
Abstract
Lysosomes are membrane-bound organelles with roles in processes involved in degrading and recycling cellular waste, cellular signalling and energy metabolism. Defects in genes encoding lysosomal proteins cause lysosomal storage disorders, in which enzyme replacement therapy has proved successful. Growing evidence also implicates roles for lysosomal dysfunction in more common diseases including inflammatory and autoimmune disorders, neurodegenerative diseases, cancer and metabolic disorders. With a focus on lysosomal dysfunction in autoimmune disorders and neurodegenerative diseases — including lupus, rheumatoid arthritis, multiple sclerosis, Alzheimer disease and Parkinson disease — this Review critically analyses progress and opportunities for therapeutically targeting lysosomal proteins and processes, particularly with small molecules and peptide drugs.
Similar content being viewed by others


Introduction
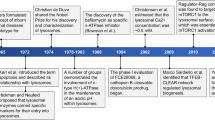
Discovered in the 1950s by Christian de Duve, lysosomes are membrane-bound vesicles containing numerous hydrolytic enzymes that can break down biological polymers such as proteins, lipids, nucleic acids and polysaccharides1,2. Lysosomes have long been known to have a key role in the degradation and recycling of extracellular material via endocytosis and phagocytosis, and intracellular material via autophagy (reviewed elsewhere2,3,4,5) (Fig. 1). The products of lysosomal degradation through these processes can be trafficked to the Golgi apparatus for reuse or for release from the cell through lysosomal exocytosis, which is important in immune system processes. In addition, it has become clear more recently that lysosomes have an important role in other cellular processes including nutrient sensing and the control of energy metabolism3,5,6,7 (Fig. 1).

a | Functional lysosomes are involved in the degradation (endocytic and autophagic) and regulation of exogenous and endogenous cellular material, including recycling processes. Extracellular material endocytosed by the endosomes and intracellular cargo internalized by the autophagosomes fuse with lysosomes for degradation, which produces energy (ATP production) and source molecules for the macromolecules. Mechanistic target of rapamycin complex 1 (mTORC1) plays a key role in lysosomal nutrient sensing signals (lysosome-to-nucleus axis) to regulate energy metabolism. Factors such as energy levels, type of pH, ion channel regulation and others decide the fate of the catabolic process. During lysosomal exocytosis, the lysosomal content favours plasma membrane (PM) repair, bone resorption, immune response and elimination of pathogenic stores. b | The lysosome is the ultimate cell compartment that digests unwanted protein materials generated by macroautophagy, microautophagy (pathways during which the cytoplasmic material is trapped in the lysosome by a process of membrane invagination) and chaperone-mediated autophagy (CMA). In general, lipid droplets (LDs) are degraded by lipophagy, a subtype of macroautophagy, which is activated by cytosolic lipases. CMA has also been demonstrated to participate in the degradation of LDs in which perilipin (PLIN2/3) proteins are phosphorylated (P) by AMP-activated protein kinase (AMPK) with the help of the HSPA8 chaperone. Mechanistic target of rapamycin complex 2 (mTORC2) and AKT (also known as protein kinase B) are negative regulators of CMA, where they exert their effect on the translocation complex of CMA. In situations of starvation, negative regulators are controlled by pleckstrin homology domain and leucine-rich repeat protein phosphatase (PHLPP). Lysosomal stability effects the transcription factor EB (TFEB) translation to the nucleus in which TFEB binds to the coordinated lysosomal expression and regulation (CLEAR) motifs to regulate the transcription of genes. EF1a, elongation factor 1a; Lys, lysosome; Rac1, Ras-related C3 botulinum toxin substrate 1.
Alterations in lysosomal functions, either in the fusion processes involved in the general pathways mentioned above or related to the function of lysosomal enzymes and non-enzymatic proteins, can result in broad detrimental effects, including failure to clear potentially toxic cellular waste, inflammation, apoptosis and dysregulation of cellular signalling8. Such defects have been implicated in many diseases, ranging from rare lysosomal storage disorders (LSDs), which are caused by the dysfunction of particular lysosomal proteins, to more common autoimmune and neurodegenerative disorders5,9,10. Despite some limitations, impressive results have been achieved in treating several LSDs through enzyme replacement therapy (ERT). In addition, substantial efforts have been focused on therapeutically targeting the autophagy processes upstream of lysosomes11,12,13,14. However, there has so far been less attention on investigating the potential to directly target lysosomes with small molecules and peptide drugs.
Nevertheless, with recent advances in understanding of lysosomal function and dysfunction in diseases, promising novel opportunities for therapeutic intervention through targeting lysosomes specifically are beginning to emerge. This Review will provide a brief overview of lysosomal biogenesis, structure and function, and describe the role of lysosomal dysfunction in LSDs as well as other, more common diseases. Specifically, the article will focus on organ-specific and non-organ-specific autoimmune diseases, including lupus, rheumatoid arthritis (RA) and multiple sclerosis (MS), as these have not been extensively reviewed elsewhere, but will also briefly highlight neurodegenerative disorders such as Alzheimer disease (AD) and Parkinson disease (PD), to further illustrate the breadth and nature of the emerging therapeutic opportunities. The current ‘toolbox’ of pharmacological agents that modulate lysosomal functions and emerging novel targets and strategies in this set of indications will be highlighted. It should be noted that therapeutic approaches to treat inflammatory and autoimmune diseases aim to inhibit the deleterious excessive lysosomal activity, whereas lysosomal activation would be the goal in the treatment of neurodegenerative diseases. Although beyond the scope of this review, such approaches may have applications in other diseases in which lysosomes may play a role, including cancer, metabolic diseases and ageing (reviewed elsewhere15,16).
Lysosomal biogenesis, structure and function
The formation of mature lysosomes is a complex process, which involves the fusion of late endosomes that contain material taken up at the cell surface with transport vesicles that bud from the trans-Golgi network5,8,17. These vesicles contain nearly 60 different hydrolytic enzymes (grouped into nucleases, proteases, phosphatases, lipases, sulfatases and others), which are synthesized in the endoplasmic reticulum and delivered to the transport vesicles via diverse systems, such as mannose-6-phosphate tags that are recognized by mannose-6-phosphate receptors (MPRs) at the membrane8,18 or glucocerebrosidase (GCase) that is transported to lysosomes by lysosomal integral membrane protein-2, an ubiquitously expressed type III transmembrane glycoprotein mainly located in endosomes and lysosomes19.
Mature lysosomes have an acidic internal pH, at which the lysosomal hydrolases are active, and a lining known as a glycocalyx that protects the internal lysosomal perimeter from the acidic environment of the lumen5,8,20. This acidic environment is maintained through the activity of a vacuolar-type proton adenosine triphosphatase (v-ATPase), which harnesses energy from hydrolysing ATP to drive the translocation of protons through a V0 membrane domain (reviewed elsewhere5,21). Other key lysosomal proteins include structural proteins such as lysosome-associated membrane protein 1 (LAMP1); proteins involved in trafficking and fusion, such as soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) and RAB GTPases; transporters such as LAMP2A, which has a key role in chaperone-mediated autophagy (CMA); and ion channels such as the chloride channel ClC7 and the cation channel mucolipin 1, a member of the transient receptor potential (TRP) family that is also known as TRPML1 (refs22,23). Most of the proteins are delivered through the clathrin adaptor protein 3-alkaline phosphatase (ALP) pathway, but some proteins are translocated through the lysosome-associated-protein transmembrane-5, a protein that is preferentially expressed in immune cells3,24.
Although the concept still remains controversial, two lysosome species — conventional or secretory — are often distinguished based on their physical, biochemical and functional properties. Catabolism is the main function of conventional lysosomes, and several other lysosome-related organelles (LROs), such as melanosomes, the late endosomal major histocompatibility complex class II (MHCII) compartment (MIIC), lytic granules from neutrophils, eosinophils, basophils, mast cells, CD8+ T cells and platelets, complement these functions8,25,26,27,28,29. Many of the LROs act as professional secretory organelles. LROs share with lysosomes the majority of typical characteristics (acidic environment, lysosomal transmembrane proteins, fusion property to phagosomes and others), in addition to particular properties resulting from their specific cargoes (for example, melanosomes contain melanosome-specific transmembrane glycoprotein, and natural killer cells and CD8+ T cells contain perforins and granzymes). The detailed mechanisms of biogenesis and secretion of LROs remain unclear, although it is known that genetic defects in LROs are involved in rare autosomal recessive disorders characterized by reduced pigmentation, such as Chediak–Higashi disease and Hermansky–Pudlak syndrome30. Secretory lysosomes contain many more proteins in addition to those contained in conventional lysosomes, and they participate in multiple cell functions such as plasma membrane repair, tissue and bone regeneration, apoptotic cell death, cholesterol homeostasis, pathogen defence and cell signalling8.
Lysosomal biogenesis and function are regulated by the basic helix–loop–helix leucine zipper transcription factor EB (TFEB) and the coordinated lysosomal expression and regulation (CLEAR) network4,31,32 (Fig. 2). For example, autophagy, a crucial process in immunity and autoimmunity33, is transcriptionally regulated by TFEB31. Interestingly, lysosomal exocytosis, which is important in many immune functions, also depends on TFEB activation31,32. Moreover, it has been demonstrated that TFEB orchestrates lysosomal Ca2+ signalling34. The fact that multiple lysosomal processes are dependent on TFEB activation strengthens its role as a master regulator in lysosomal functions. Like other transcription factors, TFEB undergoes phosphorylation and dephosphorylation via different cytosolic and lysosomal pathways (Fig. 2), processes regulated by mechanistic target of rapamycin complex 1 (mTORC1), a master controller of cell growth35,36.

After their synthesis in the rough endoplasmic reticulum (RER), the substrates (cargo) that are intended to be degraded through the endo-lysosomal pathway are transported to lysosomes via the trans-Golgi network (TGN). Among the key enzymatic systems that are involved in the lysosomal enzyme transportation of cargos from Golgi to lysosomes, the best studied is the mannose-6-phosphate (M6P) receptor (MPR) system, which binds newly synthesized lysosomal hydrolases in the TGN and delivers them to pre-lysosomal compartments. A few components synthesized in the late Golgi compartment are delivered directly to lysosomes via the 3-alkaline phosphatase (ALP) pathway. Lysosomal components, such as enzymes (lytic enzymes and kinases), membrane-bound proteins/complexes (mechanistic target of rapamycin (mTOR)), transporters and ion channels (vacuolar-type proton adenosine triphosphatase (v-ATPase), TRPML1 and osteopetrosis associated transmembrane protein 1 (Ostm1)) and chaperone-mediated transportation are the best-known targeting sites for lysosomal dysfunction. As depicted in the figure, many pharmacological antagonists and agonists exert activities that potentially correct lysosomal dysfunction and therefore represent potential effective pharmacological tools. CLEAR, coordinated lysosomal expression and regulation; CQ, chloroquine; HCQ, hydroxychloroquine; mTORC1, mTOR complex 1; PtdIns(3,5)P2, phosphatidylinositol-3,5-bisphosphate; RAPTOR, regulatory-associated protein of mTOR; SER, smooth endoplasmic reticulum; TFEB, transcription factor EB.
Lysosomes are at the crossroads of various degradative pathways, including endocytosis (phagocytosis) and autophagy (Fig. 1). Three main forms of autophagy have been described: macroautophagy (the most extensively characterized form), microautophagy and CMA. At the initiation of macroautophagy, a double-membrane sequestering compartment termed the phagophore, which contains cytoplasmic material, is formed and matures into a vesicle called the autophagosome. The cargo is degraded into vacuoles issued from the fusion of autophagic vesicles and lysosomes (called autolysosomes), and the resulting short products are released back into the cytosol for reuse or, according to sometimes contested observations, possibly dispatched into the MIIC for ultimate processing and MHCII molecule loading for presentation to CD4+ T cells37,38. In contrast to macroautophagy, microautophagy is characterized by direct lysosomal engulfment of cytosolic material into lysosomes, via the formation of characteristic invaginations of the lysosomal membrane. The third major form of autophagy is CMA, which involves the recognition of substrate proteins containing a KFERQ-like motif by a HSPA8/HSC70-containing complex (Fig. 1b). In CMA, two proteins have a key role: HSPA8 ensures the selectivity of proteins, which will be degraded via the CMA pathway; and LAMP2A translocates the targeted cytosolic proteins across the lysosomal membrane (reviewed elsewhere7). The terminal step of autophagy is called autophagic lysosome reformation, in which tubular proto-lysosomes are extruded from autolysosomes (containing lysosomal membrane components) and mature into functional lysosomes39. This step is not solely a lysosomal biogenesis process; it also includes a series of elements that are tightly correlated with the regulation of autophagy40.
In combination with autophagy, lysosomes are involved in both innate and adaptive immune functions, including foreign material recognition (bacterial, parasitic and viral), activation of pattern recognition receptors (such as Toll-like receptors (TLRs) and nucleotide oligomerization domain-like receptor), antigen processing and presentation, especially in the context of MHCII molecules, T cell homeostasis, antibody production and induction of various immune signals (co-stimulation and cytokine secretion)41. Besides being a degradative organelle, the lysosome has recently been recognized as a cellular signalling platform3,42. It plays an important role in nutrient sensing through mTORC1 and other additional protein complexes, or the so-called ‘lysosome nutrient sensing machinery’. The discovery of a stress-induced lysosome-to-nucleus signalling mechanism through TFEB further supports the key role of lysosomes in cellular signalling36.
Lysosome dysfunction in diseases
The lysosome occupies a central position in the maintenance of cellular homeostasis, being involved in the exclusion of infectious agents from penetrating host tissue and concomitantly promoting immune regulation. Lysosomes must therefore be able to respond quickly, with increased or decreased functions, to various metabolic conditions aimed at protecting cells from death or damage. Lysosomes are very diverse in size and shape. For reasons that are not totally understood — possibly according to their position in the cytosol43 and/or their composition — some lysosomes in a single cell are more prone to act and defend cells. Given the wide range of functions of lysosomes in all metabolic compartments of the cell, any dysregulation of their activity could lead to the impairment of various elements of the cellular metabolic machinery (including the transport and biogenesis of sugar (glycolysis), lipids, proteins and nucleic acids) and of metabolic pathways, phagocytosis, endocytosis and autophagy. Although the underlying mechanisms are far from being fully deciphered, it has been seen that lysosomal dysfunction or defects in fusion with vesicles containing cargo are commonly observed abnormalities in proteinopathic neurodegenerative diseases. Dysfunctions of lysosomes can affect the proper activity of other organelles such as peroxisomes and mitochondria, leading to excessive production of reactive oxygen species with pathological features associated with ageing, cancer, chronic inflammation, neurological diseases, male infertility and infections.
Such dysregulation is thus central to LSDs, and also implicated in a wide range of other disorders, including autoimmune and neurological disorders, in which the autophagy–lysosomal network under the control of TFEB has attracted considerable attention.
Lysosomal storage disorders
LSDs are a heterogeneous group of about 50 inherited metabolic disorders, which have an incidence of ~1 in 5,000 live births44. These disorders and their treatment have been reviewed extensively elsewhere45,46, and so will only be covered relatively briefly here. The mutations responsible for most LSDs have been largely elucidated (Tables 1,2), and many result in the dysfunction of a particular lysosomal hydrolase, leading to the accumulation of the substrate of that hydrolase. For example, in Gaucher disease, the sphingolipid glucocerebroside accumulates in cells (particularly macrophages) and organs, including the liver and spleen, owing to deficiency in the enzyme GCase24,66. In certain LSDs, the resultant pathology can be explained by the nature of molecules that accumulate (Tables 1,2). Thus, the abundance of cerebrosides and gangliosides that deposit in the central nervous system (CNS) of patients with sphingolipid storage disorders, such as type II (acute infantile neuronopathic) Gaucher disease, underlies the severe neurological symptoms of such disorders67,68. In patients with Pompe disease, which is caused by α-glucosidase deficiency, the high levels of non-degraded glycogen that accumulate in muscles could explain the observed myopathy69,70. However, how the undegraded material accumulates and causes the observed cellular and organ pathology in many other LSDs remains unclear.
