
Abstract
Since the discovery of AMP-activated protein kinase (AMPK) as a central regulator of energy homeostasis, many exciting insights into its structure, regulation and physiological roles have been revealed. While exercise, caloric restriction, metformin and many natural products increase AMPK activity and exert a multitude of health benefits, developing direct activators of AMPK to elicit beneficial effects has been challenging. However, in recent years, direct AMPK activators have been identified and tested in preclinical models, and a small number have entered clinical trials. Despite these advances, which disease(s) represent the best indications for therapeutic AMPK activation and the long-term safety of such approaches remain to be established.
Similar content being viewed by others

Introduction
Dramatic improvements in health care coupled with an increased standard of living, including better nutrition and education, have led to a remarkable increase in human lifespan1. Importantly, the number of years spent in good health is also increasing2. Despite these positive developments, there are substantial risks that challenge continued improvements in human health. Perhaps the greatest threat to future health is a chronic energy imbalance in which intake exceeds expenditure3. This energy imbalance contributes to a global epidemic of overweight and obesity4 that accelerates ageing and also increases the risk of developing type 2 diabetes, cardiovascular disease (CVD), non-alcoholic fatty liver disease (NAFLD), chronic kidney disease and certain cancers5. And although lifestyle interventions that target this energy imbalance through caloric restriction and/or endurance exercise can reduce ageing, obesity and related diseases, implementing and maintaining these changes for prolonged periods is challenging given the pervasiveness of calorically dense foods and sedentary lifestyle-enabling technologies. Therefore, developing new pharmacological strategies that mimic a low energy state, such as that elicited by exercise and caloric restriction, could be potentially beneficial for treating chronic metabolic diseases.
At the cellular level, the balance between energy intake and demand can be inferred by the relative levels of the adenine nucleotides AMP, ADP and ATP that are continuously produced and consumed through numerous metabolic reactions6. Given their central role as a measure of cellular energy balance, a number of enzymes were known for many years to be regulated by alterations in adenine nucleotides7. However, a pivotal step in identifying a unifying mechanism that could link these changes to multiple branches of metabolism followed the discovery that the AMP-activated protein kinase (AMPK) provided a common regulatory mechanism for inhibiting both cholesterol (through phosphorylation of HMG-CoA reductase (HMGR)) and fatty acid (through phosphorylation of acetyl-CoA carboxylase (ACC)) synthesis8 (Box 1).
Over the last 30 years since formally naming AMPK9, interest in the enzyme as a drug target has continued to grow. This interest has been stimulated in part by the discovery that AMPK is activated by physiological regulators that are associated with health and longevity such as caloric restriction10 and exercise11, hormones such as leptin12 and adiponectin13 and many natural products that have been used as traditional herbal medicines (reviewed in ref.14). Many of these herbal medicines such as resveratrol15 and berberine15,16,17 are being reformulated to enhance their bioavailability and efficacy. Two of the most widely used medicines in the world, metformin18, the mostly commonly used drug for type 2 diabetes, and salicylate19, the active ingredient in aspirin and salsalate, also activate AMPK, suggesting that some of their beneficial effects may be mediated in part by AMPK. The sodium/glucose cotransporter 2 (SGLT2) inhibitor canagliflozin, which has recently been approved for treating type 2 diabetes, also indirectly activates AMPK in a manner similar to metformin20,21. More recently, a number of small molecules have been identified that directly activate AMPK. Most of these compounds bind in a specific pocket formed by an interaction between the α and β subunits22, termed the allosteric drug and metabolite (ADaM) site23, and studies demonstrating preclinical and clinical efficacy of these activators are beginning to emerge24,25,26,27. For example, O304 has recently been shown to reduce fasting plasma glucose and blood pressure in individuals with type 2 diabetes taking metformin28. However, the most appropriate disease areas to be targeted and potential adverse effects of AMPK activation are still being clarified. Here, we assess the current state of play regarding the therapeutic potential of AMPK across distinct areas including metabolic diseases, cancer, neuromuscular disease, chronic kidney disease, pain and ageing, highlighting key drug development challenges.
Structure of AMPK
Mammalian AMPK is a heterotrimeric complex composed of three subunits: an α subunit (encoded by protein kinase AMP-activated-α (PRKAA)) harbouring a protein kinase catalytic domain and non-catalytic β (PRKAB) and γ (PRKAG) regulatory subunits. There are two isoforms of the α and β subunits and three isoforms of the γ subunit, giving rise to twelve possible combinations of the heterotrimeric αβγ AMPK complex.
The amino-terminal region of the α subunit contains a typical serine/threonine kinase domain. Within the kinase domain is a region known as the activation loop, or T-loop, which is conserved in many protein kinases and plays a pivotal role in their regulation. In AMPK, phosphorylation of Thr172 (pThr172) within the activation loop is required for maximal activity of AMPK29,30. Two upstream kinases account for the physiological phosphorylation of Thr172: liver kinase B1 (LKB1)31,32,33 and calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2; sometimes referred to as CAMKKβ)34,35,36. Numerous studies have shown that Thr172 phosphorylation increases AMPK activity both in vitro and in vivo, and regulation of Thr172 phosphorylation is a central component of the mechanism of activation of AMPK by small molecules (see below). The kinase domain is followed by a flexible region, termed the α-linker37. Within the α-linker is a region of ~60 amino acids that has been named the autoinhibitory domain (AID), so named because addition of this sequence to the isolated kinase domain reduces AMPK activity by approximately tenfold38,39,40. Closer to the carboxy-terminus than the AID is a short sequence that has been termed the α-hook37, or α-regulatory subunit interacting motif 2 (α-RIM2)41,42, which plays an important role in the nucleotide-dependent regulation of AMPK. Another region within the α subunit that may play a regulatory role is the serine/threonine-rich loop43. This region is not conserved in all eukaryotic species and so might represent a somewhat late evolutionary adaptation43.
The β subunit isoforms undergo constitutive amino-terminal myristoylation, and this modification is required for AMP-stimulated and ADP-stimulated phosphorylation of Thr172 (refs44,45). Within the β subunit is a region termed the carbohydrate binding module (CBM; sometimes referred to as the glycogen-binding domain), which shares primary sequence identity with sequences found in a number of proteins that metabolize glycogen or starch46. The CBM forms part of the binding pocket for small-molecule AMPK activators, underlining its significance as a key determinant of AMPK regulation. The carboxy-terminal region of the β subunit interacts with the α and γ subunits, and these interactions act as a scaffold for the formation of the heterotrimeric AMPK complex47.
The γ subunit isoforms share highly conserved carboxy-terminal regions that harbour four cystathionine-β-synthase (CBS) domains of ~60 amino acids48. The γ2 and γ3 isoforms contain amino-terminal extensions that are not present in the γ1 isoform; there is no obvious sequence conservation between γ2 and γ3 or significant homology with other proteins49. In addition, variant forms of γ2 and γ3 transcripts have been identified that encode predicted proteins with truncated amino-terminal regions50,51. Recent studies demonstrate that the γ isoforms confer differential regulation of AMPK to both nucleotides and small-molecule activators52,53,54. To date, the only crystal structures available for AMPK are for γ1-containing complexes, and further work is required to elucidate the precise roles of the amino-terminal regions of γ2 and γ3 on AMPK function. Nonetheless, the finding that these regions affect the regulation of AMPK suggests that different γ isoform complexes could be differentially targeted by drugs.
The existence of multiple isoforms of AMPK subunits has been known for many years55, but the biological relevance of the different isoforms remains only partially understood. One area where there appears to be a substantial difference between the isoforms is in their relative tissue distribution. An important point to note here is the possibility of differences in AMPK isoform expression between species. Most of the in vivo studies on mammalian AMPK have used the mouse as the experimental model. However, there are significant differences in the expression of AMPK isoforms between mice and humans, which has important implications for extrapolating data between species. In rodent liver, α1 and α2 are expressed to similar levels, while β1 and γ1 are the major regulatory subunit isoforms, meaning that rodent liver expresses predominantly a mixture of α1β1γ1 and α2β1γ1 complexes56,57. However, in human liver, α1 is the predominant catalytic isoform, and β2 the major β isoform, such that α1β2γ1 is the predominantly expressed AMPK complex57,58. Similarly, the γ2 isoform is highly expressed in human heart, whereas the γ1 isoform appears to be the predominant γ subunit in rodent heart57,59.
Regulation of AMPK activity
The activity of AMPK is tightly regulated in response to various hormonal and metabolic signals, involving several different mechanisms (Fig. 1). AMPK has been dubbed the fuel gauge of the cell, or nature’s energy sensor60,61, as it is activated in response to an increase in intracellular AMP and ADP levels that occurs in response to a fall in ATP, the immediate energy source for living cells. A consensus view for the regulation of AMPK by adenine nucleotides has now emerged, based in part on structural information identifying the nucleotide binding sites within AMPK. Binding of AMP, but not ADP, allosterically activates AMPK up to tenfold29,37,53, while AMP and ADP increase phosphorylation of Thr172 and protect against dephosphorylation29,37,53. The effects of AMP and ADP are antagonized by ATP, such that regulation is dependent on the AMP/ADP:ATP ratio62,63, and the isoform composition of AMPK, particularly that of the γ isoform, effects nucleotide-dependent regulation52,53. The crystal structure of the regulatory core of AMPK containing full-length γ1 revealed that three of the four CBS domains bind nucleotides47. CBS2 (also referred to as site 2 (ref.64)) does not bind nucleotides. CBS4 (site 4) contains an AMP molecule that is permanently bound under physiological conditions47, although AMP binding can be disrupted under non-physiological conditions65. The other two sites (CBS1 and CBS3) bind adenine nucleotides in an exchangeable manner, providing the basis for nucleotide sensing by AMPK47. Mutations in the γ isoforms can result in a constitutively active AMPK, and their physiological function has been explored using genetic models (Box 2).

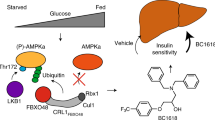
a | AMP-activated protein kinase (AMPK) is activated by phosphorylation of Thr172 (pThr172) within the activation loop of the α subunit, which is catalysed by liver kinase B1 (LKB1) or calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2). An increase in intracellular calcium activates CAMKK2, whereas AMP increases phosphorylation by LKB1 by a mechanism involving axin. Low glucose levels can lead to an increase in Thr172 phosphorylation by LKB1 via a complex signalling pathway involving a decrease in fructose 1,6-bisphosphate (FBP) levels and the interaction between aldolase, vacuolar ATPase (v-ATPase) and axin. The molecular details of the interaction between these proteins remain unclear, and this is represented by the broken arrows. Binding of AMP to the γ subunit directly activates AMPK through an allosteric mechanism. b | AMPK activity can be modulated pharmacologically by compounds that deplete ATP. One mechanism by which this can occur is through inhibition of mitochondrial respiration, for example, complex I inhibition by metformin, canagliflozin, berberine and dinitrophenol. Compounds that bind to the γ subunit (5-aminoimidazole-4-carboxamide ribonucleotide (ZMP) and 5-(5-hydroxyl-isoxazol-3-yl)-furan-2-phosphonic acid (C2)) allosterically activate AMPK and protect against pThr172 dephosphorylation. A fall in intracellular ATP levels leads to a rise in ADP and AMP, decreasing the ATP:ADP/AMP ratio, and this protects against dephosphorylation of pThr172. In addition, AMP and ADP have been reported to increase phosphorylation of AMPK by the upstream kinases LKB1 and CAMKK2. Another class of activators (forexample, 991, A769662, PF739 and MK-8722) bind at the allosteric drug and metabolite (ADaM) site, leading to allosteric activation and protection against dephosphorylation. Phosphorylation of Ser108 (pSer108) in the β1 subunit increases the affinity of binding of these ligands. Compound 0304 activates AMPK by protecting against dephosphorylation, but the binding site has not yet been determined.
AMPK is also activated by an increase in intracellular calcium ions, which is mediated by CAMKK2 (refs34,35,36). Activation by calcium appears to play an important role in regulating AMPK in some tissues in response to hormones, such as VEGF-induced activation of muscarinic receptors in endothelial cells66,67,68 or ghrelin acting on the ghrelin receptor in neuronal cells66,69. An exciting recent development is the finding that fructose 1,6-bisphosphate (FBP), a glycolytic intermediate, mediates glucose sensing by AMPK70. Acute glucose starvation activates AMPK by a mechanism that involves decreased binding of FBP to aldolase. This promotes the interaction between AMPK and LKB1 via binding to axin on the lysosome, leading to phosphorylation and activation of AMPK independently of changes in adenine nucleotides70. This model is discussed in depth in a recent review71.
Physiological functions of AMPK
AMPK activation in response to alterations in adenine nucleotides, calcium and substrate availability as detailed above leads to the phosphorylation of over 100 distinct proteins across a diverse array of metabolic pathways (Fig. 2). The optimal consensus motif of AMPK substrates has been established (Box 3). As detailed below, the phosphorylation of numerous key metabolic proteins influences lipid, cholesterol, carbohydrate and amino acid metabolism as well as mitochondrial function, autophagy and cell growth. AMPK exerts these effects acutely through covalent modifications, which can influence metabolic activity, and chronically through phosphorylation of key transcriptional programmes, altering substrate utilization and availability and ultimately restoring cellular and whole organismal homeostasis.

AMP-activated protein kinase (AMPK) phosphorylates proteins critical for regulating fatty acid, cholesterol, carbohydrate and amino acid metabolism as well as autophagy, mitochondrial function (biogenesis, fission and mitophagy) and cell growth. AMPK phosphorylation of key substrates can modulate enzyme activities, cellular localization and the activation state of transcriptional programmes to elicit both acute and chronic effects in order to match energy demand with availability. ACC, acetyl-CoA carboxylase; ACE2, angiotensin-converting enzyme 2; AKAP1, A kinase anchor protein 1; ATG9, authophagy-related protein 9; ATGL, adipose triglyceride lipase; BECN1, beclin 1; CHOP, C/EBP homologous protein; ChREBP, carbohydrate-response element-binding protein; eNOS, endothelial nitric oxide synthase; FOXO3, forkhead box protein O3; GFAT1, glutamine fructose-6-phosphate aminotransferase 1; GLUT, glucose transporter; GPAT, glycerol-3-phosphate acyl transferase; GS, glycogen synthase; HDAC, histone deacetylase; HMGR, HMG-CoA reductase; HNF4A, hepatocyte nuclear factor 4α; HSL, hormone-sensitive lipase; JAK1, Janus kinase 1; MFF, mitochondrial fission factor; NET1A, neuroepithelial cell-transforming 1A; nNOS, neuronal NOS; PAK, p21-activated kinase; PAQR3, progestin and adipoQ receptor family member 3; PD-L1, programmed cell death ligand 1; PDE4B, phosphodiesterase 4B; PGC1α, peroxisome proliferator-activated receptor-γ co-activator 1α; PFK2, 6-phosphofructo-2-kinase; PIKfyve, 1-phosphatidylinositol 3-phosphate 5-kinase; PLD1, phospholipase D1; pSer108, phosphorylated Ser108; pThr172, phosphorylated Thr172; RACK1, receptor of activated protein C kinase 1; Rb, retinoblastoma protein; SIRT1, sirtuin 1; SREBP, sterol-response element-binding protein; TBC1D1, TBC1 domain family member 1; TSC2, tuberous sclerosis complex 2; TXNIP, thioredoxin-interacting protein; ULK1, unc-51-like autophagy activating kinase 1; VEGF, vascular endothelial growth factor.
Regulation of lipid metabolism
AMPK reduces lipid storage through phosphorylation of multiple substrates across distinct pathways that collectively act to promote fatty acid oxidation while suppressing fatty acid and cholesterol synthesis. The synthesis of both cholesterol and fatty acids is dependent on the same key intracellular metabolite, acetyl-CoA. With respect to cholesterol synthesis, acetyl-CoA becomes committed to the mevalonate pathway via a series of condensation reactions followed by reduction to mevalonic acid by HMGR72. AMPK inhibits HMGR and the synthesis of cholesterol through phosphorylation at Ser872 both in vitro8 and in the liver of mice, leading to lower serum and liver cholesterol73. Acetyl-CoA is also converted to malonyl-CoA, the first committed step in fatty acid synthesis, by ACC, which exists as two distinct isoforms: ACC1, which is predominately expressed in lipogenic tissues such as liver and adipose tissue, and ACC2, which is more common in heart and skeletal muscle. The inhibitory effects of AMPK on fatty acid synthesis require phosphorylation of ACC1 and ACC2, as mice lacking these phosphorylation sites have elevated ACC activity, malonyl-CoA and fatty acid synthesis and are also insensitive to AMPK activators25,74,75.
