
Abstract
Despite recent advances in the treatment of autoimmune and inflammatory diseases, unmet medical needs in some areas still exist. One of the main therapeutic approaches to alleviate dysregulated inflammation has been to target the activity of kinases that regulate production of inflammatory mediators. Small-molecule kinase inhibitors have the potential for broad efficacy, convenience and tissue penetrance, and thus often offer important advantages over biologics. However, designing kinase inhibitors with target selectivity and minimal off-target effects can be challenging. Nevertheless, immense progress has been made in advancing kinase inhibitors with desirable drug-like properties into the clinic, including inhibitors of JAKs, IRAK4, RIPKs, BTK, SYK and TPL2. This Review will address the latest discoveries around kinase inhibitors with an emphasis on clinically validated autoimmunity and inflammatory pathways.
Similar content being viewed by others
Introduction
Inflammation is a physiological response of the immune system to injury and infection. This process signals the immune system to heal and repair damaged tissue, as well as to defend itself against infective agents, such as viruses and bacteria. However, unresolved or inappropriately activated inflammation can become pathogenic1. Chronic inflammation is the primary cause of a broad spectrum of diseases, including rheumatoid arthritis (RA), inflammatory bowel disease (IBD; including gastrointestinal conditions such as Crohn’s disease and ulcerative colitis), chronic obstructive pulmonary disease (COPD), asthma, psoriasis and idiopathic pulmonary fibrosis (IPF), among others1. Viral and bacterial infections or other insults (such toxins, chemicals and so forth) can lead to uncontrolled acute inflammatory responses and injury often seen in patients with underlying pathogenic conditions (such as COPD or asthma). Acute lung injury (ALI) is a syndrome with diagnostic criteria based on hypoxaemia and a classical radiological appearance, with acute respiratory distress syndrome (ARDS) at the severe end of the disease spectrum impairing gas exchange, leading to multiple organ failure widespread inflammation in the lungs and sepsis2. A recent example of virally induced ALI and ARDS includes SARS-CoV-2 infection, which is associated with a cytokine storm (characterized by high levels of IL-6, IL-12 and IL-1β, and tumour necrosis factor (TNF)) and defective type I interferon activity3. This inflammatory response resembles the cytokine release syndrome observed in patients receiving chimeric antigen receptor (CAR) T cell therapy and bispecific T cell-engaging antibodies, which can be treated with anti-cytokine therapy targeting the IL-6–IL-6 receptor (IL-6R) signalling pathway3. Although select biologics and kinase inhibitors (see below) are effective in treating various inflammatory diseases, a large proportion of patients are not responsive to current therapies, and effective treatment approaches for this subset of patients are needed4.
Autoimmune diseases refer to a spectrum of conditions in which the immune system mistakenly attacks one’s own body5. This autoimmune response often involves dysregulated adaptive immunity (mediated by B and T lymphocytes) towards anatomical self-antigens (such as insulin)5. Certain human leukocyte antigen (HLA) genes have also been demonstrated to be predictive of the development of autoimmune diseases. HLA molecules on antigen-presenting cells present antigens to effector T cells in an interactive process required for antigen-specific T cell activation. Effector T cells then generate local inflammation by producing inflammatory cytokines or directly damaging the tissues, whereas CD4+CD25+ regulatory T (Treg) cells counteract the inflammatory response to maintain immune homeostasis in tissues. Autoimmune diseases are on the rise and contribute to approximately 100 clinical indications affecting 3–5% of the population5. They are caused by the deregulation of such cellular dynamics resulting in organ damage, including systemic lupus erythematosus (SLE; systemic disease with many organs targeted), type 1 diabetes (affecting the pancreas), multiple sclerosis (which affects the central nervous system), coeliac disease (which affects the small intestine), primary biliary cirrhosis (affecting the liver), chronic spontaneous urticaria (which affects the skin), immune thrombocytopenic purpura (ITP; platelets), autoimmune haemolytic anaemia (which affects red blood cells) and IgA nephropathy (which affects kidney glomeruli), among others5.
Autoinflammatory syndromes are a rare set of disorders caused mostly by genetic mutations that affect innate immune cells (such as macrophages or neutrophils) and lead to uncontrolled activation of the immune system when there is no actual infection6. These patients often respond better to select anti-inflammatory drugs (such as anti-TNF or anti-IL-1β) but not broad immunosuppressives. Examples of autoinflammatory disorders include familial Mediterranean fever, TNF receptor-associated periodic syndrome, cryopyrin-associated periodic syndromes, deficiency of the IL-1 receptor (IL-1R) antagonist (IL-1Rα), deficiency of the IL-36Rα and interferonopathies6.
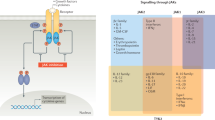
Although understanding signalling pathways in inflammatory and autoimmune diseases is challenging, preclinical and clinical research has been greatly instructive for therapeutic development. Biologics (such as antibody antagonists or fusion proteins) have validated several pathogenic pathways involved in these diseases (Fig. 1). Examples include therapies that use inhibition of cytokine receptors and/or ligands (such as TNF and IL-1), cellular depletion to reduce pathogenic cellular response (such as anti-CD20 antibodies for B cell depletion to limit autoantibody production or B cell-mediated antigen-presenting cell function) or inhibition of cellular differentiation (such as inhibition of macrophage colony-stimulating factor 1 receptor (CSF1R) to reduce macrophage differentiation). Recently, strategies to stimulate immune receptors to reset productive immunity are evolving as a powerful approach in drug development7.

Major inflammatory pathways and downstream kinases are depicted to show surface versus intracellular drug targets, highlighting drugs that are currently being clinically evaluated or already approved. All biologics that are included have mostly been evaluated in phase II trials and beyond. All small-molecule kinase inhibitors have been evaluated in phase I and beyond. BAFFR, B cell activating-factor receptor; BCMA, B cell maturation antigen; BCR, B cell receptor; BTK, Bruton’s tyrosine kinase; CD40L, CD40 ligand; CSF1R, colony-stimulating factor 1 receptor; CTLA4, cytotoxic T lymphocyte-associated protein 4; FcεR, Fcε receptor; IKKε, inhibitor of NF-κB subunit-ε; IL-1R, IL-1 receptor; IRAK, IL-1R-associated kinase; ITK, IL-2-inducible T cell kinase; JAK, Janus-associated kinase; MD2, myeloid differentiation factor 2; NF-κB, nuclear factor-κ-light-chain-enhancer of activated B cells; NIK, NF-κB-inducing kinase; RANKL, receptor activator of NF-κB ligand; RIP1, receptor-interacting protein 1; RLK, resting lymphocyte kinase; ST2, IL-1R-like 1; SYK, spleen tyrosine kinase; TACI, transmembrane activator and CAML interactor; TBK1, TANK-binding kinase 1; TCR, T cell receptor; TEC, Tec protein tyrosine kinase; TLR, Toll-like receptor; TNF, tumour necrosis factor; TNFR, TNF receptor; TPL2, tumour progression locus 2; TSLP, thymic stromal lymphopoietin; TYK2, tyrosine kinase 2.
However, there are still considerable unmet medical needs for the treatment of some inflammatory (COPD, IPF and IBD) and autoimmune (SLE, type 1 diabetes, primary biliary cirrhosis, Graves disease and multiple sclerosis) diseases, indicating the demand for effective therapeutics5. Even for diseases such as RA, for which several approved drugs exist, 74% of patients are not satisfied with their current treatment, according to surveys8. Thus, availability of effective treatments with disease-modifying potential and minimal adverse effects to reset productive immunity is crucial.
Kinases (518 encoded in genome) are enzymes that phosphorylate up to one-third of the proteome, and their utility as drugs is expanding in cancer, inflammatory and neurodegenerative diseases9. Owing to the distribution of select kinases in multiple signalling cascades in immune cells, the use of small-molecule kinase inhibitors has the potential to disable inflammation in a targeted fashion10. In addition, emerging data suggest that combination therapy with non-overlapping therapeutics (such as combinations of biologics and kinase inhibitors) may be more effective than single agents11,12. Therefore, a comprehensive understanding of signalling kinases combined with the ongoing clinical evaluations should lead to the discovery of effective therapies.
There has been tremendous progress in advancing several kinase inhibitors into preclinical and clinical investigations. Janus-associated kinase inhibitors (JAKis) have already been proven clinically beneficial for the treatment of RA and are being advanced in several other indications (Crohn’s disease, alopecia areata, psoriasis, Alzheimer disease); however, intense efforts are underway to optimize their selectivity and modes of delivery to reduce toxicity13. This Review captures the current efforts, progress and new discoveries around kinase inhibitors with an emphasis on key clinically validated pathways and targets with potential to mitigate human disease.
Inflammation and immune response
Tissue inflammation involves an influx of immune cells — including neutrophils, monocytes and macrophages — to various tissues, such as the skin, gut or lung1. This process is often regulated by a complex hierarchy of immune cells, cell surface receptors, signal transduction and the resultant gene transcription and translation of immunomodulating factors. Activation of receptors on immune cells drives signalling cascades that dictate, maintain and amplify local or systemic immune responses. Accordingly, chronic or dysregulated signalling can perpetuate inflammation and generate excessive levels of superoxide radicals, proteases, and cytokines and chemokines that can then cause tissue damage14. Importantly, the production of these pro-inflammatory mediators is subject to multiple regulatory mechanisms at the transcriptional and post-transcriptional levels. Early induction of the majority of inflammatory transcripts depends on transcription factor networks including NF-κB (canonical and non-canonical), signal transducers and activators of transcription (STATs), nuclear factor of activated T cells (NFATs) and interferon-regulatory factors (IRFs). However, the net production of the corresponding proteins depends, in part, on mitogen-activated protein kinases (MAPK) and molecular programmes that regulate transcript stability and translation14. The canonical NF-κB pathway mediates the activation of transcription factors NF-κB1 p50, transcription factor p65 (encoded by RELA) and proto-oncogene REL, whereas the non-canonical NF-κB pathway selectively activates p100-sequestered NF-κB members, predominantly NF-κB2 p52 subunit and RelB15. MAPK signalling (such as that mediated by ERK1/2 and p38) regulates RNA stability and translation of cytokines, which enable immune cells to respond promptly16. The STAT family of transcription factors integrates the signalling cascade of several cytokine receptors and ligands13. Activated STATs bind to GAS (IFNγ-activated sequence) DNA elements, and initiate transcription of target genes. Diverse outcomes of STAT signalling are not only determined by the expression of specific receptors but also by the interaction of STATs (such as STAT5) with cofactors, and by the cell-specific activity of members of the suppressor of cytokine signalling (SOCS) family, which negatively regulate STAT function13. Therefore, complex positive and negative regulatory networks orchestrate immune responses.
The physiological or pathogenic immune response involves multiple receptors on different immune cells and their cognate ligands. Host immunity is divided into innate and adaptive immune responses17. The former reacts rapidly and non-specifically to pathogens, whereas the latter responds in a slower but specific manner, with the generation of long-lived immunological memory17. Strict regulation of immune response is partly regulated by CD4+ T helper (TH) cells because they regulate the function of other immune or even non-immune cells18. Naive CD4+ T cells can differentiate into multiple distinct T cell subsets, such as TH1, TH2, TH17 and Treg cells, depending on the cytokine milieu18. Treg cells are essential in preventing autoimmune diseases and avoiding prolonged immunopathological processes and allergies acting via classic suppressive mechanisms on other immune cells as well as reparative functions19. B cells, in addition to their function in antibody production, also express a high level of MHC class II and can present antigens to TH cells to mount an immune response20. Self-reactive B cells and T cells can turn the immune system against its own body to cause various autoimmune disorders5.
The innate immune response is carried out by neutrophils and plasmacytoid dendritic cells (pDCs), basophils, natural killer cells, innate lymphoid cells and granulocytes21. These cells express various cytokines and selected receptors and ligands to mount an immune response21. Cytokines and other inflammatory mediators function as messengers that bind to specific receptors to regulate immune response1. The TNF superfamily contains 19 members that bind 29 receptors that are expressed predominantly by immune cells and function as cytokines regulating diverse cellular functions, including immune response and inflammation22. The IL-1R family comprises ten members23, and includes several ligand-binding receptors (IL-1R1, IL-1R2, IL-1R4, IL-1R5, IL-1R6), two types of accessory chain (IL-1R3, IL-1R7), molecules that act as inhibitors of IL-1 and IL-18 cytokines (IL-1R2, IL-1R8, IL-18BP) and two orphan receptors (IL-1R9, IL-1R10) with no known ligand23. The majority of the receptors from the IL-1R family promote activation, proliferation, differentiation and production of pro-inflammatory cytokines from various cell types23. IL-6 is a pleiotropic cytokine implicated in several diseases, including arthritis, sepsis, anaemia of chronic diseases, angiogenesis acute-phase response, bone and cartilage metabolism disorders and cancer24. IL-6 binds IL-6R, which has two subunits, IL-6Rα and IL-6Rβ (also known as gp130). Cells only express gp130 and are not responsive to IL-6 alone, but they can respond to a complex formed by IL-6 bound to a naturally occurring soluble form of the IL-6R, a process known as trans-signalling and that controls the pro-inflammatory responses of IL-6 (ref.24). The discovery of the IL-6–IL-6R axis provided a foundation to understand the biology of a group of related cytokines, including the IL-12 family of cytokines (IL-12, IL-23, IL-27, IL-35), which use shared receptors and cytokine subunits25. IL-12 is produced by innate cells, such as macrophages and dendritic cells, and binds to a heterodimeric receptor formed by IL-12Rβ1 and IL-12Rβ2, which promotes development of IFNγ-producing TH1 cells from naive T cells26. IL-23 is also produced by innate cells and signals through the IL-23R and the shared subunit IL-12Rβ1 (ref.27). IL-23 is a heterodimeric cytokine formed by the p19 and p40 subunits that binds the IL-12Rβ1 and IL-23R receptor complex expressed by several cells (natural killer cells, macrophages, dendritic cells, memory T cells and keratinocytes). Comparing the phenotypes of mice deficient in IL-23 or IL-12 receptor and ligand subunits established that IL-23 is a main culprit in autoimmune disease models27,28. IL-23 facilitates the production of IL-17 in TH17 cells and acts on cellular targets — including keratinocytes, neutrophils, endothelial cells and fibroblasts — to stimulate production of various chemokines and cytokines, which, in turn, promote tissue inflammation28. Correspondingly, the blockade of IL-17 or IL-23 is effective in managing the symptoms of certain diseases, such as psoriasis28.
IgG Fc receptors (FcRs) bind to antibodies to clear infected cells or invading pathogens29. This complex mediates inflammatory signalling via the immunoreceptor tyrosine-based activation motif (ITAM) in phagocytic or cytotoxic cells to destroy microbes, or in infected cells by antibody-mediated phagocytosis or antibody-dependent cell-mediated cytotoxicity29. In similar fashion, autoantibodies and autoimmune complexes (autoantibody bound to self-antigen) may serve as pathogenic factors in autoimmune or inflammatory injury, as they are responsible for the initiation of the inflammatory cascade and its resulting tissue damage29.
Toll-like receptors (TLRs) are sensors of microbial antigens that recognize pathogen-associated molecular patterns (PAMPs), which are conserved structures found on microbial cell walls that activate the host innate immune response30. TLRs can also recognize damage-associated molecular patterns (DAMPs) that are generated in the host following tissue injury or cellular activation30. There are ten TLRs identified in humans (TLR1–TLR10). Most TLRs are expressed on the cell surface and recognize antigens present on bacterial outer membranes. TLR3, TLR7, TLR8 and TLR9, however, are expressed intracellularly in endosomes and recognize nucleic acid ligands from various sources, including viruses or DAMPs30. Excessive TLR activation by DAMPs or PAMPs disrupts the immune homeostasis by sustained pro-inflammatory cytokine production, and consequently contributes to the development of several inflammatory diseases30.
Targeting kinases in immunity
Signalling from multiple cytokine receptors converges on a few kinases — such as JAK1 — which has made kinases potential targets to disable inflammation in a targeted fashion13. In cases such as JAK1, even partial target inhibition is sufficient to reduce several pathogenic pathways simultaneously in the clinic13. Other kinases with restricted or preferred immune cell function are emerging as promising drug targets to alleviate dysregulated inflammation with reduced side effects. For example, the non-canonical NF-κB pathway is less universal and integrates signalling cascades downstream of selected immune receptors that are validated as attractive drug targets, such as CD40 and BAFFR (also known as tumour necrosis factor receptor superfamily member 13C (TNFRSF13)) in immunological disorders15 or NF-κB-inducing kinase (NIK; also known as MAP3K14). In addition, select kinases (such as MAP3K8, also known as tumour progression locus 2 (TPL2)) participate in positive feedback loops in which kinase-dependent production of pathogenic mediators re-engage the original signalling cascade to activate the same kinase. TPL2 is transcriptionally induced and activated by the inflammatory receptors, including multiple TLRs, TNF receptor 1 (TNFR1), IL-1R1 and IL-1R2 (ref.31). TPL2 also amplifies local inflammation by promoting the production of TNF and IL-1, which bind and activate their corresponding receptors31. Therefore, inhibition of TPL2 may disrupt the feedback loop and dampen such pathogenesis in diseases such as RA, IBD and psoriasis31.
It should be emphasized that challenges remain for targeting kinases and the success rate for the generation of selective small-molecule inhibitors is low. This is because most kinase inhibitors are aimed to bind to the kinase pocket to compete with the ATP-binding site, which is highly conserved across the kinome. In addition, signal transduction for some kinases may extend beyond its activity, with additional roles in creating structural docking hubs (such as IRAK4 as discussed below)32,33. Another obstacle is the complexity of each signalling pathway in different cell types, which is not well understood in humans.