The accumulation of such undigested macromolecules or monomers in LSDs instigates the formation of secondary products, which ultimately escape from the endosomal–autophagic–lysosomal pathways9,71 and lead to multiple consequences that affect most organs, including the brain, liver, spleen, heart, eyes, muscles and bone (Table 2). Most, if not all, organelles are altered in LSDs, including endosomes, autophagosomes and lysosomes, and their functions in lysosomal formation/reformation and fusion of endosomes or autophagosomes to lysosomes are abnormal. Alterations in several autophagy processes have also been described in LSDs. Thus, deregulated mitophagy, which results in the accumulation of damaged mitochondria, occurs in LSDs, leading to major inflammatory consequences in specific tissues67,72. Perturbations in mitochondrial dynamics are frequently observed, which have been linked to the increased production of reactive oxygen species, ATP production and Ca2+ imbalance. In LSDs, reduced macroautophagy activity (with a decreased autophagic flux) rather than hyperactive autophagy processes, as seen in numerous autoimmune diseases, seems to be responsible for the accumulation of non-degraded cytoplasmic proteins such as α-synuclein, huntingtin (HTT) and others73. Mucolipidosis type IV (Table 2), a disease characterized by severe neurological and ophthalmological abnormalities, is caused by mutations in the MCOLN1 gene and is inherited in an autosomal recessive manner. This gene encodes a non-selective cation channel, mucolipin 1, which has recently been shown to be required for efficient fusion of both late endosomes and autophagosomes with lysosomes74,75. Impaired autophagosome degradation results in the accumulation of autophagosomes in LSDs76. Microautophagy processes that do not involve de novo synthesis of nascent vacuoles also appear to be impaired in LSDs, and were notably revealed in primary myoblasts from patients with the muscle-wasting condition Pompe disease77. Finally, defective CMA components, such as LAMP2A, could also lead to lysosomal dysfunction. For example, mutations in the LAMP2 gene have been claimed to cause Danon disease (inherited in an X-linked dominant pattern)51. Further investigations are needed to support this assertion.
Autoimmune disorders
Lysosomes are involved in pathways central to the immune system, including the degradation of intracellular and extracellular material, plasma membrane repair, cell death signalling, cell homeostasis and death. Although the direct involvement of lysosomes in immunity is far from fully understood, it has long been expected that lysosome dysfunction will have a major impact in immune diseases (Table 2). Strikingly, however, this field has not been extensively explored. However, elevated levels of lysosomal enzyme activity have been reported to occur in several autoimmune diseases, such as RA, systemic lupus erythematosus (SLE), dermatomyositis and psoriasis3,14,17,18,20,21,22,23.
As discussed, autophagosomes formed during the autophagy process must fuse with lysosomes to generate peptide epitopes for further processing, clear possibly deleterious apoptotic debris, fuel the amino acid pool and produce energy (Fig. 1). Any deviation in this complex processing will affect crucial immune cell functions, such as the control of cytokine release, autoimmune cell anergy and programmed cell death of type I (apoptosis) and type II (autophagy). Secretory lysosomes regulate the release of both pro-inflammatory and anti-inflammatory cytokines, in a process that is dependent on the type of stimulation. In addition, lysosomes degrade glucocorticoid receptors, which are essential to bind glucocorticoids, although the reasons are not known78. In this complex system, lysosomes execute anti-inflammatory action via the phospholipase A2 and cyclooxygenase-2 pathways, and also induce inflammation through the IL-1β–caspase-1 pathway. In both conditions (pro-inflammatory and anti-inflammatory), lysosomes act as indirect precursors for autoimmunity. However, induction and suppression of inflammatory signals are stimulus dependent78.
Lysosomal cathepsins have a central role in degrading biological macromolecules in the lysosomes and in the immune response. There are approximately 12 members in this large protease family, most of which are endopeptidases that can cleave peptide bonds of their protein substrates79,80. Cathepsins A and G are serine proteases, cathepsins D and E are aspartic proteases and cathepsins B, C, F, H, K, L, O, S, V, X and W are cysteine proteases. For example, cathepsin S is responsible for the degradation of antigens (and autoantigens) in antigen-presenting cells (dendritic cells, macrophages and B cells), and is therefore involved at an upstream level in the presentation of MHCII–(auto)antigenic peptide complexes to CD4+ T cells81. Cathepsin L preferentially cleaves peptide bonds with aromatic residues in the P2 position and hydrophobic residues in the P3 position. It is central in antigen processing, bone resorption, tumour invasion and metastasis, and turnover of intracellular and secreted proteins involved in growth regulation. Cathepsin L-deficient mice display less adipose tissue, lower serum glucose and insulin levels, more insulin receptor subunits, more glucose transporter type 4 and more fibronectin than wild-type controls82. Cathepsin G is primarily known for its function in killing and digestion of engulfed pathogens83. It is also involved in connective tissue remodelling at sites of inflammation84. Anti-neutrophil cytoplasmic antibodies reacting with cathepsin G have been identified in some patients with SLE85.
Lupus
Abnormal antigen processing and presentation is known to be one of the upstream events that perturb immune responses in SLE86. Because this process is mediated through lysosomes, it was rational to speculate that lysosomal functions could be altered in lupus. Interestingly, hypotheses were raised in the 1960s on the ‘lysosomal fragility’ in lupus, but without much further pursuit87. The composition and fluidity of the lysosomal membrane are effectively crucial in the regulation of lysosomal fusion with other vesicular organelles and for lysosomal uptake of macromolecules. The integrity of the lysosomal membrane also ensures the prevention of release of lysosomal enzymes into the cytoplasm. Some lysosomal enzymes released from ‘fragile’ lysosomes were regarded potentially harmful in lupus88.
Lysosomes are abnormal in splenic B cells from Fas-deficient Murphy Roths Large (MRL)/lpr mice, a mouse model of lupus, compared with B cells from healthy CBA/J mice89. TFEB expression was increased, indicating an enhanced biogenesis of lysosomes, and the lysosomal volume was raised. The expression levels of LAMP1 and cathepsin D were also increased. These results reinforce previous data showing that the expression and activity of some lysosomal enzymes (such as cathepsins S, L and B) that play important roles in antigen processing are altered in lupus and other autoimmune diseases90,91.
Substantial variations of the acidic endo-lysosomal pH also occur in MRL/lpr mice, being raised by 2 pH units in splenic B cells53,92. This pH change could dramatically influence the activity of soluble lysosomal hydrolases (such as cathepsins) as well as lysosomal membrane proteins (such as LAMPs) that are critical for lysosome activity. pH may also affect the elimination of immune complexes that accumulate in lupus as a result of deficits in complement, lower expression of scavenger receptors, increased expression of Fcγ receptors and other reasons93. These immune complexes, which contain non-selective IgG antibodies or autoantibodies associated with autoantigen (including some apoptotic debris), can initiate inflammation of tissues once deposited (for example, in the kidneys and the skin) and generate a cascade of deleterious effects, such as the release of harmful cytokines and chemokines54.
Recent studies have highlighted the key role of mammalian target of rapamycin complex 2 (mTORC2) in the disruption of lysosome acidification that occurs in this process94. In normal conditions, the regulation of lysosomal acidification requires cleavage of the RAB small GTPase RAB39a, occurring on the surface of phagocytic vesicles by locally activated caspase-194. This finely regulated process requires the association of cofilin with actin that surrounds the vesicle and recruits caspase-11, which then activates caspase-1 (ref.94). In lupus-prone macrophages, chronically active mTORC2 enhances cofilin phosphorylation, thereby hampering its association with actin and affecting the downstream cascade of events leading to the appropriate acidification of lysosomes94. The importance of mTORC1 and mTORC2 has been established earlier in lupus T cells, and in particular, in this context, mTORC1 activity was increased whereas mTORC2 activity was reduced95.
In addition, lysosomal cathepsin K was seen to contribute to the pathological events that develop in Faslpr mice, another model of lupus disease, in part through its activity in TLR-7 proteolytic processing and subsequent effects on regulatory T cells. Cathepsin K-deficiency in Faslpr mice reduced all kidney pathological manifestations (glomerulus and tubulointerstitial scores, glomerulus complement C3 fraction and IgG deposition, chemokine expression and macrophage infiltration) and decreased the levels of potentially pathogenic serum autoantibodies96.
In line with these internal alterations of lysosomes, notably those related to cathepsin functioning, deregulation of autophagy has been reported to contribute to lupus pathology92,97,98,99,100. Autophagy failures have been described in the lymphocytes of MRL/lpr mice and (NZBxNZW)F1 mice56,92,97,101 (two spontaneous murine models of systemic autoimmunity of distinct genetic origins and that display different MHC haplotypes) as well as in T and B lymphocytes of patients with SLE97,98,100. Murine and human T cells from the peripheral blood showed a significant accumulation of autophagic vacuoles compared with normal97. The underlying reasons for the dysfunctions in autophagy observed in lupus are not clearly understood, but several independent investigations have identified risk loci spanning autophagy-linked genes in patients with lupus102,103,104,105,106.
Sjögren’s syndrome
Recent studies have demonstrated an increase in the level of macroautophagy in salivary gland T lymphocytes and in tears and conjunctival epithelial cells of patients with primary Sjögren’s syndrome (SjS)107,108. Alteration of CMA activity was also recently found to occur in the salivary glands of MRL/lpr mice that develop a secondary SjS-like disease56. Lysosomes, which as discussed are mechanistically involved at the downstream level of both macroautophagy and CMA, were found to be altered in salivary glands. Flow cytometry analyses revealed that the mean pH of acidic vesicles in MRL/lpr salivary glands was significantly higher compared with those in mouse control glands and the ATP content was significantly diminished in MRL/lpr salivary gland cells56. Furthermore, amounts of several leukocyte glycosidases and proteases were revealed to be increased in leukocytes of patients with SjS in comparison with healthy controls55. Notably, raised levels of the lysosomal enzymes glucosidase, β-glucuronidase and dipeptidyl peptidase I are involved in the tissue injury in SjS55. Increased expression of lacrimal gland cathepsin S was also reported, which may have application as a diagnostic tool in SjS91. Two members of the RAS oncogene family, RAB3D and RAB27, were found to be implicated in the regulation of cathepsin S secretion levels in SjS109. In vitro studies on lacrimal gland acinar cells suggested further that secreted IFNγ from acinar cells increases cathepsin S expression and that IFNγ stimulated the MHCII-mediated antigen presentation in ocular pathogenesis of SjS110.
Rheumatoid arthritis
Lysosomal cathepsins have important roles in the induction and diagnosis of RA, and levels of several cathepsins (B, D, G, K, L and S) that are present in the serum and synovial fluid of patients have been proposed as a basis for RA diagnosis111,112,113,114,115,116. Cathepsin S and cathepsin L are highly expressed in synovial macrophages and thymic cortical cells. They each exert essential roles in the positive selection of T cells and antigen presentation, respectively, and participate in the local inflammation and matrix degradation that occurs in joints116. Cathepsin B is involved in collagen degradation, which leads to joint destruction in RA112,117. Expression of cathepsin G, which participates in joint inflammation through its chemoattractant activity, has been shown to be raised in the synovial fluid of patients with RA when compared with individuals with osteoarthritis115. Autoantibodies reacting with cathepsin G were also identified in patients with RA85. Compared with patients with osteoarthritis, cathepsin K expression was found to be elevated in RA113, and genetic deletion of this particular cathepsin was shown to reduce inflammation and bone erosion in RA conditions via TLR mediation118.
Neurological autoimmune diseases
MS, myasthenia gravis, Guillain–Barré syndrome, chronic inflammatory demyelinating polyneuropathy (CIDP), neuromyelitis optica and neuropsychiatric lupus are neurological diseases induced by abnormal autoimmunity62,119,120,121,122,123. Neurological autoimmunity against various proteins, such as myelin in MS or N-methyl-d-aspartate receptor in neuropsychiatric lupus62,123,124, can affect various structures within the CNS and peripheral nervous system, with diverse consequences. Although the exact cause of amyotrophic lateral sclerosis (ALS) still remains unknown, studies support the existence of autoimmune mechanisms, and ALS is therefore also included in this section. Indeed, autoantibodies against ganglioside GM1 and GD1a, sulfoglucuronylparagloboside, neurofilament proteins, FAS/CD95 and voltage-gated Ca2+ channels have all been reported in patients with ALS (reviewed elsewhere125).
In general, the origin of the breakdown in immune tolerance that occurs in this set of neurological diseases is not known. Only recently have investigations discovered that autophagy processes are altered in some of these diseases59,62,126,127,128,129,130. In MS and in experimental autoimmune encephalomyelitis, an experimental model of MS, upregulation of the protein kinase mTOR has been described, and treatment with rapamycin/sirolimus (an immunosuppressant that inhibits mTOR and consequently stimulates macroautophagy) ameliorates some clinical and histological signs of the disease131. Increased levels of macroautophagy markers were measured in the blood and brain of patients with MS122,132. However, impaired macroautophagy was found in the spinal cord of experimental autoimmune encephalomyelitis mice133. In a rat model mimicking human CIDP, both macroautophagy and CMA processes were found to be hyperactivated in lymphatic system cells and non-neuronal cells (sciatic nerves) of peripheral nervous system cells59. In ALS, current data are conflicted62. Some data suggest an activation of macroautophagy processes with an accumulation of autophagosomes in brain tissues of patients with ALS, or an increase of autophagic vacuoles, aggregated ubiquitin and SOD1 proteins associated with MAP1LC3B-II in motor neurons of mice developing an ALS-like disease134,135. In contrast, other data suggest a reduction of autophagy activity136,137. Mutations in SQSTM1, valosin-containing protein, dynactin (a protein complex that activates the dynein motor protein, enabling intracellular transport) and RAB7 (a member of small GTPases that is important in the process of endosomes and autophagosomes maturation) have also been described in ALS138,139,140,141. Further studies are required to better understand the type and extent of autophagy dysfunction in this family of complex diseases.
There are only a few published studies on lysosomal dysfunction in neurological autoimmune diseases (Table 2). These notably include lysosome fragility, which was observed in patients with MS in the white matter of cerebral tissue, an area of the CNS that is mainly made up of myelinated axons142. Lysosome fragility was also suspected in SLE (see above) and other rheumatic autoimmune diseases, albeit in other organs53,58,92. As noted above, significant variations in lysosomal pH have been measured in autoimmune conditions such as lupus and SjS, but to our knowledge such studies conducted in the brain or elements of the peripheral nervous system of patients or animal models with neurological autoimmune diseases have not been published78.
In CIDP, it has been shown that Schwann cells dedifferentiate into immature states and that these dedifferentiated cells activate lysosomal and proteasomal protein degradation systems143,144. Based on these observations, Schwann cells have been claimed to actively participate in demyelinating processes via this dedifferentiation process, but the mechanism involved remains undefined145. In the rat model of CIDP mentioned above, it was shown that LAMP2A expression was drastically increased in the sciatic nerve macrophages and reduced macroautophagy was observed in Schwann cells and macrophages59.
In MS, studies conducted on white matter demonstrated that lysosomes are involved in myelin sheath degeneration as well as in the fragmented protein formation. Lysosomal swelling was observed near the degenerated materials of astrocytes146, and an accumulation of lipids was found60. It has been hypothesized that lysosomal swelling/permeabilization might cause the release of hydrolases in the cytosol, where they affect native proteins147.
In ALS, patients also show dysfunctions in the endo/lysosomal pathways, which affect both lower and upper motor neurons (Table 2). Cathepsin B was particularly found to be involved in the motor neuron degeneration, whereas cathepsins H, L and D were not significantly affected148. A cDNA microarray analysis on post-mortem spinal cord specimens of four sporadic patients with ALS revealed major changes in the expression of mRNA in 60 genes including increased expression of cathepsins B and D149. Several disease-causing mutations in genes related to autophagy have been identified, such as SOD1, TDP643, FUS, UBQLN2, OPTN, SQSTM1 and C9orf72 (refs61,150), but none of them code for lysosomal proteins. So, a crucial remaining issue is to clearly determine whether the lysosomal abnormalities that are observed are linked to intrinsic defaults of lysosomes or result from upstream dysregulation in autophagosome formation and fusion61,62,151.
Neurodegenerative disorders
Insufficient clearance of neurotoxic proteins by the autophagy–lysosomal network has been implicated in numerous neurodegenerative disorders152. In disorders such as AD, Huntington disease (HD) and PD, modified or misfolded proteins abnormally accumulate in specific regions of the brain. Accumulation of aggregated proteins is also seen in ALS (see above). These abnormal proteins form deposits in intracellular inclusions or extracellular aggregates, which are characteristic for each disease153,154,155. Although there has been substantial research in this field, it is still unclear why sophisticated ‘quality-control’ systems, such as the lysosome–autophagosome system in particular, fail in certain circumstances to protect the brain against such protein accumulation156.
