
Abstract
Atherosclerosis is a chronic inflammatory disease of the arterial wall, characterized by the formation of plaques containing lipid, connective tissue and immune cells in the intima of large and medium-sized arteries. Over the past three decades, a substantial reduction in cardiovascular mortality has been achieved largely through LDL-cholesterol-lowering regimes and therapies targeting other traditional risk factors for cardiovascular disease, such as hypertension, smoking, diabetes mellitus and obesity. However, the overall benefits of targeting these risk factors have stagnated, and a huge global burden of cardiovascular disease remains. The indispensable role of immunological components in the establishment and chronicity of atherosclerosis has come to the forefront as a clinical target, with proof-of-principle studies demonstrating the benefit and challenges of targeting inflammation and the immune system in cardiovascular disease. In this Review, we provide an overview of the role of the immune system in atherosclerosis by discussing findings from preclinical research and clinical trials. We also identify important challenges that need to be addressed to advance the field and for successful clinical translation, including patient selection, identification of responders and non-responders to immunotherapies, implementation of patient immunophenotyping and potential surrogate end points for vascular inflammation. Finally, we provide strategic guidance for the translation of novel targets of immunotherapy into improvements in patient outcomes.
Key points
-
Inflammation is an important component of the pathophysiology of cardiovascular disease; an imbalance between pro-inflammatory and anti-inflammatory processes drives chronic inflammation and the formation of atherosclerotic plaques in the vessel wall.
-
Clinical trials assessing canakinumab and colchicine therapies in atherosclerotic cardiovascular disease have provided proof-of-principle of the benefits associated with therapeutic targeting of the immune system in atherosclerosis.
-
The immunosuppressive adverse effects associated with the systemic use of anti-inflammatory drugs can be minimized through targeted delivery of anti-inflammatory drugs to the atherosclerotic plaque, defining the window of opportunity for treatment and identifying more specific targets for cardiovascular inflammation.
-
Implementing immunophenotyping in clinical trials in patients with atherosclerotic cardiovascular disease will allow the identification of immune signatures and the selection of patients with the highest probability of deriving benefit from a specific therapy.
-
Clinical stratification via novel risk factors and discovery of new surrogate markers of vascular inflammation are crucial for identifying new immunotherapeutic targets and their successful translation into the clinic.
Similar content being viewed by others
Introduction
Atherosclerosis, the major cause of cardiovascular disease (CVD), is a chronic inflammatory disease triggered by the accumulation of cholesterol-containing LDL particles in the arterial wall1. The gold standard of treatment for atherosclerosis is the prevention of cardiovascular events by targeting modifiable risk factors and the re-establishment of arterial flow by percutaneous or surgical procedures2,3. However, the therapeutic benefit of these strategies on cardiovascular outcomes has stagnated and a huge global burden of CVD remains4.
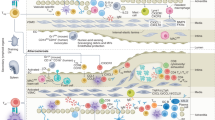
Evidence for the role of inflammation in atherosclerosis has accumulated over the past 35 years (Fig. 1). Attilio Maseri (1935–2021) was one of the first investigators to foresee the importance of inflammation as a component of the pathogenesis of acute coronary syndromes5,6. The arterial wall is populated by various immune cells, both in healthy individuals and in patients with disease7,8. The innate immune system is the first line of defence against invading pathogens and the innate immune response is usually initiated by pattern recognition receptors, including Toll-like receptors (TLRs)9,10. The innate immune response induces the activation of antigen-presenting cells such as macrophages and dendritic cells that mediate antigen presentation, co-stimulation and cytokine production in the immune synapse to trigger the adaptive immune response. The adaptive immune response involves B cells and T cells and is slower but more specific and long-lived than the innate immune response. Athero-inflammation involves the activation of both innate and adaptive immune responses, with both inherently linked8,11 (Fig. 2). Immune cells in the arteries are activated owing to persistent inflammatory stimuli or a failure in the resolution of inflammation, leading to chronic inflammation, a hallmark of CVD12. To understand atherogenesis, we must consider the interplay between cellular immunity and lipid retention13 and the complex crosstalk between and within immune and non-immune cells, as well as the advantages and disadvantages of the experimental models used in this research field (Box 1).

The timeline shows the main milestones in the past four decades of research into the role of inflammation in atherosclerosis. In the 1980s, the introduction of immunohistochemical techniques to study atherosclerotic plaques provided evidence of HLA-DR expression in human atherosclerotic plaques, followed by identification of monocytes, macrophages and T cells in the plaque29,44,263,264,265,266,267,268,269. In the 1990s, studies showed the presence of pro-inflammatory cytokines, such as tumour necrosis factor (TNF), in atherosclerotic plaques270,271,272,273,274, and the association between high plasma C-reactive protein (CRP) levels and coronary artery disease (CAD)5,275. During this decade, the first mouse models of hypercholesterolaemia with an inflammatory gene knockout were developed274,276,277 and titres of antibodies against oxidized LDL (oxLDL) in the serum were shown to predict cardiovascular disease outcomes278. In the 2000s, studies demonstrated the association between increased levels of inflammatory markers and increased risk of cardiovascular events279,280. An increased risk of cardiovascular disease was shown in patients with inflammatory diseases281,282,283, and several studies demonstrated the association between elevated levels of CRP, IL-6 and TNF in the plasma and worse clinical outcomes in patients with cardiovascular disease115,127,284,285. This finding led to the introduction of inflammation as a therapeutic target in cardiovascular disease286. In the late 2010s, studies showed that immune checkpoint inhibitor treatment increased the risk of cardiovascular disease in patients with cancer287,288. In the past decade, clinical trials investigated whether targeting inflammation in cardiovascular disease is beneficial32,34,135. Numerous studies also demonstrated the involvement of the bone marrow in atherosclerosis172,177,178 and performed single-cell analysis of plaque immune cells19,25. Preclinical discoveries are shown in blue boxes and clinical discoveries in red boxes. MI, myocardial infarction; VSMC, vascular smooth muscle cell.

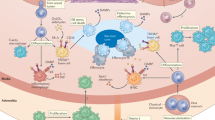
In medium and large arteries, haemodynamic forces create areas of low shear stress that are often predictors of atherosclerotic plaque location. As the atherosclerotic plaque begins to form, circulating apolipoprotein B (ApoB)-containing lipoproteins (ApoB-LP) and ApoB peptides enter the subendothelial space, where they can be modified and recognized by innate immune cells as danger signals. These danger signals activate Toll-like receptor (TLR) signalling and the inflammasome in innate immune cells, eliciting responses that drive inflammation, including production and secretion of cytokines, release of neutrophil extracellular traps (NETs), upregulation of co-stimulatory molecules and promotion of monocyte recruitment to the plaque289. Macrophages derived from monocyte differentiation, local proliferation or from transdifferentiation of vascular smooth muscle cells (VSMCs) take up lipoproteins present in the plaque and become lipid-laden foam cells that lay the foundation for the formation of the plaque necrotic core. At the immune synapse, antigen-presenting cells (APCs), including macrophages, dendritic cells and B cells, present lipid antigens to invariant natural killer T (iNKT) cells and peptide antigens to T cells, the latter engaging adaptive T cell and B cell responses. Antigen presentation occurs in the plaque and in secondary lymph organs, such as the lymph node8. Together, all these processes contribute to endothelial dysfunction, leading to further aggravation of inflammation through continued monocyte recruitment, increased uptake of lipoproteins adding to the plaque lipid burden, VSMC activation and proliferation, and fibroblast migration contributing to fibrous cap formation. ECM, extracellular matrix; IFN, interferon; MHC, major histocompatibility complex; ROS, reactive oxygen species; TCR, T cell receptor; TH1, T helper 1; TNF, tumour necrosis factor; Treg cell, regulatory T cell.
A unique aspect that sets aside atherogenesis from other chronic inflammatory diseases is the crucial role of lipid particles in the induction of atherogenesis. Modified lipoproteins, such as oxidized LDL (oxLDL), trigger the immune response through a unique property, whereby these particles can act as both antigens activating the adaptive immune response8,14 and adjuvant molecular patterns activating the innate immune response15,16. In advanced atherosclerosis, complex chronic inflammatory processes result in the generation of a plaque with a thin fibrous cap and a large necrotic core, or in plaque erosion or other plaque morphologies associated with clinical vulnerability to rupture, which lead to ischaemic events17. The complexity of inflammation in atherosclerosis has been emphasized by single-cell studies in humans and mice showing the high heterogeneity of vascular leukocytes in atherosclerotic lesions18,19,20,21,22,23,24,25,26,27. This heterogeneity underscores the importance of targeting specific cell subsets to inhibit atherosclerosis progression while maintaining tissue homeostasis. Superimposing the single-cell transcriptional landscape of leukocytes from mouse and human atherosclerotic plaques will help identify the different pathways, genes or cells that can be used in animal models to study human disease. Moreover, emerging evidence now shows that atherogenesis is a multiorgan process with contributions from organs such as the bone marrow and spleen28,29. In particular, the presence of clonal haematopoiesis of indeterminate potential (CHIP), an age-related process in which certain somatic mutations in bone marrow progenitor cells confer a competitive advantage leading to the expansion of specific cell clones, has been proposed as a risk factor for CVD30,31.
The first proof of the benefits of targeting inflammation in CVD in humans came from the 2017 CANTOS trial32, which showed improved clinical outcomes in patients with a history of myocardial infarction (MI) who received treatment with antibodies against IL-1β (canakinumab) compared with those who received placebo (Table 1). This finding was quickly followed by evidence from two clinical trials published in 2019 and 2020 showing that the anti-inflammatory effects of colchicine therapy reduced the risk of cardiovascular events in patients with recent MI33 or coronary artery disease (CAD)34. Evidence for the role of inflammation in CVD has also been described in other disease settings. Patients with chronic inflammatory diseases such as lupus or rheumatoid arthritis (RA) have an increased risk of CVD (tenfold and twofold, respectively) compared with healthy controls, and this risk significantly correlates with the magnitude of systemic inflammation35. Moreover, checkpoint inhibitor therapies used for several cancer types to improve tumour surveillance by the immune system are associated with an increased risk of CVD, adding to the challenges in the cardio-oncology field36,37. Together, these studies highlight immunotherapeutics as the next step in CVD therapy that will provide an opportunity to surpass the ceiling reached with the current management of classic risk factors for CVD to address the residual cardiovascular risk38. At present, the challenge lies in identifying crucial effectors of atherosclerosis-specific inflammation among the plethora of inflammatory mediators while sparing the host defence.
