
Abstract
Kaposi sarcoma (KS) gained public attention as an AIDS-defining malignancy; its appearance on the skin was a highly stigmatizing sign of HIV infection during the height of the AIDS epidemic. The widespread introduction of effective antiretrovirals to control HIV by restoring immunocompetence reduced the prevalence of AIDS-related KS, although KS does occur in individuals with well-controlled HIV infection. KS also presents in individuals without HIV infection in older men (classic KS), in sub-Saharan Africa (endemic KS) and in transplant recipients (iatrogenic KS). The aetiologic agent of KS is KS herpesvirus (KSHV; also known as human herpesvirus-8), and viral proteins can induce KS-associated cellular changes that enable the virus to evade the host immune system and allow the infected cell to survive and proliferate despite viral infection. Currently, most cases of KS occur in sub-Saharan Africa, where KSHV infection is prevalent owing to transmission by saliva in childhood compounded by the ongoing AIDS epidemic. Treatment for early AIDS-related KS in previously untreated patients should start with the control of HIV with antiretrovirals, which frequently results in KS regression. In advanced-stage KS, chemotherapy with pegylated liposomal doxorubicin or paclitaxel is the most common treatment, although it is seldom curative. In sub-Saharan Africa, KS continues to have a poor prognosis. Newer treatments for KS based on the mechanisms of its pathogenesis are being explored.
Similar content being viewed by others

Introduction
Kaposi sarcoma (KS) was first reported in 1872 by Moritz Kaposi, a physician and dermatologist. He described several cases of a multifocal pigmented sarcoma of the skin in elderly European men, all of who died within 2 years1. Four main epidemiological forms of KS are now widely recognized (Table 1). The form of KS originally identified by Kaposi became known as classic KS or sporadic KS. Classic KS occurs mostly in elderly men of Mediterranean or Jewish ancestry and, unlike the cases originally described by Kaposi, typically shows an indolent, protracted clinical course and primarily affects skin on the legs. Starting in 1947, several reports documented cases of KS in Africa, including a lymphadenopathic form of KS in children2,3,4; this form of KS is now generally referred to as endemic KS. KS came to the forefront of public attention at the onset of the AIDS epidemic, and the first report of highly aggressive KS affecting young men who have sex with men (MSM)5, in 1981, occurred just before the realization that these men were severely immunodeficient and affected by opportunistic infections. This type of KS is now known as AIDS-related KS or epidemic KS. Note that, although KS is more commonly associated with HIV-1 infection than with HIV-2 infection6, we refer to HIV in general in this article as it cannot be ruled out that individuals infected with HIV-2 were included in the studies discussed. KS also occurs in individuals with iatrogenic immunodeficiency, such as that seen in organ transplant recipients; this type of KS is known as iatrogenic KS7,8,9. Finally, of note, many cases of KS have been reported in MSM without HIV infection10, and KS in MSM without HIV infection is increasingly being recognized as a possible distinct fifth form of KS11,12,13.
The cause of KS was not known until 1994 when, on the basis of epidemiologic suggestions that this cancer had an infectious origin independent of HIV, a directed search led to the discovery of the KS herpesvirus (KSHV; also known as human herpesvirus-8 (HHV-8))14. It is now known that a combination of KSHV infection and impaired host immunity causes KS but, although AIDS-related KS and iatrogenic KS are associated with well-defined immunodeficiency, the impaired immune function in classic KS (thought to relate to ‘immunosenescence’; that is, an ageing immune system) and endemic KS (thought to relate to chronic infection and malnutrition) is not well characterized. In addition to KS, KSHV causes two lymphoproliferative disorders — primary effusion lymphoma (PEL)15 and multicentric Castleman disease (MCD)16 — and an inflammatory syndrome called KSHV inflammatory cytokine syndrome17 (Box 1).
In this Primer, we describe the epidemiological and clinical features of the different epidemiological forms of KS and discuss our current understanding of the pathobiology of this disease. Treatment approaches for managing KS and improving quality of life (QOL) will also be considered.
Epidemiology
KS was a rare disease before the AIDS epidemic in the early 1980s, when the reported incidence of classic KS ranged from 0.01 per 100,000 person-years for the UK18 and 0.2 per 100,000 person-years for the USA to 1.6 per 100,000 person-years for Sardinia19; incidence was 2–3-fold higher in men than in women globally. Reported estimated incidence rates for endemic KS in Africa before the AIDS epidemic were higher for Zaire, Uganda, Tanzania and Cameroon (>6 per 1,000 person-years) than in southern and north Africa (0.5–1.5 per 1,000 person-years)20. To date, most studies of KS in Africa (comprising endemic and AIDS-related KS) reported KS as a percentage of all malignancies owing to a scarcity of population-based studies.
Incidence of KS is reported to currently be ~200-fold higher in recipients of solid-organ transplants (that is, in iatrogenic KS) than in the general population9. Furthermore, rates of iatrogenic KS in transplant recipients positively correlate with the prevalence of KSHV and rates of classic KS based on the area where the transplant recipient lives, and iatrogenic KS is also associated with the male sex and increased age21. Indeed, overall, the geographical variation in the incidence of KS is now known to reflect differences in the prevalence of KSHV22 (Fig. 1).

a | The age-standardized incidence rate of Kaposi sarcoma (KS) per 100,000 males is depicted, and rates (apart from for the USA) were obtained from the International Agency for Research on Cancer (IARC) Cancer Incidence in Five Continents Volume X1 and ‘Cancer Today’ Global Cancer Observatory resources255,256. The rate provided for the USA is an average for 2000–2015 (0.7 affected individuals per 100,000 males) and rates are from Surveillance, Epidemiology, and End Results (SEER). However, rates in some regions based on the population reported are higher than others, ranging from 1.7 affected individuals per 100,000 males (for Atlanta) to 0.1 affected individuals per 100,000 males (for Iowa and Utah). Overall rates in the USA show racial disparities: among non-Hispanic white individuals, white Hispanics and black individuals, the incidence rate is 0.4, 0.7 and 1 affected individual per 100,000 males, respectively. b | Seroprevalence rates were compiled from multiple studies6,39,180,257,258,259,260,261,262,263,264,265,266,267,268,269,270,271,272,273,274,275,276,277,278,279,280,281,282,283,284,285,286,287,288,289,290,291,292,293,294. The seroprevalence of KS herpesvirus (KSHV) infection in northern Europe, Asia and the USA is <10%, but in most of sub-Saharan Africa, overall seroprevalence is >40%. The Mediterranean region (that is, Italy, Sicily and Sardinia) has intermediate seroprevalence rates of 10–30%. Figure adapted from ref.22, Springer Nature Limited.
Prevalence of KSHV is highest in sub-Saharan Africa, where in some populations the prevalence in adults is >90%. In the Mediterranean, prevalence is 20–30%, and in northern Europe, Asia and the USA, prevalence is <10%. The reasons for the geographical variation in KSHV prevalence are not understood, but there is some evidence that environmental factors, such as co-infection with malaria and other parasitic infections, may increase shedding of KSHV in saliva, thereby increasing transmission rates23. Although these co-infections were reported to increase seropositivity for KSHV23, their effect on saliva shedding has not been convincingly demonstrated. It is also possible that saliva sharing is more common in highly endemic areas, where infection is acquired in childhood through practices such as the premastication of food for infants, candy sharing among children and the sharing of toothbrushes24.
In the early 1980s, one of the first signs of the emerging AIDS epidemic was the frequent occurrence of KS in MSM5,25. In the USA, KS was reported to be 20,000 times more frequent in patients with AIDS than in the general population and to be 300 times more frequent in patients with AIDS than in other immune-suppressed patient groups26. Similar patterns of KS risk were reported in Europe and Australia27,28. Subsequent studies in the USA and Europe showed that the prevalence of KSHV in MSM is higher than the prevalence of KSHV in other HIV-infected risk groups29. In sub-Saharan Africa, the incidence of KS has increased around 20-fold since the onset of the AIDS epidemic in the early 1980s, making KS the most common cancer in men, and the second most common cancer in women, in countries such as Uganda, Malawi, Zimbabwe and Swaziland30,31.
The introduction of combination antiretroviral therapy (cART) in 1996 dramatically decreased the incidence of AIDS-related KS32. Indeed, an international study reporting cancer incidence data from 23 prospective studies from the USA, Europe and Australia revealed that the overall incidence of KS in these countries decreased from 15.2 per 1,000 person-years in 1992 to 4.9 per 1,000 person-years between 1997 and 1999 (ref.33); this decrease was driven by a reduction in the number of cases of AIDS-related KS. The effect of cART on the incidence of AIDS-related KS in sub-Saharan Africa is difficult to quantify because fewer data are available34,35.
A 2017 study, based on over 200,000 patients, reported raw KS incidence per 100,000 person-years in 42 cohorts from 57 countries, including North America (237 per 100,000 person-years), Latin America (244 per 100,000 person-years), Europe (180 per 100,000 person-years), Asia-Pacific (52 per 100,000 person-years) and South Africa (280 per 100,000 person-years)36. KS risk was approximately two times higher in heterosexual men than in women and six times higher in MSM than in women36.
Despite a decrease in the incidence of AIDS-related KS globally since the introduction of cART, KS continues to occur in patients infected with HIV37,38. Changes in the incidence and prevalence of KSHV in the HIV and cART era37,39 may result in changes in the incidence patterns of all forms of KS, but thus far few studies have addressed this. There is some evidence that risk groups of individuals presenting with classic KS may be changing, as documented by a retrospective cohort study of classic KS in Paris between 2006 and 2015 that reported that <40% of patients were of Mediterranean origin and 28% were MSM13.
Mechanisms/pathophysiology
KS is causally associated with KSHV infection, and progress has been made in our understanding of the role of this virus in KS pathogenesis. The virus was first identified in KS lesions using representational difference analysis, a method that combines subtractive hybridization with DNA amplification14.
The viral life cycle
KSHV can infect several different cell types, including endothelial cells, B cells, epithelial cells, dendritic cells, monocytes and fibroblasts40. To gain entry into endothelial cells, KSHV is thought to bind to several host cell surface receptors such as integrins (including α3β1, αVβ5 and αVβ3), the cystine–glutamate transporter xCT, heparan sulfate and the tyrosine protein kinase receptor EPHA2. This binding induces a signal transduction cascade, which results in cellular changes that allow the virus to enter the cell and traffic within the cytoplasm41,42,43. As KS tumours express endothelial cell markers, endothelial cells are thought to be the KSHV-infected cell type in KS tumours. The KSHV-associated lymphoproliferative disorders PEL and MCD involve B cell infection by KSHV (Box 1). KSHV is a linear double-stranded DNA virus with an icosahedral capsid, a tegument (the space between the envelopes and nucleocapsid which contains proteins and RNAs) and an envelope42 (Fig. 2). Gylcoproteins in the viral envelope interact with cell-type-specific cellular entry receptors43. Viral entry results in the delivery of the virion capsid into the cytoplasm, followed by its uncoating and the delivery of the KSHV genome into the nucleus. In the nucleus, the genome circularizes, remaining as an episome. The virus then enters latency (its default pathway) or undergoes sporadic bouts of lytic reactivation during the lifecycle of the virus40.

The Kaposi sarcoma herpesvirus (KSHV) virion binds to receptors present on the cell surface (such as integrins, the cystine–glutamate transporter (xCT), CD98 and heparan sulfate) via glycoproteins (such as gpK8.1, gB, gM–gN and gH–gL) on its envelope; this binding, in most cases, results in the endocytosis of the virion into the cell42. The virion uncoats itself in the cell cytoplasm, and the capsid containing the viral genome traverses to the nucleus. The viral genome enters the nucleus, where it can remain latent as a circular episome tethered to host chromosomes via its latency-associated nuclear protein (LANA), or it can enter the lytic cycle where the viral genomes are replicated and new virions are produced through a complex mechanism of envelopment and ultimately released from the cell via budding. Note that KSHV proteins can increase host signalling through the phosphoinositide 3-kinase (PI3K), mitogen-activated protein kinase (MAPK) and nuclear factor-κB (NF-κB) signalling pathways. RTA, replication and transcription activator.
Latency
Similar to other herpesviruses, infection with KSHV is lifelong because the virus can establish latency in human B cells and endothelial cells. During the latent state in cell culture studies, the virus expresses the latency locus, which includes ORF71 (also called ORFK13; encoding viral FLICE inhibitory protein (vFLIP)), ORF72 (encoding vCyclin), ORF73 (encoding latency-associated nuclear protein (LANA)), ORFK12 (encoding the kaposins, which are signalling proteins) and several microRNAs (miRNAs)44,45. Some additional genes, such as K15 and K1 (which encode transmembrane proteins), as well as viral IL-6 (vIL6), are also expressed at low levels46,47,48. The latent genes are expressed in most KSHV-infected tumour cells and are thought to promote tumorigenesis.
The latent KSHV genome forms an episome, which is tethered by the KSHV LANA to the host chromosome so that the viral genome is replicated with the host genome during normal cell division49. The protein products of other latent genes support the survival of the infected cell. For example, vFLIP activates I-κB kinase 1 (IKK1) to stimulate the nuclear factor-κB (NF-κB) pathway to increase cell survival50,51,52,53. The viral miRNAs that are encoded during KSHV latency45,54,55 help keep the infected cell alive, by inhibiting apoptosis, and the virus latent56; these viral miRNAs are expressed in KS, PEL and MCD57,58,59. Furthermore, similar to kaposin, KSHV mir-K12-10a possesses in vitro transforming abilities in NIH 3T3 cells and is contained within the open reading frame of kaposin60. Several KSHV miRNAs also promote endothelial cell reprogramming61, and KSHV miR-K12-3 induces the migration and invasion of endothelial cells by activating protein kinase B (AKT)62. Finally, KSHV miRK9* targets the transcript of the major lytic switch protein (replication and transcription activator (RTA; also known as ORF50)) to prevent reactivation from latency63.
Of note, the expression of several of the KSHV latent genes and miRNAs in B cells predisposed mice to lymphomas and hyperplasia64,65,66,67, and the protein product of at least one of these latent genes, vFLIP, promotes vascular proliferation and an inflammatory phenotype when expressed in endothelial cells68. There have been attempts to target some of these latent viral proteins to inhibit their function, as exemplified by the inhibition of vFLIP with inhibitory peptides69,70, but these have been in experimental models in cell culture that have not reached clinical feasibility. Inhibitors of vFLIP have also been sought in cell-based drug screening assays using NF-κB reporter PEL cell lines. However, these assays identified a small molecule that selectively treats PEL independently of vFLIP inhibition and does not target vFLIP in KS71. A number of studies have been aimed at targeting cellular genes that are activated by viral proteins to kill KSHV-infected cells, which is a practical approach as drugs that are already in clinical use for other diseases can be screened. An example is the use of rapamycin (also known as sirolimus), because the mechanistic target of rapamycin (mTOR) pathway is activated in latently infected tumour cells in both PEL and KS72,73,74. Another example is the use of HSP90 inhibitors, given that HSP90 chaperones vFLIP and LANA as well as members of the NF-κB, AKT and apoptosis pathways75,76.
The lytic cycle
The physiological stimuli that allow spontaneous reactivation of KSHV from latency are not well defined. However, it is clear that KSHV undergoes spontaneous lytic reactivation sporadically throughout the lifetime of the host. The lytic phase, during which viral genes are expressed in a temporal order, allows the replication of the viral genome and the production of infectious viral progeny. Immediate-early (IE) genes are expressed first. The protein products of most IE genes control transcription and the key lytic protein — RTA — is encoded by an IE gene. RTA is a viral transcription factor that activates many viral and cellular promoters42 and ensures the expression of viral genes required for viral replication. Delayed-early (DE) genes and late genes are expressed after IE genes42. Many of the DE genes encode proteins that control viral DNA replication, which takes place after the DE phase, occurs through a rolling circle mechanism and produces linear genomes that are packaged into capsids42. The late lytic phase results in the expression of all of the viral structural proteins and in the production of the infectious virus.