In AD, one of the most common neurodegenerative disorders, some alterations in the endo/lysosomal pathways have been described (reviewed elsewhere157,158). The amyloid precursor protein (APP) is cleaved by β- and γ-secretases into amyloid-β peptide (Aβ) fragments, particularly Aβ40 and Aβ42 (ref.159). These fragments are found in the amyloid plaques that are one of the hallmarks of AD (the other being neurofibrillary tangles containing phosphorylated tau), and have been widely considered to have an important role in AD pathogenesis159,160. Cell-based experiments have demonstrated that lysosomal cathepsins have a role in the generation of Aβ peptides (through cathepsins D and E) and the degradation of Aβ peptides (by cathepsin B)161. Lysosomal dysfunction has been observed in patients with AD162,163, and accumulation of the Aβ42 fragment in neuronal cells was shown to lead to lysosomal membrane alterations, which cause neuronal cell death63. In this context, it is noteworthy that inhibition of cathepsin D, which is involved in the lysosomal dysfunction and notably in the cleavage of the tau protein into tangle-like fragments, diminishes its hyperphosphorylation in the brain of patients with AD164. In addition, patients with AD with an inherited form of the disease may carry mutations in the presenilin proteins (PSEN1 and PSEN2), APP or apolipoprotein E, resulting in increased production of the longer form of the Aβ fragment (reviewed elsewhere165). Mutation of PSEN1, for instance, leads to direct disruption of the lysosomal acidification due to impaired delivery of the V0A1 subunit of v-ATPase, a proton pump responsible for controlling the intracellular and extracellular pH of cells. The acidification deficit causes excessive release of lysosomal Ca2+ through TRPML1 channels, which has numerous deleterious effects166. These findings strongly support the hypothesis that dysfunction of endo/lysosomal pathways is pivotal in AD.
Approximately 15% of patients with PD have a family history of the disorder, although the underlying molecular mechanisms remain unclear. In the context of lysosomal dysfunction, it is notable that the most common of the known PD genetic mutations are in GBA1 (encoding the lysosomal β-GCAse) — the same gene that underlies Gaucher disease — which are present in up to 10% of patients with PD in the United States167. GBA1 mutations are also associated with dementia with Lewy bodies167. Several other genes linked to PD are directly or indirectly related to the endo/lysosomal machinery, such as mutations in SNCA (coding for α-synuclein)63,168. A hallmark of PD is the presence in neurons of protein inclusions called Lewy bodies, which are mainly composed of fibrillar α-synuclein. The α-synuclein protein is normally degraded by the lysosomes through the CMA pathway, but macro-aggregates of α-synuclein mutants, which display a longer half-life compared with the non-aggregated wild-type protein, are not degraded by this pathway and, rather, would be degraded via the macroautophagy pathway169,170,171,172. It was further shown that the mutant proteins bind to LAMP2A and inhibit the translocation of other substrates and, therefore, their final degradation170. Biochemical analyses suggest that α-synuclein is mainly degraded by lysosomal proteases and notably by cathepsin D, rather than by non-lysosomal proteases (for example, calpain I)173,174. Accumulation of α-synuclein was observed in cathepsin D-deficient mice, whereas, conversely, the accumulation of α-synuclein aggregates was reduced in transgenic mice that overexpressed this cathepsin, resulting in protection of dopaminergic neuronal cells from damage175.
HD is a rare autosomal-dominant neurodegenerative disease caused by an aberrant expansion of CAG trinucleotide repeats within exon 1 of the HTT gene, which results in the production of aggregation-prone HTT mutants (mHTT) that are detrimental to neurons176,177. Whereas HTT has a protective role against neuronal apoptosis, accumulation of mHTT, however, induces pathophysiological consequences including lysosomal and autophagy dysfunctions. Thus, mHTT perturbs post-Golgi trafficking to lysosomal compartments by delocalizing the optineurin/RAB8 complex, which, in turn, affects lysosomal function177. Excessive mHTT induces accumulation of clathrin adaptor complex 1 in the Golgi and an increase of clathrin-coated vesicles in the vicinity of Golgi cisternae177. The activity of several cathepsins such as B, D, E, L and Z has also been linked to HD63,80,174,177,178,179. Cathepsin D is responsible for full degradation of HTT but is less efficient at degrading mHTT, which is processed by cathepsin L180,181. Cathepsin Z also cleaves HTT and elongated polyglutamine tracts182,183. Thus, lysosomal modulators acting on cathepsin activity might have beneficial effects in the treatment of HD. Notably, hyperexpression of cathepsin D (and cathepsin B) was shown to protect primary neurons against mHTT toxicity179. Alterations in macroautophagy, mitophagy and CMA have also been implicated in HD184,185. CMA activity was increased in response to macroautophagy failure in the early stages of HD186, a result supported by the findings that HSPA8 and LAMP2A have important roles in the clearance of HTT187 and that shRNA-mediated silencing of LAMP2A increased the aggregation of mHTT188. Other studies focusing on the HTT secretory pathway revealed that mHTT secretion is mediated by the Ca2+-dependent lysosomal exostosis mechanism via the synaptotagmin 7 sensor in neuro2A cells189. The extracellular release of mHTT was efficiently inhibited by the phosphoinositide 3-kinase and sphingomyelinase inhibitors Ly294002 and GW4869. HD-dependent perinuclear localization of lysosomes was also demonstrated190.
Increasing evidence thus implicates lysosomal (and autophagy) dysfunction in the pathogenesis of neurodegenerative disorders62,63,127,128,130,191,192. TFEB has received particular attention in this regard193,194,195, with recent data suggesting that TFEB is selectively lost in patients with AD (as well as ALS)196. Increasing TFEB activity might therefore prevent neuronal death and restore neuronal function in certain neurodegenerative diseases, including PD194.
Lysosomes as therapeutic targets
Given the evidence discussed above, the various lysosomal pathways and their components could represent potential pharmacological targets for a wide range of diseases. When considering lysosomes as targets, it is important to note the need for specificity; that is, agents that will not target all lysosomes, but will specifically target those lysosomes/lysosomal proteins that are defective in certain organs, tissues or cells. In addition, inhibitors or activators of lysosomal components may be required, depending on the disease context.
There has been considerable interest in therapeutically targeting different autophagy pathways, including lysosome-dependent pathways, and progress in the discovery and development of small molecules and biologics that target these processes has been reviewed extensively11,119,120,121,122,197,198. However, very few therapies that specifically target lysosomal components have so far been generated and found to be effective in clinical trials, with one general exception — the development of ERTs and small-molecule drugs for LSDs (Box 1). This topic has recently been comprehensively reviewed46 and so will not be discussed in depth here.
It is important to target lysosomes and not the whole autophagy process for several reasons. First, regarding safety, the integral role of lysosomes in several key physiological processes means that therapeutic windows for pharmacological intervention with unacceptable side effects may be limited. For example, azithromycin, an antibiotic with anti-inflammatory properties that is used in the treatment of patients with chronic inflammatory lung diseases such as cystic fibrosis, was found to block autophagy in macrophages, inhibiting intracellular killing of mycobacteria within them and, thereby, increasing the risk of mycobacterial infection204. Second, in some diseases, autophagy may be enhanced in certain tissues or organs but compromised in others, for example in the spleen and salivary glands of MRL/lpr mice56. This phenomenon makes it highly challenging to identify a single drug able to correct a failure, unless a cell-specific targeting molecule could be incorporated into the autophagy activator/inhibitor to enable tissue specificity205. Again, the precise targeting of lysosomes in specialized cells may circumvent the complexity of dysregulation mechanisms of autophagy processes in pathophysiological settings14,56,206,207.
As indicated, the current arsenal of lysosome-specific targeted drugs is small. In fact, many drugs claimed to target lysosomal components have also been found to be capable of interacting with several non-lysosomal receptors, limiting their efficacy and safety12. One example is provided by chloroquine (CQ), a 4-aminoquinoline compound, and its derivative hydroxychloroquine (HCQ), which are widely prescribed to patients with rheumatic diseases, and historically also for the prophylaxis and treatment of malaria (Fig. 3). CQ and HCQ are lysosomotropic agents and as such they raise intralysosomal pH, thereby affecting overall lysosomal function and impairing autophagic protein degradation (Fig. 2). Although the mechanism of action of these agents is not fully elucidated, it is well established that CQ and HCQ display pleiotropic activity208,209,210 and have important deleterious properties. In certain settings, they have been claimed to operate by interacting directly with TLR ligands and not through an effect on the lysosomal pH, for example211. Toxicity of CQ/HCQ, in particular in the eye (cornea and macula) and the occurrence of cardiomyopathies212, remains a major limitation. The observed ocular toxicity is related to the total cumulative dose rather than the daily dose; therefore, it becomes a serious potential problem in the cases of long-term use. Several HCQ analogues and mimics have been designed that aim to retain the therapeutic activity without secondary effects213,214.

Small molecules and peptides highlighted in this figure are activators and inhibitors of lysosomal constituents targeting mechanistic target of rapamycin (mTOR), vacuolar-type proton adenosine triphosphatase (v-ATPase), TRPML1, PIK kinase and HSPA8. For details, see the text and accompanying tables.
Furthermore, most, if not all, of the small molecules that have so far been identified and investigated as modulators of autophagy and/or lysosomal functions exhibit complex pleiotropic properties affecting the overall function of lysosomes, and also different autophagy pathways (for example, mTOR-dependent and mTOR-independent pathways), as well as other quality-control mechanisms that affect the cell life/death balance. As discussed below, several widely used molecules exert dual, sometimes opposite, effects on upstream and downstream molecular events of the autophagy–lysosomal network.
Several robust assays to characterize autophagy activators and inhibitors, as well as lysosomal effectors, are currently available and validated (Table 3). However, each assay has inherent biases, and so it is necessary to use several independent, in vitro and in vivo approaches to ascertain the reactivity and specificity of novel molecules able to modulate these pathways (Box 2).
In this regard, the tremendous work in recent years to establish international guidelines for standardizing research in autophagy — and, in particular, to propose relevant methodologies for monitoring autophagy that are accepted by the whole community — is unique231,232. A better definition of terms and concepts has also been adopted by the community, leading to much easier understanding between researchers worldwide233. These guidelines and definitions should be used by investigators evaluating new molecules designed to selectively target key steps of autophagy or developing new high-throughput screening methods for autophagy-modulating pharmacological molecules. However, even the more sophisticated and detailed assays will not recapitulate the full complexity of integrated living systems, which can only be established in clinical trials.
Pharmacological regulators of lysosomal activity
The pipeline of specific agonists and antagonists of autophagic activity is currently small, particularly for CMA (Tables 4,5; Figs 2,3). However, high-throughput screening programmes to identify such small molecules are ongoing, which should yield additional therapeutic targets and useful tools. Small molecules that specifically target lysosomes are even rarer (Table 4; Fig. 2). Small-molecule drugs developed specifically for particular LSDs, including substrate reduction therapies and small-molecule chaperones, have reached the market, but other small-molecule candidates for more common diseases are at an earlier stage of development. These molecules that more specifically act on lysosomes, some of which have been discovered by high-throughput screening, mostly target LAMP2A, various lysosomal enzymes such as cathepsins, acid sphingomyelinase, α-galactosidase A and acid β-glucocerebrosidase, and chaperones such as HSPA8 and β-N-acetyl hexosaminidase. Although not solely present in lysosomes, v-ATPase, a proton pump responsible for controlling the intracellular and extracellular pH of cells, and TRPML1, a cation channel located within endosomal and lysosomal membranes, are also pertinent targets.
Below and in Table 4, we summarize the availability of pharmacological tool compounds and progress in drug development, where applicable, for each broad target class.
Substrate reduction therapies and small-molecule chaperones
In addition to ERTs for LSDs (Box 1), drug discovery programmes have also focused on alternative small molecule-based approaches, which may be particularly relevant for LSDs that affect the CNS, due to the lack of blood–brain barrier penetration by ERTs283.
Small molecules used in substrate reduction therapies prevent the accumulation of substrates of the defective enzymes in LSDs by inhibiting enzymes involved in substrate production284. Miglustat was the first such drug to be approved in the early 2000s by the US Food and Drug Administration and the European Medicines Agency for Gaucher disease and in 2009 for Niemann–Pick disease type C in Europe. This iminosugar inhibits glucosylceramide synthase (GCS), which catalyses the initial step in formation of many glycosphingolipids. Within cells, glycosphingolipids tend to localize to the outer leaflet of the plasma membrane; they cycle within the cell through endocytic pathways that involve the lysosome. Inhibition of GCS therefore reduces the deleterious accumulation of glycosphingolipids within lysosomes with potential therapeutic benefits in diseases like LSDs. Miglustat also inhibits disaccharidases in the gastrointestinal tract, resulting in diarrhoea as a side effect285. Eliglustat, another GCS inhibitor that does not penetrate the CNS, was also approved for Gaucher disease in 2014. Other GCS inhibitors in clinical development include lucerastat, a miglustat analogue with an improved safety profile that is currently in a phase III trial for Fabry disease (FD)236,286, and ibiglustat, which penetrates the CNS. The latter is in clinical development for FD (phase II), for Gaucher disease type 3 (phase II) and for patients with PD who carry a mutation in GBA (phase II). Recent findings generated in a small number of patients have suggested a possible link between PD and FD287, which also exists between patients with PD and Gaucher disease who have GBA mutations (see above). Finally, genistein, a pleotropic natural product that inhibits kinases involved in the regulation of proteoglycan biosynthesis and also affects TFEB function, is in a phase III trial for Sanfilippo syndrome288.
Substrate mimetics that inhibit lysosomal enzymes have also been found to stabilize mutated enzymes in LSDs, thereby leading to restoration of some enzyme activity when suitable subinhibitory concentrations are used, as the enzyme remains stable and functional after dissociation of the inhibitor46,283. The pioneering example of this approach is migalastat, described above, that binds to the active site of α-galactosidase A, which is mutated in FD, and stabilizes the mutant enzyme. Other examples of this strategy include afegostat in Gaucher disease (which failed in a phase II clinical trial in 2009 due to lack of efficacy), pyrimethamine in Sandhoff disease and Tay–Sachs disease, and ambroxol in Gaucher disease with neurological symptoms (Table 4). Agents that are at earlier developmental stages include N-octyl-β-valienamine, a competitive inhibitor of β-glucosidase, for Gaucher disease; N-acetylcysteine for Pompe disease; α-lobeline, 3,4,7-trihydroxyisoflavone and azasugar in Krabbe disease; and N-octyl-4-epi-β-valienamine and 5N,6S-(N′-butyliminomethylidene)-6-thio-1-deoxygalactonojirimycin indicated in GM1 gangliosidosis289. The chemical structures of these pharmacological chaperones have been described recently290,291. Finally, an alternative strategy for stabilizing mutant enzymes, by binding away from the active site, is also being investigated. A promising example of this approach is NCGC607, a non-inhibitory small-molecule chaperone of GCase discovered by screening for molecules that improved the activity of the mutant enzyme46,250. Treatment with NCGC607 reduced lysosomal substrate storage and α-synuclein levels in dopaminergic neurons derived from induced pluripotent stem cells from patients with Gaucher disease with parkinsonism46,250. Further testing of NCGC607 in patients with PD and GBA mutations is awaited. Although promising, conflicting viewpoints still remain on the strength of such small molecule-based approaches, primarily because these compounds bind to the catalytic site of enzymes, which may be a risk at high concentrations if they inhibit rather than increase activity291,292. More clinical trials are therefore required in order to analyse the robustness of this approach.
