
Abstract
Interest in epicardial adipose tissue (EAT) is growing rapidly, and research in this area appeals to a broad, multidisciplinary audience. EAT is unique in its anatomy and unobstructed proximity to the heart and has a transcriptome and secretome very different from that of other fat depots. EAT has physiological and pathological properties that vary depending on its location. It can be highly protective for the adjacent myocardium through dynamic brown fat-like thermogenic function and harmful via paracrine or vasocrine secretion of pro-inflammatory and profibrotic cytokines. EAT is a modifiable risk factor that can be assessed with traditional and novel imaging techniques. Coronary and left atrial EAT are involved in the pathogenesis of coronary artery disease and atrial fibrillation, respectively, and it also contributes to the development and progression of heart failure. In addition, EAT might have a role in coronavirus disease 2019 (COVID-19)-related cardiac syndrome. EAT is a reliable potential therapeutic target for drugs with cardiovascular benefits such as glucagon-like peptide 1 receptor agonists and sodium–glucose co-transporter 2 inhibitors. This Review provides a comprehensive and up-to-date overview of the role of EAT in cardiovascular disease and highlights the translational nature of EAT research and its applications in contemporary cardiology.
Key points
-
Epicardial adipose tissue (EAT) has anatomical and functional interactions with the heart owing to the shared circulation and the absence of muscle fascia separating the two organs.
-
EAT can be clinically measured with cardiac imaging techniques that can help to predict and stratify cardiovascular risk.
-
Regional distribution of EAT is important because pericoronary EAT and left atrial EAT differently affect the risk of coronary artery diseases and atrial fibrillation, respectively.
-
EAT has a role in the development of several cardiovascular diseases through complex mechanisms, including gene expression profile, pro-inflammatory and profibrotic proteome, neuromodulation, and glucose and lipid metabolism.
-
EAT could be a potential therapeutic target for novel cardiometabolic medications that modulate adipose tissue such as glucagon-like peptide 1 receptor agonists and sodium–glucose co-transporter 2 inhibitors.
-
EAT might be a reservoir of severe acute respiratory syndrome coronavirus 2 and an amplifier of coronavirus disease 2019 (COVID-19)-related cardiac syndrome.
Similar content being viewed by others


Introduction
Epicardial adipose tissue (EAT) is a unique fat depot located between the myocardium and the visceral layer of the epicardium, with multiple implications for research and clinical practice in contemporary cardiology. Since the pioneering work by my research group in the early 2000s1,2, interest in EAT has rapidly grown among a multidisciplinary audience ranging from clinical and research cardiologists to basic scientists and internal medicine practitioners. As research in the field of cardiometabolic diseases has evolved, the focus has narrowed from general obesity to organ-specific adiposity. To date, almost 2,000 original articles describing the multifaceted aspects of EAT have been published.
The unicity of EAT lies not only in its peculiar anatomy and unobstructed proximity to the heart3 but also in its distinctive transcriptome, which is substantially different from that of other visceral and subcutaneous fat depots4. EAT can be highly protective for the adjacent myocardium through its dynamic brown fat-like thermogenic function and deeply harmful via paracrine or vasocrine secretion of pro-inflammatory and profibrotic cytokines5. Owing to its functional proximity to the heart, EAT has been suggested to have a role in the progression and development of leading causes of morbidity and mortality such as coronary artery disease (CAD), atrial fibrillation and heart failure. Of note, EAT is not equally distributed throughout the heart and its regional distribution is not randomly allocated. EAT surrounding the left atrium is not the same as that infiltrating the coronary arteries. Each local EAT depot has a distinct transcriptome and proteome and, therefore, has a different effect on the adjacent heart structures.
The proliferative and translational nature of research on EAT makes the subject of this Review very timely. In this Review, I provide a comprehensive and up-to-date overview of EAT and its role in cardiovascular diseases. I also discuss imaging techniques for the assessment of EAT and the potential for EAT to be a therapeutic target for drugs with cardiovascular benefits such as glucagon-like peptide 1 receptor (GLP1R) agonists and sodium–glucose co-transporter 2 (SGLT2) inhibitors6. Finally, I explore the hypothesis that EAT amplifies the inflammatory response and cardiac syndrome related to coronavirus disease 2019 (COVID-19)7.
Anatomy and physiology of EAT
EAT is the fat depot located between the myocardium and the epicardium3,8 supplied by branches of the coronary arteries. By contrast, pericardial adipose tissue (PAT) is located externally and is supplied by non-coronary arteries. EAT is mostly located in the atrioventricular and interventricular grooves and can be differentiated into pericoronary EAT (located directly around or on the coronary artery adventitia) and myocardial EAT (the fat depot just over the myocardium)9. Microscopically, EAT is composed mainly of adipocytes but also contains nerve cells, inflammatory cells (mainly macrophages and mast cells), stromal cells, vascular cells and immune cells. EAT is a white adipose tissue but also has brown fat-like and beige fat-like features10. No muscle fascia is present between EAT and the myocardium; therefore, the two tissues share the same microcirculation3. This feature is unique to EAT; no other visceral fat depot has this contiguity with the target organ. The lack of an anatomical barrier allows crosstalk between EAT and the contiguous myocardium.
The function of EAT in normal conditions is protective. Indeed, EAT provides the adjacent myocardium with free fatty acids and functions as a buffer, protecting the heart against high fatty acid levels11. Adipocyte fatty acid-binding protein (also known as FABP4), which is highly expressed in EAT, participates in the intracellular transport of fatty acids from epicardial fat into the myocardium12. Fatty acids can reach the myocardium through paracrine or vasocrine pathways, the latter being impaired by coronary atherosclerosis13. The EAT transcriptome is rich in genes encoding cardioprotective adipokines, such as ADIPOQ and ADM, encoding adiponectin and adrenomedullin, respectively, both of which have potential anti-inflammatory and anti-atherogenic properties14,15. Interestingly, ADIPOQ expression in EAT is locally controlled by oxidation factors released from the heart in response to myocardial oxidative stress16. The paracrine modulation and activation of EAT adiponectin can contribute to myocardial redox homeostasis. Furthermore, when compared with subcutaneous adipose tissue, the EAT transcriptome contains more genes encoding proteins related to potassium channel activity, mesoderm development, regulation of body fluid levels, wound healing, the endoplasmic reticulum nuclear signalling pathway, plasma membrane organization and biogenesis4.
EAT function and morphology change with age and under pathological conditions (Fig. 1). Interestingly, epicardial and intra-abdominal fat depots both evolve from brown adipose tissue11. EAT is thought to provide a direct source of heat to the myocardium and to protect the heart during unfavourable haemodynamic conditions such as ischaemia or hypoxia17. The processes implicated in the control of thermogenesis in EAT are complex and yet to be fully understood. In neonates, EAT has brown fat-like properties and functions, with limited physical flexibility and responsiveness to external factors18. With ageing, epicardial adipocytes become more susceptible to environmental, metabolic and haemodynamic factors, which gradually change the function of EAT from thermogenesis to energy storage18. Indeed, EAT brown fat-like activity decreases substantially with age18. The changes are not only functional but also structural. The proportion of brown adipocytes decreases in favour of more unilocular white adipocytes in older individuals10. This finding suggests that the transition from brown fat to beige fat is a feature of EAT in adults. However, chronic and long-term ischaemic conditions, such as the advanced stages of CAD, can also depress brown fat-like activity in EAT18. In patients with advanced CAD, the expression of genes encoding proteins related to adipocyte browning and thermogenic activation is downregulated in EAT, with reciprocal increases in the expression of genes encoding pro-inflammatory cytokines19. These changes in gene expression could be a consequence of fibrosis and apoptosis that can occur in EAT in end-stage organ disease4. However, EAT can be induced to resume its brown fat-like function and provide beneficial effects to the heart in patients with long-term ischaemic conditions. Pharmacological upregulation of gene expression for proteins involved in brown fat activation and mitochondrial signalling in EAT has been associated with a significant reduction in left ventricular mass and EAT inflammation20. Further studies are necessary to evaluate whether EAT can adapt to various metabolic conditions and function like a brown fat or beige fat depot as needed.

In the neonate and early years of life, epicardial adipose tissue (EAT) is morphologically and functionally similar to brown adipose tissue. Under physiological conditions, the brown fat-like properties of EAT rapidly decrease with age, from childhood to adulthood. However, EAT maintains cardioprotective functions such as providing a source of energy and heat to the heart. In pathological conditions, such as coronary artery disease, diabetes mellitus, heart failure and atrial fibrillation, EAT becomes pro-atherogenic and pro-arrhythmogenic. In patients with advanced or end-stage organ disease, such as cardiac diseases, and in elderly individuals, the thermogenic function of EAT can be further decreased, with reciprocal increases in the expression of genes encoding profibrotic and pro-apoptotic factors.
Assessment of EAT
Imaging techniques are an essential component of contemporary cardiology. EAT can be assessed with traditional and novel techniques (Table 1). The thickness of EAT can be visualized and measured with standard 2D echocardiography as first proposed by my group in 2003 (ref.1). EAT is generally identified as the echo-free space between the outer wall of the myocardium and the visceral layer of the pericardium, but EAT can also appear as an echo-dense space when inflammation or large amounts of EAT are present21. EAT thickness is measured perpendicularly on the free wall of the right ventricle at end-systole when both walls collapse and allow the widest measurement21. However, a much greater EAT thickness can be measured just to the right of the aortic annular plane owing to the steep downward turn of the free wall of the right ventricle as it approaches the proximal ascending aorta.
Echocardiographic measurement of EAT thickness is a marker of visceral adiposity, and EAT thickness variability (ranging from 1 mm to 25 mm) reflects the variation in intra-abdominal fat accumulation2. However, EAT thickness is primarily a marker of ectopic fat accumulation. Intramyocardial and intrahepatic lipid content, measured with 1H-magnetic resonance spectroscopy techniques, are correlated with EAT thickness regardless of BMI22. Increased lipid accumulation in the myocardium has been associated with myocardial disarray, fibrosis and apoptosis, leading to heart failure and atrial fibrillation23,24. Intuitively, the use of echocardiography to measure EAT thickness has several advantages, including its low cost, accessibility and reproducibility but also has several limitations. Even if excellent interobserver and intraobserver agreement is reported, echocardiography is still an operator-dependent technique.
