
Abstract
Lysosomal storage diseases (LSDs) are a group of over 70 diseases that are characterized by lysosomal dysfunction, most of which are inherited as autosomal recessive traits. These disorders are individually rare but collectively affect 1 in 5,000 live births. LSDs typically present in infancy and childhood, although adult-onset forms also occur. Most LSDs have a progressive neurodegenerative clinical course, although symptoms in other organ systems are frequent. LSD-associated genes encode different lysosomal proteins, including lysosomal enzymes and lysosomal membrane proteins. The lysosome is the key cellular hub for macromolecule catabolism, recycling and signalling, and defects that impair any of these functions cause the accumulation of undigested or partially digested macromolecules in lysosomes (that is, ‘storage’) or impair the transport of molecules, which can result in cellular damage. Consequently, the cellular pathogenesis of these diseases is complex and is currently incompletely understood. Several LSDs can be treated with approved, disease-specific therapies that are mostly based on enzyme replacement. However, small-molecule therapies, including substrate reduction and chaperone therapies, have also been developed and are approved for some LSDs, whereas gene therapy and genome editing are at advanced preclinical stages and, for a few disorders, have already progressed to the clinic.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 1 digital issues and online access to articles
$99.00 per year
only $99.00 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
Change history
17 May 2019
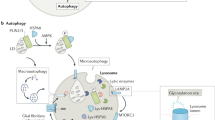
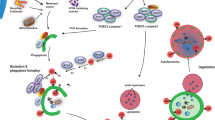
In the originally published version of Figure 3, APP was incorrectly linked to CMA. In addition, the label for NCP2 was omitted, and GlcSph was incorrectly labelled as GlcCer. This figure has now been corrected.
18 October 2018
In the version of the article originally published, in Figure 2 and the accompanying legend, LIMP 2 was incorrectly referred to as LIMP 1. The article has now been corrected.
References
Di Fruscio, G. et al. Lysoplex: an efficient toolkit to detect DNA sequence variations in the autophagy-lysosomal pathway. Autophagy 11, 928–938 (2015).
Palmieri, M. et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 20, 3852–3866 (2011).
Chapel, A. et al. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol. Cell Proteom. 12, 1572–1588 (2013).
Schroder, B. et al. Integral and associated lysosomal membrane proteins. Traffic 8, 1676–1686 (2007).
Sleat, D. E. et al. Mass spectrometry-based protein profiling to determine the cause of lysosomal storage diseases of unknown etiology. Mol. Cell Proteom. 8, 1708–1718 (2009).
Szklarczyk, D. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 (2015).
Parenti, G., Andria, G. & Ballabio, A. Lysosomal storage diseases: from pathophysiology to therapy. Annu. Rev. Med. 66, 471–486 (2015).
Meikle, P. J., Hopwood, J. J., Clague, A. E. & Carey, W. F. Prevalence of lysosomal storage disorders. JAMA. 281, 249–254 (1999).
US National Library of Medicine, Genetics Home Reference. NIH https://ghr.nlm.nih.gov/ (2018).
Nita, D. A., Mole, S. E. & Minassian, B. A. Neuronal ceroid lipofuscinoses. Epilept. Disord. 18, 73–88 (2016).
Aronson, N. N. Jr Aspartylglycosaminuria: biochemistry and molecular biology. Biochim. Biophys. Acta 1455, 139–154 (1999).
Witkop, C. J. et al. Albinism and Hermansky-Pudlak syndrome in Puerto Rico. Bol. Asoc. Med. P R. 82, 333–339 (1990).
Witkop, C. J., Almadovar, C., Pineiro, B. & Nunez Babcock, M. Hermansky-Pudlak syndrome (HPS). An epidemiologic study. Ophthalm. Paediatr. Genet. 11, 245–250 (1990).
Kishnani, P. S. et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 148, 671–676 (2006).
Kishnani, P. S. et al. Recombinant human acid α-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 68, 99–109 (2007).
van der Meijden, J. C. et al. Long-term follow-up of 17 patients with childhood Pompe disease treated with enzyme replacement therapy. J. Inherit Metab. Dis. https://doi.org/10.1007/s10545-018-0166-3 (2018).
Bley, A. E. et al. Natural history of infantile G(M2) gangliosidosis. Pediatrics 128, e1233–e1241 (2011).
Schulz, A. et al. Study of intraventricular cerliponase alfa for CLN2 disease. N. Engl. J. Med. 378, 1898–1907 (2018).
Jones, H. N. et al. Oropharyngeal dysphagia in infants and children with infantile Pompe disease. Dysphagia 25, 277–283 (2010).
Nicolino, M. et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet. Med. 11, 210–219 (2009).
Chakrapani, A., Vellodi, A., Robinson, P., Jones, S. & Wraith, J. E. Treatment of infantile Pompe disease with alglucosidase alpha: the UK experience. J. Inherit Metab. Dis. 33, 747–750 (2010).