Cathepsin modulators
Robust genetic and pharmacological preclinical investigations have consistently showed that regulating cathepsin activity can favourably improve pathological features in certain autoimmune and inflammatory diseases. Inhibitors of several cathepsins (B, D, L, K and S) have been described174,293 and their activity has been evaluated in rheumatic autoimmune diseases (such as SLE, RA and SjS) and neurodegenerative disorders, notably in AD294 (Table 4). Selective inhibition of cathepsin S with a potent active site inhibitor known as RO5461111 (Roche) mitigated disease in MRL/lpr lupus-prone mice, by reducing priming of T and B cells by dendritic cells, and plasma cell generation262. Promising data have also been generated in murine models, in the context of diabetic nephropathy and cardiovascular diseases295. Further studies based on cathepsin S inhibitors should evaluate the clinical safety and utility of treating patients affected by autoimmune and inflammatory diseases295. Cathepsin K, which is highly expressed by osteoclasts and very efficiently degrades type I collagen, the major component of the organic bone matrix, is also a potential target for modulating lysosomal dysfunction in some of the disorders discussed above, such as SLE96. Yet further investigations with selective cathepsin K inhibitors are required to determine whether this targeted strategy might apply in SLE and other inflammatory conditions in which articular manifestations are a major component (RA, ankylosing spondylitis, psoriatic arthritis and others). It should be noted, however, that various cathepsin K inhibitors have been pursued for postmenopausal osteoporosis, including odanacatib (Merck) which reached phase III trials296. Although odanacatib was effective, its development was discontinued in 2016 due to an increased risk of stroke in treated patients. Other cathepsin inhibitors and their context of clinical evaluation are listed in Table 4.
Despite multiple efforts to develop selective pharmacologic cathepsin modulators, important concerns still remain with regard to off-target effects due to activity against other cathepsins or towards cathepsins present at non-relevant or unwanted sites. Nonetheless, the underlying biology and clinical effects of certain cathepsin inhibitors or activators remain of considerable interest and could guide future therapeutic approaches.
v-ATPase inhibitors
As reported below, v-ATPase, a multisubunit ATP-driven proton pump, is best known for its role in acidification of endosomes and lysosomes. Regulating the function of v-ATPase may impact lysosomal activity and, hence, the acidification of specialized cells and diverse signalling pathways, such as autophagy. v-ATPase inhibitors like bafilomycin A1 and concanamycin A are non-selective compounds (Table 4; Fig. 3) that inhibit both mammalian and non-mammalian v-ATPases, which control the lysosomal pH of acidic vesicles via a manner that is not fully understood (Fig. 2). Through this mechanism, bafilomycin A1 inhibits autophagic flux by preventing the acidification of endosomes and lysosomes297. Bafilomycin and CQ also affect mitochondrial functions, as discovered recently using intact neurons298. Benzolactoneenamides (salicylihalamide A, lobatamides and oximidines; Table 4; Figs 2,3) are much more selective v-ATPase inhibitors299 than bafilomycin A1 and concanamycin A, but also much less potent. Further investigations into v-ATPase regulation of signalling pathways are needed to identify specific and safe molecules that regulate this vital proton pump300.
Ion channel modulators
As discussed above, lysosomal ion channels are master elements of lysosome activity and, thereby, of cell homeostasis. In the family of TRP channels, TRML1 is essential, being widely expressed in late endosomes and lysosomes, and preferentially associates with LAMP1 in the lysosomal membrane22,301,302. Genetic mutations leading to inactivation of TRPML1 cause a rare genetic disorder called mucolipidosis type IV (MLIV). Pharmacological activation of TRPML1 ameliorated some lysosomal functions that are classically associated with MLIV, NPCs and certain LSDs (Tables 2,4; Fig. 2). Thus, the small molecule SF-22 (Fig. 3), which was identified in a screen for TRPML3 activators, was defined as an activator of both TRPML3 and TRPML1 (ref.274), and displayed an additive effect in combination with the endogenous activator phosphatidylinositol-3,5-bisphosphate (PtdIns(3,5)P2)274,303. An analogue of SF-22, in which chlorine on the thiophene had been replaced by a methyl group, showed greater efficacy on TRPML1 activation303,304. Another molecule called ML-SA1 (Figs 2,3), acting as a mucolipin synthetic agonist, also showed an additive effect with endogenous PtdIns(3,5)P2 on TRPML1 channels305. It is important to note that in neurological diseases, as well as in other indications in which lysosomal acidification is defective (see above), interfering with TRML1 may have contraindications.
Phosphatidylinositol kinase modulators
A central modulator of lysosomes is the lipid kinase FYVE finger-containing phosphoinositide kinase (PIKfyve), which converts phosphatidylinositol-3-phosphate into PtdIns(3,5)P2. The latter regulates Ca2+ release from the lysosome lumen and is required for acidification by v-ATPase. Inactivation of PIKfyve leads to many pathophysiological problems including neurodegeneration and immune dysfunction, mostly related to impaired autophagic flux and alteration of lysosomes (trafficking, Ca2+ transport, biogenesis and swelling)306. The small-molecule apilimod (Fig. 3; Table 4) was originally identified as an inhibitor of TLR-induced IL-12 and IL-23, and later found to be a highly specific inhibitor of PIKfyve276. Apilimod was evaluated in clinical trials involving several hundred patients with T helper 1 and T helper 17 cell-mediated inflammatory diseases such as Crohn’s disease, RA and psoriasis277,278. It was well tolerated in more than 700 human subjects (normal healthy volunteers and patients with inflammatory disease), but the clinical trials did not meet their primary endpoints and further development was abandoned. Apilimod is currently being evaluated in a clinical trial (NCT02594384) aimed at defining a maximum tolerated dose in patients with B cell non-Hodgkin lymphoma and monitoring safety, pharmacokinetics, pharmacodynamics and preliminary efficacy307. YM-201636 is another selective inhibitor of PIKfyve (Table 4; Fig. 3). This inhibitor contains a FYVE-type zinc finger domain. YM-201636 was found to significantly reduce the survival of primary mouse hippocampal neurons in culture and reversibly impair endosomal trafficking in NIH3T3 cells, mimicking the effect produced by depleting PIKfyve with small interfering RNA. It was also found to block retroviral exit by budding from cells275. From a clinical perspective, although targeting PIKfyve is highly promising, further work is required to pave a way towards a future treatment.
Farnesyl transferase inhibitors
Several molecules with farnesyl transferase inhibitory activity have been developed. However, some earlier compounds were found to have major side effects, and their development was discontinued. Lonafarnib (SCH66336; Eiger Biopharmaceuticals), a synthetic tricyclic halogenated carboxamide, has recently shown some promise in a transgenic mouse, which expresses human tau carrying a P301L mutation282 (Table 4). These mice develop tangles in the hippocampus, amygdala, entorhinal cortex and cerebral cortex by 16 weeks, and about 60% of hippocampal neurons die at about 22 weeks. Compared with untreated mice, mice that received lonafarnib displayed less abnormal behaviour and half of the tangles in the hippocampi and cortices. Treatment also prevented brain atrophy that typically occurs in these transgenic mice, while reducing microgliosis in the hippocampus and tempering astrogliosis in the cortex. Mechanistic studies have shown in lonafarnib-treated mice that substrates were more efficiently delivered to lysosomes, their degradation products disappeared faster and the organelles were more readily degraded, specifically by improving lysosome efficiency. Knowing that lonafarnib is already approved for use in humans for other indications (cancer, and ongoing evaluation for progeria and hepatitis delta virus infection), it might therefore be repurposed for use in patients with tauopathy. In this class of farnesyl transferase inhibitors, tipifarnib (R115777; Johnson & Johnson) might also display interesting therapeutic properties as it has been seen to block lysosomal-dependent degradation of bortezomib-induced aggresomes without inhibition of the early steps of autophagy. Kura-oncology in-licenced tipifarnib in 2014.
Chaperone modulators
Molecules targeting chaperone proteins involved in lysosomal function have also been designed for potential therapeutic applications. One of these molecules is VER-155008, a small-molecule inhibitor of HSPA8, a key element of CMA308,309. VER-155008 binds to the nucleotide binding domain of HSPA8 and HSP70, and acts as an ATP-competitive inhibitor of ATPase and chaperone activity. In a mouse model of AD (5XFAD mice), intraperitoneal treatment with VER-155008 reduced the two main pathological features of AD (amyloid plaques and paired helical filament tau accumulation) and improved object recognition, location and episodic-like memory280.
Another molecule, the 21-mer phosphopeptide P140 (Table 4; Fig. 3), was also shown to interact with HSPA8 (Fig. 2)310 and lodge in the HSPA8 nucleotide binding domain92,311. P140 and VER-155008, however, do not have the same mechanism of action, and their effects were not additive312. P140 is a phosphorylated analogue of a nominal peptide that was initially spotted in a cellular screening assay using overlapping peptides covering the whole spliceosomal U1-70K protein and CD4+ T cells collected from the lymph nodes of lupus-prone MRL/lpr mice313. P140 peptide enters B cells via a clathrin coat-dependent endocytosis process to reach early endosomes and then late endosomes/lysosomes92. It affects CMA that is hyperactivated in lupus, likely by hampering the CMA-mediating chaperone HSPA8 (ref.101). P140 peptide reduces the excessive expression of HSPA8 and LAMP2A observed in lupus mice, alters the (auto)antigen presentation by MHCII molecules in the MIIC compartment and, consequently, attenuates the activation of autoreactive T cells92. A significant diminution of MHC molecule expression at the surface of antigen-presenting cells was measured in mice that received the P140 peptide intravenously and on patient’s peripheral cells treated ex vivo with the peptide92,101,314. As a downstream consequence, the activation of autoreactive B cells and their differentiation into autoantibody-secreting cells is repressed101,314. T cells from patients with lupus are no longer responders ex vivo to peptides encompassing CD4+ T cell epitopes315. The effect of P140 on CMA was demonstrated in vitro, using a fibroblast cell line that stably expresses a CMA reporter53,92. P140, which selectively targets the CMA/lysosome process and has no effect on mitophagy316, has been evaluated in murine models mimicking other rheumatic diseases with very promising results, notably in mice developing SjS features56, in mice with neuropsychiatric lupus symptoms62 and in rats that develop a CIDP-like disease with disturbance of both CMA and macroautophagy in sciatic nerves59. In clinical trials that included patients with SLE, P140 formulated in mannitol was found to be safe and non-immunogenic after several subcutaneous administrations of peptide312,317,318. P140 showed significant efficacy in a multicentre, double-blind, phase II trial317. This peptide is currently being evaluated in phase III trials in the United States, Europe and Mauritius. In continuation, an open-label trial including several hundred patients with lupus worldwide is planned.
Another peptide has been discovered that, in contrast to P140, activates CMA319. This 24-mer peptide called humanin was originally identified from surviving neurons in patients with AD, and was found to directly enhance CMA activity by increasing substrate binding and translocation into lysosomes. Humanin interacts with HSP90 and stabilizes the binding of this chaperone to CMA cargos as they bind to the lysosomal membrane. These results are important as humanin had been shown to possess some cardioprotective and neuroprotective properties in diseases such as AD, cardiovascular disease, stroke, myocardial infarction, diabetes and cancer320.
Emerging potential lysosomal therapeutic targets
In addition to the targets discussed above, there are a few emerging potential lysosomal therapeutic targets for which there is strong biological validation, but not yet any small molecules in development that target them. An example with likely pharmacological tractability is a lysosomal K+ channel called TMEM175, which is important for maintaining the membrane potential and pH stability in lysosomes321. Deficiency in TMEM175 may play a critical role in PD pathogenicity322. Importantly, the structure of TMEM175 has been recently refined323.
Another target for which ligands have not yet been validated is the KCNQ2/3 channel (also named M-channel or Kv7.2/7.3 channel). It has been shown in NPC1 disease that reduced cholesterol efflux from lysosomes aberrantly modifies neuronal firing patterns324. This disruption of lysosomal cholesterol efflux with decreases in PtdIns(4,5)P2-dependent KCNQ2/3 channel activity may lead to the aberrant neuronal activity. The cholesterol transporter and PtdIns(4,5)P2 floppase, ABCA1, is responsible for the decline in PtdIns(4,5)P2 that consequently modifies the electrical properties of NPC1 disease neurons. Dysfunction in the activity of KCNQ2/3 or altered levels of PtdIns(4,5)P2, due notably to genetic mutations, might also be involved in other neuropathies (for example, some forms of epilepsy, HD, PD, AD, ALS and Friedrich ataxia). Although further experiments are needed to validate the link discovered between hyperexcitability and cell death in NPC1 disease and other neurodegenerative diseases, small molecules such as retigabine, an anti-convulsant drug that keeps KCNQ2/3 channels open, might represent important therapeutic tools324,325. Other channel opener ligands of KCNQ2/Q3 include ICA-069673 and its derivatives.
Another promising therapeutic target is sphingomyelin phosphodiesterase 1 (SMPD1). Defects in the gene encoding SMPD1 cause Niemann–Pick disease type A and type B. SMPD1 converts sphingomyelin to ceramide, and also has phospholipase C activity. Reduced activity of acid sphingomyelinase, associated with a marked decrease in lysosomal stability, has been described in patients with Niemann–Pick disease, a phenotype that was corrected by treating cells with recombinant HSP70326.
Finally, as LAMP2A, a specific lysosomal protein that displays a decisive role in CMA, has been shown to be overexpressed in certain pathological settings such as certain cancers and inflammatory diseases (autoimmune or non-autoimmune), downregulating its expression might be therapeutically beneficial53,327. As mentioned above, however, in other indications there is a defect in LAMP2A. The latter can be due to reduced stability of the CMA receptor and not to decreased de novo synthesis (for example, in ageing)328 or can result from aggregation to the lysosomal membrane of pathogenic proteins such as α-synuclein, ubiquitin carboxy-terminal hydrolase L1 (a deubiquitinating enzyme) and mutant tau, known to amass in neurodegenerative disorders (see above). Targeting LAMP2A therefore remains a challenge, although several strategies may be envisaged, for example by controlling de novo synthesis, by hampering its multimerization into lysosomes (possibly via HSP90 and/or other chaperones) or by regulating the degradation rate of LAMP2A monomers (for reuse) into lysosomes.
Challenges and outlook
Current research into lysosomal function and dysfunction is revealing novel roles of lysosomes in disease pathogenesis and highlighting new opportunities to treat such lysosomal and autophagy-related diseases. As in the case of autophagy modulation14,56,207, lysosomal activation or inhibition must be investigated with caution, as lysosomal activity can be abnormally reduced or enhanced in some organs or tissues and not in others, and, at another scale, lysosome activity can be altered in certain lysosomes and not in others within the same cell. Biodistribution studies in vivo must be undertaken to avoid accumulation of pharmaceuticals in healthy organs or tissues. There is an obvious requirement for safety, to ensure that a drug used as a lysosome modulator for a particular type of lysosomal disease does not increase vulnerability to another disease.
There is still much to be learned about the intimate working of lysosomes. This is due to the abundance of constitutive elements that comprise these vesicles, the added complexity resulting from their plasticity (ion channels and transporters, acidification and swelling) and the vast amount of proteins and peptides that are translocated into lysosomes and digested by lytic enzymes. Sensitive analysis methods have allowed important information to be generated about lysosomal membrane proteins, a large majority of which are transporters8. However, many questions remain related to how their expression is regulated and how they regulate their translocator and chaperoning activities. For example, certain cells only contain so-called secretory lysosomes (as in cytotoxic T cells), whereas other cell subsets contain both conventional and secretory lysosomes (as in platelets). Considering the large family of endo-lysosomal vesicles, the whole notion of ‘secretory’ and ‘conventional’ lysosomes remains a matter of debate. In many instances, lysosomes act as a basal cell metabolism organelle; whereas in other cases, they assist in the regulation of homeostasis through unconventional secretory pathways, known as lysosomal exocytosis, and different signalling mechanisms.
Although several assays used to measure the activity of lysosomes have been validated worldwide (Box 2; Table 3), they have their limitations, including issues associated with reliability, performance and sensitivity, notably in vivo. Another level of complexity comes from the inherent organelle heterogeneity, which is an issue of tremendous importance. Unfortunately, with the tools and equipment we have in hand today, it is virtually impossible to examine what happens in the lysosomes of an individual patient. The introduction of microfluidic single-cell analysis technologies has enabled cellular populations to be characterized and huge advances to be performed. However, the level of precision has not yet been achieved at the level of lysosomes (0.2–0.5 μm). We know that lysosomes are heterogeneous in nature, composition and activity even in ‘normal’ settings; they are not all equally competent for autophagy or any other types of activity. Currently, this is obviously the focus of intense research.