IRAK4 functions downstream of several innate immune cell receptors, such as TLRs and IL-1Rs. Most recently, for the first time, positive clinical data have been reported with IRAK4 inhibitors in patients with RA, opening an opportunity for probing this target across several other inflammatory indications34. Receptor-interacting serine/threonine protein (RIP) kinase inhibitors are under investigation in patients with RA, ulcerative colitis and neurodegenerative diseases; multiple Bruton’s tyrosine kinase (BTK) inhibitors (BTKis) are being evaluated in lupus and RA35; and a TPL2 inhibitor is advancing in clinical studies with promising potential for multiple inflammatory diseases36. Several inhibitors of tyrosine-protein kinase SYK are currently being evaluated for the treatment of autoimmune haemolytic anaemia, IgA nephropathy and chronic spontaneous urticaria37. Beyond these advanced targets, the emerging genetic and functional data plus the availability of experimental tool kinase inhibitors support the utility of other kinases, including p38δ, p38γ, IL-2 inducible T cell kinase (ITK), NIK, TANK-binding kinase 1 (TBK1), inhibitor of NF-κB subunit-ε (IKKε), cyclin-dependent kinase 8 (CDK8) and CDK19.
In the following sections, we provide an overview of several kinases that are positioned in key inflammatory cascades and their utility as drug targets in various inflammatory diseases.
JAKs and TYK2
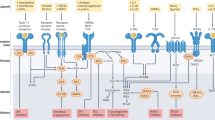
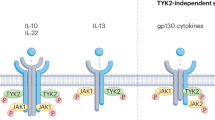
The JAK family of kinases includes JAK1, JAK2, JAK3 and non-receptor tyrosine-protein kinase TYK2 (ref.13). JAKs transduce signals from many cytokine receptors of the interleukin and interferon families as well as from growth hormone and erythropoietin (EPO)13 (Fig. 2). JAKs transduce signalling of IL-2R, IL-4R, IL-5R, IL-6R, IL-13R and type I interferons, which all have been validated as pathogenic pathways in different diseases such as RA and asthma. TYK2 is activated downstream after receptor binding by IL-23, IL-12 and type I interferons, each of which are implicated in the pathogenesis of multiple inflammatory diseases38. Dedicated combinations of STAT family members (STAT1–STAT6) unique to each receptor and the associated docking sites are recruited and phosphorylated by JAKs, leading to STAT dimerization and subsequent nuclear translocation for gene regulation13 (Fig. 2).

The binding of extracellular ligands leads to pathway activation via changes to the receptors that permit trans-phosphorylation of associated Janus-associated kinases (JAKs). Activated JAKs then phosphorylate both the receptor and cognate signal transducer and activator of transcription (STAT) proteins. Activated and dimerized STATs then enter the nucleus to bind to transcriptional regulatory sites of target genes. Receptors that use JAK2 and JAK3, JAK3 alone, tyrosine kinase 2 (TYK2) alone or JAK3 and TYK2 have not been described. EPO, erythropoietin; GH, growth hormone; GM-CSF, granulocyte–macrophage colony-stimulating factor; LIF, leukaemia inhibitory factor; OSM, oncostatin M; P, phosphorus; TPO, thrombopoietin; TSLP, thymic stromal lymphopoietin.
JAKis, both reversible and irreversible, have been advanced to clinical evaluation with varying degrees of selectivity13 (Fig. 2). Covalent inhibitors bind irreversibly to kinase pockets or to the adjacent chemically reactive amino acid (usually cysteine, lysine or aspartic acid) to form a bond and block activity (Table 1). Tofacitinib was the first JAKi approved for the treatment of RA and inhibits JAK3, JAK1 and, to a lesser degree, JAK2 (ref.13). Baricitinib is also approved for RA and inhibits JAK1 and JAK2 (ref.39). Peficitinib is a pan-JAKi awaiting more clinical data13. PF-06651600 is the only irreversible covalent JAK3i tested for the treatment of RA, alopecia areata and ulcerative colitis with breakthrough designation13,40. PF-06651600 is unique among JAKis in that it inhibits only cytokine receptors that use the common γ-chain. PF-06651600 has selectivity for JAK3 owing to its covalent interaction with a cysteine residue (Cys909) in the catalytic domain of JAK3, which, in the other JAK isoforms, is replaced by a serine residue13. Figlotininb (Gilead) is a selective JAK1i that is effective in modulating the immune response (measured by cellular and serum biomarkers) and achieving clinical response in patients with moderate to severe RA41. JAK3 may be a potential target in asthma and organ transplants based on preclinical model studies that have described its selective activation downstream of IL-4, IL-15 and IL-21 receptors, all of which use the common γ-chain42,43,44 (Fig. 1). PF-06651600 can also inhibit the tyrosine-protein kinase TEC family including BTK, bone marrow kinase (BMX), ITK, resting lymphocyte kinase (RLK) and Tec protein tyrosine kinase (TEC) by binding similarly to the Cys909 shared in the binding pocket of the kinase domain40. The inhibition of TEC kinases might expand the mechanism of action of JAK3 to other cell types, such as lymphocytes (see below). JAK2 is activated downstream of receptors for the cytokines thymic stromal lymphopoietin (TSLP), IL-13, IL-23 and IFNγ (Fig. 1). Ruxolitinib inhibits JAK2 and, to a lesser extent, JAK1 and JAK3, and is approved for myelofibrosis and polycythemia vera45. Fedratinib targets JAK2 and was recently approved for myelofibrosis, showing significant improvements in symptoms and a reduction in spleen size46.
Cytokine antagonists have provided a precedent for the utility of selective JAKis in the clinic. The use of tezepelumab, an antibody against TSLP in adults with uncontrolled asthma, suggest that selective JAK2 inhibition might be beneficial47. In addition, given the potential side effects of systemic JAKis, other localized routes of administration of JAKis with unique biophysical properties (such as those restricted to the gut, or topical or inhaled routes) may be beneficial in intestinal48, dermatological49,50 or respiratory33 diseases, although determining the dosing regimens may be challenging and it is not clear whether partial inhibition of systemic JAKi is sufficient in all of these indications49,50.
A coding variant of TYK2 that protects from multiple autoimmune diseases38 leads to the substitution of a proline residue with alanine at position 1104 (P1104A) in the catalytic domain, preventing receptor-mediated activation. This finding has enabled rationale designs to target TYK2 (refs51,52), such as BMS-986165, which blocks the receptor-mediated activation of TYK2 allosterically, in a mechanism similar to that of the deactivating TYK2-P1104A coding variant. BMS-986165 targets the pseudokinase domain of TYK2, which is promising in the discovery of a selective inhibitor of TYK2 that limits off-target effects in other kinases, particularly of the related JAK kinases. BMS-986165 blocks IL-12 and IL-23 signalling in human cells and also prevents type I interferon signalling, which showed protection from disease in mouse models of colitis or SLE38. BMS-986165 was well tolerated in healthy volunteers during a phase I trial and dampened responses to an in vivo interferon challenge38. It was also beneficial in psoriasis in a phase II study, with a large phase III programme currently ongoing53. Another selective TYK2 inhibitor, PF-06826647, is also being tested in moderate-to-severe psoriasis in an ongoing phase II clinical trial (NCT03895372). Furthermore, another inhibitor that targets both TYK2 and JAK2, PF-06826647, is also being tested in moderate-to-severe psoriasis in a phase II clinical trial (NCT03895372).
JAK inhibition may benefit the management of COVID-19 patients by reducing JAK-dependent cytokine storms (such as that mediated by IL-6 or IL-12)3. Baricitinib could also impair SARS-CoV-2 endocytosis by inhibiting AP2-associated kinase 1 (AAK1) and the cyclin G-associated kinase (GAK) kinases54. Several clinical trials assessing the efficacy of JAKis in COVID-19 are ongoing, including baricitinib, ruxolitinib, tofacitinib and the nebulized TD-0903 molecule from Theravance Biopharma3. In an initial open-label, small trial (NCT04358614, 12 patients), baricitinib-treated patients achieved significantly greater improvements in clinical symptoms, lung function and hospitalization55. If successful, JAKis might be considered for patients with non-COVID-19-induced ALI and ARDS. It is crucial to identify companion predictive and diagnostic biomarkers to improve the diagnosis and treatment of patients with ALI and/or ARDS2.
IRAK1 and IRAK4
IL-1Rs and TLRs share a conserved Toll/IL-1R receptor (TIR) domain on the cytoplasmic tails of each receptor, and therefore categorically use similar signalling pathways30. The TIR domain on all IL-1R family members (except for TLR3) recruits the TIR domain found on the carboxy terminus of myeloid differentiation primary response 88 (MyD88)30. On its amino terminus, MyD88 contains a death domain that recruits respective death domains found on IL-1R-associated kinases (IRAKs), and together they form a signalling complex called the Myddosome56,57 (Fig. 3).

IL-1 receptor (IL-1R)-associated kinase 1 (IRAK1) and IRAK4 function is regulated by protein–protein interactions and by post-translational modifications that may be uniquely regulated in different cell types to fine-tune an immune response. IL-1R or Toll-like receptor (TLR; except for TLR3) engagement causes the recruitment of adaptor protein myeloid differentiation primary response 88 (MyD88) to the intracellular Toll/IL-1R receptor (TIR) domains to initiate the Myddosome assembly. MyD88 recruits IRAK4 via death domain interactions. IRAK4 is activated via autophosphorylation and is also K63-ubiquitylated (grey circles). Phosphorylated IRAK4 can activate IRAK1, which facilitates IRAK1–tumour necrosis factor receptor-associated factor 6 (TRAF6) complex formation. K48 ubiquitylation (blue circles) of IRAK1 is required for the activation of TGFβ-activated kinase 1-binding protein 1 (TAK1), and its binding to TAB1 and TAB2 to drive the formation of the inhibitor of NF-κB (IKK) complex and subsequent NF-κB inhibitor-α (IκBα) activation, which then leads to NF-κB, mitogen-activated protein kinase (MAPK) and interferon-regulatory factor (IRF) activation to induce the transcription of pro-inflammatory cytokines and cellular processes, such as proliferation and activation61,62. The mechanism of IRF activation by MyD88 is less understood. Downstream of IRAK4 activation, TAK1 and IKKβ complex can mediate IRF5 phosphorylation, nuclear translocation and transcription in monocytes74, but IRAK1 can also directly bind and phosphorylate IRF7 in plasmacytoid dendritic cells263. The mechanism of IRAK4 kinase activity-independent action is less understood but may involve K63-ubiquitylated signalling hubs and other novel molecular scaffolds92. AP-1, activator protein 1; CREB, cAMP response element-binding protein; P, phosphorus; TAB, TGFβ-activated kinase 1 binding protein; Ub, ubiquitin.
As with most kinases, IRAK activity is modulated, in part, by conformational changes and post-translational modifications30,58. At the Myddosome, IRAK4 is activated via trans-autophosphorylation to then activate IRAK1 by phosphorylation58 (Fig. 3). Activated IRAK1 and TNFR-associated factor 6 (TRAF6) dissociate from the Myddosome and activate TGFβ-activated kinase 1-binding protein 1 (TAK1), a member of the MAPK kinase family59,60,61. TAK1 activates IKKβ in the IKK complex, which phosphorylates NF-κB inhibitor-α (IκBα), resulting in MAPK activation and NF-κB-regulated transcription61,62 (Fig. 3). IRAK2 may not be required for receptor-mediated NF-κB activation, but it has been observed that IRAK2-mediated post-translational modifications are important for mRNA stability and translation by facilitating nuclear export of NF-κB-regulated transcripts63,64,65. IRAK3 lacks kinase activity owing to the absence of an aspartate residue at the active site and, instead, inhibits IRAK signalling by binding to and arresting IRAK4, IRAK1 and IRAK2 in inactive states66,67,68,69. TLR7, TLR8 and TLR9 induce the production of both NF-κB-dependent cytokines as well as type I interferons70. In pDCs, MyD88 forms a complex with IRAK1, TRAF6, TRAF3, IKKα and IRF7. In this complex, IRAK1 may directly activate IRF7 to drive the expression of type I interferons. In conventional dendritic cells, activation of TLR7 and TLR9 results in IRF1-mediated IFNβ gene expression71,72. Mutations in MYD88, IRAK4 or IRAK1 found in patients have revealed essential roles of these proteins in host defence. Patients with an autosomal-recessive disorder who are deficient in IRAK4 or MYD88 are equally susceptible to a subset of pyogenic bacterial infections, but are resistant to other infections, including other bacteria, most viruses, fungi and parasites73. As the first kinase in the receptor signalling cascade, IRAK4 kinase activity is most critical in activating pathways downstream of IL-1R family members (Fig. 3) and, therefore, is a prime target candidate for the treatment of several inflammatory diseases59,60,61,74. Mutations in the kinase domain in IRAK4 that abrogate its activity protect mice in several inflammatory disease models, including septic shock63,75,76,77, SLE78,79,80, acute liver injury81, cardiovascular disease82 and the APPPS1 Alzheimer disease model83.
The endosomal receptors TLR3, TLR7, TLR8 and TLR9 cannot discriminate between self and foreign nucleic acids, and therefore can pose serious threats to the development of autoimmunity. SLE development is attributed to the activation of endosomal TLRs. IRAK4 inhibition using BMS-986126 in preclinical models of lupus (MRL/lpr and NZB/NZW) demonstrated strong attenuation of disease symptoms and minimal off-target effects78. Similarly, IRAK4 inhibition using PF-06650833 in patients with RA showed significant improvements in disease severity34. Interestingly, deletion of IRAK1 resulted in only a partial loss of signalling in immune cells in vitro76,84. Only one IRAK1-deficient immunocompromised patient with deletions of several nearby genes has been reported85. In contrast to IRAK4, IRAK1 seems to be redundant downstream of the IL-1R in human fibroblasts in vitro. However, IRAK1 is essential for signalling downstream of TLR2, TLR6, TLR4, TLR7 and TLR8 in fibroblasts as well as in mature B cells. Therefore, the functions of IRAK4 and IRAK1 may be cell type and receptor specific86,87. Interestingly, compared with IRAK4 deficiency in humans, which confers susceptibility to a few bacterial infection and decreases with age, Irak4-deficient mice are susceptible to multiple bacterial, viral, fungal and parasitic infections at all ages88.
IRAK2 may partially compensate for a loss in activity of other IRAKs63,76,89. IRAK2 is required for the production of pro-inflammatory cytokines89,90 where IRAK2–TRAF6 interaction becomes rate-limiting after IRAK1 is degraded from the cells after prolonged TLR stimulation89. Allosteric inhibitors that disrupt the interaction between IRAK2 and TRAF6 may hold potential for therapeutic development in inflammatory diseases such as RA and SLE.
The precise mechanisms of IRAK4 signal transduction are still being studied. Leukocytes and fibroblasts from several IRAK4-deficient patients showed that IRAK4 kinase activity was more essential for TLRs and IL-1R-mediated signalling in innate immune cells than in fibroblasts91, suggesting that IRAK4 may be regulated differently in these cell types. One mechanism that facilitates IRAK1 and IRAK4 oligomerization is K63 ubiquitylation92 (Fig. 3). During ubiquitylation, the C terminus of ubiquitin (Ub) is covalently attached to the lysine residue of substrate proteins. First, the Ub moiety is activated by E1 enzymes (also known as Ub-activating enzymes). Following activation, an E2 Ub-conjugating enzyme (UBC) transfers Ub from E1 to an E3 enzyme (also known as Ub ligases) to which the substrate protein is specifically bound. The Ub moiety contains several lysine (K) residues (such as K48 and K63) and a methionine at the N terminus (M1), which can attach another Ub to form a polyUb chain. K48-linked polyUb chains control targeting of a substrate to 26S proteasomes for degradation. K63-linked polyUb chains provide additional structural scaffolding for protein–protein interactions, which can facilitate signalling. IRAK1 has 31% sequence identity to IRAK4, and contains two important lysine residues in the linker domain that are required for K63-linked polyUb chains, which is essential for activation of NF-κB92. Receptor engagement has been shown to induce K63-linked polyUb chains on several subunits of the Myddosome, suggesting that this type of modification may be of particular importance in this pathway92. Because IRAK4 provides structural integrity to the Myddosome, it is possible that IRAK4 kinase activity-dependent and activity-independent mechanisms work in parallel to facilitate cytokine production downstream of the Myddosome76,93,94. Therefore, further characterization of post-translational modifications of IRAK4 may yield kinase-independent and cell-specific mechanisms of signalling.
It is unclear why TLRs and IL-1R have evolved to be uniquely complex across cell types and species. It is possible that different mechanisms of activation can facilitate the fine-tuning of a complex immune response. Immune cells may be more sensitive to catalytic kinase activity in order to trigger, as well as terminate, the immune response, and other cell types such as epithelial cells or fibroblasts might rely less on kinase activation to transiently control inflammation and avoid collateral tissue damage. Several IRAK4 inhibitors are currently being evaluated in the clinic, including PF-06650833, BAY-1834845, CA-4948 and R835 (Fig. 1). PF-06650833 was shown to improve clinical scores in a phase II trial in patients with active arthritis that is not responding to methotrexate34. There are no reports on IRAK1 or IRAK4 ubiquitylation-specific modules that alter TLR or IL-1R signalling in the clinical stage.
Receptor-interacting protein kinases
Receptor-interacting protein kinases (RIPKs) are critical regulators of cell death and inflammation with important roles in the maintenance of tissue homeostasis95 (Fig. 4). RIPKs exert multiple signalling functions through their kinase activity, protein binding and post-translational modifications96. Dysregulation of RIPK functions can lead to unbalance in multiple signalling pathways and cause severe inflammatory conditions, suggesting that these kinases are important sentinels of human health95.