Similar to the protein products of latency genes, the protein products of lytic genes can contribute to tumorigenesis. However, in contrast to latent genes that are expressed in all tumour cells, lytic genes and the encoded viral proteins are produced only by a very low proportion of tumour cells42. Some lytic proteins may act in a paracrine fashion to stimulate tumour growth. For example, KSHV vIL-6 protein has been detected in the sera of patients with KS, PEL and MCD77, and the KSHV G protein-coupled receptor (vGPCR; a chemokine transmembrane receptor homologue) can constitutively (that is, in the absence of a ligand) induce cellular signalling in cell culture and mouse models, leading to the expression of pro-inflammatory and angiogenic factors, including vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF)78,79,80. Notably, vGPCR can induce vascular lesions in mice, even when expressed in small numbers of cells81,82,83, which is consistent with a model of paracrine oncogenesis84. However, expression of this protein has never been documented in human KS, although the K1 protein is thought to be expressed in a subset of KS tumours85.
The expression of KSHV lytic genes has been clinically targeted in both KS and MCD. In MCD, lytic viral replication can be prevented with zidovudine and valganciclovir. These inhibitors are prodrugs that are phosphorylated by the viral kinases ORF21 and ORF36 to produce their active forms86. Treatment with a combination of zidovudine and valganciclovir has yielded a clinical response of >80% in MCD, in which lytic viral replication appears to be consequential86. However, evidence that this approach is effective in patients with KS is limited. In patients with AIDS given ganciclovir to treat cytomegalovirus retinitis, the subsequent chance of developing KS was decreased by 75% compared with patients with cytomegalovirus retinitis not treated with ganciclovir87. However, another study showed that valganciclovir did not induce the regression of classic KS, highlighting that inhibiting the lytic replication of KS has not universally been useful in the treatment of KS88.
Modulating host signalling pathways
As alluded to above, to survive and persist in its host, KSHV has evolved to modulate many host cell signalling pathways, including the phosphoinositide 3-kinase (PI3K)–AKT–mTOR pathway, the mitogen-activated protein kinase (MAPK) pathway and the NF-κB pathway42. These pathways are activated by multiple KSHV viral proteins (see below), which suggests that they are critically important for the virus. Furthermore, all of these pathways promote cell survival and cell proliferation and are upregulated in many different cancers. In short, the virus has chosen to target pathways that presumably allow for the survival of virus-infected cells.
The PI3K–AKT–mTOR pathway is activated in patients with KS or PEL72,73 by viral proteins including KSHV vIL-6, vGPCR, K1, K15, ORF45 and ORF36 (refs89,90,91) (Fig. 3). KSHV K1 and vGPCR can immortalize and transform endothelial cells and fibroblasts, respectively79,85,92. Furthermore, KSHV K1 can activate Syk-related tyrosine kinase (SRK) and PI3K–AKT signalling85,93,94 to increase the survival of KSHV-infected cells95, and vGPCR can activate PI3K–AKT signalling96,97 to induce KS-like lesions in a number of mouse models81,82,83. KSHV K15 activates the MAPK/ERK kinase 1 (MEK1)/MEK2–extracellular-signal-regulated kinase 1 (ERK1)/ERK2 pathway in a TNF receptor-associated factor 2 (TRAF2)-dependent manner98, and it can interact and activate phospholipase C, γ1 (PLCγ1) to induce angiogenesis99. KSHV vGPCR can also activate MEK1/MEK2–ERK1/ERK2 signalling100.

A graphic representation illustrates how Kaposi sarcoma herpesvirus (KSHV) representative viral proteins (in orange) modulate host cellular signalling proteins (in blue) to promote cellular survival and the inhibition of apoptosis. Viral proteins including KSHV K15, K1, viral G protein-coupled receptor (vGPCR) and viral IL-6 (vIL-6) can activate the phosphoinositide 3-kinase (PI3K)–protein kinase B (AKT)–mechanistic target of rapamycin (mTOR) pathway, thereby increasing cell survival and promoting protein synthesis. KSHV ORF36 acts further downstream and directly phosphorylates cellular ribosomal protein S6, which in turn leads to increased global protein synthesis. KSHV ORF36 also phosphorylates Jun N-terminal kinase (JNK). Some of the KSHV viral proteins, such as vGPCR, K1 and K15, also activate the extracellular-signal-regulated kinase 1 (ERK1)–ERK2 pathway to promote protein synthesis through the activation of ribosomal S6 kinase (RSK; which in turn phosphorylates S6), and KSHV ORF45 directly binds to RSK, leading to its activation. Note that there is a certain degree of crosstalk between both the PI3K–AKT–mTOR and ERK1–ERK2 pathways. Finally, KSHV viral FLICE inhibitory protein (vFLIP) potently activates nuclear factor-κB (NF-κB) signalling by directly binding NF-κB essential modulator (NEMO). This signalling leads to degradation of the inhibitor of NF-κB, IκBα, and translocation of the NF-κB transcription factors into the nucleus. Dashed lines indicate indirect activation. 4EBP1, eukaryotic translation initiation factor 4E-binding protein 1; eIF4B, eukaryotic translation initiation factor 4B; gp130, membrane glycoprotein 130; MEK, MAPK/ERK kinase; mTORC1, mechanistic TOR complex 1; PLCγ1, phospholipase C, γ1; SRK, Syk-related tyrosine kinase; S6KB1, ribosomal protein S6 kinase.
KSHV vIL-6 is a viral homologue of human IL-6. Human IL-6 (hIL-6) must bind both membrane glycoprotein 130 (gp130) (one subunit of the type I cytokine receptor) and IL-6α chain to activate B cell signalling pathways whereas vIL-6 can activate these pathways by binding gp130 alone101. KSHV vIL-6 also activates the JAK–STAT, MAPK–ERK, and PI3K–AKT pathways upon binding to gp130 (ref.102). Interestingly, transgenic mice expressing vIL-6 develop an MCD-like disease103.
KSHV ORF36 encodes a serine/threonine viral protein kinase (vPK) that is expressed under conditions of hypoxia to activate Jun N-terminal kinase (JNK), a stress kinase that belongs to the MAPK family90,91,104. vPK appears to resemble cellular ribosomal protein S6 kinase (S6KB1; a kinase downstream of AKT–mTOR complex 1 (mTORC1) signalling) and to phosphorylate ribosomal protein S6 in vitro to increase protein synthesis and augment anchorage-independent growth, angiogenesis and cell proliferation91. In addition, KSHV vPK transgenic mice develop lymphomas105. Another protein, KSHV ORF45, binds to cellular ribosomal S6 kinase 1 (RSK1) and RSK2, stabilizing its interaction with ERK and preventing its dephosphorylation, which is important for lytic replication90.
As mentioned above, KSHV vFLIP is a potent activator of the pro-survival NF-κB pathway52,106,107; it activates NF-κB by interacting directly with NF-κB essential modulator (NEMO; also known as IKKγ)51,53. Indeed, small interfering RNA (siRNA)-mediated depletion of vFLIP in PEL induces apoptosis, suggesting that vFLIP enhances cell survival52,108. Mice expressing vFLIP in the B cell compartment displayed MCD-like abnormalities67, whereas those expressing vFLIP in the endothelial compartment had a pro-inflammatory phenotype and some vascular abnormalities68.
Thus, KSHV encodes a multitude of proteins that modulate host cell signalling pathways to allow cell survival and cell proliferation and to augment viral replication. Although a number of these viral proteins are expressed, or highly induced, only during lytic replication, their expression leads to the production of secreted cytokines and growth factors that can influence neighbouring cells to induce angiogenesis and inflammation, thereby contributing to KS pathogenesis84.
KSHV and the immune system
Similar to other herpesviruses, KSHV establishes a delicate balance between activating and suppressing the immune response to establish a latent infection that lasts for the entire life of the infected host. Humoral and cellular immune responses to KSHV are evident, as reflected by the much higher incidence of KSHV infection in patients with immunodeficiencies than in individuals with an uncompromised immune system. Humoral responses were described soon after KSHV was discovered as the causative agent of KS and PEL. Specifically, PEL cell lines were positive for immunofluorescence when stained using patient sera; this nuclear staining was later shown to be LANA. Furthermore, when cells were induced to undergo lytic replication, patient sera strongly stained the cytoplasm of PEL cell lines consistent with it recognizing lytic antigens109. Subsequently, a large number of seroepidemiologic studies using enzyme-linked immunosorbent assay (ELISA) relied on the presence of antibodies to a number of recombinant viral proteins29. Among the latent KSHV proteins, LANA seems to be most immunogenic, and among the lytic proteins, K8.1 has been used in most serologic assays. Systemic analysis of antibodies to all KSHV proteins showed that ORF38, ORF61, ORF59 and K5 elicited detectable responses in individuals with KSHV-associated diseases110. However, antibodies to KSHV are rarely neutralizing111.
To assess cellular responses to KSHV, a systematic approach was recently used in which the whole KSHV proteome was examined by IFNγ enzyme-linked immunospot (an immunoassay that measures the frequency of cytokine-secreting cells at the single-cell level)32. This study found variable responses of both CD4 and CD8 cells to a wide variety of viral antigens, indicating a lack of shared immunodominance among individuals.
In this subsection, we review the cell-intrinsic mechanisms used by KSHV to induce an immune response while, at least temporarily, simultaneously avoiding immune recognition to establish lifelong infection.
Triggering an antiviral immune response
Following viral entry or reactivation, the host mounts an immune response to KSHV via innate immune receptors such as Toll-like receptors (TLRs), retinoic acid-inducible gene I protein (RIG-I)-like receptors (RLRs), nucleotide-binding and leucine rich repeat or Nod-like receptors (NLRs), AIM2-like receptors (ALRs) and cyclic GMP-AMP synthase (cGAS)–stimulator of interferon genes protein (STING)112. TLRs are the first line of defence against many viruses; accordingly, KSHV activates TLR3 (ref.113), TLR4 (ref.114) and TLR9 (ref.115) in a cell-type-dependent manner, as well as the NLR proteins NOD-, LRR- and pyrin domain-containing 1 (NLRP1) and NLRP3, upon primary infection116. Moreover, the AIM2 family member IFI16 induces activation of the inflammasome upon KSHV infection117, and KSHV activates the cGAS–STING pathway during primary infection and reactivation118. Activation of TLRs and RLRs generally leads to the induction of type I interferon, which is considered antiviral. Furthermore, the activation of NLR-dependent inflammasomes leads to the production of pro-inflammatory cytokines such as IL-1β and IL-18. However, despite activating multiple innate immune receptors, KSHV persists for a lifetime in the infected host by encoding both lytic and latent proteins that inhibit host innate and adaptive immunity (Fig. 4).

Kaposi sarcoma herpesvirus (KSHV) encodes an arsenal of viral proteins that modulate multiple innate immune pathways. KSHV viral interferon regulatory factor 1 (vIRF1), viral G protein-coupled receptor (vGPCR) and replication and transcription activator (RTA) can downregulate Toll-like receptor 2 (TLR2) and TLR4, and TRIF (also known as TIR domain-containing adaptor molecule 1 (TICAM1)), a TLR adaptor protein, is further inhibited by KSHV RTA. Inhibition of these TLR signalling pathways results in the downregulation of the type I interferon response. Ubiquitylation of the RNA sensor, retinoic acid-inducible gene I protein (RIG-I), which is crucial for its activation, is inhibited by KSHV ORF64, resulting in the dampening of RIG-I activity and inhibition of the interferon response. KSHV vIRFs and ORF45 can inhibit activation of cellular IRFs such as IRF3 and IRF7. Cyclic GMP-AMP synthase (cGAS)–stimulator of interferon genes protein (STING) is a cytosolic DNA sensing pathway, and cGAS activation is inhibited by KSHV ORF52 and KSHV latency-associated nuclear protein (LANA), whereas KSHV vIRF1 prevents STING association with the serine/threonine-protein kinase TBK1, thereby inhibiting interferon production through the pathway mediated by IRF3 and IRF7. The NOD-, LRR- and pyrin domain-containing 1 (NLRP1) inflammasome, which comprises NLRP1, apoptosis-associated speck-like protein containing a CARD (ASC) and pro-caspase-1, is inhibited by KSHV ORF63, resulting in inhibition of IL-1β and IL-18 activation. 5ʹppp, 5ʹ-triphosphate; dsRNA, double-stranded RNA; MAVS, mitochondrial antiviral-signalling protein; MDA5, melanoma differentiation-associated protein 5; MyD88, myeloid differentiation primary response protein MyD88; NF-κB, nuclear factor-κB; NLR, Nod-like receptor; TRAF3, TNF receptor-associated factor 3; Ub, ubiquitin.
Evading the immune response
KSHV K3 and K5 are lytic genes that encode modulator of immune recognition 1 (MIR1) and MIR2, respectively, both of which inhibit major histocompatibility complex (MHC) class I antigen presentation to prevent the immune system from detecting KSHV-infected cells119. MIR1 downregulates all four of the human leukocyte antigen (HLA) gene alleles or allotypes (HLA-A, HLA-B, HLA-C and HLA-E) and MIR2 downregulates HLA-A and HLA-B120.
KSHV homologues of interferon regulatory factors (IRFs), viral IRFs (vIRFs), are lytic proteins that inhibit type I interferons. The KSHV genome encodes four vIRFs: vIRF1, vIRF2, vIRF3 and vIRF4 (ref.121). At the transcriptional level, vIRF1 binds cellular IRF1 and IRF3 to prevent them from transactivating the promoters of interferon genes122 and binds STING to inhibit cGAS–STING signalling and IFNβ induction123. KSHV ORF52 and LANA also inhibit the cGAS–STING pathway, but by targeting cGAS instead of STING124,125. vIRF2 impairs the induction of interferon gene expression by binding IRF1 and IRF3 to inhibit cellular IRF1-mediated and IRF3-mediated transcription126. Finally, vIRF3 binds IRF3 and IRF7 and prevents them from transactivating the promoters of IFNα4 and IFNα6 (ref.127). vIRF3 also dampens IFNγ-mediated activation of the GAS promoter128. KSHV vIRF1 and vGPCR downregulate expression of TLR4 (ref.114), and RTA was reported to inhibit TLR2-dependent NF-κB activation129; RTA can also induce the ubiquitylation and degradation of IRF7 (ref.130). KSHV vIRF1 and vIRF2 expression reduced the level of IFNβ, both at the mRNA and protein level, following TLR3 activation121. KSHV ORF45 impairs IRF7 phosphorylation131 and KSHV vIL-6 can be directly activated by IFNα to block interferon induction132.
In terms of RLRs and NLRs, KSHV ORF64 is a viral deubiquitinase that can deubiquitinate RIG-I to prevent RIG-I-mediated interferon induction133, and the KSHV tegument protein ORF63 inhibits the NLR inflammasomes, NLRP1 and NLRP3 (ref.116).
KSHV also encodes three CC-chemokine ligands (CCLs; formerly known as vMIPs): vCCL-1 (encoded by ORFK6), vCCL2 (encoded by ORFK4) and vCCL3 (encoded by ORFK4.1)134, which can negatively regulate inflammation. vCCL2 interacts with host CC-chemokine receptors (CCRs), including CCR1, CCR2, CCR5, CXC-chemokine receptor 1 (CXCR1), CXCR2 and CXCR4, to inhibit signalling from them. For example, vCCL2 prevents host CCL5 (also known as RANTES) and host CCL3 (also known as MIP1α) from binding to CCR5 (refs135,136). vCCL2 also inhibits CD8 T cell migration and hinders cytotoxic T lymphocyte-mediated rejection of corneal and cardiac allografts in an experimental model137. vCCL1 and vCCL2 also promote endothelial cell survival and virus replication138, which could play a role in KS pathogenesis.