Settembre, C., Fraldi, A., Medina, D. L. & Ballabio, A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296 (2013).
Medina, D. L. & Ballabio, A. Lysosomal calcium regulates autophagy. Autophagy 11, 970–971 (2015).
Todkar, K., Ilamathi, H. S. & Germain, M. Mitochondria and lysosomes: discovering bonds. Front. Cell Dev. Biol. 5, 106 (2017).
Kilpatrick, B. S. et al. An endosomal NAADP-sensitive two-pore Ca(2+) channel regulates ER-endosome membrane contact sites to control growth factor signaling. Cell Rep. 18, 1636–1645 (2017).
Annunziata, I., Sano, R. & d’Azzo, A. Mitochondria-associated ER membranes (MAMs) and lysosomal storage diseases. Cell Death Dis. 9, 328 (2018).
Wong, Y. C., Ysselstein, D. & Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554, 382–386 (2018).
Grabowski, G. A. Overview of inflammation in neurometabolic diseases. Semin. Pediatr. Neurol. 24, 207–213 (2017).
Rigante, D., Cipolla, C., Basile, U., Gulli, F. & Savastano, M. C. Overview of immune abnormalities in lysosomal storage disorders. Immunol. Lett. 188, 79–85 (2017).
Vitner, E. B., Platt, F. M. & Futerman, A. H. Common and uncommon pathogenic cascades in lysosomal storage diseases. J. Biol. Chem. 285, 20423–20427 (2010).
Vitner, E. B. et al. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat. Med. 20, 204–208 (2014).
Wada, R., Tifft, C. J. & Proia, R. L. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is supressed by bone marrow transplantation. Proc. Natl Acad. Sci. USA 97, 10954–10959 (2000).
Jeyakumar, M. et al. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 126, 974–987 (2003).
Jeyakumar, M. et al. NSAIDs increase survival in the Sandhoff disease mouse: synergy with N-butyldeoxynojirimycin. Ann. Neurol. 56, 642–649 (2004).
Bosch, M. E. & Kielian, T. Neuroinflammatory paradigms in lysosomal storage diseases. Front. Neurosci. 9, 417 (2015).
Smith, D., Wallom, K. L., Williams, I. M., Jeyakumar, M. & Platt, F. M. Beneficial effects of anti-inflammatory therapy in a mouse model of Niemann-Pick disease type C1. Neurobiol. Dis. 36, 242–251 (2009).
Yamaguchi, A. et al. Possible role of autoantibodies in the pathophysiology of GM2 gangliosidoses. J. Clin. Invest. 113, 200–208 (2004).
Ballabio, A. & Gieselmann, V. Lysosomal disorders: from storage to cellular damage. Biochim. Biophys. Acta 1793, 684–696 (2009).
Saftig, P. in Fabry Disease: Perspectives from 5 Years of FOS (eds Mehta, A., Beck, M. & Sunder-Plassmann, G.) 21–31 (2006).
Sidransky, E. & Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 11, 986–998 (2012). This review summarizes the link between mutations in a rare disease-causing gene, GBA , and a common neurological disorder, PD.
Annunziata, I. et al. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-beta secretion via deregulated lysosomal exocytosis. Nat. Commun. 4, 2734 (2013).
Zanoteli, E. et al. Muscle degeneration in neuraminidase 1-deficient mice results from infiltration of the muscle fibers by expanded connective tissue. Biochim. Biophys. Acta 1802, 659–672 (2010).
Machado, E. et al. Regulated lysosomal exocytosis mediates cancer progression. Sci. Adv. 1, e1500603 (2015).
Bellettato, C. M. & Scarpa, M. Pathophysiology of neuropathic lysosomal storage disorders. J. Inherit. Metab. Dis. 33, 347–362 (2010).
Sidransky, E. et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661 (2009). This study proves a strong genetic relationship between GBA mutations and risk of PD.
Borger, D. K., Aflaki, E. & Sidransky, E. Applications of iPSC-derived models of Gaucher disease. Ann. Transl Med. 3, 295 (2015).
De Filippis, L., Zalfa, C. & Ferrari, D. Neural stem cells and human induced pluripotent stem cells to model rare CNS diseases. CNS Neurol Disord Drug Targets. https://doi.org/10.2174/1871527316666170615121753 (2017).
Lojewski, X. et al. Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum. Mol. Genet. 23, 2005–2022 (2014).
Panicker, L. M. et al. Induced pluripotent stem cell model recapitulates pathologic hallmarks of Gaucher disease. Proc. Natl Acad. Sci. USA 109, 18054–18059 (2012).
Aflaki, E. et al. A new glucocerebrosidase chaperone reduces alpha-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and parkinsonism. J. Neurosci. 36, 7441–7452 (2016).
Mistry, P. K. et al. Gaucher disease: progress and ongoing challenges. Mol. Genet. Metab. 120, 8–21 (2017).
Beutler, E. Gaucher’s disease. N. Engl. J. Med. 325, 1354–1360 (1991).
