–1036 (2007).
Nanda, S. K. et al. Distinct signals and immune cells drive liver pathology and glomerulonephritis in ABIN1[D485N] mice. Life Sci. Alliance 2, e201900533 (2019).
Seganish, W. M. Inhibitors of interleukin-1 receptor-associated kinase 4 (IRAK4): a patent review (2012–2015). Expert. Opin. Ther. Pat. 26, 917–932 (2016).
Picard, C., Casanova, J. L. & Puel, A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IκBα deficiency. Clin. Microbiol. Rev. 24, 490–497 (2011).
Picard, C. et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine 89, 403–425 (2010). Review of the remarkable long-term clinical study of IRAK4-deficient patients, which revealed why IRAK4 inhibition is unlikely to cause life-threatening microbial infections in adults, and de-risked this drug target.
Danto, S. I. et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of PF-06650833, a selective interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitor, in single and multiple ascending dose randomized phase 1 studies in healthy subjects. Arthritis Res. Ther. 21, 269 (2019).
Wiese, M. D., Manning-Bennett, A. T. & Abuhelwa, A. Y. Investigational IRAK-4 inhibitors for the treatment of rheumatoid arthritis. Expert Opin. Investig. Drugs 29, 475–482 (2020).
Darling, N. J., Toth, R., Arthur, J. S. & Clark, K. Inhibition of SIK2 and SIK3 during differentiation enhances the anti-inflammatory phenotype of macrophages. Biochem. J. 474, 521–537 (2017).
Galapagos. Galapagos’ R&D Roundtable showcases Toledo program. GlobeNewswire https://www.globenewswire.com/news-release/2020/10/27/2115341/0/en/Galapagos-R-D-Roundtable-showcases-Toledo-program.html (2020).
Mujahid, N. et al. A UV-independent topical small-molecule approach for melanin production in human skin. Cell Rep. 19, 2177–2184 (2017).
Wein, M. N. et al. SIKs control osteocyte responses to parathyroid hormone. Nat. Commun. 7, 13176 (2016).
Kim, M. K. et al. Salt-inducible kinase 1 regulates bone anabolism via the CRTC1-CREB-Id1 axis. Cell Death Dis. 10, 826 (2019).
Lee, J. C. et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372, 739–746 (1994).
Cuenda, A. et al. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 364, 229–233 (1995).
Eyers, P. A., van den, I. P., Quinlan, R. A., Goedert, M. & Cohen, P. Use of a drug-resistant mutant of stress-activated protein kinase 2a/p38 to validate the in vivo specificity of SB 203580. FEBS Lett. 451, 191–196 (1999).
Hammaker, D. & Firestein, G. S. “Go upstream, young man”: lessons learned from the p38 saga. Ann. Rheum. Dis. 69, i77–i82 (2010).
Alam, J. J. Selective brain-targeted antagonism of p38 MAPKα reduces hippocampal IL-1β levels and improves Morris water maze performance in aged rats. J. Alzheimers Dis. 48, 219–227 (2015).
Alam, J., Blackburn, K. & Patrick, D. Neflamapimod: clinical phase 2b-ready oral small molecule inhibitor of p38α to reverse synaptic dysfunction in early Alzheimer’s disease. J. Prev. Alzheimers Dis. 4, 273–278 (2017).
Fagiani, F., Lanni, C., Racchi, M. & Govoni, S. Targeting dementias through cancer kinases inhibition. Alzheimers Dement. 6, e12044 (2020).
Cohen, P. & Goedert, M. GSK3 inhibitors: development and therapeutic potential. Nat. Rev. Drug Discov. 3, 479–487 (2004).
Martinez, A., Alonso, M., Castro, A., Pérez, C. & Moreno, F. J. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J. Med. Chem. 45, 1292–1299 (2002).
Serenó, L. et al. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 35, 359–367 (2009).
Paisán-Ruiz, C., Lewis, P. A. & Singleton, A. B. LRRK2: cause, risk, and mechanism. J. Parkinsons Dis. 3, 85–103 (2013).