Tumour necrosis factor (TNF) signalling can lead to receptor-interacting protein 1 (RIP1)-dependent apoptosis (mediated by caspase 8) or necroptosis (mediated by RIP3 and mixed-lineage kinase domain-like protein (MLKL)) to cause tissue damage and an inflammatory milieu. RIP1 inhibition can block RIP1-mediated apoptosis and necroptosis, and reduce inflammation by inhibiting inflammatory cell death. The kinase domain of RIP2 allows the binding of E3 ligase X-linked inhibitor of apoptosis protein (XIAP) and subsequent RIP2 ubiquitylation, which is a critical mediator of nucleotide-binding oligomerization domain-containing protein 2 (NOD2) inflammatory signalling. Consequently, RIP2 kinase inhibitors prevent XIAP binding and RIP2 ubiquitylation to inhibit NOD2 pathway-activated NF-κB and mitogen-activated protein kinase (MAPK) signalling, and consequent production and release of pro-inflammatory cytokines, thus blocking inflammation. FADD, FAS-associated death domain; IAP1/2, inhibitor of apoptosis 1 and 2; LUBAC, linear ubiquitin chain assembly complex; MDP, muramyl dipeptide; P, phosphorus; TNFR1, TNF receptor 1; TRADD, TNFR-associated death domain; TRAF2, TNFR-associated factor 2; Ub, ubiquitin.
RIP1 (also known as RIPK1) is a seminal component of TNF signalling that mediates proliferative NF-κB and MAPK activation as well as apoptotic and necroptotic cell death pathways97 (Fig. 4). Necroptosis is referred to as a regulated form of necrosis or inflammatory cell death. Typically, necrosis or cellular injury is associated with unprogrammed cell death that results from cellular damage or invasion by pathogens, in contrast to orderly, programmed cell death via apoptosis95. Binding of TNF to the TNFR1 triggers the recruitment of adaptor proteins TNFR-associated death domain (TRADD), TRAF2 and RIP1, and the Ub ligases inhibitors of apoptosis 1 and 2 (IAP1/2)98. Ubiquitination of RIP1 mediated by IAP1/2 by addition of K63-linked and K11-linked polyUb chains leads to recruitment of additional signalling complexes, including the IKK complex (which includes NF-κB essential modulator (NEMO)), TAK1 associated with TAB2 and TAB3, and the linear ubiquitin chain assembly complex (LUBAC; which consists of E3 ubiquitin-protein ligase HOIP and cofactors HOIL-1 and Sharpin)96. Linear ubiquitylation of RIP1 and several other TNFR1-associated proteins further enhance TNF-stimulated NF-κB and MAPK signalling. Conversely, deubiquitinases A20 and CYLD function to disassemble such polyUb chains to limit NF-κB and MAPK activation in order to promote cell death99,100,101.
TNF induces cell death via apoptosis and necroptosis, which largely depends on RIP1 and involves one translocation to the cytosol, where it binds FAS-associated death domain (FADD) and caspase 8 (and caspase 10 in some cases)98 (Fig. 4). Caspases are effectors of apoptotic cell death; however, in cases in which caspase 8 is inhibited or insufficiently activated, RIP1 can engage RIP3 in a kinase-dependent and RIP homology-interaction motif (RHIM)-dependent manner to form a necrosome98. Autophosphorylation of RIP1 and then RIP3 results in their full activation and leads to RIP3-mediated phosphorylation of the pseudokinase mixed-lineage kinase domain-like protein (MLKL), triggering its oligomerization and translocation to the plasma membrane to induce necroptotic cell death102.
RIP1, and to a lesser extent RIP3, are also polyubiquitylated within the necrosome and dynamic changes in their ubiquitination status influence cell death and inflammatory responses103,104. Overall, differential phosphorylation and ubiquitylation of RIP1 by numerous kinases (TAK1, IKK2 and MK2; also known as MAPKAPK2) and E3 ligases (IAP1/2 and LUBAC) allows for spatial and temporal regulation of the transition from TNFR1-associated complexes, in which RIP1 is inactive, to the multiple cytoplasmic complexes where RIP1 kinase activity is crucial for activating programmes of cell death96.
Both the discovery of RIP1-specific kinase inhibitors (necrostatins, GNE684 and other classes) and the lack of detrimental phenotypes observed in RIP1 kinase-dead mice have suggested that RIP1 is a promising clinical target105,106,107. Such is not the case with RIP3, as RIP3 inhibitors can activate apoptotic cell death and genetic RIP3 kinase inactivation is lethal106,107. Although targeting RIP1 activity represents an appealing strategy for the treatment of inflammatory pathologies and tissue damage, RIP1 relevance for various diseases must first be better characterized, as tissue-specific and/or cellular-specific processes that lead to RIP1 kinase activation do not always behave similarly108. For example, ischaemia–reperfusion kidney injury or myocardial infarction can be ameliorated by RIP1 kinase inhibition or inactivation in rodents108. Similarly, in Rip3-knockout mice, and to a lesser extent in Mlkl-knockout mice, the absence of RIP3 and MLKL promotes survival and reduces nephropathy in kidney injury models, indicating a critical role for necroptosis in kidney tissue damage108,109. Similarly, in mice, RIP1 inhibition ameliorates collagen antibody-induced arthritis, skin inflammation caused by mutant Sharpin or colitis caused by intestinal deletion of Nemo105. In skin inflammation models, necroptosis also plays an important part in RIP1 inhibition as RIP1 inhibition reduces skin inflammation and phosphorylation of necroptosis markers RIP3 and MLKL110,111. Conversely, RIP1 inhibition does not affect tumour growth or survival in pancreatic tumour models driven by mutant KRAS, or lung metastasis in a B16 melanoma model112.
Another interesting feature of RIP1 kinase activity is its involvement in the interplay of multiple cell death and survival pathways. Although kinase activity of RIP1 is clearly instrumental for necroptotic cell death, it has also been implicated in TNF-induced apoptosis when key NF-κB signalling kinases (such as TAK1 or IKK) are inhibited or deleted113,114. Caspase 8 negatively impacts RIP1 activation115,116,117 but it is still unclear how RIP1 kinase activity regulates caspase activation. It is possible that a threshold of caspase activation determines which regulated cell death pathway is activated, but RIP1-mediated apoptosis and necroptosis are unlikely to be mutually exclusive. Additionally, defective autophagy caused by deletion of the gene encoding for autophagy-related protein 16-1 (ATG16L1) in intestinal organoids or the intestinal epithelium of mice results in TNF-driven necroptotic death, which can be rescued by RIP1 kinase inhibitors105,118. Therefore, RIP1 kinase activity can aggregate multiple signalling inputs that induce regulated cell death to mediate tissue damage and inflammation. This is evident in both acute and chronic animal disease models. In the acute kidney injury model or the TNF-induced shock model (systemic inflammatory response syndrome), genetic or chemical inhibition of RIP1 kinase efficiently blocks tissue damage105,108,119. Furthermore, blocking both apoptosis and necroptosis pathways by ablation of caspase 8 and RIP3, or of caspase 8 and MLKL, provides complete protection against kidney injury108. Similarly, in a Sharpin-mutant mouse model of skin inflammation, RIP1 inhibition blocks both apoptotic caspase 3 cleavage and necroptotic RIP3 phosphorylation in the epidermis of mutant mice110. Currently, GSK and Denali are evaluating the utility of RIP1 inhibitors (GSK2982772, DNL747) in the clinic in neurodegenerative and systemic inflammatory diseases (ALS, RA and ulcerative colitis)120,121 (Fig. 1).
RIP2 does not participate in cell death signalling but, instead, is a mediator of NOD1 and NOD2 inflammatory signalling122 (Fig. 4). NOD2 is mutated in numerous inflammatory diseases such as Crohn’s disease, Blau syndrome and very early-onset sarcoidosis123. NOD2 recognizes bacterial peptidoglycans, such as muramyl dipeptide (MDP) that results in NOD2 activation and subsequent recruitment of RIP2 and its E3 ligase, XIAP122,124. XIAP (and potentially other E3 ligases) promotes K63-linked ubiquitylation of RIP2, which enables LUBAC-mediated linear ubiquitylation of RIP2 and subsequent activation of NF-κB and MAPK to promote cytokine and chemokine production96. The kinase domain of RIP2 serves as a docking module that enables XIAP to bind125. Mutational inactivation of RIP2 kinase activity had no effect on RIP2-mediated NOD2 signalling as it did not prevent RIP2 binding to XIAP125. However, RIP2 kinase inhibitors that disrupted RIP2–XIAP interactions were successful in blocking NOD2 signalling125,126. This is a rare example of a kinase whose enzymatic activity is not needed for its biological role. Instead, RIP2 ubiquitylation by XIAP enables the assembly of signalling complexes and stimulation of inflammatory responses124,125. Therefore, targeting RIP2 kinase to disrupt the interaction between RIP2 and XIAP may be effective in NOD2-mediated diseases.
ITK and BTK kinases
Tec family kinases are primarily expressed in haematopoietic cells and have important roles in the development and function of leukocytes downstream of SRC and SYK127. Among Tec kinases, BTK and ITK are attractive drug targets given their established roles in B cell and T cell activation, respectively128. In addition, BTK also regulates Fcε receptor (FcεR) signalling in mast cells, positing it as an attractive drug in IgE-mediated diseases such as allergy, asthma and atopic dermatitis128.
In T cells, ITK positively regulates TCR signalling to induce the production of IL-2, IL-4 and IL-13 (refs127,129) (Fig. 5a). When peptide-MHC binds to the cognate TCR, ITK is directly phosphorylated by the tyrosine-protein kinase LCK, and subsequently undergoes autophosphorylation. ITK associates with the LAT–SLP76 complex through its two SRC homology domains SH2 and SH3, creating a signalling complex that is dependent on upstream LCK and Zap70 signalling127. ITK is then able to phosphorylate phospholipase Cγ (PLCγ), which cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) at the plasma membrane, generating the secondary messengers inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 and DAG primarily activate NFAT and calcium signalling, which targets gene promoter activation including IL-2, IL-4 and IL-13 (Fig. 5a).

a | In T cells, IL-2 inducible T cell kinase (ITK) phosphorylates secondary messengers to activate nuclear factor of activated T cells, cytoplasmic 1 (NFATc) and calcium signalling to positively regulate cytokines (such as IL-2) or negatively regulate certain genes such as FASLG. Following T cell receptor (TCR) engagement, active lymphocyte-specific protein tyrosine kinase (LCK) phosphorylates the immunoreceptor tyrosine-based activation motifs (ITAMs), Zap70 and IL-2-inducible T cell kinase (ITK). Zap70 phosphorylates SLP76 and linker for activation of T cells (LAT); ITK phosphorylates phospholipase Cγ1 (PLCγ1). Activated PLCγ1 then hydrolyses phosphatidylinositol 4,5-bisphosphate (PIP2) to produce diacylglcycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), leading to increased calcium flux. b | Bruton’s tyrosine kinase (BTK) regulates multiple receptors including B cell receptor (BCR) or Toll-like receptors (TLRs) in B cells, Fcγ receptor (FCγR) or TLRs in macrophages or plasmacytoid dendritic cells (pDCs) and FCγR or TLRs in basophils and mast cells. Upon BCR engagement, BTK is activated by Src kinases and phosphorylates PLCγ2. PLCγ2 activates NFAT and enhances Ca2+ flux, and activates mitogen-activated protein kinase (MAPK) and NF-κB. BTK has also been implicated in TLR, IC and FCγR or FCεR signalling in multiple cell types via Ca2+ or as yet unknown pathways. AICD, activation-induced cell death; AP-1, activator protein 1; FasL, Fas ligand; IC, immune complex; IRAK4, IL-1 receptor-associated kinase 4; LYN, Lck/Yes-related novel tyrosine kinase; MHC, major histocompatibility complex; MyD88, myeloid differentiation primary response 88; P, phosphorus; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PKC, protein kinase C; SYK, spleen tyrosine kinase.
Comparing Itk knockout versus ITK inhibitor studies has revealed novel insights into ITK function130. ITK plays a critical part in priming T cells; however, in rechallenged antigen-experienced T cells, ITK regulates activation-induced cell death130, highlighting differences between Itk knockout and kinase inhibitor studies130. Activation-induced cell death is a mechanism of programmed cell death evolved to dampen an ongoing immune response and involves interactions of TNFRSF6 (also known as FAS and CD95) and its ligand TNFL6 (also known as FASL or CD95L) on neighbouring cells131. ITK inhibitors can reduce FASL expression on T cells to promote expansion of activated T cells130. This surprising finding has drawn a new paradigm for the utility of this target130. Therefore, although the inhibition of ITK may not be beneficial in the treatment of asthma, this mechanism of ITK activity may be beneficial in cancer immunotherapy to promote T cell survival132. JTE-051 is the only ITK inhibitor under clinical evaluation in the treatments of RA and psoriasis (NCT03358290).
BTK integrates BCR signalling to regulate B cell development (Fig. 3b). Mutations that inactivate BTK block B cell development causing X-linked agammaglobulinaemia133. Types I and III interferon production are impaired in BTK-deficient patients during viral infections such as polio, but not during influenza134. Different isotypes of immunoglobulins exert their effector functions, in part, by binding to the respective FcRs135. IgE antibodies bind FcεR on mast cells and basophils to trigger degranulation and acute inflammation135, whereas IgG binds FcγR on macrophages, pDCs and natural killer cells to promote cellular activation or phagocytosis135. In certain autoimmune diseases, self-reactive IgG binds to self-antigens, such as nucleic acids in SLE, and forms immune complexes135. BTK positively regulates FcεR signalling in mast cells and basophils136,137,138 and FcγR signalling in macrophages or pDCs to internalize and deliver immune complexes139 (Fig. 3b). TLR-induced B cell differentiation and proliferation is dependent on BTK, whereas the function of BTK in TLR signalling in myeloid cells is not well understood140 (Fig. 3b). In B cells, BCR activation exposes the ITAM to LYN/SYK/BTK, which activates PLCγ2 and phosphatidylinositol 3-kinase (PI3K). Active PLCγ2 and PI3K allow for calcium signalling, which is required for the activation of transcription factors, such as NFAT and NF-κB, that regulate proliferation, survival and cytokine expression141,142 (Fig. 5b). Similar to BTK-deficient mice, PLCγ2-deficient mice have defects in B cell development143. These mice also have defective FCεR and FCγRII/III signalling in mast cells and natural killer cells, respectively, but macrophage function and numbers are not altered143. The function of BTK in pDCs is less understood; however, one report suggests that BTK regulates TLR9, but not TLR7, signalling in human pDCs144. These overlapping phenotypes support the idea that BTKis could effectively target B cell differentiation, mast cell and basophil-associated immune pathologies but no other myeloid cells.
Cell-specific BTK activity may be determined by post-translational modifications. Phosphorylation at Y551 is important in FCεR and FCγR signalling whereas Y223 activation is essential for BCR signalling145 (Fig. 5b). In preclinical rodent models, BTK inhibition is protective against the development of arthritis or SLE by reducing autoantibody production and inflammatory cytokines139,140,146,147,148; encouragingly, BTK inhibition was more efficient than BAFF blockade or SYK inhibition140. However, animal models are, unfortunately, often not representative of human disease and are likely to emphasize limited pathways in disease progression. For example, NZW/NZB F1 mice are an SLE-like model that is highly B cell dependent11. Not surprisingly, treatment with either anti-CD20 B cell depletion or BTK is protective in this model140. However, further investigation into the function of BTK in macrophages and pDCs (additional pathogenic cell types in autoimmune diseases such as RA and SLE) will be needed to dissect its role in myeloid cells. Recent data demonstrate that BTK kinase activity is more critical in B cells or downstream of FCεR signalling than in FCγR signalling149. In this study, the authors show that in pDCs IRAK4 inhibition is more effective at blocking immune complex-mediated inflammation downstream of endosomal TLRs.
Several BTKis are being investigated in the clinic. In phase II trials for the treatment of relapsing multiple sclerosis, evobrutinib (a covalent BTKi) reduces enhancing brain lesion plaques after 3 months150. Branebrutinib (BMS-986195, a covalent BTKi) has advanced into clinical studies for the treatment of RA, Sjögren’s syndrome and SLE151. Fenebrutinib (GDC-0853, a reversible BTKi) reduces disease activity in combination with methotrexate in patients with RA with an inadequate response to TNF-based therapy152. PRN1008 is a novel reversible covalent BTKi that has shown promising results in phase II in pemphigus (NCT02704429) and is being further investigated in a phase III global trial (NCT03762265). Covalent BTKis often block the activities of other kinases, namely RLK, ITK and TEC132. Although off-target effects are generally considered to increase the risk of adverse effects, select cases have demonstrated that they can be beneficial. For example, it was recently shown that specific inhibition of ITK by ibrutinib was beneficial in cancer by promoting the expansion of T cells via reducing activation-induced cell death and FASL expression130,132. Although the tolerance of less selective but efficacious molecules such as ibrutinib is higher in oncology, the bar for safety on daily medicines for chronic inflammatory indications such as RA and IBD is much higher, and for kinase drugs this safety is linked to selectivity. Additional clinical data that evaluate the generation of covalent or reversible BTKis should help us to better understand the function of BTK in different cell types to determine which drugs provide the optimal therapeutic index with minimal safety liability. Given the uncertainties around BTK activity in inflammation, combination therapy with BTK or other anti-inflammatory molecules might be desired to explore in RA or SLE. BTKis are also being tested for their ability to interfere with the cytokine storm in severe COVID-19 patients; preclinical studies and case series have suggested that the BTKi ibrutinib may provide protection against severe lung injury153.