Borger, D. K., Sidransky, E. & Aflaki, E. New macrophage models of Gaucher disease offer new tools for drug development. Macrophage (Houst) 2, e712 (2015).
Aflaki, E. et al. Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 15, 77–88 (2016).
Aflaki, E., Westbroek, W. & Sidransky, E. The complicated relationship between Gaucher disease and parkinsonism: insights from a rare disease. Neuron 93, 737–746 (2017).
Wong, K. et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 82, 192–207 (2004).
Vitner, E. B. & Futerman, A. H. in Sphingolipids in Disease. Handbook of Experimental Pharmacology, vol 216 (eds Gulbins E. & Petrache I.) 405–419 (Springer, Vienna 2013).
Vitner, E. B., Farfel-Becker, T., Eilam, R., Biton, I. & Futerman, A. H. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain 135, 1724–1735 (2012).
Vitner, E. B. et al. Induction of the type I interferon response in neurological forms of Gaucher disease. J. Neuroinflamm. 13, 104 (2016).
Allen, M. J., Myer, B. J., Khokher, A. M., Rushton, N. & Cox, T. M. Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: increased release of interleukin-6 and interleukin-10. Q. J. Med. 90, 19–25 (1997).
Cox, T. M. Gaucher disease: understanding the molecular pathogenesis of sphingolipidoses. J. Inherit. Metab. Dis. 24, 106–121 (2001).
Pandey, M. K. et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 543, 108–112 (2017).
Aker, M., Zimran, A., Abrahamov, A., Horowitz, M. & Matzner, Y. Abnormal neutrophil chemotaxis in Gaucher disease. Br. J. Haematol. 83, 187–191 (1993).
Deganuto, M. et al. Altered intracellular redox status in Gaucher disease fibroblasts and impairment of adaptive response against oxidative stress. J. Cell. Physiol. 212, 223–235 (2007).
Thomas, A. S., Mehta, A. & Hughes, D. A. Gaucher disease: haematological presentations and complications. Br. J. Haematol. 165, 427–440 (2014).
Enquist, I. B. et al. Murine models of acute neuronopathic Gaucher disease. Proc. Natl Acad. Sci. USA 104, 17483–17488 (2007).
Farfel-Becker, T., Vitner, E. B. & Futerman, A. H. Animal models for Gaucher disease research. Dis. Model. Mech. 4, 746–752 (2011).
Tayebi, N. et al. Gaucher disease and parkinsonism: a phenotypic and genotypic characterization. Mol. Genet. Metab. 73, 313–321 (2001).
Aflaki, E. et al. Efferocytosis is impaired in Gaucher macrophages. Haematologica 102, 656–665 (2017).
Pringsheim, T., Jette, N., Frolkis, A. & Steeves, T. D. The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov Disord. 29, 1583–1590 (2014).
Mazzulli, J. R. et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52 (2011).
Cullen, V. et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann. Neurol. 69, 940–953 (2011).
Zunke, F. et al. Reversible conformational conversion of alpha-synuclein into toxic assemblies by glucosylceramide. Neuron 97, 92–107 (2018).
Robak, L. A. et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140, 3191–3203 (2017).
Suzuki, Y., Oshima, A. & Nanba, E. in The Metabolic and Molecular Bases of Inherited Disease Vol. 3 (eds Scriver, C. R., Beadet, A. L., Valle, D. & Sly, W. S.) 3775–3809 (McGraw Hill, 2001).
d’Azzo, A., Tessitore, A. & Sano, R. Gangliosides as apoptotic signals in ER stress response. Cell Death Differ. 13, 404–414 (2006).
Ledeen, R. W. & Wu, G. GM1 ganglioside: another nuclear lipid that modulates nuclear calcium. GM1 potentiates the nuclear sodium-calcium exchanger. Can. J. Physiol. Pharmacol. 84, 393–402 (2006).
Ledeen, R. W. & Wu, G. The multi-tasked life of GM1 ganglioside, a true factotum of nature. Trends Biochem. Sci. 40, 407–418 (2015).
Hahn, C. N. et al. Generalized CNS disease and massive GM1-ganglioside accumulation in mice defective in lysosomal acid beta-galactosidase. Hum. Mol. Genet. 6, 205–211 (1997).
Tessitore, A. et al. A GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol. Cell. 15, 753–766 (2004).
Sano, R. et al. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol. Cell 36, 500–511 (2009).
Yanagisawa, K., Odaka, A., Suzuki, N. & Ihara, Y. GM1 ganglioside-bound amyloid beta-protein (A beta): a possible form of preamyloid in Alzheimer’s disease. Nat. Med. 1, 1062–1066 (1995).
Yanagisawa, K. GM1 ganglioside and Alzheimer’s disease. Glycoconj. J. 32, 87–91 (2015).
Hirano-Sakamaki, W. et al. Alzheimer’s disease is associated with disordered localization of ganglioside GM1 molecular species in the human dentate gyrus. FEBS Lett. 589, 3611–3616 (2015).