Alessi, D. R. & Sammler, E. LRRK2 kinase in Parkinson’s disease. Science 360, 36–37 (2018).
Di Maio, R. et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl Med. 10, eaar5429 (2018).
Wong, M. M. K. et al. Neurodegeneration in SCA14 is associated with increased PKCγ kinase activity, mislocalization and aggregation. Acta Neuropathol. Commun. 6, 99 (2018).
Mabillard, H. & Sayer, J. A. The molecular genetics of Gordon syndrome. Genes 10, 986 (2019).
Di Maira, G. et al. The protein kinase CK2 contributes to the malignant phenotype of cholangiocarcinoma cells. Oncogenesis 8, 61 (2019).
D’Amore, C., Borgo, C., Sarno, S. & Salvi, M. Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy - potential clinical relevance. Cell. Oncol. 43, 1003–1016 (2020).
Boumahdi, S. & de Sauvage, F. J. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 19, 39–56 (2020). Interesting review of the emerging concept of drug-tolerant cells (also referred to as minimal residual disease) that can drive a drug-refractory phenotypic state that no longer depends on the drug-targeted pathway.
Oikkonen, J. et al. Prospective longitudinal ctDNA workflow reveals clinically actionable alterations in ovarian cancer. JCO Precis. Oncol. 3, 1–12 (2019).
Jiang, J. et al. Plasma-based longitudinal mutation monitoring as a potential predictor of disease progression in subjects with adenocarcinoma in advanced non-small cell lung cancer. BMC Cancer 20, 885 (2020).
Msaouel, P., Genovese, G., Gao, J., Sen, S. & Tannir, N. M. TAM kinase inhibition and immune checkpoint blockade- a winning combination in cancer treatment? Expert Opin. Ther. Targets 25, 141–151 (2021).
Xun, Q., Wang, Z., Hu, X., Ding, K. & Lu, X. Small-molecule CSF1R inhibitors as anticancer agents. Curr. Med. Chem. 27, 3944–3966 (2020).
Moen, M. D., McKeage, K., Plosker, G. L. & Siddiqui, M. A. A. Imatinib. Drugs 67, 299–320 (2007).
Joensuu, H. et al. Adjuvant imatinib for high-risk GI stromal tumor: analysis of a randomized trial. J. Clin. Oncol. 34, 244–250 (2016).
Wu, Y. L. et al. Osimertinib in resected EGFR-mutated non-small-cell lung cancer. N. Engl. J. Med. 383, 1711–1723 (2020). First approval of a kinase inhibitor for reducing risk of recurrence in early-stage resectable NSCLC.
Abbosh, C., Birkbak, N. J. & Swanton, C. Early stage NSCLC - challenges to implementing ctDNA-based screening and MRD detection. Nat. Rev. Clin. Oncol. 15, 577–586 (2018). Overview of the technical challenges and feasibility of low-frequency mutation detection using NGS-based ctDNA profiling to deliver the potential paradigm shifting approach needed for early disease and minimal residual disease detection.
Whyte, J. L., Smith, A. A. & Helms, J. A. Wnt signaling and injury repair. Cold Spring Harb. Perspect. Biol. 4, a008078 (2012).
Lu, X., Yang, J., Zhao, S. & Liu, S. Advances of Wnt signalling pathway in dental development and potential clinical application. Organogenesis 15, 101–110 (2019).
Neves, V. C., Babb, R., Chandrasekaran, D. & Sharpe, P. T. Promotion of natural tooth repair by small molecule GSK3 antagonists. Sci. Rep. 7, 39654 (2017). An interesting and unexpected example of the repurposing of a protein kinase inhibitor for a completely different clinical application.
Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M. & Hemmings, B. A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789 (1995).
Hudis, C. A. Trastuzumab–mechanism of action and use in clinical practice. N. Engl. J. Med. 357, 39–51 (2007).