In addition to the acute regulation of fatty acid and cholesterol synthesis through phosphorylation of HMGR and ACC, AMPK may also play a role in inhibiting this pathway through repression of transcriptional programmes. Lipid synthesis is largely governed by the sterol-response element-binding proteins (SREBPs), which are synthesized as precursor forms that reside in the endoplasmic reticulum (ER) and must undergo subsequent processing before translocating to the nucleus. Although cholesterol and fatty acids are both synthesized from a common substrate (acetyl-CoA), their biosynthetic pathways are largely regulated by distinct SREBPs, with cholesterol metabolism (for example, HMGR and low-density lipoprotein (LDL) receptor) being regulated by the activity of SREBP2 and fatty acid metabolism (ATP-citrate lyase (ACLY), ACC and fatty acid synthase (FASN)) being regulated by SREBP1A and SREBP1C76. While the activity of the SREBP proteins is primarily regulated through intracellular concentrations of unsaturated fatty acids and cholesterol76, AMPK phosphorylation of Ser372 and Ser374 on SREBP1C and SREBP2, respectively, may also inhibit their activity by preventing proteolytic processing and thus transcriptional activity45. However, the relative importance of AMPK phosphorylation of SREBP1C and SREBP2 for controlling fatty acid and cholesterol synthesis has not yet been established in vivo. The inhibitory effects of AMPK on SREBP may also occur through indirect mechanisms involving the regulation of p53, sirtuins and forkhead box protein (FOXO)77,78. In addition to SREBPs, the suppression of the lipogenic programme by AMPK can also occur through inhibition and phosphorylation of the carbohydrate-response element-binding protein (ChREBP) at Ser568 (ref.79). Thus, the inhibition of transcriptional programmes may further reinforce the effects of AMPK on ACC and HMGR to repress lipid synthesis.
As well as de novo lipogenesis, cellular lipid content is regulated through a balance of uptake, oxidation and lipolysis. The key rate-limiting enzymes regulating lipolysis are adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL). Early studies indicated that the AMPK activator 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR, the pro-drug of ZMP) inhibited β-adrenergic-induced lipolysis and that this involved inhibitory phosphorylation of HSL at Ser565 (refs80,81); however, more recent studies in mice lacking adipose tissue AMPK have shown that this is likely an off-target effect82 potentially related to the inhibition of adenylyl cyclase, as described previously in the liver83. And while genetic reductions in adipose tissue AMPK are associated with increases in β-adrenergic-stimulated lipolysis in in some studies84,85,86, this inhibitory role for AMPK on lipolysis is inconsistent with observations that AMPK is activated in adipose tissue under conditions of enhanced lipolytic flux, such as occurs with β-adrenergic stimuli, cold, exercise or fasting82,88,89 and studies in other genetic models which show no effect of AMPK on lipolysis82,87. Interestingly, the activation of AMPK by β-adrenergic stimuli is dependent on the lipase ATGL88, which also becomes activated by AMPK under some conditions90. Further studies investigating the role of adipose tissue AMPK in regulating lipolysis under physiological conditions are required.
Non-esterified fatty acids released by adipose tissue lipolysis are an important substrate for many tissues and are taken up from the circulation by facilitative transporters such as fatty acid translocase (FAT; also known as CD36). Pharmacological and hormonal activation of AMPK can increase the translocation of FAT to the plasma membrane of muscle, leading to greater fatty acid uptake91,92. Once inside the cell, the entry of fatty acyl-CoA into mitochondria requires carnitine palmitoyl-transferase 1 (CPT1). Malonyl-CoA produced by ACC is an allosteric inhibitor of CPT1, and consistent with the inhibitory role of AMPK phosphorylation of ACC, pharmacological activators of AMPK increase fatty acid oxidation in liver and skeletal muscle through a pathway requiring ACC phosphorylation74,75,93. However, under conditions of elevated energetic demands such as occurs in contracting skeletal94 or cardiac95 muscle or in activated brown adipose tissue (BAT)82, AMPK phosphorylation of ACC is not always essential for mediating increases in fatty acid oxidation, suggesting that alternative pathways may be important.
Regulation of mitochondrial function
AMPK may also control the rates of fatty acid oxidation indirectly by regulating mitochondrial function. Under conditions of heightened energetic demands, mitochondrial fusion occurs to maximize ATP production. A regulator of mitochondrial fusion is the A kinase anchor protein 1 (AKAP1), which is a mitochondrial scaffold protein that binds mitochondrial-targeted protein kinase A (PKA)96. In vitro, AMPK phosphorylates AKAP1 at Ser103, and mutation of this site blocks increases in fatty acid oxidation96. Although not fully understood, AKAP1-mediated increased fatty acid oxidation may involve the translocation of AKAP1 to the mitochondria, where it then provides a scaffold for PKA to promote the subsequent phosphorylation of dynamin-related protein 1 (DRP1), which is important for mitochondrial fission96. Consistent with this concept, both the circadian clock97 and caloric restriction98 enhance mitochondrial fusion through pathways involving AMPK, potentially through changes in DRP1 phosphorylation and activity. Future studies investigating the importance of AMPK phosphorylation of AKAP1 for regulating fatty acid oxidation under distinct physiological conditions will be important.
A consistent finding across many different systems is that the activation of AMPK increases mitochondrial content, while genetic loss of function has the opposite effect (for reviews see refs99,100). These alterations in mitochondrial content may be due to changes in either the synthesis (biogenesis) and/or degradation (mitophagy) of mitochondria, and it appears that AMPK plays a role in regulating both aspects. AMPK increases mitochondrial biogenesis through multiple complementary pathways. In skeletal muscle and adipose tissue, a key pathway regulating mitochondrial biogenesis is the transcriptional co-activator peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α), and consistent with changes in AMPK activity, there are often parallel changes in PGC1α and mitochondrial biogenesis99,101,102, effects that are largely eliminated when AMPK is activated in the absence of PGC1α103,104. AMPK-induced upregulation of PGC1α likely involves multiple complementary mechanisms including the activation of p53, SIRT1 and histone deacetylase 4 (HDAC4)99. Collectively, this activation of AMPK enhances mitochondrial content and subsequently the capacity of the cell to respond to future energetic challenges100.
Increases in mitochondrial biogenesis must be balanced by the removal of damaged mitochondria100. Severe energetic stress can be harmful to mitochondria; however, over time, all mitochondria become damaged and need to be replaced, a process that is initiated by mitochondrial fission. The fission of mitochondria is mediated by DRP1, which is recruited to the outer mitochondrial membrane by mitochondrial fission factor (MFF), fission mitochondrial 1 (FIS1) and MID49 and/or MID51. AMPK phosphorylates MFF (Ser129)105,106, and this is essential for initiating mitochondrial fission106. Once fission occurs, AMPK further promotes the degradation of damaged mitochondria through a selective form of autophagy called mitophagy65,66. The activation of unc-51-like autophagy activating kinase 1 (ULK1) is a key signal for the formation of a mature phagophore, a newly formed membrane that encapsulates cytosolic constituents, delivering them to the lysosome69. AMPK binds to and directly phosphorylates ULK1 (refs107,108) to promote mitophagy109,110. AMPK also phosphorylates authophagy-related protein 9 (ATG9)111 and beclin 1 (ref.112), which activates the pro-autophagy Vps34 complex.
AMPK also indirectly regulates mitophagy through several mechanisms. The first involves the inhibition of mechanistic target of rapamycin complex 1 (mTORC1), which phosphorylates and inhibits ULK1 (ref.100). AMPK inhibition of mTORC1 involves phosphorylation of tuberous sclerosis complex 2 (TSC2)113 and the mTOR subunit Raptor114. AMPK also phosphorylates acetyl-CoA synthetase (ACSS) at Ser656, which promotes nuclear acetate uptake and may result in the acetylation and activation of transcription factors such as TFEB, which promotes lysosome biogenesis115. AMPK inhibition of mTORC1 may also contribute to TFEB activation116. Last, AMPK promotes autophagy by phosphorylating and increasing the transcriptional activity of FOXO3 (ref.117), which in turn increases the expression of several key autophagy genes including those encoding LC3, beclin 1, VPS34 and BNIP3 (refs118,119). In addition, AMPK phosphorylation has recently been reported to promote import of FOXO3 into mitochondria, where it is cleaved, allowing it to regulate mitochondrial transcription120. Thus, AMPK promotes mitophagy both through direct phosphorylation and activation of ULK1, FOXO3 and ACSS and by antagonizing mTORC1.
Regulation of carbohydrate metabolism
In addition to regulating fatty acid metabolism, which in many cases may indirectly influence carbohydrate utilization owing to the Randle cycle, AMPK also directly regulates pathways controlling carbohydrate metabolism through multiple mechanisms, as detailed below.
In skeletal muscle of rodents, activation of AMPKα2β2γ3 heterotrimers increases glucose uptake by enhancing glucose transporter type 4 (GLUT4) translocation121,122,123,124,125. The mechanisms by which AMPK promotes skeletal muscle glucose uptake likely involve multiple pathways including the phosphorylation and inhibition of the RAB GTPase-activating protein (GAP) TBC1 domain family member 1 (TBC1D1), which normally sequesters GLUT4 to the Golgi126,127. AMPK also phosphorylates 1-phosphatidylinositol 3-phosphate 5-kinase (PIKfyve)128, a phosphoinositide phosphate kinase, as well as phospholipase D1 (ref.129), which activates ERK, and this further promotes GLUT4 translocation to the plasma membrane. In addition to stimulating the acute translocation of GLUT4 to the plasma membrane, AMPK phosphorylation of HDAC4 results in subsequent nuclear translocation that increases myocyte enhancer factor and GLUT4 expression130,131. However, many cells do not express GLUT4 and instead rely on GLUT1 to control glucose uptake. AMPK has been reported to increase GLUT1-dependent glucose uptake through activation of GLUT1 transporters at the plasma membrane132 and increasing GLUT1 expression133. In addition, AMPK phosphorylation of thioredoxin-interacting protein (TXNIP) promotes rapid degradation of TXNIP, which increases both GLUT1 plasma membrane localization and mRNA expression134. TXNIP expression is also reduced in response to AMPK phosphorylation and inhibition of ChREBP135. Collectively, this activation of AMPK promotes cellular glucose uptake by enhancing the translocation and expression of GLUT proteins.
Once glucose enters the cell, it is rapidly converted to glucose-6-phosphate (G6P), which is then directed towards glycolysis or glycogen synthesis depending on energetic demands. In some cells, activation of AMPK stimulates glycolysis through phosphorylation and activation of 6-phosphofructo-2-kinase (PFK2), which leads to a decrease in fructose 2,6-bisphosphate, an allosteric activator of 6-phosphofructo-1-kinase (PFK1)136. PFK2 exists in several isoforms, but only the cardiac136 and inducible137 isoforms (present in haemopoietic cells and in some tumour cells) are regulated by AMPK, accounting for the cell-type-specific effect of AMPK on glycolysis. One of the initial substrates of AMPK to be identified was glycogen synthase. AMPK inhibited glycogen synthase activity in vitro through phosphorylation of Ser7 (ref.138); however, this inhibitory effect was later found to be inconsistent with observations that both genetic and pharmacological activation of AMPK enhanced glycogen storage139. Subsequent studies using genetic knock-in mice established that allosteric activation of glycogen synthase by G6P could override the inhibitory effects of AMPK phosphorylation in response to pharmacological AMPK activators140. AMPK also promotes inhibitory phosphorylation of glycogen synthase in liver, which is important for mediating the effects of pharmacological AMPK activators, but not glucagon, to reduce glycogen synthesis141. In addition to glycogen synthesis, elevations in G6P can result in hexosamine biosynthesis. Glutamine fructose-6-phosphate aminotransferase 1 (GFAT1) is the rate-limiting enzyme in the hexosamine biosynthesis pathway, and AMPK phosphorylates GFAT1 (Ser243) to inhibit its activity142. This inhibition of glycogen synthase and GFAT1 helps to ensure that increases in G6P are directed towards glycolysis rather than storage.
Early studies using nonspecific AMPK activators such as AICAR143,144 indicated that AMPK repressed key gluconeogenic genes such as those encoding phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase), both of which have been shown to contribute to aberrant hepatic glucose production in metabolic disorders145. However, AMPK becomes activated in the liver during exercise, an effect associated with increased, not lower, hepatic glucose output146. Studies using a combination of genetic mouse models lacking AMPK and direct AMPK activators (see below) have found that acute AMPK activation does not inhibit hepatic gluconeogenesis and that previous observations using nonspecific activators such as AICAR or metformin may have been related to the inhibition of adenylyl cyclase or alterations in mitochondrial redox status25,83,147,148. Despite not directly inhibiting gluconeogenic gene expression, the activation of AMPK in hepatocytes by metformin and A769662 indirectly suppresses gluconeogenesis by reducing lipid-induced insulin resistance, thereby allowing insulin to lower hepatic glucose production more effectively74. Recently, AMPK has been shown to phosphorylate phosphodiesterase 4B, leading to its activation149, and this antagonizes glucagon-stimulated cAMP accumulation, an effect that would lead to decreased hepatic gluconeogenesis and may also have implications for the regulation of adipose tissue lipolysis discussed above. Mice lacking liver AMPK also have lower levels of liver glycogen, which reduces glycogenic flux and blood glucose during exercise146. Similarly, during prolonged fasting, AMPK activation in muscle is important for promoting autophagy, muscle breakdown and the release of essential gluconeogenic precursors such as alanine150. Thus, while AMPK in the liver does not directly inhibit liver gluconeogenesis, AMPK in liver and muscle is important for maintaining glucose homeostasis during energetic challenges such as exercise and caloric restriction and regulating liver insulin sensitivity by suppressing lipogenesis.
Pharmacological activation of AMPK
A number of pharmacological compounds that increase AMPK activity indirectly (Table 1) or through direct binding (Table 2) have been identified, and in many cases, their mode of action has been elucidated (Fig. 1). A small number of these compounds have also been approved for use or are in clinical trials.
Indirect AMPK activators
AMPK is activated in response to an increase in AMP and ADP levels, and this underpins the mechanism of action of a wide number of compounds that indirectly activate AMPK (Fig. 1). Essentially, any treatment that leads to a rise in the AMP/ADP:ATP ratio leads to activation of AMPK. It is not surprising, therefore, that many compounds that activate AMPK in cells do so by inhibition of the mitochondrial electron transport chain or by causing mitochondrial uncoupling, both of which would be expected to reduce mitochondrial ATP synthesis.
The biguanide metformin (Table 1), a widely used oral anti-diabetic agent, inhibits complex I in the mitochondria, leading to a reduction in mitochondrial respiration and ATP production151, and has been shown to activate AMPK18. Metformin-induced activation of AMPK is dependent on uptake from the circulation by organic cation transporters, and consistent with this response, the activity of these transporters is important for dictating therapeutic effects between individuals152. Organic cation transporters are highly expressed in the liver152, where the activation of AMPK by metformin requires the upstream kinase LKB1 (ref.153). However, the closely related and lipophilic biguanide phenformin has a much greater propensity to enter cells and activate AMPK outside of hepatocytes, but this compound was withdrawn from most countries owing to increased risk of lactic acidosis. To promote AMPK activation outside the liver, a number of groups have embarked on developing metformin-like complex I inhibitors such as R419 (ref.154) (Table 1).
A number of plant-derived natural products, which are used in traditional herbal medicine, also activate AMPK indirectly by inhibition of mitochondrial respiration14 (for example, berberine and quercetin (see Table 1)). Most widely studied is the polyphenol resveratrol, which activates AMPK through multiple mechanisms including the inhibition of mitochondrial function155 and phosphodiesterases156, as well as through activation of SIRT1 (ref.157). PT-1, a small-molecule AMPK activator, was initially reported to activate AMPK by binding between the kinase domain and AID region of the α subunit, relieving auto-inhibition158. A subsequent study, however, showed that in cells, PT-1 activates AMPK by inhibiting the mitochondrial respiratory chain159. More recently, the sodium/glucose cotransporter inhibitor canagliflozin, but not the closely related compounds dapagliflozin and empagliflozin, was shown to activate AMPK via inhibition of complex I20,21. In addition to electron transport chain inhibition, compounds that cause mitochondrial uncoupling, such as dinitrophenol, increase the AMP/ADP:ATP ratio and activate AMPK160. However, as these compounds activate AMPK indirectly, it is likely that some of their effects are not mediated by AMPK83,147,161,162.
Direct AMPK activators
The initial efforts to identify direct activators of AMPK focused on compounds that would mimic nucleotide-dependent activation, although at the time of these studies, the nature of the nucleotide binding sites was not known. The first such activator to be identified was 5-aminoimidazole-4-carboxamide ribonucleotide (also known as ZMP), the monophosphate derivative of the cell permeable precursor AICAR80,81,163 (Table 2). This finding led to the wide use of AICAR as a pharmacological tool to investigate the effect of activation of AMPK in cells. The levels of ZMP in the cell can also be increased by methotrexate, which inhibits the folate-dependent enzyme AICA ribotide transformylase/IMP cyclohydrolase (ATIC), which is responsible for converting ZMP to IMP164. However, a problem with approaches that increase ZMP is that this affects other AMP-sensitive enzymes, such as fructose 1,6-bisphosphatase and glycogen phosphorylase165,166. It is now clear that some of the effects of AICAR on metabolic pathways are AMPK-independent82,147,167, cautioning the use of AICAR as a sole means for demonstrating the involvement of AMPK in a particular pathway.