Calamai, M. et al. Single molecule experiments emphasize GM1 as a key player of the different cytotoxicity of structurally distinct Aβ1-42 oligomers. Biochim. Biophys. Acta 1858, 386–392 (2016).
Calamai, M. & Pavone, F. S. Partitioning and confinement of GM1 ganglioside induced by amyloid aggregates. FEBS Lett. 587, 1385–1391 (2013).
d’Azzo, A., Machado, E. & Annunziata, I. Pathogenesis, emerging therapeutic targets and treatment in sialidosis. Expert Opin. Orphan Drugs 3, 491–504 (2015).
Bonten, E. J., Annunziata, I. & d’Azzo, A. Lysosomal multienzyme complex: pros and cons of working together. Cell. Mol. Life Sci. 71, 2017–2032 (2014).
de Geest, N. et al. Systemic and neurologic abnormalities distinguish the lysosomal disorders sialidosis and galactosialidosis in mice. Hum. Mol. Genet. 11, 1455–1464 (2002).
Yogalingam, G. et al. Neuraminidase 1 is a negative regulator of lysosomal exocytosis. Dev. Cell 15, 74–86 (2008). This study is the first demonstration that excessive lysosomal exocytosis downstream of NEU1 deficiency is responsible for the pathogenesis of sialidosis.
Wu, X. et al. Vacuolization and alterations of lysosomal membrane proteins in cochlear marginal cells contribute to hearing loss in neuraminidase 1-deficient mice. Biochim. Biophys. Acta 1802, 259–268 (2010).
Reddy, A., Caler, E. V. & Andrews, N. W. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 106, 157–169 (2001).
Vanier, M. T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 5, 16 (2010).
Lloyd-Evans, E. et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14, 1247–1255 (2008).
Lee, H. et al. Bone marrow-derived mesenchymal stem cells prevent the loss of Niemann-Pick type C mouse Purkinje neurons by correcting sphingolipid metabolism and increasing sphingosine-1-phosphate. Stem Cells 28, 821–831 (2010).
Visentin, S. et al. The stimulation of adenosine A2A receptors ameliorates the pathological phenotype of fibroblasts from Niemann-Pick type C patients. J. Neurosci. 33, 15388–15393 (2013).
Xu, M. et al. δ-Tocopherol reduces lipid accumulation in Niemann-Pick type C1 and Wolman cholesterol storage disorders. J. Biol. Chem. 287, 39349–39360 (2012).
Liscum, L. Niemann-Pick type C mutations cause lipid traffic jam. Traffic 1, 218–225 (2000).
Ioannou, Y. A. Multidrug permeases and subcellular cholesterol transport. Nat. Rev. Mol. Cell Biol. 2, 657–668 (2001).
Davies, J. P., Chen, F. W. & Ioannou, Y. A. Transmembrane molecular pump activity of Niemann-Pick C1 protein. Science 290, 2295–2298 (2000).
Goldman, S. D. & Krise, J. P. Niemann-Pick C1 functions independently of Niemann-Pick C2 in the initial stage of retrograde transport of membrane-impermeable lysosomal cargo. J. Biol. Chem. 285, 4983–4994 (2010).
Auer, I. A. et al. Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer’s disease. Acta Neuropathol. 90, 547–551 (1995).
Huizing, M., Helip-Wooley, A., Westbroek, W., Gunay-Aygun, M. & Gahl, W. A. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu. Rev. Genom. Hum. Genet. 9, 359–386 (2008).
Raposo, G., Fevrier, B., Stoorvogel, W. & Marks, M. S. Lysosome-related organelles: a view from immunity and pigmentation. Cell Struct. Funct. 27, 443–456 (2002).
Neudorfer, O. et al. Late-onset Tay-Sachs disease: phenotypic characterization and genotypic correlations in 21 affected patients. Genet. Med. 7, 119–123 (2005).
Veys, K. R., Elmonem, M. A., Arcolino, F. O., van den Heuvel, L. & Levtchenko, E. Nephropathic cystinosis: an update. Curr. Opin. Pediatr. 29, 168–178 (2017).
Patterson, M. C. et al. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol. Genet. Metab. 106, 330–344 (2012).
Evans, W. R. & Hendriksz, C. J. Niemann-Pick type C disease - the tip of the iceberg? A review of neuropsychiatric presentation, diagnosis and treatment. BJPsych Bull. 41, 109–114 (2017).
Cooper, J. D., Tarczyluk, M. A. & Nelvagal, H. R. Towards a new understanding of NCL pathogenesis. Biochim. Biophys. Acta 1852, 2256–2261 (2015).
Preising, M. N., Abura, M., Jager, M., Wassill, K. H. & Lorenz, B. Ocular morphology and function in juvenile neuronal ceroid lipofuscinosis (CLN3) in the first decade of life. Ophthalm. Genet. 38, 252–259 (2017).