In this Review, we discuss the therapeutic potential of targeting the immune system in atherosclerosis. First, we provide an overview of immune cells involved in CVD. Next, we summarize the published and ongoing clinical trials targeting the immune system in atherosclerosis and identify important challenges that need to be addressed to advance the translation of novel immunotherapeutics into the clinic. Finally, we highlight the new therapeutic targets emerging from preclinical studies with the biggest potential for translational pay-off in the medium term.
Immune cells involved in atherosclerosis
In this section, we summarize the functional diversity of innate and adaptive immune cells in atherosclerosis and refer to previous reviews for further in-depth discussion. The role of platelets and other non-immune cells in inflammation have been previously reviewed39,40,41.
Monocytes
Monocytes are present in the blood, bone marrow and spleen during homeostasis. Monocytes can be classified into two main populations: classical monocytes (Ly6Chigh in mice and CD14+CD16− in humans) and non-classical monocytes (Ly6Clow in mice and CD14lowCD16+ in humans). In atherosclerosis, classical monocytes are recruited to atherosclerotic plaques after engagement of the chemokine receptors CCR2, CCR5 and CX3CR1 (refs11,42). In the plaque, monocytes differentiate into dendritic cells and macrophages that show high functional and phenotypic heterogeneity43. In both mice42,44 and humans45, an increase in the blood monocyte pool is associated with increased severity of atherosclerosis. Preclinical studies in mice have demonstrated that splenic Ly6Chigh monocytes contribute to both the growing atheroma and plaque instability29,46. However, monocyte recruitment also has an important role in atherosclerosis regression47, and ‘patrolling’ Ly6Clow monocytes, which are derived from Ly6Chigh monocytes, are important for endothelial cell maintenance48. Hypercholesterolaemia, stress, inflammation and other risk factors for atherosclerosis can induce emergency haematopoiesis, including extramedullary haematopoiesis in the spleen29, and contribute to disease progression by skewing haematopoietic stem cells in the bone marrow towards monopoiesis29,44,49.
Macrophages
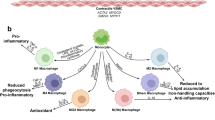
Two distinct resident macrophage populations are found in mouse arteries, one in the intima and the other in the adventitia50. Both macrophage populations originate from embryonic precursors and their survival depends on the presence of colony-stimulating factor 1. Resident adventitial macrophages are replenished by bone-marrow-derived monocytes in the period immediately after birth and are maintained by local proliferation in adulthood51. In atherogenesis, monocytes reconstitute the population of resident macrophages in the arterial intima during early stages of atherosclerosis50, whereas local proliferation of lesional macrophages contributes to macrophage accumulation in advanced lesions52. In both health and disease, adventitial macrophages expressing lymphatic vessel endothelial hyaluronic acid receptor 1 (LYVE1) prevent unfavourable arterial remodelling, largely through the regulation of collagen production in medial vascular smooth muscle cells (VSMCs)53. Arterial intima-resident macrophages have a pro-atherogenic function, and ablation of these macrophages prevents lesion formation50. A subset of LYVE1+ vascular macrophages expressing the innate immune receptor C-type lectin CLEC4A2 has anti-atherogenic functions and the ablation of this macrophage population increases lesion formation54.
Arterial macrophages have distinct functional and ontogenetic signatures and this plasticity reflects the heterogeneous environment of atherosclerotic plaques, which is increasingly being appreciated. Genetic lineage tracing and monocyte fate mapping studies have started exploring the contributions of monocytes to specific macrophage subpopulations in atherosclerosis20,47 and have helped to understand how local progenitor cells and proliferation of resident macrophages contribute to plaque progression50,52,55. Three main macrophage populations with different inflammatory properties have been identified in single-cell studies of human19 and mouse18,27 atherosclerotic plaques, suggesting that macrophage heterogeneity in the plaques cannot be explained simply by the M1–M2 macrophage polarization paradigm56. Strikingly, a pro-inflammatory macrophage population found in mice and humans expresses high levels of IL-1β18,19, a well-recognized immune target in atherosclerosis, further highlighting the relevance of this cytokine for atherosclerosis progression. Another population of the identified macrophage subsets has a more resident-like phenotype and is enriched in transcripts of proteins involved in antigen presentation and endocytosis18,25.
Foam cells are a hallmark of atherosclerosis. These cells are derived from macrophages, dendritic cells and VSMCs57. Foam cells drive necrotic core formation through uptake of intraplaque lipids, which leads to increased endoplasmic reticulum stress and cell death57. A single-cell study of mouse atherosclerotic lesions showed that plaque Trem2high macrophages, a subset that has also been identified in adipose tissue, express genes associated with lipid handling and have a profile consistent with a foamy macrophage phenotype27. TREM2high macrophages in human and mouse atherosclerotic lesions do not express genes encoding inflammatory factors, suggesting that these subsets have a homeostatic lipid-handling role in the plaques18,22,25,58. The profile of this macrophage subset is consistent with evidence showing that intracellular accumulation of desmosterol, a precursor in cholesterol biosynthesis, maintains macrophage homeostasis through the activation of transcription of liver X receptor target genes and the suppression of inflammation18,22,25,58. This discovery draws important parallels between the pathophysiology of CVD and obesity, highlighting a common blueprint between the two most prevalent metabolic diseases at present59,60. At the same time, these findings call into question the concept of lipid-driven inflammation. Further studies are warranted to reconcile inflammatory and lipid drivers of the disease. Another aspect of plaque macrophage biology to consider is the role of these cells in plaque rupture and thrombosis through the production of matrix metalloproteinases and tissue factor61, and the coordination of intraplaque efferocytosis, a crucial mechanism for resolving inflammation in atherosclerosis62 (Box 2).
Dendritic cells
Dendritic cells are another crucial cell type driving atherosclerotic plaque inflammation that bridges the innate and adaptive immune responses. Dendritic cells can be classified into three main subsets: plasmacytoid dendritic cells, type 1 conventional dendritic cells (cDC1s) and type 2 conventional dendritic cells (cDC2s). Plasmacytoid dendritic cells are generally located in blood and lymphoid tissues. After encountering pathogens, these cells produce large amounts of type I interferon (IFN). By contrast, conventional dendritic cells are found in lymphoid and non-lymphoid sites. cDC1s are involved in cross-presentation of antigens and drive cytotoxic immune responses, whereas cDC2s are involved in T cell priming63.
In humans, plaque dendritic cell numbers positively correlate with plaque vulnerability64. Dendritic cells have been found to have both pro-atherogenic and anti-atherogenic functions in mouse models, as reviewed previously8. Dendritic cells elicit an adaptive immune response that encompasses both T cells and B cells8. During atherosclerosis regression in mice, dendritic cells can leave the lesions and migrate to the lymphatic tissue in a process mediated by the chemokine ligands CCL19 and CCL21 and their receptor CCR7 on the surface of dendritic cells65. Dendritic cells expressing CCL17 have a pro-atherogenic role in mice66. CD103+ cDC1s can promote atheroprotective regulatory T (Treg) cell responses67. Loss of myeloid differentiation factor 88 (MyD88) signalling in CD11c+ dendritic cells leads to loss of Treg cells and increased atherogenesis in mice68. By contrast, plasmacytoid dendritic cells have been reported to have both pro-atherogenic and anti-atherogenic roles in mice, possibly owing to subtle cellular heterogeneity in this subset69,70.
Neutrophils
Neutrophils are involved in all stages of atherosclerosis71. In mice, neutrophil depletion reduces atherosclerosis, whereas increased levels of circulating neutrophils exacerbate plaque formation, suggesting a role of this cell type in lesion development72. Neutrophils promote vascular inflammation through the secretion of reactive oxygen species, which leads to increased permeability of the endothelial cell barrier73. Neutrophils attract monocytes via secretion of chemotactic molecules and can activate macrophages via extrusion of their nuclear material as neutrophil extracellular traps (NETs)74. NETs contain histone H4, which binds to VSMCs and induces cell lysis, resulting in plaque destabilization75. In addition, NETs induce plaque erosion and platelet aggregation, leading to thrombosis76. Overall, neutrophils have a pro-atherogenic role. However, during thrombotic events, neutrophils have reparative functions through the promotion of endothelial repair and angiogenesis77.
T cells
T cells are important for atherosclerosis initiation and progression, as reviewed previously78,79. A mass cytometry study revealed that T cells outnumber macrophages in human carotid artery plaques25, in contrast to plaques in mice, in which the overall proportion of T cells is lower24. T cells in human atherosclerotic plaques show more activation-related and exhaustion-related gene expression than peripheral blood T cells. High expression of the inhibitory molecule PD1 as a consequence of chronic antigen stimulation can result in inefficient T cell effector function and dysregulation of the immune response within the plaque19,25. Once activated, T cells directly mediate effector functions in the arterial wall or help B cells produce antibodies. CD4+ T cells are the most abundant T cells in mouse atherosclerotic plaques, and are polarized predominantly towards a pro-inflammatory phenotype (T helper 1 (TH1) cells)79. CD4+ T cells have been shown to both protect against and promote atherogenesis depending on the subset involved. TH1 cells have been consistently shown to have pro-atherogenic roles, whereas Treg cells are thought to have atheroprotective roles via IL-10 and TGFβ secretion78. The role of TH2 cells and TH17 cells in atherosclerosis is controversial78. Phenotyping of CD4+ T cells in a mouse model of atherosclerosis with the use of single-cell RNA sequencing revealed a CD4+ T cell population80 that shared transcriptional similarities with apolipoprotein B (ApoB)-reactive CD4+ T cells81. During atherosclerosis progression, ApoB-reactive CD4+ T cells undergo a transition from a Treg cell to a pro-inflammatory phenotype, which might contribute to further disease progression81.
CD8+ T cells in atherosclerotic lesions have also been found to have dual functions, with pro-atherogenic effects mediated by IFNγ production and macrophage activation, and atheroprotective effects via B cell modulation78. CD8+ T cells in mice have been identified as drivers of plaque inflammation and apoptosis, promoting unstable plaque phenotypes and plaque erosion82,83. CD8+ T cells outnumber CD4+ T cells in advanced human atherosclerotic plaques25,82, and an increase in CD8+ T cell numbers in blood is associated with the presence of CAD84,85.
Invariant natural killer T (iNKT) cells are a distinct subset of T cells that express unique invariant T cell receptors and natural killer cell surface molecules, such as CD161 (also known as NK1.1 in mice) and killer cell immunoglobulin-like receptors (analogous to the Ly49 family in mice)86. Given the central role of lipids in atherosclerosis, iNKT cells are a relevant cell type because they respond to lipid antigens presented by CD1d on antigen-presenting cells. In mice, iNKT cells are considered to be pro-atherogenic owing to their production of pro-inflammatory cytokines such as IFNγ86. In humans, rupture-prone plaques have higher numbers of iNKT cells than stable plaques87 but the exact mechanism underlying this observation is unknown.