SYK
SYK is an Src family member essential in FcR and BCR signalling, and functions in parallel to its homologue, tyrosine-protein kinase Zap70, in TCR signalling154, which makes it an attractive drug target in the treatments of chronic inflammation and autoimmunity (Fig. 3b). SYK contains two SH2 domains and a C-terminal kinase domain connected by linkers A and B, respectively. These linkers are bound together, rendering the SH2 domains inactive at steady state154. Receptor activation causes the release and autophosphorylation of the SH2 domains that enable docking at receptor ITAMs155,156. Further phosphorylation of SYK causes it to dissociate from ITAMs and activate context-specific signalling cascades (including its own degradation) via Tec-family tyrosine kinases, lipid kinases, phospholipases and guanine-mediated exchange factors154,157,158,159. SYK plays an important part in various immunologically relevant pathways, including the PI3K–AKT, Ras–ERK, PLCγ–NFAT, Vav1–Rac and IKK–NF-κB pathways. Accordingly, targeting SYK has implications in several of the cellular processes that these pathways regulate, such as phagocytosis, cytokine production, degranulation, proliferation, B cell maturation, osteoclastogenesis and platelet activation160 (Fig. 5b).
FcγRs expressed on myeloid cells internalize IgG-opsonized particles through SYK-dependent phagocytosis and have a critical role in protective inflammatory responses161 (Fig. 5b). However, activation of FcγR by IgG immune complexes against autoantigens represents a key hallmark of RA, chronic spontaneous urticaria, ITP and SLE. Syk-deficient mice are perinatally lethal and lack mature B cells in utero, which suggests that SYK has significant roles in general development and in the immune system162,163. Syk-deficient bone marrow chimaera mouse models, however, were viable and resistant to the arthritogenic serum-induced model of arthritis, likely caused by both a lack of mature B cells and impaired FcγR internalization164.
Numerous small-molecule inhibitors generated by Rigel Pharmaceuticals have entered clinical investigation (Fig. 1). R406 (and its prodrug R788 or fostamatinib) showed efficacy in the prevention of arthritis in mice165,166. In clinical trials, R406 and R788 have shown moderate efficacy in achieving the American College of Rheumatology 20% criterion, although they are less robust compared with anti-inflammatory drugs such as TNF antagonists167,168. Fostamatinib resulted in improved symptoms of RA likely owing to both on-target and off-target effects169; it also resulted in several adverse side effects, and thus the development of this drug in RA was discontinued167,170,171. Fostamatinib is currently approved for the treatment of ITP, an autoimmune disease in which autoreactive IgG antibodies target and destroy platelets via macrophages through SYK-dependent, FcγR-mediated phagocytosis172,173. Fostamatinib did not meet its primary end point in IgA nephropathy (NCT02112838) and is being further developed to treat autoimmune haemolytic anaemia (NCT04138927) and renal transplantation (NCT03991780).
Developing highly selective inhibitors is critical but is the most challenging facet of small-molecule discovery. R406 is a good example of how off-target activities that were not appreciated early on complicated its clinical development. In an initial in vitro selectivity assessment, fostamatinib bound to other targets (such as FLT3, LYN and LCK), but showed 5-fold to 100-fold greater inhibition of SYK than other tyrosine kinases when tested in a phosphorylation assay169. A decade later, the comprehensive pharmacological profile of fostamatinib, using a broad range of in vitro assays followed by functional and cellular assays, challenged key targets at therapeutically relevant concentrations174. Using a larger kinase selectivity panel, fostamatinib inhibited 117 kinases, 100 of which had half-maximal inhibitory concentration (IC50) values within 3-fold of the IC50 value for SYK, including FLT1, KDR (also known as vascular endothelial growth factor receptor 2 (VEGFR2)), SRC and KIT, which are associated with increased blood pressure, based on the analysis of published literature174. Among non-kinases, antagonist activity was found against adenosine A3 receptor174. Therefore, investment in generating a selective molecule using robust pharmacological profiling facilitates assessment of each target in the clinic with confidence. By contrast, polypharmacological effects complicate the interpretations of the clinical data to evaluate the desired target and, often, it is too costly to repeat such trials with new molecules.
IgE is commonly induced during allergic reactions and causes the cross-linking of the high-affinity FCεRI expressed on mast cells. Following stimulation of FCεRI, SYK is immediately recruited and activated. SYK-dependent activation of PI3K and AKT was shown to cause mast cell degranulation of histamine and the production of leukotrienes, prostaglandins and cytokines175. A study of B cell lymphomas demonstrated that a subset of malignant B cells with receptor hyperstimulation have linked SYK activity to cell survival and proliferation176. Gilead has developed GS-9973 (entospletinib), which has greater selectivity for SYK over R406 and is currently in clinical trials for the treatment of chronic lymphocytic lymphoma177,178,179 (Fig. 1).
MAPK: TPL2, p38γ, p38δ and ERK5
MAPKs are a highly conserved family of serine/threonine protein kinases that are induced in response to stress and inflammation and regulate proliferation, differentiation, survival, apoptosis and other cellular processes16. MAPKs are downstream of several immune and cytokine receptors, such as TLRs, IL-1R, TNFR, CSF1R and IL-17R, as well as growth receptors, such as EGF, FGF and VEGF. Several inhibitors for the major MAPKs such as p38α/β, MEK1/2 and ERK1/2 have been advanced into the clinic and reviewed extensively10,16. Of particular interest are MAPK kinases that have tissue or cell-specific expression and prominent function in inflammation, including TPL2, p38γ and p38δ.
TPL2 is activated by several receptors, including TNFR, IL-1R, TLR, CD40R, IL-17R and Gαi2-transduced GPCR signals180 (Fig. 6). At steady state, TPL2 forms a complex with A20-binding inhibitor of NF-κB (ABIN2) and only a small fraction (5% in the case of macrophages) of cellular NF-κB1 (p105 subunit)181,182. The kinase domain of TPL2 interacts with the death domain of p105 and this interaction blocks substrate access to the TPL2 active site183. Full-length p105 is an REL protein-specific transcription inhibitor, whereas its processed form is a 50-kDa protein (p50), DNA-binding subunit of the NF-κB complex16. Upon receptor engagement, IKKβ phosphorylates p105 at Ser927 and Ser932, leading to its degradation16. As a result, TPL2 is released from the complex, then autophosphorylated and/or trans-phosphorylated180 and activates targeting of its substrates MEK1 and MEK2 (ref.184) as well as MEK3 and MEK6 (ref.31) to regulate ERK1 and ERK2 or p38α and p38δ in inflammation. The TPL2 action on downstream ERK and p38 has a profound effect on the net outcome of an inflammatory response via several transcriptional and post-transcriptional mechanisms180. First, it can regulate gene transcription via CREB/AP-1 activation185. The TPL2/ERK/p38 axis can determine the stability and abundance of the AU-rich element (ARE) mRNAs, which is a feature of many cytokines and chemokines (such as TNF or IL-6)184. Second, nucleocytoplasmic localization of select genes (such as TNF) can be modulated184. In addition, other regulatory processes, such as CAP-dependent RNA translation and protein export and processing via disintegrin and metalloproteinase domain-containing protein 17 (ADAM17; also known as TACE), are regulated by TPL2 (ref.186). Thus, the net effect of TPL2 inhibition has a profound impact on inflammatory outputs without compromising the NF-κB pathway (Fig. 6). Each subunit of the TPL2 complex is essential for its stability. This was demonstrated by p105 deficiency, which resulted in reduced protein levels but not transcript levels of both TPL2 and ABIN2 (ref.187), whereas TPL2 protein levels were greatly reduced in ABIN2-deficient mouse cells16. TPL2 deficiency also reduces ABIN2 protein levels16. Accordingly, it has been challenging to determine the exact functions of TPL2, ABIN2 or p105 in disease models until recently. TPL2 inhibition or mice expressing kinase dead mutant TPL2 have since been developed in order to probe the function of TPL2 catalytic activity in preclinical models31,188. Recent studies have shown that inhibiting TPL2 catalytic activity is protective in preclinical models of multiple sclerosis188, as well as arthritis31 and psoriasis31.

The action of both mitogen-activated protein kinase (MAPK) and NF-κB orchestrates the transcription of target genes. In the resting state, p105 prevents and masks tumour progression locus 2 (TPL2) kinase effector function. Agonist stimulation activates the inhibitor of NF-κB (IKK)/NF-κB essential modulator (NEMO) complex264. Subsequently, IKKβ phosphorylates IKKα, targeting IKKα for proteasomal degradation. Then, released RelA/p50 dimers are translocated to the nucleus and modulate target gene expression. IKKβ also phosphorylates the target residues S927 and S932 in p105, leading to its proteasomal degradation that results in TPL2 liberation. IKKβ phosphorylates TPL2 at residue S400 to enhance its kinase activity. Free TPL2 then activates MEK1 and MEK2 as well as MEK3 and MEK6 to positively regulate ERK1/2 or p38α/δ to regulate gene transcription via cAMP response element-binding protein (CREB)/activator protein 1 (AP-1) as well as mRNA stability and protein production. The function of A20-binding inhibitor of NF-κB (ABIN2) is not completely understood and involves regulation of protaglandin E2 (PGE2) and cyclooxygenase 2 (COX2) in fibroblasts. TPL2 binds to a small fraction of p105 but the p50 domain of processed p105 can directly impact gene transcription16. Post-transcriptional regulation of cytokines and chemokines by MAPKs involves AU-rich elements (AREs) on messenger RNAs to dictate their stability (in the case of tumour necrosis factor (TNF) or IL-6, for instance) or cellular localization (TNF). In addition, other regulatory processes such as CAP-dependent RNA translation and protein export via TNFα-converting enzyme (TACE) (in the case of TNF) are regulated by the TPL2/MAPK axis. Therefore, the net effect of TPL2 inhibition has a profound effect on inflammatory outputs without compromising the NF-κB pathway. IL-1R, IL-1 receptor; P, phosphorus; TLR, Toll-like receptor; TNFR, TNF receptor; Ub, ubiquitin.
Tpl2-deficient mice are protected from numerous inflammatory and autoimmune diseases180. Analysis of mice expressing kinase-dead TPL2 (D270A), in which ABIN2 expression is unaltered, showed that TPL2 kinase activity is a critical mediator in TNFR, TLR and IL-1R signalling by positively regulating MAPK31. When challenged with a TLR agonist (such as lipopolysaccharide (LPS)), these mice produced significantly fewer inflammatory cytokines and showed fewer immune cell infiltrates31. At the molecular level, TPL2 not only activates MEK1 and MEK2 but also activates isoforms p38γ and p38δ, which are key regulators of mRNA stability and translation machinery for inflammation-related proteins31,180. This role of TPL2 is most profound in neutrophils, monocytes and macrophages31. This selective action of TPL2 on specific MAPKs is intriguing and should expand the utility of this target for several indications (see below). Recent studies with catalytically inactive ABIN2 (D310N) in gut inflammation showed that ABIN2 regulates IL-1β-dependent induction of cyclooxygenase 2 (COX2) and PGE2 secretion189, functions previously thought to be regulated by TPL2 (ref.190). Pan-kinase inhibitors, such as p38 or MEK inhibitors, suggest that the complex and often overlapping nature of different kinases in signalling would likely lead to problems with toxicity or inhibitor specificity16. Targeting an immune-specific MAP3K such as TPL2 will likely maximize safety margins over broad MAPK inhibitors (such as p38i) in the clinic. Gilead has advanced its TPL2 inhibitor (GS-4875) to the clinic for the treatment of ulcerative colitis36 (Fig. 1).
p38α, p38β, p38γ and p38δ isoforms are uniquely expressed throughout mammalian tissues191. Activation of p38 is cell type-specific, receptor-specific and signal strength-specific192,193. All p38 family members share the Thr-Gly-Tyr (TGY) activation motif, which is dually phosphorylated by MKK3 and MKK6 (and, in some cases, MKK4)192. p38α and p38β have been extensively characterized, but much less is known about p38γ and p38δ. p38γ (also known as ERK6, SAPK3 or MAPK12) and p38δ (also known as SAPK4 or MAPK13) are similar to each other (70% identity), and are both not as similar to p38α (~60% identity)191. Intriguingly, p38γ and p38δ regulate protein stability of TPL2 in both macrophages and dendritic cells194. As TPL2–ABIN2 signalling is required for LPS-induced TNF and IL-1β production, mice lacking p38γ or p38δ were less sensitive to septic shock and hepatitis after LPS treatment194,195. Furthermore, TPL2 positively regulates p38δ, suggesting a feedback loop between inflammation and homeostatic conditions31,196. In mouse models of collagen-induced arthritis, deficiency of p38γ or p38δ caused decreased serum IL-17, IFNγ and autoantibody production197. p38δ was found to be highly expressed in neutrophils, and its deficiency in myeloid cells caused impaired neutrophil recruitment in a murine model of peritonitis198,199. p38 MAPKs may also create structural scaffolds independent of kinase activity. p38γ was required for p38γ–ERK complex formation and Ras-mediated oncogenic transformation200.
Several inhibitors against p38 have been investigated in the clinic10. However, most p38 inhibitors to date (such as SB203580 and SB202190, among others) target isoforms p38α and p38β and lack inhibitory activity for p38γ and p38δ201,202,203. Targeting of p38α/β has not been very successful owing to toxicity, pleiotropic effects on various cell types, poor predictability of animal models or lack of efficacy in humans204,205. A unique inhibitor of p38α, CDD-450, was recently reported to selectively block p38α activation of the pro-inflammatory kinase MK2 while sparing p38α activation of MAPK-activated protein kinase 5 (also known as PRAK) and cAMP-dependent transcription factor ATF2 (ref.206). CDD-450 promotes IL1B, TNF and IL6 mRNA decay attenuating arthritis in rats206. CDD-450 offers the potential to avoid tachyphylaxis associated with global p38α inhibitors that may result from their inhibition of non-MK2 substrates involved in anti-inflammatory and housekeeping responses206. Thus far, only three inhibitors that effectively inhibit p38γ and p38δ have been considered for clinical evaluation. BIRB796 (which inhibits all p38 isoforms) was evaluated for RA, psoriasis and Crohn’s disease, but generated hepatotoxicity and, therefore, clinical evaluation was discontinued207. RV568 — a p38α and p38γ inhibitor — was more effective than BIRB796 at suppressing inflammation in cell-based assays, in vivo with rodent smoke emphysema models of COPD112,208. Pirfenidone may target p38γ and has been approved for the treatment of IPF but is continuing to be evaluated in scleroderma-associated interstitial lung disease, kidney disease, wound healing, fibrosis, cardiomyopathy and fibroids209,210,211,212,213,214. SU-002 and SU-005 are newly identified molecules with better specificity for p38γ and p38δ but not p38α and p38β (ref.215). Although degrees of compensation exist, understanding the regulation and functions unique to each isoform will reveal cell-specific and tissue-specific mechanisms in inflammation and malignancy.
ERK5 (also known as MAPK7) is a ubiquitously expressed MAPK that is activated by MEKK2, MEKK3 and MEK5 (ref.216). ERK5 functions downstream of cellular stress, several immune receptors (such as TLRs, IL-1R, TNFR or IL-17R), CSF1R and growth receptors (EGF, FGF or VEGF)216. Among these receptors, CSF1R signalling has gained attention given its role in macrophage differentiation and the important function of macrophages in cancer or inflammation217. Several CSF1R inhibitors (PLX-3397, JNJ-40346527, ARRY-382, ABT-869, BLZ-945) have moved to clinical trials218 (Fig. 1). ERK5 has a kinase domain located at the N-terminal half of the protein that is homologous to the ERK2 kinase domain219. In contrast to other MAPKs, ERK5 has a unique C-terminal transcriptional activation domain220,221. Therefore, ERK5 is able to activate transcription not only by phosphorylating transcription factors but also by acting as a transcriptional coactivator itself. The role of ERK5 kinase activity in regulating inflammation is controversial. One study suggests that ERK5 kinase activity regulates cytokine production and the recruitment of immune cells222, but this finding has been challenged and this effect was attributed to off-target BET bromodomain activity using biochemical and cellular assays223. Intriguingly, both studies show that ERK5 knockdown in epithelial cells and primary human vascular endothelial cells reduced inflammatory cytokine production222,223. These data suggest that the non-catalytic activity of ERK5 positively regulates the inflammatory response. Consistent with this, in mice with ERK5-deficient keratinocytes, ERK5 was shown to be an essential component of skin inflammation224. Germline Mapk7-deficient mice are embryonic lethal at embryonic day 10.5 owing to loss of vasculature integrity225. Similar phenotypes were seen in mice lacking MEK5 and MEKK3, which suggests that this pathway is linear and important in both vasculogenesis and angiogenesis216. Initial developmental defects may primarily be due to the function of ERK5 in endothelial cells. Induced genetic ablation of Mapk7 is lethal in mice up to 3 weeks of age, which suggests that full ablation of this pathway may not be safe. However, given the complex functional domains of ERK5, including its action as a kinase or transcription factor, knockout studies could be misleading to determine the outcome of its kinase inhibition. Correspondingly, mice treated with ERK5 inhibitors are viable at least for the duration of published studies, but the selectivity and potency of this ERK5 inhibitor requires further investigation222. Generation of selective ERK5 (allosteric or degraders) or MEK5 inhibitors for chronic dosing should help to better evaluate this target in cancer and acute or chronic inflammation.