Williams, R. E. et al. Management strategies for CLN2 disease. Pediatr. Neurol. 69, 102–112 (2017).
Introne, W. J. et al. Neurologic involvement in patients with atypical Chediak-Higashi disease. Neurology 88, e57–e65 (2017).
Suzuki, K. Enzymic diagnosis of sphingolipidoses. Methods Enzymol. 50, 456–488 (1978).
Kaback, M. et al. Tay-Sachs disease—carrier screening, prenatal diagnosis, and the molecular era. An international perspective, 1970 to 1993. The International TSD Data Collection Network. JAMA. 270, 2307–2315 (1993). This seminal paper discusses the high-risk population screening that reduced the incidence of a uniformly fatal disease by 90% within 10 years.
Regier, D. S., Proia, R. L., D’Azzo, A. & Tifft, C. J. The GM1 and GM2 Gangliosidoses: natural history and progress toward therapy. Pediatr. Endocrinol. Rev. 13, 663–673 (2016).
van der Tol, L. et al. A systematic review on screening for Fabry disease: prevalence of individuals with genetic variants of unknown significance. J. Med. Genet. 51, 1–9 (2014).
Hoffman, J. D. et al. Next-generation DNA sequencing of HEXA: a step in the right direction for carrier screening. Mol. Genet. Genom. Med. 1, 260–268 (2013).
[No authors listed]. Committee Opinion Number 691. American College of Obstetricians and Gynecologists https://www.acog.org/Clinical-Guidance-and-Publications/Committee-Opinions/Committee-on-Genetics/Carrier-Screening-for-Genetic-Conditions (2017).
Schielen, P., Kemper, E. A. & Gelb, M. H. Newborn screening for lysosomal storage diseases: a concise review of the literature on screening methods, therapeutic possibilities and regional programs. Int. J. Neonatal Screen. https://doi.org/10.3390/ijns3020006 (2017).
Cox, T. M. Innovative treatments for lysosomal diseases. Best Pract. Res. Clin. Endocrinol. Metab. 29, 275–311 (2015).
Hobbs, J. R. et al. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 2, 709–712 (1981). This study is the first demonstration that Hurler syndrome is a treatable disease.
Aldenhoven, M. & Kurtzberg, J. Cord blood is the optimal graft source for the treatment of pediatric patients with lysosomal storage diseases: clinical outcomes and future directions. Cytotherapy 17, 765–774 (2015).
Boelens, J. J. et al. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood 121, 3981–3987 (2013).
Poe, M. D., Chagnon, S. L. & Escolar, M. L. Early treatment is associated with improved cognition in Hurler syndrome. Ann. Neurol. 76, 747–753 (2014).
Sly, W. S., Kaplan, A., Achord, D. T., Brot, F. E. & Bell, C. E. Receptor-mediated uptake of lysosomal enzymes. Prog. Clin. Biol. Res. 23, 547–551 (1978). This paper reviews the evidence for recognition of lysosomal enzymes by receptors for mannose and for mannose 6-phosphate.
Kan, S. H. et al. Delivery of an enzyme-IGFII fusion protein to the mouse brain is therapeutic for mucopolysaccharidosis type IIIB. Proc. Natl Acad. Sci. USA 111, 14870–14875 (2014).
Grabowski, G. A. Gaucher disease and other storage disorders. Hematol. Am. Soc. Hematol. Educ. Program 2012, 13–18 (2012).
Zimran, A. How I treat Gaucher disease. Blood 118, 1463–1471 (2011).
Boado, R. J., Lu, J. Z., Hui, E. K., Lin, H. & Pardridge, W. M. Insulin receptor Antibody-alpha-N-Acetylglucosaminidase fusion protein penetrates the primate blood-brain barrier and reduces glycosoaminoglycans in Sanfilippo Type B fibroblasts. Mol. Pharm. 13, 1385–1392 (2016).
Markham, A. Cerliponase Alfa: first global approval. Drugs 77, 1247–1249 (2017).
Broomfield, A., Jones, S. A., Hughes, S. M. & Bigger, B. W. The impact of the immune system on the safety and efficiency of enzyme replacement therapy in lysosomal storage disorders. J. Inherit Metab. Dis. 39, 499–512 (2016).
Afroze, B. & Brown, N. Ethical issues in managing Lysosomal storage disorders in children in low and middle income countries. Pak. J. Med. Sci. 33, 1036–1041 (2017).
McGill, J. J. et al. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age — a sibling control study. Clin. Genet. 77, 492–498 (2010).
Aerts, J. M., Hollak, C. E., Boot, R. G., Groener, J. E. & Maas, M. Substrate reduction therapy of glycosphingolipid storage disorders. J. Inherit Metab. Dis. 29, 449–456 (2006).
Lachmann, R. H. & Platt, F. M. Substrate reduction therapy for glycosphingolipid storage disorders. Expert Opin. Investig. Drugs 10, 455–466 (2001).