Finally, the KSHV K14 gene encodes for a viral OX2 (vOX2), an immunoglobulin superfamily member with homology to the cellular OX2 membrane glycoprotein (OX2; also known as CD200) that binds to the receptor CD200R139. Both stimulatory and tolerogenic roles for cellular OX2 in presentation of the antigen have been proposed140,141. KSHV vOX2 fused to a crystallizable fragment (Fc) antibody domain suppressed neutrophil activation, decreased CCL2 (also known as MCP1) and IL-8 production and inhibited oxidative burst in neutrophils stimulated to undergo phagocytosis142. However, purified glycosylated vOX2 protein stimulated primary monocytes, macrophages and dendritic cells to express inflammatory cytokines including IL-1β, IL-6, monocyte chemoattractant protein 1 and TNF143. This induction of inflammatory cytokines may contribute to the inflammatory infiltrates seen in KS if vOX2 is expressed in cells in the lesions that are undergoing lytic replication.
Diagnosis, screening and prevention
Clinical manifestations
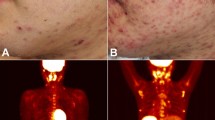
The clinical manifestations of the epidemiological forms of KS overlap, although certain characteristics are more commonly associated with one form of KS than with others. In all forms of KS, cutaneous lesions usually present as multiple, pigmented, raised or flat, painless lesions that do not blanch (that is, they do not lose colour with pressure) (Fig. 5). The earliest cutaneous lesions are often asymptomatic, innocuous-looking, pigmented macules or small papules that vary in colour from pale pink to vivid purple. Although KS is often diagnosed on the basis of the characteristic appearance of lesions alone, the diagnosis should be confirmed histologically because even experienced clinicians can misdiagnose KS144. Larger plaques on the trunk often follow the skin creases as oblong lesions. Occasionally, lesions form exophytic, ulcerated and bleeding nodules that can be associated with painful oedema.

Different manifestations of Kaposi sarcoma (KS) include (part a) macular lesions on the back and nodules on the arm; (part b) extensive KS plaques on the legs with tumour-associated oedema; (part c) exophytic KS lesions on the foot; (part d) extensive gingival KS nodules; and (part e) flat, violaceous lesions on the hard palate. The image in part c is of a patient with classic KS; all other images are of patients with AIDS-related KS.
In AIDS-related KS, oral lesions (that is, in the palate and on gums) are common and may lead to dysphagia and secondary infection. Endemic KS is frequently associated with lymphoedema in African children and young adults, regardless of HIV status, and is difficult to control145. Finally, visceral lesions frequently can occur in the lungs and gastrointestinal tract and do so mostly in individuals with AIDS-related KS. Pulmonary lesions, which usually present with dyspnoea, dry cough and sometimes haemoptysis, with or without fever, are life-threatening. These lesions typically appear as a diffuse reticulo-nodular infiltrate and/or pleural effusion on chest radiography. Gastrointestinal lesions are usually asymptomatic, but may bleed or cause obstruction, and their presence is usually confirmed at endoscopy. Nevertheless, visceral lesions with KS are uncommon (in one study, only 15% of 469 patients had visceral lesions upon diagnosis with AIDS-related KS)146, and CT scans, bronchoscopy and endoscopy are not warranted in patients in the absence of symptoms indicative of visceral lesions.
To date, the staging of KS has not been unified or incorporated into the American Joint Committee on Cancer (AJCC) tumour, node and metastasis (TNM) staging system. Instead, the modified AIDS Clinical Trials Group (ACTG) staging classification, which is based on tumour, immune status and systemic illness (TIS), is used for AIDS-related KS147,148 (Table 2), and the classification of classic KS focuses only on the tumour and originated from a case series of 300 patients149 (Table 3). There are no specific staging systems for endemic or iatrogenic KS.
Diagnosis
Pathologic diagnosis
When there is clinical suspicion of KS, a biopsy sample is taken to confirm the diagnosis histologically. Although this is straightforward in resource-rich settings, the process can be challenging in resource-limited settings, such as in Africa, where KS is most common, and macroscopic clinical visualization is often the only available means for diagnosing KS. Indeed, in one study from East Africa, visual diagnosis alone had only 80% positive predictive value for KS150; some patients were falsely diagnosed with KS, giving them an indication for needless chemotherapy and missing the correct, often easily treatable, diagnoses (for example, bacillary angiomatosis)151. To remedy this, several efforts have been aimed at increasing the histologic diagnosis of KS, including task shifting the performance of biopsies to non-physicians152 as well as teledermatology and telepathology153.
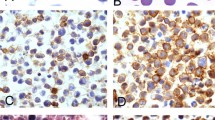
Pathologic diagnosis of KS can often be made using conventional haematoxylin and eosin (H&E) staining alone to assess for several basic features of KS that are present, to varying degrees, in all cases of the disease. These features include vascular proliferation in the dermis (with the formation of slit-like spaces that are not lined by endothelium), an increased number of vessels without an endothelial cell lining, the presence of extravasated blood resulting in the formation of hyaline globules and haemosiderin accumulation and an inflammatory infiltrate (Fig. 6). Spindle cell proliferation is also a typical feature of KS. These spindle cells, which are characterized by elongated cytoplasm and nuclei and sometimes contain haemosiderin and hyaline inclusions, express endothelial markers and are considered to be the KS tumour cell. Although spindle cells are usually seen in sheets or fascicles (Fig. 6b), they may be difficult to distinguish in early lesions (Fig. 6c).

a | Haematoxylin and eosin (H&E) staining of a skin biopsy sample showing characteristic features of Kaposi sarcoma (KS) is shown. Note the presence of spindle cells replacing dermal collagen, vascular spaces containing red blood cells, extravasated red blood cells and haemosiderin in macrophages (that is, siderophages). The original magnification is 20× (left panel) and 60× (right panels). b | H&E staining showing sheets of spindle cells in a biopsy sample of a nodular KS lesion. Original magnification 60×. c | A biopsy sample of a patch-stage KS lesion, stained with H&E, which has a sparse cellular infiltrate with many inflammatory cells (seen as dark, small cells). Original magnification is 20×. d | Immunohistochemistry for KS herpesvirus (KSHV) latency-associated nuclear protein (LANA) in a skin biopsy sample of patch-stage KS, in which cells with brown nuclei line vascular spaces. e | H&E staining of a nodular-stage KS lesion is shown at low power (4×), with a large nodule formed by swirling fascicles of spindle cells. Original magnification 20×. f | Immunohistochemistry for KSHV LANA in an area of a nodular KS lesion biopsy sample, showing immunoreactivity indicated by brown staining with a characteristic punctate pattern in the nuclei of spindle cells. Original magnification 60×.
Pathologic classification has been described as including the following stages of tumour progression, which roughly correspond to the clinical appearance of the skin lesions (that is, morphotypes); however, frequent overlap between the stages exists, and it is unclear whether these stages occur in sequential order (note that pathologic stages are different from the clinical staging described in Table 2 for AIDS-related KS)154.
Macule or patch KS lesions may be the most difficult to distinguish histologically from other conditions as, although many of their features are evident on H&E staining alone, they can mimic other inflammatory skin disorders such as minor vascular anomalies and inflammatory conditions (Fig. 6c,d). Macule or patch KS lesions are characterized by a patchy, sparse perivascular infiltrate composed of lymphocytes and plasma cells; red blood cell (RBC) extravasation and siderophages (macrophages containing iron (that is, haemosiderin)); narrow cords of cells between collagen bundles; and, sometimes, fascicles of spindle cells.
Plaque KS lesions are characterized by a diffuse infiltrate of vessels throughout the dermis, with fascicles of spindle cells replacing the dermal collagen. Vascular spaces usually have jagged outlines and separate collagen bundles. There is commonly extravasation of RBCs with siderophages. Inflammatory infiltrate includes numerous macrophages, lymphocytes and, frequently, plasma cells.
Nodular KS lesions have the most distinct histology. They are characterized by well-defined nodules composed of sheets of spindle cells that replace the dermal collagen (Fig. 6e). A honeycomb-like pattern of vascular spaces filled with RBCs is frequently seen closely associated with interweaving spindle cells. Pseudovascular spaces, where RBCs appear to directly contact spindle cells, are common. There is RBC extravasation, with siderophages and hyaline globules that are eosinophilic spheres 1–7 μm in diameter. Advanced cases of KS, such as the anaplastic variant, can display sheets of atypical spindle cells that mimic other sarcomas; in these cases, immunohistochemistry can aid diagnosis154.
The role of immunohistochemistry in diagnosing KS
KS lesions have a heterogeneous cellular composition. Immunohistochemistry of the spindle cells using antibodies against vascular endothelial markers such as CD34 revealed that they have a vascular nature, and the subsequent detection of lymphatic endothelial markers in the spindle cells, such as podoplanin, LYVE1 and VEGF receptor 3, suggested that KS has a lymphatic endothelial cell origin155,156. However, KS lesions also express mesenchymal markers, such as vimentin157, and a mesenchymal origin of KS was recently proposed158. Most immunohistochemical evidence and gene expression and experimental data currently suggest that the spindle cells are lymphatic endothelial, vascular endothelial and/or mesenchymal cells that undergo reprogramming following KSHV infection to produce cells with an aberrant combined immunophenotye158,159. In addition to lymphocytes and plasma cells, histiocytes (that is, tissue macrophages) are abundant in KS lesions, and these can be identified with immunohistochemistry160,161. These immunohistochemical markers have aided our understanding of the cellular composition of KS and may still help in the differential diagnosis of KS in rare and complicated cases.
Immunohistochemical stains for antigens of KSHV (in addition to conventional H&E stains) are very useful in diagnosing KS. Specifically, antibodies that recognize KSHV LANA can be used in routine histopathology to confirm a KS diagnosis in difficult cases, and these are routinely used in many resource-rich pathology settings. Immunohistochemistry for LANA is particularly useful in diagnosing KS presenting with early macular or patch KS lesions or lesions that resemble other sarcomas. However, although LANA is thought to be expressed in every infected cell, the proportion of infected cells is variable, ranging from <10% to >90% of the total cell population in the lesional areas (Fig. 6d,f). Furthermore, LANA immunohistochemistry should be considered positive only when a distinct punctate nuclear pattern is seen, which prevents LANA staining from being confused with cytoplasmic haemosiderin or melanin when using a brown chromogen. Although LANA positivity confirms a diagnosis of KS, a negative stain may not rule out KS as sampling errors or false negatives can result from poor tissue preservation or other technical artefacts. Thus, whether a positive LANA stain is required to establish a diagnosis of KS is controversial and depends on specific circumstances.
Molecular diagnosis
When examined with sensitive nucleic acid amplification techniques, KS lesions almost always contain KSHV DNA. Indeed, a review in 2009 of >25 studies found that KSHV DNA is detected by PCR in >95% of all epidemiologic forms of KS29. Whether the pathologic evaluation of the KS cases deemed negative for KSHV by PCR would result in 100% of KS cases being diagnosed is unknown. Regardless, the detection of KSHV DNA by PCR is highly, if not optimally, sensitive for KS, and the absence of KSHV DNA in a well-prepared sample essentially excludes the diagnosis. Except for in research laboratories, however, PCR for KSHV DNA is currently available only in a few highly specialized clinical molecular pathology laboratories. The specificity of detecting KSHV DNA is less clear, especially in KSHV-endemic geographic regions (for example, Africa), where up to 60% of persons in the general community are positive for KSHV antibodies. Data in this regard are very limited (14% of archival specimens from individuals without KS in Uganda were positive for KSHV DNA162), but they suggest that a quantitative threshold of KSHV DNA may distinguish KSHV-DNA-positive individuals with KS from KSHV-DNA-positive individuals without KS. If so, the automated objective molecular diagnosis of KS could conceivably be performed at the point of clinical care and, in large part, remove the expense and subjectivity of histopathologic diagnosis.
A similar approach has been implemented for tuberculosis with the GeneXpert platform (from Cepheid). A novel point-of-care device for KS diagnosis, which features multiple energy sources for use in areas where electricity is limited, is under development9,163,164,165. The latest stage of this device, which is called TINY (Tiny Isothemal Nucleic acid quantifications sYstem), relies on isothermal amplification and can be heated via electricity, sunlight or flame165. Pilot testing of this device in Uganda showed that KSHV DNA could be detected in KS biopsy samples in <3 hours after applying anaesthesia to the patient165. Broad implementation of this device could enable a definite diagnosis of the patient while still in the clinic rather than weeks later when relying on a pathologic diagnosis. A device such as TINY may lead to earlier diagnosis and prevent loss to follow-up.
Screening
The current status of KS screening
Unlike for some cancers, there is no pre-neoplastic stage of KS for which screening can be performed. Furthermore, conventional screening by health-care practitioners for established KS before the development of clinical symptoms is of limited use because KS usually occurs first on visible skin and/or mucous membranes166 and is thus initially observed by patients themselves. The exception is KS in the oral cavity, which could benefit from screening as it is often the first anatomic site of involvement in AIDS-related KS and might be missed by the patient. Other than screening for oral KS, the only other conceivable means of screening for preclinical KS would be to evaluate the lower respiratory tract or gastrointestinal tract for visceral lesions. Methods to perform such screening, however, are nonspecific (chest radiography or chest CT), expensive or associated with potential adverse events (bronchoscopy and endoscopy). Given that it is rare for KS to be present in visceral organs and not on visible mucous membranes or skin166, screening of the lower respiratory tract and gastrointestinal tract is not recommended.
Benefit of early diagnosis
Although a role for the formal screening for preclinical KS is limited (that is, it is useful only in the detection of lesions in the oral cavity), much could be gained by early detection of clinically apparent KS; preclinical screening and early detection of clinically apparent KS can be thought of as one intervention. Despite not being universally defined, early KS is generally agreed to be mild to moderate disease without symptomatic visceral symptoms, lymphatic obstruction or function-altering oedema, difficulty swallowing or chewing or any other functionally disabling manifestation167. An important rationale behind early detection is that early KS has a better clinical outcome than KS that is detected at a later stage. Indeed, even before cART, the stage at which KS was diagnosed was predictive of survival; patients with AIDS-related KS in the USA diagnosed with T0 tumours had a 1-year greater median survival than patients diagnosed with T1 tumours147 (Table 2). The availability of cART has markedly improved the overall prognosis of KS, but the stage of KS at presentation remains similarly, if not more, important prognostically. In one of the largest studies of prognosis in the cART era, among 211 patients with AIDS-related KS in Italy, those with T1 disease at diagnosis had a 2.6-fold greater rate of death than those with T0 disease at diagnosis168. Among Swiss patients with AIDS-related KS, those with T1 disease at diagnosis had a 5.2 fold greater rate of death or need of chemotherapy than patients with T0 disease at diagnosis169. One study from South Africa found that individuals with T1 disease had a 2.4-fold higher mortality than individuals with T0 disease170.
Although the observational studies discussed suggest that the early detection of KS is clinically beneficial, and this is known to be the case for other cancers, these benefits have not been tested in randomized trials. This lack of randomized trials might explain why the evidence-based clinical guidelines of most national and international normative bodies for KS have not emphasized or mentioned early detection of the disease (although there are exceptions to this, such as in Uganda171). Formally proving the clinical benefits of early detection of KS experimentally would be time consuming and costly. In addition, the areas where the evidence would be most useful (that is, in sub-Saharan Africa) have the fewest resources to perform such studies. Therefore, individual practitioners and public health bodies must continue to make their own decisions on the utility of promoting early KS detection without gold-standard evidence. Inattention to KS has likely resulted in very few policy groups or individual practitioners ever considering the benefits of early detection of the disease. Also unproved is whether patients diagnosed with the fewest number and size of lesions (that is, with the smallest biological involvement) have the best prognosis, although observational research suggests that this is the case172.
In sum, detecting KS early will require the involvement of medical practitioners and patients. Medical providers can identify early-stage KS inexpensively by macroscopic examination of the oral cavity and complete examination of mucous membranes and skin. Although these examinations are neither time nor resource intensive, they are hindered by tight scheduling in resource-rich settings and by overwhelmed clinics, a lack of private space and cultural avoidance in many resource-limited settings. Even if medical providers always examined patients for KS, it would not mean that affected patients seek appropriate medical care earlier. In particular, patients in resource-limited settings are unlikely to recognize KS as a possibility when painless lesions appear on their skin. Indeed, a health-care-facility-based randomized trial of an intervention to improve early detection of KS failed to improve outcomes in Zimbabwe173. The use of traditional healers by patients with KS in resource-limited settings may also delay early detection174. Thus, it is likely that the benefits of early KS detection will be realized only through community-based public education campaigns. Efforts in this regard have started in Uganda175 but will require investment and scale-up to achieve impact.