More recently, a screen of AMP mimetics identified 5-(5-hydroxyl-isoxazol-3-yl)-furan-2-phosphonic acid (compound 2 (C2)) as a potent allosteric AMPK activator168. C2 appears to selectively activate α1-containing AMPK complexes and has no effect on γ3-containing complexes169,170. The crystal structure of AMPK bound to C2 has been resolved, revealing two molecules of C2 bound to the γ subunit170. Intriguingly, these binding sites differ from the nucleotide binding sites, suggesting that the γ subunit could be exploited for drug targeting independently of nucleotide binding170. In this regard, a recent study identified 2-(2-(4-(trifluoromethyl)phenylamino)thiazol-4-yl)acetic acid as a potent AMPK activator that acts as an AMP mimetic171. Another small molecule, O304, has recently been shown to protect against pThr172 dephosphorylation by protein phosphatase 2 C without allosteric activation of AMPK, thus mimicking the effect of ADP but not AMP28. The exact site of binding of O304 has not been reported.
In 2006, Abbott Laboratories identified a small-molecule activator of AMPK from a screen of ~700,000 compounds172. Subsequent optimization led to A-769662 (Table 2), which was shown to allosterically activate purified AMPK and to activate AMPK in primary hepatocytes172. In contrast to AMP, A-769662 had no direct effect on fructose 1,6-bisphosphatase or glycogen phosphorylase. Activation of AMPK by A-769662 was additive with AMP, suggesting that A-769662 bound at a site distinct from the nucleotide binding sites172. For several years, A-769662 remained the only direct AMPK activator of its type. In 2012, a paper reviewed the existing patent databases for AMPK activators, reporting 26 patents that disclosed 10 classes of direct AMPK activators173. Strikingly, many of the compounds listed were closely related to A-769662, suggesting a common mode of action. Indeed, further studies indicated that many of the listed compounds appeared to activate AMPK by binding to a single site within AMPK. The crystal structure of full-length AMPK (α2β1γ1) in complex with one of these small-molecule activators (termed 991, see Table 2) revealed that the compound binds in a pocket formed between the amino-terminal lobe of the kinase domain and the CBM of the β subunit22. Several structures of AMPK bound to different activators have now been published24,27,174, allowing the molecular basis for activation to be determined. The binding pocket is formed by the interface generated by one end of the five stranded β-sheet of the amino-terminal lobe of the kinase packing against a pair of anti-parallel β-strands from the CBM. A key feature of the structure was the finding that phosphorylated Ser108 in β1 is involved in a network of electrostatic interactions with Lys31 and Thr21 (both from the kinase domain) and Asn111 (from the CBM)22. Mutation of Ser108 to Ala decreased binding of either 991 or A769662 by 50-fold to 100-fold22, providing an explanation for an earlier study showing that phosphorylation of Ser108 in β1 was important for the activation of AMPK by A-769662 (ref.175). The nature of the binding pocket suggests that, in addition to drugs, a natural ligand could bind at the ADaM site, and while an endogenous metabolite has not been identified, salicylate, a natural product derived from willow bark, activates AMPK by directly binding to the ADaM site19. In vivo, aspirin (acetylsalicylate) and salsalate (a dimer of salicylate) are converted to salicylate, and so activation of AMPK may play a role in mediating some of the beneficial effects of these compounds in tissues where concentrations of salicylate reach sufficiently high levels.
Ligand binding at the ADaM site allosterically activates AMPK and protects against dephosphorylation39,175. Protection against dephosphorylation is mediated by stabilization of the interaction of the CBM with the kinase domain in the ligand-bound state, promoting interaction of the kinase domain with the regulatory core of the enzyme22. Most of the contacts with the regulatory core are formed by residues in the activation loop of the kinase, stabilizing the activation loop structure. In this conformation, although the phosphate group of pThr172 is partially exposed to the solvent, it is not accessible to protein phosphatases. Allosteric activation following ligand binding at the ADaM site is dependent on the isoform composition of the AMPK complex22,52,54 and involves the interaction between an α-helix immediately carboxy-terminal to the CBM (referred to as the C-interacting helix) and the αC helix of the kinase domain22. Mutation of Leu166 to Glu within the C-interacting helix, which is predicted to block interaction with the αC helix, significantly reduces allosteric activation of AMPK by 991 but has no effect on allosteric activation by AMP22. The amino acid sequence of the C-interacting helix differs between β1 and β2, and the sequence of the αC helix differs between α1 and α2 (ref.22). It is possible, therefore, that these isoform-specific sequence variations could account for the differences in allosteric activation of the AMPK complexes by binding at the ADaM site.
All the ADaM site activators identified to date bind more tightly to β1-containing than to β2-containing AMPK complexes. The difference in β-isoform binding is compound-dependent, varying from less than 10-fold (for example, 991 (ref.22)) to over 1,000-fold (for example, PF249 and PF06409577 (ref.27)). In some cases, binding to β2-containing complexes is so weak that the compound is effectively a β1-specific activator, for example, PF249 and PF06409577 (ref.27), whereas in other cases, the compound binds sufficiently strongly to both β isoforms to activate β1 and β2 AMPK complexes, for example, PF739 (ref.24) and MK-8722 (ref.26). Using a recently identified activator, SC4, it was shown that Asp111 in β2 interacts with the imidazopyridine 4ʹ-nitrogen of the compound, which could provide increased stabilization of binding to β2-containing AMPK complexes176. The availability of pan-β activators in addition to highly selective β1 activators has provided a useful tool for investigating β isoform-specific effects in vivo.
A recent study reported that in contrast to β1, Ser108 phosphorylation is not involved in binding of 991 to β2-containing AMPK complexes54. There is no high-resolution structural information available for AMPKβ2 complexes bound to ADaM site ligands, and so the molecular basis for the difference in requirement for Ser108 phosphorylation between the β-isoforms remains unknown. Nonetheless, the dramatic increase in binding affinity for ADaM site ligands following phosphorylation of Ser108 in β1 raises the possibility that the modulation of Ser108 phosphorylation could provide a potential strategy for targeting drugs to β1-containing AMPK complexes. The recent finding that ULK1 phosphorylates Ser108 in β1 under conditions that favour an increase in the AMP/ADP:ATP ratio provides a potential way to exploit this possibility177. By increasing Ser108 phosphorylation, through ULK1, it might be possible to increase the potency of AMPK β1 activation. However, Ser108 is an autophosphorylation site178,179, and in recombinant AMPK preparations expressed in Escherichia coli, Ser108 is efficiently phosphorylated even in the absence of Thr172 phosphorylation180, so whether it would be possible to exploit Ser108 phosphorylation in vivo remains speculative.
Dual activation of AMPK (nucleotide and ADaM site)
The fact that AMPK can be activated by ligands that bind to distinct sites within the enzyme provides an attractive rationale for using dual treatments to activate AMPK. In cell-free assays, there is synergistic activation of AMPK when AMP and salicylate or A769662 are provided in combination, effects that are dependent on phosphorylation of Ser108 within the β1 subunit181,182. Surprisingly, AMP and A-769662 synergistically activate recombinant AMPK in the absence of Thr172 phosphorylation182. Subsequent studies confirmed these findings using different AMPK activators170,180. One study resolved the crystal structure of non-phosphorylated Thr172 AMPK complex (α2β1γ1) bound to AMP and 991, showing that the activation loop adopts a similar conformation to that seen in the phosphorylated AMPK complex180. The structure also revealed that Ser108 in β1 was phosphorylated, explaining the finding that 991 binding was the same with both the phosphorylated and non-phosphorylated Thr172 complexes180. These studies indicate that the binding energies of 991 (or A-769662) and AMP are capable of inducing an active-like conformation in the non-phosphorylated activation loop, and this accounts for the activity of the non-phosphorylated complex in vitro. The possibility of synergistically activating AMPK, while bypassing the requirement for upstream kinase activity, has major implications for potential therapeutic strategies targeting AMPK where the upstream kinase may be lacking (for example, LKB1-deficient tumours). However, in vitro studies indicate that this strategy is unlikely to be feasible. In human cells engineered to lack expression of both LKB1 and CAMKK2, Thr172 phosphorylation was almost completely abolished. Treatment of these cells with 991 and conditions that increased AMP failed to lead to increased phosphorylation of ACC, suggesting that in human cells Thr172 phosphorylation is essential for AMPK function180. However, outside of tumours, under most conditions, upstream kinase activity does not appear to be altered, suggesting that combination therapies may be an effective means to enhance AMPK activity in tissues. For example, combined treatment of hepatocytes and skeletal or cardiac muscle with AMPKβ-directed activators (A769662, 991 and salicylate) and AICAR (where ZMP binds to the γ isoform) or metformin (where metformin increases AMP and/or ADP) leads to a synergistic increase in Thr172 phosphorylation and activation of AMPK183,184,185,186. These findings support the hypothesis that dual activation of AMPK through the nucleotide-binding and ADaM-binding sites may be an effective means for maximally activating AMPK.
Therapeutically targeting AMPK
The role of AMPK in regulating a diverse range of cellular processes has revealed the potential of AMPK-activating therapies to treat a number of different diseases (Fig. 3).

AMP-activated protein kinase (AMPK) regulates a diverse range of cellular processes that are linked to human disease. The major pathways known to be regulated by AMPK in different tissues are highlighted for different organs and cell types within the body. Those that are positively impacted by AMPK are shown in green boxes, while those that are inhibited by AMPK are in red boxes. Diseases that have the potential to be treated by AMPK modulation are shown in yellow boxes. CVD, cardiovascular disease; FA, fatty acid; HCC, hepatocellular carcinoma; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; NO, nitric oxide; T2D, type 2 diabetes.
Metabolic diseases
The epidemics of obesity and type 2 diabetes have prompted the need to develop new therapies that can reduce their incidence and related comorbidities including NAFLD, non-alcoholic steatohepatitis (NASH) and CVD. The findings that AMPK activation promotes fatty acid oxidation, mitochondrial function, autophagy and skeletal muscle glucose uptake, while suppressing inflammation as well as fatty acid and cholesterol synthesis, provides a strong rationale for therapeutic targeting. Notably, AMPK activity is lower in mesenchymal stem cells from human infants born from obese mothers187, an effect that is also observed in multiple tissues of individuals with obesity and insulin resistance188. Importantly, these reductions in AMPK are largely restored with treatments that exert beneficial metabolic effects such as endurance exercise, caloric restriction and bariatric surgery, suggesting that AMPK may play an important role in mediating these effects188.
Obesity
Reducing obesity through a combination of lowering caloric intake and increasing energy expenditure, both of which are regulated by AMPK, is one means to effectively treat multiple aspects of cardiometabolic disease. Consistent with its role in increasing energy supply, hypothalamic AMPK activation in response to ghrelin or low glucose stimulates appetite, while reduced AMPK activity in response to leptin and insulin suppresses appetite69,189. Similarly, genetic inactivation of AMPK in the hypothalamus reduces appetite in mice87,190, while a constitutively active AMPK mutation is associated with increased food intake and adiposity in both mice and humans191. The ability of AMPK to increase appetite, at least with respect to ghrelin, is dependent on the phosphorylation and inhibition of ACC, which reduces malonyl-CoA, a known regulator of food intake and neuropeptide expression192, while activation of autophagy193 and p21-activated kinase (PAK) may be important in the response to fasting194. Despite substantial evidence suggesting that activation of hypothalamic AMPK increases appetite, it should be noted this is not always observed, as mice expressing a germline constitutively active AMPK γ2 mutation have reduced food intake195, suggesting that there may be differential responses to AMPK between distinct neuronal populations. Further studies examining the specific neuronal populations that are important for AMPK regulation of appetite and the mechanisms mediating these effects will be important to establish whether this pathway may be pharmacologically manipulated for the treatment of obesity.
In addition to suppressing appetite, reductions in hypothalamic AMPK may also promote weight loss by enhancing energy expenditure. An important regulator of energy expenditure is BAT. The hypothalamus controls sympathetic outflows to BAT, which enhances the activity of uncoupling protein 1 (UCP1), decoupling oxidative phosphorylation from ATP synthesis, thereby generating a futile cycle that enhances ATP turnover. Importantly, in humans with obesity and type 2 diabetes, the metabolic activity of BAT is reduced, suggesting that finding ways to enhance the metabolic capacity of this tissue may increase energy expenditure. Interestingly, factors that enhance BAT thermogenesis and energy expenditure such as 3,3ʹ,5-triiodothyronine (T3), nicotine, oestradiol and bone morphogenetic protein 8 all suppress hypothalamic AMPK196,197. Some studies have used compound C as a pharmacological inhibitor of AMPK, but it is now well established that this compound inhibits a wide variety of kinases198 and so the results of these studies need to be interpreted with caution. With the recent identification of more specific AMPK inhibitors, such as SBI-0206965 (ref.199), it will be interesting to examine whether delivery of AMPK inhibitors into the central nervous system might suppress appetite and promote weight loss.
While reductions in hypothalamic AMPK activity may enhance sympathetic drive to BAT, AMPK expression is very high within BAT itself compared with other tissues and is further activated in response to cold or β-adrenergic stimuli89,200, suggesting an important role for the kinase in controlling BAT function. Supporting this idea, when either the AMPK α or β subunits are genetically removed in adipose tissue of adult mice, defects in BAT mitochondrial function rapidly develop, resulting in an impaired ability to enhance energy expenditure in response to cold or β-adrenergic stimuli82,201. Impaired BAT thermogenesis in mice lacking AMPK has been linked to reduced BAT development104,202, as well as reductions in mitophagy that result in impaired mitochondrial function82. Reductions in adipose tissue AMPK also limit the browning of white adipose tissue and the generation of beige and brite adipose tissue in response to β-adrenergic stimuli82 or direct AMPK activators such as A769662 (ref.201) through mechanisms that are not yet fully understood. Importantly, these defects in brown and beige adipose tissue in the absence of AMPK translate into reductions in whole body energy expenditure and modest increases in obesity when mice are fed a high-fat diet82,201. These findings are consistent with studies showing that pharmacological and indirect genetic activation of AMPK promotes the browning of white fat102,204,205,206,207. Intriguingly, activation of AMPK by 0304 also increases adipose tissue energy expenditure, independently of changes in UCP1 expression, when mice are housed under thermoneutral conditions28. Genetic activation of AMPK also protects against diet-induced obesity through a mechanism that involves increased UCP1-independent thermogenesis in subcutaneous white adipose tissue208. Although the precise molecular details of these effects remain to be fully established, these studies provide evidence of new pathways that might be exploited for therapeutic strategies for treating obesity. And while it is known that AMPK activity is reduced in several different adipose tissue depots of individuals with obesity and insulin resistance209, potentially owing to inflammation210, ubiquitylation211 or microRNAs104, future studies are needed to determine whether activating adipose tissue AMPK under thermoneutral conditions in humans is sufficient for increasing adipose tissue energy expenditure. It will also be interesting to evaluate whether the activation of AMPK in adipose tissue in combination with appetite-suppressing therapies, such as GLP1 receptor agonists, may yield synergies towards weight loss and the treatment of obesity.
Non-alcoholic fatty liver disease, non-alcoholic steatohepatitis and type 2 diabetes
NAFLD is defined as hepatic fat accumulation (>5% of liver weight) and includes a wide spectrum of liver pathology ranging from steatosis without inflammation to NASH, which increases the risk of type 2 diabetes, CVD, liver failure, cirrhosis and hepatocellular carcinoma212. There are currently limited pharmacotherapies that are effective for treating NAFLD and NASH212. An important and defining feature of NAFLD is elevated rates of liver de novo lipogenesis (DNL) and adipose tissue insulin resistance. Consistent with the role for AMPK in inhibiting DNL, activation of liver AMPK using A-769662 reduces NAFLD in rodent models172; however, the poor solubility, limited bioavailability and modest potency of A-769662 limited its use to preclinical studies. Since this time, multiple studies have established that genetically or pharmacologically activating AMPK reduces NAFLD, liver insulin resistance and markers of inflammation and liver fibrosis74,186,213, effects that are primarily mediated through the phosphorylation of ACC and subsequent reductions in malonyl-CoA and liver DNL74. Consistent with their different mechanisms of AMPK activation discussed above, metformin and salicylate synergistically activate AMPK in vitro and in vivo, causing a greater suppression of DNL, liver triglycerides and insulin resistance than either treatment alone181. Whether these beneficial effects are also observed in humans remains to be determined.