B cells
B cell subpopulations make different contributions to atherogenesis88. B cells are central to humoral immunity and mediate the production of antibodies against oxidation-specific epitopes to help dampen inflammation. B cells are classified into two lineages: B1 cells, which are mainly produced in the fetal liver, and B2 cells, which originate in the bone marrow. B1 cells are further subdivided into B1a and B1b subsets. B2 cells can differentiate into transitional (T1 and T2 marginal zone progenitor) B cells, marginal zone B cells, follicular B cells and antibody-secreting plasma cells88. In atherosclerosis, B cells are not always found in the plaque and are more commonly localized in the adventitia or in node-like structures, referred to as tertiary lymphoid organs, that form in the adventitia as a result of chronic inflammation79. B1 cells have been described as atheroprotective in mice owing to the production of IgM antibodies that block the uptake of oxLDL by macrophages in lesions16,89. By contrast, B2 cells have been shown overall to be pro-atherogenic, through antibody responses formed via germinal centre B cell reactions that further drive adaptive immunity88. In mice fed a high-cholesterol diet, subsets of B2 cells with atheroprotective functions arise in secondary lymphoid organs, such as the lymph node (T2 marginal zone progenitor B cells)90 and the spleen (marginal zone B cells)91. These subsets act either through PDL1-mediated suppression of T follicular helper cells91 or via IL-10, although the role of IL-10 varies in different mouse models (IL-10 was shown to have a role in Apoe−/− mice90 but not in Ldlr−/− chimeric mice92) and is dependent on the microbiome93 and the radioresistance of B cell subsets94.
Clinical trials of immunotherapies in CVD
Over the past 5 years, promising results from clinical trials targeting inflammation in CVD have been reported. In this section, we summarize the positive phase III trials, promising phase II studies, ongoing trials and trials with neutral results, and the lessons learnt from these studies (Fig. 3).

a–d | Immunotherapies for the treatment of atherosclerosis that showed benefit (green), no benefit (red) or potential benefit (yellow) in reducing inflammation or cardiovascular events in clinical trials or currently being tested in ongoing clinical trials (blue) are shown. Therapeutics targeting innate immunity include IL-1 inhibitors, IL-6 inhibitors, tumour necrosis factor (TNF) blockers and p38 inhibitors (panel a). Therapeutics targeting adaptive immunity include local proliferation inhibitors in drug-eluting stents and low-dose IL-2 targeting regulatory T (Treg) cells (panel b). Therapeutics targeting lipoproteins to reduce inflammation include antibodies against oxidized LDL (oxLDL), lipoprotein-associated phospholipase A2 (Lp-PLA2), secretory phospholipase A2 (sPLA2) and lectin-like oxidized LDL receptor 1 (LOX1) (panel c). Therapeutics with broad immunosuppressive effects include colchicine, low-dose methotrexate, glucocorticoids and hydroxychloroquine (panel d). See Tables 1,2 and 3 and Supplementary Table 1 for further details. e–g | Overview of therapeutics in preclinical development targeting innate immunity (panel e), co-stimulation pathways (panel f) and B cell and T cell regulation (panel g). APC, antigen-presenting cell; ApoB, apolipoprotein B; BAFF, B cell activating factor; BCMA, B cell maturation antigen; BTLA, B and T lymphocyte attenuator; CCR, C-C chemokine receptor; CD30L, CD30 ligand; CD40L, CD40 ligand; CTLA4, cytotoxic T lymphocyte antigen 4; CVD, cardiovascular disease; GLUT1, glucose transporter 1; HDAC, histone deacetylase; HSPC, haematopoietic stem and progenitor cell; IRF5, interferon regulatory factor 5; OX40L, OX40 ligand; PPARγ, peroxisome proliferator-activated receptor-γ; siRNA, small interfering RNA; SPM, specialized pro-resolving mediators; TLR, Toll-like receptor; TRAF6, tumour necrosis factor receptor-associated factor 6.
Phase III clinical trials showing cardiovascular benefits
Two immunotherapeutics have been successful in improving the cardiovascular outcomes of patients with CVD: canakinumab32 and colchicine33,34,95 (Table 1).
Canakinumab
The CANTOS trial32 was a double-blind, randomized, controlled trial investigating the effects of canakinumab, a monoclonal antibody against the pro-inflammatory cytokine IL-1β, in patients with recent MI. In total, 10,061 patients with a history of MI who were receiving optimal management for cardiovascular risk factors and had high-sensitivity C-reactive protein (hsCRP) levels of >2 mg/l were randomly assigned to receive canakinumab or placebo. Canakinumab was administered subcutaneously at doses of 50 mg, 150 mg or 300 mg every 3 months. Patients were followed up for a median of 3.7 years. The 150-mg canakinumab dose led to a significantly lower rate of recurrent cardiovascular events than placebo, independently of lipid-level lowering (HR 0.85, 95% CI 0.74–0.98; P = 0.021)32. No effect was observed on total mortality, owing to a small but significant increased risk of infection with canakinumab. Notably, among patients receiving canakinumab, those with a reduction in on-treatment hsCRP levels to <2 mg/l benefited the most from the treatment, and the effect of canakinumab at reducing hsCRP levels was dose-dependent96. A subanalysis extended the scope of the effects of canakinumab beyond IL-1β by showing that the modulation of plasma IL-6 levels is associated with the beneficial effects of canakinumab in reducing the risk of cardiovascular events97. Moreover, canakinumab reduced cancer mortality98. The CANTOS trial demonstrated for the first time the proof-of-principle that therapeutic targeting of the immune system can be beneficial for cardiovascular outcomes in patients.
Colchicine
Colchicine, which is widely used for the treatment of gout and pericarditis, decreases inflammation by inhibiting cytoskeletal microtubule formation99,100. Colchicine has broad cellular effects, including reduction of monocyte and neutrophil motility and inhibition of inflammasome assembly in vitro101. The LoDoCo2 trial34 included 5,522 patients with stable chronic CAD. After 1 month of open-label use of colchicine (0.5 mg once daily), patients were randomly assigned to receive colchicine or placebo and followed up for a median of 28.6 months. Patients receiving colchicine had a 31% reduction in the incidence of the primary composite end point of cardiovascular death, MI, ischaemic stroke and ischaemia-driven coronary revascularization compared with patients receiving placebo (HR 0.69, 95% CI 0.57–0.83; P < 0.001). Unfortunately, data on the effects of colchicine on inflammatory markers are not available. The results of this trial are consistent with those of two phase II trials investigating colchicine, LoDoCo95 (in patients with stable chronic CAD) and COLCOT33 (in patients with MI), and provide further support for the potential benefits of anti-inflammatory therapy in patients with acute coronary disease. Taken together, these trials demonstrated that anti-inflammatory therapies are efficacious in reducing cardiovascular events in patients with stable CVD. Although CANTOS and LoDoCo2 have not yet changed the treatment strategy in cardiovascular risk management in clinical practice, these trials are a crucial milestone for the clinical translation of immunomodulatory therapeutics in CVD. Both treatments target innate immunity, offering proof in humans of the importance of the innate response of the immune system in triggering inflammation in atherosclerosis.
Promising phase II clinical trials
Several cytokine blockers have shown promising results in phase II trials (Table 2). Cytokine blockers are the first line of biologics for the treatment of chronic inflammatory diseases, including RA, inflammatory bowel disease and psoriasis102,103,104. Therefore, an arsenal of potential therapeutics for CVD is available, some of which will soon be available as generic drugs (such as tumour necrosis factor (TNF) blockers).
IL-1 blockade
IL-1 is a pro-inflammatory cytokine that drives inflammation in atherosclerosis105. Both isoforms of IL-1, IL-1α and IL-1β, are involved in atherosclerosis. Studies in mice have shown that IL-1α has a role in the remodelling of arteries during early atherogenesis, whereas IL-1β mainly drives vascular inflammation in later stages of atherosclerosis106. However, IL-1β had a protective role in advanced atherosclerosis in mice through the promotion and maintenance of a fibrous cap rich in VSMCs and collagen107. Additionally, IL-1α forms a link between the immune system and coagulation through the activation of IL-1α by thrombin, underscoring the importance of this isoform in the pathogenesis of adverse cardiovascular events108. In humans, the levels of IL-1β in the coronary arteries are higher in patients with CAD than in patients with non-ischaemic cardiomyopathy109, and this cytokine is considered to be therapeutically tractable. Several options are available for IL-1 blockade, including canakinumab (selective IL-1β targeting), anakinra (an IL-1 receptor antagonist, which thereby targets IL-1α and IL-1β) and xilonix (a monoclonal antibody specifically targeting IL-1α). In two separate studies, therapy with anakinra significantly reduced hsCRP levels in the acute setting in patients with ACS compared with placebo110,111. Therapy with xilonix plus standard of care showed a non-significant trend towards a reduction in restenosis and the incidence of major adverse cardiovascular events compared with standard of care only in patients undergoing percutaneous femoral artery revascularization112. Whereas the CANTOS trial highlighted the relevance of targeting IL-1β in stable CAD, these studies illustrate the importance of IL-1 as a target in the acute setting of thrombotic events. Additional studies in larger patient groups should be performed to further assess the effect of these therapeutics on cardiovascular outcomes.
IL-6 blockade
IL-6 is a pro-inflammatory cytokine involved in the innate immune response and a downstream mediator of a cytokine cascade featuring TNF and IL-1. IL-6 is a central stimulus for the acute phase response. In particular, IL-6 stimulates the production of CRP, among other acute phase reactants, in hepatocytes113. IL-6 signalling contributes to atherosclerosis and plaque destabilization in mice114. Data from humans show that elevated IL-6 levels in the plasma are associated with an increased risk of MI, and genetic studies have provided evidence of a causal role for IL-6 receptor signalling in CVD115,116,117. Therapy with tocilizumab, a monoclonal antibody targeting the IL-6 receptor, reduced hsCRP levels in patients with ST-segment elevation MI (STEMI)118 or non-STEMI119 compared with placebo. Tocilizumab therapy also significantly increased the myocardial salvage index in patients with STEMI118; however, the absolute difference between the tocilizumab and placebo groups was only 5.6%, meaning that this increase might be of limited clinical relevance. In a phase II trial published in 2021, IL-6 blocking with the antibody ziltivekimab reduced hsCRP levels in patients with chronic kidney disease, who are at high risk of atherosclerosis120. These studies demonstrate the efficacy of IL-6 blockade for inflammation reduction. Follow-up studies, including the ZEUS trial121, will provide a more complete picture of the clinical relevance of IL-6-targeted therapies in CVD.