Cardiac multidetector CT and cardiac MRI can provide volumetric measurement of EAT and additional functional information by detecting deep regional EAT that is not accessible with transthoracic echocardiography25,26. Visualizing peri-atrial and pericoronary EAT is important in understanding, predicting and possibly preventing the effects of EAT in atrial fibrillation and CAD27,28. Both contrast-enhanced and non-contrast-enhanced, cardiac-gated multidetector CT are used to quantify EAT25. The combination of high spatial resolution, volume coverage of the entire heart and increasing availability of software analysis tools makes the use of CT to measure EAT ideal. However, differences exist in the CT attenuation (a measure of EAT density, expressed in Hounsfield units (HU)) characteristics of EAT depending on the presence or absence of iodinated contrast and inflammatory status29. EAT density is a marker of both EAT and general inflammation30,31. EAT attenuation ranges between –45 HU and –195 HU, where a lower negative means higher density29,30,31. Radiographic fat density is determined by adipocyte hypertrophy, hyperplasia and fibrosis that oppositely influence fat CT signal attenuation. Hypertrophic and hyperplastic fat depots usually have low density29,30,31. The increased EAT density reported in patients with CAD or severe COVID-19 could be caused by inflammation and fibrosis that mitigate the expected effect of hypertrophic or hyperplasic fat cells on fat CT attenuation32.
Although 18F-FDG-PET–CT can detect EAT inflammatory activity, this modality is not cost-effective or readily available. Therefore, the need for imaging biomarkers to directly assess the interaction between adipose tissue and inflammation is compelling. An innovative imaging metric — the CT fat attenuation index (FAI) — has been proposed as a marker of perivascular fat inflammation33. FAI reflects transcriptomic, metabolic and phenotypic changes in perivascular fat. FAI is significantly higher around culprit lesions than around non-culprit lesions in individual patients with CAD34. FAI can detect the inflammatory burden around vulnerable plaques and predict early subclinical CAD in vivo. Further studies evaluating FAI assessment of regional EAT depots, such as peri-atrial and pericoronary EAT, are warranted. Artificial intelligence and radiomic analysis to process and elaborate on images, including those of fat depots obtained by common non-invasive imaging methods, could be used to improve the assessment of EAT physiology and pathophysiology35.
Role of EAT in cardiovascular disease
Coronary artery disease
The pathogenesis of CAD is multifactorial and includes established and novel mechanisms. EAT was first suggested to be a factor in the multifaceted pathways causing coronary atherosclerosis in the early 2000s. Undoubtedly, the anatomical and unobstructed contiguity of EAT with the coronary arteries supports the argument for a local effect. However, vicinity is not the only factor, because the quantity and activity of EAT have more important roles27. The mechanisms through which EAT can cause atherosclerosis are complex and include inflammation, exaggerated innate immunity response, oxidative stress, endothelial damage, adipocyte stress, lipid accumulation and glucotoxicity5 (Figs 2,3).

The epicardial adipose tissue (EAT) is distributed as localized depots lying between the myocardium and the visceral layer of the pericardium. EAT can infiltrate the left atrium (left atrial EAT) and surround the coronary arteries (coronary EAT). By contrast, pericardial adipose tissue (PAT) is located more externally, within the visceral and parietal layers of the pericardium. EAT contributes to the development and progression of coronary artery disease and atrial fibrillation through complex and multifactorial pathways. The regional distribution of EAT has an important role because each EAT depot is anatomically, genetically and functionally different. a | Left atrial EAT has a high expression of genes encoding pro-arrhythmogenic factors. Left atrial EAT can contribute to atrial fibrillation through the local secretion of profibrotic factors (matrix metalloproteinases (MMPs), transforming growth factor-β1 (TGFβ1) and TGFβ2, connective tissue growth factor (cTGF) and activin A) and inflammatory factors (IL-6 and tumour necrosis factor (TNF)) as well as free fatty acid (FFA) infiltration and increased autonomic control via ganglionated plexi. b | The coronary EAT has a high expression of genes encoding pro-inflammatory adipokines and factors regulating glucose and lipid metabolism. Coronary EAT can influence the development and progression of coronary artery disease through increased infiltration of pro-inflammatory M1 macrophages from EAT into the adjacent myocardium, the paracrine or vasocrine release of several pro-inflammatory cytokines (CCL2, IL-6 and TNF) and adipokines (chemerin, intelectin 1 (also known as omentin 1), resistin and serglycin), and the activation of innate immune response factors such as JUN N-terminal kinase (JNK), nuclear factor-κB (NF-κB) and Toll-like receptors (TLRs). Upregulation of signalling via advanced glycation end products (AGE) binding to their receptor RAGE in EAT can contribute to the oxidative stress and endothelial damage associated with coronary atherosclerosis in patients with diabetes mellitus. The excessive influx of FFAs from EAT into the coronary arteries is mediated by enzymes such as group II secretory phospholipase A2 (sPLA2-II) and adipocyte fatty acid-binding protein (also known as FABP4). GLUT4, glucose transporter type 4.

In patients with coronary artery disease, coronary epicardial adipose tissue (EAT) has a dense inflammatory infiltrate with a high prevalence of pro-inflammatory M1 macrophages. Coronary EAT secretes pro-inflammatory cytokines (such as CCL2, IL-6 and tumour necrosis factor (TNF)) and adipokines (such as chemerin, intelectin 1 (also known as omentin 1), resistin and serglycin) into the coronary lumen, thereby contributing to systemic inflammation. Coronary EAT inflammation also contributes locally to coronary atherosclerotic plaque inflammation. The upregulation in the coronary EAT of innate immune response signalling, such as JUN N-terminal kinase (JNK), nuclear factor-κB (NF-κB) and Toll-like receptor (TLR) signalling, can also induce the secretion of inflammatory mediators from the coronary EAT. The excessive influx of free fatty acids (FFAs) mediated by group II secretory phospholipase A2 (sPLA2-II) and adipocyte fatty acid-binding protein (also known as FABP4) from epicardial adipocytes might infiltrate the adventitia and contribute to the lipid build-up in coronary artery atherosclerotic plaques. The co-occurrence of coronary artery disease with chronic hyperglycaemia can upregulate signalling via advanced glycation end products (AGE) binding to their receptor RAGE and reduce levels of glucose transporter type 4 (GLUT4), thereby contributing to oxidative stress and endothelial cell damage.
Inflammation is the major feature of EAT in patients with CAD, with dense infiltrates of macrophages, mast cells and CD8+ T cells36. Pro-inflammatory M1 macrophages are significantly more prevalent than anti-inflammatory M2 macrophages in EAT from individuals with CAD37. The presence of macrophages in EAT has been argued to be reactive to coronary artery plaque rupture and instability rather than the result of intrinsic inflammation36. However, a study using microarray analysis demonstrated that EAT has a pro-atherogenic transcriptional profile in CAD4. The genes encoding many pro-inflammatory cytokines (such as IL-6, CCL2 (also known as MCP1) and tumour necrosis factor (TNF)), chemokine ligands and receptors as well as several novel pro-inflammatory adipokines (such as chemerin, resistin, serglycin and intelectin 1 (also known as omentin 1))38,39,40 are upregulated in EAT of patients with CAD. The level of inflammation is not only greater in EAT than in the subcutaneous adipose tissue in these patients but is also greater than in any other visceral fat depot. For example, levels of CD45 (a marker of haematopoietic cells) have been reported to be significantly higher in EAT than in omental fat depots, indicating substantial macrophage infiltration in EAT41. Given the proximity of EAT to the coronary arteries, this rich pro-inflammatory proteasome surrounds the coronary adventitia and goes directly into the coronary lumen following paracrine or vasocrine pathways. The thicker the layer of EAT and the closer it is to the coronary artery, the greater the inflammatory activity and, consequently, the more severe the coronary atherosclerosis36. The disequilibrium between anti-inflammatory and pro-inflammatory EAT adipokine secretion has a significant effect on the progression and severity of coronary atherosclerosis41,42. Whereas the production of EAT pro-inflammatory adipokines is significantly higher in patients with CAD than in individuals without CAD, gene and protein expression of adiponectin (a cytokine with anti-inflammatory properties) are lower14. The peculiar pro-atherogenic transcriptome of EAT4 can influence adipokine production. The proximity of EAT to the coronary arteries makes the defective paracrine secretion of adiponectin an important contributor to coronary atherosclerosis.
The adaptive and innate immune responses also contribute to EAT inflammation in CAD. High concentrations of adaptive immune cells, particularly CD4+ T cells, in EAT are frequently observed in individuals with obesity or diabetes mellitus43,44,45. The activation of mediators of the EAT innate response, such as nuclear factor-κB (NF-κB), JUN N-terminal kinase (JNK) and Toll-like receptors, in patients with CAD can lead to the upregulation of inflammatory cytokine expression in EAT41.
EAT also secretes factors that regulate endothelial function, such as resistin, which is associated with increased endothelial cell permeability46. EAT is also implicated in oxidative stress. Increased levels of reactive oxygen species and reduced expression of antioxidant enzymes trigger inflammation and atherogenicity in epicardial adipocytes47. In patients with CAD, the EAT transcriptome is rich in genes involved in haemostasis and coagulation, including tissue plasminogen activator, which links fibrinolysis and inflammation in human adipose tissue4. The epicardial adipocytes of individuals with CAD overexpress markers of cellular stress (such as the kinases MAP2K3 and MAP3K5), which are linked to coronary inflammation, as well as multiple proteases involved in lysosomal degradation and cellular apoptosis4.