Although a certain number of preclinical studies involving lysosomal regulators have been conducted over the years, only a small number of lysosome-targeted therapeutics have so far moved into clinical development. One of the biggest advances in developing such strategies would be the identification of a genetic signature that would allow those patients most likely to respond to a specific therapy to be selected. However, at this stage of our knowledge of specific lysosome-directed drugs and intrinsic lysosomal failures, genetic features that might predict potential responders are still lacking (with the exception of LSDs). Further investigations are required to achieve this level of knowledge, which obviously will also depend on the type of disease, heterogeneity and frequency.
Another issue associated with the development of lysosome-targeted therapeutics relates to delivery. The use of nanovectors represents an attractive delivery method, owing, in particular, to their unique ability to penetrate across cell barriers and, via the endo-lysosomal pathway, to preferentially home in on organelles such as lysosomes. Several nanoscale galenic forms have been developed to serve as vectors or carriers of proteins, peptides or nucleic acids, and a vast literature describes the many advantages of using such nanostructures in nanomedicine. However, safety is a concern as some carbon nanostructures have been claimed to induce nanotoxicity, accompanied by the induction of autophagy and lysosomal dysfunction329,330,331,332 (reviewed elsewhere333,334,335,336).
The purpose of this Review is to gain awareness of the importance of lysosomes in disease, and to encourage the development of novel lysosomal targeted drugs. However, more research is needed to characterize components that are specifically linked to the lysosome, such as LAMP2A and HSPA8, and to more clearly define their specific involvement in lysosome biogenesis and metabolism. Special attention should be given to the mode of administration of lysosome-targeted medications in order to minimize toxicity and promote specific targeting. It is our hope that a large field of therapeutic applications could emerge from such investigations, encompassing rare and common autoimmune, neurodegenerative and metabolic diseases, as well as cancer, senescence and ageing.
References
De Duve, C., Pressman, B. C., Gianetto, R., Wattiaux, R. & Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 60, 604–617 (1955).
Wang, F., Gomez-Sintes, R. & Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 19, 918–931 (2018).
Settembre, C., Fraldi, A., Medina, D. L. & Ballabio, A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296 (2013).
Xu, H. & Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 77, 57–80 (2015). This article is an encyclopaedia of lysosomal physiology.
Perera, R. M. & Zoncu, R. The lysosome as a regulatory hub. Annu. Rev. Cell. Dev. Biol. 32, 223–253 (2016).
Pous, C. & Codogno, P. Lysosome positioning coordinates mTORC1 activity and autophagy. Nat. Cell Biol. 13, 342–344 (2011).
Kaushik, S. & Cuervo, A. M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19, 365–381 (2018).
Saftig, P. & Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 10, 623–635 (2009). This article presents a comprehensive review of lysosomal function.
Ballabio, A. & Gieselmann, V. Lysosomal disorders: from storage to cellular damage. Biochim. Biophys. Acta 1793, 684–696 (2009).
Desnick, R. J. & Schuchman, E. H. Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu. Rev. Genom. Hum. Genet. 13, 307–335 (2012).
Fleming, A., Noda, T., Yoshimori, T. & Rubinsztein, D. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 7, 9–17 (2011).
Gros, F. & Muller, S. Pharmacological regulators of autophagy and their link with modulators of lupus disease. Br. J. Pharmacol. 171, 4337–4359 (2014). Together with that by Fleming et al. (2011), this article lists chemical modulators of autophagy processes and lysosome activity.
Dikic, I. & Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19, 349–364 (2018).
Bonam, S. R., Wang, F. & Muller, S. Autophagy: a new concept in autoimmunity regulation and a novel therapeutic option. J. Autoimmun. 94, 16–32 (2018).
Davidson, S. M. & Vander Heiden, M. G. Critical functions of the lysosome in cancer biology. Annu. Rev. Pharmacol. Toxicol. 57, 481–507 (2017).
Lawrence, R. E. & Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 21, 133–142 (2019).
Di Ronza, A. et al. CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis. Nat. Cell Biol. 20, 1370–1377 (2018).
Griffiths, G., Hoflack, B., Simons, K., Mellman, I. & Kornfeld, S. The mannose 6-phosphate receptor and the biogenesis of lysosomes. Cell 52, 329–341 (1988).
Malini, E. et al. Role of LIMP-2 in the intracellular trafficking of β-glucosidase in different human cellular models. FEBS J. 29, 3839–3852 (2015).
Mindell, J. A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 74, 69–86 (2012).
Forgac, M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8, 917–929 (2007).
Samie, M. et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev. Cell 26, 511–524 (2013).
Zhang, X. et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 7, 12109 (2016).
Parenti, G., Andria, G. & Ballabio, A. Lysosomal storage diseases: from pathophysiology to therapy. Annu. Rev. Med. 66, 471–486 (2015).
Jancic, C. et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat. Cell Biol. 9, 367–378 (2007). This article describes the role of Rab27a, increased phagosome acidification, alteration of subsets of LROs and antigen degradation defects in antigen cross-presentation.
Raposo, G. & Marks, M. S. Melanosomes—dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 8, 786–797 (2007).
Luzio, J. P., Pryor, P. R. & Bright, N. A. Lysosomes: fusion and function. Nat. Rev. Mol. Cell Biol. 8, 622–632 (2007). This review is one of the must-read introductions to the complex and dynamic systems of the lysosomal network.
Marks, M. S., Heijnen, H. F. & Raposo, G. Lysosome-related organelles: unusual compartments become mainstream. Curr. Opin. Cell Biol. 25, 495–505 (2013).
Patwardhan, A. et al. Routing of the RAB6 secretory pathway towards the lysosome related organelle of melanocytes. Nat. Commun. 8, 15835 (2017).
Huizing, M., Helip-Wooley, A., Westbroek, W., Gunay-Aygun, M. & Gahl, W. A. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu. Rev. Genomics Hum. Genet. 9, 359–386 (2008).
Settembre, C. et al. TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433 (2011). This article is the first research demonstrating that TFEB controls not only lysosomal biogenesis but also the autophagy network, making TFEB a highly attractive therapeutic target.
Raben, N. & Puertollano, R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu. Rev. Cell. Dev. Biol. 32, 255–278 (2016).
Levine, B., Mizushima, N. & Virgin, H. W. Autophagy in immunity and inflammation. Nature 469, 323–335 (2011). This review brought scientists’ attention to the role of autophagy in immunity and inflammation, which prompted intense research into this topic.
Medina, D. et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299 (2015).
Martina, J. A., Chen, Y., Gucek, M. & Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914 (2012).
Settembre, C. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108 (2012).
Aichinger, M., Wu, C., Nedjic, J. & Klein, L. Macroautophagy substrates are loaded onto MHC class II of medullary thymic epithelial cells for central tolerance. J. Exp. Med. 210, 287–300 (2013).
Blum, J. S., Wearsch, P. A. & Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 31, 443–473 (2013).
Yu, L. et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–946 (2010).
Chen, Y. & Yu, L. Development of research into autophagic lysosome reformation. Mol. Cells 41, 45–49 (2018).
Levine, B. & Deretic, V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 7, 767–777 (2007).
Lim, C. Y. & Zoncu, R. The lysosome as a command-and-control center for cellular metabolism. J. Cell Biol. 214, 653–664 (2016).
Johnson, D. E., Ostrowski, P., Jaumouillé, V. & Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 212, 677–692 (2016). This article highlights the heterogeneity of lysosomes in terms of cell positioning, motility and luminal pH, which all affect lysosomal functionality.
Kroemer, G. & Jaattela, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 5, 886–897 (2005).
Boustany, R. Lysosomal storage diseases—the horizon expands. Nat. Rev. Neurol. 9, 583–598 (2013).
Platt, F. M. Emptying the stores: lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 17, 133–150 (2018).
Malm, D. & Nilssen, Ø. Alpha-mannosidosis. Orphanet J. Rare Dis. 3, 21 (2008).
Mikulka, C. R. & Sands, M. S. Treatment for Krabbe’s disease: finding the combination. J. Neurosci. Res. 94, 1126–1137 (2016).
Sabourdy, F. et al. Natural disease history and characterisation of SUMF1 molecular defects in ten unrelated patients with multiple sulfatase deficiency. Orphanet J. Rare Dis. 10, 31 (2015).
Fidzianska, A., Walczak, E. & Walski, M. Abnormal chaperone-mediated autophagy (CMA). Folia Neuropathol. 45, 133–139 (2007).
Nishino, I. et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406, 906–910 (2000).
Zielonka, M., Garbade, S. F., Kolker, S., Hoffmann, G. F. & Ries, M. A cross-sectional quantitative analysis of the natural history of free sialic acid storage disease—an ultra-orphan multisystemic lysosomal storage disorder. Genet. Med. 21, 347–352 (2019).
Wang, F. & Muller, S. Manipulating autophagic processes in autoimmune diseases: a special focus on modulating chaperone-mediated autophagy, an emerging therapeutic target. Front. Immunol. 6, 252 (2015).
Monteith, A. J. et al. Defects in lysosomal maturation facilitate the activation of innate sensors in systemic lupus erythematosus. Proc. Natl Acad. Sci. USA 113, E2142–E2151 (2016).
Sohar, N., Sohar, I. & Hammer, H. Lysosomal enzyme activities: new potential markers for Sjögren’s syndrome. Clin. Biochem. 38, 1120–1126 (2005).
Li, B., Wang, F., Schall, N. & Muller, S. Rescue of autophagy and lysosome defects in salivary glands of MRL/lpr mice by a therapeutic phosphopeptide. J. Autoimmun. 90, 132–145 (2018).
Lassen, K. G. et al. Genetic coding variant in GPR65 alters lysosomal pH and links lysosomal dysfunction with colitis risk. Immunity 44, 1392–1405 (2016).
Weissmann, G. Lysosomes and joint disease. Arthritis Rheum. 9, 834–840 (1966).
Brun, S. et al. An autophagy-targeting peptide to treat chronic inflammatory demyelinating polyneuropathies. J. Autoimmun. 92, 114–125 (2018).
Kim, I., DeBartolo, D., Ramanan, S., Ponath, G. & Pitt, D. Excess lipid accumulation in cortical neurons in multiple sclerosis may lead to autophagic dysfunction and neurodegeneration. Neurology 84, P5.237 (2015).
Ramesh, N. & Pandey, U. B. Autophagy dysregulation in ALS: when protein aggregates get out of hand. Front. Mol. Neurosci. 10, 263 (2017).
Muller, S. et al. Autophagy in neuroinflammatory diseases. Autoimmun. Rev. 16, 856–874 (2017).
Zhang, L., Sheng, R. & Qin, Z. The lysosome and neurodegenerative diseases. Acta Biochim. Biophys. Sin. 41, 437–445 (2009).
Murphy, K. et al. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 137, 834–848 (2014).
Cortes, C. J. & La Spada, A. R. The many faces of autophagy dysfunction in Huntington’s disease: from mechanism to therapy. Drug Discov. Today 19, 963–971 (2014).
Neufeld, E. F. Lysosomal storage diseases. Annu. Rev. Biochem. 60, 257–280 (1991).
Plotegher, N. & Duchen, M. R. Mitochondrial dysfunction and neurodegeneration in lysosomal storage disorders. Trends Mol. Med. 23, 116–134 (2017).
Lee, J. S. et al. Diagnostic challenge for the rare lysosomal storage disease: late infantile GM1 gangliosidosis. Brain Dev. 40, 383–390 (2018).
Perez-Lopez, J. et al. Delayed diagnosis of late-onset Pompe disease in patients with myopathies of unknown origin and/or hyperCKemia. Mol. Genet. Metab. 114, 580–583 (2015).
Lukacs, Z. et al. Prevalence of Pompe disease in 3,076 patients with hyperCKemia and limb-girdle muscular weakness. Neurology 87, 295–298 (2016).
Lee, J. & Ye, Y. The roles of endo-lysosomes in unconventional protein secretion. Cells 7, 198 (2018).
Ezaki, J., Wolfe, L. S. & Kominami, E. Specific delay in the degradation of mitochondrial ATP synthase subunit c in late infantile neuronal ceroid lipofuscinosis is derived from cellular proteolytic dysfunction rather than structural alteration of subunit c. J. Neurochem. 67, 1677–1687 (1996).
Settembre, C. et al. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 17, 119–129 (2008).
Vergarajauregui, S. & Puertollano, R. Mucolipidosis type IV: the importance of functional lysosomes for efficient autophagy. Autophagy 4, 832–834 (2008).
Takikita, S., Myerowitz, R., Zaal, K., Raben, N. & Plotz, P. H. Murine muscle cell models for Pompe disease and their use in studying therapeutic approaches. Mol. Genet. Metab. 96, 208–217 (2009).
Lieberman, A. P. et al. Autophagy in lysosomal storage disorders. Autophagy 8, 719–730 (2012).
Takikita, S. et al. The values and limits of an in vitro model of Pompe disease: the best laid schemes o’mice an’men …. Autophagy 5, 729–731 (2009).
Ge, W., Li, D., Gao, Y. & Cao, X. The roles of lysosomes in inflammation and autoimmune diseases. Int. Rev. Immunol. 34, 415–431 (2015).
Turk, V. et al. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim. Biophys. Acta 1824, 68–88 (2012).
Stoka, V., Turk, V. & Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 32, 22–37 (2016).
Tato, M. et al. Cathepsin S inhibition combines control of systemic and peripheral pathomechanisms of autoimmune tissue injury. Sci. Rep. 7, 2775 (2017).
Yang, M. et al. Cathepsin L activity controls adipogenesis and glucose tolerance. Nat. Cell Biol. 9, 970–977 (2007).
Shafer, W., Pohl, J., Onunka, V., Bangalore, N. & Travis, J. Human lysosomal cathepsin G and granzyme B share a functionally conserved broad spectrum antibacterial peptide. J. Biol. Chem. 266, 112–116 (1991).
Janoff, A. & Scherer, J. Mediators of inflammation in leukocyte lysosomes: IX. Elastinolytic activity in granules of human polymorphonuclear leukocytes. J. Exp. Med. 128, 1137–1155 (1968).
Tamiya, H. et al. Defensins-and cathepsin G-ANCA in systemic lupus erythematosus. Rheumatol. Int. 27, 147–152 (2006).
Zhu, J. et al. T cell hyperactivity in lupus as a consequence of hyperstimulatory antigen-presenting cells. J. Clin. Invest. 115, 1869–1878 (2005).
Weissmann, G. & Thomas, L. Steroids, lyosomes and systemic lupus erythematosus. Bull. N. Y. Acad. Med. 38, 779–787 (1962).
Kallenberg, C. et al. Autoimmunity to lysosomal enzymes: new clues to vasculitis and glomerulonephritis? Immunol. Today 12, 61–64 (1991).
Wang, F., Li, B., Schall, N., Wilhelm, M. & Muller, S. Assessing autophagy in mouse models and patients with systemic autoimmune diseases. Cells 6, 16 (2017).
Li, X. et al. Increased expression of cathepsins and obesity-induced proinflammatory cytokines in lacrimal glands of male NOD mouse. Invest. Ophthalmol. Vis. Sci. 51, 5019–5029 (2010).
Hamm-Alvarez, S. F. et al. Tear cathepsin S as a candidate biomarker for Sjögren’s syndrome. Arthritis Rheumatol. 66, 1872–1881 (2014).
Macri, C. et al. Modulation of deregulated chaperone-mediated autophagy by a phosphopeptide. Autophagy 11, 472–486 (2015). This study together with that of Wang et al. (2015) were the first to demonstrate in vivo that regulating CMA and lysosomal dysfunctions could provide potential therapeutic benefit for autoimmune diseases.
Pickering, M., Botto, M., Taylor, P., Lachmann, P. & Walport, M. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv. Immunol. 76, 227–324 (2000).
Monteith, A. J. et al. mTORC2 activity disrupts lysosome acidification in systemic lupus erythematosus by impairing caspase-1 cleavage of Rab39a. J. Immunol. 201, 371–382 (2018).
Kato, H. & Perl, A. Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4–CD8-– double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus. J. Immunol. 192, 4134–4144 (2014).
Zhou, Y. et al. Cathepsin K deficiency ameliorates systemic lupus erythematosus-like manifestations in Faslpr mice. J. Immunol. 198, 1846–1854 (2017).
Gros, F. et al. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy 8, 1113–1123 (2012).
Alessandri, C. et al. T lymphocytes from patients with systemic lupus erythematosus are resistant to induction of autophagy. FEBS J. 26, 4722–4732 (2012).