Blockade of other cytokines
Alternatives to IL-1 and IL-6 blockade include TNF or IL-23 blockers, given that preclinical and clinical research has demonstrated a pro-atherogenic role for these cytokines122,123,124. TNF is a pro-inflammatory cytokine and is produced by several cells involved in atherosclerosis, including macrophages and VSMCs125. In mice, TNF deficiency reduced atherogenesis126. In humans, TNF is present in atherosclerotic plaques and the levels of TNF in peripheral blood predict future coronary events in patients with MI125,127. In observational studies in patients with arthritis, inflammation was a strong risk factor for cardiovascular events and TNF blockade resulted in reduced atherogenesis and lower incidence of cardiovascular events compared with patients with arthritis who did not receive TNF-blocking therapy35. However, in clinical trials in patients with heart failure, TNF blockade had no efficacy or even worsened the clinical outcome128,129. Therefore, TNF blockers might not be suitable for patients with substantial deterioration of left ventricular systolic function.
IL-23 is present in human atherosclerotic plaques, and high plasma levels of IL-23 are associated with increased mortality in patients with carotid artery stenosis123. Studies in mice have shown that IL-23 drives TH17 cell function, contributing to the aggravation of atherosclerosis130,131,132. Despite the pro-atherogenic role of IL-23 in mice, several meta-analyses of studies in patients with psoriasis showed either no effect or possible worsening of cardiovascular outcomes after treatment with IL-23 blockers (ustekinumab and briakinumab) compared with placebo133,134. These studies were primarily designed to assess the effect of the IL-23 blockers on psoriasis and, therefore, conclusions cannot be drawn about their effect on inflammation in atherosclerosis. Other alternative therapeutic targets currently being tested in trials, including hydroxychloroquine and low-dose IL-2, are discussed in Box 3 and Table 3.
Challenges
Several strategies for targeting inflammation in CVD have been tested in clinical trials but have not resulted in the reduction of inflammation markers and/or cardiovascular events (Supplementary Table 1). Notable examples are methotrexate and a p38 inhibitor, which did not reduce cardiovascular events or mortality in patients with CVD135,136. The majority of the trials that did not show efficacy of the drug being tested included unselected patient cohorts; therefore, a potential explanation for the lack of efficacy might be the heterogeneity of the patient group. The CANTOS trial32 was the first trial to take a step towards the use of precision medicine by specifically selecting patients with an increased residual inflammatory risk (measured as hsCRP >2 mg/l). However, the trials investigating colchicine also included unselected patient groups and did show beneficial effects on cardiovascular outcomes33,34. This finding illustrates that failure to demonstrate efficacy might also be mechanism-based and that inhibiting inflammation in CVD is effective provided the correct inflammatory target or drug is chosen.
The variability of disease settings in clinical trials of CVD might explain the lack of beneficial effects of p38 inhibitors. p38 is an intracellular kinase that is activated in CVD by several stressors, such as oxLDL and hypertension, and is involved in the stabilization of mRNA encoding several inflammatory mediators that are crucial in CVD137,138. The first study of the p38 inhibitor losmapimod in CVD included patients with stable atherosclerosis139. Vascular inflammation was assessed with fluorodeoxyglucose (FDG) PET–CT imaging. Losmapimod therapy did not significantly reduce the overall uptake of FDG in the index vessel compared with placebo but reduced inflammation in the most inflamed regions139. However, losmapimod had no effect on clinical outcomes in subsequent trials that included larger cohorts of patients with acute MI136,140, suggesting that p38 might have a selective role in chronic stable CVD, which is consistent with the role of p38 in prolonging inflammatory responses via modulation of mRNA stability138.
Other studies have also used FDG PET–CT imaging to assess vascular inflammation, such as the GLACIER trial141. The trial included 147 patients with stable atherosclerotic disease who were randomly assigned to receive a single dose of the anti-oxLDL antibody MLDL1278A, multiple doses of MLDL1278A or placebo. None of the MLDL1278A regimens had a significant effect on carotid plaque inflammation, possibly owing to the concomitant use of lipid-lowering medication, which might have masked the effect of passive vaccination with MLDL1278A141. This study also raises questions about the use of imaging as a surrogate end point for cardiovascular events. New PET–CT imaging tracers that can detect meaningful cardiovascular inflammation more accurately than FDG are needed142. An imaging technique developed in the past 4 years that is based on CT angiography showed that changes in the CT attenuation index of perivascular adipose tissue might be a marker of coronary perivascular inflammation associated with cardiovascular outcomes143,144. Further improvements in the imaging of atherosclerosis will facilitate the development of valid surrogate end points of cardiovascular outcomes. Although cardiovascular surrogate end points are at present not sufficiently specific and, therefore, have not reached the benchmark of a clinical trial, developments in the field of machine learning could be used to combine multiple surrogate end points for a more accurate prediction of clinical outcomes145,146.
Considering the above-mentioned successes in therapeutic targeting of the immune system in atherosclerosis, the number of ongoing trials in this setting is surprisingly low. One reason could be the high costs of clinical trials in CVD, which make this area less attractive for industry investments. Trials in CVD are event-driven rather than symptom-driven and, therefore, require high patient numbers and long follow-up. Therefore, identifying reliable surrogate markers of vascular inflammation is crucial to facilitate the design of small proof-of-principle trials, allowing rapid innovation and reduced risks. One crucial need is the early identification of patients who are likely to respond to a specific treatment and patients who would not benefit from the interruption of a specific inflammatory pathway. This concept is well exemplified by the CANTOS trial32, which demonstrated that patients with the larger reductions in hsCRP levels with canakinumab therapy derived the largest clinical benefit from the treatment. Patients with a decrease in hsCRP levels greater than the median percentage reduction had a 27% reduction in cardiovascular events compared with a reduction of only 5% in those patients with a decrease in hsCRP levels that was lower than the median96. Moreover, the fall in hsCRP levels has so far gone hand in hand with outcome benefits in the majority of clinical trials of anti-inflammatory therapies in CVD. In the future, new surrogate end points that are based on immunophenotyping and/or imaging could be used in clinical trials, provided that an association with cardiovascular outcomes is demonstrable.
Looking to the future, the secondary effects of anti-inflammatory therapies should be carefully considered. Canakinumab administration was associated with a major reduction in the incidence of lung cancer compared with placebo in the CANTOS trial98. By contrast, in the CIRT trial135,147, methotrexate was linked to a small increase in the incidence of skin cancer compared with placebo, emphasizing the complexity of the effects of immunotherapy on CVD and cancer. Immunosuppression and chronic inflammation can both increase the risk of cancer147. Furthermore, preclinical studies have spotlighted the existence of an immune-mediated link between MI and breast cancer that can accelerate cancer progression148. An increasing number of studies have also shown that immune checkpoint inhibitor therapies might increase the risk of CVD in patients with cancer36,37, whereas inhibition of adaptive immunity increases the risk of cancer through disruption of antitumour immunity149. Now that anti-inflammatory therapies in CVD are close to implementation in clinical practice, unravelling the complex immunological relationship between cancer and CVD is crucial.
Finally, the pathogenesis of CVD is multifactorial, and several types of coronary culprit lesions lead to the same clinical presentation and syndromes17. Different disease settings have distinct immune signatures, as illustrated by the different signatures in plaque erosion and rupture150, which calls for the identification of the disease setting in which a therapy will be most successful. Implementing deep immunophenotyping strategies can improve the selection of patients with the highest likelihood of benefiting from a specific therapy and facilitate rapid identification of responders and non-responders to therapy151. Immunophenotyping of patients with CVD is still in its infancy; however, a few of the currently available markers could guide patient selection, such as hsCRP and IL-6 levels in the plasma96,97,152. The discovery of CHIP as a novel risk factor of atherosclerosis will potentially enable further risk stratification of patients153. For example, a re-analysis of CANTOS data suggested that anti-inflammatory treatment might be more effective in patients carrying CHIP-associated gene variants154. Taken together, extensive immunophenotyping and immune-based risk stratification might facilitate patient selection and stratification and identification of treatment responders, allowing efficient design of clinical trials and realizing the potential of targeted immunomodulatory therapies for CVD.
In summary, the challenges in addressing the low-grade inflammation associated with CVD are manifold and encompass the need for careful risk–benefit assessment, the existence of several coronary syndromes with potentially different endotypes and pathogenesis, our current inability to identify responders to treatment early, and our reliance on ‘hard’ clinical end points in trial design owing to the limitations of our current imaging techniques. Further understanding of the immune signature of CVD together with the evolution of cardiovascular imaging technologies will accelerate the translation of therapies targeting inflammation from the preclinical to the clinical arena.
New targets for clinical translation
Advances in our understanding of the pathogenesis of atherosclerosis have highlighted several potential cellular and molecular therapeutic targets. In this section, we focus on a selection of the most promising areas supported by the convergence of several lines of evidence from CVD and other diseases, and which are, therefore, closer to translation to patient therapies in the medium term (Fig. 3).
Immunometabolism and trained immunity
Targeting immunometabolic processes is a promising strategy for modulating inflammation and immunity. Atherosclerosis-associated changes in blood and bone marrow are regulated by immunometabolic events155. In mice, a Western diet and hyperglycaemia have been shown to induce epigenetic reprogramming of myeloid progenitors, which resulted in sustained monocyte and macrophage pro-inflammatory priming, thereby driving tissue inflammation and CVD156,157,158. These effects persisted even after restoring lipid and glucose levels to normal levels owing to the phenomenon of ‘trained immunity’, whereby transcriptomic, epigenetic and metabolic rewiring of innate immune cells leads to an altered response towards a second challenge159.
Epigenetic regulation is of particular interest because of the potential for pharmacological inhibition. Histone deacetylases (HDACs) repress gene expression by removing open-chromatin acetylation marks. Broad HDAC inhibition in atherosclerotic mice showed mixed results160,161,162, whereas inhibition or genetic deletion of HDAC3 or HDAC9 reduced atherosclerosis in mice163,164,165. Variants in HDAC9 have been associated with abdominal aortic calcification and ischaemic stroke in genome-wide association studies in humans166,167, highlighting the clinical potential of specific HDAC targeting in CVD.
Targeting metabolic rewiring is an alternative strategy because increased glucose metabolism in human and mouse haematopoietic stem and progenitor cells (HSPCs) dictates myeloid lineage commitment168. Glucose transporter 1 (GLUT1), a ubiquitously expressed glucose transporter, is a well-recognized target in other inflammatory conditions169. GLUT1 deficiency in bone marrow cells resulted in reduced HSPC proliferation, myelopoiesis and atherogenesis in mice170. However, further investigation of the effects of GLUT1 inhibition in humans is necessary, because patients with GLUT1 deficiency syndrome have neurological symptoms, such as epilepsy171.