While numerous AMPK activators exert positive effects in mouse models of NAFLD (reviewed in ref.214), a key challenge with respect to the development of direct AMPK activators for treating NAFLD in humans has been that, in contrast to mice87, human liver consists of predominately the AMPK β2 isoform57,58,181. Recent studies testing an AMPK activator (PF-06409577) that has greater selectivity towards β1-containing complexes but also activates β2-containing complexes has shown positive effects on NAFLD and NASH in non-human primates, effects that were also observed in mice and shown to be completely dependent on liver AMPK and the phosphorylation of ACC25. Furthermore, novel small-molecule inhibitors of ACC, which mimic the effects of AMPK phosphorylation, are currently in clinical development for the treatment of NASH215, non-small-cell lung carcinoma216 and hepatocellular carcinoma217. Interestingly, this activation of liver AMPK promoted a dramatic increase in SREBP1C, consistent with previous observations obtained with small-molecule inhibitors of ACC218. However, in contrast to ACC inhibition218, AMPK activation did not increase triglyceride levels (hypertriglyceridaemia)25. These data suggest that AMPK regulation of additional substrates involved in triglyceride synthesis or packaging, such as glycerol-3-phosphate acyl transferase (GPAT)219, may be important for restraining this therapeutic liability that has been observed in response to ACC inhibition. Future studies examining additional mechanisms by which AMPK reduces DNL without causing hypertriglyceridaemia and whether AMPK activation is also effective for reducing liver DNL in humans, will be important for determining whether AMPK activation is effective for treating NAFLD.
As well as directly phosphorylating ACC, reductions in liver DNL can also be achieved by starving ACC of its key substrate acetyl-CoA through several mechanisms214. Both salicylate, when delivered to mice as salsalate, and dinitrophenol cause mitochondrial uncoupling and reduce acetyl-CoA and NAFLD in mouse models, effects that are likely largely independent of AMPK activation161,162. Fatty acids derived from adipose tissue lipolysis also contribute to liver acetyl-CoA and can be reduced by promoting fatty acid oxidation within adipocytes through the browning of white fat (increasing the amount of beige and brite adipose tissue)212,214. Consistent with an important role for AMPK in regulating this process, mice lacking AMPK in adipose tissue have decreased beige adipose tissue in response to a β3 agonist and develop NAFLD independently of substantial alterations in adiposity82. Activation of AMPK with O304 also protects mice from developing NAFLD, effects that are associated with increases in white adipose tissue energy expenditure and reductions in lipolysis28. However, further studies with adipose tissue-targeted AMPK activators will be important to establish the therapeutic importance of adipose tissue AMPK for potentially treating NAFLD.
Improving adipose tissue insulin sensitivity by suppressing macrophage inflammation also reduces lipolysis and liver acetyl-CoA212. Genetic and pharmacological studies have established that the activation of macrophage AMPK is associated with the suppression of inflammation220,221,222. In addition, anti-inflammatory agents, such as salicylate and methotrexate, activate AMPK19,223, while pro-inflammatory stimuli (for example, lipopolysaccharide and TNF) suppress AMPK activity by increasing protein phosphatase activity and through inhibitory phosphorylation210,224,225,226, collectively supporting the concept that AMPK plays a vital role in mediating the balance between pro-inflammatory and anti-inflammatory stimuli227,228. The ability of AMPK to suppress multiple inflammatory pathways (for example, NF-κB, the NLRP3 inflammasome and ER stress) in response to numerous distinct stimuli suggests that AMPK suppression of inflammatory programmes may be mediated in part through regulation of key tenets controlling cellular metabolism such as mitochondrial function, mitophagy and/or autophagy and fatty acid oxidation, all of which have been shown to regulate inflammatory pathways220,221,222,229. Additional levels of control may also involve the circadian clock, which is vital for regulating immune cell function203 and is controlled through AMPK phosphorylation of cryptochrome 1 (ref.230). In addition, AMPK suppression of inflammatory pathways may also involve phosphorylation of C/EBP homologous protein (CHOP; also known as DDIT3)231, a critical component of the ER stress response, as well as Janus kinase (JAK), which is required for activation of the signal transducer and activator of transcription (STAT) pathway232. Further studies examining whether AMPK also directly phosphorylates and inhibits key components of the NF-κB and NLRP3 inflammasome pathway are warranted. Importantly, mice lacking AMPK in macrophages have greater inflammation in liver and adipose tissue and elevated free fatty acids when fed a high-fat diet220, supporting a potentially important role for macrophage AMPK in reducing the progression of NAFLD to NASH. However, further studies in more advanced models of NASH are required to confirm that activation of macrophage AMPK may be effective for reversing established disease, especially where substantial cirrhosis may have developed. Importantly, inflamed M1 macrophages can reduce liver fibrosis233, an effect that might be impaired because of AMPK promoting a switch to anti-inflammatory M2 macrophages227.
Another approach to help lower blood glucose and free fatty acids and potentially divert these substrates away from liver DNL involves enhancing skeletal muscle glucose uptake and fatty acid oxidation214. The activation of skeletal muscle AMPK using indirect AMPK activators such as AICAR or genetic mutations in the γ isoform enhances glucose uptake, glycogen synthesis, mitochondrial biogenesis, fatty acid oxidation and exercise capacity in rodent models234. With respect to glucose uptake, fatty acid oxidation and mitochondrial biogenesis, TBC1D1126,127, acetyl-CoA carboxylase 2 (ACC2)75 and PGC1α103 appear to be important for the effects of AICAR in mice. AICAR also acutely increases skeletal muscle glucose uptake in healthy participants235; however, these effects are blunted in older individuals236 and those with hyperinsulinaemia237, suggesting that it may not be of substantial utility for increasing glucose uptake in many patients with insulin resistance. Other AMPK activators such as R419, a complex I inhibitor (Table 1), enhanced muscle mitochondrial content, exercise capacity and insulin sensitivity in control but not AMPK-muscle-null mice238; however, the potential for development of lactic acidosis precluded clinical development.
Given the positive effects of AMPK activators on glucose uptake, fatty acid oxidation, mitochondrial biogenesis and insulin sensitivity in preclinical models, more specific AMPK agonists targeting both the β1 and importantly the β2 isoform, which is highly expressed in skeletal muscle and important for stimulating glucose uptake124, have recently been developed. These pan-AMPK activators, 991 (ref.183), PF739 (ref.24), MK-8722 (ref.26) and O304 (ref.28), all enhance skeletal muscle glucose uptake and lower blood glucose in a variety of models including obese mice, dogs and non-human primates. Although speculative, this reduction in blood glucose independent of insulin might be anticipated to allow pancreatic β-cells to recover, potentially alleviating or reversing type 2 diabetes24. Despite these positive effects, mice and non-human primates treated with MK-8722 also developed cardiac hypertrophy with glycogen accumulation, but without cardiac dysfunction26. This led to the speculation that the cardiac adaptations with MK-8722 may be similar to those observed with chronic endurance exercise26. Given the known effects of AMPK activators to improve exercise capacity and spontaneous activity238,239,240, it is interesting to speculate that perhaps this may have been a contributing factor to the cardiac hypertrophy observed with AMPK activation. Importantly, a recent phase IIa trial with O304 in individuals with type 2 diabetes treated with metformin has also shown glucose-lowering effects, however, without cardiac hypertrophy28. Future studies examining the mechanisms by which these new-generation AMPK activators reduce blood glucose and in some cases cause cardiac hypertrophy and whether they may also be effective for treating type 2 diabetes, NAFLD and/or NASH will be important.
Cardiovascular disease
Type 2 diabetes is an independent risk factor for CVD and accounts for most deaths in people with type 2 diabetes. The majority of heart attacks and strokes are caused by thrombus formation superimposed on disrupted atherosclerotic plaques241. This process is accelerated with type 2 diabetes and is linked to both elevations in low-density lipoprotein cholesterol (LDL-C) and chronic low-grade inflammation, both of which are regulated by AMPK. Given that the activation of AMPK catalyses the inhibitory phosphorylation on HMGR (Ser872), it could be anticipated that the corresponding suppression of cholesterol synthesis would promote the activation of SREBP1C, resulting in increased LDL receptor expression in the liver and subsequent reductions in LDL-C, similar to the effects observed with statin therapy76. Supporting this hypothesis, recent studies using a mouse model in which the AMPK phosphorylation site on HMGR is mutated to Ala have demonstrated that this phosphorylation event inhibits cholesterol synthesis and is important for suppressing serum and liver cholesterol levels73. Interestingly, these mice also develop fatty liver disease and insulin resistance, effects that are attributed to increases in SREBP1C and enhanced expression of lipogenic enzymes such as ACC and FASN. Although initial studies in mouse models using a variety of AMPK activators reported equivocal results in relation to plasma LDL-C or atherosclerosis progression242,243,244,245,246,247,248, recent studies in a mouse model that has cholesterol profiles more closely resembling those seen in humans and in non-human primate models found that PF-06409577 lowered liver cholesterol, leading to the activation of SREBP1C and reductions in LDL-C25. Consistent with an inhibitory role for AMPK on cholesterol synthesis, metformin has a modest effect on lowering LDL-C in patients with type 2 diabetes249. Future studies investigating whether the effects of AMPK activators are mediated through the phosphorylation of HMGR and whether this may be effective in reducing atherosclerosis in preclinical models and possibly in humans are now required. Supporting the therapeutic potential of targeting this pathway, the prodrug bempedoic acid (ETC-1002) is converted to ETC-1002-CoA in the liver, which allosterically activates AMPK and inhibits ACLY, resulting in lower LDL-C and atherosclerosis in mice248. The mechanisms by which ETC-1002-CoA directly activates AMPK are currently undefined but appear to involve direct interactions with the β1 isoform248. Phase III trials with ETC-1002 for CVD are currently underway, with early studies indicating reductions in LDL-C and inflammation in patients taking statins250.
Reducing lipid-laden and inflamed macrophages within atherosclerotic plaques may also be an important strategy for reducing cardiovascular events241. In addition to lowering LDL-C, AMPK activation in response to salicylate and A769662 enhances reverse cholesterol efflux from macrophages owing to upregulation of the transport proteins ATP-binding cassette subfamily A member 1 (ABCA1) and ABCG1 (refs251,252), or by increasing scavenger receptor class B type 1 (SRB1)-mediated hepatic delivery247. AMPK suppression of macrophage inflammation within plaques229,231,253 and adipose tissue220 may also be important for suppressing atherosclerosis development. In addition to regulating macrophage polarization, increased proliferation of monocytes to macrophages and reductions in autophagy are important for atherosclerosis254, and in this regard, AMPK inhibition of cellular proliferative pathways such as mTOR and p53 as well as the induction of autophagy would be expected to reduce atherosclerosis progression255; however, this has not been observed in all studies256. Similarly, the induction of autophagy may reduce atherosclerosis and improve plaque stability, further supporting the possible beneficial role of activating macrophage AMPK257. However, recent clinical trials with salsalate in combination with statin therapy for 30 weeks reported no reductions in atherosclerosis compared with statin therapy alone258; however, there were improvements in glycaemia, supporting the potential role for this therapy in people at high risk of developing type 2 diabetes259. Further studies in mice with AMPK-activating and AMPK-inactivating mutations are now needed to determine whether targeting this pathway may be effective in reducing atherosclerosis and blood glucose.
Besides elevations in LDL-C and inflammation, an important risk factor for CVD is hypertension. Numerous studies have indicated that AICAR lowers blood pressure in both rodents260 and humans237, findings that have more recently also been observed using PF-06409577 (ref.27) and O304 (ref.28). While the exact mechanisms mediating these hypotensive effects are not entirely clear, activation of AMPK using pharmacological stimuli (for example, AICAR28,260 or O304 (ref.28)) consistently promotes vasodilation. AMPK may facilitate vasodilation by increasing endothelial nitric oxide through phosphorylation of endothelial nitric oxide synthase (eNOS) (Ser633 and Ser1177)261 and angiotensin-converting enzyme 2 (Ser680)262, and by promoting an increase in calcium within vascular smooth muscle cells263. Importantly, these anti-hypertensive effects have also been observed in a recent phase IIa study in people with type 2 diabetes using O304, suggesting that this may be an important mechanism by which AMPK activation could reduce cardiovascular events.
Much of the mortality associated with CVD is the result of heart failure. Cardiac AMPK is increased in most cases of heart failure, consistent with lower levels of ATP and reductions in oxidative metabolism (reviewed in ref.264), suggesting that the activation of AMPK in this context is a consequence and not a cause of heart failure. Loss of cardiac AMPK reduces systolic and diastolic function, an effect that results in ventricular shortening in the absence of changes in fatty acid or glucose metabolism or cardiac hypertrophy95,265. However, hypertrophy is also observed with chronic genetic and pharmacological activation of AMPK, effects that, in contrast to Wolff–Parkinson–White syndrome, have been associated with increased stroke volume26,195,266 and subsequent reductions in heart rate267, suggesting that activation of cardiac AMPK may be potentially beneficial in people with heart failure. However, the pathways controlling cardiac hypertrophy are complex, and under some conditions, the activation of AMPK has been shown to be protective against cardiac hypertrophy268. Activating cardiac AMPK may also help protect against cardiac reperfusion injury by enhancing glucose uptake269,270 and suppressing ER stress271. Future studies investigating the effects of direct pharmacological AMPK activators in the context of heart failure and cardiac reperfusion injury are required.
Summary
As detailed above, the pharmacological activation of AMPK exerts positive effects on many aspects of cardiometabolic disease including hyperglycaemia, hyperlipidaemia, NAFLD, insulin resistance, hypertension and chronic low-grade inflammation. These effects are likely mediated through the simultaneous modulation of multiple molecular targets in several different tissues including adipose tissue, liver, immune cells such as macrophages, skeletal and cardiac muscle and the kidney (discussed below). In aggregate, these diverse disease-modifying activities might be expected to exert substantial positive effects on cardiometabolic risk in a more effective manner than existing standards of care such as metformin, GLP1 agonists and SGLT2 inhibitors. Clinical studies investigating these actions are currently underway with some AMPK-activating therapies.
Cancer
Mutations in tumour suppressors and oncogenes such as phosphatase and tensin homologue (PTEN), LKB1, mTOR, p53, RAS and MYC are often associated with reductions in AMPK activity owing to alterations in transcription272, phosphorylation32,43,273,274 or ubiquination275,276. Similarly, increases in AMPK expression and activity have been linked to improved survival in multiple tumour types in humans277, suggesting a potentially important role for AMPK in cancer.
When mice lacking AMPK α1 (ref.278) and β1 (ref.279) subunits are crossed with MYC-overexpressing mice or p53-null mice, respectively, there is an increase in the appearance of T cell lymphomas and reduced survival. A defining feature of the tumours lacking AMPK was reductions in ACC phosphorylation and an increased rate of de novo lipogenesis278,279. In both cells and mice in which ACC is insensitive to AMPK phosphorylation, lipogenesis, cell proliferation and tumorigenesis are also increased217. Similarly, the inhibition of de novo lipogenesis in response to indirect (metformin, phenformin or canagliflozin) and direct (salicylate, MT-6378 or 991) AMPK activators correlates strongly with decreased proliferation and colony formation in many tumour types21,280,281,282, effects that are enhanced when direct and indirect activators are used in combination280 and could involve the suppression of mitosis283. However, it should be noted that some of these effects on lipogenesis may be related to alterations in mitochondrial function that do not require AMPK21,162,282,284. Further supporting the concept that inhibiting lipogenesis may be important for limiting cancer cell proliferation, small-molecule inhibitors of ACC, which mimic the effects of AMPK phosphorylation of ACC to prevent dimerization, suppress the growth of both non-small-cell lung carcinoma216 and hepatocellular carcinoma217 in mice. In addition to directly inhibiting tumour lipogenesis, AMPK activation would also be expected to exert multiple beneficial systemic effects to inhibit tumour growth. This includes the phosphorylation and degradation of programmed cell death ligand 1 (PD-L1), which enhances immune destruction of tumours285, along with lowering of blood glucose and insulin, which would be expected to restore AMPK activity, stabilizing the epigenetic modifying enzyme TET2 (ref.286), and enhance the effectiveness of PI3K inhibitors287. Collectively, these tumour-directed and systemic roles for AMPK have laid the foundation for the initiation of many clinical trials examining the effects of metformin, phenformin or salicylates in preventing many cancers or for enhancing the effects of radiation and chemotherapeutics. Future studies examining whether similar beneficial effects are also observed in preclinical models in vivo with the new generation of direct AMPK activators are warranted.