Prevention
No specific intervention is currently recommended in routine clinical practice to prevent KS. An anti-herpesvirus agent, ganciclovir, decreased the incidence of KS among individuals with HIV infection treated for cytomegalovirus retinitis in a trial mostly conducted before the cART era87. However, routine use of this drug in patients with HIV infection (or other populations with KSHV infection) is not indicated because of its toxicity. Therefore, interventions aimed at evading HIV infection (including pre-exposure prophylaxis)176, or suppressing HIV replication and maintaining the immune function of patients with HIV infection177, are currently the most practical ways to avoid developing KS. Avoidance of KSHV infection would also prevent KS, but there are no rigorously examined interventions for prevention of this infection, mainly because the specific routes of KSHV transmission are not understood. Saliva is the body fluid that most commonly harbours KSHV178 and is thus likely the most important conduit for transmission. Therefore, in resource-rich settings, MSM (the group with the highest seroprevalence for KSHV infection)179,180 should be counselled about the possible spread of KSHV through saliva181. Population-based studies of MSM in California, however, indicate very little awareness of KSHV182. Furthermore, the use of saliva as a lubricant in anal sexual practices concentrated among MSM could play a role in KSHV transmission183. Considering the avoidance of this practice should at least be part of an educational message to at-risk populations. In Africa, non-sexual horizontal transmission in childhood is the principal route of spread in the highest prevalence areas184, but children are exposed to saliva in many ways24 and a main form of exposure has not been identified. As such, there is currently no role for a broad recommendation to avoid saliva exposure in the general population.
Management
In patients with forms of KS in which immunosuppression is potentially reversible, the first-line approach is to bolster the immune system; for example, the treatment of HIV with cART in patients with AIDS-related KS may cause regression of T0 tumours. Similarly, patients with iatrogenic KS may be treated by reducing the level of immunosuppression or by changing the immunosuppressive agents used, for example, by swapping calcineurin inhibitors for inhibitors of the PI3K–AKT–mTOR pathway, such as rapamycin. However, reducing immunosuppression in patients with iatrogenic KS may risk graft rejection. Treatments directed at the tumours are necessary in patients with AIDS-related KS and iatrogenic KS in whom agents targeting the immune system are insufficient, and in patients with endemic KS, classic KS or KS in MSM without HIV infection. High-quality evidence for the clinical management of KS is confined to the management of AIDS-related KS; the clinical approach for treating patients with other forms of KS is generally based on small retrospective case series and clinician experience rather than trial data.
For example, prospectively designed phase II trials in classic KS are scarce, usually include few patients, do not use standardized objective methods to document response and are rarely prospectively randomized. Despite these shortcomings, the current treatment options for classic KS include the observation of patients with a limited number of asymptomatic lesions that do not impair function; the management of symptoms from lower-extremity oedema with elastic compression stockings and various local and systemic tumour-directed therapies similar to those used for AIDS-related KS (see below). Of note, the choice of tumour-directed therapies for treating patients with classic KS depends on the number and anatomic distribution of the lesions, the pace at which the disease is progressing and the severity of other comorbidities; comorbidities are often present in elderly individuals presenting with classic KS (reviewed in ref.185).
AIDS-related KS
Cytotoxic therapy
The clinical management of AIDS-related KS is largely determined by clinical staging. Patients with T0 early-stage disease should commence cART (if not already receiving this treatment for HIV), to which KS will often respond (that is, lesions will shrink by ≥50% in size and/or number) within 6–12 months. Indeed, up to 80% of patients with T0 stage KS that were not previously treated with cART will require no other treatment for KS than continued cART over 10 years146.
The management of T1 advanced-stage or progressive AIDS-related KS was established before effective cART was available and largely remains based on clinical trials from that time. Three sizeable randomized controlled trials conducted in the USA and the UK demonstrated the superiority of single-agent liposomal anthracyclines for treating patients with advanced-stage or progressive AIDS-related KS compared with conventional combination chemotherapy186,187,188. The safety and tolerability of liposomal anthracyclines in patients on cART has since been established189,190. Only one study has directly compared the efficacy of liposomal daunorubicin (DaunoXome) and pegylated liposomal doxorubicin (Caelyx or Doxil) in treating advanced-stage or disfiguring AIDS-related KS; as this study was underpowered, there is insufficient evidence to favour one agent over the other191. Nonetheless, on the basis of patient response rates and durations and on toxicity profiles, liposomal anthracyclines are considered the standard first-line chemotherapy for advanced AIDS-related KS, and most clinicians favour liposomal doxorubicin over liposomal daunorubicin as it is more widely available and has a less frequent administration schedule. Over 80% of patients with advanced AIDS-related KS have been reported to show tumour regression using the current approach of combining cART with liposomal doxorubicin189, although a minority of patients have anthracycline-refractory KS or relapse soon after completing chemotherapy and may be eligible for second-line therapy. A few patients with AIDS-related KS who are well established on cART and have an undetectable plasma HIV viral load and good CD4 cell counts still develop progressive or visceral KS192,193. These patients are generally treated with systemic chemotherapy using the same regimens.
Several phase II studies have demonstrated the efficacy of paclitaxel in treating patients with advanced-stage AIDS-related KS, including those with anthracycline-refractory disease194,195,196. Although the only head-to-head comparison of pegylated liposomal doxorubicin and paclitaxel in the treatment of advanced AIDS-related KS showed no significant differences in response rate (P = 0.49), progression-free survival (P = 0.66) or overall survival (P = 0.49), there was significantly greater neurotoxicity (P = 0.045) and alopecia (P < 0.001) in the paclitaxel arm, making paclitaxel a less attractive first-line treatment option than pegylated liposomal doxorubicin197. Thus, in high-income settings, expensive liposomal anthracyclines are favoured for treating patients with AIDS-related KS, and paclitaxel, which is more affordable, is generally reserved for patients with recurrent or refractory AIDS-related KS. In low-income and middle-income settings, however, paclitaxel is becoming more widely available at a reasonable cost and may be preferred as a first-line regimen for treating patients with AIDS-related KS (Box 2). Finally, a study conducted primarily in sub-Saharan Africa, where liposomal anthracyclines are generally unaffordable, showed that a routinely used combination of bleomycin and vincristine was inferior to paclitaxel for treating patients with advanced AIDS-related KS198.
Pathogenesis-directed therapy
Two pathogenesis-directed therapies, IFNα and alitretinoin (a retinoid receptor pan-agonist), are approved for AIDS-related KS, but their use is largely historical. IFNα is a cytokine with direct antiproliferative and antiviral effects that can inhibit angiogenesis and modulate host cellular and humoral immune responses. The parenteral administration of recombinant IFNα was studied extensively in the 1980s and 1990s and induced regression of AIDS-related KS, particularly when combined with single inhibitors of HIV-1 nucleotide reverse transcriptase199. Although approved for AIDS-related KS treatment by various regulatory authorities in resource-rich countries, it is now rarely used owing to the availability of alternative agents that are more easily administered and have more favourable adverse event profiles. Topical alitretinoin gel200,201 is thought to inhibit cell proliferation and promote cellular differentiation, and it induces the apoptosis of KS-infected cells in vitro.
Our growing understanding of KS pathogenesis has suggested multiple potential treatment targets for this disease, although none of the agents directed at these targets have yet been approved for treating patients. Nevertheless, the following treatments have shown promise in small clinical trials: imatinib, which inhibits tyrosine kinase-mediated transmembrane receptor signalling to prevent KS cell proliferation and angiogenesis202; bevacizumab, a monoclonal antibody against VEGF, an angiogenic growth factor that is highly expressed in KS lesions203; IL-12, a cytokine that enhances type 1 immune responses, mediates antiangiogenic effects and downregulates vGPCR activity204; immunomodulatory imide drugs, including thalidomide, lenalidomide and pomalidomide, all of which possess anti-inflammatory, antiangiogenic and immunomodulatory properties205,206; proteasome inhibitors, such as bortezomib207, which may promote the KSHV lytic cycle and/or inhibit NF-κB signalling; inhibitors of the constitutively activated PI3K–AKT–TOR pathway, such as rapamycin, which have activity against iatrogenic and AIDS-related KS73,74; and agents such as timolol and propranolol that inhibit autocrine β-adrenergic-receptor-mediated signalling (through which KSHV usually drives the proliferation of transformed cells and represses the expression of viral lytic genes)208.
KS immune reconstitution inflammatory syndrome
KS immune reconstitution inflammatory syndrome (KS-IRIS) refers to the clinical worsening of existing KS or, less often, to the development or ‘unmasking’ of previously undiagnosed KS, following cART-mediated reconstitution of the immune system. Estimates of KS-IRIS frequency range from <10% to nearly 40% depending on the precise case definition applied, the case-detection approach and the clinical setting209,210,211. Moreover, there is no standard for distinguishing KS progression as an immune reconstitution-associated event from the natural history of KS. Most current definitions of KS-IRIS include evidence of progressive KS within 12 weeks of initiating cART in parallel with the suppression of HIV RNA levels by ≥0.5–1 log10 and/or an increase in CD4 T cell counts by at least 50 cells µl–1 compared with pre-cART levels; however, these definitions do not specify that ‘inflammatory’ characteristics of KS progression must be present. There is evidence that KS-IRIS occurs more frequently in sub-Saharan Africa than in the UK210; is more common among persons with T1 stage KS, high plasma HIV-1 RNA levels and detectable plasma KSHV DNA levels (than in patients with T0 stage KS, low plasma HIV-1 RNA levels and undetectable plasma KSHV DNA levels); and is less likely to be diagnosed among individuals receiving concomitant KS-specific chemotherapy and cART210,212.
There is no standard approach for managing KS-IRIS. In some cases, KS progression subsides and may reverse without additional treatment and without stopping cART. In other cases, immediate addition of chemotherapy to cART may be lifesaving and has been associated with improved patient survival210. Of note, KS-IRIS, unlike other manifestations of immune reconstitution, may be exacerbated by the addition of corticosteroids213, which should be avoided. Similar progression or unmasking of KS may occur following the treatment of KSHV-associated MCD with the monoclonal antibody rituximab214, even if the patients are on cART.
Quality of life
Many physical and psychosocial problems associated with KS negatively influence QOL. For example, although gastrointestinal KS lesions are often asymptomatic, some may cause pain, bleeding, difficulty with feeding, diarrhoea, intestinal obstruction, malabsorption and weight loss. Pain, severe oedema and cellulitis can accompany ulcerated skin lesions, and oedema can also be present in the absence of skin lesions. Oedema of the lower extremities may impede or prevent ambulation, as can oedema of the external genitalia, which may also obstruct urination. Facial and periorbital oedema is disfiguring and, in extreme cases, may obstruct vision. Pulmonary lesions and effusions may be associated with dyspnoea, cough, haemoptysis and restricted activity. Skin lesions, particularly those on the face that are hard to camouflage but also those on the torso and extremities, may lead to self-imposed social isolation, ostracism by others and psychological distress. Although many of these problems have most commonly been associated with AIDS-related KS, they may occur with all epidemiological forms of the disease.
Given that current treatments for KS are not curative, symptom palliation is often a major objective of KS therapy. In one study investigating preference techniques to value the potential health gains from different KS treatments215, 44% of respondents rated the presence of cutaneous lesions on the face and trunk (even in the absence of visceral involvement or oedema) as equivalent to death and indicated that fairly modest treatment effects greatly improved QOL. A number of studies have measured changes in QOL during the treatment of AIDS-related KS195,197,216,217,218. However, the methods used to assess QOL, including assessments of both general health and KS-specific signs and symptoms, have been inconsistent, as have the specific treatment interventions used, the prior KS treatment status of participants and the availability and use of antiretroviral therapy. Nonetheless, there is essentially unanimous agreement that chemotherapy for KS often improves QOL, despite the side effects of treatment. In particular, KS-associated pain and oedema were most likely to be positively influenced by chemotherapy195,197,216,217,218. Although improved QOL measures sometimes positively correlated with a measured objective response of KS to treatment (for example, shrinkage of the tumour by ≥50%)216,217, symptom palliation occurred in many individuals in the absence of the objective response197,216. These findings highlight the relevance of integrating measurements of QOL and patient benefit into the evaluation of the therapeutic efficacy of treatments for KS219. Cases of highly symptomatic AIDS-related KS have been reduced in high-resource settings but remain common in resource-constrained settings that account for the majority of new cases worldwide. Thus, using QOL as an integrated measure of therapeutic effectiveness in evaluating KS treatments in resource-constrained settings remains highly relevant.
Outlook
Basic science and pathophysiology
KSHV encodes viral proteins that are observed in KS lesions, where they can induce cellular changes by activating cellular pathways. Additionally, some KSHV proteins (for example vGPCR, vFLIP and LANA) may drive KS pathogenesis, thus representing potential therapeutic targets in KS; inhibitors of these proteins remain at the experimental stage and, even if they do become clinically available, this may take ≥10 years.
The cellular oncogenome of KS is poorly understood, and it is unclear whether KS is a polyclonal or monoclonal disease entity. In general terms, cancers originate from a single cell, therefore monoclonality is a feature of neoplastic diseases, whereas an inflammatory condition would arise from a number of different cells reacting to a stimulus and be polyclonal in origin. Indeed, most evidence suggests that KS can be both entities, sometimes being polyclonal220 with other cases showing monoclonality221,222. These molecular findings may be consistent with some cases behaving more aggressively than others, although a correlation of clonality with clinical behaviour has not been documented. This scenario is not unprecedented for a herpesvirus-driven disease, as Epstein–Barr-virus-associated post-transplant lymphoproliferative disorders range from reactive polyclonal proliferations to true lymphomas223. Cellular genetic alterations — namely, in the cancer-related genes TP53 and KRAS — have been reported only in a few cases of KS224,225,226. Current genomic techniques and biocomputational methods in cohorts with annotated clinical information should provide a much broader understanding of the pathophysiology of KS, beyond KSHV infection, in the coming years.
Clinical presentation
One of the difficulties in understanding KS is that its clinical presentation varies widely. Some patients with KS have indolent disease, which has led some researchers to conclude that KS may not result from a transformation event that leads to autonomously growing tumour cells; instead, it might represent a hyperplastic proliferative disease due to ongoing viral stimulation that drives angiogenesis and local and systemic inflammation227,228. By contrast, however, some patients with KS have aggressive, disseminated disease, with malignant behaviour. This heterogeneity is also seen at the histological level, where lesions that are largely composed of inflammatory infiltrates, including lymphocytes, plasma cells and macrophages or sheets of spindle cells, can be observed. The proportion of KSHV-infected cells also varies among lesions in patients with KS, ranging from a few to the majority of cells being positive for KSHV. The clinical or prognostic implication of these histological differences is not clear, and correlative analysis of the proportion of infected cells with the clinical features present is ongoing in AIDS Malignancy Consortium trials for KS.
Prevention
The prevention of KS should be possible given that it is an infectious disease. An HIV vaccine would eliminate AIDS-related KS, and a KSHV vaccine would eliminate all cases of KS. Although progress is being made towards developing a KSHV vaccine (for example, by bettering our understanding of T cell responses to KSHV in humans32 and testing, in mice, virus-like particles containing the gpK8.1, gB and gH–gL KSHV glycoproteins that are involved in virus entry into host cells229), a vaccine for use in humans is unlikely to be available in the near future. The development of a KSHV vaccine has been hampered by specific biological features of KSHV, such as latency and its ability to evade the host immune system230.
In addition, only a fraction of individuals infected with both HIV and KSHV develop KS38, suggesting that there are other causal factors in the aetiology of KS. The importance of an inflammatory milieu in the development of KS has received attention both at an anecdotal clinical macroscopic level231,232 and in basic science investigation233. Moreover, emerging evidence suggests that biomarkers of inflammation are associated with the occurrence of KS in human studies. For example, a higher ratio of plasma kynurenine to tryptophan levels, reflective of tryptophan metabolism, is associated with lower occurrence of KS234. Another study showed that CXCL10 (also known as IP10), IL-1 receptor type 2, soluble form (sIL-1RII), IL-2 receptor-α (IL-2RA) and CCL3 were markedly associated with KS after adjustment for age and smoking status235. Understanding which of these biomarkers are causal (and hence candidates for intervention), rather than markers of biological processes, is at the forefront of translational research.