Targeting CHIP
The discovery of CHIP has led to the identification of new potential targets. The most commonly occurring variants associated with CHIP are loss-of-function variants in DNTM3A, ASXL1 and TET2 and gain-of-function variants in JAK2 (JAK2V617F), that all result in growth and survival advantages in the cells carrying the gene variant172. Mice with TET2 deficiency or carrying the Jak2V617F variant showed accelerated atherogenesis30,153,173,174. Both macrophages from Tet2-knockout mice and peripheral blood monocytes from patients with aortic valve stenosis carrying a DNTMA3 or TET2 variant produce high levels of IL-1β and show NLRP3 inflammasome priming30,153,175. NLRP3 inflammasome inhibition by administration of MCC950 prevented TET2-dependent atherosclerosis progression in mice in vivo30,153. Similarly, clonal haematopoiesis driven by TET2 deficiency aggravated heart failure, cardiac dysfunction and obesity in mice, whereas NLRP3 inhibition with MCC950 protected against the development of heart failure and insulin sensitivity176,177,178. Activation of the absent in melanoma 2 (AIM2) inflammasome has been associated with Jak2V617F-driven atherosclerosis in mice. In a mouse model of Jak2V617F-driven atherosclerosis, deletion of the genes encoding for essential components that act downstream of the AIM2 inflammasome, such as caspase 1, caspase 11 and gasdermin D, induced a more stable plaque phenotype179 (Box 4). Taken together, the findings of these studies highlight the potential of targeting CHIP-driven inflammation with the use of NLRP3 or AIM2 inflammasome inhibitors.
JAK2 inhibitors could represent an alternative strategy for targeting inflammation in atherogenesis. Ruxolitinib and fedratinib are FDA-approved drugs for the treatment of myeloproliferative neoplasms and are currently being tested for use in other inflammatory conditions, such as RA180. Both drugs were effective in reducing inflammation and atherosclerosis in mouse and rabbit models of atherosclerosis174,181. Although treatment with the JAK1–JAK2 inhibitor ruxolitinib reduced atherosclerotic plaque size in mice with Jak2V617F-dependent atherosclerosis174,179, the treatment also increased necrotic core size and reduced cap thickness, resulting in an unstable plaque phenotype179. Therefore, a more specific JAK2 inhibitor, such as fedratinib, might be of interest in CVD.
Targeting monocyte recruitment
Monocyte recruitment in atherosclerosis depends on the CCR2, CCR5 and CX3CR1 chemokine receptors182. Genetic deletion of Ccr2 or its ligand Ccl2 reduced bone marrow-derived monocytosis and atherosclerotic lesion size in mice42,183,184,185. Similarly, mice with MI treated with a small interfering RNA (siRNA) targeting Ccr2 had decreased monocyte recruitment to the infarct area and reduced disease severity186. In humans, genetic predisposition to elevated plasma CCL2 levels is associated with an increased risk of stroke, MI and CAD, and increased CCL2 levels in the blood and atherosclerotic plaques correlate with a higher risk of stroke and with markers of plaque destabilization187. MLN1202, a CCR2-blocking antibody, reduced hsCRP levels in patients at risk of atherosclerotic CVD188. Pharmacological inhibition of CCR5 with the FDA-approved CCR5 antagonist maraviroc reduced atherosclerosis in Ldlr−/− mice189,190. Interestingly, treatment with maraviroc also led to reduced atheroprogression in patients with HIV infection and high risk of CVD compared with baseline, as assessed by intima–media thickness191,192. However, given that circulating monocytes traffic into tissues during homeostasis, inflammation and inflammation resolution47,193, the effect of targeting monocyte recruitment on these processes will need monitoring.
Reprogramming inflammatory macrophages
Macrophage polarization is orchestrated by key master regulators, including nuclear factor-κB, the STAT family, peroxisome proliferator-activated receptor-γ (PPARγ) and the interferon regulatory factor (IRF) family194. Reprogramming pro-inflammatory macrophage populations that drive vascular inflammation towards homeostatic pro-resolving phenotypes could reduce disease burden. Pioglitazone is an FDA-approved PPARγ agonist that induces a pro-resolving macrophage phenotype by reducing pro-inflammatory cytokine production and promoting monocyte differentiation into alternatively activated macrophages195,196,197. In mice with atherosclerosis, administration of pioglitazone reduced macrophage content and increased plaque stability198,199. Clinical studies investigating the role of pioglitazone in patients with CVD and/or type 2 diabetes mellitus showed atheroprotective effects and a reduction of cardiovascular events with pioglitazone therapy200,201,202,203, highlighting the therapeutic potential of this drug in CVD.
In mouse models of CVD, global or myeloid-specific IRF5 deficiency reduced atherosclerosis and improved plaque stability204,205, and IRF5 inhibition with nanoparticles decreased myocardial infarct size206. The transcription factor IRF5 induces a pro-inflammatory phenotype in mouse and human macrophages207. Therefore, IRF5 is a promising therapeutic target in CVD. Inhibitors of IRF5 have proven to be therapeutically effective in mouse models of systemic lupus erythematosus208,209.
Targeting the inflammasomes
Selective inhibition of the NLRP3 inflammasome with MCC950 reduced atherosclerosis in hypercholesterolaemic or diabetic mice210,211. MCC950 has been tested in phase II trials in patients with RA, but the trials had to be discontinued owing to liver toxicity212. The interest in using NLRP3 inflammasome inhibitors for the treatment of chronic inflammatory and neuroinflammatory diseases is increasing and these agents are being tested in clinical trials213. The NLRP3 inflammasome inhibitor OLT1177 has been assessed in phase I–II clinical trials in patients with osteoarthritis214, acute gout213 or heart failure215 and has shown high tolerability. OLT1177 is also currently being tested in a study in patients with COVID-19 (ref.216).
Alternative approaches to targeting the inflammasome in atherosclerosis include the prevention of inflammasome priming with the use of TLR inhibitors217, targeting the AIM2 inflammasome179 and inhibition of caspase 1 (Box 4). The catalytic activity of caspase 1 is required to convert pro-IL-1β into its active form downstream of NLRP3 and AIM2. The caspase 1 inhibitor VX-765 reduces atherosclerosis in mice218. However, phase II trials of the caspase 1 inhibitors VX-740 and VX-765 in patients with psoriasis or epilepsy revealed drug-induced hepatotoxicity and further development was stopped219, highlighting the challenges presented by inhibition of inflammasomes.
Targeting the adaptive immune system
Immune recognition of LDL and oxLDL moieties leads to the generation of autoantibodies and oxLDL-reactive T cells14,220. Immunization with ApoB-derived antigens induces atheroprotective effects in mice and rabbits via diverse mechanisms including the induction of a humoral antibody response, Treg cell activation, suppression of CD4+ T cells and reduction of dendritic cell numbers in the plaque221,222,223. However, passive immunization with MLDL1278A, an anti-oxLDL antibody, added to lipid-lowering therapies did not reduce cardiovascular events in patients with stable atherosclerotic disease, as discussed above141. To improve the translation of ApoB-based immunization therapies from the preclinical to the clinical setting, Wolf and colleagues used in silico prediction methods to identify ApoB peptides that would bind to various major histocompatibility complex class II variants81. Using the in silico methods, the investigators identified 30 ApoB peptides that successfully induced a response in human T cells in vitro81.
Another approach to targeting adaptive immune cells is the direct targeting of atherogenic B cell subsets224. B cell depletion therapies are already in clinical use for the treatment of RA and multiple sclerosis, and studies in mice have shown that preferential B2 cell depletion with the use of an anti-CD20 antibody reduces atherosclerosis225,226. A single dose of rituximab, a B cell-depleting anti-CD20 antibody, was safe and efficiently depleted B cells in patients with acute STEMI227. Antibodies for B cell depletion targeting CD19 (blinatumomab and inebilizumab), CD22 (inotuzumab ozogamicin) or B cell maturation antigen (belantamab mafodotin and AMG420) have been approved or are currently in clinical development for the treatment of multiple sclerosis and cancer. Other promising strategies targeting B cells include: impairment of B cell survival and proliferation (with atacicept, belimumab, blisibimod and ianalumab), modulation of B cell receptor signalling (with acalabrutinib, epratuzumab and ibrutinib), antibody neutralization (with omalizumab), and the modulation of B cell co-stimulation (with abatacept)224,228.
Targeting co-stimulation pathways
Immune checkpoints are immune regulatory co-stimulatory molecules that provide stimulatory or inhibitory signals to adaptive and innate immune cells229. Immune checkpoints modulate the immune response in CVD229. In vivo studies in mice identified crucial co-stimulatory axes in atherosclerosis with the use of genetic deletion and agonistic and antagonist antibodies: activation of CD27–CD70, B and T lymphocyte attenuator (BTLA), CD200 receptor (CD200R)–CD200 and CD80/CD86–CTLA4 (cytotoxic T lymphocyte antigen 4) pathways or inhibition of CD40–CD40 ligand, OX40–OX40 ligand and CD30–CD30 ligand pathways might be beneficial therapeutic strategies in atherosclerosis230,231,232,233,234,235,236,237. Multiple immune checkpoint inhibitors and agonists targeting the above pathways are in clinical development for the treatment of cancer and RA238,239. In preclinical models, specific inhibition of tumour necrosis factor receptor-associated factor 6 (TRAF6), downstream of the pro-inflammatory CD40 signalling pathway, with small-molecule inhibitors resulted in plaque stabilization without inducing adverse effects and sparing host defence240. Similarly, CD200R expression is restricted to the myeloid compartment, making the CD200–CD200R pathway amenable for selective targeting of the monocyte–macrophage axis locally and in the bone marrow in CVD234.
Targeting the atherosclerotic plaque
Long-term immunosuppression might disrupt cardiovascular homeostasis and host defence241. Local delivery of drugs has been used in the clinic in the vascular field for many years with the use of drug-eluting stents containing sirolimus or paclitaxel, both of which have anti-inflammatory properties242. Furthermore, microneedle injections of dexamethasone in the adventitia prevents restenosis in patients who have undergone percutaneous transluminal angioplasty243.
An alternative strategy for minimizing the systemic adverse effects of off-target cell activation with systemic immunosuppressive approaches and improving accessibility to the cell type of interest is the use of cell-targeted delivery approaches. Nanoparticles have a high engagement with myeloid cells and can be modified to target specific subsets with ligand-decorated nanomaterials244. Nanoparticles have been used to target macrophages in several trials in patients with CVD245,246. Flores and colleagues used PEGylated, single-wall carbon nanotubes to deliver a downstream inhibitor of the anti-phagocytic CD47 pathway to lesional macrophages in mice, which resulted in a reduced plaque burden without toxic effects247. Administration of macrophage-targeted nanoparticles carrying siRNA against Camk2g increased plaque stability in mice owing to improved efferocytosis, leading to rebalancing of the immune system in atherosclerosis248 (Box 2). Nanoparticles decorated with collagen type IV accumulate in the atherosclerotic lesion shoulder and the use of these nanoparticles for the targeted delivery of IL-10 or the anti-inflammatory annexin A1 biomimetic Ac2-26 peptide stabilized atherosclerotic lesions in mice249,250. TRAF6 inhibitors or pioglitazone delivered with nanoparticles was also effective in increasing plaque stability in atherosclerotic mice198,240. These studies highlight the potential of modulating the immune system in CVD by specifically targeting atherosclerotic plaques to avoid toxic effects associated with systemic immunosuppression approaches.