AMPK may also exert anti-neoplastic activities though both direct and indirect regulation of many other important pathways vital for regulating cell growth and proliferation. AMPK inhibits mTOR activity through direct phosphorylation of TSC and Raptor114,273, which in turn suppresses protein synthesis and translation through inhibition of 4EBP1 and S6 kinase, respectively. Cell growth is further limited through AMPK inhibition of both the Hedgehog and Hippo pathways, effects that are mediated through phosphorylation of GLI1 and YAP288,289. AMPK might also indirectly inhibit the Hippo pathway through phosphorylation of HMGR, which would be expected to reduce cholesterol synthesis, protein prenylation and RHO GTPase activity, which is essential for activating YAP. Similarly, in some tumour types, AMPK may also inhibit RAS activity through both direct phosphorylation and inhibition of BRAF (Ser729)290 and indirectly through inhibition of HMGR and subsequent protein prenylation73,76, which is required for activation of this pathway and subsequent increases in MAPK–ERK signalling. AMPK also promotes cell cycle arrest through activation of tumour suppressors such as p53 (refs291,292), retinoblastoma protein293 and p27 (ref.294), effects that may be mediated through direct phosphorylation or indirectly through the phosphorylation of proteins critical for controlling acetylation such as SIRT1 (Thr344)77. Further fine tuning of cell cycle progression may involve phosphorylation of key components of the mitotic machinery such as protein phosphatase 1 regulatory subunit 12 C, p21-activated protein kinase, SNX17 (Ser437) and CDC42EP1 (Ser192)295. Consistent with a role in inhibiting cell division, AMPK also phosphorylates and inhibits proteins critical for cell invasion, migration and/or motility and adhesion such as neuroepithelial cell-transforming 1 (NET1) (Ser46), cingulin (Ser137) and CLIP170 (ref.296) while at the same time stabilizing existing cell junctions to maintain cell polarity through phosphorylation of Gα-interacting vesicle-associated protein (Ser245)297. AMPK activation therefore modulates numerous distinct pathways, which collectively limits cell proliferation and growth, suggesting that therapies targeting AMPK may be effective for preventing disease or slowing disease progression. However, activation of these pathways is unlikely to induce apoptosis or cell death, indicating that AMPK activation may not be sufficient for treating established disease.
There are several contexts where increasing or maintaining cellular AMPK activity may be undesirable. For example, reductions in LKB1 and/or AMPK increase tumour sensitivity to mitochondrial complex I inhibitors such as phenformin298. Similarly, in KRAS and p53 lung tumours, a deficiency in AMPK reduces tumour load299. The treatment of tumour cells with a direct AMPK activator (A769662) has also been shown to promote proliferation under hypoxic conditions284. The mechanisms by which AMPK promotes survival are likely multifaceted and tumour-dependent. For example, under hypoxic conditions or in the presence of complex I inhibitors (that is, phenformin and/or metformin), AMPK activation may enhance mitochondrial biogenesis and/or spare respiratory capacity284,300 and glucose uptake134, together promoting cell survival. AMPK may also promote cell growth, metastasis and anchorage-dependent growth through AMPK phosphorylation of GFAT, which enhances angiogenesis142, and ACC phosphorylation, which enhances fatty acid oxidation301. Lastly, as the activation of autophagy is an important mechanism by which cells avoid apoptosis, activation of this pathway by AMPK may lead to resistance to therapeutics such as RAS inhibitors302. Therefore, while activating AMPK may exert many positive effects with respect to cancer prevention, it will be important that therapeutics targeting AMPK are carefully considered and evaluated in the context of tumour type, microenvironment and treatment regimens such as radiation, chemotherapeutics and immunotherapy.
Emerging indications for AMPK
Neuromuscular disorders
AMPK exerts many effects on muscle function, which may be beneficial for the treatment of neuromuscular disorders303. Muscles from mice lacking AMPK rapidly fatigue, an effect that is associated with an increased number of glycolytic fibres, centrally located nuclei and split and necrotic myofibres123,304. By contrast, the chronic activation of AMPK promotes the development of slow oxidative fibres240,305, which are important for improving muscle function in a mouse model of Duchenne muscular dystrophy and may involve the upregulation of utrophin A306. AMPK activation also increases expression of the dystrophin-associated protein complex (DAPC), MYOD and myogenin307 while also enhancing autophagic flux150, all of which are impaired in many neuromuscular disorders303. AMPK is also important for maintaining capillary density and vascular perfusion effects associated with the phosphorylation (Ser1446) of neuronal NOSμ (nNOSμ)308. Interestingly, several years before this finding, AMPK had been shown to phosphorylate nNOS in muscle in response to exercise309. Furthermore, mice and humans treated with pharmacological AMPK activators have increased blood perfusion into muscle, which may also help improve muscle function237,310,311. Future studies examining the effects of muscle-specific AMPK activators in mouse models of myopathic disease such as Duchenne muscular dystrophy will be important.
Kidney disease
Chronic hypertension, obesity and type 2 diabetes accelerate the progression of chronic kidney disease and autosomal dominant polycystic kidney disease towards end-stage renal disease. Metformin-induced activation of AMPK improves renal function, an effect that has recently been linked to reductions in fibrosis and the phosphorylation of ACC312. Recent studies have also examined the effects of a direct pharmacological AMPK β1 activator in a mouse model of diabetes and hypertension and found marked reductions in proteinuria, improvements in kidney histology and reductions in S6 phosphorylation, suggesting that activation of AMPK may reduce the development of diabetic nephropathy by suppressing mTOR27.
Chronic pain
A number of studies have revealed that AMPK activation might counteract pathways that promote neuronal activity involved in signalling pathological pain313. In preclinical models, the activation of AMPK can reduce the excitability of nociceptors, effects that are blocked in mouse models lacking AMPK α2 (ref.314). In the setting of chronic pain, AMPK activation inhibits the growth and plasticity of the neuronal circuits that detect pain313 while also reducing interleukin-1β and increasing astrocyte glutamate clearance315. Mechanistically, these effects are attenuated in mice lacking AMPK α1 and may involve the inhibition of mTOR or MAPK. And although the precise role of AMPK in nociception is currently unclear, it provides a potentially attractive target for treatment of chronic pain.
Ageing
It has been known for many years that AMPK activity is diminished with ageing in tissues of humans and rodents316,317,318; however, the mechanisms by which this occurred were unclear. Recent studies have indicated that DNA-dependent protein kinase (DNA-PK), whose activity is increased in response to DNA double-strand breaks that are increased with ageing, inhibits AMPK activity by repressing Thr172 phosphorylation through upregulation of a protein chaperone that inhibits LKB1 activity319. Many therapies that activate AMPK such as endurance exercise, caloric restriction, resveratrol and metformin also improve longevity and health span98,320. The mechanisms by which AMPK elicits these effects are likely complex; however, one important and obvious link involves the induction of autophagy, which is reduced in numerous organs with ageing and has been identified as a critical factor that may reduce longevity321,322,323. Of particular importance may be AMPK regulation of mitochondrial fusion and autophagy or mitophagy97,98,324, as ageing is often associated with the accumulation of giant, defective mitochondria that have a decreased respiratory capacity and are unable to take part in fusion, which is required to replenish contents and initiate mitophagy325. The inability to remove these large mitochondria perpetuates further growth and an increased population of damaged mitochondria that contribute poorly to ATP production, which may further exacerbate ageing-related declines326. Consistent with this idea, mice lacking skeletal muscle AMPK have defects in mitophagy that lead to large dysfunctional mitochondria and sarcopenia150. Interestingly, skeletal muscle AMPK is also important for controlling the ageing of skin, effects that are mediated through transcriptional control of interleukin-15, which is an important cytokine regulating mitochondrial biogenesis327. Activation of AMPK may also be beneficial for ageing owing to its suppressive effects on multiple inflammatory pathways227,228, which may involve the control of mitochondrial content220. Indeed, metformin increases AMPK and mitochondrial function in multiple tissues, effects that are associated with reductions in oxidative damage, chronic low-grade inflammation and improved health span and lifespan in mice320. The AMPK activator quercetin when used in combination with dasatinib has also been shown to exert anti-ageing effects by delaying senescent cell accumulation or reducing senescent cell burden in numerous tissues of rodents328. Whether these beneficial effects are replicated in humans and/or are observed with the new generation of direct AMPK activators that do not inhibit mitochondrial function remains to be determined.
Challenges and outlook
There is now overwhelming evidence supporting the hypothesis that AMPK activation is beneficial for both the prevention and treatment of a wide variety of chronic diseases. The identification of small-molecule activators, coupled with detailed mechanistic and structural information regarding the nature of their binding and mode of action, as well as unique tissue-specific genetic tools, now provides the basis for trials that will directly address the efficacy of AMPK activation across distinct disease conditions. Indeed, studies have already demonstrated that in cardiometabolic disease AMPK activation may have substantial beneficial effects in lowering plasma glucose and lipids, NAFLD and blood pressure in preclinical models, findings that have been confirmed in recent phase II clinical trials with direct AMPK activators such as O304. Future studies examining whether these effects are additive or perhaps synergistic with metformin, SGLT2 inhibitors or GLP1 receptor agonists will be important.
However, like all therapies for chronic diseases, there remain important safety issues that need to be carefully considered and examined, such as the potential for AMPK activation to promote cardiac hypertrophy or the survival of cancer cells under hypoxic conditions. To avoid these potential liabilities, the development of AMPK activators that can be targeted to specific tissues (for example, liver, muscle and macrophages) by taking advantage of isoform-specific selectivity may be beneficial. For example, the availability of AMPK γ3-selective activators could restrict AMPK activation to skeletal muscle, reducing the risk of adverse effects of AMPK activation in other tissues and cell types. Another important consideration would be the optimal pharmacokinetics and pharmacodynamics of a small molecule designed to activate AMPK. For example, both acute endurance exercise and metformin exert positive health benefits associated with transient activation of AMPK329. Therefore, although speculative, it is possible to envision that, unlike typical drug design, where prolonged target engagement is often desired, the optimal strategy to maximize benefits and minimize liabilities may be to develop a therapy that acutely switches on AMPK once or twice per day following a meal.
Despite the potential challenges, ultimately, the only way to determine the long-term safety and efficacy of AMPK activators will be to conduct more extensive clinical trials. Until these studies are completed, the field will be poised for the answer to the long-awaited question of whether AMPK will provide the target for a metabolic wonder drug.
References
Vaupel, J. W. et al. Biodemographic trajectories of longevity. Science 280, 855–860 (1998).
Christensen, K., Doblhammer, G., Rau, R. & Vaupel, J. W. Ageing populations: the challenges ahead. Lancet 374, 1196–1208 (2009).
World Health Organization. Obesity and overweight. WHO http://www.who.int/Mediacentre/Factsheets/fs311/en/ (updated 16 Feb 2018).
Hall, K. D. et al. Energy balance and its components: implications for body weight regulation. Am. J. Clin. Nutr. 95, 989–994 (2012).
Centers for Disease Control and Prevention. The health effects of overweight and obesity. CDC https://www.cdc.gov/healthyweight/effects/index.html (updated 5 Jun 2015).
Boyer, P. D. et al. Oxidative phosphorylation and photophosphorylation. Annu. Rev. Biochem. 46, 955–1026 (1977).
Atkinson, D. E. The energy charge of the adenylate pool as a regulatory parameter. Interaction with modifiers. Biochemistry 7, 4030–4034 (1968).
Carling, D., Zammit, V. A. & Hardie, D. G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 223, 217–222 (1987). This is the first paper to demonstrate that the same protein kinase activity (AMPK) phosphorylates and inactivates ACC and HMGR.
Munday, M. R., Campbell, D. G., Carling, D. & Hardie, D. G. Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur. J. Biochem. 175, 331–338 (1988). This is the first paper to formally cite AMPK.
Witters, L. A., Gao, G., Kemp, B. E. & Quistorff, B. Hepatic 5ʹ-AMP-activated protein kinase: zonal distribution and relationship to acetyl-CoA carboxylase activity in varying nutritional states. Arch. Biochem. Biophys. 308, 413–419 (1994). This is the fist paper indicating that AMPK is activated by caloric restriction.
Winder, W. W. & Hardie, D. G. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am. J. Physiol. 270, E299–E304 (1996). This is the first paper indicating that AMPK is activated by exercise.
Minokoshi, Y. et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415, 339–343 (2002).
Yamauchi, T. et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 8, 1288–1295 (2002). References 12 and 13 are the first papers linking AMPK with endocrine factors critical for controlling insulin sensitivity and fatty acid metabolism.
Hardie, D. G. AMPK: a target for drugs and natural products with effects on both diabetes and cancer. Diabetes 62, 2164–2172 (2013).
Zang, M. et al. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes 55, 2180–2191 (2006).
Brusq, J. M. et al. Inhibition of lipid synthesis through activation of AMP kinase: an additional mechanism for the hypolipidemic effects of berberine. J. Lipid Res. 47, 1281–1288 (2006).
Lee, Y. S. et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 55, 2256–2264 (2006).
Zhou, G. et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174 (2001).
Hawley, S. A. et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 336, 918–922 (2012). This paper provides evidence demonstrating that salicylate activates AMPK through direct interactions involving Ser108 within the β1 subunit.
Hawley, S. A. et al. The Na+/glucose co-transporter inhibitor canagliflozin activates AMP-activated protein kinase by inhibiting mitochondrial function and increasing cellular AMP levels. Diabetes 65, 2784–2794 (2016).
Villani, L. A. et al. The diabetes medication Canagliflozin reduces cancer cell proliferation by inhibiting mitochondrial complex-I supported respiration. Mol. Metab. 5, 1048–1056 (2016).
Xiao, B. et al. Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 4, 3017 (2013). This paper provides the full-length structure of mammalian AMPK and identifies the binding site for 991 and A769662 (ADaM site).
Langendorf, C. G. & Kemp, B. E. Choregraphy of AMPK activation. Cell Res. 25, 5–6 (2015).
Cokorinos, E. C. et al. Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab. 25, 1147–1159 (2017).
Esquejo, R. M. et al. Activation of liver AMPK with PF-06409577 corrects NAFLD and lowers cholesterol in rodent and primate preclinical models. EBioMedicine 31, 122–132 (2018).
Myers, R. W. et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 357, 507–511 (2017).
Salatto, C. T. et al. Selective activation of AMPK b1-containing isoforms improves kidney function in a rat model of diabetic nephropathy. J. Pharmacol. Exp. Ther. 361, 303–311 (2017). References 24–27 are the first papers describing the generation and characterization of potent ADaM site binding agents that increase AMPK and are effective for improving kidney function and lowering blood glucose, serum cholesterol and liver lipids.
Steneberg, P. et al. PAN-AMPK activator O304 improves glucose homeostasis and microvascular perfusion in mice and type 2 diabetes patients. JCI Insight 3, 99114 (2018). This paper describes the activity of O304 to protect against AMPK Thr172 dephosphorylation and to lower blood glucose and blood pressure in patients with type 2 diabetes taking metformin.
Sanders, M. J., Grondin, P. O., Hegarty, B. D., Snowden, M. A. & Carling, D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem. J. 403, 139–148 (2007).
Suter, M. et al. Dissecting the role of AMP for allosteric stimulation, activation and deactivation of AMP-activated protein kinase. J. Biol. Chem. 281, 32207–32216 (2006).
Hawley, S. A. et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2, 28 (2003).
Shaw, R. J. et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl Acad. Sci. USA 101, 3329–3335 (2004).
Woods, A. et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008 (2003). References 31–33 are a series of papers showing that the tumour suppressor LKB1 is the upstream kinase phosphorylating AMPK at Thr172.
Hawley, S. A. et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2, 9–19 (2005).
Hurley, R. L. et al. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 280, 29060–29066 (2005).
Woods, A. et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2, 21–33 (2005). References 34–36 are a series of papers showing that CAMKK2 can act as an upstream kinase phosphorylating AMPK at Thr172 in some cell types.
Xiao, B. et al. Structure of mammalian AMPK and its regulation by ADP. Nature 472, 230–233 (2011). This paper provides a partial AMPK structure, revealing mechanisms for nucleotide protection against dephosphorylation and showing that ADP, as well as AMP, activates AMPK.
Crute, B. E., Seefeld, K., Gamble, J., Kemp, B. E. & Witters, L. A. Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. J. Biol. Chem. 273, 35347–35354 (1998).
Goransson, O. et al. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J. Biol. Chem. 282, 32549–32560 (2007).
Pang, T. et al. Conserved a-helix acts as an autoinhibitory sequence in AMP-activated protein kinase a subunits. J. Biol. Chem. 282, 495–506 (2007).
Chen, L. et al. Conserved regulatory elements in AMPK. Nature 498, E8–E10 (2013).
Xin, F. J., Wang, J., Zhao, R. Q., Wang, Z. X. & Wu, J. W. Coordinated regulation of AMPK activity by multiple elements in the a subunit. Cell Res. 23, 1237–1240 (2013).
Hawley, S. A. et al. Phosphorylation by Akt within the ST loop of AMPK-alpha1 down-regulates its activation in tumour cells. Biochem. J. 459, 275–287 (2014).
Oakhill, J. S. et al. β-subunit myristoylation is the gatekeeper for initaiting metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc. Natl Sci. Acad. USA 107, 19237–19241 (2010).
Oakhill, J. S. et al. AMPK is a direct adenylate charge-regulated protein kinase. Science 332, 1433–1435 (2011). Together with reference 37, this paper describes the activation of AMPK by ADP.
Machovic, M. & Janecek, S. The evolution of putative starch-binding domains. FEBS Lett. 580, 6349–6356 (2006).
Xiao, B. et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449, 496–500 (2007).
Bateman, A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 22, 12–13 (1997).
Cheung, P. C. F., Salt, I. P., Davies, S. P., Hardie, D. G. & Carling, D. Characterization of AMP-activated protein kinase g-subunit isoforms and their role in AMP binding. Biochem. J. 346, 659–669 (2000).