Li, B., Yue, Y., Dong, C., Shi, Y. & Xiong, S. Blockade of macrophage autophagy ameliorates activated lymphocytes-derived DNA induced murine lupus possibly via inhibition of proinflammatory cytokine production. Clin. Exp. Rheumatol. 32, 705–714 (2014).
Clarke, A. J. et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann. Rheum. Dis. 74, 912–920 (2015).
Page, N. et al. HSC70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann. Rheum. Dis. 70, 837–843 (2011).
Lettre, G. & Rioux, J. D. Autoimmune diseases: insights from genome-wide association studies. Hum. Mol. Genet. 17, R116–R121 (2008).
Orozco, G. et al. Study of the common genetic background for rheumatoid arthritis and systemic lupus erythematosus. Ann. Rheum. Dis. 70, 463–468 (2011).
Ramos, P., Shaftman, S., Ward, R. & Langefeld, C. Genes associated with SLE are targets of recent positive selection. Autoimmune Dis. 2014, 203435 (2014).
Yang, Z., Goronzy, J. J. & Weyand, C. M. Autophagy in autoimmune disease. J. Mol. Med. 93, 707–717 (2015).
Lessard, C. J. et al. Identification of a systemic lupus erythematosus risk locus spanning ATG16L2, FCHSD2, and P2RY2 in Koreans. Arthritis Rheumatol. 68, 1197–1209 (2016).
Alessandri, C. et al. CD4 T lymphocyte autophagy is upregulated in the salivary glands of primary Sjögren’s syndrome patients and correlates with focus score and disease activity. Arthritis Res. Ther. 19, 178 (2017).
Byun, Y. S., Lee, H. J., Shin, S. & Chung, S. H. Elevation of autophagy markers in Sjögren syndrome dry eye. Sci. Rep. 7, 17280 (2017).
Meng, Z. et al. Imbalanced Rab3D versus Rab27 increases cathepsin S secretion from lacrimal acini in a mouse model of Sjögren’s syndrome. Am. J. Physiol. Cell Physiol. 310, C942–C954 (2016).
Meng, Z., Klinngam, W., Edman, M. C. & Hamm-Alvarez, S. F. Interferon-γ treatment in vitro elicits some of the changes in cathepsin S and antigen presentation characteristic of lacrimal glands and corneas from the NOD mouse model of Sjögren’s syndrome. PLOS ONE 12, e0184781 (2017).
Artmann, G., Fehr, K. & Boni, A. Cathepsin D agglutinators in rheumatoid arthritis. Arthritis Rheumatol. 20, 1105–1113 (1977).
Hashimoto, Y. et al. Significance of cathepsin B accumulation in synovial fluid of rheumatoid arthritis. Biochem. Biophys. Res. Commun. 283, 334–339 (2001).
Hou, W. S. et al. Comparison of cathepsins K and S expression within the rheumatoid and osteoarthritic synovium. Arthritis Rheumatol. 46, 663–674 (2002).
Skoumal, M. et al. Serum cathepsin K levels of patients with longstanding rheumatoid arthritis: correlation with radiological destruction. Arthritis Res. Ther. 7, R65–R70 (2005).
Miyata, J. et al. Cathepsin G: the significance in rheumatoid arthritis as a monocyte chemoattractant. Rheumatol. Int. 27, 375–382 (2007).
Weitoft, T. et al. Cathepsin S and cathepsin L in serum and synovial fluid in rheumatoid arthritis with and without autoantibodies. Rheumatology 54, 1923–1928 (2015).
Trabandt, A., Gay, R. E., Fassbender, H. G. & Gay, S. Cathepsin B in synovial cells at the site of joint destruction in rheumatoid arthritis. Arthritis Rheumatol. 34, 1444–1451 (1991).
Hao, L. et al. Deficiency of cathepsin K prevents inflammation and bone erosion in rheumatoid arthritis and periodontitis and reveals its shared osteoimmune role. FEBS Lett. 589, 1331–1339 (2015).
Yan, W. X., Taylor, J., Andrias-Kauba, S. & Pollard, J. D. Passive transfer of demyelination by serum or IgG from chronic inflammatory demyelinating polyneuropathy patients. Ann. Neurol. 47, 765–775 (2000).
Cleveland, D. W. & Rothstein, J. D. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2, 806–819 (2001).
Vincent, A. Unravelling the pathogenesis of myasthenia gravis. Nat. Rev. Immunol. 2, 797–804 (2002).
Alirezaei, M. et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy 5, 152–158 (2009).
Jeltsch-David, H. & Muller, S. Neuropsychiatric systemic lupus erythematosus: pathogenesis and biomarkers. Nat. Rev. Neurol. 10, 579–596 (2014).
DeGiorgio, L. A. et al. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat. Med. 7, 1189–1193 (2001).
Lall, D. & Baloh, R. H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Invest. 127, 3250–3258 (2017).
Schneider, J. L. & Cuervo, A. M. Autophagy and human disease: emerging themes. Curr. Opin. Genet. Dev. 26, 16–23 (2014).
Colacurcio, D. J. & Nixon, R. A. Disorders of lysosomal acidification—the emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev. 32, 75–88 (2016).
Menzies, F. M. et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034 (2017).
Brun, S., Schall, N., Jeltsch-David, H., Seze, J. D. & Muller, S. Assessing autophagy in sciatic nerves of a rat model that develops inflammatory autoimmune peripheral neuropathies. Cells 6, 30 (2017).
Mc Donald, J. M. & Krainc, D. Lysosomal proteins as a therapeutic target in neurodegeneration. Annu. Rev. Med. 68, 445–458 (2017).
Nicoletti, F., Fagone, P., Meroni, P., McCubrey, J. & Bendtzen, K. mTOR as a multifunctional therapeutic target in HIV infection. Drug Discov. Today 16, 715–721 (2011).
Patergnani, S. et al. Autophagy and mitophagy elements are increased in body fluids of multiple sclerosis-affected individuals. J. Neurol. Neurosurg. Psychiatry 89, 439–441 (2018).
Feng, X., Hou, H., Zou, Y. & Guo, L. Defective autophagy is associated with neuronal injury in a mouse model of multiple sclerosis. Bosn. J. Basic Med. Sci. 17, 95–103 (2017).
Chen, S., Zhang, X., Song, L. & Le, W. Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol. 22, 110–116 (2012).
Le, W. & Zhang, X. Autophagy dysregulation in amyotrophic lateral sclerosis. J. Neurol. Sci. 357, e69–e71 (2015).
Farg, M. A. et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 23, 3579–3595 (2014).
Soo, K. Y. et al. Rab1-dependent ER–Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP-43 and FUS-associated ALS. Acta Neuropathol. 130, 679–697 (2015).
Otomo, A., Pan, L. & Hadano, S. Dysregulation of the autophagy–endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurol. Res. Int. 2012, 12 (2012).
Barmada, S. J. et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 10, 677–685 (2014).
Nassif, M. et al. Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy 10, 1256–1271 (2014). This pioneering work describes the role of Beclin 1 in the development of amyotrophic lateral sclerosis and highlights the complexity of predicting the effects of manipulating autophagy in a disease context.
Bettencourt, C. & Houlden, H. Exome sequencing uncovers hidden pathways in familial and sporadic ALS. Nat. Neurosci. 18, 611–613 (2015).
McKeown, S. R. & Allen, I. V. The fragility of cerebral lysosomes in multiple sclerosis. Neuropathol. Appl. Neurobiol. 5, 405–415 (1979).
Pollard, J. D. & Armati, P. J. CIDP—the relevance of recent advances in Schwann cell/axonal neurobiology. J. Peripher. Nerv. Syst. 16, 15–23 (2011).
Boerboom, A., Dion, V., Chariot, A. & Franzen, R. Molecular mechanisms involved in Schwann cell plasticity. Front. Mol. Neurosci. 10, 38 (2017).
Kim, J., Lee, H. & Park, H. Two faces of Schwann cell dedifferentiation in peripheral neurodegenerative diseases: pro-demyelinating and axon-preservative functions. Neural Regen. Res. 9, 1952–1954 (2014).
Arstila, A., Riekkinen, P., Rinne, U. & Laitinen, L. Studies on the pathogenesis of multiple sclerosis. Eur. Neurol. 9, 1–20 (1973).
Smith, C. M., Mayer, J. A. & Duncan, I. D. Autophagy promotes oligodendrocyte survival and function following dysmyelination in a long-lived myelin mutant. J. Neurosci. 33, 8088–8100 (2013).
Kikuchi, H. et al. Involvement of cathepsin B in the motor neuron degeneration of amyotrophic lateral sclerosis. Acta Neuropathol. 105, 462–468 (2003).
Offen, D. et al. Spinal cord mRNA profile in patients with ALS: comparison with transgenic mice expressing the human SOD-1 mutant. J. Mol. Neurosci. 38, 85–93 (2009).
Balch, W. E., Morimoto, R. I., Dillin, A. & Kelly, J. W. Adapting proteostasis for disease intervention. Science 319, 916–919 (2008).
Wu, H. et al. Caspases: a molecular switch node in the crosstalk between autophagy and apoptosis. Int. J. Biol. Sci. 10, 1072 (2014).
Boland, B. et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 17, 660–688 (2018).
Ross, C. & Poirier, M. Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17 (2004).
Takalo, M., Salminen, A., Soininen, H., Hiltunen, M. & Haapasalo, A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2, 1–14 (2013).
Kumar, V. et al. Protein aggregation and neurodegenerative diseases: from theory to therapy. Eur. J. Med. Chem. 124, 1105–1120 (2016).
Eskelinen, E. L. & Saftig, P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 1793, 664–673 (2009).
Whyte, L. S., Lau, A. A., Hemsley, K. M., Hopwood, J. J. & Sargeant, T. J. Endo-lysosomal and autophagic dysfunction: a driving factor in Alzheimer’s disease? J. Neurochem. 140, 703–717 (2017).
Oikawa, N. & Walter, J. Presenilins and γ-secretase in membrane proteostasis. Cells 8, 209 (2019).
Hampel, H. et al. Biomarkers for Alzheimer’s disease: academic, industry and regulatory perspectives. Nat. Rev. Drug Discov. 9, 560–574 (2010).
Corbett, A. et al. Drug repositioning for Alzheimer’s disease. Nat. Rev. Drug Discov. 11, 833–846 (2012).
Siman, R. et al. Processing of the beta-amyloid precursor. Multiple proteases generate and degrade potentially amyloidogenic fragments. J. Biol. Chem. 268, 16602–16609 (1993).
Cataldo, A. M., Paskevich, P. A., Kominami, E. & Nixon, R. A. Lysosomal hydrolases of different classes are abnormally distributed in brains of patients with Alzheimer disease. Proc. Natl Acad. Sci. USA 88, 10998–11002 (1991).
Cataldo, A. M. et al. Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron 14, 671–680 (1995).
Bi, X. et al. Novel cathepsin D inhibitors block the formation of hyperphosphorylated tau fragments in hippocampus. J. Neurochem. 74, 1469–1477 (2000).
Lauritzen, I., Pardossi-Piquard, R., Bourgeois, A., Bécot, A. & Checler, F. Does intraneuronal accumulation of carboxyl terminal fragments of the amyloid precursor protein trigger early neurotoxicity in Alzheimer’s disease? Curr. Alzheimer Res. 16, 453-457 (2019).
Lee, J.-H. et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158 (2010).
Aflaki, E., Westbroek, W. & Sidransky, E. The complicated relationship between Gaucher disease and parkinsonism: insights from a rare disease. Neuron 93, 737–746 (2017).
Dehay, B. et al. Lysosomal impairment in Parkinson’s disease. Mov. Disord. 28, 725–732 (2013).
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N. & Rubinsztein, D. C. α-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013 (2003).
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T. & Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295 (2004). This article discovered that α-synuclein, a protein associated with several neurodegenerative diseases, is degraded at least partially through CMA, making the latter a very attractive therapeutic target.
Xilouri, M., Vogiatzi, T., Vekrellis, K., Park, D. & Stefanis, L. Abberant α-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLOS ONE 4, e5515 (2009).
Mak, S., McCormack, A., Manning-Bog, A., Cuervo, A. & Di Monte, D. A. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 285, 13621–13629 (2010).
Sevlever, D., Jiang, P. & Yen, S. H. Cathepsin D is the main lysosomal enzyme involved in the degradation of α-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 47, 9678–9687 (2008).
Vidoni, C., Follo, C., Savino, M., Melone, M. A. & Isidoro, C. The role of cathepsin D in the pathogenesis of human neurodegenerative disorders. Med. Res. Rev. 36, 845–870 (2016).
Cullen, V. et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol. Brain 2, 5–5 (2009).
Caron, N. S., Dorsey, E. R. & Hayden, M. R. Therapeutic approaches to Huntington disease: from the bench to the clinic. Nat. Rev. Drug Discov. 17, 729–750 (2018).
Del Toro, D. et al. Mutant huntingtin impairs post-Golgi trafficking to lysosomes by delocalizing optineurin/Rab8 complex from the Golgi apparatus. Mol. Biol. Cell 20, 1478–1492 (2009).
Nakanishi, H. et al. Age-related changes in activities and localizations of cathepsins D, E, B, and L in the rat brain tissues. Exp. Neurol. 126, 119–128 (1994).
Liang, Q., Ouyang, X., Schneider, L. & Zhang, J. Reduction of mutant huntingtin accumulation and toxicity by lysosomal cathepsins D and B in neurons. Mol. Neurodegener. 6, 37 (2011).
Qin, Z. H. et al. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 12, 3231–3244 (2003).
Kim, Y. J. et al. Lysosomal proteases are involved in generation of N-terminal huntingtin fragments. Neurobiol. Dis. 22, 346–356 (2006).
Ratovitski, T., Chighladze, E., Waldron, E., Hirschhorn, R. & Ross, C. Cysteine proteases bleomycin hydrolase and cathepsin Z mediate N-terminal proteolysis and toxicity of mutant huntingtin. J. Biol. Chem. 286, 12578–12589 (2011).
Bhutani, S., Das, A., Maheshwari, M., Lakhotia, S. & Jana, N. Dysregulation of core components of SCF complex in poly-glutamine disorders. Cell Death Dis. 3, e428 (2012).
Ravikumar, B., Imarisio, S., Sarkar, S., O’Kane, C. J. & Rubinsztein, D. C. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J. Cell Sci. 121, 1649–1660 (2008).
Qi, L. & Zhang, X. D. Role of chaperone-mediated autophagy in degrading Huntington’s disease-associated huntingtin protein. Acta Biochim. Biophys. Sin. 46, 83–91 (2014).
Koga, H., Martinez-Vicente, M., Macian, F., Verkhusha, V. & Cuervo, A. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat. Commun. 2, 386 (2011).
Qi, L. et al. The role of chaperone-mediated autophagy in huntingtin degradation. PLOS ONE 7, e46834 (2012).
Bauer, P. et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 28, 256–263 (2010).
Trajkovic, K., Jeong, H. & Krainc, D. Mutant Huntingtin is secreted via a late endosomal/lysosomal unconventional secretory pathway. J. Neurosci. 37, 9000–9012 (2017).
Erie, C., Sacino, M., Houle, L., Lu, M. L. & Wei, J. Altered lysosomal positioning affects lysosomal functions in a cellular model of Huntington’s disease. Eur. J. Neurosci. 42, 1941–1951 (2015).
Usenovic, M. & Krainc, D. Lysosomal dysfunction in neurodegeneration: the role of ATP13A2/PARK9. Autophagy 8, 987–988 (2012).
Sweeney, P. et al. Protein misfolding in neurodegenerative diseases: implications and strategies. Transl. Neurodegener. 6, 6 (2017).
Martini-Stoica, H., Xu, Y., Ballabio, A. & Zheng, H. The autophagy–lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci. 39, 221–234 (2016).
Torra, A. et al. Overexpression of TFEB drives a pleiotropic neurotrophic effect and prevents Parkinson’s disease-related neurodegeneration. Mol. Ther. 26, 1552–1567 (2018).
Cortes, C. J. & La Spada, A. R. TFEB dysregulation as a driver of autophagy dysfunction in neurodegenerative disease: molecular mechanisms, cellular processes, and emerging therapeutic opportunities. Neurobiol. Dis. 122, 83–93 (2019).