EAT is also a local source of ectopic lipids. The excessive secretion and release of fatty acids from epicardial adipocytes infiltrating the adventitia could contribute to lipid accumulation in the coronary arteries. Group II secretory phospholipase A2, the rate-limiting enzyme in the synthesis of pro-inflammatory lipids, is present in high concentrations in EAT of patients with CAD compared with healthy individuals48. FABP4 is expressed in epicardial adipocytes and might act as a local contributor to ectopic fat accumulation in atherosclerotic plaque49. The lipogenic effect of epicardial fat can also be attributed to the high content of conjugated fatty acids in EAT50. The innate immune response in EAT mediated by Toll-like receptors is upregulated by excessive fatty acid release from EAT41,51. Of note, the expression of genes encoding proteins involved in lipid metabolism, such as endothelial lipase (also known as lipase G) and large neutral amino acids transporter small subunit 1 (also known as SLC7A5), is upregulated in EAT of patients with CAD and type 2 diabetes compared with patients with CAD without diabetes52. Insulin-stimulated lipogenesis is greater in EAT than in other visceral fat depots, whereas glucose uptake is extremely low in EAT11. Therefore, EAT can contribute to local insulin resistance in the coronary arteries53,54. Interestingly, in patients with CAD, levels of GLUT4 mRNA (which encodes glucose transporter type 4 (GLUT4)) are lower in EAT than in subcutaneous fat55. Lower GLUT4 levels affect insulin-mediated glucose uptake into EAT and the adjacent myocardium. Studies suggest that mechanisms underlying coronary atherosclerosis in patients with diabetes include upregulation of signalling between advanced glycation end products (AGEs) and their receptors (RAGEs) in EAT52. Upregulated AGE–RAGE signalling can contribute to oxidative stress and endothelial damage associated with diabetic coronary atherosclerosis. The overexpression of genes related to inflammation in EAT of patients with CAD and diabetes is primarily the result of the increased activity of transcription factors such as those in the NF-κB and FOS families51. The increased atherogenicity of EAT in patients with diabetes can also be linked to a high concentration of unsaturated fatty acids, sphingolipids and ceramides in EAT56.
The complex mechanisms underlying the role of EAT in coronary atherosclerosis can be translated into clinical practice, for example, in early diagnosis and risk stratification. EAT volume and thickness are greater in patients with CAD than in individuals without atherosclerosis57. A coronary artery calcium (CAC) score >10 is associated with higher EAT volume, which can predict the risk of atherosclerosis with a sensitivity and specificity of 72% and 70%, respectively58. In the EPICHEART study59, a high EAT volume was independently associated with a high CAC score in men but not in women. In the Heinz Nixdorf Recall cohort study56, a high EAT volume was associated with the progression of coronary artery calcification, particularly in younger (age <55 years) individuals and in those with mild obesity. Other studies seem to confirm the role of EAT volume in predicting the early stages of atherosclerosis in asymptomatic individuals, often independent of obesity60. This observation can be explained by the visceral fat phenotype of EAT and the poor sensitivity of BMI-defined obesity in representing body fat distribution2,23. The role of EAT volume in predicting early atherosclerosis in individuals at high risk of atherosclerotic cardiovascular disease has also been confirmed in patients with asymptomatic diabetes61,62. Although calcification is a key component of atherosclerotic plaques, EAT volume can predict the risk of CAD independently of the CAC score63,64,65. An increased EAT volume is associated with the presence of obstructive and vulnerable plaques in patients with symptomatic atherosclerosis and a CAC score of 0 (ref.66). EAT volume is higher in patients with non-calcified, vulnerable unstable plaques than in those with stable and calcified lesions63,64,65. Therefore, EAT might contribute to the development of early and not yet calcified coronary atherosclerotic plaques, which are highly unstable and vulnerable to rupture67.
EAT is not equally distributed through the heart and, therefore, has regional effects. Pericoronary EAT affects the proximal coronary arteries owing to their anatomical proximity27,68,69. Unlike echocardiography, cardiac CT and MRI allow the detection and measurement of regional EAT. A greater pericoronary EAT volume is associated with more severe coronary artery stenosis and CAC score in women27. The portion of EAT infiltrating the left atrioventricular groove has a stronger association with coronary atherosclerosis than the total EAT volume68. The inflammatory activity of EAT is also dependent on its location as confirmed by 18F-FDG-PET–CT studies70. The proteasome derived from pericoronary EAT produces inflammation in the underlying coronary atherosclerotic plaques, and EAT inflammation levels correlate with plaque burden and plaque necrotic core area69. EAT volume can also predict major cardiovascular events. In the Heinz Nixdorf Recall cohort study71, the incidence of fatal or non-fatal coronary events significantly increased by quartile of EAT volume increase and the association remained significant even after adjustment for CAC score. The MESA study72 (and other large, population studies) showed the independent association between EAT volume and the incidence of major adverse cardiac events. EAT assessment can, therefore, help to predict the risk of major coronary events before the accumulation of calcium in the atherosclerotic plaque occurs and in individuals with asymptomatic atherosclerosis who are not obese. The use of imaging techniques for the assessment of EAT could be implemented as routine procedures for effective prediction and stratification of CAD.
Atrial fibrillation
Atrial fibrillation increases the risk of heart failure, stroke and all-cause death73. Obesity is a known risk factor for atrial fibrillation, and weight loss and lifestyle modification can reduce this risk74. EAT has emerged as a risk factor and independent predictor of atrial fibrillation development and recurrence after ablation75,76. Importantly, however, EAT has not always been measured in studies as a fat depot separate and distinct from PAT. This issue is not a trivial matter of terminology because EAT is anatomically and functionally different from PAT. Although PAT is a paracardiac visceral fat depot, an excess of which can affect the heart, it is not contiguous to the atrial myocardium77. In the Framingham Heart Study cohort, PAT volume was an independent predictor of atrial fibrillation even after adjusting for other risk factors78. An association has also been reported between EAT volume or thickness and atrial conduction delays such as prolonged P-wave duration, interatrial conduction block and longer P–R interval76. CT-derived posterior left atria adiposity, including peri-atrial EAT thickness, is associated with atrial fibrillation burden independently of left atrium area and BMI28. Increased atrial PAT volume was also associated with increased prevalence and severity of atrial fibrillation even after adjusting for body weight74. Of note, these studies all showed that the association between cardiac fat and atrial fibrillation was partially or totally independent of obesity.
Several mechanisms for how altered EAT can cause or contribute to atrial fibrillation have been proposed, including genetic and neural factors, inflammation, fibrosis, fatty infiltration, and atrial electrical or structural remodelling (Fig. 2). The pathogenic role of EAT in atrial fibrillation could begin with its embryogenesis and development. Embryonic epicardium can generate coronary smooth muscle cells and cardiac fibroblast or undergo adipogenic differentiation79. Atrial EAT adipocytes originate from the differentiation of progenitor cells resident in the epicardium and from the secretome of atrial myocytes79. Interestingly, atrial natriuretic factor secreted by atrial myocytes in response to mechanical stress has adipogenic properties that can contribute to atrial EAT development80. Of note, the adipogenic potential of the atrial cell secretome is greater in patients with atrial fibrillation than in those without80. Importantly, the epicardium is reactivated during the development of atrial cardiomyopathy and contributes to the fibro-fatty infiltration of subepicardium81. Under pathological conditions, the atria could be postulated to contribute to peri-atrial EAT expansion and myocardial fibrosis and, therefore, to the development of atrial fibrillation substrate.
As in CAD, the location of EAT is important in atrial fibrillation, and regional EAT distribution has emerged as an important factor in atrial fibrillation. The epicardial fat pad surrounding the left atrium, namely peri-atrial EAT, has a unique transcriptome and secretome with potential arrhythmogenic properties that are different from those detected in other EAT depots82. Peri-atrial EAT has a specific gene expression signature compared with periventricular and pericoronary EAT82. EAT infiltrating the atrium has increased expression of genes encoding proteins involved in oxidative phosphorylation, muscular contraction and calcium signalling compared with periventricular and pericoronary EAT82. The absence of a fascia separating peri-atrial EAT from the underlying left atrial myocardium and a shared blood supply provide a milieu for bidirectional communication. Pro-inflammatory and profibrotic cytokines, such as interleukins and TNF, and profibrotic factors, such as matrix metalloproteinases (MMPs) and activin A, can diffuse from EAT into the adjacent atrial myocardium and promote arrhythmias82. Fibrosis also has an important pathogenic role in the development of atrial fibrillation83. MMPs, which are abundantly produced in EAT, are regulators of extracellular matrix homeostasis and their overexpression can cause fibrosis84. In rat atria, EAT-conditioned medium upregulates the expression of transforming growth factor-β1 (TGFβ1) and TGFβ2 and promotes fibrosis in vitro, which is mediated by EAT secretion of activin A84. Connective tissue growth factor (cTGF) can also contribute to atrial fibrosis. cTGF expression is significantly higher in EAT than in subcutaneous fat or PAT from patients with atrial fibrillation and in EAT from patients with sinus rhythm85. High EAT volume is associated with increased fibrosis, lateralization of connexin 40 and slow conduction in patients with CAD86. Interestingly, EAT-derived extracellular vesicles collected from EAT from patients with atrial fibrillation contain profibrotic cytokines and microRNAs87. This finding supports the paracrine and local interaction between peri-atrial EAT and the adjacent left atrium. EAT can serve as a source of lipids infiltrating the contiguous atrium. Free fatty acids can also be transported from EAT to the myocardium and lead to electromechanical changes in atrial tissue. Free fatty acid infiltration can separate cardiomyocytes, resulting in conduction slowing, loss of side-to-side cell connections88 and myocardial disorganization that leads to conduction delay and re-entry (Fig. 4). EAT can also influence the local electrophysiological properties of the atrial and pulmonary veins, such as the refractory period, and therefore sustain atrial fibrillation89. Investigations with cultured human induced pluripotent stem cell-derived cardiomyocytes indicate that local peri-atrial EAT accumulation, rather than global cardiac adiposity, contributes to conduction abnormalities underlying the atrial fibrillation substrate86. Peri-atrial EAT accumulation can slow conduction and prolong cardiomyocyte field potential duration through two mechanisms: by physical conduction block caused by extensive fibrosis, and by local EAT infiltration of the adjacent atrial myocardium, which causes conduction heterogeneity and electrophysiological changes through the paracrine release of cytokines that induce inter-cardiomyocyte adhesion disruption and abnormal cell coupling, alter ionic currents and myocardial metabolism, and promote inflammation86,90.