Garcia, J. et al. Bevacizumab (Avastin®) in cancer treatment: a review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 86, 102017 (2020).
Yun, J. et al. Antitumor activity of amivantamab (JNJ-61186372), an EGFR–MET bispecific antibody, in diverse models of EGFR exon 20 insertion–driven NSCLC. Cancer Discov. 10, 1194–1209 (2020).
Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N. Engl. J. Med. 382, 610–621 (2020).
Verma, S. et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 367, 1783–1791 (2012).
Hashimoto, Y. et al. A novel HER3-targeting antibody-drug conjugate, U3-1402, exhibits potent therapeutic efficacy through the delivery of cytotoxic payload by efficient internalization. Clin. Cancer Res. 25, 7151–7161 (2019).
Zeng, S. et al. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: recent progress and future challenges. Eur. J. Med. Chem. 210, 112981 (2020).
Henning, R. K. et al. Degradation of Akt using protein-catalyzed capture agents. J. Pept. Sci. 22, 196–200 (2016).
Tovell, H. et al. Design and characterization of SGK3-PROTAC1, an isoform specific SGK3 kinase PROTAC degrader. ACS Chem. Biol. 14, 2024–2034 (2019).
Burslem, G. M. et al. Targeting BCR-ABL1 in chronic myeloid leukemia by PROTAC-mediated targeted protein degradation. Cancer Res. 79, 4744–4753 (2019).
Lu, X., Smaill, J. B. & Ding, K. New promise and opportunities for allosteric kinase inhibitors. Angew. Chem. Int. Ed. 59, 13764–13776 (2020).
Wylie, A. A. et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 543, 733–737 (2017).
Hughes, T. P. et al. Asciminib in chronic myeloid leukemia after ABL kinase inhibitor failure. N. Engl. J. Med. 381, 2315–2326 (2019). The first clinical data demonstrating efficacy of an allosteric ABL inhibitor in patients with CML who had failed prior ATP-competitive ABL inhibitors.
To, C. et al. Single and dual targeting of mutant EGFR with an allosteric inhibitor. Cancer Discov. 9, 926–943 (2019).
Eide, C. A. et al. Combining the allosteric inhibitor asciminib with ponatinib suppresses emergence of and restores efficacy against highly resistant BCR-ABL1 mutants. Cancer Cell 36, 431–443.e435 (2019).
Acknowledgements
The authors thank R. Ward at AstraZeneca for valuable input, including help in generating Figs. 4 and 5, and R. Marais for valuable discussions.
Author information
Authors and Affiliations
Contributions
P.C., D.C. and P.J. researched, wrote and edited this Review.
Corresponding authors
Ethics declarations
Competing interests
P.C. has shares in Alliance Pharma, AstraZeneca and GlaxoSmithKline and is a member of the Scientific Advisory Boards of Mission Therapeutics, Ubiquigent and Biocatalyst International. D.C. is an employee and shareholder of AstraZeneca. P.A.J. has received consulting fees from AstraZeneca, Boehringer-Ingelheim, Pfizer, Roche/Genentech, Takeda Oncology, ACEA Biosciences, Eli Lilly and Company, Araxes Pharma, Ignyta, Mirati Therapeutics, Novartis, LOXO Oncology, Daiichi Sankyo, Sanofi Oncology, Voronoi, SFJ Pharmaceuticals, Biocartis, Novartis Oncology, Nuvalent, Esai, Bayer, Transcenta and Silicon Therapeutics; receives post-marketing royalties from DFCI-owned intellectual property on EGFR mutations licensed to Lab Corp; has sponsored research agreements with AstraZeneca, Daichi-Sankyo, PUMA, Boehringer-Ingelheim, Eli Lilly and Company, Revolution Medicines, and Astellas Pharmaceuticals; and has stock ownership in Gatekeeper Pharmaceuticals.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- Protein kinases
-
Enzymes that catalyse transfer of the γ-phosphate of ATP to amino acid side chains in substrate proteins, such as serine, threonine and tyrosine residues.