Finally, as we increase our understanding of the risk factors for cART-resistant KS and for the development of KS-IRIS, particularly in low-resource environments, it may be possible to devise pre-emptive strategies, such as early initiation of KS-specific therapy, to prevent these adverse outcomes.
Management
KS management in the near term is likely to capitalize on the growing understanding of KS pathogenesis, which has already provided a rationale for targeted treatments that have induced the regression of KS lesions and ameliorated disease symptoms. In addition to conducting larger efficacy trials of these drugs, especially orally bioavailable agents such as pomalidomide and bortezomib, there is a strong rationale to assess their efficacy as part of combination therapies that target multiple steps in KS development and progression and as adjuncts to established chemotherapy regimens. However, it has been difficult to mount large-scale efficacy trials of promising agents and drug combinations because of the declining incidence of AIDS-related KS in high-resource settings and the limited cancer research infrastructure in lower-resource settings where AIDS-related KS is still common.
Other novel therapeutic approaches being studied for treating other neoplasms — in particular, the blockade of inhibitory receptors (for example, programmed cell death protein 1 (PD1), programmed cell death 1 ligand 1 (PDL1) and cytotoxic T lymphocyte protein 4 (CTLA4)) that might otherwise prevent effective immune responses to virally induced neoplasms236 — are being explored in KS. Indeed, PDL1 and PD1 are expressed in HIV-positive KS tissue samples237, including samples derived from patients on cART with well-controlled HIV and high CD4 cell counts238. Pilot studies of the use of immune checkpoint inhibitors in KS have been promising both in patients with HIV239 and in patients who are HIV seronegative240.
Finally, of note, targeted approaches that utilize orally bioavailable drugs with acceptable safety profiles and that can be easily integrated into outpatient treatment regimens may be studied more intensively in high-incidence, low-resource settings than intravenous chemotherapy and radiation therapy.
Integration
KS is a complex and heterogeneous disease, and, although the discovery of KSHV as its causal agent 24 years ago led to an improved understanding of its transmission and pathogenesis, much remains unclear. We do now have both epidemiologic and molecular evidence of causality, and it is clear that KSHV infection is necessary, although not sufficient, for KS to develop. However, a number of questions that have been speculated upon, but not fully experimentally validated, are outstanding. For example, it is unclear why KSHV is more highly seroprevalent in sub-Saharan Africa; one possible explanation for this is a greater propensity to share saliva in this area (see above), but genetic, nutritional and other factors may also contribute. Another interesting and outstanding question is why KS is more common in patients living near volcanoes than in patients living elsewhere241. It has been proposed that localized immunodeficiency in the skin favours the development of KS, and this may be induced in the extremities by exposure to volcanic soil leads242 and by the use of topical steroids243. The much higher incidence of KS in men than in women remains poorly understood. A possible explanation is that there are gender disparities in the immune response to KSHV and that disease susceptibility is related to sex steroid hormones244.
Clinically there have been major strides in the management of KS, in particular with respect to AIDS-related KS in line with the improved control of HIV infection; AIDS-related KS incidence rates have declined with good HIV control, and effectively treating patients with KS with cART improves the outcome of early KS. However, the current treatment of AIDS-related KS that does not respond to HIV control is largely based on chemotherapy. More targeted biological treatments, such as those directly inhibiting vascular proliferation, or the targeting of latent viral proteins such as vFLIP, are only just being tested in experimental models or in clinical trials, and it is not clear whether any of these will be curative.
A preventive approach would be ideal to greatly reducing the global incidence of KS, and studies are underway to achieve this. In the meantime, as with other cancers in which a personalized, targeted approach has been used to tailor therapy, researchers will need to determine whether distinctive biomarkers or genetic features can inform clinicians on the best therapeutic interventions for KS. In contrast to most other cancers, the geographic location of the majority of KS patients in low-resource countries makes these studies challenging. Nevertheless, our increased understanding of the pathobiology of KS and our increased armamentarium of targeted agents and immunomodulators makes this an achievable goal.
References
Kaposi, M. Idiopatisches multiples pigmentsarkom der haut [German]. Arch. Dermatol. Syph. 4, 265–273 (1872).
Montpellier, J. & Mussini-Montpellier, J. Le Cancer en France d’outre-mer: Considérations Pathogéniques (Libraire Ferraris, 1947).
D’Oliveira, J. J. & Torres, F. O. Kaposi’s sarcoma in the Bantu of Mozambique. Cancer 30, 553–561 (1972).
Thijs, A. L’angiosarcomatose de Kaposi au Congo Beige et au Ruanda-Urundi [French]. Ann. Soc. Belg. Med. Trop. 37, 295–307 (1957).
Gottlieb, G. J. et al. A preliminary communication on extensively disseminated Kaposi’s sarcoma in young homosexual men. Am. J. Dermatopathol. 3, 111–114 (1981). This paper presents the first report of KS in MSM as a harbinger of the AIDS epidemic.
Ariyoshi, K. et al. Kaposi’s sarcoma in the Gambia, West Africa is less frequent in human immunodeficiency virus type 2 than in human immunodeficiency virus type 1 infection despite a high prevalence of human herpesvirus 8. J. Hum. Virol. 1, 193–199 (1998).
Siegel, J. H. et al. Disseminated visceral Kaposi’s sarcoma. Appearance after human renal homograft operation. JAMA 207, 1493–1496 (1969).
Fahey, J. L. Cancer in the immunosuppressed patient. Ann. Intern. Med. 75, 310–312 (1971).
Grulich, A. E. & Vajdic, C. M. The epidemiology of cancers in human immunodeficiency virus infection and after organ transplantation. Semin. Oncol. 42, 247–257 (2015).
Friedman-Kien, A. E. et al. Kaposi’s sarcoma in HIV-negative homosexual men. Lancet 335, 168–169 (1990).
Lanternier, F. et al. Kaposi’s sarcoma in HIV-negative men having sex with men. AIDS 22, 1163–1168 (2008).
Rashidghamat, E., Bunker, C. B., Bower, M. & Banerjee, P. Kaposi sarcoma in HIV-negative men who have sex with men. Br. J. Dermatol. 171, 1267–1268 (2014).
Denis, D. et al. A fifth subtype of Kaposi’s sarcoma, classic Kaposi’s sarcoma in men who have sex with men: a cohort study in Paris. J. Eur. Acad. Dermatol. Venereol. 32, 1377–1384 (2018). This study recognizes a fifth type of KS occurring in MSM without HIV infection.
Chang, Y. et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266, 1865–1869 (1994). This study discovers KSHV in a KS lesion.
Cesarman, E., Chang, Y., Moore, P. S., Said, J. W. & Knowles, D. M. Kaposi’s Sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body cavity-based lymphomas. N. Eng. J. Med. 332, 1186–1191 (1995).
Soulier, J. et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 86, 1275–1280 (1995).
Uldrick, T. S. et al. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without Multicentric Castleman disease. Clin. Infect. Dis. 51, 350–358 (2010).
Grulich, A. E., Beral, V. & Swerdlow, A. J. Kaposi’s sarcoma in England and Wales before the AIDS epidemic. Br. J. Cancer 66, 1135–1137 (1992).
Cottoni, F., De Marco, R. & Montesu, M. A. Classical Kaposi’s sarcoma in north-east Sardinia: an overview from 1977 to 1991. Br. J. Cancer 73, 1132–1133 (1996).
Cook-Mozaffari, P., Newton, R., Beral, V. & Burkitt, D. P. The geographical distribution of Kaposi’s sarcoma and of lymphomas in Africa before the AIDS epidemic. Br. J. Cancer 78, 1521–1528 (1998).
Lebbe, C., Legendre, C. & Frances, C. Kaposi sarcoma in transplantation. Transplant Rev. (Orlando) 22, 252–261 (2008).
Mesri, E. A., Cesarman, E. & Boshoff, C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 10, 707–719 (2010).
Wakeham, K. et al. Parasite infection is associated with Kaposi’s sarcoma associated herpesvirus (KSHV) in Ugandan women. Infect. Agent Cancer 6, 15 (2011).
Butler, L. M., Neilands, T. B., Mosam, A., Mzolo, S. & Martin, J. N. A population-based study of how children are exposed to saliva in KwaZulu-Natal Province, South Africa: implications for the spread of saliva-borne pathogens to children. Trop. Med. Int. Health 15, (442–453 (2010).
Hymes, K. B. et al. Kaposi’s sarcoma in homosexual men-a report of eight cases. Lancet 2, 598–600 (1981).
Beral, V., Peterman, T. A., Berkelman, R. L. & Jaffe, H. W. Kaposi’s sarcoma among persons with AIDS: a sexually transmitted infection? Lancet 335, 123–128 (1990).
Hermans, P. et al. Epidemiology of AIDS-related Kaposi’s sarcoma in Europe over 10 years. AIDS in Europe Study Group. Aids 10, 911–917 (1996).
Elford, J., McDonald, A. & Kaldor, J. Kaposi’s sarcoma as a sexually transmissible infection: an analysis of Australian AIDS surveillance data. The National HIV Surveillance Committee. Aids 7, 1667–1671 (1993).
International Agency for Reseach on Cancer. in IARC Monographs on the Evaluation of Carcinogenic Risks to Humans Vol. 100B (International Agency for Reseach on Cancer, 2012).
Wabinga, H. R., Parkin, D. M., Wabwire-Mangen, F. & Mugerwa, J. W. Cancer in Kampala, Uganda, in 1989-91: changes in incidence in the era of AIDS. Int. J. Cancer 54, 26–36 (1993).
Parkin, D. M. et al. Part I: cancer in indigenous Africans—burden, distribution, and trends. Lancet Oncol. 9, 683–692 (2008).
Roshan, R. et al. T-cell responses to KSHV infection: a systematic approach. Oncotarget 8, 109402–109416 (2017). This study assesses T cell responses to all of the KSHV proteins, finding that responses are heterogenous and occur in response to diverse antigens.
International Collaboration on HIV and Cancer. Highly active antiretroviral therapy and incidence of cancer in human immunodeficiency virus-infected adults. J. Natl Cancer Inst. 92, 1823–1830 (2000).
Semeere, A. S., Busakhala, N. & Martin, J. N. Impact of antiretroviral therapy on the incidence of Kaposi’s sarcoma in resource-rich and resource-limited settings. Curr. Opin. Oncol. 24, 522–530 (2012).
Chaabna, K. et al. Kaposi sarcoma trends in Uganda and Zimbabwe: a sustained decline in incidence? Int. J. Cancer 133, 1197–1203 (2013).
The AIDS-defining Cancer Project Working Group for IeDEA and COHERE in EuroCoord. Comparison of Kaposi sarcoma risk in human immunodeficiency virus-positive adults across 5 continents: a multiregional multicohort study. Clin. Infect. Dis. 65, 1316–1326 (2017).
Labo, N., Miley, W., Benson, C. A., Campbell, T. B. & Whitby, D. Epidemiology of Kaposi’s sarcoma-associated herpesvirus in HIV-1-infected US persons in the era of combination antiretroviral therapy. AIDS 29, 1217–1225 (2015).
Yarchoan, R. & Uldrick, T. S. HIV-associated cancers and related diseases. N. Engl. J. Med. 378, 1029–1041 (2018).
Newton, R. et al. Kaposi sarcoma-associated herpesvirus in a rural Ugandan cohort, 1992-2008. J. Infect. Dis. 217, 263–269 (2018).
Bechtel, J. T., Liang, Y., Hvidding, J. & Ganem, D. Host range of Kaposi’s sarcoma-associated herpesvirus in cultured cells. J. Virol. 77, 6474–6481 (2003).
Kumar, B., Roy, A., Veettil, M. V. & Chandran, B. Insight into the roles of E3 ubiquitin ligase c-Cbl, ESCRT machinery, and host cell signaling in Kaposi’s sarcoma-associated herpesvirus entry and trafficking. J. Virol. 92, e01376-17 (2018).
Damania, B. & Cesarman, E. in Field’s Virology Vol. 2 (eds Knipe, D. M. et al.) 2080–2128 (Lippincott Williams & Wilkins, 2013).
Kumar, B. & Chandran, B. KSHV entry and trafficking in target cells-hijacking of cell signal pathways, actin and membrane dynamics. Viruses 8, 305 (2016).
Sarid, R., Flore, O., Bohenzky, R. A., Chang, Y. & Moore, P. S. Transcription mapping of the Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 72, 1005–1012 (1998).
Cai, X. et al. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl Acad. Sci. USA 102, 5570–5575 (2005). This article presents the first discovery of KSHV-encoded viral miRNAs.
Chandriani, S. & Ganem, D. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 84, 5565–5573 (2010).
Hosseinipour, M. C. et al. Viral profiling identifies multiple subtypes of Kaposi’s sarcoma. mBio 5, e01633–01614 (2014).
Abere, B. et al. The Kaposi’s sarcoma-associated herpesvirus (KSHV) non-structural membrane protein K15 is required for viral lytic replication and may represent a therapeutic target. PLOS Pathog. 13, e1006639 (2017).
Ballestas, M. E., Chatis, P. A. & Kaye, K. M. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284, 641–644 (1999).
Chaudhary, P. M., Jasmin, A., Eby, M. T. & Hood, L. Modulation of the NF-κB pathway by virally encoded death effector domains-containing proteins. Oncogene 14, 5738–5746 (1999).
Field, N. et al. KSHV vFLIP binds to IKK-γ to activate IKK. J. Cell Sci. 116, 3721–3728 (2003).
Guasparri, I., Keller, S. A. & Cesarman, E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 199, 993–1003 (2004).
Guasparri, I., Wu, H. & Cesarman, E. The KSHV oncoprotein vFLIP contains a TRAF-interacting motif and requires TRAF2 and TRAF3 for signalling. EMBO Rep. 7, 114–119 (2006).
Samols, M. A., Hu, J., Skalsky, R. L. & Renne, R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 79, 9301–9305 (2005).
Pfeffer, S. et al. Identification of microRNAs of the herpesvirus family. Nat. Methods 2, 269–276 (2005).
Dittmer, D. P. & Damania, B. Kaposi sarcoma-associated herpesvirus: immunobiology, oncogenesis, and therapy. J. Clin. Invest. 126, 3165–3175 (2016).
O’Hara, A. J. et al. Pre-micro RNA signatures delineate stages of endothelial cell transformation in Kaposi sarcoma. PLOS Pathog. 5, e1000389 (2009).
O’Hara, A. J. et al. Tumor suppressor microRNAs are underrepresented in primary effusion lymphoma and Kaposi sarcoma. Blood 113, 5938–5941 (2009).
Ray, A. et al. Sequence analysis of Kaposi sarcoma-associated herpesvirus (KSHV) microRNAs in patients with multicentric Castleman disease and KSHV-associated inflammatory cytokine syndrome. J. Infect. Dis. 205, 1665–1676 (2012).
Forte, E. et al. MicroRNA-mediated transformation by the Kaposi’s sarcoma-associated herpesvirus Kaposin locus. J. Virol. 89, 2333–2341 (2015).
Hansen, A. et al. KSHV-encoded miRNAs target MAF to induce endothelial cell reprogramming. Genes Dev. 24, 195–205 (2010).
Hu, M. et al. A KSHV microRNA directly targets G protein-coupled receptor kinase 2 to promote the migration and invasion of endothelial cells by inducing CXCR2 and activating AKT signaling. PLOS Pathog. 11, e1005171 (2015).
Bellare, P. & Ganem, D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 6, 570–575 (2009).
Verschuren, E. W., Klefstrom, J., Evan, G. I. & Jones, N. The oncogenic potential of Kaposi’s sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell 2, 229–241 (2002).