Given the anatomical contiguity of left atrial epicardial adipose tissue (EAT) with the adjacent left atrium, profibrotic factors (such as activin A, connective tissue growth factor (cTGF), matrix metalloproteinases (MMPs), and transforming growth factor-β1 (TGFβ1) and TGFβ2) released by EAT, via secretion or through extracellular vesicles (EVs), can cause atrial myocardial fibrosis. Left atrial EAT can also contribute to atrial fibrillation through the local secretion of pro-inflammatory factors such as IL-6 and tumour necrosis factor (TNF). Excessive influx of free fatty acids (FFAs) from the left atrial EAT affects the continuity of cardiomyocytes, causing ‘zig-zag’ conduction and facilitating the development of re-entrant circuits. The increased activity of the ganglionated plexi in EAT can increase the autonomic effects of atrial cardiomyocytes and prolong the action potential duration. ECM, extracellular matrix.
EAT contains sympathetic and parasympathetic nerve fibres that contribute to overall cardiac autonomic neuronal output. EAT is the site of the ganglionated plexi, which are responsible for the initiation and maintenance of atrial fibrillation3. Activation of these ganglia can lead to shortening of action potential duration and to an increase in the calcium transient amplitude in the atrial myocardium91,92. Interestingly, botulinum injection into EAT during cardiac surgery can suppress ganglionated plexi, reduce autonomic nervous activity and have long-term beneficial effects on atrial fibrillation93. A large peri-atrial EAT pad can also mechanically affect the left atrium and cause dilatation94. The infiltration of adipocytes into the atrial myocardium disorganizes the depolarization wavefront, inducing micro re-entry circuits and local conduction blocks90.
EAT thickness and volume are greater in patients with chronic, persistent atrial fibrillation than in those with paroxysmal atrial fibrillation independent of obesity, age, sex, or presence of CAD, diabetes, dyslipidaemia or hypertension74,75,78,94. Several studies have highlighted the use of EAT measurement in predicting outcomes after catheter ablation for paroxysmal or persistent atrial fibrillation95,96. Peri-atrial EAT volume is greater in patients with atrial fibrillation and is associated with recurrence after catheter ablation95,96,97,98. EAT volume is associated with atrial fibrillation persistence independent of other risk factors or BMI99. EAT is, therefore, a potential substrate for the pathogenesis of atrial fibrillation. The ease with which EAT can be measured and its responsiveness to drugs currently under investigation in the context of atrial fibrillation (such as GLP1R agonists and SGLT2 inhibitors), raises the possibility of novel therapeutic approaches for atrial fibrillation treatment and prevention of atrial fibrillation recurrence after catheter ablation.
Heart failure
Heart failure is a complex clinical condition that can result from diastolic or systolic dysfunction100. If left ventricular filling and relaxation are affected but the heart maintains good systolic function, the condition is defined as heart failure with preserved ejection fraction (HFpEF), whereas heart failure with reduced ejection fraction (HFrEF) indicates an impairment in systolic performance with an ejection fraction <40%. Patients with either HFpEF or HFrEF have a poor quality of life and increased risks of arrhythmias and premature death100. Overall, heart failure includes abnormalities in various components of the heart, although the mechanisms are poorly understood.
EAT has been suggested to have a role in heart failure, particularly in patients with HFpEF101,102,103,104. The volume of EAT is significantly higher in patients with HFpEF than in healthy individuals although few studies ruled out potential confounders such as CAD or obesity101,102. The association between EAT thickness or volume and HFrEF is controversial, because they have been shown to be either higher or, more frequently, lower than in healthy individuals99,103,104. The lower burden of EAT observed in patients with HFrEF is attributed to the left ventricular remodelling that occurs in heart failure102,103,104. This variability can be explained by the presence of comorbidities, such as CAD, obesity and diabetes, which can affect the volume of EAT in HFrEF. The overall metabolic and haemodynamic status of patients with HFrEF can also modulate EAT volume101,102,103,104,105. Severely ill patients with HFrEF can present with diffuse systematic fat loss and, therefore, with reduced EAT volume106.
EAT can affect cardiac function in the setting of heart failure via several mechanisms, such as increased inflammation, fibrosis and autonomic dysregulation (Fig. 5), as well as through the mechanical effects of a large, fibrotic fat pad. The EAT proteome can also contribute to the pathogenesis of heart failure107,108,109. Inflammatory proteins, such as α1-antichymotrypsin (also known as serpin A3), creatine kinase B‐type and MMP14, are upregulated in patients with heart failure107. α1-Antichymotrypsin could function as a modulator of the inflammatory status, although its role in heart failure needs further investigation. TP53 mRNA expression levels have also been shown to be higher in EAT than in subcutaneous fat in patients with heart failure108. p53 is a marker of inflammation and its levels are inversely correlated with the levels of adiponectin, specifically in patients with heart failure109. The EAT secretome might also affect biochemical processes involved in diastole. Increased EAT volume shows a strong correlation with worsening left ventricular diastolic relaxation and filling103,104. As described earlier, the location of EAT and the lack of fascia enable lipids to infiltrate the myocardium. Excessive EAT-derived fatty acids can be taken up by cardiomyocytes and lead to ectopic myocardial lipid accumulation110, which contributes to the development of heart failure by causing cardiomyocyte disarray, dysfunction and apoptosis110. Patients with HFpEF have significantly more intramyocardial fat than patients with HFrEF or individuals without heart failure111. Increased intramyocardial fat content correlates with left ventricular dysfunction parameters in patients with HFpEF112.

Epicardial adipose tissue (EAT) can affect heart function in the setting of heart failure via inflammation, fibrosis and neural dysregulation as observed in coronary artery disease and atrial fibrillation. However, several specific mechanisms link EAT with heart failure. The EAT proteome can contribute to the pathogenesis of heart failure through the paracrine secretion of profibrotic factors, such as α1-antichymotrypsin (ACT; also known as serpin A3) and matrix metalloproteinase 14 (MMP14), inflammatory markers, such as p53, and free fatty acids (FFAs). Large and fibrotic EAT can also exert mechanical effects on both diastolic and systolic function. EAT can also be involved in the pathogenesis of heart failure through neurohormonal mechanisms. The increased catecholamine biosynthetic activity of EAT can increase noradrenaline accumulation in the myocardium and worsen systolic performance. ECM, extracellular matrix.
EAT can also be involved in the pathogenesis of heart failure through neurohormonal mechanisms via the intrinsic adrenergic and cholinergic nerves, which interact with the extrinsic cardiac sympathetic and parasympathetic nervous systems113,114,115. EAT is, therefore, an important source of catecholamines, both noradrenaline and adrenaline116. The production of these molecules is relevant to the heart because EAT secretory abnormalities are implicated in the development of pathological conditions, including heart failure. In patients with heart failure, noradrenaline levels were increased 5.6-fold in EAT compared with subcutaneous adipose tissue and twofold compared with plasma116. In addition, the levels of the catecholamine biosynthetic enzymes tyrosine hydroxylase and dopamine β-hydroxylase were upregulated in EAT compared with subcutaneous fat in patients with heart failure116. The increased catecholamine biosynthetic activity of EAT might contribute to the increased prevalence of atrial fibrillation in patients with HFpEF. By contrast, in HFrEF, this increased activity might increase total catecholamine accumulation in the myocardium and worsen systolic performance. A biopsy study in patients with heart failure showed that, after treatment with isoprenaline (an agonist of the catecholaminergic β-adrenergic receptor), EAT releases molecules involved in the inflammatory response or extracellular matrix117. Interestingly, EAT expression of CD5L, a macrophage apoptosis inhibitor stimulated by isoprenaline, was higher in patients with heart failure who developed atrial fibrillation during follow-up, although circulating levels of CD5L were not correlated with the risk of atrial fibrillation118. Of note, in animal models, lipolysis stimulated by isoprenaline is decreased in EAT compared with subcutaneous fat, leading to lipid storage and inflammation119.
Targeting EAT in cardiovascular disease
EAT is a modifiable cardiovascular risk factor and a potential novel therapeutic target owing to its responsiveness to drugs with pleiotropic effects such as GLP1R agonists and SGLT2 inhibitors (Fig. 6). Cardiovascular outcomes trials have shown that GLP1R agonist and SGLT2 inhibitor therapies reduce the incidence of major cardiovascular events, with effect sizes suggesting mechanisms beyond improvements in glycaemic control, although the mechanisms are not fully elucidated120,121,122,123,124,125.

Potential beneficial cardiometabolic effects of sodium–glucose co-transporter 2 (SGLT2) inhibitor and glucagon-like peptide 1 receptor (GLP1R) agonist therapies beyond their glycaemic and haemodynamic effects. SGLT2 inhibitors and GLP1R agonists can target both left atrial epicardial adipose tissue (EAT) and coronary EAT for the treatment and prevention of atrial fibrillation (part a) and coronary artery disease (part b), respectively. Both SGLT2 inhibitors and GLP1R agonists can reduce EAT inflammation and increase free fatty acid (FFA) oxidation as fuel for the myocardium, and GLP1R agonists induce fat browning (white to brown fat differentiation and pre-adipocyte differentiation, leading to improved myocardial insulin sensitivity), all of which improve myocardial metabolism. SGLT2 inhibitors can induce sympatholytic and lipolytic effects in EAT to increase ketogenesis and reduce oxygen consumption in the setting of heart failure.
GLP1R agonists are injectable medications for the treatment of type 2 diabetes and obesity that provide cardiovascular benefits beyond glucose control120,121,122. Visceral fat reduction has been suggested as one of the non-glycaemic effects of the GLP1R agonist liraglutide126. In patients with type 2 diabetes and obesity, the GLP1R agonists liraglutide (daily dose), semaglutide (weekly dose) and dulaglutide (weekly dose) reduce EAT thickness to a greater extent than overall weight loss127,128,129,130. Notably, EAT expresses GLP1R, whereas subcutaneous fat does not131. Therefore, the presence of GLP1R in EAT supports the hypothesis of a direct effect on the fat depot. Activation of EAT GLP1R can reduce local adipogenesis, improve fat utilization, induce brown fat differentiation and modulate the renin–angiotensin–aldosterone system132,133,134. These metabolic changes might contribute to the beneficial effects of GLP1R agonists on the cardiovascular system125. Interestingly, GLP1R is also expressed in human cardiomyocytes135.