TBK1 and IKKε
TBK1 and its homologue IKKε are two serine/threonine protein kinases that activate IRFs to induce type I interferon genes and interferon-stimulated genes (ISGs)226, which are associated with several autoinflammatory or autoimmune diseases such as interferonopathies227 and SLE228. In contrast to SLE, interferonopathies comprise monogenic Mendelian diseases characterized by disturbance of the homeostatic control of interferon-mediated immune responses227 with various genetic and molecular features. These pathologies include Aicardi–Goutières syndrome (an encephalopathy that affects newborn brains), familial chilblain lupus (childhood lupus), spondyloenchondrodysplasia (skeletal anomalies), interferon-stimulated gene 15 (ISG15) deficiency and stimulator of interferon genes (STING)-associated vasculopathy with disease onset during infancy229. The disturbance of both the innate and the adaptive immune system may potentiate autoimmunity in some cases, for example SLE, in which a systemic immune response is triggered against self-antigens (such as nuclear antigens) in multiple organs such as the kidney and skin227. TBK1 and IKKε are also induced in the liver and adipose tissue by high-fat diet-mediated NF-κB activation230.
TBK1 and IKKε regulate many innate immune receptors, including TLRs, RIG-I-like receptors (RLRs; which sense cytosolic nucleic acids) and STING (which is important for establishing an immediate antiviral state during acute infection)30 (Fig. 7). AAK1 and GAK are host kinases that regulate clathrin adaptor protein (AP)-mediated trafficking in the endocytic and secretory pathways231. This pathway is important in the assembly and entry of certain viruses that hijack clathrin-mediated pathways, and is being investigated for antiviral therapeutics232. Given the central role of TBK1 and IKKε in the production of type I interferons, both TBK1 and IKKε are attractive drug targets especially in interferonopathies. Mutations in TBK1 result in neuroinflammatory and neurodegenerative disorders of the central nervous system, such as amyotrophic lateral sclerosis, which in part might be a consequence of dysregulated interferon signalling233. Whether TBK1 kinase inhibition results in amyotrophic lateral sclerosis pathology is unclear.

Nucleic acid sensors include the endosomal Toll-like receptors (TLRs) TLR3, TLR7, TLR8 and TLR9; the cytosolic DNA sensor cyclic GMP–AMP synthase (cGAS); and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs). RLRs are RNA sensors that include RIG-I and melanoma differentiation-associated protein 5 (MDA5). Unusual RNAs (double-strand RNA (dsRNA), 5′-phosphorylated (5′P) mRNA) can activate RIG-I or MDA5. Multimerized RIG-I and MDA5 can then bind to create a mitochondrial antiviral-signalling protein (MAVS), mitochondrial-associated membranes and peroxisomes, which in turn activate TANK-binding kinase 1 (TBK1) and inhibitor of NF-κB subunit-ε (IKKε) to activate interferon-regulatory factor 3 (IRF3) and IRF7. Double-stranded DNA (dsDNA; such as self-DNA) can induce an allosteric structural change in cGAS that, in turn, activates second messengers to promote stimulator of interferon genes (STING) to undergo dimerization to form a complex with TBK1 and IKKε that phosphorylates IRF3 to activate gene transcription. DNA-dependent activator of interferon-regulatory factors (DAI; also known as Z-DNA-binding protein 1 (ZBP1)) can also act as a sensor of Z-dsDNA (left-handed double-helical structure), often acquired during viral infection in a cell type-specific manner. DAI recruits TBK1 and IRF3 upon DNA binding and may get further phosphorylated to amplify the DAI/TBK1/IRF3 circuit265. Numb-associated kinases (adaptor protein 2 (AP2)-associated protein kinase 1 (AAK1) and cyclin G-associated kinase (GAK)) regulate intracellular viral trafficking during entry, assembly and release of unrelated viruses. Disruption of endocytosis by inhibiting AAK1/GAK can prevent the virus passage into cells. Endosomal TLRs (TLR7/8/9) recruit myeloid differentiation primary response 88 (MyD88) and signal through IL-1 receptor-associated kinase 4 (IRAK4) to activate IRFs similar to cell surface TLRs (see Fig. 3). TIR-domain-containing adapter-inducing IFNβ (TRIF)-dependent and MyD88-independent signalling could also activate TBK1/IKKε to activate IRF-mediated gene transcription. P, phosphorus; ssDNA, single-strand DNA.
TBK1 is constitutively and broadly expressed, but IKKε expression is inducible and limited to specific cell types, including lymphocytes234. Both phosphorylation and ubiquitylation regulate TBK1 signalling. Deubiquitination of K63-linked polyubiquitin chains from TBK1 terminates TBK1 activation and negatively regulates the antiviral immune response235. Mutation studies of TBK1 in cells suggest that TBK1 has a dominant role in interferon production and may be an essential component of antiviral immunity236. IKKε-deficient mice are susceptible to few viral infections, including γ-herpesvirus (γHV68) DNA virus235. Intriguingly, IKKε may positively regulate IL-17R signalling via its interaction with NF-κB activator 1 (ACT1) to induce expression of pro-inflammatory cytokines237. IKKε-deficient mice are viable whereas TBK1 deficiency is embryonic lethal, which raises concerns about systemic inhibition of TBK1 (refs238,239). Multiple TBK inhibitors are currently under development mainly at the preclinical stage, including the original BX795 molecule (developed by Amgen with suboptimal potency of 1 μM), MRT67307 (developed by the University of Dundee, similar to BX795 with much improved potency of 19 nM), AZ13102909 (developed by Astrazeneca with an IC50 of 5 nM), Domainex TBK1i (IC50 of 2 nM), and Myrexis MPI-0485520 (potency of 0.5 nM) and compound II (developed by University of Texas Southwestern Medical Center with potency of 13 nM)240,241. Finally, amlexanox is an anti-inflammatory drug approved by the FDA to treat recurrent aphthous ulcers of the mouth and asthma that was later shown to inhibit TBK1 and IKKε, although at much lower potency of 1000 nM.
The generation of selective IKKε or TBK1 inhibitors has been extremely challenging, owing to the high degree of homology within the kinase domains of the two proteins240. Studies with current IKKε or TBK1 inhibitors in preclinical models of interferonopathies (such as compound II in three prime repair exonuclease 1 (Trex1)-knockout mice)241 as well as neuroinflammatory mouse models (such as MRT67307 inhibitor in experimental autoimmune encephalomyelitis)242 suggest that targeting IKKε and TBK1 might be safe and beneficial in certain autoimmune or autoinflammatory disorders.
NIK
NIK is an integral component of non-canonical NF-κB signalling, and is found downstream of a subset of TNFR superfamily members including BAFF, CD40, TWEAK, RANK, TNFR2, FN14, CD27, CD30 and OX40 (ref.243) (Fig. 8). NIK deficiency results in combined immunodeficiency syndrome accompanied by B cell lymphopenia, impaired differentiation of memory B cells, abnormal natural killer cell development and function as well as aberrant T cell responses244. NIK-mutant mice have disorganized lymph nodes, Peyer’s patches and splenic and thymic structures that lead to reduced B cell numbers and immunoglobulins15. Conditional NIK-mutant mice and bone marrow chimeric mice have revealed the cell-specific functions of NIK245,246,247. Broad deletion of Map3k14 in adult mice recapitulates the B cell effects observed with germline deletion247. B cells from mice in which Map3k14 is deleted fail to respond to BAFF stimulation, which supports the essential role of NIK in BAFF receptor signalling248. Intriguingly, T cell-specific deletion of Map3k14 revealed non-redundant functions of NIK in T cells245. Furthermore, deletion of Map3k14 in myeloid cells compromises CD40 signalling and cross-priming in dendritic cells246.

NF-κB-inducing kinase (NIK) itself is regulated at the basal level by a destruction complex, and signal-induced non-canonical NF-κB signalling involves NIK stabilization. The canonical NF-κB pathway involves activation of inhibitor of NF-κB (IKK) complex by TGFβ-activated kinase 1-binding protein 1 (TAK1), IKK-mediated NF-κB inhibitor-α (IκBα) phosphorylation and subsequent degradation, resulting in rapid and transient nuclear translocation of the NF-κB heterodimer RelA/p50. NIK protein levels remain low via active degradation using ubiquitin ligase complex, comprising tumour necrosis factor receptor-associated factor 3 (TRAF3), TRAF2 and inhibitor of apoptosis 1 and 2 (IAP1/2). Receptor activation by agonists recruits this complex to the receptor where activated IAP mediates K48 ubiquitylation and proteasomal degradation of TRAF3, resulting in stabilization and accumulation of NIK. Subsequently, NIK activates IKKα to trigger p100 phosphorylation and processing to enforce persistent activation of RelB/p52 complex to activate gene transcription. BAFF, B cell-activating factor; BCR, B cell receptor; LTβ, lymphotoxin-β; P, phosphorus; TCR, T cell receptor; TLR, Toll-like receptor; TWEAK, tumour necrosis factor-related weak inducer of apoptosis; Ub, ubiquitin.
NIK activation is partly regulated by intricate mechanisms that regulate its protein stability (Fig. 8). At steady state, NIK protein levels remain low as it is actively degraded by the proteasome via a Ub ligase complex comprising TRAF3, TRAF2 and IAP1/2 (ref.15) (Fig. 8). When the non-canonical NF-κB pathway is activated, NIK activates IKKα, which in turn regulates the processing of NF-κB p100 to p52. p52 dimerizes with RelB, and the complex translocates to the nucleus to trigger transcription of target genes15 (Fig. 8).
Systemic canonical NF-κB inhibition (and proteosomal inhibitors) may not offer a sufficient therapeutic index in chronic indications given the broad requirements for NF-κB signalling in biological processes. However, inhibiting immune-regulatory nodes such as NIK offer plausible therapeutic approaches for the treatment of diseases in which non-canonical NF-κB signalling is aberrantly activated, including RA, IBD, SLE, IgA nephropathy, metabolic syndrome and multiple sclerosis. Inhibition of NIK with the inhibitor NIK SMI1 provided evidence that NIK kinase activity positively regulates several important pathways, such as BAFF, OX40, CD40, inducible T cell co-stimulator (ICOS), IL-21 and TNFRSF12A (also known as TWEAK receptor) signalling, and its inhibition was protective in preclinical models of lupus247. This finding corroborates the findings from the NIK-mutant mice, in which its action downstream of the same immune receptors had been established244,247. In a 4-week NZB/W mouse model, NIK inhibition decreased the frequency and numbers of splenic B cells, germinal centre B cells and plasma cells247. NIK inhibition was superior to BAFF blockade in the suppression of autoantibody titre247. In addition, NIK SMI1 significantly reduced the frequency and numbers of splenic T effector memory and T follicular helper as well as the production of cytokines: IL-21, P40 subunit of IL-12 and IL-23. In a 9-week NZB/W efficacy study, NIK SMI1 improved survival and renal function247. NIK SMI1 is fairly selective, inhibiting only 3 out of 222 off-target kinases (KHS1, LRRK2 and PKD1 (PKCμ)) to an extent >75% at a concentration of 1 μM247. In rodent models, the chronic dosing of NIK SMI1 is safe, but additional toxicity studies with higher doses of NIK inhibitors with improved potency and pharmacokinetic properties are needed to ensure that these molecules are safe before they are moved into the clinic. NIK SMI1 should inhibit several pathogenic pathways that are each individually validated in the clinic, including BAFFR and CD40/CD40L. Belimumab is an approved drug in SLE. Several modulatory antibodies or fusion proteins — anti-CD40 (CFZ533 (NCT02291029), leselumab (NCT01585233), I-655064 (NCT03385564) and FFP104 (NCT02465944)) and anti-CD40L (dapirolizumab (NCT02804763) and letolizumab (NCT02273960)) — are being investigated in the clinic for various indications such as psoriasis, RA, Crohn’s disease, SLE, ITP, primary biliary cirrhosis and transplantation249.
CDK8 and CDK19
CDK8 and its paralog CDK19 (which share 97% protein homology with each other) regulate RNA polymerase II (RNAP II) activity250. Certain subsets of CDKs (CDK1, CDK2, CDK4 and CDK6) and the corresponding cyclins are directly involved in cell cycle regulation251. Overexpression of CDK8 or CDK19 was found in several cancers and led to the discovery of multiple CDK8 inhibitors250. Numerous potent inhibitors targeting the kinase activities of CDK8 and CDK19 have been developed, such as Cortistatin A252, Senexin A253, CCT251921 (ref.254), MSC2530818 (ref.255) and BRD6989 (ref.256). Most studies focus mainly on the oncogenic function of CDK8 and CDK19, but emerging data suggest that CDK8 and CDK19 may also have a function in cellular reprogramming. In dendritic cells and macrophages, BRD6989, an inhibitor of both CDK8 and CDK19, upregulates IL-10 production by enhancing AP-1 activity256, which indicates that CDK8 and CDK19 have roles in innate immunity. Therefore, the function of the CDK module in transcription seems to be context-dependent, such that its biological function may vary among different cell types or in response to distinct stimuli.
Two independent studies have demonstrated that inhibitors against both CDK8 and CDK19 promoted Treg cell differentiation257,258. Akamatsu et al. elegantly carried out a functional screen using a compound library of close to 5,000 inhibitors with different molecular scaffolds to assess their effects on the differentiation of naive CD4+ T cells into FOXP3+ Treg cells258. The authors showed that inhibition of CDK8 and CDK19 can activate STAT5, which positively regulates FOXP3 expression, in a TGFβ-independent manner258. Inhibition of CDK8 and CDK19 in vivo enhanced the development of antigen-specific Treg cells, which dampened autoimmunity in preclinical models of experimental autoimmune encephalomyelitis and non-obese diabetes258. In a separate study, Guo et al. used CDK8 and CDK19 inhibitors (CCT251921 or Senexin A), which enhanced TGFβ signalling and drove Treg cell differentiation. This pathway depends partially on the attenuation of IFNγ–STAT1 signalling and on elevated SMAD2 phosphorylation257. Although the mechanisms of CDK8 and CDK19 inhibition elucidated in these studies differ, the net effects seem to promote Treg cell differentiation from effector T cells. More studies are needed to understand how inhibition of CDK8 and CDK9 can reprogramme conventional T cells to Treg cells. Pharmacological inhibition of CDK8 and CDK19 may have potential in T cell conversion of differentiated, antigen-specific effector memory T cells into FOXP3+ Treg cells for the treatment of autoimmune or inflammatory diseases. Currently, a CDK8 inhibitor, BCD-115, has been used in clinical trials to treat advanced-stage and metastatic breast cancer via directly blocking cancer cell differentiation (NCT03065010). In tumours, pathogenic tissue-resident Treg cells counteract productive immunity by inhibitory mechanisms, such as the downregulation of HLA and CD80/86 on dendritic cells and the modulation of the cytokine milieu such as TGFβ and IL-2 production259. Therefore, CDK8 and CDK19 inhibitors may be a powerful tool to induce Treg cell differentiation in vitro applicable to Treg cell-based cellular therapies. However, given the likelihood of the broad action of these kinases in other cell types, these inhibitors may not be safe for chronic dosing.
Future directions and conclusions
Tremendous progress has been made to advance various drugs, including inhibitors of JAKs, TYK2, IRAK4, BTK, SYK, RIPs and TPL2, into the clinic. The diversity of immunological pathways targeted by these molecules provides a golden opportunity to understand human immunology and to better design targeted therapeutics for multiple debilitating inflammatory and autoimmune diseases.
Potent kinase inhibitors are designed to be selective with minimal off-target effects, often by targeting the ATP-binding site of the kinase domain. However, as the ATP-binding site is relatively conserved among kinases, the design and discovery of selective kinase inhibitors still remain challenging. Furthermore, because many kinases induced during inflammation also regulate non-inflammatory pathways, kinase inhibition may also result in unknown on-target effects. It will be critical to identify several independent lead chemical scaffolds. The continued availability of extensive and diverse small-molecule libraries increases the likelihood of finding multiple candidates. The use of bioinformatics combined with machine learning can further mine the information provided by such libraries to expedite alignments and capture molecules with pharmacological and selectivity potential. Artificial intelligence with a large repository of structured medical information — including numerous connections extracted from scientific literature by machine learning — holds great potential to rapidly nominate rational targets260. One example is the identification of JAKis as a possible treatment for COVID-19 patients, after the finding that bariticinib inhibits clathrin-mediated endocytosis54 of the virus. In addition, advances in structural methodologies (such as cryo-electron microscopy) will broaden structure-based design for molecule development. Adaptation of innovative strategies, including platforms that determine biochemical and cellular targets as well as profiling selectivity against proteome, should help to prioritize better molecules. Novel developments in kinase inhibitor design to target allosteric pseudo-kinase domains (such as TYK2) may broaden possibilities for target selectivity. TYK2 pseudo-kinase inhibitors have shown that using human genetics with the analysis of rare coding variants enables both the identification and the design of novel drug targets51. Improving organ-specific (such as gut, lung or skin)48 and/or cell-specific261 delivery of kinase inhibitors should also improve therapeutic efficacy with reduced side effects.
Emerging evidence suggests that kinase function is not limited to catalytic activity and may also serve as structural scaffolds. Thus, the function of some kinases (such as IRAK4) may be only partially susceptible to activity-based small-molecule inhibition, suggesting that full suppression can only be achieved by additive modalities such as targeting protein–protein interactions, conformational antagonism or protein degradation. Depending on the pathway, partial or full suppression of kinase function may be advantageous to calibrate desired safety and efficacy outcomes for the disease indication. However, this also creates uncertainties around the efficacy of the molecule in the clinic, as, traditionally, full suppression of the pathways is the most desired outcome in order to tune optimal dose adjustments. Recent positive clinical data with IRAK4 inhibitors argue that, ultimately, clinical assessment of the target is needed to determine its net effect on disease outcomes34.