Conclusions
Cardiovascular research lags behind oncology and rheumatology in recognizing the effects of chronic inflammation on CVD and translating inflammatory targets to human cardiovascular therapy. Although our understanding of the role of inflammation in atherosclerosis has improved substantially over the past two decades, the nuanced balance between pro-inflammatory and anti-inflammatory cells required for homeostasis remains elusive. To identify new therapeutic targets in atherosclerosis, we need to improve our interpretation of the determinants of this equilibrium. Single-cell biology approaches can accelerate clinical translation by facilitating the examination of immune signatures in patients with CVD. Identification of culprit cell types with the use of multiomics approaches could help identify the most suitable patient population for clinical trials and support target selection and informed decision-making in a clinical setting, moving towards personalized medicine. In addition, it is imperative to determine the window of opportunity for anti-inflammatory therapy in atherosclerosis, in which the benefits of immune system inhibition outweigh the systemic immunosuppressive effects. More targeted approaches using biologics or vaccination might allow specific targeting of atherosclerotic inflammation and thus minimize off-target effects. The development of mRNA vaccines has revolutionized the field of RNA-based therapeutics, extending the toolkit for vaccines against atherosclerosis and for previously ‘undruggable’ targets251. The association of CHIP with CVD risk exemplifies the importance of patient stratification beyond the use of traditional risk factors to define the patient population that will benefit from treatment. It is time to take inflammation seriously as a pathogenic driver of CVD and direct resources towards mechanistic and translational studies to find the cause of and a remedy for inflammation in this context. There has never been a more exciting time for research in cardiovascular inflammation.
References
Libby, P., Ridker, P. M. & Maseri, A. Inflammation and atherosclerosis. Circulation 105, 1135–1143 (2002).
Arnett, D. K. et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 140, e596–e646 (2019).
Neumann, F. J. et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 41, 407–477 (2020).
Timmis, A. et al. European Society of Cardiology: cardiovascular disease statistics 2019. Eur. Heart J. 41, 12–85 (2020).
Liuzzo, G. et al. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N. Engl. J. Med. 331, 417–424 (1994).
Biasucci, L. M. et al. Elevated levels of interleukin-6 in unstable angina. Circulation 94, 874–877 (1996).
Galkina, E. et al. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J. Exp. Med. 203, 1273–1282 (2006).
Roy, P., Orecchioni, M. & Ley, K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-021-00584-1 (2021).
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
O’Neill, L. A. J., Golenbock, D. & Bowie, A. G. The history of Toll-like receptors–redefining innate immunity. Nat. Rev. Immunol. 13, 453–460 (2013).
Tabas, I. & Lichtman, A. H. Monocyte-macrophages and T cells in atherosclerosis. Immunity 47, 621–634 (2017).
Moore, K. J. & Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355 (2011).
Tsiantoulas, D. et al. APRIL limits atherosclerosis by binding to heparan sulfate proteoglycans. Nature 597, 92–96 (2021).
Stemme, S. et al. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc. Natl Acad. Sci. USA 92, 3893–3897 (1995).
Stewart, C. R. et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161 (2010).
Binder, C. J., Papac-Milicevic, N. & Witztum, J. L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 16, 485–497 (2016).
Naghavi, M. et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part I. Circulation 108, 1664–1672 (2003).
Zernecke, A. et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ. Res. 127, 402–426 (2020).
Depuydt, M. A. C. et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ. Res. 127, 1437–1455 (2020).
Lin, J. D. et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight 4, e124574 (2019).
Winkels, H. et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Circ. Res. 122, 1675–1688 (2018).
Kim, K. et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ. Res. 123, 1127–1142 (2018).
McArdle, S. et al. Migratory and dancing macrophage subsets in atherosclerotic lesions. Circ. Res. 125, 1038–1051 (2019).
Cole, J. E. et al. Immune cell census in murine atherosclerosis: cytometry by time of flight illuminates vascular myeloid cell diversity. Cardiovasc. Res 114, 1360–1371 (2018).
Fernandez, D. M. et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 25, 1576–1588 (2019).
Gu, W. et al. Adventitial cell atlas of wt (wild type) and ApoE (apolipoprotein E)-deficient mice defined by single-cell RNA sequencing. Arterioscler. Thromb. Vasc. Biol. 39, 1055–1071 (2019).
Cochain, C. et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ. Res. 122, 1661–1674 (2018).
McAlpine, C. S. et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature 566, 383–387 (2019).
Robbins, C. S. et al. Extramedullary hematopoiesis generates Ly-6C high monocytes that infiltrate atherosclerotic lesions. Circulation 125, 364–374 (2012).
Fuster, J. J. et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 (2017).
Soehnlein, O. & Libby, P. Targeting inflammation in atherosclerosis–from experimental insights to the clinic. Nat. Rev. Drug Discov. 20, 589–610 (2021).
Ridker, P. M. et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med 377, 1119–1131 (2017).
Tardif, J.-C. et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 381, 2497–2505 (2019).
Nidorf, S. M. et al. Colchicine in patients with chronic coronary disease. N. Engl. J. Med 383, 1838–1847 (2020).
Full, L. E. & Monaco, C. Targeting inflammation as a therapeutic strategy in accelerated atherosclerosis in rheumatoid arthritis. Cardiovasc. Ther. 29, 231–242 (2011).
Drobni, Z. D. et al. Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation 142, 2299–2311 (2020).
Poels, K. et al. Immune checkpoint inhibitor therapy aggravates T cell-driven plaque inflammation in atherosclerosis. JACC Cardiovasc. Oncol. 2, 599–610 (2020).
Ridker, P. M. How common is residual inflammatory risk? Circ. Res. 120, 617–619 (2017).
Allahverdian, S., Chaabane, C., Boukais, K., Francis, G. A. & Bochaton-Piallat, M.-L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 114, 540–550 (2018).
Doran, A. C., Meller, N. & McNamara, C. A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 28, 812–819 (2008).
Lievens, D. & von Hundelshausen, P. Platelets in atherosclerosis. Thromb. Haemost. 106, 827–838 (2011).
Combadière, C. et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6Chi and Ly6Clo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 117, 1649–1657 (2008).
Tacke, F. et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Invest. 117, 185–194 (2007).
Swirski, F. K. et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Invest. 117, 195–205 (2007).
Shimizu, Y. et al. Radiation exposure and circulatory disease risk: Hiroshima and Nagasaki atomic bomb survivor data, 1950-2003. BMJ 340, b5349 (2010).
Swirski, F. K. et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325, 612–616 (2009).
Rahman, K. et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J. Clin. Invest. 127, 2904–2915 (2017).
Woollard, K. J. & Geissmann, F. Monocytes in atherosclerosis: subsets and functions. Nat. Rev. Cardiol. 7, 77–86 (2010).
Schloss, M. J., Swirski, F. K. & Nahrendorf, M. Modifiable cardiovascular risk, hematopoiesis, and innate immunity. Circ. Res. 126, 1242–1259 (2020).
Williams, J. W. et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat. Immunol. 21, 1194–1204 (2020).
Ensan, S. et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1+ precursors and circulating monocytes immediately after birth. Nat. Immunol. 17, 159–168 (2016).
Robbins, C. S. et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 19, 1166–1172 (2013).
Lim, H. Y. et al. Hyaluronan receptor LYVE-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity 49, 326–341.e7 (2018).
Park, I. et al. C-type lectin receptor CLEC4A2 promotes tissue adaptation of macrophages and protects against atherosclerosis. Nat. Commun. 13, 215 (2022).
Weinberger, T. et al. Ontogeny of arterial macrophages defines their functions in homeostasis and inflammation. Nat. Commun. 11, 4549 (2020).
Murray, P. J. Macrophage polarization. Annu. Rev. Physiol. 79, 541–566 (2017).
Owsiany, K. M., Alencar, G. F. & Owens, G. K. Revealing the origins of foam cells in atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 39, 836–838 (2019).
Spann, N. J. et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 151, 138–152 (2012).
Voisin, M. et al. Inhibiting LXRα phosphorylation in hematopoietic cells reduces inflammation and attenuates atherosclerosis and obesity in mice. Commun. Biol. 4, 420 (2021).
Jaitin, D. A. et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell 178, 686–698.e14 (2019).
Deguchi, J. O. et al. Inflammation in atherosclerosis: visualizing matrix metalloproteinase action in macrophages in vivo. Circulation 114, 55–62 (2006).
Kojima, Y., Weissman, I. L. & Leeper, N. J. The role of efferocytosis in atherosclerosis. Circulation 135, 476–489 (2017).
Merad, M., Sathe, P., Helft, J., Miller, J. & Mortha, A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 31, 563–604 (2013).
Yilmaz, A. et al. Emergence of dendritic cells in rupture-prone regions of vulnerable carotid plaques. Atherosclerosis 176, 101–110 (2004).
Trogan, E. et al. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc. Natl Acad. Sci. USA 103, 3781–3786 (2006).
Weber, C. et al. CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J. Clin. Invest. 121, 2898–2910 (2011).
Choi, J. H. et al. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity 35, 819–831 (2011).
Subramanian, M., Thorp, E., Hansson, G. K. & Tabas, I. Treg-mediated suppression of atherosclerosis requires MYD88 signaling in DCs. J. Clin. Invest. 123, 179–188 (2013).
Niessner, A. et al. Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-α. Circulation 114, 2482–2489 (2006).
MacRitchie, N. et al. Plasmacytoid dendritic cells play a key role in promoting atherosclerosis in apolipoprotein e-deficient mice. Arterioscler. Thromb. Vasc. Biol. 32, 2569–2579 (2012).
Silvestre-Roig, C., Braster, Q., Ortega-Gomez, A. & Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 17, 327–340 (2020).
Zernecke, A. et al. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ. Res. 102, 209–217 (2008).
Wang, L. et al. ROS-producing immature neutrophils in giant cell arteritis are linked to vascular pathologies. JCI Insight 5, e139163 (2020).
Warnatsch, A., Ioannou, M., Wang, Q. & Papayannopoulos, V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 349, 316–320 (2015).
Silvestre-Roig, C. et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 569, 236–240 (2019).