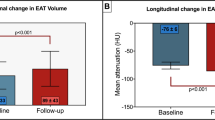
Selective SGLT2 inhibitors are oral antidiabetic agents that are indicated for the treatment of both HFpEF and HFrEF, irrespective of diabetes status. Cardiovascular outcomes trials have shown that SGLT2 inhibitor therapy can reduce the risk of major adverse cardiovascular events, cardiovascular death and heart failure123,124. SGLT2 inhibitors, such as dapagliflozin and empagliflozin, reduce EAT thickness or volume136,137,138,139,140,141 to a clinically significant degree, partially independent of weight loss136. The cardiovascular benefits of SGLT2 inhibitors can be exerted throughout glycosuric and non-glycaemic effects, including targeting EAT. In response to the decreased plasma glucose level caused by glycosuria, SGLT2 inhibitor therapy promotes a shift to fatty acid substrate utilization, leading to increased fatty acid oxidation, lipolysis, ketogenesis and improved myocardial glucose metabolism142. In heart failure, myocardial insulin-mediated glucose uptake and mitochondrial oxidative metabolism are impaired143. The failing heart reduces fatty acid and glucose oxidation, with an adaptive increase in myocardial ketone utilization. The oxidation of ketone bodies, such as β-hydroxybutyrate, induced by SGLT2 inhibitor therapy becomes the preferential and alternative energy source to both glucose and fatty acid oxidation144. This substrate selection improves oxygen consumption, translating to better cardiac performance at the mitochondrial level because the energy cost for β-hydroxybutyrate oxidation is reduced compared with oxidation of glucose and pyruvate144. EAT might serve as a mediator of the non-glycosuric cardiovascular effects of SGLT2 inhibitors. Indeed, SGLT2 inhibitors could induce EAT lipolysis and contribute to the improvement of myocardial metabolism. EAT is a major source of fatty acids and lipids that, if excessive and stored, can infiltrate the underlying myocardium and contribute to heart failure111. Therefore, SGLT2 inhibitors, such as dapagliflozin and empagliflozin, could reduce intramyocardial lipid content by increasing EAT lipolysis and ketone body oxidation. Although the cardiovascular beneficial effects of EAT lipolysis induced by SGLT2 inhibitor therapy remain to be demonstrated145, some potential mechanisms can be hypothesized on the basis of existing data. The expression of heart fatty acid-binding protein (also known as FABP3) is upregulated in EAT of patients with heart failure146. FABP3 mobilizes elevated circulating fatty acids that are released during EAT lipolysis and transports them to the adjacent myocardium. Fatty acid oxidation is greatly influenced by insulin sensitivity, and dapagliflozin has been shown to improve insulin sensitivity and glucose uptake142. Therefore, the ameliorated myocardial glucose metabolism and insulin sensitivity induced by SGLT2 inhibitors can improve fatty acid utilization147. However, if the myocardium becomes oversaturated with ectopic fatty acids and is unable to utilize them, the oxidation of ketone bodies induced by SGLT2 inhibitors would become the alternative source of fuel143. In addition to these metabolic changes, the mass reduction of EAT induced by SGLT2 inhibitors might contribute to improved systolic and diastolic function. Further studies are warranted to elucidate the independent effects of SGLT2 inhibitors on EAT.
A reduction in EAT thickness induced by statins has been reported in a few studies148,149,150, potentially through modulation of peroxisome proliferator-activated receptors (PPARs). Activation of PPARα and PPARγ can improve EAT insulin sensitivity and glucose uptake150. However, statins have fewer effects on EAT than GLP1R agonists and SGLT2 inhibitors126,136. Interestingly, in patients with metabolic syndrome, the addition of pioglitazone (an antidiabetic thiazolidinedione) to simvastatin therapy results in a significant reduction in EAT inflammation150. Clinical trials testing the efficacy of drugs used for cardiometabolic disease in reducing EAT volume or thickness are summarized in Table 2.
EAT and COVID-19
The novel COVID-19 caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is associated with cardiac involvement, mainly characterized by myocarditis, pericarditis and thrombosis151. Visceral fat, such as EAT, has been suggested to serve as a functional reservoir and amplifier of SARS-CoV-2 (ref.7). The intrinsic inflammatory milieu of visceral fat depots might amplify the inflammatory response in patients with COVID-19, leading to serious cardiovascular complications. Owing to its contiguity to the myocardium and high inflammatory secretome, EAT has been suggested to be implicated in the pathophysiology of COVID-19-related myocarditis7.
Angiotensin-converting enzyme 2 (ACE2), which is widely recognized as the entry receptor for SARS-CoV-2 into host cells152, is expressed in human EAT133. The downregulation of ACE2 levels increases EAT inflammation, whereas treatment with angiotensin 1–7 reduced EAT inflammatory cytokines in a mouse model153. The modulation of ACE in EAT might, therefore, have a role in COVID-19-related myocardial and perivascular inflammation. ACE inhibitors could be a potential component of therapy for these sequelae of COVID-19, although data are still insufficient and controversial154.
EAT of patients hospitalized with severe or critical COVID-19 shows signs of increased inflammation on CT, irrespective of whether CAD is present32,155,156,157. In patients with COVID-19, EAT density on CT is markedly elevated at hospital admission and decreases to normal at discharge, whereas subcutaneous fat shows no signs of inflammation32. EAT inflammation decreased in patients with COVID-19 who received oral or intravenous dexamethasone, whereas no significant changes in inflammation were observed with other COVID-19 therapies157. Therefore, EAT might have a role in COVID-19-related cardiac syndrome, and CT-measured EAT attenuation could be a marker of inflammation and severity of COVID-19.
Conclusions
The physiology and pathophysiology of EAT and their clinical implications form a fast-moving and productive field of research. EAT is a measurable and modifiable cardiovascular risk factor that adds qualitative value to the stratification of cardiovascular risk. Assessment of EAT, with commonly used imaging techniques, such as echocardiography, CT and MRI, should be readily accessible to contemporary cardiologists.
EAT provides a novel and unconventional perspective on the pathophysiology of major cardiovascular diseases. EAT directly contributes to the development and progression of CAD, mainly by causing inflammation but also by endothelial damage and oxidative stress as well as the accumulation of glucose and lipids in the proximal coronary arteries. In the context of atrial fibrillation, EAT represents a new pathogenic substrate through the regional secretion of factors that induce fibrosis and neurohormonal disarray of the atrial myocytes. The role of EAT in heart failure is mediated through several pathways, including the excessive release of fatty acids leading to intracardiac cell ectopic lipid accumulation, overexpression of local pro-inflammatory and profibrotic cytokines with pro-arrhythmogenic properties, and increased β-adrenergic receptor activation.
Pharmacological modulation of EAT induces previously unexpected beneficial cardiometabolic effects. The potential to restore the cardioprotective function of EAT with targeted agents, such as GLP1R agonists and SGLT2 inhibitors, can open new avenues in pharmacotherapy for cardiovascular diseases. Several challenges remain for research on EAT. Further investigations are needed to determine whether reducing the mass of EAT can help to improve or eliminate atherosclerosis or prevent the development of atrial fibrillation and heart failure. The potential for pharmacological manipulation of the EAT transcriptome to restore its physiological and protective properties is a fascinating concept but is yet to be demonstrated.
References
Iacobellis, G. et al. Epicardial fat from echocardiography: a new method for visceral adipose tissue prediction. Obes. Res. 11, 304–310 (2003).
Iacobellis, G. et al. Echocardiographic epicardial adipose tissue is related to anthropometric and clinical parameters of metabolic syndrome: a new indicator of cardiovascular risk. J. Clin. Endocrinol. Metab. 388, 5163–5168 (2003).
Iacobellis, G., Corradi, D. & Sharma, A. M. Epicardial adipose tissue: anatomic, biomolecular and clinical relationships with the heart. Nat. Clin. Pract. Cardiovasc. Med. 2, 536–543 (2005).
McAninch, E. A. et al. Epicardial adipose tissue has a unique transcriptome modified in severe coronary artery disease. Obesity 23, 1267–1278 (2015).
Iacobellis, G. Local and systemic effects of the multifaceted epicardial adipose tissue depot. Nat. Rev. Endocrinol. 11, 363–371 (2015).
Iacobellis, G. Epicardial fat: a new cardiovascular therapeutic target. Curr. Opin. Pharmacol. 27, 13–18 (2016).
Malavazos, A. E., Goldberger, J. J. & Iacobellis, G. Does epicardial fat contribute to COVID-19 myocardial inflammation? Eur. Heart J. 41, 233 (2020).
Corradi, D. et al. The ventricular epicardial fat is related to the myocardial mass in normal, ischemic and hypertrophic hearts. Cardiovasc. Pathol. 13, 313–316 (2014).
Company, J. M. et al. Epicardial fat gene expression after aerobic exercise training in pigs with coronary atherosclerosis: relationship to visceral and subcutaneous fat. J. Appl. Physiol. 109, 1904–1912 (2010).
Sacks, H. S. et al. Human epicardial fat exhibits beige features. J. Clin. Endocrinol. Metab. 98, E1448–E1455 (2013).
Marchington, J. M. & Pond, C. M. Site-specific properties of pericardial and epicardial adipose tissue: the effects of insulin and high-fat feeding on lipogenesis and the incorporation of fatty acids in vitro. Int. J. Obes. 14, 1013–1022 (1990).
Vural, B. et al. Presence of fatty-acid-binding protein 4 expression in human epicardial adipose tissue in metabolic syndrome. Cardiovasc. Pathol. 17, 392–398 (2008).
Judkin, J. S., Eringa, E. & Stehouwer, C. D. A. “Vasocrine signalling” from perivascular fat: a mechanism linking insulin resistance to vascular disease. Lancet 365, 1817–1820 (2005).
Iacobellis, G. et al. Adiponectin expression in human epicardial adipose tissue in vivo is lower in patients with CAD. Cytokine 29, 251–255 (2005).