Identifying the appropriate patient populations within a given disease indication is an important consideration as many inflammatory diseases are heterogeneous in nature, and, therefore, require differential treatments for effective symptom amelioration. For example, BTK inhibition may only be a suitable treatment for a fraction of patients as RA is a heterogeneous disease and different subsets of patients are responsive to different treatments, including B cell depletion, TNF blockade or JAKi therapies262. Defining such subpopulations requires predictive disease biomarkers and a deeper understanding of the molecular mechanisms of kinase activity and the pathways they participate in. Identifying biomarker profiles predictive of efficacy of inhibitor-specific treatments remains a significant challenge.
As we gain confidence in the safety and efficacy of novel small molecules, an additional therapeutic strategy will be to use them in combination therapy. There is a strong scientific rationale to consider therapeutics with non-overlapping mechanisms of action. Oral treatment with kinase inhibitors may be advantageous given that these drugs can be dosed to partially inhibit a pathway and often are taken on a daily basis owing to their short half-life. By contrast, biologics often ablate downstream signalling and persist for a few days to weeks. The use of multiple inhibitors, or both inhibitors and biologics, holds promise in treating chronic inflammation, in part, to address molecules that did not meet expectations in the clinic when used as single agents. Although this is a promising route to explore, combination therapy will require completion of safety studies with single agents as well as careful trial design in order to monitor undesired safety effects.
References
Fullerton, J. N. & Gilroy, D. W. Resolution of inflammation: a new therapeutic frontier. Nat. Rev. Drug Discov. 15, 551–567 (2016).
Matthay, M. A. et al. Acute respiratory distress syndrome. Nat. Rev. Dis. Prim. 5, 18 (2019).
Spinelli, F. R., Conti, F. & Gadina, M. HiJAKing SARS-CoV-2? The potential role of JAK inhibitors in the management of COVID-19. Sci. Immunol. 5, eabc5367 (2020).
Patterson, H., Nibbs, R., McInnes, I. & Siebert, S. Protein kinase inhibitors in the treatment of inflammatory and autoimmune diseases. Clin. Exp. Immunol. 176, 1–10 (2014).
Wang, L., Wang, F. S. & Gershwin, M. E. Human autoimmune diseases: a comprehensive update. J. Intern. Med. 278, 369–395 (2015).
Krainer, J., Siebenhandl, S. & Weinhausel, A. Systemic autoinflammatory diseases. J. Autoimmun. 109, 102421 (2020).
Paluch, C., Santos, A. M., Anzilotti, C., Cornall, R. J. & Davis, S. J. Immune checkpoints as therapeutic targets in autoimmunity. Front. Immunol. 9, 2306 (2018).
Radawski, C. et al. Patient perceptions of unmet medical need in rheumatoid arthritis: a cross-sectional survey in the USA. Rheumatol. Ther. 6, 461–471 (2019).
Ferguson, F. M. & Gray, N. S. Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov. 17, 353–377 (2018). This comprehensive review on various kinases and their utility also describes the technologies that are enabling efficient generation of highly optimized kinase inhibitors.
Cohen, P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell Biol. 21, 317–324 (2009).
Lin, W. et al. Dual B cell immunotherapy is superior to individual anti-CD20 depletion or BAFF blockade in murine models of spontaneous or accelerated lupus. Arthritis Rheumatol. 67, 215–224 (2015).
Boleto, G., Kanagaratnam, L., Drame, M. & Salmon, J. H. Safety of combination therapy with two bDMARDs in patients with rheumatoid arthritis: a systematic review and meta-analysis. Semin. Arthritis Rheum. 49, 35–42 (2018).
Schwartz, D. M. et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 16, 843–862 (2017). This review focuses on the biology of JAK family members and various inhibitors that are in the clinic.
Medzhitov, R. & Horng, T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 9, 692–703 (2009).
Sun, S. C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 17, 545–558 (2017).
Arthur, J. S. & Ley, S. C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 13, 679–692 (2013).
Farber, D. L., Netea, M. G., Radbruch, A., Rajewsky, K. & Zinkernagel, R. M. Immunological memory: lessons from the past and a look to the future. Nat. Rev. Immunol. 16, 124–128 (2016).
Bluestone, J. A., Mackay, C. R., O’Shea, J. J. & Stockinger, B. The functional plasticity of T cell subsets. Nat. Rev. Immunol. 9, 811–816 (2009).
Boothby, I. C., Cohen, J. N. & Rosenblum, M. D. Regulatory T cells in skin injury: at the crossroads of tolerance and tissue repair. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aaz9631 (2020).
Adler, L. N. et al. The other function: class II-restricted antigen presentation by B cells. Front. Immunol. 8, 319 (2017).
Netea, M. G., Schlitzer, A., Placek, K., Joosten, L. A. B. & Schultze, J. L. Innate and adaptive immune memory: an evolutionary continuum in the host’s response to pathogens. Cell Host Microbe 25, 13–26 (2019).
Vanamee, E. S. & Faustman, D. L. Structural principles of tumor necrosis factor superfamily signaling. Sci. Signal. https://doi.org/10.1126/scisignal.aao4910 (2018).
Boraschi, D., Italiani, P., Weil, S. & Martin, M. U. The family of the interleukin-1 receptors. Immunol. Rev. 281, 197–232 (2018).
Scheller, J., Chalaris, A., Schmidt-Arras, D. & Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 1813, 878–888 (2011).
Wojno, E. D. & Hunter, C. A. New directions in the basic and translational biology of interleukin-27. Trends Immunol. 33, 91–97 (2012).
Hsieh, C. S. et al. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science 260, 547–549 (1993).
Cua, D. J. et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421, 744–748 (2003).
Teng, M. W. et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 21, 719–729 (2015).
Clynes, R. et al. Modulation of immune complex-induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J. Exp. Med. 189, 179–185 (1999).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Senger, K. et al. The kinase TPL2 activates ERK and p38 signaling to promote neutrophilic inflammation. Sci Signal. 10, eaah4273 (2017). This report shows that TPL2 activates both ERK and p38 signalling to impact neutrophilic inflammation.
Kung, J. E. & Jura, N. Structural basis for the non-catalytic functions of protein kinases. Structure 24, 7–24 (2016).
Rauch, J., Volinsky, N., Romano, D. & Kolch, W. The secret life of kinases: functions beyond catalysis. Cell Commun. Signal. 9, 23 (2011).
Danto, S. I. et al. Efficacy and safety of the selective interleukin-1 receptor associated kinase 4 inhibitor, PF-06650833, in patients with active rheumatoid arthritis and inadequate response to methotrexate [Abstract 2909]. Arthritis Rheumatol. 70 (Suppl. 10) (2019).
Owen, C., Berinstein, N. L., Christofides, A. & Sehn, L. H. Review of Bruton tyrosine kinase inhibitors for the treatment of relapsed or refractory mantle cell lymphoma. Curr. Oncol. 26, e233–e240 (2019).
Warr, M. et al. GS-4875, a first-in-class TPL2 inhibitor suppresses MEK-ERK inflammatory signaling and proinflammatory cytokine production in primary human monocytes [Abstract 33]. Arthritis Rheumatol. 71 (Suppl. 10) (2019). This is the first report of clinical application of TPL2 inhibitors in inflammatory diseases.
Mullard, A. FDA approves first-in-class SYK inhibitor. Nat. Rev. Drug Discov. 17, 385 (2018).
Burke, J. R. et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci. Transl Med. https://doi.org/10.1126/scitranslmed.aaw1736 (2019). This remarkable study shows that inhibiting the pseudo-kinase activity of TYK2 with the novel molecule BMS-986165 can reduce kinase activity with much greater selectivity for TYK2.
Mayence, A. & Vanden Eynde, J. J. Baricitinib: a 2018 novel FDA-approved small molecule inhibiting janus kinases. Pharmaceuticals https://doi.org/10.3390/ph12010037 (2019).
Xu, H. et al. PF-06651600, a dual JAK3/TEC family kinase inhibitor. ACS Chem. Biol. 14, 1235–1242 (2019).
Tarrant, J. M. et al. Filgotinib, a JAK1 inhibitor, modulates disease-related biomarkers in rheumatoid arthritis: results from two randomized, controlled phase 2b trials. Rheumatol. Ther. 7, 173–190 (2020).
Changelian, P. S. et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 302, 875–878 (2003).
Kudlacz, E., Conklyn, M., Andresen, C., Whitney-Pickett, C. & Changelian, P. The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur. J. Pharmacol. 582, 154–161 (2008).
Kudlacz, E. et al. The novel JAK-3 inhibitor CP-690550 is a potent immunosuppressive agent in various murine models. Am. J. Transpl. 4, 51–57 (2004).
Vainchenker, W. et al. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Research 7, 82 (2018).
Bewersdorf, J. P., Jaszczur, S. M., Afifi, S., Zhao, J. C. & Zeidan, A. M. Beyond ruxolitinib: fedratinib and other emergent treatment options for myelofibrosis. Cancer Manag. Res. 11, 10777–10790 (2019).
Corren, J. et al. Tezepelumab in adults with uncontrolled asthma. N. Engl. J. Med. 377, 936–946 (2017).
Sandborn, W. J. et al. Development of gut-selective pan-Janus kinase inhibitor TD-1473 for ulcerative colitis: a translational medicine program. J. Crohns Colitis https://doi.org/10.1093/ecco-jcc/jjaa049 (2020).
Jones, P. et al. Design and synthesis of a pan-Janus kinase inhibitor clinical candidate (PF-06263276) suitable for inhaled and topical delivery for the treatment of inflammatory diseases of the lungs and skin. J. Med. Chem. 60, 767–786 (2017). This paper describes the generation of the inhaled or topical JAK inhibitors with restricted exposure in specific organs, such as the lung or skin.
Dengler, H. S. et al. Lung-restricted inhibition of Janus kinase 1 is effective in rodent models of asthma. Sci. Transl Med. https://doi.org/10.1126/scitranslmed.aao2151 (2018).
Dendrou, C. A. et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci. Transl Med. 8, 363ra149 (2016).
Couturier, N. et al. Tyrosine kinase 2 variant influences T lymphocyte polarization and multiple sclerosis susceptibility. Brain 134, 693–703 (2011).
Papp, K. et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N. Engl. J. Med. 379, 1313–1321 (2018). This paper shows that BMS-986165 works better than placebo to clear psoriasis over 12 weeks.
Richardson, P. et al. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 395, e30–e31 (2020).
Cantini, F. et al. Baricitinib therapy in COVID-19: a pilot study on safety and clinical impact. J. Infect. https://doi.org/10.1016/j.jinf.2020.04.017 (2020).
Motshwene, P. G. et al. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J. Biol. Chem. 284, 25404–25411 (2009).
Lin, S. C., Lo, Y. C. & Wu, H. Helical assembly in the MyD88–IRAK4–IRAK2 complex in TLR/IL-1R signalling. Nature 465, 885–890 (2010).
Ferrao, R. et al. IRAK4 dimerization and trans-autophosphorylation are induced by Myddosome assembly. Mol. Cell 55, 891–903 (2014).
Kawagoe, T. et al. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat. Immunol. 9, 684–691 (2008).
Li, S., Strelow, A., Fontana, E. J. & Wesche, H. IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc. Natl Acad. Sci. USA 99, 5567–5572 (2002).
Cao, Z., Xiong, J., Takeuchi, M., Kurama, T. & Goeddel, D. V. TRAF6 is a signal transducer for interleukin-1. Nature 383, 443–446 (1996).
Jiang, Z., Ninomiya-Tsuji, J., Qian, Y., Matsumoto, K. & Li, X. Interleukin-1 (IL-1) receptor-associated kinase-dependent IL-1-induced signaling complexes phosphorylate TAK1 and TAB2 at the plasma membrane and activate TAK1 in the cytosol. Mol. Cell Biol. 22, 7158–7167 (2002).
Wan, Y. et al. Interleukin-1 receptor-associated kinase 2 is critical for lipopolysaccharide-mediated post-transcriptional control. J. Biol. Chem. 284, 10367–10375 (2009).
Ye, H. et al. Distinct molecular mechanism for initiating TRAF6 signalling. Nature 418, 443–447 (2002).
Zhou, H. et al. IRAK2 directs stimulus-dependent nuclear export of inflammatory mRNAs. eLife https://doi.org/10.7554/eLife.29630 (2017).
Wesche, H. et al. IRAK-M is a novel member of the Pelle/interleukin-1 receptor-associated kinase (IRAK) family. J. Biol. Chem. 274, 19403–19410 (1999).
Kobayashi, K. et al. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110, 191–202 (2002).
Zhou, H. et al. IRAK-M mediates Toll-like receptor/IL-1R-induced NFκB activation and cytokine production. EMBO J. 32, 583–596 (2013).
Su, J., Zhang, T., Tyson, J. & Li, L. The interleukin-1 receptor-associated kinase M selectively inhibits the alternative, instead of the classical NFκB pathway. J. Innate Immun. 1, 164–174 (2009).
Chiang, C. Y. et al. Cofactors required for TLR7- and TLR9-dependent innate immune responses. Cell Host Microbe 11, 306–318 (2012).
Negishi, H. et al. Evidence for licensing of IFN-γ-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc. Natl Acad. Sci. USA 103, 15136–15141 (2006).
Schmitz, F. et al. Interferon-regulatory-factor 1 controls Toll-like receptor 9-mediated IFN-β production in myeloid dendritic cells. Eur. J. Immunol. 37, 315–327 (2007).
Ku, C. L. et al. IRAK4 and NEMO mutations in otherwise healthy children with recurrent invasive pneumococcal disease. J. Med. Genet. 44, 16–23 (2007).
Cushing, L. et al. IRAK4 kinase activity controls Toll-like receptor-induced inflammation through the transcription factor IRF5 in primary human monocytes. J. Biol. Chem. 292, 18689–18698 (2017).
Kawagoe, T. et al. Essential role of IRAK-4 protein and its kinase activity in Toll-like receptor-mediated immune responses but not in TCR signaling. J. Exp. Med. 204, 1013–1024 (2007).
Swantek, J. L., Tsen, M. F., Cobb, M. H. & Thomas, J. A. IL-1 receptor-associated kinase modulates host responsiveness to endotoxin. J. Immunol. 164, 4301–4306 (2000).
Koziczak-Holbro, M. et al. IRAK-4 kinase activity is required for interleukin-1 (IL-1) receptor- and Toll-like receptor 7-mediated signaling and gene expression. J. Biol. Chem. 282, 13552–13560 (2007).
Dudhgaonkar, S. et al. Selective IRAK4 inhibition attenuates disease in murine lupus models and demonstrates steroid sparing activity. J. Immunol. 198, 1308–1319 (2017). This paper is the first demonstration that selective IRAK4 inhibitor can dampen disease in preclinical models of lupus.
Murphy, M., Pattabiraman, G., Manavalan, T. T. & Medvedev, A. E. Deficiency in IRAK4 activity attenuates manifestations of murine lupus. Eur. J. Immunol. 47, 880–891 (2017).
Nanda, S. K., Lopez-Pelaez, M., Arthur, J. S., Marchesi, F. & Cohen, P. Suppression of IRAK1 or IRAK4 catalytic activity, but not type 1 IFN signaling, prevents lupus nephritis in mice expressing a ubiquitin binding-defective mutant of ABIN1. J. Immunol. 197, 4266–4273 (2016).
Kim, S. H. et al. The dietary flavonoid kaempferol mediates anti-inflammatory responses via the Src, Syk, IRAK1, and IRAK4 molecular targets. Mediators Inflamm. 2015, 904142 (2015).
Rekhter, M. et al. Genetic ablation of IRAK4 kinase activity inhibits vascular lesion formation. Biochem. Biophys. Res. Commun. 367, 642–648 (2008).
Cameron, B. et al. Loss of interleukin receptor-associated kinase 4 signaling suppresses amyloid pathology and alters microglial phenotype in a mouse model of Alzheimer’s disease. J. Neurosci. 32, 15112–15123 (2012).
Thomas, J. A. et al. Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J. Immunol. 163, 978–984 (1999).
Della Mina, E. et al. Inherited human IRAK-1 deficiency selectively impairs TLR signaling in fibroblasts. Proc. Natl Acad. Sci. USA 114, E514–E523 (2017). This paper reports that IRAK1 deficiency dampens the immune response, although the effect is milder than the deficiency of IRAK4.
Sun, J. et al. Comprehensive RNAi-based screening of human and mouse TLR pathways identifies species-specific preferences in signaling protein use. Sci. Signal. 9, ra3 (2016).
Perkins, D. J. & Vogel, S. N. Inflammation: species-specific TLR signalling — insight into human disease. Nat. Rev. Rheumatol. 12, 198–200 (2016).
von Bernuth, H., Picard, C., Puel, A. & Casanova, J. L. Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur. J. Immunol. 42, 3126–3135 (2012).
Pauls, E. et al. Two phases of inflammatory mediator production defined by the study of IRAK2 and IRAK1 knock-in mice. J. Immunol. 191, 2717–2730 (2013).
Flannery, S. M., Keating, S. E., Szymak, J. & Bowie, A. G. Human interleukin-1 receptor-associated kinase-2 is essential for Toll-like receptor-mediated transcriptional and post-transcriptional regulation of tumor necrosis factor α. J. Biol. Chem. 286, 23688–23697 (2011).
Alsina, L. et al. A narrow repertoire of transcriptional modules responsive to pyogenic bacteria is impaired in patients carrying loss-of-function mutations in MYD88 or IRAK4. Nat. Immunol. 15, 1134–1142 (2014).
Emmerich, C. H. et al. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc. Natl Acad. Sci. USA 110, 15247–15252 (2013).
De, S. et al. Mechanism of dysfunction of human variants of the IRAK4 kinase and a role for its kinase activity in interleukin-1 receptor signaling. J. Biol. Chem. 293, 15208–15220 (2018).