Pinter, K. et al. Embryonic expression of AMPK γ subunits and the identification of a novel γ2 transcript variant in adult heart. J. Mol. Cell Cardiol. 53, 342–349 (2012).
Yu, H., Fujii, N., Hirshman, M. F., Pomerleau, J. M. & Goodyear, L. J. Cloning and characterization of mouse 5ʹ-AMP-activated protein kinase gamma3 subunit. Am. J. Physiol. Cell Physiol. 286, C283–C292 (2004).
Rajamohan, F. et al. Probing the enzyme kinetics, allosteric modulation and activation of α1- and α2-subunit-containing AMP-activated protein kinase (AMPK) heterotrimeric complexes by pharmacological and physiological activators. Biochem. J. 473, 581–592 (2016).
Ross, F. A., Jensen, T. E. & Hardie, D. G. Differential regulation by AMP and ADP of AMPK complexes containing different gamma subunit isoforms. Biochem. J. 473, 189–199 (2016).
Willows, R., Navaratnam, N., Lima, A., Read, J. & Carling, D. Effect of different γ-subunit isoforms on the regulation of AMPK. Biochem. J. 474, 1741–1754 (2017).
Carling, D. The AMP-activated protein kinase cascade-a unifying system for energy control. Trends Biochem. Sci. 29, 18–24 (2004).
Woods, A., Salt, I., Scott, J., Hardie, D. G. & Carling, D. The α1 and α2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro. FEBS Lett. 397, 347–351 (1996).
Wu, J. et al. Chemoproteomic analysis of intertissue and interspecies isoform diversity of AMP-activated protein kinase (AMPK). J. Biol. Chem. 288, 35904–35912 (2013).
Stephenne, X. et al. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia 54, 3101–3110 (2011).
Carling, D. AMPK signalling in health and disease. Curr. Opin. Cell Biol. 45, 31–37 (2017).
Carling, D., Mayer, F. V., Sanders, M. J. & Gamblin, S. J. AMP-activated protein kinase: nature’s energy sensor. Nat. Chem. Biol. 7, 512–518 (2011).
Hardie, D. G. & Carling, D. The AMP-activated protein kinase: fuel gauge of the mammalian cell. Eur. J. Biochem. 246, 259–273 (1997).
Davies, S. P., Helps, N. R., Cohen, P. T. & Hardie, D. G. 5ʹ-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 377, 421–425 (1995).
Hardie, D. G., Salt, I. P., Hawley, S. A. & Davies, S. P. AMP-activated protein kinase: an ultrasensitive system for monitoring cellular energy charge. Biochem. J. 338, 717–722 (1999).
Kemp, B., Oakhill, J. S. & Scott, J. W. AMPK structure and regulation from three angles. Structure 15, 1161–1163 (2007).
Chen, L. et al. AMP-activated protein kinase undergoes nucleotide-dependent conformational changes. Nat. Struct. Mol. Biol. 19, 716–718 (2012).
Anderson, K. A. et al. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 7, 377–388 (2008).
Stahmann, N., Woods, A., Carling, D. & Heller, R. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase beta. Mol. Cell. Biol. 26, 5933–5945 (2006).
Thornton, C., Sardini, A. & Carling, D. Muscarinic receptor activation of AMP-activated protein kinase inhibits orexigenic neuropeptide mRNA expression. J. Biol. Chem. 283, 17116–17122 (2008).
Andersson, U. et al. AMP-activated protein kinase plays a role in the control of food intake. J. Biol. Chem. 279, 12005–12008 (2004).
Zhang, C. S. et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112–116 (2017). This study reveals a mechanism involving FBP binding to aldolase for activation of AMPK in response to low glucose.
Lin, S. C. & Hardie, D. G. AMPK: sensing glucose as well as cellular energy status. Cell Metab. 27, 299–313 (2018).
Ingebritsen, T. S., Geelen, M. J., Parker, R. A., Evenson, K. J. & Gibson, D. M. Modulation of hydroxymethylglutaryl-CoA reductase activity, reductase kinase activity, and cholesterol synthesis in rat hepatocytes in response to insulin and glucagon. J. Biol. Chem. 254, 9986–9989 (1979).
Loh, K. et al. Inhibition of AMPK-HMGCR signaling leads to hypercholesterolemia, hepatic steatosis and insulin resistance. Hepatol. Commun. 3, 84–98 (2018).
Fullerton, M. D. et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 19, 1649–1654 (2013). This study provides genetic evidence in mice demonstrating that AMPK phosphorylation of both ACC1 and ACC2 is vital for controlling lipid synthesis and fatty acid oxidation and is effective for reducing NAFLD and insulin resistance.
O’Neill, H. M. et al. AMPK phosphorylation of ACC2 is required for skeletal muscle fatty acid oxidation and insulin sensitivity in mice. Diabetologia 57, 1693–1702 (2014).
Ye, J. & DeBose-Boyd, R. A. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb. Perspect. Biol. 3, a004754 (2011).
Lee, C. W. et al. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 72, 4394–4404 (2012).
Haeusler, R. A. et al. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat. Commun. 5, 5190 (2014).
Kawaguchi, T., Osatomi, K., Yamashita, H., Kabashima, T. & Uyeda, K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J. Biol. Chem. 277, 3829–3835 (2002).
Corton, J. M., Gillespie, J. G., Hawley, S. A. & Hardie, D. G. 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 229, 558–565 (1995).
Sullivan, J. E. et al. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 353, 33–36 (1994).
Mottillo, E. P. et al. Lack of adipocyte AMPK exacerbates insulin resistance and hepatic steatosis through brown and beige adipose tissue function. Cell Metab. 24, 118–129 (2016). This paper provides genetic evidence establishing an important role for AMPK in controlling brown and beige adipose tissue thermogenesis in mice and that this can be effective for reducing NAFLD and insulin resistance.
Miller, R. A. et al. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 494, 256–260 (2013).
Wu, Y. et al. Activation of AMPKalpha2 in adipocytes is essential for nicotine-induced insulin resistance in vivo. Nat. Med. 21, 373–382 (2015).
Rohm, M. et al. An AMP-activated protein kinase-stabilizing peptide ameliorates adipose tissue wasting in cancer cachexia in mice. Nat. Med. 22, 1120–1130 (2016).
Daval, M. et al. Anti-lipolytic action of AMP-activated protein kinase in rodent adipocytes. J. Biol. Chem. 280, 25250–25257 (2005).
Dzamko, N. et al. AMPK β1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. J. Biol. Chem. 285, 115–122 (2010).
MacPherson, R. E. et al. Reduced ATGL-mediated lipolysis attenuates beta-adrenergic-induced AMPK signaling, but not the induction of PKA-targeted genes, in adipocytes and adipose tissue. Am. J. Physiol. Cell Physiol. 311, C269–C276 (2016).
Mulligan, J. D., Gonzalez, A. A., Stewart, A. M., Carey, H. V. & Saupe, K. W. Upregulation of AMPK during cold exposure occurs via distinct mechanisms in brown and white adipose tissue of the mouse. J. Physiol. 580, 677–684 (2007).
Kim, S. J. et al. AMPK phosphorylates desnutrin/ATGL and hormone-sensitive lipase to regulate lipolysis and fatty acid oxidation within adipose tissue. Mol. Cell. Biol. 36, 1961–1976 (2016).
Jeppesen, J. et al. Contraction-induced skeletal muscle FAT/CD36 trafficking and FA uptake is AMPK independent. J. Lipid Res. 52, 699–711 (2011).
Momken, I. et al. A new leptin-mediated mechanism for stimulating fatty acid oxidation: a pivotal role for sarcolemmal FAT/CD36. Biochem. J. 474, 149–162 (2017).
Fentz, J. et al. AMPKα is critical for enhancing skeletal muscle fatty acid utilization during in vivo exercise in mice. FASEB J. 29, 1725–1738 (2015).
O’Neill, H. M. et al. Skeletal muscle ACC2 S212 phosphorylation is not required for the control of fatty acid oxidation during exercise. Physiol. Rep. 3, e12444 (2015).
Zordoky, B. N. et al. AMPK-dependent inhibitory phosphorylation of ACC is not essential for maintaining myocardial fatty acid oxidation. Circ. Res. 115, 518–524 (2014).
Hoffman, N. J. et al. Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise-regulated kinases and AMPK substrates. Cell Metab. 22, 922–935 (2015).
Schmitt, K. et al. Circadian control of DRP1 activity regulates mitochondrial dynamics and bioenergetics. Cell Metab. 27, 657–666 (2018).
Weir, H. J. et al. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. 26, 884–896 (2017).
O’Neill, H. M., Holloway, G. P. & Steinberg, G. R. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: implications for obesity. Mol. Cell Endocrinol. 366, 135–151 (2013).
Garcia, D. & Shaw, R. J. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 66, 789–800 (2017).
Lee, W. J. et al. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARα and PGC-1. Biochem. Biophys. Res. Commun. 340, 291–295 (2006).
Wan, Z. et al. Evidence for the role of AMPK in regulating PGC-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity (Silver Spring) 22, 730–738 (2014).
Leick, L. et al. PGC-1α is required for AICAR-induced expression of GLUT4 and mitochondrial proteins in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 299, E456–E465 (2010).
Zhang, H. et al. MicroRNA-455 regulates brown adipogenesis via a novel HIF1an-AMPK-PGC1α signaling network. EMBO Rep. 16, 1378–1393 (2015).
Ducommun, S. et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell. Signal. 27, 978–988 (2015).
Toyama, E. Q. et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281 (2016). References 105 and 106 are the first papers describing that AMPK phosphorylates MFF.
Lee, J. W., Park, S., Takahashi, Y. & Wang, H. G. The association of AMPK with ULK1 regulates autophagy. PLOS ONE 5, e15394 (2010).
Behrends, C., Sowa, M. E., Gygi, S. P. & Harper, J. W. Network organization of the human autophagy system. Nature 466, 68–76 (2010).
Kim, J., Kundu, M., Viollet, B. & Guan, K. L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 (2011).
Egan, D. F. et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461 (2011). References 109 and 110 are the first papers identifying how AMPK directly increases autophagy and mitophagy (independently of inhibiting mTOR) through phosphorylation of ULK1.
Weerasekara, V. K. et al. Metabolic-stress-induced rearrangement of the 14-3-3zeta interactome promotes autophagy via a ULK1- and AMPK-regulated 14-3-3zeta interaction with phosphorylated Atg9. Mol. Cell. Biol. 34, 4379–4388 (2014).
Zhang, D. et al. AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy 12, 1447–1459 (2016).
Inoki, K., Zhu, T. & Guan, K. L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590 (2003).
Gwinn, D. M. et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 (2008). References 113 and 114 are the first papers directly linking AMPK with the inhibition of mTOR signalling through phosphorylation of TSC2 and Raptor, thereby providing important connections with cell proliferation and growth.
Li, X. et al. Nucleus-translocated ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Mol. Cell 66, 684–697 (2017).
Young, N. P. et al. AMPK governs lineage specification through Tfeb-dependent regulation of lysosomes. Genes Dev. 30, 535–552 (2016).
Greer, E. L. et al. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 282, 30107–30119 (2007).
Zhao, J. et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 6, 472–483 (2007).
Mammucari, C. et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471 (2007).
Celestini, V. et al. Uncoupling FoxO3A mitochondrial and nuclear functions in cancer cells undergoing metabolic stress and chemotherapy. Cell Death Dis. 9, 231 (2018).
Barnes, B. R. et al. The 5ʹ-AMP-activated protein kinase γ3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J. Biol. Chem. 279, 38441–38447 (2004).
Jorgensen, S. B. et al. Knockout of the α2 but not α1 5ʹ-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J. Biol. Chem. 279, 1070–1079 (2004).
O’Neill, H. M. et al. AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl Acad. Sci. USA 108, 16092–16097 (2011).
Steinberg, G. R. et al. Whole body deletion of AMP-activated protein kinase β 2 reduces muscle AMPK activity and exercise capacity. J. Biol. Chem. 285, 37198–37209 (2010). References 121–124 identify the key AMPK isoforms required to stimulate glucose uptake in skeletal muscle and demonstrate an important role for AMPK in regulating exercise capacity.
Kurth-Kraczek, E. J., Hirshman, M. F., Goodyear, L. J. & Winder, W. W. 5’ AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes 48, 1667–1671 (1999).
Taylor, E. B. et al. Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J. Biol. Chem. 283, 9787–9796 (2008).
Treebak, J. T. et al. AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes 55, 2051–2058 (2006).
Liu, Y. et al. Phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) is an AMPK target participating in contraction-stimulated glucose uptake in skeletal muscle. Biochem. J. 455, 195–206 (2013).
Kim, J. H. et al. Phospholipase D1 mediates AMP-activated protein kinase signaling for glucose uptake. PLOS ONE 5, e9600 (2010).
Mihaylova, M. M. et al. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 145, 607–621 (2011).
McGee, S. L. et al. Compensatory regulation of HDAC5 in muscle maintains metabolic adaptive responses and metabolism in response to energetic stress. FASEB J. 28, 3384–3395 (2014).
Abbud, W. et al. Stimulation of AMP-activated protein kinase (AMPK) is associated with enhancement of Glut1-mediated glucose transport. Arch. Biochem. Biophys. 380, 347–352 (2000).
Fryer, L. G. et al. Characterization of the role of the AMP-activated protein kinase in the stimulation of glucose transport in skeletal muscle cells. Biochem. J. 363, 167–174 (2002).
Wu, N. et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 49, 1167–1175 (2013).
Shaked, M., Ketzinel-Gilad, M., Cerasi, E., Kaiser, N. & Leibowitz, G. AMP-activated protein kinase (AMPK) mediates nutrient regulation of thioredoxin-interacting protein (TXNIP) in pancreatic beta-cells. PLOS ONE 6, e28804 (2011).
Marsin, A. S. et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol. 10, 1247–1255 (2000).
Marsin, A. S., Bouzin, C., Bertrand, L. & Hue, L. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J. Biol. Chem. 277, 30778–30783 (2002).
Carling, D. & Hardie, D. G. The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim. Biophys. Acta 1012, 81–86 (1989).
Aschenbach, W. G. et al. Effect of AICAR treatment on glycogen metabolism in skeletal muscle. Diabetes 51, 567–573 (2002).
Hunter, R. W., Treebak, J. T., Wojtaszewski, J. F. & Sakamoto, K. Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes 60, 766–774 (2011).
Bultot, L. et al. AMP-activated protein kinase phosphorylates and inactivates liver glycogen synthase. Biochem. J. 443, 193–203 (2012).
Zibrova, D. et al. GFAT1 phosphorylation by AMPK promotes VEGF-induced angiogenesis. Biochem. J. 474, 983–1001 (2017).
Bergeron, R. et al. Effect of 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes 50, 1076–1082 (2001).
Lochhead, P. A., Salt, I. P., Walker, K. S., Hardie, D. G. & Sutherland, C. 5-Aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes 49, 896–903 (2000).
O’Brien, R. M. & Granner, D. K. Regulation of gene expression by insulin. Physiol. Rev. 76, 1109–1161 (1996).
Hughey, C. C. et al. Loss of hepatic AMP-activated protein kinase impedes the rate of glycogenolysis but not gluconeogenic fluxes in exercising mice. J. Biol. Chem. 292, 20125–20140 (2017).
Foretz, M. et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 120, 2355–2369 (2010). This paper provides evidence indicating that AMPK does not play a direct role in regulating hepatic glucose production.
Hasenour, C. M. et al. 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) effect on glucose production, but not energy metabolism, is independent of hepatic AMPK in vivo. J. Biol. Chem. 289, 5950–5959 (2014).
Johanns, M. et al. AMPK antagonizes hepatic glucagon-stimulated cyclic AMP signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4B. Nat. Commun. 7, 10856 (2016).
Bujak, A. L. et al. AMPK activation of muscle autophagy prevents fasting-induced hypoglycemia and myopathy during aging. Cell Metab. 21, 883–890 (2015).
Owen, M. R., Doran, E. & Halestrap, A. P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 348, 607–614 (2000).
Shu, Y. et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Invest. 117, 1422–1431 (2007).
Shaw, R. J. et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 (2005).
Jenkins, Y. et al. AMPK activation through mitochondrial regulation results in increased substrate oxidation and improved metabolic parameters in models of diabetes. PLOS ONE 8, e81870 (2013).
Hawley, S. A. et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 11, 554–565 (2010).
Park, S. J. et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 148, 421–433 (2012).
Price, N. L. et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 15, 675–690 (2012).
Pang, T. et al. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J. Biol. Chem. 283, 16051–16060 (2008).
Jensen, T. E. et al. PT-1 selectively activates AMPK-gamma1 complexes in mouse skeletal muscle, but activates all three gamma subunit complexes in cultured human cells by inhibiting the respiratory chain. Biochem. J. 467, 461–472 (2015).
Fryer, L. G., Parbu-Patel, A. & Carling, D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 277, 25226–25232 (2002).
Perry, R. J., Zhang, D., Zhang, X. M., Boyer, J. L. & Shulman, G. I. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347, 1253–1256 (2015).