- Polypharmacology
-
A term to describe a feature of kinase inhibitors that can inhibit multiple kinase targets at clinically achievable concentrations. This feature can lead to a broader range of clinical activity and/or increased toxicity.
- Next-generation inhibitor
-
A kinase inhibitor that is improved sufficiently compared with the original first-generation drug that it is effective in preventing disease progression in patients who are no longer responsive to the original drug.
- Bromodomain
-
(BRD). A particular type of protein domain comprising approximately 110 amino acids that recognizes acetylated lysine residues in other proteins, frequently leading to the remodelling of chromatin and changes in gene transcription. Many drugs initially developed as protein kinase inhibitors also bind to and inhibit the functions of BRDs.
- Pseudokinase
-
Pseudokinases are proteins that possess a domain that closely resembles the catalytic domains of protein kinases, but that lack one or more amino acid residues essential for catalysis by other kinases, and are therefore presumed to be catalytically inactive. Some pseudokinases have subsequently been found to display kinase catalytic activity for interesting reasons or to have acquired entirely novel catalytic functions.
- Oncogenic addiction
-
A process in which cancers with genetic, epigenetic or chromosomal irregularities become dependent on one or a few genes for maintenance and survival. Such cancers can be exquisitely sensitive to killing by particular kinase inhibitors if one of the genes required for cancer survival encodes a particular protein kinase or a regulator of kinase activation.
- Blood–brain barrier
-
(BBB). A highly selective semipermeable border of endothelial cells that prevents solutes in the circulating blood from non-selectively crossing into the extracellular fluid of the central nervous system.
- Acquired drug resistance
-
Clinical drug resistance, characterized by tumour growth while on treatment, that develops in patients following an initial clinical benefit (a clinical response or prolonged stable disease).
- Gatekeeper mutation
-
The gatekeeper is a conserved amino acid residue near the ATP-binding site that, when occupied by an amino acid with a small side chain (typically threonine), creates a small hydrophobic pocket that is targeted by many protein kinase inhibitors. The mutation of the gatekeeper to an amino acid with a bulkier side chain fills the hydrophobic pocket, making it inaccessible to the kinase inhibitor, but not ATP. Such mutations are a frequent cause of drug resistance.
- Solvent-front mutations
-
The solvent front is a region of the ATP pocket in the catalytic domain that has a relatively high solvent exposure, and where multiple kinase inhibitors typically make contact. Mutation of residues in this region frequently cause resistance to kinase inhibitors.
- Kinase profiling
-
A technology for assessing which of the 500 plus protein kinases encoded by the human genome is inhibited by a particular kinase inhibitor. Some technologies used for kinase profiling can identify targets of such drugs that are not protein kinases.
- Covalent inhibitors
-
Kinase inhibitors that bind irreversibly to their targets, most commonly by making a covalent chemical bond with a cysteine residue of the kinase.
Rights and permissions
About this article

Cite this article
Cohen, P., Cross, D. & Jänne, P.A. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat Rev Drug Discov 20, 551–569 (2021). https://doi.org/10.1038/s41573-021-00195-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-021-00195-4
This article is cited by
-
Application of nanotechnology in the treatment of glomerulonephritis: current status and future perspectives
Journal of Nanobiotechnology (2024)
-
DBPP-Predictor: a novel strategy for prediction of chemical drug-likeness based on property profiles
Journal of Cheminformatics (2024)
-
CircHAS2 activates CCNE2 to promote cell proliferation and sensitizes the response of colorectal cancer to anlotinib
Molecular Cancer (2024)
-
Design, synthesis, biological assessment and molecular modeling studies of novel imidazothiazole-thiazolidinone hybrids as potential anticancer and anti-inflammatory agents
Scientific Reports (2024)
-
The molecular interaction pattern of lenvatinib enables inhibition of wild-type or kinase-mutated FGFR2-driven cholangiocarcinoma
Nature Communications (2024)