Iacobellis, G. et al. Epicardial adipose tissue and intracoronary adrenomedullin levels in coronary artery disease. Horm. Metab. Res. 41, 855–860 (2009).
Antonopoulos, A. S. et al. Mutual regulation of epicardial adipose tissue and myocardial redox state by PPAR-γ/adiponectin signaling. Circ. Res. 118, 842–855 (2016).
Sacks, H. S. et al. Uncoupling protein-1 and related mRNAs in human epicardial and other adipose tissues: epicardial fat functioning as brown fat. J. Clin. Endocrinol. Metab. 94, 3611–3615 (2009).
Fainberg, H. P. et al. Transcriptional analysis of adipose tissue during development reveals depot-specific responsiveness to maternal dietary supplementation. Sci. Rep. 8, 9628 (2018).
Sacks, H. S. et al. Depot-specific overexpression of proinflammatory, redox, endothelial cell, and angiogenic genes in epicardial fat adjacent to severe stable coronary atherosclerosis. Metab. Syndr. Relat. Disord. 9, 433–439 (2011).
Peterson, S. J., Yadav, R. & Iacobellis, G. Cardioprotective heme oxygenase 1-PGC1α signaling in epicardial fat attenuates cardiovascular risk in humans as in obese mice. Obesity 27, 1560–1561 (2019).
Iacobellis, G. & Willens, H. J. Echocardiographic epicardial fat: a review of research and clinical applications. J. Am. Soc. Echocardiogr. 22, 1311–1319 (2009).
Malavazos, A. E. et al. Relation of echocardiographic epicardial fat thickness and myocardial fat. Am. J. Cardiol. 105, 1831–1835 (2010).
Neeland, I. J., Poirier, P. & Després, J. P. Cardiovascular and metabolic heterogeneity of obesity: clinical challenges and implications for management. Circulation 137, 1391–1406 (2018).
Oikonomou, E. K. & Antoniades, C. The role of adipose tissue in cardiovascular health and disease. Nat. Rev. Cardiol. 16, 83–99 (2019).
Spearman, J. V. et al. Prognostic value of epicardial fat volume measurements by computed tomography: a systematic review of the literature. Eur. Radiol. 25, 3372–3381 (2015).
Nelson, A. J. et al. Validation of cardiovascular magnetic resonance assessment of pericardial adipose tissue volume. J. Cardiovasc. Magn. Reson. 11, 15–18 (2009).
de Vos, A. M. et al. Peri-coronary epicardial adipose tissue is related to cardiovascular risk factors and coronary artery calcification in post-menopausal women. Eur. Heart J. 29, 777–783 (2008).
Batal, O. et al. Left atrial epicardial adiposity and atrial fibrillation. Circ. Arrhythm. Electrophysiol. 3, 230–236 (2010).
Liu, Z. et al. Association of epicardial adipose tissue attenuation with coronary atherosclerosis in patients with a high risk of coronary artery disease. Atherosclerosis 284, 230–236 (2019).
Iacobellis, G. & Mahabadi, A. A. Is epicardial fat attenuation a novel marker of coronary inflammation? Atherosclerosis 284, 212–213 (2019).
Franssens, B. T., Nathoe, H. M., Leiner, T., van der Graaf, Y. & Visseren, F. L. Relation between cardiovascular disease risk factors and epicardial adipose tissue density on cardiac computed tomography in patients at high risk of cardiovascular events. Eur. J. Prev. Cardiol. 24, 660–670 (2017).
Iacobellis, G. et al. Epicardial fat inflammation in severe COVID-19. Obesity 28, 2260–2262 (2020).
Antonopoulos, A. S. et al. Detecting human coronary inflammation by imaging perivascular fat. Sci. Transl Med. 9, 398 (2017).
Oikonomou, E. K. et al. Non-invasive detection of coronary inflammation using computed tomography and prediction of residual cardiovascular risk (the CRISP CT study): a post-hoc analysis of prospective outcome data. Lancet 392, 929–939 (2018).
Attanasio, S. et al. Artificial intelligence, radiomics and other horizons in body composition assessment. Quant. Imaging Med. Surg. 10, 1650–1660 (2020).
Mazurek, T. et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation 108, 2460–2466 (2003).
Hirata, Y. et al. Coronary atherosclerosis is associated with macrophage polarization in epicardial adipose tissue. J. Am. Coll. Cardiol. 58, 248–255 (2011).
Langheim, S. et al. Increased expression and secretion of resistin in epicardial adipose tissue of patients with acute coronary syndrome. Am. J. Physiol. Heart Circ. Physiol. 298, H746–H753 (2010).
Gao, X. et al. Association of chemerin mRNA expression in human epicardial adipose tissue with coronary atherosclerosis. Cardiovasc. Diabetol. 10, 8 (2011).
Imoto-Tsubakimoto, H. et al. Serglycin is a novel adipocytokine highly expressed in epicardial adipose tissue. Biochem. Biophys. Res. Commun. 432, 105–110 (2013).
Baker, A. R. et al. Epicardial adipose tissue as a source of nuclear factor-kappaB and c-Jun N-terminal kinase mediated inflammation in patients with coronary artery disease. J. Clin. Endocrinol. Metab. 94, 261–267 (2009).
Du, Y. et al. Association between omentin-1 expression in human epicardial adipose tissue and coronary atherosclerosis. Cardiovasc. Diabetol. 15, 90 (2016).
Karastergiou, K. et al. Epicardial adipokines in obesity and coronary artery disease induce atherogenic changes in monocytes and endothelial cells. Arterioscler. Thromb. Vasc. Biol. 30, 1340–1346 (2010).
Kremen, J. et al. Increased subcutaneous and epicardial adipose tissue production of proinflammatory cytokines in cardiac surgery patients: possible role in postoperative insulin resistance. J. Clin. Endocrinol. Metab. 91, 4620–4627 (2006).
Cheng, K. H. et al. Adipocytokines and proinflammatory mediators from abdominal and epicardial adipose tissue in patients with coronary artery disease. Int. J. Obes. 32, 268–274 (2008).
Wang, J. et al. Vasodilator-stimulated phosphoprotein: regulators of adipokines resistin and phenotype conversion of epicardial adipocytes. Med. Sci. Monit. 24, 6010–6020 (2018).
Salgado-Somoza, A., Teijeira-Fernández, E., Fernández, A. L., González-Juanatey, J. R. & Eiras, S. Proteomic analysis of epicardial and subcutaneous adipose tissue reveals differences in proteins involved in oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 299, H202–H209 (2010).
Dutour, A. et al. Secretory type II phospholipase A2 is produced and secreted by epicardial adipose tissue and overexpressed in patients with CAD. J. Clin. Endocrinol. Metab. 95, 963–967 (2010).
Furuhashi, M. et al. Local production of fatty acid-binding protein 4 in epicardial/perivascular fat and macrophages is linked to coronary atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 36, 825–834 (2016).
Pezeshkian, M. & Mahtabipour, M. R. Epicardial and subcutaneous adipose tissue fatty acids profiles in diabetic and non-diabetic patients candidate for coronary artery bypass graft. Bioimpacts 3, 83–89 (2013).
Suganami, T. et al. Role of the Toll-like receptor 4/NF-κB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler. Thromb. Vasc. Biol. 27, 84–91 (2007).
Camarena, V. et al. Novel atherogenic pathways from the differential transcriptome analysis of diabetic epicardial adipose tissue. Nutr. Metab. Cardiovasc. Dis. 27, 739–750 (2017).
Dozio, E. et al. Expression of the receptor for advanced glycation end products in epicardial fat: link with tissue thickness and local insulin resistance in coronary artery disease. J. Diabetes Res. 2016, 2327341 (2016).
Vyas, V. et al. Obesity and diabetes are major risk factors for epicardial adipose tissue inflammation. JCI Insight 6, e145495 (2021).
Salgado-Somoza, A. et al. Coronary artery disease is associated with higher epicardial retinol-binding protein 4 (RBP4) and lower glucose transporter (GLUT) 4 levels in epicardial and subcutaneous adipose tissue. Clin. Endocrinol. 76, 51–58 (2012).
Błachnio-Zabielska, A. U. et al. Epicardial adipose tissue lipid content/ceramide. Lipids 47, 1131 (2012).
Mahabadi, A. A. et al. Association of epicardial adipose tissue with progression of coronary artery calcification is more pronounced in the early phase of atherosclerosis: results from the Heinz Nixdorf Recall Study. JACC Cardiovasc. Imaging 7, 909–916 (2014).
Gorter, P. M. et al. Relation of epicardial and pericoronary fat to coronary atherosclerosis and coronary artery calcium in patients undergoing coronary angiography. Am. J. Cardiol. 102, 380–385 (2008).
Djaberi, R. et al. Relation of epicardial adipose tissue to coronary atherosclerosis. Am. J. Cardiol. 102, 1602–1607 (2008).
Mancio, J. et al. Gender differences in the association of epicardial adipose tissue and coronary artery calcification: EPICHEART study: EAT and coronary calcification by gender. Int. J. Cardiol. 249, 419–425 (2017).
Iacobellis, G., Lonn, E., Lamy, A., Singh, N. & Sharma, A. M. Epicardial fat thickness and CAD correlate independently of obesity. Int. J. Cardiol. 146, 452–454 (2011).
Yerramasu, A. et al. Increased volume of epicardial fat is an independent risk factor for accelerated progression of sub-clinical coronary atherosclerosis. Atherosclerosis 220, 223–230 (2012).
Bachar, G. N., Dicker, D., Kornowski, R. & Atar, E. Epicardial adipose tissue as a predictor of coronary artery disease in asymptomatic subjects. Am. J. Cardiol. 110, 534–538 (2012).
Oka, T. et al. Association between epicardial adipose tissue volume and characteristics of non-calcified plaques assessed by coronary computed tomographic angiography. Int. J. Cardiol. 161, 45–49 (2012).
Alexopoulos, N. et al. Epicardial adipose tissue and coronary artery plaque characteristics. Atherosclerosis 210, 150–154 (2010).