De Nardo, D. et al. Interleukin-1 receptor-associated kinase 4 (IRAK4) plays a dual role in Myddosome formation and Toll-like receptor signaling. J. Biol. Chem. 293, 15195–15207 (2018). This study shows that IRAK4 has a scaffold function in Myddosome formation and that its kinase activity is dispensable for Myddosome assembly.
Silke, J., Rickard, J. A. & Gerlic, M. The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 16, 689–697 (2015).
Witt, A. & Vucic, D. Diverse ubiquitin linkages regulate RIP kinases-mediated inflammatory and cell death signaling. Cell Death Differ. 24, 1160–1171 (2017).
Newton, K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 25, 347–353 (2015).
Varfolomeev, E. & Vucic, D. Intracellular regulation of TNF activity in health and disease. Cytokine 101, 26–32 (2018).
Bertrand, M. J. et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 (2008).
Varfolomeev, E. et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor α (TNFα)-induced NF-κB activation. J. Biol. Chem. 283, 24295–24299 (2008).
Dynek, J. N. et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 29, 4198–4209 (2010).
Newton, K. Multitasking kinase RIPK1 regulates cell death and inflammation. Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a036368 (2019).
de Almagro, M. C. et al. Coordinated ubiquitination and phosphorylation of RIP1 regulates necroptotic cell death. Cell Death Differ. 24, 26–37 (2017).
Lawlor, K. E. et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6, 6282 (2015).
Patel, S. et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 27, 161–175 (2020). This study describes the instrumental and protective role for RIP1 kinase inhibition in inflammatory disease despite its lack of relevance in tumour progression and metastasis.
Newton, K. et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360 (2014).
Mandal, P. et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 56, 481–495 (2014).
Newton, K. et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 23, 1565–1576 (2016).
Mulay, S. R. et al. Cytotoxicity of crystals involves RIPK3–MLKL-mediated necroptosis. Nat. Commun. 7, 10274 (2016).
Webster, J. D. et al. RIP1 kinase activity is critical for skin inflammation but not for viral propagation. J. Leukoc. Biol. (2020).
Duan, X. et al. Inhibition of keratinocyte necroptosis mediated by RIPK1/RIPK3/MLKL provides a protective effect against psoriatic inflammation. Cell Death Dis. 11, 134 (2020).
Alevy, Y. G. et al. IL-13-induced airway mucus production is attenuated by MAPK13 inhibition. J. Clin. Invest. 122, 4555–4568 (2012).
Dondelinger, Y. et al. NF-κB-independent role of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60, 63–76 (2015).
Geng, J. et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 8, 359 (2017).
Tao, P. et al. A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature 577, 109–114 (2020).
Newton, K. et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 574, 428–431 (2019).
Lalaoui, N. et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 577, 103–108 (2020).
Matsuzawa-Ishimoto, Y. et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med. 214, 3687–3705 (2017).
Linkermann, A. et al. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 81, 751–761 (2012).
Degterev, A., Ofengeim, D. & Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl Acad. Sci. USA 116, 9714–9722 (2019).
Harris, P. A. et al. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J. Med. Chem. 60, 1247–1261 (2017). This paper describes the first RIP1 inhibitor that entered clinical trials.
Caruso, R., Warner, N., Inohara, N. & Nunez, G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 41, 898–908 (2014).
Caso, F. et al. Autoinflammatory granulomatous diseases: from Blau syndrome and early-onset sarcoidosis to NOD2-mediated disease and Crohn’s disease. RMD Open 1, e000097 (2015).
Damgaard, R. B. et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol. Cell 46, 746–758 (2012).
Goncharov, T. et al. Disruption of XIAP–RIP2 association blocks NOD2-mediated inflammatory signaling. Mol. Cell 69, 551–565 (2018). This study describes a critical role for the XIAP–RIP2 interaction in NOD2 inflammatory signalling and provides a molecular basis for the design of novel therapeutic strategies based on XIAP antagonists and RIP2 kinase inhibitors.
Hrdinka, M. et al. Small molecule inhibitors reveal an indispensable scaffolding role of RIPK2 in NOD2 signaling. EMBO J. https://doi.org/10.15252/embj.201899372 (2018).
Schwartzberg, P. L., Finkelstein, L. D. & Readinger, J. A. TEC-family kinases: regulators of T-helper-cell differentiation. Nat. Rev. Immunol. 5, 284–295 (2005).
Vargas, L., Hamasy, A., Nore, B. F. & Smith, C. I. Inhibitors of BTK and ITK: state of the new drugs for cancer, autoimmunity and inflammatory diseases. Scand. J. Immunol. 78, 130–139 (2013).
Schaeffer, E. M. et al. Tec family kinases modulate thresholds for thymocyte development and selection. J. Exp. Med. 192, 987–1000 (2000).
Sun, Y. et al. Inhibition of the kinase ITK in a mouse model of asthma reduces cell death and fails to inhibit the inflammatory response. Sci. Signal. 8, ra122 (2015).
Nagata, S. Apoptosis by death factor. Cell 88, 355–365 (1997).
Long, M. et al. Ibrutinib treatment improves T cell number and function in CLL patients. J. Clin. Invest. 127, 3052–3064 (2017).
Khan, W. N. et al. Defective B cell development and function in Btk-deficient mice. Immunity 3, 283–299 (1995).
Luk, A. D. W. et al. Type I and III interferon productions are impaired in X-linked agammaglobulinemia patients toward poliovirus but not influenza virus. Front. Immunol. 9, 1826 (2018).
Nimmerjahn, F. & Ravetch, J. V. Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34–47 (2008).
Kawakami, Y. et al. Tyrosine phosphorylation and activation of Bruton tyrosine kinase upon Fcε RI cross-linking. Mol. Cell Biol. 14, 5108–5113 (1994).
Hata, D. et al. Involvement of Bruton’s tyrosine kinase in FcεRI-dependent mast cell degranulation and cytokine production. J. Exp. Med. 187, 1235–1247 (1998).
Smiljkovic, D. et al. BTK inhibition is a potent approach to block IgE-mediated histamine release in human basophils. Allergy 72, 1666–1676 (2017).
Di Paolo, J. A. et al. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat. Chem. Biol. 7, 41–50 (2011).
Katewa, A. et al. Btk-specific inhibition blocks pathogenic plasma cell signatures and myeloid cell-associated damage in IFNα-driven lupus nephritis. JCI Insight 2, e90111 (2017).
Takata, M., Homma, Y. & Kurosaki, T. Requirement of phospholipase C-γ 2 activation in surface immunoglobulin M-induced B cell apoptosis. J. Exp. Med. 182, 907–914 (1995).
Fluckiger, A. C. et al. Btk/Tec kinases regulate sustained increases in intracellular Ca2+ following B-cell receptor activation. EMBO J. 17, 1973–1985 (1998).
Wang, D. et al. Phospholipase Cγ2 is essential in the functions of B cell and several Fc receptors. Immunity 13, 25–35 (2000).
Wang, J., Lau, K. Y., Jung, J., Ravindran, P. & Barrat, F. J. Bruton’s tyrosine kinase regulates TLR9 but not TLR7 signaling in human plasmacytoid dendritic cells. Eur. J. Immunol. 44, 1130–1136 (2014).
Bender, A. T. et al. Ability of Bruton’s tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of fc receptor but not B-cell receptor signaling. Mol. Pharmacol. 91, 208–219 (2017).
Gillooly, K. M. et al. Bruton’s tyrosine kinase inhibitor BMS-986142 in experimental models of rheumatoid arthritis enhances efficacy of agents representing clinical standard-of-care. PLoS ONE 12, e0181782 (2017).
Chalmers, S. A. et al. Therapeutic blockade of immune complex-mediated glomerulonephritis by highly selective inhibition of Bruton’s tyrosine yinase. Sci. Rep. 6, 26164 (2016).
Bender, A. T. et al. Btk inhibition treats TLR7/IFN driven murine lupus. Clin. Immunol. 164, 65–77 (2016).
Corzo, C. A. et al. The kinase IRAK4 promotes endosomal TLR and immune complex signaling in B cells and plasmacytoid dendritic cells. Sci. Signal. https://doi.org/10.1126/scisignal.aaz1053 (2020). This paper compares IRAK inhibition versus BTK inhibition and shows that IRAK4 has a major role in both TLR and immune-complex signalling, therefore providing a rationale to target it in SLE.
Montalban, X. et al. Placebo-controlled trial of an oral BTK inhibitor in multiple sclerosis. N. Engl. J. Med. 380, 2406–2417 (2019).
Watterson, S. H. et al. Discovery of branebrutinib (BMS-986195): a strategy for identifying a highly potent and selective covalent inhibitor providing rapid in vivo unactivation of Bruton’s tyrosine kinase (BTK). J. Med. Chem. 62, 3228–3250 (2019).
Cohen, S. et al. Efficacy and safety of fenebrutinib, a BTK inhibitor, compared to placebo in rheumatoid arthritis patients with active disease despite TNF inhibitor treatment: randomized, double blind, phase 2 study [Abstract 929]. Arthritis Rheumatol. 71 (Suppl. 10) (2019).
Treon, S. P. et al. The BTK-inhibitor ibrutinib may protect against pulmonary injury in COVID-19 infected patients. Blood https://doi.org/10.1182/blood.2020006288 (2020).
Mocsai, A., Ruland, J. & Tybulewicz, V. L. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat. Rev. Immunol. 10, 387–402 (2010).
Gradler, U. et al. Structural and biophysical characterization of the Syk activation switch. J. Mol. Biol. 425, 309–333 (2013).
Deindl, S. et al. Structural basis for the inhibition of tyrosine kinase activity of ZAP-70. Cell 129, 735–746 (2007).
Keshvara, L. M., Isaacson, C., Harrison, M. L. & Geahlen, R. L. Syk activation and dissociation from the B-cell antigen receptor is mediated by phosphorylation of tyrosine 130. J. Biol. Chem. 272, 10377–10381 (1997).
Lupher, M. L. Jr et al. Cbl-mediated negative regulation of the Syk tyrosine kinase. A critical role for Cbl phosphotyrosine-binding domain binding to Syk phosphotyrosine 323. J. Biol. Chem. 273, 35273–35281 (1998).
Yankee, T. M., Keshvara, L. M., Sawasdikosol, S., Harrison, M. L. & Geahlen, R. L. Inhibition of signaling through the B cell antigen receptor by the protooncogene product, c-Cbl, requires Syk tyrosine 317 and the c-Cbl phosphotyrosine-binding domain. J. Immunol. 163, 5827–5835 (1999).
Geahlen, R. L. Getting Syk: spleen tyrosine kinase as a therapeutic target. Trends Pharmacol. Sci. 35, 414–422 (2014).
Ozaki, N. et al. Syk-dependent signaling pathways in neutrophils and macrophages are indispensable in the pathogenesis of anti-collagen antibody-induced arthritis. Int. Immunol. 24, 539–550 (2012).
Cheng, A. M. et al. Syk tyrosine kinase required for mouse viability and B-cell development. Nature 378, 303–306 (1995).
Turner, M. et al. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature 378, 298–302 (1995).
Jakus, Z., Simon, E., Balazs, B. & Mocsai, A. Genetic deficiency of Syk protects mice from autoantibody-induced arthritis. Arthritis Rheum. 62, 1899–1910 (2010).
Pine, P. R. et al. Inflammation and bone erosion are suppressed in models of rheumatoid arthritis following treatment with a novel Syk inhibitor. Clin. Immunol. 124, 244–257 (2007).
Coffey, G. et al. Specific inhibition of spleen tyrosine kinase suppresses leukocyte immune function and inflammation in animal models of rheumatoid arthritis. J. Pharmacol. Exp. Ther. 340, 350–359 (2012).
Genovese, M. C. et al. A phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study of 2 dosing regimens of fostamatinib in patients with rheumatoid arthritis with an inadequate response to a tumor necrosis factor-alpha antagonist. J. Rheumatol. 41, 2120–2128 (2014).
Weinblatt, M. E. et al. Treatment of rheumatoid arthritis with a Syk kinase inhibitor: a twelve-week, randomized, placebo-controlled trial. Arthritis Rheum. 58, 3309–3318 (2008).
Braselmann, S. et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J. Pharmacol. Exp. Ther. 319, 998–1008 (2006).
Kunwar, S., Devkota, A. R. & Ghimire, D. K. Fostamatinib, an oral spleen tyrosine kinase inhibitor, in the treatment of rheumatoid arthritis: a meta-analysis of randomized controlled trials. Rheumatol. Int. 36, 1077–1087 (2016).
Weinblatt, M. E. et al. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N. Engl. J. Med. 363, 1303–1312 (2010).
Markham, A. Fostamatinib: first global approval. Drugs 78, 959–963 (2018).
Newland, A., Lee, E. J., McDonald, V. & Bussel, J. B. Fostamatinib for persistent/chronic adult immune thrombocytopenia. Immunotherapy 10, 9–25 (2018).
Rolf, M. G. et al. In vitro pharmacological profiling of R406 identifies molecular targets underlying the clinical effects of fostamatinib. Pharmacol. Res. Perspect. 3, e00175 (2015). This paper reports the utility of various biochemical and cellular assays to identify several off-target effects of R406/fostamatinib.
Simon, M., Vanes, L., Geahlen, R. L. & Tybulewicz, V. L. Distinct roles for the linker region tyrosines of Syk in FcεRI signaling in primary mast cells. J. Biol. Chem. 280, 4510–4517 (2005).
Chen, L. et al. ZAP-70 directly enhances IgM signaling in chronic lymphocytic leukemia. Blood 105, 2036–2041 (2005).
Currie, K. S. et al. Discovery of GS-9973, a selective and orally efficacious inhibitor of spleen tyrosine kinase. J. Med. Chem. 57, 3856–3873 (2014). This article describes a recently developed small-molecule inhibitor, GS-9973, which is highly specific for SYK, and its clinical application in autoimmunity and oncology.
Sharman, J. et al. An open-label phase 2 trial of entospletinib (GS-9973), a selective spleen tyrosine kinase inhibitor, in chronic lymphocytic leukemia. Blood 125, 2336–2343 (2015).
Barr, P. M. et al. Phase 2 study of idelalisib and entospletinib: pneumonitis limits combination therapy in relapsed refractory CLL and NHL. Blood 127, 2411–2415 (2016).
Xu, D., Matsumoto, M. L., McKenzie, B. S. & Zarrin, A. A. TPL2 kinase action and control of inflammation. Pharmacol. Res. https://doi.org/10.1016/j.phrs.2017.11.031 (2017).
Beinke, S., Robinson, M. J., Hugunin, M. & Ley, S. C. Lipopolysaccharide activation of the TPL-2/MEK/extracellular signal-regulated kinase mitogen-activated protein kinase cascade is regulated by IκB kinase-induced proteolysis of NF-κB1 p105. Mol. Cell Biol. 24, 9658–9667 (2004).
Lang, V. et al. ABIN-2 forms a ternary complex with TPL-2 and NF-κB1 p105 and is essential for TPL-2 protein stability. Mol. Cell Biol. 24, 5235–5248 (2004).
Beinke, S. et al. NF-κB1 p105 negatively regulates TPL-2 MEK kinase activity. Mol. Cell Biol. 23, 4739–4752 (2003).
Dumitru, C. D. et al. TNF-α induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell 103, 1071–1083 (2000).
Das, S. et al. Tpl2/cot signals activate ERK, JNK, and NF-κB in a cell-type and stimulus-specific manner. J. Biol. Chem. 280, 23748–23757 (2005).
Rousseau, S. et al. TPL2-mediated activation of ERK1 and ERK2 regulates the processing of pre-TNFα in LPS-stimulated macrophages. J. Cell Sci. 121, 149–154 (2008).
Waterfield, M. R., Zhang, M., Norman, L. P. & Sun, S. C. NF-κB1/p105 regulates lipopolysaccharide-stimulated MAP kinase signaling by governing the stability and function of the Tpl2 kinase. Mol. Cell 11, 685–694 (2003).
Sriskantharajah, S. et al. Regulation of experimental autoimmune encephalomyelitis by TPL-2 kinase. J. Immunol. 192, 3518–3529 (2014).
Nanda, S. K. et al. ABIN2 function is required to suppress DSS-induced colitis by a Tpl2-independent mechanism. J. Immunol. 201, 3373–3382 (2018).
Roulis, M. et al. Intestinal myofibroblast-specific Tpl2–Cox-2–PGE2 pathway links innate sensing to epithelial homeostasis. Proc. Natl Acad. Sci. USA 111, E4658–E4667 (2014).
Cuenda, A. & Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 1773, 1358–1375 (2007).
Remy, G. et al. Differential activation of p38MAPK isoforms by MKK6 and MKK3. Cell Signal. 22, 660–667 (2010).
Jiang, Y. et al. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38δ. J. Biol. Chem. 272, 30122–30128 (1997).
Risco, A. et al. p38γ and p38δ kinases regulate the Toll-like receptor 4 (TLR4)-induced cytokine production by controlling ERK1/2 protein kinase pathway activation. Proc. Natl Acad. Sci. USA 109, 11200–11205 (2012). This study reports the function of p38γ and p38δ isoforms in innate immunity via ERK activation and preferential distribution of expression.
Gonzalez-Teran, B. et al. Eukaryotic elongation factor 2 controls TNF-α translation in LPS-induced hepatitis. J. Clin. Invest. 123, 164–178 (2013).
Menon, M. B. & Gaestel, M. TPL2 meets p38MAPK: emergence of a novel positive feedback loop in inflammation. Biochem. J. 473, 2995–2999 (2016).
Criado, G. et al. Alternative p38 MAPKs are essential for collagen-induced arthritis. Arthritis Rheumatol. 66, 1208–1217 (2014).
Ittner, A. et al. Regulation of PTEN activity by p38δ–PKD1 signaling in neutrophils confers inflammatory responses in the lung. J. Exp. Med. 209, 2229–2246 (2012).