Sin, S. H. & Dittmer, D. P. Viral latency locus augments B-cell response in vivo to induce chronic marginal zone enlargement, plasma cell hyperplasia, and lymphoma. Blood 121, 2952–2963 (2013).
Ahmad, A. et al. Kaposi’s sarcoma associated herpesvirus-encoded viral FLICE inhibitory protein (vFLIP) K13 cooperates with Myc to promote lymphoma in mice. Cancer Biol. Ther. 10, 1033–1040 (2010).
Ballon, G., Chen, K., Perez, R., Tam, W. & Cesarman, E. Kaposi sarcoma herpesvirus (KSHV) vFLIP oncoprotein induces B cell transdifferentiation and tumorigenesis in mice. J. Clin. Invest. 121, 1141–1153 (2011). This paper reports that vFLIP is an oncoprotein in vivo.
Ballon, G., Akar, G. & Cesarman, E. Systemic expression of Kaposi sarcoma herpesvirus (KSHV) Vflip in endothelial cells leads to a profound proinflammatory phenotype and myeloid lineage remodeling in vivo. PLOS Pathog. 11, e1004581 (2015).
Lee, J. S. et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 11, 1355–1362 (2009).
Briggs, L. C. et al. IKKγ-mimetic peptides block the resistance to apoptosis associated with Kaposi’s sarcoma-associated herpesvirus infection. J. Virol. 91, e01170-17 (2017).
Nayar, U. et al. Identification of a nucleoside analog active against adenosine kinase-expressing plasma cell malignancies. J. Clin. Invest. 127, 2066–2080 (2017).
Sin, S. H. et al. Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood 109, 2165–2173 (2007).
Stallone, G. et al. Sirolimus for Kaposi’s sarcoma in renal-transplant recipients. N. Engl. J. Med. 352, 1317–1323 (2005).
Krown, S. E. et al. Rapamycin with antiretroviral therapy in AIDS-associated Kaposi sarcoma. J. Acquir. Immune Defic. Syndr. 59, 447–454 (2012).
Nayar, U. et al. Targeting the Hsp90-associated viral oncoproteome in gammaherpesvirus-associated malignancies. Blood 122, 2837–2847 (2013).
Chen, W., Sin, S. H., Wen, K. W., Damania, B. & Dittmer, D. P. Hsp90 inhibitors are efficacious against Kaposi Sarcoma by enhancing the degradation of the essential viral gene LANA, of the viral co-receptor EphA2 as well as other client proteins. PLOS Pathog. 8, e1003048 (2012).
Aoki, Y. et al. Detection of viral interleukin-6 in Kaposi sarcoma-associated herpesvirus-linked disorders. Blood 97, 2173–2176 (2001).
Arvanitakis, L., Geras-Raaka, E., Gershengorn, M. C. & Cesarman, E. Human herpesvirus KSHV encodes a constitutively active G protein-coupled receptor linked to cell proliferation. Nature 385, 347–350 (1997).
Bais, C. et al. G-protein-coupled receptor of Kaposi’s sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 391, 86–89 (1998). This article shows that vGPCR is a viral oncogene.
Cavallin, L. E. et al. KSHV-induced ligand mediated activation of PDGF receptor-alpha drives Kaposi’s sarcomagenesis. PLOS Pathog. 14, e1007175 (2018). This paper shows that PDGFRA is a mediator of KSHV sarcoma genesis, which is important because there are inhibitors of this tyrosine kinase receptor.
Yang, T.-Y. et al. Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi’s sarcoma. J. Exp. Med. 191, 445–454 (2000).
Montaner, S. et al. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi’s sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell 3, 23–36 (2003).
Mutlu, A. D. et al. In vivo-restricted and reversible malignancy induced by human herpesvirus-8 KSHV: a cell and animal model of virally induced Kaposi’s sarcoma. Cancer Cell 11, 245–258 (2007).
Cesarman, E., Mesri, E. A. & Gershengorn, M. C. Viral G protein-coupled receptor and Kaposi’s sarcoma: a model of paracrine neoplasia? J. Exp. Med. 191, 417–422 (2000).
Wang, L., Dittmer, D. P., Tomlinson, C. C., Fakhari, F. D. & Damania, B. Immortalization of primary endothelial cells by the K1 protein of Kaposi’s sarcoma-associated herpesvirus. Cancer Res. 66, 3658–3666 (2006).
Uldrick, T. S. et al. High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus-associated multicentric Castleman disease: a pilot study of virus-activated cytotoxic therapy. Blood 117, 6977–6986 (2011).
Martin, D. F. et al. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. N. Engl. J. Med. 340, 1063–1070 (1999).
Krown, S. E., Dittmer, D. P. & Cesarman, E. Pilot study of oral valganciclovir therapy in patients with classic Kaposi sarcoma. J. Infect. Dis. 203, 1082–1086 (2011).
Bhatt, A. P. & Damania, B. AKTivation of PI3K/AKT/mTOR signaling pathway by KSHV. Front. Immunol. 3, 401 (2012).
Avey, D., Tepper, S., Li, W., Turpin, Z. & Zhu, F. Phosphoproteomic analysis of KSHV-infected cells reveals roles of ORF45-activated RSK during lytic replication. PLOS Pathog. 11, e1004993 (2015).
Bhatt, A. P. et al. A viral kinase mimics S6 kinase to enhance cell proliferation. Proc. Natl Acad. Sci. USA 113, 7876–7881 (2016).
Lee, H. et al. Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi’s sarcoma-associated herpesvirus. Mol. Cell. Biol. 18, 5219–5228 (1998).
Tomlinson, C. C. & Damania, B. The K1 protein of Kaposi’s sarcoma-associated herpesvirus activates the Akt signaling pathway. J. Virol. 78, 1918–1927 (2004).
Zhang, Z. et al. The K1 protein of Kaposi’s sarcoma-associated herpesvirus augments viral lytic replication. J. Virol. 90, 7657–7666 (2016).
Anders, P. M., Zhang, Z., Bhende, P. M., Giffin, L. & Damania, B. The KSHV K1 protein modulates AMPK function to enhance cell survival. PLOS Pathog. 12, e1005985 (2016).
Bais, C. et al. Kaposi’s sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/ KDR. Cancer Cell 3, 131–143 (2003).
Sodhi, A. et al. The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell 10, 133–143 (2006).
Brinkmann, M. M. et al. Activation of mitogen-activated protein kinase and NF-κB pathways by a Kaposi’s sarcoma-associated herpesvirus K15 membrane protein. J. Virol. 77, 9346–9358 (2003).
Gramolelli, S. et al. Inhibiting the recruitment of PLCγ1 to Kaposi’s sarcoma herpesvirus K15 protein reduces the invasiveness and angiogenesis of infected endothelial cells. PLOS Pathog. 11, e1005105 (2015).
Cannon, M., Philpott, N. J. & Cesarman, E. The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor has broad signaling effects in primary effusion lymphoma cells. J. Virol. 77, 57–67 (2003).
Li, H., Wang, H. & Nicholas, J. Detection of direct binding of human herpesvirus 8-encoded interleukin-6 (vIL-6) to both gp130 and IL-6 receptor (IL-6R) and identification of amino acid residues of vIL-6 important for IL-6R-dependent and -independent signaling. J. Virol. 75, 3325–3334 (2001).
Molden, J., Chang, Y., You, Y., Moore, P. S. & Goldsmith, M. A. A. Kaposi’s sarcoma-associated herpesvirus-encoded cytokine homolog (vIL-6) activates signaling through the shared gp130 receptor subunit. J. Biol. Chem. 272, 19625–19631 (1997).
Suthaus, J. et al. HHV-8-encoded viral IL-6 collaborates with mouse IL-6 in the development of multicentric Castleman disease in mice. Blood 119, 5173–5181 (2012).
Hamza, M. S. et al. ORF36 protein kinase of Kaposi’s sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J. Biol. Chem. 279, 38325–38330 (2004).
Anders, P. et al. Human herpesvirus encoded kinase induces B cell lymphomas in vivo. J. Clin. Invest. 128, 2519–2534 (2018).
Matta, H., Sun, Q., Moses, G. & Chaudhary, P. M. Molecular genetic analysis of human herpes virus 8-encoded viral FLICE inhibitory protein-induced NF-κB activation. J. Biol. Chem. 278, 52406–52411 (2003).
Sun, Q., Matta, H. & Chaudhary, P. M. The human herpes virus 8-encoded viral FLICE inhibitory protein protects against growth factor withdrawal-induced apoptosis via NF-κ B activation. Blood 101, 1956–1961 (2003).
Godfrey, A., Anderson, J., Papanastasiou, A., Takeuchi, Y. & Boshoff, C. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood 105, 2510–2518 (2005).
Moore, P. S. et al. Primary characterization of a herpesvirus agent associated with Kaposi’s sarcoma. J. Virol. 70, 549–558 (1996).
Labo, N. et al. Heterogeneity and breadth of host antibody response to KSHV infection demonstrated by systematic analysis of the KSHV proteome. PLOS Pathog. 10, e1004046 (2014).
Olp, L. N. et al. Longitudinal analysis of the humoral response to Kaposi’s sarcoma-associated herpesvirus after primary infection in children. J. Med. Virol. 88, 1973–1981 (2016).
Hopcraft, S. E. & Damania, B. Tumour viruses and innate immunity. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 372, 20160267 (2017).
West, J. & Damania, B. Upregulation of the TLR3 pathway by Kaposi’s sarcoma-associated herpesvirus during primary infection. J. Virol. 82, 5440–5449 (2008).
Lagos, D. et al. Toll-like receptor 4 mediates innate immunity to Kaposi sarcoma herpesvirus. Cell Host Microbe 4, 470–483 (2008).
West, J. A., Gregory, S. M., Sivaraman, V., Su, L. & Damania, B. Activation of plasmacytoid dendritic cells by Kaposi’s sarcoma-associated herpesvirus. J. Virol. 85, 895–904 (2011).
Gregory, S. M. et al. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331, 330–334 (2011). This article demonstrates that KSHV encodes a viral protein, ORF63, that inhibits inflammasomes.
Kerur, N. et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375 (2011).
Li, W. et al. Kaposi’s sarcoma-associated herpesvirus inhibitor of cGAS (KicGAS), encoded by ORF52, is an abundant tegument protein and is required for production of infectious progeny viruses. J. Virol. 90, 5329–5342 (2016).
Coscoy, L. & Ganem, D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl Acad. Sci. USA 97, 8051–8056 (2000).
Ishido, S., Wang, C., Lee, B. S., Cohen, G. B. & Jung, J. U. Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 74, 5300–5309 (2000).
Jacobs, S. R. et al. The viral interferon regulatory factors of kaposi’s sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by toll-like receptor 3. J. Virol. 87, 798–806 (2013).
Burysek, L. et al. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J. Virol. 73, 7334–7342 (1999).
Ma, Z. et al. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl Acad. Sci. USA 112, E4306–E4315 (2015).
Wu, J. J. et al. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18, 333–344 (2015).
Zhang, G. et al. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl Acad. Sci. USA 113, E1034–E1043 (2016).
Fuld, S., Cunningham, C., Klucher, K., Davison, A. J. & Blackbourn, D. J. Inhibition of interferon signaling by the Kaposi’s sarcoma-associated herpesvirus full-length viral interferon regulatory factor 2 protein. J. Virol. 80, 3092–3097 (2006).
Joo, C. H. et al. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol. 81, 8282–8292 (2007).
Lubyova, B., Kellum, M. J., Frisancho, A. J. & Pitha, P. M. Kaposi’s sarcoma-associated herpesvirus-encoded vIRF-3 stimulates the transcriptional activity of cellular IRF-3 and IRF-7. J. Biol. Chem. 279, 7643–7654 (2004).
Bussey, K. A. et al. The gammaherpesviruses Kaposi’s sarcoma-associated herpesvirus and murine gammaherpesvirus 68 modulate the Toll-like receptor-induced proinflammatory cytokine response. J. Virol. 88, 9245–9259 (2014).
Yu, Y., Wang, S. E. & Hayward, G. S. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22, 59–70 (2005).
Zhu, F. X., King, S. M., Smith, E. J., Levy, D. E. & Yuan, Y. A. Kaposi’s sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl Acad. Sci. USA 99, 5573–5578 (2002).
Chatterjee, M., Osborne, J., Bestetti, G., Chang, Y. & Moore, P. S. Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science 298, 1432–1435 (2002).
Inn, K. S. et al. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 85, 10899–10904 (2011).
Nicholas, J. et al. Kaposi’s sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat. Med. 3, 287–292 (1997). This paper reports the presence of several viral homologues of cellular proteins involved in the immune response.
Kledal, T. N. et al. A broad-spectrum chemokine antagonist encoded by Kaposi’s sarcoma-associated herpesvirus. Science 277, 1656–1659 (1997).
Yamin, R. et al. The viral KSHV chemokine vMIP-II inhibits the migration of Naive and activated human NK cells by antagonizing two distinct chemokine receptors. PLOS Pathog. 9, e1003568 (2013).
DeBruyne, L. A., Li, K., Bishop, D. K. & Bromberg, J. S. Gene transfer of virally encoded chemokine antagonists vMIP-II and MC148 prolongs cardiac allograft survival and inhibits donor-specific immunity. Gene Ther. 7, 575–582 (2000).
Choi, Y. B. & Nicholas, J. Autocrine and paracrine promotion of cell survival and virus replication by human herpesvirus 8 chemokines. J. Virol. 82, 6501–6513 (2008).
Foster-Cuevas, M., Wright, G. J., Puklavec, M. J., Brown, M. H. & Barclay, A. N. Human herpesvirus 8 K14 protein mimics CD200 in down-regulating macrophage activation through CD200 receptor. J. Virol. 78, 7667–7676 (2004).
Gorczynski, R. M. et al. An immunoadhesin incorporating the molecule OX-2 is a potent immunosuppressant that prolongs allo- and xenograft survival. J. Immunol. 163, 1654–1660 (1999).
Borriello, F., Lederer, J., Scott, S. & Sharpe, A. H. MRC OX-2 defines a novel T cell costimulatory pathway. J. Immunol. 158, 4548–4554 (1997).
Rezaee, S. A., Gracie, J. A., McInnes, I. B. & Blackbourn, D. J. Inhibition of neutrophil function by the Kaposi’s sarcoma-associated herpesvirus vOX2 protein. AIDS 19, 1907–1910 (2005).
Chung, Y. H., Means, R. E., Choi, J. K., Lee, B. S. & Jung, J. U. Kaposi’s sarcoma-associated herpesvirus OX2 glycoprotein activates myeloid-lineage cells to induce inflammatory cytokine production. J. Virol. 76, 4688–4698 (2002).
Amerson, E. et al. Diagnosing Kaposi’s sarcoma (KS) in East Africa: how accurate are clinicians and pathologists? Infect. Agents Cancer 7 (Suppl. 1), P6 (2012).
Pantanowitz, L. & Duke, W. H. Lymphoedematous variants of Kaposi’s sarcoma. J. Eur. Acad. Dermatol. Venereol. 22, 118–120 (2008).
Bower, M. et al. Prospective stage-stratified approach to AIDS-related Kaposi’s sarcoma. J. Clin. Oncol. 32, 409–414 (2014).
Krown, S. E., Testa, M. A. & Huang, J. AIDS-related Kaposi’s sarcoma: prospective validation of the AIDS Clinical Trials Group staging classification. J. Clin. Oncol. 15, 3085–3092 (1997). The classification of KS is described and reported to be relevant in this paper.
Krown, S. E., Metroka, C. & Wernz, J. C. Kaposi’s sarcoma in the acquired immune deficiency syndrome: a proposal for uniform evaluation, response, and staging criteria. AIDS Clinical Trials Group Oncology Committee. J. Clin. Oncol. 7, 1201–1207 (1989).
Brambilla, L., Boneschi, V., Taglioni, M. & Ferrucci, S. Staging of classic Kaposi’s sarcoma: a useful tool for therapeutic choices. Eur. J. Dermatol. 13, 83–86 (2003).