Nakanishi, K. et al. Persistent epicardial adipose tissue accumulation is associated with coronary plaque vulnerability and future acute coronary syndrome in non-obese subjects with coronary artery disease. Atherosclerosis 237, 353–360 (2014).
Ito, T. Impact of epicardial fat volume on coronary artery disease in symptomatic patients with a zero calcium score. Int. J. Cardiol. 167, 2852–2858 (2013).
Alam, M. S., Green, R., de Kemp, R., Beanlands, R. S. & Chow, B. J. Epicardial adipose tissue thickness as a predictor of impaired microvascular function in patients with non-obstructive coronary artery disease. J. Nucl. Cardiol. 20, 804–812 (2013).
Wang, T. D. et al. Association of epicardial adipose tissue with coronary atherosclerosis is region-specific and independent of conventional risk factors and intra-abdominal adiposity. Atherosclerosis 213, 279–287 (2010).
Mazurek, T. et al. PET/CT evaluation of 18F-FDG uptake in pericoronary adipose tissue in patients with stable coronary artery disease: independent predictor of atherosclerotic lesions’ formation? J. Nucl. Cardiol. 24, 1075–1084 (2017).
Mahabadi, A. A. et al. Association of epicardial fat with cardiovascular risk factors and incident myocardial infarction in the general population: the Heinz Nixdorf recall study. J. Am. Coll. Cardiol. 61, 1388–1395 (2013).
Ding, J. et al. The association of pericardial fat with incident coronary heart disease: the multi-ethnic study of atherosclerosis (MESA). Am. J. Clin. Nutr. 90, 499–504 (2009).
Magnani, J. W. et al. Atrial fibrillation: current knowledge and future directions in epidemiology and genomics. Circulation 124, 1982–1993 (2011).
Pathak, R. K. et al. Aggressive risk factor reduction study for atrial fibrillation and implications for the outcome of ablation: the ARREST-AF cohort study. J. Am. Coll. Cardiol. 64, 2222–2231 (2014).
Wong, C. X., Ganesan, A. N. & Selvanayagam, J. B. Epicardial fat and atrial fibrillation: current evidence, potential mechanisms, clinical implications, and future directions. Eur. Heart J. 38, 1294–1302 (2017).
Jhuo, S. J. et al. The association of the amounts of epicardial fat, P wave duration, and PR interval in electrocardiogram. J. Electrocardiol. 51, 645–651 (2018).
Iacobellis, G. Epicardial and pericardial fat: close, but very different. Obesity 17, 625 (2009).
Thanassoulis, G. et al. Pericardial fat is associated with prevalent atrial fibrillation: the Framingham heart study. Circ. Arrhythm. Electrophysiol. 3, 345–350 (2010).
Yamaguchi, Y. et al. Adipogenesis and epicardial adipose tissue: a novel fate of the epicardium induced by mesenchymal transformation and PPARγ activation. Proc. Natl Acad. Sci. USA 112, 2070–2075 (2015).
Suffee, N. et al. Atrial natriuretic peptide regulates adipose tissue accumulation in adult atria. Proc. Natl Acad. Sci. USA 114, E771–E780 (2017).
Suffee, N. et al. Reactivation of the epicardium at the origin of myocardial fibro-fatty infiltration during the atrial cardiomyopathy. Circ. Res. 126, 1330–1342 (2020).
Gaborit, B. et al. Human epicardial adipose tissue has a specific transcriptomic signature depending on its anatomical peri-atrial, peri-ventricular, or peri-coronary location. Cardiovasc. Res. 108, 62–73 (2015).
Goldberger, J. et al. Evaluating the atrial myopathy underlying atrial fibrillation: identifying the arrhythmogenic and thrombogenic substrate. Circulation 132, 278–291 (2015).
Venteclef, N. et al. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur. Heart J. 36, 795–805 (2015).
Wang, Q. et al. Human epicardial adipose tissue cTGF expression is an independent risk factor for atrial fibrillation and highly associated with atrial fibrosis. Sci. Rep. 8, 3585 (2018).
Nalliah, C. J. et al. Epicardial adipose tissue accumulation confers atrial conduction abnormality. J. Am. Coll. Cardiol. 76, 1197 (2020).
Shaihov-Teper, O. et al. Extracellular vesicles from epicardial fat facilitate atrial fibrillation. Circulation 143, 2475–2493 (2021).
Wong, C. X. et al. Direction-dependent conduction in lone atrial fibrillation. Heart Rhythm 9, 1192–1199 (2010).
Munger, T. M. et al. Electrophysiological and hemodynamic characteristics associated with obesity in patients with atrial fibrillation. J. Am. Coll. Cardiol. 60, 851–860 (2012).
Lin, Y. K., Chen, Y. C., Chen, J. H., Chen, S. A. & Chen, Y. J. Adipocytes modulate the electrophysiology of atrial myocytes: implications in obesity-induced atrial fibrillation. Basic Res. Cardiol. 107, 293 (2012).
Po, S. S., Nakagawa, H. & Jackman, W. M. Localization of left atrial ganglionated plexi in patients with atrial fibrillation. J. Cardiovasc. Electrophysiol. 20, 1186–1189 (2009).
Balcioglu, A. S. et al. Arrhythmogenic evidence for epicardial adipose tissue: heart rate variability and turbulence are influenced by epicardial fat thickness. Pacing Clin. Electrophysiol. 38, 99–106 (2015).
Pokushalov, E. et al. Long-term suppression of atrial fibrillation by botulinum toxin injection into epicardial fat pads in patients undergoing cardiac surgery: one-year follow-up of a randomized pilot study. Circ. Arrhythm. Electrophysiol. 8, 1334–1341 (2015).
Nagashima, K. et al. Association between epicardial adipose tissue volumes on 3-dimensional reconstructed CT images and recurrence of atrial fibrillation after catheter ablation. Circulation 75, 2559–2565 (2011).
Tsao, H. M. et al. Quantitative analysis of quantity and distribution of epicardial adipose tissue surrounding the left atrium in patients with atrial fibrillation and effect of recurrence after ablation. Am. J. Cardiol. 107, 1498–1503 (2011).
Chao, T.-F. et al. Epicardial adipose tissue thickness and ablation outcome of atrial fibrillation. PLoS ONE 8, e74926 (2013).
Kocyigit, D. et al. Periatrial epicardial adipose tissue thickness is an independent predictor of atrial fibrillation recurrence after cryoballoon-based pulmonary vein isolation. J. Cardiovasc. Comput. Tomogr. 9, 295–302 (2015).
Masuda, M. et al. Abundant epicardial adipose tissue surrounding the left atrium predicts early rather than late recurrence of atrial fibrillation after catheter ablation. J. Interv. Card. Electrophysiol. 44, 31–37 (2015).
Iacobellis, G., Zaki, M. C., Garcia, D. & Willens, H. J. Epicardial fat in atrial fibrillation and heart failure. Horm. Metab. Res. 46, 587–590 (2014).
Yancy, C. W. et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J. Am. Coll. Cardiol. 70, 776–803 (2017).
van Woerden, G. et al. Epicardial fat in heart failure patients with mid-range and preserved ejection fraction. Eur. J. Heart Fail. 20, 1559–1566 (2018).
Fontes-Carvalho, R. et al. Influence of epicardial and visceral fat on left ventricular diastolic and systolic functions in patients after myocardial infarction. Am. J. Cardiol. 114, 1663–1669 (2014).
Doesch, C. et al. Epicardial adipose tissue in patients with heart failure. J. Cardiovasc. Magn. Reason. 12, 40 (2010).
Doesch, C. et al. Epicardial adipose tissue assessed by cardiac magnetic resonance imaging in patients with heart failure due to dilated cardiomyopathy. Obesity 21, E253–E261 (2013).
Khawaja, T. et al. Epicardial fat volume in patients with left ventricular systolic dysfunction. Am. J. Cardiol. 108, 397–401 (2011).
Pocock, S. J. et al. Weight loss and mortality risk in patients with chronic heart failure in the candesartan in heart failure: assessment of reduction in mortality and morbidity (CHARM) programme. Eur. Heart J. 29, 2641–2650 (2008).
Zhao, L. et al. Proteomics of epicardial adipose tissue in patients with heart failure. J. Cell. Mol. Med. 4, 511–520 (2020).
Agra, R. M. et al. Adiponectin and p53 mRNA in epicardial and subcutaneous fat from heart failure patients. Eur. J. Clin. Invest. 44, 29–37 (2014).
Krstic, J., Reinisch, I., Schupp, M., Schulz, T. J. & Prokesch, A. p53 functions in adipose tissue metabolism and homeostasis. Int. J. Mol. Sci. 19, 2622 (2018).
Wu, C. K. et al. Evolutional change in epicardial fat and its correlation with myocardial diffuse fibrosis in heart failure patients. J. Clin. Lipidol. 11, 1421–1431 (2017).
Kankaanpää, M. et al. Myocardial triglyceride content and epicardial fat mass in human obesity: relationship to left ventricular function and serum free fatty acid levels. J. Clin. Endocrinol. Metab. 91, 4689–4695 (2006).
Wu, C. K. et al. Myocardial adipose deposition and the development of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 22, 445–445 (2020).
White, A. Cardiac sympathetic denervation in the failing heart: a role for epicardial adipose tissue. Circ. Res. 118, 1189–1191 (2016).
Lymperopoulos, A., Rengo, G., Funakoshi, H., Eckhart, A. D. & Koch, W. J. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat. Med. 13, 315–323 (2007).
Lymperopoulos, A. et al. Reduction of sympathetic activity via adrenal-targeted GRK2 gene deletion attenuates heart failure progression and improves cardiac function after myocardial infarction. J. Biol. Chem. 285, 16378–16386 (2010).
Parisi, V. et al. Increased epicardial adipose tissue volume correlates with cardiac sympathetic denervation in patients with heart failure. Circ. Res. 118, 1244–1253 (2016).
Agra-Bermejo, R. M. et al. CD5L, macrophage apoptosis inhibitor, was identified in epicardial fat-secretome and regulated by isoproterenol from patients with heart failure. Front. Physiol. 11, 620 (2020).