Sumara, G. et al. Regulation of PKD by the MAPK p38δ in insulin secretion and glucose homeostasis. Cell 136, 235–248 (2009).
Tang, J., Qi, X., Mercola, D., Han, J. & Chen, G. Essential role of p38γ in K-Ras transformation independent of phosphorylation. J. Biol. Chem. 280, 23910–23917 (2005).
Eyers, P. A., Craxton, M., Morrice, N., Cohen, P. & Goedert, M. Conversion of SB 203580-insensitive MAP kinase family members to drug-sensitive forms by a single amino-acid substitution. Chem. Biol. 5, 321–328 (1998).
Fitzgerald, C. E. et al. Structural basis for p38α MAP kinase quinazolinone and pyridol-pyrimidine inhibitor specificity. Nat. Struct. Biol. 10, 764–769 (2003).
Gum, R. J. et al. Acquisition of sensitivity of stress-activated protein kinases to the p38 inhibitor, SB 203580, by alteration of one or more amino acids within the ATP binding pocket. J. Biol. Chem. 273, 15605–15610 (1998).
Gangwal, R. P., Bhadauriya, A., Damre, M. V., Dhoke, G. V. & Sangamwar, A. T. p38 mitogen-activated protein kinase inhibitors: a review on pharmacophore mapping and QSAR studies. Curr. Top. Medicinal Chem. 13, 1015–1035 (2013).
Coulthard, L. R., White, D. E., Jones, D. L., McDermott, M. F. & Burchill, S. A. p38MAPK: stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 15, 369–379 (2009).
Wang, C. et al. Selective inhibition of the p38α MAPK–MK2 axis inhibits inflammatory cues including inflammasome priming signals. J. Exp. Med. 215, 1315–1325 (2018).
Schreiber, S. et al. Oral p38 mitogen-activated protein kinase inhibition with BIRB 796 for active Crohn’s disease: a randomized, double-blind, placebo-controlled trial. Clin. Gastroenterol. Hepatol. 4, 325–334 (2006).
Charron, C. E. et al. RV568, a narrow-spectrum kinase inhibitor with p38 MAPK-α and -γ selectivity, suppresses COPD inflammation. Eur. Respir. J. https://doi.org/10.1183/13993003.00188-2017 (2017).
Armendariz-Borunda, J. et al. A controlled clinical trial with pirfenidone in the treatment of pathological skin scarring caused by burns in pediatric patients. Ann. Plastic Surg. 68, 22–28 (2012).
Margaritopoulos, G. A., Vasarmidi, E. & Antoniou, K. M. Pirfenidone in the treatment of idiopathic pulmonary fibrosis: an evidence-based review of its place in therapy. Core Evid. 11, 11–22 (2016).
Khanna, D. et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: the LOTUSS trial. J. Rheumatol. 43, 1672–1679 (2016).
Rodriguez-Castellanos, M., Tlacuilo-Parra, A., Sanchez-Enriquez, S., Velez-Gomez, E. & Guevara-Gutierrez, E. Pirfenidone gel in patients with localized scleroderma: a phase II study. Arthritis Res. Ther. 16, 510 (2015).
Sharma, K. et al. Pirfenidone for diabetic nephropathy. J. Am. Soc. Nephrol. 22, 1144–1151 (2011).
Flores-Contreras, L. et al. Treatment with pirfenidone for two years decreases fibrosis, cytokine levels and enhances CB2 gene expression in patients with chronic hepatitis C. BMC Gastroenterol. 14, 131 (2014).
Kondoh, Y. et al. Comparative chemical array screening for p38γ/δ MAPK inhibitors using a single gatekeeper residue difference between p38α/β and p38γ/δ. Sci. Rep. 6, 29881 (2016). This paper describes small molecules in development with high specificity for p38γ and p38δ isoforms over p38α and p38β.
Nithianandarajah-Jones, G. N., Wilm, B., Goldring, C. E., Muller, J. & Cross, M. J. ERK5: structure, regulation and function. Cell Signal. 24, 2187–2196 (2012).
Lin, W. et al. Function of CSF1 and IL34 in macrophage homeostasis, inflammation, and cancer. Front. Immunol. 10, 2019 (2019).
Kumari, A., Silakari, O. & Singh, R. K. Recent advances in colony stimulating factor-1 receptor/c-FMS as an emerging target for various therapeutic implications. Biomed. Pharmacother. 103, 662–679 (2018).
Yan, C., Luo, H., Lee, J. D., Abe, J. & Berk, B. C. Molecular cloning of mouse ERK5/BMK1 splice variants and characterization of ERK5 functional domains. J. Biol. Chem. 276, 10870–10878 (2001).
Buschbeck, M. & Ullrich, A. The unique C-terminal tail of the mitogen-activated protein kinase ERK5 regulates its activation and nuclear shuttling. J. Biol. Chem. 280, 2659–2667 (2005).
Kasler, H. G., Victoria, J., Duramad, O. & Winoto, A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell Biol. 20, 8382–8389 (2000).
Wilhelmsen, K. et al. Extracellular signal-regulated kinase 5 promotes acute cellular and systemic inflammation. Sci. Signal. 8, ra86 (2015).
Lin, E. C. et al. ERK5 kinase activity is dispensable for cellular immune response and proliferation. Proc. Natl Acad. Sci. USA 113, 11865–11870 (2016).
Finegan, K. G. et al. ERK5 is a critical mediator of inflammation-driven cancer. Cancer Res. 75, 742–753 (2015).
Hayashi, M. et al. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J. Clin. Invest. 113, 1138–1148 (2004).
Platanias, L. C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386 (2005).
Eleftheriou, D. & Brogan, P. A. Genetic interferonopathies: an overview. Best Pract. Res. Clin. Rheumatol. 31, 441–459 (2017).
Baechler, E. C. et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl Acad. Sci. USA 100, 2610–2615 (2003).
Volpi, S., Picco, P., Caorsi, R., Candotti, F. & Gattorno, M. Type I interferonopathies in pediatric rheumatology. Pediatric Rheumatol. 14, 35 (2016).
Reilly, S. M. et al. An inhibitor of the protein kinases TBK1 and IKK-ε improves obesity-related metabolic dysfunctions in mice. Nat. Med. 19, 313–321 (2013). This paper shows that amlexanox inhibits TBK1 and, in obese mice, can elevate energy expenditure through increased thermogenesis, producing weight loss, improved insulin sensitivity and decreased steatosis.
Neveu, G. et al. AP-2-associated protein kinase 1 and cyclin G-associated kinase regulate hepatitis C virus entry and are potential drug targets. J. Virol. 89, 4387–4404 (2015). This paper reveals that by regulating HCV entry and its life cycle, AAK1 and GAK represent potential targets for antiviral strategies.
Schor, S. & Einav, S. Repurposing of kinase inhibitors as broad-spectrum antiviral drugs. DNA Cell Biol. 37, 63–69 (2018).
Ahmad, L., Zhang, S. Y., Casanova, J. L. & Sancho-Shimizu, V. Human TBK1: a gatekeeper of neuroinflammation. Trends Mol. Med. 22, 511–527 (2016).
Peters, R. T. & Maniatis, T. A new family of IKK-related kinases may function as IκB kinase kinases. Biochim. Biophys. Acta 1471, M57–M62 (2001).
Zhang, L., Zhao, X., Zhang, M., Zhao, W. & Gao, C. Ubiquitin-specific protease 2b negatively regulates IFN-β production and antiviral activity by targeting TANK-binding kinase 1. J. Immunol. 193, 2230–2237 (2014). This report shows that USP2b deubiquitinates K63-linked polyubiquitin chains from TBK1 to terminate TBK1 activation.
McWhirter, S. M. et al. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc. Natl Acad. Sci. USA 101, 233–238 (2004).
Bulek, K. et al. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat. Immunol. 12, 844–852 (2011).
Hemmi, H. et al. The roles of two IκB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 199, 1641–1650 (2004).
Tenoever, B. R. et al. Multiple functions of the IKK-related kinase IKKε in interferon-mediated antiviral immunity. Science 315, 1274–1278 (2007).
Hasan, M. & Yan, N. Therapeutic potential of targeting TBK1 in autoimmune diseases and interferonopathies. Pharmacol. Res. 111, 336–342 (2016).
Hasan, M. et al. Cutting edge: inhibiting TBK1 by compound II ameliorates autoimmune disease in mice. J. Immunol. 195, 4573–4577 (2015). This report demonstrates that TBK inhibitor is efficacious in interferonopathy mouse models (such as TREX-KO) with a high interferon signature.
Yu, J. et al. Regulation of T-cell activation and migration by the kinase TBK1 during neuroinflammation. Nat. Commun. 6, 6074 (2015). This paper shows that TBK1 regulates AKT1/mTORC1 to control T cell activation and egress from draining lymph nodes using conditional T cell-specific knockout mice and TBK1 inhibitors.
Zarnegar, B. J. et al. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 9, 1371–1378 (2008).
Willmann, K. L. et al. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat. Commun. 5, 5360 (2014).
Li, Y. et al. Cell intrinsic role of NF-κB-inducing kinase in regulating T cell-mediated immune and autoimmune responses. Sci. Rep. 6, 22115 (2016).
Katakam, A. K. et al. Dendritic cells require NIK for CD40-dependent cross-priming of CD8+ T cells. Proc. Natl Acad. Sci. USA 112, 14664–14669 (2015).
Brightbill, H. D. et al. NF-κB inducing kinase is a therapeutic target for systemic lupus erythematosus. Nat. Commun. 9, 179 (2018). This study shows that selective NIK inhibition can inhibit multiple immunological pathways, including BAFF, OX40, CD40, ICOS, IL-21 and TWEAK, and provide efficacy in preclinical SLE models.
Brightbill, H. D. et al. Conditional deletion of NF-κB-inducing kinase (NIK) in adult mice disrupts mature B cell survival and activation. J. Immunol. 195, 953–964 (2015).
Karnell, J. L., Rieder, S. A., Ettinger, R. & Kolbeck, R. Targeting the CD40–CD40L pathway in autoimmune diseases: humoral immunity and beyond. Adv. Drug Deliv. Rev. 141, 92–103 (2019).
Tsai, K. L. et al. A conserved mediator-CDK8 kinase module association regulates mediator–RNA polymerase II interaction. Nat. Struct. Mol. Biol. 20, 611–619 (2013).
Malumbres, M. & Barbacid, M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 (2009).
Cee, V. J., Chen, D. Y., Lee, M. R. & Nicolaou, K. C. Cortistatin A is a high-affinity ligand of protein kinases ROCK, CDK8, and CDK11. Angew Chem. Int. Ed. Engl. 48, 8952–8957 (2009).
Porter, D. C. et al. Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc. Natl Acad. Sci. USA 109, 13799–13804 (2012).
Mallinger, A. et al. Discovery of potent, selective, and orally bioavailable small-molecule modulators of the mediator complex-associated kinases CDK8 and CDK19. J. Med. Chem. 59, 1078–1101 (2016).
Czodrowski, P. et al. Structure-based optimization of potent, selective, and orally bioavailable CDK8 inhibitors discovered by high-throughput screening. J. Med. Chem. 59, 9337–9349 (2016).
Johannessen, L. et al. Small-molecule studies identify CDK8 as a regulator of IL-10 in myeloid cells. Nat. Chem. Biol. 13, 1102–1108 (2017). This paper screens a chemical library and identifies CDK8/19 as a target to promote Treg cell reprogramming.
Guo, Z., Wang, G., Lv, Y., Wan, Y. Y. & Zheng, J. Inhibition of Cdk8/Cdk19 activity promotes Treg cell differentiation and suppresses autoimmune diseases. Front. Immunol. 10, 1988 (2019). This paper reports that CDK8/19 inhibition can potentiate Treg cell differentiation.
Akamatsu, M. et al. Conversion of antigen-specific effector/memory T cells into Foxp3-expressing Treg cells by inhibition of CDK8/19. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aaw2707 (2019).
Han, S., Toker, A., Liu, Z. Q. & Ohashi, P. S. Turning the tide against regulatory T cells. Front. Oncol. 9, 279 (2019).
Sellwood, M. A., Ahmed, M., Segler, M. H. & Brown, N. Artificial intelligence in drug discovery. Future Med. Chem. 10, 2025–2028 (2018).
van Beuge, M. M., Poelstra, K. & Prakash, J. Specific delivery of kinase inhibitors in nonmalignant and malignant diseases. Expert Opin. Drug Deliv. 9, 59–70 (2012).
Cho, J. H. & Feldman, M. Heterogeneity of autoimmune diseases: pathophysiologic insights from genetics and implications for new therapies. Nat. Med. 21, 730–738 (2015).
Uematsu, S. et al. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-α induction. J. Exp. Med. 201, 915–923 (2005).
Vallabhapurapu, S. & Karin, M. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733 (2009).
Dempsey, A. & Bowie, A. G. Innate immune recognition of DNA: a recent history. Virology 479–480, 146–152 (2015).
Acknowledgements
The authors thank Allison Bruce for superb graphic design and figure illustrations.
Author information
Authors and Affiliations
Contributions
A.A.Z. conceived the review topic with major contributions to writing and revisions of the entire manuscript. K.B. contributed to IRAK4, Syk, p38 sections and revisions. D.V. contributed to RIPK section and revisions. P.L. contributed to Table 1.
Corresponding author
Ethics declarations
Competing interests
A.A.Z. is an employee of TRexBio and holds stock in TRexBio and the Roche Group. K.B. is an employee of Genentech. P.L. is an employee of Synthekine and holds stock in Synthekine and the Roche Group. D.V. is an employee of Genentech and holds stock and options in the Roche Group.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Rheumatoid arthritis
-
(RA). A progressive autoimmune and inflammatory disease manifested by joint pain and swelling in the feet and hands that can cause permanent joint destruction and deformity.
- Inflammatory bowel disease
-
(IBD). A group of disorders that involve chronic inflammation of the digestive tract, including ulcerative colitis and Crohn’s disease.
- Crohn’s disease
-
An inflammatory disease that causes inflammation of the lining of the digestive tract, which often spreads deep into affected tissues.
- Ulcerative colitis
-
An inflammatory disease that causes inflammation and sores (ulcers) in the innermost lining of the colon and rectum.
- Chronic obstructive pulmonary disease
-
(COPD). A chronic inflammatory lung disease that causes obstructed airflow from the lungs accompanied by breathing difficulty, cough, mucus production and wheezing.
- Idiopathic pulmonary fibrosis
-
(IPF). A chronic and progressive fibrotic lung disease with unknown aetiology accompanied by scarring, resulting in persistent dry, hacking cough.
- Hypoxaemia
-
An abnormally low concentration of oxygen in the blood.
- Plasmacytoid dendritic cells
-
(pDCs). Specialized, immunomodulatory dendritic cells that produce large quantities of type I interferons in response to viral antigens, with limited capacity to present such antigens to T cells.
- Autoantibodies
-
Antibodies reactive against an individual’s own tissues or organs.
- Breakthrough designation
-
A process designed to expedite the development and review of drugs that are intended to treat a serious condition and for which preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant end point(s).
- Pseudokinase domain
-
A kinase-like domain that lacks at least one of the conserved catalytic residues.
- Death domain
-
A protein domain that contains six α-helices that facilitate interactions with death domains in other proteins in multisubunit complex formation.
- Pyogenic bacterial infections
-
A condition characteristic of primary immunodeficiency due to myeloid differentiation primary response 88 deficiency in which patients have increased susceptibility to infections owing to their inability to signal through Toll-like receptors to activate inflammation.
- X-linked agammaglobulinaemia
-
A genetic immunosuppressive condition that results in a severe reduction in the number of B cells and, thereby, in the production of protective antibodies. Patients with this condition are predominantly males and are at greater risk of recurrent opportunistic infections.
- Pemphigus
-
Skin disorders that cause blisters or pus-filled bumps. Lesions can also form in the mucus membranes (soft linings of the eyes, nose, mouth, throat and genitals).
- American College of Rheumatology 20%
-
A standard criterion to measure the effectiveness of various arthritis medications or treatments. It means a 20% improvement in tender or swollen joint counts and other parameters.
- Tachyphylaxis
-
A rapidly diminishing response to successive doses of a drug, rendering it less effective. The effect is common with drugs acting on the nervous system.
- Amyotrophic lateral sclerosis
-
A disease of the central nervous system that affects nerves in the brain and spinal cord, causing the progressive loss of muscle control.
- Combined immunodeficiency syndrome
-
Rare disorders caused by mutations in different genes involved in the development and function of B cells and T cells.
- Peyer’s patches
-
Lymphoid nodules located on the outer lining of the small intestine, serving as monitors of the intestinal contents, often bacteria of the microbiome, to prevent the outgrowth of pathogenic bacteria in the gut.
Rights and permissions
About this article

Cite this article
Zarrin, A.A., Bao, K., Lupardus, P. et al. Kinase inhibition in autoimmunity and inflammation. Nat Rev Drug Discov 20, 39–63 (2021). https://doi.org/10.1038/s41573-020-0082-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-020-0082-8
This article is cited by
-
ERK pathway agonism for cancer therapy: evidence, insights, and a target discovery framework
npj Precision Oncology (2024)
-
Immunosuppressive Cyclotides: A Promising Approach for Treating Autoimmune Diseases
The Protein Journal (2024)
-
Plant-derived bioactive compounds as key players in the modulation of immune-related conditions
Phytochemistry Reviews (2024)
-
Treatment Options for Alopecia Areata in Children and Adolescents
Pediatric Drugs (2024)
-
CXCR1 drives the pathogenesis of EAE and ARDS via boosting dendritic cells-dependent inflammation
Cell Death & Disease (2023)