Wang, H., Wang, R., Xu, S. & Lakshmana, M. Transcription factor EB Is selectively reduced in the nuclear fractions of Alzheimer’s and amyotrophic lateral sclerosis brains. Neurosci. J. 2016, 4732837 (2016).
Vakifahmetoglu-Norberg, H., Xia, H. G. & Yuan, J. Pharmacologic agents targeting autophagy. J. Clin. Invest. 125, 5–13 (2015).
Clarke, A. J. & Simon, A. K. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol. 19, 170–183 (2019).
Brady, R. O. Enzyme replacement for lysosomal diseases. Annu. Rev. Med. 57, 283–296 (2006).
Jurecka, A. & Tylki-Szymanska, A. Enzyme replacement therapy: lessons learned and emerging questions. Expert Opin. Orphan Drugs 3, 293–305 (2015).
Safary, A., Akbarzadeh Khiavi, M., Mousavi, R., Barar, J. & Rafi, M. A. Enzyme replacement therapies: what is the best option? BioImpacts 8, 153–157 (2018).
Spada, M. et al. Early higher dosage of alglucosidase alpha in classic Pompe disease. J. Pediatr. Endocrinol. Metab. 31, 1343–1347 (2018).
Lee, B. H. et al. A multicenter, open-label, phase III study of Abcertin in Gaucher disease. Medicine (Baltimore) 96, e8492 (2017).
Renna, M. et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J. Clin. Invest. 121, 3554–3563 (2011).
Rubinsztein, D. C., Codogno, P. & Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 11, 709–730 (2012).
Yin, H. et al. The therapeutic and pathogenic role of autophagy in autoimmune diseases. Front. Immunol. 9 (2018).
Retnakumar, S. V. & Muller, S. Pharmacological autophagy regulators as therapeutic agents for inflammatory bowel diseases. Trends Mol. Med. 25, 516–537 (2019).
Ziegler, H. K. & Unanue, E. R. Decrease in macrophage antigen catabolism caused by ammonia and chloroquine is associated with inhibition of antigen presentation to T cells. Proc. Natl Acad. Sci. USA 79, 175–178 (1982).
Xiu, Y. et al. Chloroquine reduces osteoclastogenesis in murine osteoporosis by preventing TRAF3 degradation. J. Clin. Invest. 124, 297–310 (2014).
Vomero, M. et al. Autophagy and rheumatoid arthritis: current knowledges and future perspectives. Front. Immunol. 9, 1577 (2018).
Kuznik, A. et al. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J. Immunol. 186, 4794–4804 (2011).
Sumpter, M. D., Tatro, L. S., Stoecker, W. V. & Rader, R. K. Evidence for risk of cardiomyopathy with hydroxychloroquine. Lupus 21, 1594–1596 (2012).
McAfee, Q. et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl Acad. Sci. USA 109, 8253–8258 (2012).
Maiuri, M. C. & De Stefano, D. Autophagy Networks in Inflammation (Springer, 2016).
Xu, M. et al. A phenotypic compound screening assay for lysosomal storage diseases. J. Biomol. Screen. 19, 168–175 (2014).
Wang, F. et al. The biomolecular corona is retained during nanoparticle uptake and protects the cells from the damage induced by cationic nanoparticles until degraded in the lysosomes. Nanomedicine 9, 1159–1168 (2013).
Bandyopadhyay, D., Cyphersmith, A., Zapata, J. A., Kim, Y. J. & Payne, C. K. Lysosome transport as a function of lysosome diameter. PLOS ONE 9, e86847 (2014).
Valdor, R. et al. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol. 15, 1046–1054 (2014). This article is the first demonstration that CMA plays an important role in T cell immune regulation in vivo.
Sardiello, M. et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009). This article reveals that a highly coordinated gene network, regulated by the master regulator TFEB, exists in lysosomes, and that regulating this gene network via TFEB may provide a potential therapeutic strategy.
Martina, J. A., Diab, H. I., Li, H. & Puertollano, R. Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell. Mol. Life Sci. 71, 2483–2497 (2014).
Diwu, Z., Chen, C. S., Zhang, C., Klaubert, D. H. & Haugland, R. P. A novel acidotropic pH indicator and its potential application in labeling acidic organelles of live cells. Chem. Biol. 6, 411–418 (1999).
Johnson, D. E., Ostrowski, P., Jaumouille, V. & Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 212, 677–692 (2016).
Ma, L., Ouyang, Q., Werthmann, G. C., Thompson, H. M. & Morrow, E. M. Live-cell microscopy and fluorescence-based measurement of luminal pH in intracellular organelles. Front. Cell Dev. Biol. 5, 71 (2017).
Aits, S. et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 11, 1408–1424 (2015). This article elegantly decsribes a novel, specific and practical tool to detect lysosomal membrane permeabilization.
Zhou, J. et al. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 23, 508–523 (2013).
Ono, K., Kim, S. O. & Han, J. Susceptibility of lysosomes to rupture is a determinant for plasma membrane disruption in tumor necrosis factor alpha-induced cell death. Mol. Cell. Biol. 23, 665–676 (2003).
Pierzyńska-Mach, A., Janowski, P. A. & Dobrucki, J. W. Evaluation of acridine orange, LysoTracker Red, and quinacrine as fluorescent probes for long-term tracking of acidic vesicles. Cytometry Part A 85, 729–737 (2014).
Kaushik, S. & Cuervo, A. Chaperone-mediated autophagy. Methods Mol. Biol. 445, 227–244 (2008).
Kaushik, S. & Cuervo, A. Methods to monitor chaperone-mediated autophagy. Methods Enzymol. 452, 297–324 (2009).
Patel, B. & Cuervo, A. Methods to study chaperone-mediated autophagy. Methods 75, 133–140 (2015).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 (2012). This article is a comprehensive and critical overview of the numerous methods and tools that are used for evaluating autophagy and lysosomal activity, and deciphers the complex mechanisms and pathways involved in the regulation of these processes.
Bowes, J. et al. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 11, 909–922 (2012).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018).
Andrade, M. M. et al. Substrate reduction therapy with miglustat in type 1 Gaucher disease in Spain. Nine years outcomes update on ZAGAL Study. Blood 122, 4713–4713 (2013).
Smid, B. E. et al. Biochemical response to substrate reduction therapy versus enzyme replacement therapy in Gaucher disease type 1 patients. Orphanet J. Rare Dis. 11, 28 (2016).
Guerard, N. et al. Lucerastat, an iminosugar for substrate reduction therapy: tolerability, pharmacodynamics, and pharmacokinetics in patients with Fabry disease on enzyme replacement. Clin. Pharmacol. Ther. 103, 703–711 (2018).
Guerard, N., Morand, O. & Dingemanse, J. Lucerastat, an iminosugar with potential as substrate reduction therapy for glycolipid storage disorders: safety, tolerability, and pharmacokinetics in healthy subjects. Orphanet J. Rare Dis. 12, 9 (2017).
Marshall, J. et al. CNS-accessible inhibitor of glucosylceramide synthase for substrate reduction therapy of neuronopathic Gaucher disease. Mol. Ther. 24, 1019–1029 (2016).
Piotrowska, E. et al. Genistin-rich soy isoflavone extract in substrate reduction therapy for Sanfilippo syndrome: an open-label, pilot study in 10 pediatric patients. Curr. Ther. Res. 69, 166–179 (2008).
Moskot, M. et al. The phytoestrogen genistein modulates lysosomal metabolism and transcription factor EB (TFEB) activation. J. Biol. Chem. 289, 17054–17069 (2014).
Entchev, E. V. et al. Odiparcil is a promising substrate reduction therapy in MPS VI murine model. Mol. Genet. Metab. 123, S42–S43 (2018).
Steet, R. A. et al. The iminosugar isofagomine increases the activity of N370S mutant acid β-glucosidase in Gaucher fibroblasts by several mechanisms. Proc. Natl Acad. Sci. USA 103, 13813–13818 (2006).
Magalhaes, J., Gegg, M. E., Migdalska-Richards, A. & Schapira, A. H. Effects of ambroxol on the autophagy–lysosome pathway and mitochondria in primary cortical neurons. Sci. Rep. 8, 1385 (2018).
Porto, C. et al. Pharmacological enhancement of α-glucosidase by the allosteric chaperone N-acetylcysteine. Mol. Ther. 20, 2201–2211 (2012).
Xiao, J. et al. Discovery of a novel noniminosugar acid α glucosidase chaperone series. J. Med. Chem. 55, 7546–7559 (2012).
Flanagan, J. J. et al. The pharmacological chaperone 1-deoxynojirimycin increases the activity and lysosomal trafficking of multiple mutant forms of acid alpha-glucosidase. Hum. Mutat. 30, 1683–1692 (2009).
Berardi, A. S. et al. Pharmacological chaperones increase residual β-galactocerebrosidase activity in fibroblasts from Krabbe patients. Mol. Genet. Metab. 112, 294–301 (2014).
Matsuda, J. et al. Chemical chaperone therapy for brain pathology in GM1-gangliosidosis. Proc. Natl Acad. Sci. USA 100, 15912–15917 (2003).
Takai, T. et al. A bicyclic 1-deoxygalactonojirimycin derivative as a novel pharmacological chaperone for GM1 gangliosidosis. Mol. Ther. 21, 526–532 (2013).
Aflaki, E. et al. A new glucocerebrosidase chaperone reduces α-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and parkinsonism. J. Neurosci. 36, 7441–7452 (2016).
Thome, R. et al. Dendritic cells treated with chloroquine modulate experimental autoimmune encephalomyelitis. Immunol. Cell Biol. 92, 124–132 (2014).
Danza, A., Grana, D., Goni, M., Vargas, A. & Ruiz-Irastorza, G. Hydroxychloroquine for autoimmune diseases. Rev. Med. Chil. 144, 232–240 (2016).
Lai, Z. W. et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet 391, 1186–1196 (2018).
Shao, P., Ma, L., Ren, Y. & Liu, H. Modulation of the immune response in rheumatoid arthritis with strategically released rapamycin. Mol. Med. Report. 16, 5257–5262 (2017).
Matarrese, P. et al. Cathepsin B inhibition interferes with metastatic potential of human melanoma: an in vitro and in vivo study. Mol. Cancer 9, 207 (2010).
Fox, C. et al. Inhibition of lysosomal protease cathepsin D reduces renal fibrosis in murine chronic kidney disease. Sci. Rep. 6, 20101 (2016).
Ikeda, Y. et al. Cathepsins B and L in synovial fluids from patients with rheumatoid arthritis and the effect of cathepsin B on the activation of pro-urokinase. J. Med. Invest. 47, 61–75 (2000).
Yamada, A., Ishimaru, N., Arakaki, R., Katunuma, N. & Hayashi, Y. Cathepsin L inhibition prevents murine autoimmune diabetes via suppression of CD8+ T cell activity. PLOS ONE 5, e12894 (2010).
Shah, P. P. et al. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol. Pharmacol. 78, 319–324 (2010).
Saegusa, K. et al. Cathepsin S inhibitor prevents autoantigen presentation and autoimmunity. J. Clin. Invest. 110, 361–369 (2002).
Nakagawa, T. Y. et al. Impaired invariant chain degradation and antigen presentation and diminished collagen-induced arthritis in cathepsin S null mice. Immunity 10, 207–217 (1999).
Rupanagudi, K. V. et al. Cathepsin S inhibition suppresses systemic lupus erythematosus and lupus nephritis because cathepsin S is essential for MHC class II-mediated CD4 T cell and B cell priming. Ann. Rheum. Dis. 74, 452–463 (2015).
Hargreaves, P. et al. FRI0295 inhibition of cathepsin s leads to suppression of antigen specific T cells from patients with primary Sjögren syndrome. Ann. Rheum. Dis. 77, 684–684 (2018).
Edman, M. C. et al. Increased cathepsin S activity associated with decreased protease inhibitory capacity contributes to altered tear proteins in Sjögren’s syndrome patients. Sci. Rep. 8, 11044 (2018).
Katunuma, N. et al. Structure-based development of pyridoxal propionate derivatives as specific inhibitors of cathepsin K in vitro and in vivo. Biochem. Biophys. Res. Commun. 267, 850–854 (2000).
Stroup, G. B. et al. Potent and selective inhibition of human cathepsin K leads to inhibition of bone resorption in vivo in a nonhuman primate. J. Bone Miner. Res. 16, 1739–1746 (2001).
Nwosu, L. N. et al. Analgesic effects of the cathepsin K inhibitor L-006235 in the monosodium iodoacetate model of osteoarthritis pain. PAIN Rep. 3, e685 (2018).
Viswanathan, K. et al. Nonpeptidic lysosomal modulators derived from Z-Phe-Ala-diazomethylketone for treating protein accumulation diseases. ACS Med. Chem. Lett. 3, 920–924 (2012).
Yuan, N. et al. Bafilomycin A1 targets both autophagy and apoptosis pathways in pediatric B-cell acute lymphoblastic leukemia. Haematologica 100, 345–356 (2015).
Niikura, K., Takeshita, N. & Takano, M. A vacuolar ATPase inhibitor, FR167356, prevents bone resorption in ovariectomized rats with high potency and specificity: potential for clinical application. J. Bone Miner. Res. 20, 1579–1588 (2005).
Lebreton, S., Jaunbergs, J., Roth, M. G., Ferguson, D. A. & De Brabander, J. K. Evaluating the potential of vacuolar ATPase inhibitors as anticancer agents and multigram synthesis of the potent salicylihalamide analog saliphenylhalamide. Bioorg. Med. Chem. Lett. 18, 5879–5883 (2008).
Visentin, L. et al. A selective inhibitor of the osteoclastic V-H(+)-ATPase prevents bone loss in both thyroparathyroidectomized and ovariectomized rats. J. Clin. Invest. 106, 309–318 (2000).
Aldrich, L. N. et al. Discovery of a small-molecule probe for V-ATPase function. J. Am. Chem. Soc. 137, 5563–5568 (2015).
Grimm, C. et al. Small molecule activators of TRPML3. Chem. Biol. 17, 135–148 (2010).
Martin, S. et al. Inhibition of PIKfyve by YM-201636 dysregulates autophagy and leads to apoptosis-independent neuronal cell death. PLOS ONE 8, e60152 (2013).
Cai, X. et al. PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in Toll-like receptor signaling. Chem. Biol. 20, 912–921 (2013).
Sands, B. E. et al. Randomized, double-blind, placebo-controlled trial of the oral interleukin-12/23 inhibitor apilimod mesylate for treatment of active Crohn’s disease. Inflamm. Bowel Dis. 16, 1209–1218 (2010).
Krausz, S. et al. Brief Report: A phase IIa, randomized, double-blind, placebo-controlled trial of apilimod mesylate, an interleukin-12/interleukin-23 inhibitor, in patients with rheumatoid arthritis. Arthritis Rheum. 64, 1750–1755 (2012).
Wen, W., Liu, W., Shao, Y. & Chen, L. VER-155008, a small molecule inhibitor of HSP70 with potent anti-cancer activity on lung cancer cell lines. Exp. Biol. Med. 239, 638–645 (2014).
Yang, X. & Tohda, C. Heat shock cognate 70 inhibitor, VER-155008, reduces memory deficits and axonal degeneration in a mouse model of Alzheimer’s disease. Front. Pharmacol. 9, 48 (2018).
Gong, Z. et al. Humanin is an endogenous activator of chaperone-mediated autophagy. J. Cell Biol. 217, 635–647 (2018).
Hernandez, I. et al. A farnesyltransferase inhibitor activates lysosomes and reduces tau pathology in mice with tauopathy. Sci. Transl. Med. 11, eaat3005 (2019).
Lachmann, R. Treatments for lysosomal storage disorders. Biochem. Soc. Trans. 38, 1465–1468 (2010).
Radin, N. S. Treatment of Gaucher disease with an enzyme inhibitor. Glycoconjugate J. 13, 153–157 (1996).
Pineda, M., Walterfang, M. & Patterson, M. Miglustat in Niemann–Pick disease type C patients: a review. Orphanet J. Rare Dis. 13, 140–140 (2018).
Hughes, D. A. et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 54, 288–296 (2017).
Wise, A. H. et al. Parkinson’s disease prevalence in Fabry disease: a survey study. Mol. Genet. Metab. Rep. 14, 27–30 (2018).