Pabon, M. A., Manocha, K., Cheung, J. W. & Lo, J. C. Linking arrhythmias and adipocytes: insights, mechanisms, and future directions. Front. Physiol. 9, 1752 (2018).
Burgeiro, A. et al. Glucose uptake and lipid metabolism are impaired in epicardial adipose tissue from heart failure patients with or without diabetes. Am. J. Physiol. Endocrinol. Metab. 310, E550–E564 (2016).
Marso, S. P. et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 375, 311–322 (2016).
Marso, S. P. et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 375, 1834–1844 (2016).
Gerstein, H. C. et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet 394, 121–130 (2019).
Zinman, B. et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 373, 2117–2128 (2015).
Wiviott, S. D. et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 380, 347–357 (2019).
Neeland, I. J. et al. Effects of liraglutide on visceral and ectopic fat in adults with overweight and obesity at high cardiovascular risk: a randomised, double-blind, placebo-controlled, clinical trial. Lancet Diabetes Endocrinol. 9, 595–605 (2021).
Iacobellis, G., Mohsenim, M., Bianco, S. & Banga, P. K. Liraglutide causes large and rapid epicardial fat reduction. Obesity 25, 311–316 (2017).
Iacobellis, G. & Villasante Fricke, A. C. Effects of semaglutide versus dulaglutide on epicardial fat thickness in subjects with type 2 diabetes and obesity. J. Endocr. Soc. 4, bvz042 (2020).
Morano, S. et al. Short-term effects of glucagon-like peptide 1 (GLP-1) receptor agonists on fat distribution in patients with type 2 diabetes mellitus: an ultrasonography study. Acta Diabetol. 52, 727–732 (2015).
Li, Y. et al. Effect of liraglutide on epicardial adipose tissue thickness with echocardiography in patients with obese type 2 diabetes mellitus. Int. J. Diabetes Dev. Ctries 40, 500–506 (2020).
Dutour, A. et al. Exenatide decreases liver fat content and epicardial adipose tissue in patients with obesity and type 2 diabetes: a prospective randomized clinical trial using magnetic resonance imaging and spectroscopy. Diabetes Obes. Metab. 18, 882–891 (2016).
Iacobellis, G., Camarena, V., Sant, D. W. & Wang, G. Human epicardial fat expresses glucagon-like peptide 1 and 2 receptors genes. Horm. Metab. Res. 49, 625–630 (2017).
Dozio, E. et al. Epicardial adipose tissue GLP-1 receptor is associated with genes involved in fatty acid oxidation and white-to-brown fat differentiation: a target to modulate cardiovascular risk? Int. J. Cardiol. 292, 218–224 (2019).
Couselo-Seijas, M. et al. Higher ACE2 expression levels in epicardial cells than subcutaneous stromal cells from patients with cardiovascular disease: diabetes and obesity as possible enhancer. Eur. J. Clin. Invest. 51, e13463 (2020).
Beiroa, D. et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes 63, 3346–3358 (2014).
Baggio, L. L. et al. GLP-1 receptor expression within the human heart. Endocrinology 159, 1570–1584 (2018).
Iacobellis, G. & Gra-Menendez, S. Effects of dapagliflozin on epicardial fat thickness in patients with type 2 diabetes and obesity. Obesity 28, 1068–1074 (2020).
Requena-Ibanez, J. A. et al. Mechanistic insights of empagliflozin in nondiabetic patients with HFrEF: from the EMPA-TROPISM Study. JACC Heart Fail. 9, 578–589 (2021).
Sato, T. et al. The effect of dapagliflozin treatment on epicardial adipose tissue volume. Cardiovasc. Diabetol. 17, 6 (2018).
Yagi, S. et al. Canagliflozin reduces epicardial fat in patients with type 2 diabetes mellitus. Diabetol. Metab. Syndr. 9, 78 (2017).
Fukuda, T. et al. Ipragliflozin reduces epicardial fat accumulation in nonobese type 2 diabetic patients with visceral obesity: a pilot study. Diabetes Ther. 8, 851–861 (2017).
Bouchi, R. et al. Luseogliflozin reduces epicardial fat accumulation in patients with type 2 diabetes: a pilot study. Cardiovasc. Diabetol. 16, 32 (2017).
Díaz-Rodríguez, E. et al. Effects of dapagliflozin on human epicardial adipose tissue: modulation of insulin resistance, inflammatory chemokine production, and differentiation ability. Cardiovasc. Res. 114, 336–346 (2018).
Ferrannini, E., Mark, M. & Mayoux, E. CV protection in the EMPA-REG OUTCOME trial: a “thrifty substrate” hypothesis. Diabetes Care 39, 1108–1114 (2016).
Lopaschuk, G. D. et al. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 90, 207–258 (2010).
Verma, S. et al. Empagliflozin increases cardiac energy production in diabetes: novel translational insights into the heart failure benefits of SGLT2 inhibitors. JACC Basic Transl Sci. 3, 575–587 (2018).
Fosshaug, L. E. et al. Altered levels of fatty acids and inflammatory and metabolic mediators in epicardial adipose tissue in patients with systolic heart failure. J. Card. Fail. 21, 916–923 (2015).
Sorice, G. P. et al. Effect of dapagliflozin on myocardial insulin sensitivity and perfusion: rationale and design of the DAPAHEART trial. Diabetes Ther. 12, 2101–2113 (2021).
Park, J. H. et al. Effects of statins on the epicardial fat thickness in patients with coronary artery stenosis underwent percutaneous coronary intervention: comparison of, atorvastatin with simvastatin/ezetimibe. J. Cardiovasc. Ultrasound 18, 121–126 (2010).
Alexopoulos, N. et al. Effect of intensive versus moderate lipid-lowering therapy on epicardial adipose tissue in hyperlipidemic post-menopausal women: a substudy of the BELLES trial (Beyond Endorsed Lipid Lowering with EBT Scanning). J. Am. Coll. Cardiol. 61, 1956–1961 (2013).
Grosso, A. F. et al. Synergistic anti-inflammatory effect: simvastatin and pioglitazone reduce inflammatory markers of plasma and epicardial adipose tissue of coronary patients with metabolic syndrome. Diabetol. Metab. Syndr. 6, 47 (2014).
Zheng, Y. Y., Ma, Y. T., Zhang, J. Y. & Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 17, 259–260 (2020).
Lan, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020).
Patel, V. B. et al. ACE2 deficiency worsens epicardial adipose tissue inflammation and cardiac dysfunction in response to diet-induced obesity. Diabetes 65, 85–95 (2016).
Vaduganathan, M. et al. Renin-angiotensin-aldosterone system inhibitors in patients with COVID-19. N. Engl. J. Med. 382, 1653–1659 (2020).
Deng, M. et al. Obesity as a potential predictor of disease severity in young COVID-19 patients: a retrospective study. Obesity 28, 1815–1825 (2020).
Grodecki, K. et al. Epicardial adipose tissue is associated with extent of pneumonia and adverse outcomes in patients with COVID-19. Metabolism 115, 154436 (2021).
Iacobellis, G. et al. Epicardial fat inflammation response to COVID-19 therapies. Obesity 29, 1427–1433 (2021).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Peer review
Peer review information
Nature Reviews Cardiology thanks Charalambos Antoniades, Sonia Eiras and Stephane Hatem for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Review criteria
A search for original articles published between 1984 and 2021 with a focus on the past 10 years was performed in MEDLINE and PubMed. The search terms used were “epicardial fat” and “epicardial adipose tissue”. A total of 1,903 articles were found. The articles identified for this Review were restricted to English language, full-text papers. The reference lists of identified articles were searched for additional relevant papers.
Glossary
- Epicardial adipose tissue
-
(EAT). The fat depot located between the myocardium and the visceral layer of the epicardium.
- Glucagon-like peptide 1 receptor (GLP1R) agonists
-
Medications indicated for the treatment of type 2 diabetes mellitus, obesity or both, with pleiotropic effects.
- Sodium–glucose co-transporter 2 (SGLT2) inhibitors
-
Antidiabetic agents that reduce hyperglycaemia in patients with type 2 diabetes mellitus by increasing urinary glucose excretion.
- Pericardial adipose tissue
-
(PAT). The fat depot located between the visceral and parietal layers of the epicardium.
- Brown fat
-
Brown fat generates heat and non-shivering thermogenesis in response to cold and activation of the autonomic nervous system but is lost before adulthood in humans.
- Beige fat
-
Beige fat originates from white adipose tissue but has anatomical features akin to both white and brown fat and can function like brown fat under certain circumstances.
- Ectopic fat
-
The accumulation or infiltration of lipids in non-adipose tissues such as the heart, liver and muscles.
- Cardiac multidetector CT
-
Multiple-row detector CT that provides greater detail and additional views of the heart and coronary arteries than standard CT.
- CT attenuation
-
A measure of tissue density proportionate to the attenuation of X-rays that pass through the tissue; EAT attenuation ranges between –45 and –195 Housefield units.
- CT fat attenuation index
-
(FAI). A novel imaging metric that describes adipocyte lipid content and size; FAI correlates with perivascular adipose tissue inflammation.
- Coronary artery calcium (CAC) score
-
A CT-based score that measures calcium content in the coronary arteries.
Rights and permissions
About this article

Cite this article
Iacobellis, G. Epicardial adipose tissue in contemporary cardiology. Nat Rev Cardiol 19, 593–606 (2022). https://doi.org/10.1038/s41569-022-00679-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-022-00679-9
This article is cited by
-
AI-derived epicardial fat measurements improve cardiovascular risk prediction from myocardial perfusion imaging
npj Digital Medicine (2024)
-
Gut microbiota in overweight and obesity: crosstalk with adipose tissue
Nature Reviews Gastroenterology & Hepatology (2024)
-
Gastric Submucosal Fat Accumulation Is Associated with Insulin Resistance in Patients with Obesity
Obesity Surgery (2024)
-
Endothelial dysfunction and risk factors for atherosclerosis in psoriatic arthritis: overview and comparison with rheumatoid arthritis
Rheumatology International (2024)
-
Body Fat Depletion: the Yin Paradigm for Treating Type 2 Diabetes
Current Atherosclerosis Reports (2024)