Amerson, E. et al. Accuracy of clinical suspicion and pathologic diagnosis of Kaposi sarcoma in east Africa. J. Acquir. Immune. Defic. Syndr. 71, 295–301 (2016). This paper reports that visual diagnosis of KS in Africa is often nonspecific and can lead to inappropriate treatment with chemotherapy, and that pathological evaluation in Africa is also often inaccurate.
Forrestel, A. K. et al. Bacillary angiomatosis masquerading as Kaposi’s sarcoma in East Africa. J. Int. Assoc. Provid. AIDS Care 14, 21–25 (2015).
Laker-Oketta, M. O. et al. Task shifting and skin punch for the histologic diagnosis of Kaposi’s sarcoma in Sub-Saharan Africa: a public health solution to a public health problem. Oncology 89, 60–65 (2015). This study illustrates that skin biopsy, which is often performed only by physicians in settings such as the USA, can be performed safely by non-physicians in sub-Saharan Africa, thereby expanding access to the procedure.
Nguyen, A., Tran, D., Uemura, M., Bardin, R. L. & Shitabata, P. K. Practical and sustainable teledermatology and teledermatopathology: specialty care in Cameroon Africa. J. Clin. Aesthet. Dermatol. 10, 47–56 (2017).
Grayson, W. & Pantanowitz, L. Histological variants of cutaneous Kaposi sarcoma. Diagn. Pathol. 3, 31 (2008).
Kahn, H. J., Bailey, D. & Marks, A. Monoclonal antibody D2-40, a new marker of lymphatic endothelium, reacts with Kaposi’s sarcoma and a subset of angiosarcomas. Mod. Pathol. 15, 434–440 (2002).
Pyakurel, P. et al. Lymphatic and vascular origin of Kaposi’s sarcoma spindle cells during tumor development. Int. J. Cancer 119, 1262–1267 (2006).
Massarelli, G., Scott, C. A., Ibba, M., Tanda, F. & Cossu, A. Immunocytochemical profile of Kaposi’s sarcoma cells: their reactivity to a panel of antibodies directed against different tissue cell markers. Appl. Pathol. 7, 34–41 (1989).
Li, Y. et al. Evidence for Kaposi sarcoma originating from mesenchymal stem cell through KSHV-induced mesenchymal-to-endothelial transition. Cancer Res. 78, 230–245 (2018).
Wang, H. W. et al. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat. Genet. 36, 687–693 (2004).
Kaaya, E. E. et al. Heterogeneity of spindle cells in Kaposi’s sarcoma: comparison of cells in lesions and in culture. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 10, 295–305 (1995).
Hussein, M. R. Immunohistological evaluation of immune cell infiltrate in cutaneous Kaposi’s sarcoma. Cell Biol. Int. 32, 157–162 (2008).
Chang, Y. et al. Kaposi’s sarcoma-associated herpesvirus and Kaposi’s sarcoma in Africa. Arch. Intern. Med. 156, 202–204 (1996).
Jiang, L. et al. Solar thermal polymerase chain reaction for smartphone-assisted molecular diagnostics. Sci. Rep. 4, 4137 (2014).
Snodgrass, R. et al. KS-detect - validation of solar thermal PCR for the diagnosis of Kaposi’s sarcoma using pseudo-biopsy samples. PLOS ONE 11, e0147636 (2016).
Snodgrass, R. et al. A portable device for nucleic-acid quantification powered by sunlight, flame or electricity. Nat. Biomed. Engineer. 2, 657–665 (2018). This article provides a demonstration of a prototype device for how KS could conceivably be diagnosed with a point-of-care test not requiring histopathological evaluation.
Stebbing, J. et al. The presentation and survival of patients with non-cutaneous AIDS-associated Kaposi’s sarcoma. Ann. Oncol. 17, 503–506 (2006).
World Health Organization. Guidelines on the treatment of skin and oral HIV-associated conditions in children and adults. WHO https://www.who.int/maternal_child_adolescent/documents/skin-mucosal-and-hiv/en/ (2014).
Nasti, G. et al. AIDS-related Kaposi’s sarcoma: evaluation of potential new prognostic factors and assessment of the AIDS Clinical Trial Group staging system in the Haart Era—the Italian Cooperative Group on AIDS and Tumors and the Italian Cohort of Patients Naive From Antiretrovirals. J. Clin. Oncol. 21, 2876–2882 (2003).
El Amari, E. B. et al. Predicting the evolution of Kaposi sarcoma, in the highly active antiretroviral therapy era. AIDS 22, 1019–1028 (2008).
Chu, K. M. et al. AIDS-associated Kaposi’s sarcoma is linked to advanced disease and high mortality in a primary care HIV programme in South Africa. J. Int. AIDS Soc. 13, 23 (2010).
Uganda Ministry of Health. Integrated National Guidelines on Antiretroviral Therapy, Prevention of Mother to Child Transmission of HIV, and Infant & Young Child Feeding. 1st edn (Uganda Ministry of Health, October 2011).
Laker-Oketta, M. et al. in 22nd Conf. on Retroviruses and Opportunistic Infections (CROI, Seattle, WA, 2015).
Borok, M. in 16th Int. Conf. on Malignancies in HIV/AIDS (National Cancer Institute, Bethesda, MD, 2017).
De Boer, C., Niyonzima, N., Orem, J., Bartlett, J. & Zafar, S. Y. Prognosis and delay of diagnosis among Kaposi’s sarcoma patients in Uganda: a cross-sectional study. Infect. Agent Cancer 9, 17 (2014).
Laker-Oketta, M. et al. Developing media to promote community awareness of early detection of Kaposi’s sarcoma in Africa. Presented at the 6th Annual Symposium on Global Cancer Research 2018. This paper presents a demonstration of community-based communication strategies to increase awareness of the importance of early detection of KS in sub-Saharan Africa.
World Health Organization. Consolidated guidelines on HIV prevention, diagnosis, treatment, and care for key populations: policy brief - 2016 update (WHO, 2017).
U.S. Department of Health and Human Services. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV. AIDSinfo https://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-treatment-guidelines (2018).
You, H. et al. in 16th Int. Conf. on Malignancies in HIV/AIDS (National Cancer Institute, Bethesda, MD, 2017).
Martin, J. N. et al. Sexual transmission and the natural history of human herpesvirus 8 infection. N. Engl. J. Med. 338, 948–954 (1998).
Engels, E. A. et al. Risk factors for human herpesvirus 8 infection among adults in the United States and evidence for sexual transmission. J. Infect. Dis. 196, 199–207 (2007).
National Cancer Institute, National Institutes of Health. About Cancer: Infectious Agents. National Cancer Institute https://www.cancer.gov/about-cancer/causes-prevention/risk/infectious-agents (2017).
Phillips, A. M. et al. Awareness of Kaposi’s sarcoma-associated herpesvirus among men who have sex with men. Sex. Transm. Dis. 35, 1011–1014 (2008). This paper discusses that MSM, the group most at risk of KSHV in North America, have scant awareness of the virus or their risk.
Butler, L. M., Osmond, D. H., Jones, A. G. & Martin, J. N. Use of saliva as a lubricant in anal sexual practices among homosexual men. J. Acquir. Immune. Defic. Syndr. 50, 162–167 (2009).
Butler, L. M. et al. Kaposi sarcoma-associated herpesvirus (KSHV) seroprevalence in population-based samples of African children: evidence for at least 2 patterns of KSHV transmission. J. Infect. Dis. 200, 430–438 (2009).
Krown, S. & Singh, J. Classic Kaposi sarcoma: clinical features, staging, diagnosis, and treatment. UpToDate https://www.uptodate.com/contents/classic-kaposi-sarcoma-clinical-features-staging-diagnosis-and-treatment?source=history_widget (2018).
Gill, P. S. et al. Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi’s sarcoma. J. Clin. Oncol. 14, 2353–2364 (1996).
Northfelt, D. W. et al. Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi’s sarcoma: results of a randomized phase III clinical trial. J. Clin. Oncol. 16, 2445–2451 (1998).
Stewart, S. et al. Randomized comparative trial of pegylated liposomal doxorubicin versus bleomycin and vincristine in the treatment of AIDS-related Kaposi’s sarcoma. International Pegylated Liposomal Doxorubicin Study Group. J. Clin. Oncol. 16, 683–691 (1998). This paper, and reference 187, are the first to demonstrate superiority of a liposomal anthracycline over standard combination chemotherapy regimens in high-resource settings (USA and UK) prior to modern-day cART.
Lichterfeld, M. et al. Treatment of HIV-1-associated Kaposi’s sarcoma with pegylated liposomal doxorubicin and HAART simultaneously induces effective tumor remission and CD4+ T cell recovery. Infection 33, 140–147 (2005).
Esdaile, B. et al. The immunological effects of concomitant highly active antiretroviral therapy and liposomal anthracycline treatment of HIV-1-associated Kaposi’s sarcoma. AIDS 16, 2344–2347 (2002).
Cooley, T. et al. A randomized, double-blind study of pegylated liposomal doxorubicin for the treatment of AIDS-related Kaposi’s sarcoma. Oncologist 12, 114–123 (2007).
Maurer, T., Ponte, M. & Leslie, K. HIV-associated Kaposi’s sarcoma with a high CD4 count and a low viral load. N. Engl. J. Med. 357, 1352–1353 (2007). This report indicates that AIDS-related KS occurs in the era of cART, even in well-controlled patients.
Krown, S. E., Lee, J. Y. & Dittmer, D. P. More on HIV-associated Kaposi’s sarcoma. N. Engl. J. Med. 358, 535–536 (2008).
Gill, P. S. et al. Paclitaxel is safe and effective in the treatment of advanced AIDS-related Kaposi’s sarcoma. J. Clin. Oncol. 17, 1876–1883 (1999).
Tulpule, A. et al. Multicenter trial of low-dose paclitaxel in patients with advanced AIDS-related Kaposi sarcoma. Cancer 95, 147–154 (2002).
Stebbing, J. et al. Paclitaxel for anthracycline-resistant AIDS-related Kaposi’s sarcoma: clinical and angiogenic correlations. Ann. Oncol. 14, 1660–1666 (2003).
Cianfrocca, M. et al. Randomized trial of paclitaxel versus pegylated liposomal doxorubicin for advanced human immunodeficiency virus-associated Kaposi sarcoma. Cancer 116, 3969–3977 (2010).
Borok, M. et al. in 22nd Int. AIDS Conf. 1009 (International AIDS Society, Amsterdam, 2018). This article presents the first randomized clinical trial of different types of chemotherapy with cART conducted primarily in sub-Saharan Africa in advanced KS and shows the superiority of paclitaxel compared with the regional standard regimen of bleomycin and vincristine.
Krown, S. in Molecular Basis for Therapy of AIDS-defining Cancers (eds Dittmer, D. P. & Krown, S. E.) 181–198 (Springer, 2010).
Bodsworth, N. J. et al. Phase III vehicle-controlled, multi-centered study of topical alitretinoin gel 0.1% in cutaneous AIDS-related Kaposi’s sarcoma. Am. J. Clin. Dermatol. 2, 77–87 (2001).
Duvic, M. et al. Topical treatment of cutaneous lesions of acquired immunodeficiency syndrome-related Kaposi sarcoma using alitretinoin gel: results of phase 1 and 2 trials. Arch. Dermatol. 136, 1461–1469 (2000).
Koon, H. B. et al. Phase II trial of imatinib in AIDS-associated Kaposi’s sarcoma: AIDS malignancy consortium protocol 042. J. Clin. Oncol. 32, 402–408 (2014).
Uldrick, T. S. et al. Phase II study of bevacizumab in patients with HIV-associated Kaposi’s sarcoma receiving antiretroviral therapy. J. Clin. Oncol. 30, 1476–1483 (2012).
Yarchoan, R. et al. PART IV. Cytokine and hormone immunotherapy treatment of AIDS-related Kaposi’s sarcoma with interleukin-12: rationale and preliminary evidence of clinical activity. Crit. Rev. Immunol. 27, 401–414 (2007).
Shimabukuro, K. in 16th Int. Conf. on Malignancies in HIV/AIDS 37 (National Cancer Institute, Bethesda, MD, 2017).
Polizzotto, M. N. et al. Pomalidomide for symptomatic Kaposi’s sarcoma in people with and without HIV infection: a phase I/II study. J. Clin. Oncol. 34, 4125–4131 (2016). This study demonstrates that orally administered pomalidomide induces KS regression in a large percentage of patients with both classic and AIDS-associated KS and is well tolerated. Particularly notable are improvements in KS-associated lymphoedema.
Reid, E. et al. in 16th Int. Conf. on Malignancies in HIV/AIDS 96 (National Cancer Institute, Bethesda, MD, 2017).
McAllister, S. C., Hanson, R. S. & Manion, R. D. Propranolol decreases proliferation of endothelial cells transformed by Kaposi’s sarcoma-associated herpesvirus and induces lytic viral gene expression. J. Virol. 89, 11144–11149 (2015).
Letang, E., Naniche, D., Bower, M. & Miro, J. M. Kaposi sarcoma-associated immune reconstitution inflammatory syndrome: in need of a specific case definition. Clin. Infect. Dis. 55, 157–158 (2012).
Letang, E. et al. Immune reconstitution inflammatory syndrome associated with Kaposi sarcoma: higher incidence and mortality in Africa than in the UK. AIDS 27, 1603–1613 (2013).
Ablanedo-Terrazas, Y., Alvarado-De La Barrera, C. & Reyes-Teran, G. Towards a better understanding of Kaposi sarcoma-associated immune reconstitution inflammatory syndrome. AIDS 27, 1667–1669 (2013).
Hosseinipour, M. C. et al. As-needed versus immediate etoposide chemotherapy in combination with antiretroviral therapy for mild or moderate AIDS-associated Kaposi sarcoma in resource-limited settings: A5264/AMC-067 randomized clinical trial. Clin. Infect. Dis. 67, 251–260 (2018).
Volkow, P. F., Cornejo, P., Zinser, J. W., Ormsby, C. E. & Reyes-Teran, G. Life-threatening exacerbation of Kaposi’s sarcoma after prednisone treatment for immune reconstitution inflammatory syndrome. AIDS 22, 663–665 (2008).
Bower, M. et al. Clinical features and outcome in HIV-associated multicentric Castleman’s disease. J. Clin. Oncol. 29, 2481–2486 (2011).
Harris, A. H., Osborne, R. H., Streeton, C. L. & McNeil, H. Quality of life and Kaposi sarcoma: using preference techniques to value the health gains from treatment. Support Care Cancer 10, 486–493 (2002).
Northfelt, D. W. et al. Efficacy of pegylated-liposomal doxorubicin in the treatment of AIDS-related Kaposi’s sarcoma after failure of standard chemotherapy. J. Clin. Oncol. 15, 653–659 (1997).
Evans, S. R., Krown, S. E., Testa, M. A., Cooley, T. P. & Von Roenn, J. H. Phase II evaluation of low-dose oral etoposide for the treatment of relapsed or progressive AIDS-related Kaposi’s sarcoma: an AIDS Clinical Trials Group clinical study. J. Clin. Oncol. 20, 3236–3241 (2002).
Olweny, C. L. et al. Treatment of AIDS-associated Kaposi’s sarcoma in Zimbabwe: results of a randomized quality of life focused clinical trial. Int. J. Cancer 113, 632–639 (2005).
Little, R. F., Pluda, J. M., Feigal, E. & Yarchoan, R. The challenge of designing clinical trials for AIDS-related Kaposi’s sarcoma. Oncology (Williston Park, N.Y.) 12, 871–877 (1998).
Delabesse, E. et al. Molecular analysis of clonality in Kaposi’s sarcoma. J. Clin. Pathol. 50, 664–668 (1997).
Rabkin, C. S. et al. AIDS-related Kaposi’s sarcoma is a clonal neoplasm. Clin. Cancer Res. 1, 257–260 (1995).
Rabkin, C. S. et al. Monoclonal origin of multicentric Kaposi’s sarcoma lesions. N. Engl. J. Med. 336, 988–993 (1997).
Knowles, D. M. et al. Correlative morphologic and molecular genetic analysis demonstrates three distinct categories of posttransplantation lymphoproliferative disorders. Blood 85, 552–565 (1995).