Cox, T. et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 355, 1481–1485 (2000). This study provides the first clinical evidence of the efficacy of SRT using the first approved SRT drug, miglustat, in Gaucher disease type I.
Hollak, C. E., Hughes, D., van Schaik, I. N., Schwierin, B. & Bembi, B. Miglustat (Zavesca) in type 1 Gaucher disease: 5-year results of a post-authorisation safety surveillance programme. Pharmacoepidemiol. Drug Safety 18, 770–777 (2009).
Poole, R. M. Eliglustat: first global approval. Drugs 74, 1829–1836 (2014).
Belmatoug, N. et al. Management and monitoring recommendations for the use of eliglustat in adults with type 1 Gaucher disease in Europe. Eur. J. Intern. Med. 37, 25–32 (2016).
Patterson, M. C. et al. Oral miglustat in Niemann-Pick type C (NPC) disease. Rev. Neurol. (Separata) 43, 8 (2006).
Patterson, M. C., Vecchio, D., Prady, H., Abel, L. & Wraith, J. E. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 6, 765–772 (2007).
Shayman, J. A. ELIGLUSTAT TARTRATE: glucosylceramide synthase inhibitor treatment of type 1 Gaucher disease. Drugs Future 35, 613–620 (2010).
Platt, F. M., Neises, G. R., Dwek, R. A. & Butters, T. D. N-Butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J. Biol. Chem. 269, 8362–8365 (1994).
Walterfang, M. et al. Dysphagia as a risk factor for mortality in Niemann-Pick disease type C: systematic literature review and evidence from studies with miglustat. Orphanet J. Rare Dis. 7, 76 (2012).
Lyseng-Williamson, K. A. Miglustat: a review of its use in niemann-pick disease type C. Drugs 74, 61–74 (2014).
Cox, T. M. Eliglustat tartrate, an orally active glucocerebroside synthase inhibitor for the potential treatment of Gaucher disease and other lysosomal storage diseases. Curr. Opin. Investig. Drugs 11, 1169–1181 (2010).
Cox, T. M. & Mistry, P. K. Therapeutic position of eliglustat. Blood Cells Mol. Dis. 69, 117–118 (2018).
Lachmann, R. H. Miglustat. Oxford GlycoSciences/Actelion. Curr. Opin. Investig. Drugs 4, 472–479 (2003).
Belmatoug, N. et al. Management and monitoring recommendations for the use of eliglustat in adults with type 1 Gaucher disease in Europe. Eur. J. Intern. Med. 37, 25–32 (2017).
Clayton, N. P. et al. Antisense oligonucleotide-mediated suppression of muscle glycogen synthase 1 synthesis as an approach for substrate reduction therapy of Pompe disease. Mol. Ther. Nucleic Acids 3, e206 (2014).
Parenti, G., Andria, G. & Valenzano, K. J. Pharmacological chaperone therapy: preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol. Ther. 23, 1138–1148 (2015).
Markham, A. Migalastat: first global approval. Drugs 76, 1147–1152 (2016).
Hughes, D. A. et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 54, 288–296 (2017). This clinical study compares the first-in-class approved small-molecule chaperone drug migalastat with standard-of-care ERT, showing its potential as an alternative to ERT in patients with Fabry disease with chaperonable mutations.
Germain, D. P. et al. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N. Engl. J. Med. 375, 545–555 (2016).
Schiffmann, R. et al. Migalastat improves diarrhea in patients with Fabry disease: clinical-biomarker correlations from the phase 3 FACETS trial. Orphanet J. Rare Dis. 13, 68 (2018).
Citro, V. et al. Identification of an allosteric binding site on human lysosomal alpha-galactosidase opens the way to new pharmacological chaperones for Fabry disease. PLOS One 11, e0165463 (2016).
Huang, I. C. et al. Quality of life information and trust in physicians among families of children with life-limiting conditions. Patient Relat. Outcome Meas. 2010, 141–148 (2010).
Somanadhan, S. & Larkin, P. J. Parents’ experiences of living with, and caring for children, adolescents and young adults with Mucopolysaccharidosis (MPS). Orphanet J. Rare Dis. 11, 138 (2016).
Pelentsov, L. J., Laws, T. A. & Esterman, A. J. The supportive care needs of parents caring for a child with a rare disease: a scoping review. Disabil Health J. 8, 475–491 (2015).
Besier, T. et al. Anxiety, depression, and life satisfaction in parents caring for children with cystic fibrosis. Pediatr. Pulmonol 46, 672–682 (2011).
McConkie-Rosell, A. et al. Psychosocial profiles of parents of children with undiagnosed diseases: managing well or just managing? J. Genet. Couns. 27, 935–946 (2018).
Bolsover, F. E., Murphy, E., Cipolotti, L., Werring, D. J. & Lachmann, R. H. Cognitive dysfunction and depression in Fabry disease: a systematic review. J. Inherit Metab. Dis. 37, 177–187 (2014).