Smith, B. K. et al. Salsalate (salicylate) uncouples mitochondria, improves glucose homeostasis, and reduces liver lipids independent of AMPK-β1. Diabetes 65, 3352–3361 (2016).
Sullivan, J. E., Carey, F., Carling, D. & Beri, R. K. Characterization of 5ʹ-AMP-activated protein kinase in human liver using specific peptide substrates and the effects of 5ʹ-AMP analogs on enzyme activity. Biochem. Biophys. Res. Commun. 200, 1551–1556 (1994).
Beckers, A. et al. Methotrexate enhances the antianabolic and antiproliferative effects of 5-aminoimidazole-4-carboxamide riboside. Mol. Cancer Ther. 5, 2211–2217 (2006).
Vincent, M., Marangos, P. & Gruber, H. & Van den Berghe, G. Inhibition by AICA riboside of gluconeogenesis in isolated rat hepatocytes. Diabetes 40, 1259–1266 (1991).
Longnus, S. L., Wambolt, R. B., Parsons, H. L., Brownsey, R. W. & Allard, M. F. 5-Aminoimidazole-4-carboxamide 1-beta-D-ribofuranoside (AICAR) stimulates myocardial glycogenolysis by allosteric mechanisms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 284, R936–R944 (2003).
Guigas, B. et al. AMP-activated protein kinase-independent inhibition of hepatic mitochondrial oxidative phosphorylation by AICA riboside. Biochem. J. 404, 499–507 (2007).
Gomez-Galeno, J. E. et al. A potent and selective AMPK activator that inhibits de novo lipogenesis. ACS Med. Chem. Lett. 1, 478–482 (2010).
Hunter, R. W. et al. Mechanism of action of compound-13: an alpha1-selective small molecule activator of AMPK. Chem. Biol. 21, 866–879 (2014).
Langendorf, C. G. et al. Structural basis of allosteric and synergistic activation of AMPK by furan-2-phosphonic derivative C2 binding. Nat. Commun. 7, 10912 (2016).
Bung, N. et al. 2-[2-(4-(trifluoromethyl)phenylamino)thiazol-4-yl]acetic acid (Activator-3) is a potent activator of AMPK. Sci. Rep. 8, 9599 (2018).
Cool, B. et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 3, 403–416 (2006). This is the first report of a small molecule (non-nucleotide) direct activator of AMPK.
Giordanetto, F. & Karis, D. Direct AMP-activated protein kinase activators: a review of evidence from the patent literature. Expert Opin. Ther. Pat. 22, 1467–1477 (2012).
Calabrese, M. F. et al. Structural basis for AMPK activation: natural and synthetic ligands regulate kinase activity from opposite poles by different molecular mechanisms. Structure 22, 1161–1172 (2014).
Sanders, M. J. et al. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J. Biol. Chem. 282, 32539–32548 (2007). This paper identifies Ser108 within the β1 subunit as an important modulator of AMPK activation by ADaM site activators.
Ngoei, K. R. W. et al. Structural determinants for small-molecule activation of skeletal muscle AMPK α2β2γ1 by the glucose importagog SC4. Cell Chem. Biol. 25, 728–737 (2018).
Dite, T. A. et al. The autophagy initiator ULK1 sensitizes AMPK to allosteric drugs. Nat. Commun. 18, 571 (2017).
Mitchelhill, K. I. et al. Posttranslational modifications of the 5ʹ-AMP-activated protein kinase b1 subunit. J. Biol. Chem. 272, 24475–24479 (1997).
Woods, A. et al. Identification of phosphorylation sites in AMP-activated protein kinase (AMPK) for upstream AMPK kinases and study of their roles by site-directed mutagenesis. J. Biol. Chem. 278, 28434–28442 (2003).
Willows, R. et al. Phosphorylation of AMPK by upstream kinases is required for activity in mammalian cells. Biochem. J. 474, 3059–3073 (2017).
Ford, R. J. et al. Metformin and salicylate synergistically activate liver AMPK, inhibit lipogenesis and improve insulin sensitivity. Biochem. J. 468, 125–132 (2015).
Scott, J. W. et al. Small molecule drug A-769662 and AMP synergistically activate naive AMPK independent of upstream kinase signaling. Chem. Biol. 21, 619–627 (2014).
Bultot, L. et al. Benzimidazole derivative small-molecule 991 enhances AMPK activity and glucose uptake induced by AICAR or contraction in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 311, E706–E719 (2016).
Timmermans, A. D. et al. A-769662 potentiates the effect of other AMP-activated protein kinase activators on cardiac glucose uptake. Am. J. Physiol. Heart Circ. Physiol. 306, H1619–H1630 (2014).
Ducommun, S. et al. Enhanced activation of cellular AMPK by dual-small molecule treatment: AICAR and A769662. Am. J. Physiol. Endocrinol. Metab. 306, E688–E696 (2014). References 181–185 establish the synergy for activating AMPK through both direct (β1 Ser108) and indirect (adenine nucleotide) mechanisms.
Boudaba, N. et al. AMPK re-activation suppresses hepatic steatosis but its downregulation does not promote fatty liver development. EBioMedicine 28, 194–209 (2018).
Boyle, K. E. et al. Maternal obesity alters fatty acid oxidation, AMPK activity, and associated DNA methylation in mesenchymal stem cells from human infants. Mol. Metab. 6, 1503–1516 (2017).
Ruderman, N. B., Carling, D., Prentki, M. & Cacicedo, J. M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Invest. 123, 2764–2772 (2013).
Minokoshi, Y. et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428, 569–574 (2004).
Claret, M. et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J. Clin. Invest. 117, 2325–2336 (2007).
Yavari, A. et al. Chronic activation of γ2 AMPK induces obesity and reduces β cell function. Cell Metab. 23, 821–836 (2016).
Galic, S. et al. AMPK signaling to acetyl-CoA carboxylase is required for fasting- and cold-induced appetite but not thermogenesis. eLife 7, e32656 (2018).
Oh, T. S., Cho, H., Cho, J. H., Yu, S. W. & Kim, E. K. Hypothalamic AMPK-induced autophagy increases food intake by regulating NPY and POMC expression. Autophagy 12, 2009–2025 (2016).
Kong, D. et al. A postsynaptic AMPK→p21-activated kinase pathway drives fasting-induced synaptic plasticity in AgRP neurons. Neuron 91, 25–33 (2016).
Yang, X. et al. Physiological expression of AMPKγ2RG mutation causes Wolff-Parkinson-White syndrome and induces kidney injury in mice. J. Biol. Chem. 291, 23428–23439 (2016).
Lopez, M. EJE PRIZE 2017: hypothalamic AMPK: a golden target against obesity? Eur. J. Endocrinol. 176, R235–R246 (2017).
Whittle, A. J. et al. BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell 149, 871–885 (2012).
Bain, J. et al. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315 (2007).
Dite, T. A. et al. AMP-activated protein kinase selectively inhibited by the type II inhibitor SBI-0206965. J. Biol. Chem. 293, 8874–8885 (2018).
Hutchinson, D. S., Chernogubova, E., Dallner, O. S., Cannon, B. & Bengtsson, T. Beta-adrenoceptors, but not alpha-adrenoceptors, stimulate AMP-activated protein kinase in brown adipocytes independently of uncoupling protein-1. Diabetologia 48, 2386–2395 (2005).
Wu, L. et al. AMP-activated protein kinase (AMPK) regulates energy metabolism through modulating thermogenesis in adipose tissue. Front. Physiol. 9, 122 (2018).
Yang, Q. et al. AMPK/α-ketoglutarate axis dynamically mediates DNA demethylation in the Prdm16 promoter and brown adipogenesis. Cell Metab. 24, 542–554 (2016).
Man, K., Loudon, A. & Chawla, A. Immunity around the clock. Science 354, 999–1003 (2016).
Abdul-Rahman, O. et al. AMP-activated kinase (AMPK) activation by AICAR in human white adipocytes derived from pericardial white adipose tissue stem cells induces a partial beige-like phenotype. PLOS ONE 11, e0157644 (2016).
Yan, M. et al. Chronic AMPK activation via loss of FLCN induces functional beige adipose tissue through PGC-1α/ERRα. Genes Dev. 30, 1034–1046 (2016).
Wang, S. et al. Resveratrol induces brown-like adipocyte formation in white fat through activation of AMP-activated protein kinase (AMPK) α1. Int. J. Obes. (Lond.) 39, 967–976 (2015).
Shan, T., Liang, X., Bi, P. & Kuang, S. Myostatin knockout drives browning of white adipose tissue through activating the AMPK-PGC1α-Fndc5 pathway in muscle. FASEB J. 27, 1981–1989 (2013).
Pollard, A. E. et al. AMPK activation protects against diet-induced obesity through Ucp1-independent thermogenesis in subcutaneous white adipose tissue. Nat. Metab. https://doi.org/10.1038/s42255-019-0036-9 (2019).
Gauthier, M. S. et al. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem. Biophys. Res. Commun. 404, 382–387 (2011).
Steinberg, G. R. et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 4, 465–474 (2006).
Qi, J. et al. Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue. EMBO J. 27, 1537–1548 (2008).
Samuel, V. T. & Shulman, G. I. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 27, 22–41 (2018).
Woods, A. et al. Liver-specific activation of AMPK prevents steatosis on a high-fructose diet. Cell Rep. 18, 3043–3051 (2017).
Smith, B. K. et al. Treatment of nonalcoholic fatty liver disease: role of AMPK. Am. J. Physiol. Endocrinol. Metab. 311, E730–E740 (2016).
Harriman, G. et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl Acad. Sci. USA 113, E1796–1805 (2016).
Svensson, R. U. et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 22, 1108–1119 (2016).
Lally, J. S. V. et al. Inhibition of acetyl-CoA carboxylase (ACC) by phosphorylation or by the liver-specific inhibitor, ND-654, suppresses lipogenesis and hepatocellular carcinoma. Cell. Metab. 29, 174–182 (2018).
Kim, C. W. et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 26, 394–406 (2017).
Muoio, D. M., Seefeld, K., Witters, L. A. & Coleman, R. A. AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem. J. 338, 783–791 (1999).
Galic, S. et al. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Invest. 121, 4903–4915 (2011).
Mounier, R. et al. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 18, 251–264 (2013).
Sag, D., Carling, D., Stout, R. D. & Suttles, J. Adenosine 5ʹ-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J. Immunol. 181, 8633–8641 (2008). References 219 and 221 are two independent studies showing that AMPK has an important anti-inflammatory role.
Pirkmajer, S. et al. Methotrexate promotes glucose uptake and lipid oxidation in skeletal muscle via AMPK activation. Diabetes 64, 360–369 (2015).
Wu, Y., Song, P., Xu, J., Zhang, M. & Zou, M. H. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J. Biol. Chem. 282, 9777–9788 (2007).
Suzuki, T. et al. Inhibition of AMPK catabolic action by GSK3. Mol. Cell. 50, 407–419 (2013).
Dagon, Y. et al. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab. 16, 104–112 (2012).
Steinberg, G. R. & Schertzer, J. D. AMPK promotes macrophage fatty acid oxidative metabolism to mitigate inflammation: implications for diabetes and cardiovascular disease. Immunol. Cell Biol. 92, 340–345 (2014).
O’Neill, L. A. & Hardie, D. G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493, 346–355 (2013).
Cao, Q. et al. Myeloid deletion of α1AMPK exacerbates atherosclerosis in LDL receptor knockout (LDLRKO) mice. Diabetes 65, 1565–1576 (2016).
Lamia, K. A. et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 326, 437–440 (2009).
Dai, X., Ding, Y., Liu, Z., Zhang, W. & Zou, M. H. Phosphorylation of CHOP (C/EBP homologous protein) by the AMP-activated protein kinase alpha 1 in macrophages promotes CHOP degradation and reduces injury-induced neointimal disruption in vivo. Circ. Res. 119, 1089–1100 (2016).
Rutherford, C. et al. Phosphorylation of Janus kinase 1 (JAK1) by AMP-activated protein kinase (AMPK) links energy sensing to anti-inflammatory signaling. Sci. Signal 9, ra109 (2016).
Ma, P. F. et al. Cytotherapy with M1-polarized macrophages ameliorates liver fibrosis by modulating immune microenvironment in mice. J. Hepatol. 67, 770–779 (2017).
Kjobsted, R. et al. AMPK in skeletal muscle function and metabolism. FASEB J. 32, 1741–1777 (2018).
Cuthbertson, D. J. et al. 5-Aminoimidazole-4-carboxamide 1-beta-D-ribofuranoside acutely stimulates skeletal muscle 2-deoxyglucose uptake in healthy men. Diabetes 56, 2078–2084 (2007).
Babraj, J. A. et al. Blunting of AICAR-induced human skeletal muscle glucose uptake in type 2 diabetes is dependent on age rather than diabetic status. Am. J. Physiol. Endocrinol. Metab. 296, E1042–E1048 (2009).
Bosselaar, M., Smits, P., van Loon, L. J. & Tack, C. J. Intravenous AICAR during hyperinsulinemia induces systemic hemodynamic changes but has no local metabolic effect. J. Clin. Pharmacol. 51, 1449–1458 (2011).
Marcinko, K. et al. The AMPK activator R419 improves exercise capacity and skeletal muscle insulin sensitivity in obese mice. Mol. Metab. 4, 643–651 (2015).
Barre, L. et al. Genetic model for the chronic activation of skeletal muscle AMP-activated protein kinase leads to glycogen accumulation. Am. J. Physiol. Endocrinol. Metab. 292, E802–E811 (2007).
Narkar, V. A. et al. AMPK and PPARδ agonists are exercise mimetics. Cell 134, 405–415 (2008).
Libby, P., Ridker, P. M. & Hansson, G. K. & Leducq Transatlantic Network on Atherothrombosis. Inflammation in atherosclerosis: from pathophysiology to practice. J. Am. Coll. Cardiol. 54, 2129–2138 (2009).
Dong, Y. et al. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes 59, 1386–1396 (2010).
Ding, Y. et al. AMP-activated protein kinase alpha 2 deletion induces VSMC phenotypic switching and reduces features of atherosclerotic plaque stability. Circ. Res. 119, 718–730 (2016).
Cai, Z. et al. Ablation of adenosine monophosphate-activated protein kinase alpha1 in vascular smooth muscle cells promotes diet-induced atherosclerotic calcification in vivo. Circ. Res. 119, 422–433 (2016).
Dong, Y. et al. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 121, 792–803 (2010).
Wang, Q. et al. Activation of AMP-activated protein kinase is required for berberine-induced reduction of atherosclerosis in mice: the role of uncoupling protein 2. PLOS ONE 6, e25436 (2011).
Ma, A., Wang, J., Yang, L., An, Y. & Zhu, H. AMPK activation enhances the anti-atherogenic effects of high density lipoproteins in apoE−/− mice. J. Lipid Res. 58, 1536–1547 (2017).
Pinkosky, S. L. et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat. Commun. 7, 13457 (2016).
Xu, T. et al. Effects of metformin on metabolite profiles and LDL cholesterol in patients with type 2 diabetes. Diabetes Care 38, 1858–1867 (2015).
Ballantyne, C. M. et al. Efficacy and safety of bempedoic acid added to ezetimibe in statin-intolerant patients with hypercholesterolemia: a randomized, placebo-controlled study. Atherosclerosis 277, 195–203 (2018).
Fullerton, M. D. et al. Salicylate improves macrophage cholesterol homeostasis via activation of Ampk. J. Lipid Res. 56, 1025–1033 (2015).
Mo, C. et al. Fat mass and obesity-associated protein attenuates lipid accumulation in macrophage foam cells and alleviates atherosclerosis in apolipoprotein E-deficient mice. J. Hypertens. 35, 810–821 (2017).
Ouimet, M. et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J. Clin. Invest. 125, 4334–4348 (2015).
Robbins, C. S. et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 19, 1166–1172 (2013).
Wang, J., Ma, A., Zhao, M. & Zhu, H. AMPK activation reduces the number of atheromata macrophages in ApoE deficient mice. Atherosclerosis 258, 97–107 (2017).
Zhang, M. et al. AMP-activated protein kinase alpha1 promotes atherogenesis by increasing monocyte-to-macrophage differentiation. J. Biol. Chem. 292, 7888–7903 (2017).
Martinet, W., De Loof, H. & De Meyer, G. R. mTOR inhibition: a promising strategy for stabilization of atherosclerotic plaques. Atherosclerosis 233, 601–607 (2014).
Hauser, T. H. et al. Effect of targeting inflammation with salsalate: the TINSAL-CVD randomized clinical trial on progression of coronary plaque in overweight and obese patients using statins. JAMA Cardiol. 1, 413–423 (2016).
Salastekar, N. et al. Salsalate improves glycaemia in overweight persons with diabetes risk factors of stable statin-treated cardiovascular disease: a 30-month randomized placebo-controlled trial. Diabetes Obes. Metab. 19, 1458–1462 (2017).