Scinicariello, F., Dolan, M. J., Nedelcu, I., Tyring, S. K. & Hilliard, J. K. Occurrence of human papillomavirus and p53 gene mutations in Kaposi’s sarcoma. Virology 203, 153–157 (1994).
Li, J. J. et al. Expression and mutation of the tumor suppressor gene p53 in AIDS-associated Kaposi’s sarcoma. Am. J. Dermatopathol. 19, 373–378 (1997).
Nicolaides, A., Huang, Y. Q., Li, J. J., Zhang, W. G. & Friedman-Kien, A. E. Gene amplification and multiple mutations of the K-ras oncogene in Kaposi’s sarcoma. Anticancer Res. 14, 921–926 (1994).
Ensoli, B. & Sturzl, M. Kaposi’s sarcoma: a result of the interplay among inflammatory cytokines, angiogenic factors and viral agents. Cytokine Growth Factor Rev. 9, 63–83 (1998).
Ojala, P. M. & Schulz, T. F. Manipulation of endothelial cells by KSHV: implications for angiogenesis and aberrant vascular differentiation. Sem. Cancer Biol. 26, 69–77 (2014).
Barasa, A. K. et al. BALB/c mice immunized with a combination of virus-like particles incorporating Kaposi sarcoma-associated herpesvirus (KSHV) envelope glycoproteins gpK8.1, gB, and gH/gL induced comparable serum neutralizing antibody activity to UV-inactivated KSHV. Oncotarget 8, 34481–34497 (2017).
Wu, T. T., Qian, J., Ang, J. & Sun, R. Vaccine prospect of Kaposi sarcoma-associated herpesvirus. Curr. Opin. Virol. 2, 482–488 (2012).
Niedt, G. W. & Prioleau, P. G. Kaposi’s sarcoma occurring in a dermatome previously involved by herpes zoster. J. Am. Acad. Dermatol. 18, 448–451 (1988).
French, P. D., Harris, J. R. & Mercey, D. E. The Koebner phenomenon and AIDS-related Kaposi’s sarcoma. Br. J. Dermatol. 131, 746–747 (1994).
Ganem, D. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J. Clin. Invest. 120, 939–949 (2010).
Byakwaga, H. et al. The kynurenine pathway of tryptophan catabolism and AIDS-associated Kaposi sarcoma in Africa. J. Acquir. Immune. Defic. Syndr. 70, 296–303 (2015).
Aka, P. V. et al. A multiplex panel of plasma markers of immunity and inflammation in classical kaposi sarcoma. J. Infect. Dis. 211, 226–229 (2015).
Trautmann, L. et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 12, 1198–1202 (2006).
Paydas, S., Bagir, E. K., Deveci, M. A. & Gonlusen, G. Clinical and prognostic significance of PD-1 and PD-L1 expression in sarcomas. Med. Oncol. 33, 93 (2016).
Mletzko, S. et al. Programmed death ligand 1 (PD-L1) expression influences the immune-tolerogenic microenvironment in antiretroviral therapy-refractory Kaposi’s sarcoma: a pilot study. Oncoimmunology 6, e1304337 (2017).
Galanina, N., Goodman, A. M., Cohen, P. R., Frampton, G. M. & Kurzrock, R. Successful treatment of HIV-associated Kaposi sarcoma with immune checkpoint blockade. Cancer Immunol. Res. 6, 1129–1135 (2018).
Delyon, J. et al. PD-1 blockade with nivolumab in endemic Kaposi sarcoma. Ann. Oncol. 29, 1067–1069 (2018).
Montella, M., Franceschi, S., Geddes, M., Arniani, S. & Cocchiarella, G. Classic Kaposi’s sarcoma and volcanic soil in southern Italy. Lancet 347, 905–905 (1996).
Ziegler, J. L. Endemic Kaposi’s sarcoma in Africa and local volcanic soils. Lancet 342, 1348–1351 (1993).
Goedert, J. J. et al. Risk factors for classical Kaposi’s sarcoma. J. Natl Cancer Inst. 94, 1712–1718 (2002).
Giefing-Kroll, C., Berger, P., Lepperdinger, G. & Grubeck-Loebenstein, B. How sex and age affect immune responses, susceptibility to infections, and response to vaccination. Aging Cell 14, 309–321 (2015).
Nador, R. G. et al. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpesvirus. Blood 88, 645–656 (1996).
Chadburn, A. et al. KSHV-positive solid lymphomas represent an extra-cavitary variant of primary effusion lymphoma. Am. J. Surg. Pathol. 28, 1401–1416 (2004).
Goncalves, P. H., Uldrick, T. S. & Yarchoan, R. HIV-associated Kaposi sarcoma and related diseases. AIDS 31, 1903–1916 (2017).
Bhatt, S. et al. Efficacious proteasome/HDAC inhibitor combination therapy for primary effusion lymphoma. J. Clin. Invest. 123, 2616–2628 (2013).
Polizzotto, M. N. et al. Human and viral interleukin-6 and other cytokines in Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Blood 122, 4189–4198 (2013).
Dupin, N. et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 95, 1406–1412 (2000).
Totonchy, J. et al. KSHV induces immunoglobulin rearrangements in mature B lymphocytes. PLOS Pathog. 14, e1006967 (2018).
Said, J., Isaacson, P. G., Campo, E. & Harris, N. L. in WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues: WHO Classification of Tumours, Revised 4th Edition Vol. 2 (eds Swerdlow, S. H. et al.) (International Agency for Research on Cancer, 2017).
Polizzotto, M. N. et al. Clinical features and outcomes of patients with symptomatic Kaposi sarcoma herpesvirus (KSHV)-associated Inflammation: prospective characterization of KSHV inflammatory cytokine syndrome (KICS). Clin. Infect. Dis. 62, 730–738 (2016).
National Comprehensive Cancer Network. NCCN Harmonized Guidelines™ for Sub-Saharan Africa: Kaposi Sarcoma. NCCN https://www.nccn.org/professionals/physician_gls/pdf/kaposi_harmonized-africa.pdf (2018).
North, A. & South, C. Cancer Incidence in Five Continents, Vol. XI (eds Bray, F. et al.) (International Agency for Research on Cancer, 2017).
International Agency for Research on Cancer. Cancer Today. IARC https://gco.iarc.fr/today/ (2018).
Mbulaiteye, S. M. et al. Seroprevalence and risk factors for human herpesvirus 8 infection, rural Egypt. Emerg. Infect. Dis. 14, 586–591 (2008).
Stolka, K. et al. Risk factors for Kaposi’s sarcoma among HIV-positive individuals in a case control study in Cameroon. Cancer Epidemiol. 38, 137–143 (2014).
Adjei, A. A. et al. Seroprevalence of HHV-8, CMV, and EBV among the general population in Ghana, West Africa. BMC Infect. Dis. 8, 111 (2008).
Collenberg, E. et al. Seroprevalence of six different viruses among pregnant women and blood donors in rural and urban Burkina Faso: a comparative analysis. J. Med. Virol. 78, 683–692 (2006).
Lemma, E. et al. Human herpesvirus 8 infection in HIV-1-infected and uninfected pregnant women in Ethiopia. Ethiop Med. J. 47, 205–211 (2009).
de Sanjose, S. et al. Geographic variation in the prevalence of Kaposi sarcoma-associated herpesvirus and risk factors for transmission. J. Infect. Dis. 199, 1449–1456 (2009).
Baeten, J. M. et al. Correlates of human herpesvirus 8 seropositivity among heterosexual men in Kenya. AIDS 16, 2073–2078 (2002).
Engels, E. A. et al. Latent class analysis of human herpesvirus 8 assay performance and infection prevalence in sub-saharan Africa and Malta. Int. J. Cancer 88, 1003–1008 (2000).
DeSantis, S. M. et al. Demographic and immune correlates of human herpesvirus 8 seropositivity in Malawi. Africa. Int. J. Infect. Dis. 6, 266–271 (2002).
Caterino-de-Araujo, A. et al. Seroprevalence of human herpesvirus 8 infection in individuals from health care centers in Mozambique: potential for endemic and epidemic Kaposi’s sarcoma. J. Med. Virol. 82, 1216–1223 (2010).
Dollard, S. C. et al. Substantial regional differences in human herpesvirus 8 seroprevalence in sub-Saharan Africa: insights on the origin of the “Kaposi’s sarcoma belt”. Int. J. Cancer 127, 2395–2401 (2010).
Malope, B. I. et al. Transmission of Kaposi sarcoma-associated herpesvirus between mothers and children in a South African population. J. Acquir. Immune Defic. Syndr. 44, 351–355 (2007).
Klaskala, W. et al. Epidemiological characteristics of human herpesvirus-8 infection in a large population of antenatal women in Zambia. J. Med. Virol. 75, 93–100 (2005).
de-The, G., Bestetti, G., van Beveren, M. & Gessain, A. Prevalence of human herpesvirus 8 infection before the acquired immunodeficiency disease syndrome-related epidemic of Kaposi’s sarcoma in East Africa. J. Natl Cancer Inst. 91, 1888–1889 (1999).
Kouri, V. et al. Seroprevalence of Kaposi’s sarcoma-associated herpesvirus in various populations in Cuba. Rev. Panam. Salud Publica 15, 320–325 (2004).
Zago, A. et al. Seroprevalence of human herpesvirus 8 and its association with Kaposi sarcoma in Brazil. Sex. Transm. Dis. 27, 468–472 (2000).
Perez, C. et al. Seroprevalence of human herpesvirus-8 in blood donors from different geographical regions of Argentina, Brazil, and Chile. J. Med. Virol. 72, 661–667 (2004).
Sosa, C. et al. Human herpesvirus 8 as a potential sexually transmitted agent in Honduras. J. Infect. Dis. 178, 547–551 (1998).
Mohanna, S. et al. Human herpesvirus-8 in Peruvian blood donors: a population with hyperendemic disease? Clin. Infect. Dis. 44, 558–561 (2007).
Zhang, T. et al. Human herpesvirus 8 seroprevalence. China. Emerg. Infect. Dis. 18, 150–152 (2012).
Katano, H. et al. Identification of antigenic proteins encoded by human herpesvirus 8 and seroprevalence in the general population and among patients with and without Kaposi’s sarcoma. J. Virol. 74, 3478–3485 (2000).
Sachithanandham, J. et al. Human herpes virus-8 infections among subjects with human immunodeficiency virus infection and normal healthy individuals in India. Intervirology 56, 253–257 (2013).
Davidovici, B. et al. Seroepidemiology and molecular epidemiology of Kaposi’s sarcoma-associated herpesvirus among Jewish population groups in Israel. J. Natl Cancer Inst. 93, 194–202 (2001).
Ahmadi Ghezeldasht, S. et al. Oncogenic virus infections in the general population and end-stage renal disease patients with special emphasis on Kaposi’s sarcoma associated herpes virus (KSHV) in northeast of Iran. Jundishapur J. Microbiol. 8, e14920 (2015).
Alzahrani, A. J. et al. Increased seroprevalence of human herpes virus-8 in renal transplant recipients in Saudi Arabia. Nephrol. Dial. Transplant. 20, 2532–2536 (2005).
Simpson, G. R. et al. Prevalence of Kaposi’s sarcoma associated herpesvirus infection measured by antibodies to recombinant capsid protein and latent immunofluorescence antigen. Lancet 348, 1133–1138 (1996).
Whitby, D. et al. Human herpesvirus 8 seroprevalence in blood donors and lymphoma patients from different regions of Italy. J. Natl Cancer Inst. 90, 395–397 (1998).
Benavente, Y. et al. Antibodies against lytic and latent Kaposi’s sarcoma-associated herpes virus antigens and lymphoma in the European EpiLymph case-control study. Br. J. Cancer 105, 1768–1771 (2011).
Hjalgrim, H. et al. Prevalence of human herpesvirus 8 antibodies in young adults in Denmark (1976–1977). J. Natl Cancer Inst. 93, 1569–1571 (2001).
Tedeschi, R. et al. Epidemiology of Kaposi’s sarcoma herpesvirus (HHV8) in Västerbotten County, Sweden. J. Med. Virol. 78, 372–378 (2006).
Deborska, D. et al. Human herpesvirus-6 and human herpesvirus-8 seroprevalence in kidney transplant recipients. Transplant Proc. 34, 673–674 (2002).
Vitale, F. et al. Kaposi’s sarcoma herpes virus and Kaposi’s sarcoma in the elderly populations of 3 Mediterranean islands. Int. J. Cancer 91, 588–591 (2001).
Cattani, P. et al. Age-specific seroprevalence of human herpesvirus 8 in Mediterranean regions. Clin. Microbiol. Infect. 9, 274–279 (2003).
Rode, O. D., Lepej, S. Z. & Begovac, J. Low seroprevalence of human herpesvirus 8 infection in Croatia. Clin. Infect. Dis. 40, 1208 (2005).
Zavitsanou, A. et al. Human herpesvirus 8 (HHV-8) infection in healthy urban employees from Greece: seroprevalence and associated factors. J. Med. Virol. 79, 591–596 (2007).
Gurtsevich, V. E. et al. [Antibodies to herpesvirus type 8 in Kaposi’s sarcoma patients and controls in Russia]. Vopr. Virusol. 48, 19–22 (2003).
Regamey, N. et al. High human herpesvirus 8 seroprevalence in the homosexual population in Switzerland. J. Clin. Microbiol. 36, 1784–1786 (1998).
Juhasz, A. et al. Prevalence and age distribution of human herpesvirus-8 specific antibodies in Hungarian blood donors. J. Med. Virol. 64, 526–530 (2001).
Karnofsky, D. A. & Burchenal, J. H. in Evaluation of Chemotherapeutic Agents (ed. MacLeod, C. M.) 191–205 (Columbia Univ. Press, NY, 1949).
Acknowledgements
The authors apologize to all the authors they could not cite owing to space constraints but whose contributions they greatly value. D.W. is supported by US federal funds from the National Cancer Institute, NIH, under contract HHSN261200800001E. E.C. and S.E.K. are partially supported by AIDS Malignancy Consortium (AMC) grant U01 CA121947. E.C. and J.M. are partially supported by the National Institutes of Health (NIH) grants UH2CA202723 and UH3CA202723. B.D. is supported by the NIH grants CA019014, CA096500, DE028211.
Author information
Authors and Affiliations
Contributions
Introduction (E.C. and S.E.K.); Epidemiology (E.C. and D.W.); Mechanisms/pathophysiology (E.C. and B.D.); Diagnosis, screening and prevention (E.C., J.M., S.E.K. and M.B.); Management (E.C., M.B. and S.E.K.); Quality of life (M.B. and S.E.K.); Outlook (E.C., M.B. and S.E.K.); Overview of the Primer (E.C.).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
The Global Cancer Observatory: http://gco.iarc.fr/
Surveillance, Epidemiology, and End Results (SEER): https://seer.cancer.gov/
Rights and permissions
About this article

Cite this article
Cesarman, E., Damania, B., Krown, S.E. et al. Kaposi sarcoma. Nat Rev Dis Primers 5, 9 (2019). https://doi.org/10.1038/s41572-019-0060-9
Published:
DOI: https://doi.org/10.1038/s41572-019-0060-9
This article is cited by
-
Interferon induced circRNAs escape herpesvirus host shutoff and suppress lytic infection
EMBO Reports (2024)
-
Hijacking of nucleotide biosynthesis and deamidation-mediated glycolysis by an oncogenic herpesvirus
Nature Communications (2024)
-
Cause-specific mortality in a population-level cohort of diffuse large B-cell lymphoma following chemotherapy in the early 21st century
Annals of Hematology (2024)
-
Transcriptional landscape of Kaposi sarcoma tumors identifies unique immunologic signatures and key determinants of angiogenesis
Journal of Translational Medicine (2023)
-
Comparison of Epstein–Barr virus and Kaposi’s sarcoma-associated herpesvirus viral load in peripheral blood mononuclear cells and oral fluids of HIV-negative individuals aged 3–89 years from Uganda
Infectious Agents and Cancer (2023)