Arends, M., Hollak, C. E. & Biegstraaten, M. Quality of life in patients with Fabry disease: a systematic review of the literature. Orphanet J. Rare Dis. 10, 77 (2015).
Kloesel, B. & Holzman, R. S. Anesthetic management of patients with inborn errors of metabolism. Anesth Analg. 125, 822–836 (2017).
Goldstein, R. (ed.) Cameron’s Arc: Creating a Full Life (American Academy of Paediatrics, 2007).
Petersen, N. H. & Kirkegaard, T. HSP70 and lysosomal storage disorders: novel therapeutic opportunities. Biochem. Soc. Trans. 38, 1479–1483 (2010).
Kirkegaard, T. et al. Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med. 8, 355ra118 (2016).
Keeling, K. M., Xue, X., Gunn, G. & Bedwell, D. M. Therapeutics based on stop codon readthrough. Annu. Rev. Genom. Hum. Genet. 15, 371–394 (2014).
Keeling, K. M. Nonsense suppression as an approach to treat lysosomal storage diseases. Diseases 4, 32 (2016).
Hein, L. K. et al. alpha-L-iduronidase premature stop codons and potential read-through in mucopolysaccharidosis type I patients. J. Mol. Biol. 338, 453–462 (2004).
Ory, D. S. et al. Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1–2 trial. Lancet 390, 1758–1768 (2017).
Chen, F. W., Li, C. & Ioannou, Y. A. Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLOS One 5, e15054 (2010).
Dai, S. et al. Methyl-β-cyclodextrin restores impaired autophagy flux in Niemann-Pick C1-deficient cells through activation of AMPK. Autophagy 13, 1435–1451 (2017).
Davidson, C. D. et al. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLOS One 4, e6951 (2009).
Sands, M. S. & Davidson, B. L. Gene therapy for lysosomal storage diseases. Mol. Ther. 13, 839–849 (2006).
Aronovich, E. L. et al. Prolonged expression of secreted enzymes in dogs after liver-directed delivery of sleeping beauty transposons: implications for non-viral gene therapy of systemic disease. Hum. Gene Ther. 28, 551–564 (2017).
Sandrin, V., Russell, S. J. & Cosset, F. L. Targeting retroviral and lentiviral vectors. Curr. Top. Microbiol. Immunol. 281, 137–178 (2003).
Davidson, B. L. et al. Recombinant adeno-associated virus type 2, 4, and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proc. Natl Acad. Sci. USA 97, 3428–3432 (2000).
Gilkes, J. A., Bloom, M. D. & Heldermon, C. D. Preferred transduction with AAV8 and AAV9 via thalamic administration in the MPS IIIB model: a comparison of four rAAV serotypes. Mol. Genet. Metab. Rep. 6, 48–54 (2016).
Foust, K. D. et al. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 27, 59–65 (2009).
Bevan, A. K. et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol. Ther. 19, 1971–1980 (2011).
Deverman, B. E. et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 34, 204–209 (2016).
Mendell, J. R. et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 377, 1713–1722 (2017).
Biffi, A. et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341, 1233158 (2013).
Goina, E., Peruzzo, P., Bembi, B., Dardis, A. & Buratti, E. Glycogen reduction in myotubes of late-onset pompe disease patients using antisense technology. Mol. Ther. 25, 2117–2128 (2017).
van der Wal, E., Bergsma, A. J., Pijnenburg, J. M., van der Ploeg, A. T. & Pijnappel, W. Antisense oligonucleotides promote exon inclusion and correct the common c.-32-13T>G GAA splicing variant in pompe disease. Mol. Ther. Nucleic Acids 7, 90–100 (2017).
Siva, K., Covello, G. & Denti, M. A. Exon-skipping antisense oligonucleotides to correct missplicing in neurogenetic diseases. Nucleic Acid. Ther. 24, 69–86 (2014).
Corey, D. R. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat. Neurosci. 20, 497–499 (2017).
Khvorova, A. & Watts, J. K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 35, 238–248 (2017).
Schneller, J. L., Lee, C. M., Bao, G. & Venditti, C. P. Genome editing for inborn errors of metabolism: advancing towards the clinic. BMC Med. 15, 43 (2017).
Sharma, R. et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 126, 1777–1784 (2015).
Yin, H., Kauffman, K. J. & Anderson, D. G. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 16, 387–399 (2017).
Pu, J., Guardia, C. M., Keren-Kaplan, T. & Bonifacino, J. S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 129, 4329–4339 (2016).
Carette, J. E. et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477, 340–343 (2011).
Hulseberg, C. E., Feneant, L., Szymanska, K. M. & White, J. M. Lamp1 increases the efficiency of lassa virus infection by promoting fusion in less acidic endosomal compartments. MBio. https://doi.org/10.1128/mBio.01818-17 (2018).
Fineran, P. et al. Pathogenic mycobacteria achieve cellular persistence by inhibiting the Niemann-Pick Type C disease cellular pathway. Wellcome Open Res. 1, 18 (2016).