Fuchs, T. A. et al. Extracellular DNA traps promote thrombosis. Proc. Natl Acad. Sci. USA 107, 15880–15885 (2010).
Soehnlein, O. et al. Atherosclerosis: neutrophil-derived cathelicidin protects from neointimal hyperplasia. Sci. Transl. Med. 3, 103ra98 (2011).
Saigusa, R., Winkels, H. & Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 17, 387–401 (2020).
Ketelhuth, D. F. J. & Hansson, G. K. Adaptive response of T and B cells in atherosclerosis. Circ. Res. 118, 668–678 (2016).
Winkels, H. & Wolf, D. Heterogeneity of T cells in atherosclerosis defined by single-cell RNA-sequencing and cytometry by time of flight. Arterioscler. Thromb. Vasc. Biol. 41, 549–563 (2021).
Wolf, D. et al. Pathogenic autoimmunity in atherosclerosis evolves from initially protective apolipoprotein B100-reactive CD4+ T-regulatory cells. Circulation 142, 1279–1293 (2020).
Kyaw, T. et al. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in ApoE-deficient mice. Circulation 127, 1028–1039 (2013).
van Duijn, J., Kuiper, J. & Slütter, B. The many faces of CD8+ T cells in atherosclerosis. Curr. Opin. Lipidol. 29, 411–416 (2018).
Hwang, Y. et al. Expansion of CD8+ T cells lacking the IL-6 receptor α chain in patients with coronary artery diseases (CAD). Atherosclerosis 249, 44–51 (2016).
Bergström, I., Backteman, K., Lundberg, A., Ernerudh, J. & Jonasson, L. Persistent accumulation of interferon-γ-producing CD8+CD56+ T cells in blood from patients with coronary artery disease. Atherosclerosis 224, 515–520 (2012).
Getz, G. S. & Reardon, C. A. Natural killer T cells in atherosclerosis. Nat. Rev. Cardiol. 14, 304–314 (2017).
Bobryshev, Y. V. & Lord, R. S. A. Co-accumulation of dendritic cells and natural killer T cells within rupture-prone regions in human atherosclerotic plaques. J. Histochem. Cytochem. 53, 781–785 (2005).
Sage, A. P., Tsiantoulas, D., Binder, C. J. & Mallat, Z. The role of B cells in atherosclerosis. Nat. Rev. Cardiol. 16, 180–196 (2019).
Kyaw, T. et al. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ. Res. 109, 830–840 (2011).
Strom, A. C. et al. B regulatory cells are increased in hypercholesterolaemic mice and protect from lesion development via IL-10. Thromb. Haemost. 114, 835–847 (2015).
Nus, M. et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat. Med. 23, 601–610 (2017).
Sage, A. P. et al. Regulatory B cell-specific interleukin-10 is dispensable for atherosclerosis development in mice. Arterioscler. Thromb. Vasc. Biol. 35, 1770–1773 (2015).
Rosser, E. C. et al. Regulatory B cells are induced by gut microbiota-driven interleukin-1β and interleukin-6 production. Nat. Med. 20, 1334–1339 (2014).
Riggs, J. E., Lussier, A. M., Lee, S. K., Appel, M. C. & Woodland, R. T. Differential radiosensitivity among B cell subpopulations. J. Immunol. 141, 1799–1807 (1988).
Nidorf, S. M., Eikelboom, J. W., Budgeon, C. A. & Thompson, P. L. Low-dose colchicine for secondary prevention of cardiovascular disease. J. Am. Coll. Cardiol. 61, 404–410 (2013).
Ridker, P. M. et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet 391, 319–328 (2018).
Ridker, P. M. et al. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Eur. Heart J. 39, 3499–3507 (2018).
Ridker, P. M. et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842 (2017).
Daskalov, I. & Valova-Ilieva, T. Management of acute pericarditis: treatment and follow-up. ESC https://www.escardio.org/Journals/E-Journal-of-Cardiology-Practice/Volume-15/Management-of-acute-pericarditis-treatment-and-follow-up (2017).
Hui, M. et al. The British Society for Rheumatology guideline for the management of gout. Rheumatology 56, 1056–1059 (2017).
Paschke, S. et al. Technical advance: inhibition of neutrophil chemotaxis by colchicine is modulated through viscoelastic properties of subcellular compartments. J. Leukoc. Biol. 94, 1091–1096 (2013).
Aaltonen, K. J. et al. Systematic review and meta-analysis of the efficacy and safety of existing TNF blocking agents in treatment of rheumatoid arthritis. PLoS ONE 7, e30275 (2012).
Yamamoto-Furusho, J. K. Inflammatory bowel disease therapy: blockade of cytokines and cytokine signaling pathways. Curr. Opin. Gastroenterol. 34, 187–193 (2018).
Reich, K. et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet 390, 276–288 (2017).
Grebe, A., Hoss, F. & Latz, E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ. Res. 122, 1722–1740 (2018).
Vromman, A. et al. Stage-dependent differential effects of interleukin-1 isoforms on experimental atherosclerosis. Eur. Heart J. 40, 2482–2491 (2019).
Gomez, D. et al. Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 24, 1418–1429 (2018).
Burzynski, L. C. et al. The coagulation and immune systems are directly linked through the activation of interleukin-1α by thrombin. Immunity 50, 1033–1042.e6 (2019).
Galea, J. et al. Interleukin-1β in coronary arteries of patients with ischemic heart disease. Arterioscler. Thromb. Vasc. Biol. 16, 1000–1006 (1996).
Abbate, A. et al. Interleukin-1 blockade inhibits the acute inflammatory response in patients with ST-segment-elevation myocardial infarction. J. Am. Heart Assoc. 9, e014941 (2020).
Morton, A. C. et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA heart study. Eur. Heart J. 36, 377–384 (2015).
El Sayed, H., Kerensky, R., Stecher, M., Mohanty, P. & Davies, M. A randomized phase II study of Xilonix, a targeted therapy against interleukin 1α, for the prevention of superficial femoral artery restenosis after percutaneous revascularization. J. Vasc. Surg. 63, 133–141.e1 (2016).
Ridker, P. M. Anticytokine agents: targeting interleukin signaling pathways for the treatment of atherothrombosis. Circ. Res. 124, 437–450 (2019).
Zhang, K. et al. Interleukin 6 destabilizes atherosclerotic plaques by downregulating prolyl-4-hydroxylase α1 via a mitogen-activated protein kinase and c-Jun pathway. Arch. Biochem. Biophys. 528, 127–133 (2012).
Ridker, P. M., Rifai, N., Stampfer, M. J. & Hennekens, C. H. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation 101, 1767–1772 (2000).
Sarwar, N. et al. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet 379, 1205–1213 (2012).
Swerdlow, D. I. et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet 379, 1214–1224 (2012).
Broch, K. et al. Randomized trial of interleukin-6 receptor inhibition in patients with acute ST-segment elevation myocardial infarction. J. Am. Coll. Cardiol. 77, 1845–1855 (2021).
Kleveland, O. et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: a double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 37, 2406–2413 (2016).
Ridker, P. M. et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 397, 2060–2069 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT05021835 (2021).
Tousoulis, D. I., Oikonomou, E., Economou, E. K., Crea, F. & Kaski, J. C. Inflammatory cytokines in atherosclerosis: current therapeutic approaches. Eur. Heart J. 37, 1723–1735 (2016).
Abbas, A. et al. Sinterleukin 23 levels are increased in carotid atherosclerosis possible role for the interleukin 23/interleukin 17 axis. Stroke 46, 793–799 (2015).
Ohta, H. et al. Disruption of tumor necrosis factor-α gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis 180, 11–17 (2005).
Barath, P. et al. Detection and localization of tumor necrosis factor in human atheroma. Am. J. Cardiol. 65, 297–302 (1990).
Brånén, L. et al. Inhibition of tumor necrosis factor-α reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 24, 2137–2142 (2004).
Ridker, P. M. et al. Elevation of tumor necrosis factor-α and increased risk of recurrent coronary events after myocardial infarction. Circulation 101, 2149–2153 (2000).
Chung, E. S., Packer, M., Lo, K. H., Fasanmade, A. A. & Willerson, J. T. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-α, in patients with moderate-to-severe heart failure: results of the Anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 107, 3133–3140 (2003).
Mann, D. L. et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the randomized etanercept worldwide evaluation (RENEWAL). Circulation 109, 1594–1602 (2004).
Gao, Q. et al. A critical function of Th17 proinflammatory cells in the development of atherosclerotic plaque in mice. J. Immunol. 185, 5820–5827 (2010).
Langrish, C. L. et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201, 233–240 (2005).
Ma, S. et al. The immunomodulatory effect of bone marrow stromal cells (BMSCs) on interleukin (IL)-23/IL-17-mediated ischemic stroke in mice. J. Neuroimmunol. 257, 28–35 (2013).
Tzellos, T., Kyrgidis, A. & Zouboulis, C. C. Re-evaluation of the risk for major adverse cardiovascular events in patients treated with anti-IL-12/23 biological agents for chronic plaque psoriasis: a meta-analysis of randomized controlled trials. J. Eur. Acad. Dermatol. Venereol. 27, 622–627 (2013).
Ryan, C. et al. Association between biologic therapies for chronic plaque psoriasis and cardiovascular events: a meta-analysis of randomized controlled trials. JAMA 306, 864–871 (2011).
Ridker, P. M. et al. Low-dose methotrexate for the prevention of atherosclerotic events. N. Engl. J. Med 380, 752–762 (2019).
O’Donoghue, M. L. et al. Effect of losmapimod on cardiovascular outcomes in patients hospitalized with acute myocardial infarction: a randomized clinical trial. JAMA 315, 1591–1599 (2016).
Martin, E. D., Felice De Nicola, G. & Marber, M. S. New therapeutic targets in cardiology: p38 alpha mitogen-activated protein kinase for ischemic heart disease. Circulation 126, 357–368 (2012).
Dean, J. L. E., Brook, M., Clark, A. R. & Saklatvala, J. p38 Mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J. Biol. Chem. 274, 264–269 (1999).
Elkhawad, M. et al. Effects of p38 mitogen-activated protein kinase inhibition on vascular and systemic inflammation in patients with atherosclerosis. Imaging 5, 911–922 (2012).
Newby, L. K. et al. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: a randomised phase 2 trial. Lancet 384, 1187–1195 (2014).
Lehrer Graiwer, J. et al. FDG-PET imaging for oxidized LDL in stable atherosclerotic disease: a phase II study of safety, tolerability, and anti-inflammatory activity. JACC Cardiovasc. Imaging 8, 493–494 (2015).
Ćorović, A., Wall, C., Mason, J. C., Rudd, J. H. F. & Tarkin, J. M. Novel positron emission tomography tracers for imaging vascular inflammation. Curr. Cardiol. Rep. 22, 119 (2020).