Ford, R. J. et al. AMP-activated protein kinase activator AICAR acutely lowers blood pressure and relaxes isolated resistance arteries of hypertensive rats. J. Hypertens. 30, 725–733 (2012).
Chen, Z. P. et al. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 443, 285–289 (1999).
Zhang, J. et al. AMPK phosphorylation of ACE2 in endothelium mitigates pulmonary hypertension. Am. J. Respir. Crit. Care Med. 198, 509–520 (2018).
Schneider, H. et al. AMPK dilates resistance arteries via activation of SERCA and BKCa channels in smooth muscle. Hypertension 66, 108–116 (2015).
Kim, T. T. & Dyck, J. R. Is AMPK the savior of the failing heart? Trends Endocrinol. Metab. 26, 40–48 (2015).
Sung, M. M. et al. AMPK deficiency in cardiac muscle results in dilated cardiomyopathy in the absence of changes in energy metabolism. Cardiovasc. Res. 107, 235–245 (2015).
Xie, C. et al. Genome editing with CRISPR/Cas9 in postnatal mice corrects PRKAG2 cardiac syndrome. Cell Res. 26, 1099–1111 (2016).
Yavari, A. et al. Mammalian gamma2 AMPK regulates intrinsic heart rate. Nat. Commun. 8, 1258 (2017).
Gelinas, R. et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat. Commun. 9, 374 (2018).
Russell, R. R. 3rd et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Invest. 114, 495–503 (2004).
Xing, Y. et al. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J. Biol. Chem. 278, 28372–28377 (2003).
Cao, Y. et al. Activation of γ2-AMPK suppresses ribosome biogenesis and protects against myocardial ischemia/reperfusion injury. Circ. Res. 121, 1182–1191 (2017).
Li, J., Jiang, P., Robinson, M., Lawrence, T. S. & Sun, Y. AMPK-β1 subunit is a p53-independent stress responsive protein that inhibits tumor cell growth upon forced expression. Carcinogenesis 24, 827–834 (2003).
Shaw, R. J. et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6, 91–99 (2004).
Banskota, S., Regmi, S. C. & Kim, J. A. NOX1 to NOX2 switch deactivates AMPK and induces invasive phenotype in colon cancer cells through overexpression of MMP-7. Mol. Cancer 14, 123 (2015).
Pineda, C. T. et al. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 160, 715–728 (2015).
Vila, I. K. et al. A UBE2O-AMPKα2 axis that promotes tumor initiation and progression offers opportunities for therapy. Cancer Cell 31, 208–224 (2017).
He, X., Li, C., Ke, R., Luo, L. & Huang, D. Down-regulation of adenosine monophosphate-activated protein kinase activity: a driver of cancer. Tumour Biol. https://doi.org/10.1177/1010428317697576 (2017).
Faubert, B. et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 17, 113–124 (2013). This is the first paper using genetic loss of function indicating that AMPK reduces tumour growth in mice.
Houde, V. P. et al. AMPK β1 reduces tumor progression and improves survival in p53-null mice. Mol. Oncol. 11, 1143–1155 (2017).
O’Brien, A. J. et al. Salicylate activates AMPK and synergizes with metformin to reduce the survival of prostate and lung cancer cells ex vivo through inhibition of de novo lipogenesis. Biochem. J. 469, 177–187 (2015).
Zadra, G. et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 6, 519–538 (2014).
Griss, T. et al. Metformin antagonizes cancer cell proliferation by suppressing mitochondrial-dependent biosynthesis. PLOS Biol. 13, e1002309 (2015).
Scaglia, N., Tyekucheva, S., Zadra, G., Photopoulos, C. & Loda, M. De novo fatty acid synthesis at the mitotic exit is required to complete cellular division. Cell Cycle 13, 859–868 (2014).
Vincent, E. E. et al. Differential effects of AMPK agonists on cell growth and metabolism. Oncogene 34, 3627–3639 (2015).
Cha, J. H. et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol. Cell 71, 606–620 (2018).
Wu, D. et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 559, 637–641 (2018). This paper identifies AMPK as a potential link between glucose metabolism and epigenetic regulation.
Hopkins, B. D. et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503 (2018).
Wang, W. et al. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 17, 490–499 (2015).
Li, Y. H. et al. AMP-activated protein kinase directly phosphorylates and destabilizes hedgehog pathway transcription factor GLI1 in medulloblastoma. Cell Rep. 12, 599–609 (2015).
Shen, C. H. et al. Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation. Mol. Cell 52, 161–172 (2013).
Imamura, K., Ogura, T., Kishimoto, A., Kaminishi, M. & Esumi, H. Cell cycle regulation via p53 phosphorylation by a 5ʹ-AMP activated protein kinase activator, 5-aminoimidazole- 4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem. Biophys. Res. Commun. 287, 562–567 (2001).
Jones, R. G. et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293 (2005).
Dasgupta, B. & Milbrandt, J. AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Dev. Cell 16, 256–270 (2009).
Liang, J. et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 9, 218–224 (2007).
Banko, M. R. et al. Chemical genetic screen for AMPKα2 substrates uncovers a network of proteins involved in mitosis. Mol. Cell 44, 878–892 (2011).
Schaffer, B. E. et al. Identification of AMPK phosphorylation sites reveals a network of proteins involved in cell invasion and facilitates large-scale substrate prediction. Cell Metab. 22, 907–921 (2015).
Aznar, N. et al. AMP-activated protein kinase fortifies epithelial tight junctions during energetic stress via its effector GIV/Girdin. eLife 5, 20795 (2016).
Shackelford, D. B. et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 23, 143–158 (2013).
Eichner, L. J. et al. Genetic analysis reveals AMPK is required to support tumor growth in murine kras-dependent lung cancer models. Cell Metab. https://doi.org/10.1016/j.cmet.2018.10.005 (2018).
Liu, L. et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature 483, 608–612 (2012).
Jeon, S. M., Chandel, N. S. & Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665 (2012).
Sanduja, S. et al. AMPK promotes tolerance to Ras pathway inhibition by activating autophagy. Oncogene 35, 5295–5303 (2016).
Dial, A. G., Ng, S. Y., Manta, A. & Ljubicic, V. The role of AMPK in neuromuscular biology and disease. Trends Endocrinol. Metab. 29, 300–312 (2018).
Lantier, L. et al. AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB J. 28, 3211–3224 (2014).
Rockl, K. S. et al. Skeletal muscle adaptation to exercise training: AMP-activated protein kinase mediates muscle fiber type shift. Diabetes 56, 2062–2069 (2007).
Al-Rewashdy, H., Ljubicic, V., Lin, W., Renaud, J. M. & Jasmin, B. J. Utrophin A is essential in mediating the functional adaptations of mdx mouse muscle following chronic AMPK activation. Hum. Mol. Genet. 24, 1243–1255 (2015).
Dial, A. G. et al. The role of AMP-activated protein kinase in the expression of the dystrophin-associated protein complex in skeletal muscle. FASEB J. 32, 2950–2965 (2018).
Thomas, M. M. et al. Muscle-specific AMPK β1β2-null mice display a myopathy due to loss of capillary density in nonpostural muscles. FASEB J. 28, 2098–2107 (2014).
Chen, Z. P. et al. AMPK signaling in contracting human skeletal muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. Am. J. Phys. Endocrinol. Metab. 279, E1202–E1206 (2000).
Bradley, E. A. et al. Activation of AMP-activated protein kinase by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside in the muscle microcirculation increases nitric oxide synthesis and microvascular perfusion. Arterioscler. Thromb. Vasc. Biol. 30, 1137–1142 (2010).
Baltgalvis, K. A. et al. Exercise performance and peripheral vascular insufficiency improve with AMPK activation in high-fat diet-fed mice. Am. J. Physiol. Heart Circ. Physiol. 306, H1128–H1145 (2014).
Lee, M. et al. Phosphorylation of Acetyl-CoA carboxylase by AMPK reduces renal fibrosis and is essential for the anti-fibrotic effect of metformin. J. Am. Soc. Nephrol. 29, 2326–2336 (2018).
Megat, S. & Price, T. J. Therapeutic opportunities for pain medicines via targeting of specific translation signaling mechanisms. Neurobiol. Pain 4, 8–19 (2018).
Russe, O. Q. et al. Activation of the AMP-activated protein kinase reduces inflammatory nociception. J. Pain 14, 1330–1340 (2013).
Maixner, D. W., Yan, X., Gao, M., Yadav, R. & Weng, H. R. Adenosine monophosphate-activated protein kinase regulates interleukin-1β expression and glial glutamate transporter function in rodents with neuropathic pain. Anesthesiology 122, 1401–1413 (2015).
Li, M. et al. Reduced AMPK-ACC and mTOR signaling in muscle from older men, and effect of resistance exercise. Mech. Ageing Dev. 133, 655–664 (2012).
Reznick, R. M. et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 5, 151–156 (2007).
Qiang, W., Weiqiang, K., Qing, Z., Pengju, Z. & Yi, L. Aging impairs insulin-stimulated glucose uptake in rat skeletal muscle via suppressing AMPKα. Exp. Mol. Med. 39, 535–543 (2007).
Park, S. J. et al. DNA-PK promotes the mitochondrial, metabolic, and physical decline that occurs during aging. Cell Metab. 25, 1135–1146 (2017).
Martin-Montalvo, A. et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 4, 2192 (2013).
Cuervo, A. M. & Dice, J. F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 275, 31505–31513 (2000).
Kim, Y. A., Kim, Y. S., Oh, S. L., Kim, H. J. & Song, W. Autophagic response to exercise training in skeletal muscle with age. J. Physiol. Biochem. 69, 697–705 (2013).
Lipinski, M. M. et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl Acad. Sci. USA 107, 14164–14169 (2010).
Chen, J. et al. Metformin extends C. elegans lifespan through lysosomal pathway. eLife 6, 31268 (2017).
Navratil, M., Terman, A. & Arriaga, E. A. Giant mitochondria do not fuse and exchange their contents with normal mitochondria. Exp. Cell Res. 314, 164–172 (2008).
Terman, A., Kurz, T., Navratil, M., Arriaga, E. A. & Brunk, U. T. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 12, 503–535 (2010).
Crane, J. D. et al. Exercise-stimulated interleukin-15 is controlled by AMPK and regulates skin metabolism and aging. Aging Cell 14, 625–634 (2015).
Schafer, M. J., Miller, J. D. & LeBrasseur, N. K. Cellular senescence: implications for metabolic disease. Mol. Cell. Endocrinol. 455, 93–102 (2017).
Erickson, M. L., Little, J. P., Gay, J. L., McCully, K. K. & Jenkins, N. T. Postmeal exercise blunts postprandial glucose excursions in people on metformin monotherapy. J. Appl. Physiol. 123, 444–450 (2017).
Brown, M. S., Brunschede, G. Y. & Goldstein, J. L. Inactivation of 3-hyroxy-3-methylglutaryl coenzyme A reductase in vitro. J. Biol. Chem. 250, 2502–2509 (1975).
Harwood, H. J. Jr., Brandt, K. G. & Rodwell, V. W. Allosteric activation of rat liver cytosolic 3-hydroxy-3-methylglutaryl coenzyme A reductase kinase by nucleoside diphosphates. J. Biol. Chem. 259, 2810–2815 (1984).
Beg, Z. H., Stonik, J. A. & Brewer, H. B. Jr. Characterization and regulation of reductase kinase, a protein kinase that modulates the enzymic activity of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. Proc. Natl Acad. Sci. USA 76, 4375–4379 (1979).
Ingebritsen, T. S., Parker, R. A. & Gibson, D. M. Regulation of liver hydroxymethylglutaryl-CoA reductase by a bicyclic phosphorylation system. J. Biol. Chem. 256, 1138–1144 (1981).
Yeh, L., Lee, K. & Kim, K. Regulation of rat liver acetyl-CoA carboxylase. Regulation of phosphorylation and inactivation of acetyl-CoA carboxylase by adenylate energy charge. J. Biol. Chem. 255, 2308–2134 (1980).
Carling, D., Clarke, P. R., Zammit, V. A. & Hardie, D. G. Purification and characterisation of the AMP-activated protein kinase. Eur. J. Biochem. 186, 129–136 (1989).
Davies, S. P., Carling, D. & Hardie, D. G. Tissue distribution of AMP-activated protein kinase, and lack of activation by cyclic AMP-dependent protein kinase, studied using a specific and sensitive peptide assay. Eur. J. Biochem. 186, 123–128 (1989).
Guigas, B. et al. 5-Aminoimidazole-4-4carboxamide-1-beta-D-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on translocation. Diabetes 55, 865–874 (2006).
Milan, D. et al. A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 288, 1248–1251 (2000).
Arad, M., Seidman, C. E. & Seidman, J. G. AMP-activated protein kinase in the heart: role during health and disease. Circ. Res. 100, 474–488 (2007).
Kim, M. et al. Mutation in the γ2-subunit of AMP-activated protein kinase stimulates cardiomyocyte proliferation and hypertrophy independent of glycogen storage. Circ. Res. 114, 966–975 (2014).
Schönke, M., Myers, M. G., Zierath, J. R. & Björnholm, M. Skeletal muscle AMP-activated protein kinase g1H151R overexpression enhances whole body energy homeostasis and insulin sensitivity. Am. J. Physiol. 309, E679–E690 (2015).
Hardie, D. G., Schaffer, B. E. & Brunet, A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 26, 190–201 (2016).
Ahn, J., Lee, H., Kim, S., Park, J. & Ha, T. The anti-obesity effect of quercetin is mediated by the AMPK and MAPK signaling pathways. Biochem. Biophys. Res. Commun. 373, 545–549 (2008).
Acknowledgements
This work was supported by grants from the Canadian Institutes of Health Research (201709FDN-CEBA-116200 to G.R.S.), Diabetes Canada (DI-5-17-5302-GS) and the Medical Research Council UK (grant MC-A654-5QB10 to D.C.). G.R.S. is supported by a Canada Research Chair and a J. Bruce Duncan Chair in Metabolic Diseases.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
G.R.S. has received research funding from Esperion Therapeutics and Rigel Pharmaceuticals, reagents from Pfizer and Merck, and honoraria and/or consulting fees from Astra Zeneca, Eli-Lilly, Esperion Therapeutics, Novo Nordisk, Poxel, Pfizer, Merck, Rigel and Terns.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Scansite: http://scansite.mit.edu
Glossary
- Cardiovascular disease
-
(CVD). A term encompassing diseases affecting the heart or circulatory system.
- Non-alcoholic fatty liver disease
-
(NAFLD). A very common disease in humans in which there is an excessive accumulation of fat in the liver (steatosis) in individuals who are not alcoholic.
- Cystathionine-β-synthase (CBS) domains
-
Small protein domains (typically ~60 amino acids) that usually occur as tandem repeats, sometimes referred to as a Bateman domain, and that often bind to nucleotide or nucleotide-like molecules. AMP-activated protein kinase γ subunits contain four CBS domains, three of which bind adenine nucleotides.
- Autophagy
-
A process by which organisms degrade organelles and macromolecules including proteins and recycle nutrients in response to starvation.
- Lipogenesis
-
A metabolic pathway for the synthesis of fatty acids and triglycerides.
- Mitophagy
-
Similar to autophagy, but refers specifically to the process by which cells turnover mitochondria.
- Allosteric activator
-
A molecule that activates an enzyme by binding at a site distinct from the active site.
- Gluconeogenesis
-
A metabolic pathway for the synthesis of glucose from precursor substrates such as lactate and amino acids.
- Non-alcoholic steatohepatitis
-
(NASH). A severe form of non-alcoholic fatty liver disease in which the liver becomes inflamed.
- Thermogenesis
-
A process by which cells generate heat.
- Atherosclerosis
-
The formation of solid plaques within arteries that block normal blood flow.
- Wolff–Parkinson–White syndrome
-
An electrical conductance abnormality that leads to increased heart rate (tachycardia); mutations in AMP-activated protein kinase γ2 are often associated with this syndrome.
- Nociceptors
-
Neurons that respond to damaging stimuli and transmit a response to the brain that is perceived as pain.
- Pharmacokinetics
-
How an organism processes a drug.
- Pharmacodynamics
-
How drugs affect an organism.
Rights and permissions
About this article

Cite this article
Steinberg, G.R., Carling, D. AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov 18, 527–551 (2019). https://doi.org/10.1038/s41573-019-0019-2
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-019-0019-2
This article is cited by
-
Regulation of autophagy by perilysosomal calcium: a new player in β-cell lipotoxicity
Experimental & Molecular Medicine (2024)
-
The rapid proximity labeling system PhastID identifies ATP6AP1 as an unconventional GEF for Rheb
Cell Research (2024)
-
Direct effects of adipocyte lipolysis on AMPK through intracellular long-chain acyl-CoA signaling
Scientific Reports (2024)
-
Diet-inducing hypercholesterolemia show decreased O-GlcNAcylation of liver proteins through modulation of AMPK
Journal of Physiology and Biochemistry (2024)
-
AMPK pathway: an emerging target to control diabetes mellitus and its related complications
Journal of Diabetes & Metabolic Disorders (2024)