Adam, M. P. et al. (eds) GeneReviews®. NCBI https://www.ncbi.nlm.nih.gov/books/NBK1116/ (2018).
Sinigerska, I. et al. Founder mutation causing infantile GM1-gangliosidosis in the Gypsy population. Mol. Genet. Metab. 88, 93–95 (2006).
Holve, S., Hu, D. & McCandless, S. E. Metachromatic leukodystrophy in the Navajo: fallout of the American-Indian wars of the nineteenth century. Am. J. Med. Genet. 101, 203–208 (2001).
Ausems, M. G. et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur. J. Hum. Genet. 7, 713–716 (1999).
Kalatzis V. et al. Characterization of a putative founder mutation that accounts for the high incidence of cystinosis in Brittany. J. Am. Soc. Nephrol. 12, 2170–2174 (2001).
Aula, N. et al. The spectrum of SLC17A5-gene mutations resulting in free sialic acid storage diseases indicates some genotype-phenotype correlation. Am. J. Hum. Genet. 67, 832–840 (2000).
Greer, W. L. et al. The Nova Scotia (Type D) form of Niemann-Pick disease is caused by a G3097 to T transversion in NPC1. Am. J. Hum. Genet. 63, 52–54 (1998).
Acknowledgements
F.M.P. is a Royal Society Wolfson Merit award holder and a Wellcome Trust Investigator in Science. A.d.A. is supported by NIH grants DK095169 and GM104981, the Assisi Foundation of Memphis, Ultragenyx Pharmaceutical and the American Lebanese Syrian Associated Charities (ALSAC) and holds the Jewellers for Children Endowed Chair in Genetics and Gene Therapy. A.d.A. thanks B. Stelter (St. Jude Biomedical Communication) for help with the graphic design of Fig. 3 and I. Annunziata for her assistance with literature screening and building the End Note library for the Mechanisms/pathophysiology section. B.L.D. is supported by the Children’s Hospital of Philadelphia Research Institute and the NIH (NS76631, NS090390 and NS94355) and holds the Arthur V. Meigs Chair in Pediatrics. C.J.T. is supported by the Division of Intramural Research of the National Human Genome Research Institute of the NIH, US Department of Health and Human Services. C.J.T. thanks M. Huizing for design of Fig. 4 and organization of the section on disorders of LROs.
Reviewer information
Nature Reviews Disease Primers thanks M. Beck, J. Cooper, R. Giugliani, C. Hollak, G. Pastores and other anonymous referee(s) for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
Introduction (all); Epidemiology (C.J.T.); Mechanisms/pathophysiology (A.d.A. and F.M.P.); Diagnosis, screening and prevention (C.J.T.); Management (E.F.N., B.L.D. and F.M.P.); Quality of life (C.J.T.); Outlook (all); Overview of Primer (F.M.P.).
Corresponding author
Ethics declarations
Competing interests
F.M.P. is a trustee of Gordon Research Conferences, Chair of the Scientific Advisory Board of the National Tay-Sachs & Allied Diseases Association (NTSAD), a cofounder of and consultant to IntraBio and a consultant to Actelion and Orphazyme. A.d.A. has received research support from Ultragenyx Pharmaceutical. B.L.D. founded Talee Bio, Inc. and Spark Therapeutics and is on the Scientific Advisory Board of Homology Medicines, Intellia Therapeutics, Prevail Therapeutics, Inc and Sarepta Therapeutics. E.F.N. has previously received funding from BioMarin and is a member of the Scientific Advisory Board of the National MPS Society. C.J.T. is supported by the Division of Intramural Research of the National Human Genome Research Institute of the NIH, US Department of Health and Human Services and is a member of the Scientific Advisory Board of NTSAD.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ClinicalTrials.gov: https://clinicaltrials.gov/
Genetics Home Reference: https://ghr.nlm.nih.gov/
Human Fertilization and Embryology Authority: https://www.hfea.gov.uk/pgd-conditions/
Rights and permissions
About this article

Cite this article
Platt, F.M., d’Azzo, A., Davidson, B.L. et al. Lysosomal storage diseases. Nat Rev Dis Primers 4, 27 (2018). https://doi.org/10.1038/s41572-018-0025-4
Published:
DOI: https://doi.org/10.1038/s41572-018-0025-4
This article is cited by
-
GBA1 as a risk gene for osteoporosis in the specific populations and its role in the development of Gaucher disease
Orphanet Journal of Rare Diseases (2024)
-
What should rheumatologists know about Gaucher disease and Fabry disease? Connecting the dots for an overview
Advances in Rheumatology (2024)
-
Lysosomes as coordinators of cellular catabolism, metabolic signalling and organ physiology
Nature Reviews Molecular Cell Biology (2024)
-
Evidence of epigenetic landscape shifts in mucopolysaccharidosis IIIB and IVA
Scientific Reports (2024)
-
Tracing genetic diversity captures the molecular basis of misfolding disease
Nature Communications (2024)