Gaffke, L., Pierzynowska, K., Piotrowska, E. & Wegrzyn, G. How close are we to therapies for Sanfilippo disease? Metab. Brain Dis. 33, 1–10 (2018).
Mohamed, F. E., Al-Gazali, L., Al-Jasmi, F. & Ali, B. R. Pharmaceutical chaperones and proteostasis regulators in the therapy of lysosomal storage disorders: current perspective and future promises. Front. Pharmacol. 8, 448 (2017).
Ortolano, S., Vieitez, I., Navarro, C. & Spuch, C. Treatment of lysosomal storage diseases: recent patents and future strategies. Recent Pat. Endocr. Metab. Immune Drug Discov. 8, 9–25 (2014).
Pereira, D. M., Valentao, P. & Andrade, P. B. Tuning protein folding in lysosomal storage diseases: the chemistry behind pharmacological chaperones. Chem. Sci. 9, 1740–1752 (2018).
Beck, M. Treatment strategies for lysosomal storage disorders. Dev. Med. Child Neurol. 60, 13–18 (2018).
Siklos, M., BenAissa, M. & Thatcher, G. R. Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 5, 506–519 (2015).
Lowry, J. R. & Klegeris, A. Emerging roles of microglial cathepsins in neurodegenerative disease. Brain Res. Bull. 139, 144–156 (2018).
Sena, B. F., Figueiredo, J. L. & Aikawa, E. Cathepsin S as an inhibitor of cardiovascular inflammation and calcification in chronic kidney disease. Front. Cardiovasc. Med. 4, 88 (2018).
Duong, L. T., Leung, A. T. & Langdahl, B. Cathepsin K inhibition: a new mechanism for the treatment of osteoporosis. Calcif. Tissue Int. 98, 381–397 (2016).
Shacka, J. et al. Bafilomycin A1 inhibits chloroquine-induced death of cerebellar granule neurons. Mol. Pharmacol. 69, 1125–1136 (2006).
Redmann, M. et al. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 11, 73–81 (2017).
Boyd, M. R. et al. Discovery of a novel antitumor benzolactone enamide class that selectively inhibits mammalian vacuolar-type (H+)-ATPases. J. Pharmacol. Exp. Ther. 297, 114–120 (2001).
Pamarthy, S., Kulshrestha, A., Katara, G. K. & Beaman, K. D. The curious case of vacuolar ATPase: regulation of signaling pathways. Mol. Cancer 17, 41 (2018).
Sterea, A., Almasi, S. & El Hiani, Y. The hidden potential of lysosomal ion channels: a new era of oncogenes. Cell Calcium 72, 91–103 (2018).
Di Paola, S., Scotto-Rosato, A. & Medina, D. L. TRPML1: the Ca (2+) retaker of the lysosome. Cell Calcium 69, 112–121 (2018).
Chen, C. C. et al. A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nat. Commun. 5, 4681 (2014).
Waller-Evans, H. & Lloyd-Evans, E. Regulation of TRPML1 function. Biochem. Soc. Trans. 43, 442–446 (2015).
Feng, X., Xiong, J., Lu, Y., Xia, X. & Zhu, M. X. Differential mechanisms of action of the mucolipin synthetic agonist, ML-SA1, on insect TRPML and mammalian TRPML1. Cell Calcium 56, 446–456 (2014).
Choy, C. et al. Lysosome enlargement during inhibition of the lipid kinase PIKfyve proceeds through lysosome coalescence. J. Cell Sci. 131 (2018).
Gayle, S. et al. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood 129, 1768–1778 (2017).
Massey, A. J. et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother. Pharmacol. 66, 535–545 (2010).
Schlecht, R. et al. Functional analysis of Hsp70 inhibitors. PLOS ONE 8, e78443 (2013).
Page, N. et al. The spliceosomal phosphopeptide P140 controls the lupus disease by interacting with the HSC70 protein and via a mechanism mediated by γδ T cells. PLOS ONE 4, e5273 (2009).
Wang, F. et al. Blocking nuclear export of HSPA8 after heat shock stress severely alters cell survival. Sci. Rep. 8, 16820 (2018).
Schall, N. & Muller, S. Resetting the autoreactive immune system with a therapeutic peptide in lupus. Lupus 24, 412–418 (2015).
Monneaux, F., Lozano, J. M., Patarroyo, M. E., Briand, J. P. & Muller, S. T cell recognition and therapeutic effect of a phosphorylated synthetic peptide of the 70K snRNP protein administered in MRL/lpr mice. Eur. J. Immunol. 33, 287–296 (2003).
Wilhelm, M. et al. Lupus regulator peptide P140 represses B cell differentiation by reducing HLA class II molecule overexpression. Arthritis Rheumatol. 70, 1077–1088 (2018).
Monneaux, F. et al. Selective modulation of CD4+ T cells from lupus patients by a promiscuous, protective peptide analog. J. Immunol. 175, 5839–5847 (2005).
Bendorius, M. et al. The mitochondrion–lysosome axis in adaptive and innate immunity: effect of lupus regulator peptide P140 on mitochondria autophagy and NETosis. Front. Immunol. 9, 2158 (2018).
Zimmer, R., Scherbarth, H., Rillo, O., Gomez-Reino, J. & Muller, S. Lupuzor/P140 peptide in patients with systemic lupus erythematosus: a randomised, double-blind, placebo-controlled phase IIb clinical trial. Ann. Rheum. Dis. 72, 1830–1835 (2013).
Muller, S. & Wallace, D. The importance of implementing proper selection of excipients in lupus clinical trials. Lupus 23, 609–614 (2014).
Gong, Z. et al. Humanin is an endogenous activator of chaperone-mediated autophagy. J. Cell Biol. 217, 635–647 (2018).
Gong, Z. et al. Central effects of humanin on hepatic triglyceride secretion. Am. J. Physiol. Endocrinol. Metab. 309, E283–E292 (2015).
Cang, C., Aranda, K., Seo, Y.-j., Gasnier, B. & Ren, D. TMEM175 is an organelle K+ channel regulating lysosomal function. Cell 162, 1101–1112 (2015).
Jinn, S. et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc. Natl Acad. Sci. USA 114, 2389–2394 (2017).
Lee, C. et al. The lysosomal potassium channel TMEM175 adopts a novel tetrameric architecture. Nature 547, 472–475 (2017).
Vivas, O., Tiscione, S. A., Dixon, R. E., Ory, D. S. & Dickson, E. J. Niemann–Pick type C disease reveals a link between lysosomal cholesterol and PtdIns(4,5)P2 that regulates neuronal excitability. Cell Rep. 27, 2636–2648.e2634 (2019).
Ghezzi, F., Monni, L. & Nistri, A. Functional up-regulation of the M-current by retigabine contrasts hyperexcitability and excitotoxicity on rat hypoglossal motoneurons. J. Physiol. 596, 2611–2629 (2018).
Kirkegaard, T. et al. Hsp70 stabilizes lysosomes and reverts Niemann–Pick disease-associated lysosomal pathology. Nature 463, 549–553 (2010).
Kon, M. et al. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 3, 109ra117 (2011).
Zhang, C. & Cuervo, A. M. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 14, 959–965 (2008).
Stern, S. T. & Johnson, D. N. Role for nanomaterial–autophagy interaction in neurodegenerative disease. Autophagy 4, 1097–1100 (2008).
Liu, H. et al. A functionalized single-walled carbon nanotube-induced autophagic cell death in human lung cells through Akt–TSC2-mTOR signaling. Cell Death Dis. 2, e159 (2011).
Wu, L. et al. Tuning cell autophagy by diversifying carbon nanotube surface chemistry. ACS Nano 8, 2087–2099 (2014).
Duan, J. et al. Silica nanoparticles enhance autophagic activity, disturb endothelial cell homeostasis and impair angiogenesis. Part. Fibre Toxicol. 11, 50 (2014).
Stern, S. T., Adiseshaiah, P. P. & Crist, R. M. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part. Fibre Toxicol. 9, 20 (2012).
Peynshaert, K. et al. Exploiting intrinsic nanoparticle toxicity: the pros and cons of nanoparticle-induced autophagy in biomedical research. Chem. Rev. 114, 7581–7609 (2014).
Bianco, A. & Muller, S. Nanomaterials, autophagy, and lupus disease. ChemMedChem 11, 166–174 (2016).
Mohammadinejad, R., Ahmadi, Z., Tavakol, S. & Ashrafizadeh, M. Berberine as a potential autophagy modulator. J. Cell. Physiol. https://doi.org/10.1002/jcp.28325 (2019).
Acknowledgements
The authors apologize to all those whose work is not cited due to space limitations. They gratefully acknowledge Hélène Jeltsch-David for critically reading the manuscript. This research was funded by the French Centre National de la Recherche Scientifique, Région Alsace, the Laboratory of Excellence Medalis (ANR-10-LABX-0034), Initiative of Excellence (IdEx), Strasbourg University, and ImmuPharma France. S.M. is grateful to the University of Strasbourg Institute for Advanced Study (USIAS) for funding F.W., and acknowledges the support of the TRANSAUTOPHAGY COST Action (CA15138), the Club francophone de l’autophagie (CFATG) and the European Regional Development Fund of the European Union in the framework of the INTERREG V Upper Rhine programme.
Author information
Authors and Affiliations
Contributions
All authors made substantial, direct and intellectual contribution to the work and approved it for publication.
Corresponding author
Ethics declarations
Competing interests
S.M. discloses the following conflicts of interest: research funding (paid to institution) and a past consultant for ImmuPharma; co-inventor of CNRS-ImmuPharma patents on P140 peptide; owns ImmuPharma shares. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. S.R.B. and F.W. declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Endocytosis
-
A vesicle-mediated process by which cells engulf membrane and extracellular materials. Several endocytic pathways — phagocytosis, pinocytosis and receptor-mediated endocytosis — utilize different mechanisms to internalize material. Clathrin-mediated endocytosis is the major endocytic pathway in mammalian cells.
- Phagocytosis
-
An endocytic process by which certain cells called phagocytes (for example, macrophages) internalize large particles (>0.5 µm) such as bacteria, other microorganisms, foreign particles or aged red blood cells, for example, to form a phagosome.
- Autophagy
-
A vital, finely-regulated and evolutionarily-conserved intracellular pathway that continuously degrades, recycles and clears unnecessary or dysfunctional cellular components. Autophagy is crucial for cell adaptation to the environment and to maintain cell homeostasis, especially under stress conditions.
- Golgi apparatus
-
Cytosolic apparatus, meant for the regulation of proteins (modification, storing and transportation) and some forms of lipids to the other cytosolic compartments via the trans-Golgi network or outside the cell.
- Lysosomal exocytosis
-
A process of the secretory pathway in which lysosomes are fused with the plasma membrane and empty their contents outside the cell. This process plays an important role in plasma membrane repair, bone resorption, immune response and elimination of pathogenic stores (mainly in lysosomal storage disorders).
- Lysosomal storage disorders
-
(LSDs). A group of heterogeneous disorders caused by defects in the lysosomal enzymes leading to the accumulation of unmodified or unprocessed components in the lysosomes, which ultimately influence other vital pathways in the cells. LSDs implicate various vital systems of the human body including the skeleton, brain, skin, heart and central nervous system, which are connected with different metabolic pathways.
- Rheumatoid arthritis
-
(RA). An autoimmune disease involving inflammation and degeneration of the joints that affects an estimated 1% of the population, making it the most common inflammatory arthritis.
- Multiple sclerosis
-
(MS). A demyelinating disease in which the myelin sheaths wrapped around nerve fibres in the central nervous system are progressively destroyed by immune cells and possibly also by autoantibodies.
- Parkinson disease
-
(PD). A neurodegenerative disorder with symptoms including slowness of movement and a loss of fine motor control, owing to the degeneration of dopamine-producing neurons in the substantia nigra.
- Chaperone-mediated autophagy
-
(CMA). A selective autophagy pathway in which proteins that contain a signal KFERQ-like sequence are targeted by HSAP8/HSC70 chaperones and translocated into lysosomes via LAMP2A.
- Transcription factor EB
-
(TFEB). A protein that plays a pivotal role in the regulation of basic cellular processes, such as lysosomal biogenesis and autophagy. It controls lysosomal function via the coordinated lysosomal expression and regulation (CLEAR) gene network (including genes coding for hydrolases, lysosomal membrane proteins and the proton pump v-ATPase complex), and additional lysosome-related processes such as autophagy, endocytosis and exocytosis.
- Macroautophagy
-
A finely-regulated process during which the cell forms a double-membrane sequestering compartment named the phagophore, which matures into the autophagosome.
- Autophagosome
-
A double membrane-bound vesicle, which encloses cellular constituents and fuses with lysosomes to form phagolysosomes where the engulfed material is digested or degraded and either released extracellularly via exocytosis or released intracellularly to undergo further processing.
- Mitophagy
-
A key process that selectively disrupts damaged mitochondria by autolysosomal degradation, preventing excessive reactive oxygen species and activation of cell death.
- Huntingtin
-
(HTT). Discovered in 1993, HTT is a protein of 348 kDa that is widely expressed within the central nervous system. Its structure has been elucidated recently by cryo-electron microscopy. The protein is essential for embryonic development and neurogenesis. It is involved in transcription, vesicle transport, protein trafficking, endocytosis and autophagy.
- Systemic lupus erythematosus
-
(SLE). A chronic, relapsing–remitting autoinflammatory syndrome that has multiple and heterogeneous symptoms, including arthralgia, swollen joints, fever, fatigue, chest pain, kidney inflammation, cardiovascular disease and neuropsychiatric complications. Its aetiology is mostly unknown.
- Sjögren’s syndrome
-
(SjS). A multifactorial systemic autoimmune disorder characterized by lymphocytic infiltrates in exocrine organs. Symptoms include dry eyes, dry mouth and parotid enlargement, and serious complications include fatigue, chronic pain, neuropathies and lymphomas.
- Myasthenia gravis
-
Caused by antibodies targeting the muscle acetylcholine receptor or other neuromuscular junction proteins such as muscle-specific kinase. These antibodies compromise communication between nerves and muscles, leading to muscular weakness and fatigue.
- Chronic inflammatory demyelinating polyneuropathy
-
(CIDP). A progressive autoimmune disorder in which peripheral nerves (roots and trunks) and brachial plexuses are damaged owing to demyelination. It causes muscle weakness, sensory loss and reduced reflexes.
- Neuromyelitis optica
-
Also known as Devic’s syndrome, this disease is characterized by an inflammation and demyelination of the optic nerve (optic neuritis) and the spinal cord (myelitis). Antibodies reacting with aquaporin-4 water channels in the brains of patients are implicated in neuromyelitis optica.
- Amyotrophic lateral sclerosis
-
(ALS). Also known as motor neuron disease, this disease generally starts with muscle twitching and weakness in a limb, or slurred speech. It can affect control of the muscles needed to move, speak, eat and breathe, and can be fatal.
- Tau
-
A major microtubule-associated protein of a mature neuron. Hyperphosphorylated tau accumulates with ubiquitin in ageing neurons as the neurofibrillary tangles that were identified in numerous neurodegenerative diseases called tauopathies that include Alzheimer disease.
- Fabry disease
-
(FD). A progressive, X-linked inherited, multisystemic lysosomal storage disorder caused by GLA mutations, resulting in α-galactosidase deficiency and accumulation of lysosomal substrate.
Rights and permissions
About this article

Cite this article
Bonam, S.R., Wang, F. & Muller, S. Lysosomes as a therapeutic target. Nat Rev Drug Discov 18, 923–948 (2019). https://doi.org/10.1038/s41573-019-0036-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-019-0036-1
This article is cited by
-
Drosophila TMEM63 and mouse TMEM63A are lysosomal mechanosensory ion channels
Nature Cell Biology (2024)
-
Innate immune sensing of lysosomal dysfunction drives multiple lysosomal storage disorders
Nature Cell Biology (2024)
-
Lysosomes as coordinators of cellular catabolism, metabolic signalling and organ physiology
Nature Reviews Molecular Cell Biology (2024)
-
Fluorescence-based multifunctional light sheet imaging flow cytometry for high-throughput optical interrogation of live cells
Communications Physics (2024)
-
A SPLICS reporter reveals \({{{{{\boldsymbol{\alpha }}}}}}\)-synuclein regulation of lysosome-mitochondria contacts which affects TFEB nuclear translocation
Nature Communications (2024)