Oikonomou, E. K. et al. Non-invasive detection of coronary inflammation using computed tomography and prediction of residual cardiovascular risk (the CRISP CT study): a post-hoc analysis of prospective outcome data. Lancet 392, 929–939 (2018).
Antonopoulos, A. S. et al. Detecting human coronary inflammation by imaging perivascular fat. Sci. Transl. Med. 9, eaal2658 (2017).
Krittanawong, C. et al. Machine learning prediction in cardiovascular diseases: a meta-analysis. Sci. Rep. 10, 16057 (2020).
Padmanabhan, S., Tran, T. Q. B. & Dominiczak, A. F. Artificial intelligence in hypertension: seeing through a glass darkly. Circ. Res. 128, 1100–1118 (2021).
Greten, F. R. & Grivennikov, S. I. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity 51, 27–41 (2019).
Koelwyn, G. J. et al. Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nat. Med. 26, 1452–1458 (2020).
Egen, J. G., Ouyang, W. & Wu, L. C. Human anti-tumor immunity: insights from immunotherapy clinical trials. Immunity 52, 36–54 (2020).
Leistner, D. M. et al. Differential immunological signature at the culprit site distinguishes acute coronary syndrome with intact from acute coronary syndrome with ruptured fibrous cap: results from the prospective translational OPTICO-ACS study. Eur. Heart J. 41, 3549–3560 (2020).
Hamers, A. A. J. et al. Human monocyte heterogeneity as revealed by high-dimensional mass cytometry. Arterioscler. Thromb. Vasc. Biol. 39, 25–36 (2019).
Kott, K. A. et al. Single-cell immune profiling in coronary artery disease: the role of state-of-the-art immunophenotyping with mass cytometry in the diagnosis of atherosclerosis. J. Am. Heart Assoc. 9, e017759 (2020).
Jaiswal, S. et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121 (2017).
Svensson, E. et al. TET2-driven clonal hematopoiesis predicts enhanced response to canakinumab in the CANTOS trial: an exploratory analysis [abstract]. Circulation 138 (Suppl. 1), 15111 (2019).
Pålsson-McDermott, E. M. & O’Neill, L. A. J. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 30, 300–314 (2020).
Edgar, L. et al. Hyperglycemia induces trained immunity in macrophages and their precursors and promotes atherosclerosis. Circulation 144, 961–982 (2021).
Seijkens, T. et al. Hypercholesterolemia-induced priming of hematopoietic stem and progenitor cells aggravates atherosclerosis. FASEB J. 28, 2202–2213 (2014).
Christ, A. et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell 172, 162–175.e14 (2018).
Netea, M. G. et al. Trained immunity: a program of innate immune memory in health and disease. Science 352, 427 (2016).
Bowes, A. J., Khan, M. I., Shi, Y., Robertson, L. & Werstuck, G. H. Valproate attenuates accelerated atherosclerosis in hyperglycemic ApoE-deficient mice: evidence in support of a role for endoplasmic reticulum stress and glycogen synthase kinase-3 in lesion development and hepatic steatosis. Am. J. Pathol. 174, 330–342 (2009).
Manea, S. A. et al. Pharmacological inhibition of histone deacetylase reduces NADPH oxidase expression, oxidative stress and the progression of atherosclerotic lesions in hypercholesterolemic apolipoprotein E-deficient mice; potential implications for human atherosclerosis. Redox Biol. 28, 101338 (2020).
Choi, J. H. et al. Trichostatin A exacerbates atherosclerosis in low density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 25, 2404–2409 (2005).
Hoeksema, M. A. et al. Targeting macrophage histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol. Med. 6, 1124–1132 (2014).
Cao, Q. et al. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler. Thromb. Vasc. Biol. 34, 1871–1879 (2014).
Asare, Y. et al. Histone deacetylase 9 activates IKK to regulate atherosclerotic plaque vulnerability. Circ. Res. 127, 811–823 (2020).
Malhotra, R. et al. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat. Genet. 51, 1580–1587 (2019).
Bellenguez, C. et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat. Genet. 44, 328–333 (2012).
Oburoglu, L. et al. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell 15, 169–184 (2014).
Chen, Z., Dudek, J., Maack, C. & Hofmann, U. Pharmacological inhibition of GLUT1 as a new immunotherapeutic approach after myocardial infarction. Biochem. Pharmacol. 190, 114597 (2021).
Sarrazy, V. et al. Disruption of Glut1 in hematopoietic stem cells prevents myelopoiesis and enhanced glucose flux in atheromatous plaques of ApoE−/− mice. Circ. Res. 118, 1062–1077 (2016).
Klepper, J. et al. Glut1 deficiency syndrome (Glut1DS): state of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open 5, 354–365 (2020).
Jaiswal, S. & Libby, P. Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat. Rev. Cardiol. 17, 137–144 (2020).
Wang, W. et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in JAK2V617F mice. Circ. Res. 123, E35–E47 (2018).
Tang, Y. et al. Inhibition of JAK2 suppresses myelopoiesis and atherosclerosis in ApoE−/− mice. Cardiovasc. Drugs Ther. 34, 145–152 (2020).
Abplanalp, W. T. et al. Association of clonal hematopoiesis of indeterminate potential with inflammatory gene expression in patients with severe degenerative aortic valve stenosis or chronic postischemic heart failure. JAMA Cardiol. 5, 1170–1175 (2020).
Sano, S. et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 INFLAMMASOME. J. Am. Coll. Cardiol. 71, 875–886 (2018).
Wang, Y. et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight 5, e135204 (2020).
Fuster, J. J. et al. TET2-loss-of-function-driven clonal hematopoiesis exacerbates experimental insulin resistance in aging and obesity. Cell Rep. 33, 108326 (2020).
Fidler, T. P. et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 592, 296–301 (2021).
Schwartz, D. M. et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 16, 843–862 (2017).
Yang, X. et al. Inhibition of JAK2/STAT3/SOCS3 signaling attenuates atherosclerosis in rabbit. BMC Cardiovasc. Disord. 20, 133 (2020).
Hilgendorf, I., Swirski, F. K. & Robbins, C. S. Monocyte fate in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 35, 272–279 (2015).
Soehnlein, O. et al. Distinct functions of chemokine receptor axes in the atherogenic mobilization and recruitment of classical monocytes. EMBO Mol. Med. 5, 471–481 (2013).
Boring, L., Gosling, J., Cleary, M. & Charo, I. F. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 394, 894–897 (1998).
Gu, L. et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell 2, 275–281 (1998).
Majmudar, M. D. et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 127, 2038–2046 (2013).
Georgakis, M. K. et al. Monocyte-chemoattractant protein-1 levels in human atherosclerotic lesions associate with plaque vulnerability. Arterioscler. Thromb. Vasc. Biol. 41, 2038–2048 (2021).
Gilbert, J. et al. Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am. J. Cardiol. 107, 906–911 (2011).
Cipriani, S. et al. Efficacy of the CCR5 antagonist maraviroc in reducing early, ritonavir-induced atherogenesis and advanced plaque progression in mice. Circulation 127, 2114–2124 (2013).
Veillard, N. R. et al. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ. Res. 94, 253–261 (2004).
Maggi, P. et al. Effects of therapy with maraviroc on the carotid intima media thickness in HIV-1/HCV co-infected patients. In Vivo 31, 125–132 (2017).
Francisci, D. et al. Maraviroc intensification modulates atherosclerotic progression in HIV-suppressed patients at high cardiovascular risk. A randomized, crossover pilot study. Open Forum Infect. Dis. 6, ofz112 (2019).
Shi, C. & Pamer, E. G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 11, 762–774 (2011).
Lawrence, T. & Natoli, G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 11, 750–761 (2011).
Smith, U. Pioglitazone: mechanism of action. Int. J. Clin. Pract. Suppl. (121), 13–18 (2001).
Rigamonti, E., Chinetti-Gbaguidi, G. & Staels, B. Regulation of macrophage functions by PPAR-α, PPAR-γ, and LXRs in mice and men. Arterioscler. Thromb. Vasc. Biol. 28, 1050–1059 (2008).
Bouhlel, M. A. et al. PPARγ activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 6, 137–143 (2007).
Nakashiro, S. et al. Pioglitazone-incorporated nanoparticles prevent plaque destabilization and rupture by regulating monocyte/macrophage differentiation in ApoE−/− mice. Arterioscler. Thromb. Vasc. Biol. 36, 491–500 (2016).
Chang, K. et al. Pioglitazone suppresses inflammation in vivo in murine carotid atherosclerosis: novel detection by dual-target fluorescence molecular imaging. Arterioscler. Thromb. Vasc. Biol. 30, 1933–1939 (2010).
Pfützner, A. et al. Improvement of cardiovascular risk markers by pioglitazone is independent from glycemic control: results from the pioneer study. J. Am. Coll. Cardiol. 45, 1925–1931 (2005).
Erdmann, E. et al. The effect of pioglitazone on recurrent myocardial infarction in 2,445 patients with type 2 diabetes and previous myocardial infarction. results from the PROactive (PROactive 05) study. J. Am. Coll. Cardiol. 49, 1772–1780 (2007).
Langenfeld, M. R. et al. Pioglitazone decreases carotid intima-media thickness independently of glycemic control in patients with type 2 diabetes mellitus: results from a controlled randomized study. Circulation 111, 2525–2531 (2005).
de Jong, M., van der Worp, H. B., van der Graaf, Y., Visseren, F. L. J. & Westerink, J. Pioglitazone and the secondary prevention of cardiovascular disease. A meta-analysis of randomized-controlled trials. Cardiovasc. Diabetol. 16, 134 (2017).
Seneviratne, A. N. et al. Interferon regulatory factor 5 controls necrotic core formation in atherosclerotic lesions by impairing efferocytosis. Circulation 136, 1140–1154 (2017).
Leipner, J. et al. Myeloid cell-specific Irf5 deficiency stabilizes atherosclerotic plaques in Apoe−/− mice. Mol. Metab. 53, 101250 (2021).
Courties, G. et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J. Am. Coll. Cardiol. 63, 1556–1566 (2014).
Krausgruber, T. et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 12, 231–238 (2011).
Song, S. et al. Inhibition of IRF5 hyperactivation protects from lupus onset and severity. J. Clin. Invest. 130, 6700–6717 (2020).
Ban, T. et al. Genetic and chemical inhibition of IRF5 suppresses pre-existing mouse lupus-like disease. Nat. Commun. 12, 4379 (2021).
Sharma, A. et al. Specific NLRP3 inhibition protects against diabetes-associated atherosclerosis. Diabetes 70, 772–787 (2021).
Van Der Heijden, T. et al. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein e-deficient mice–brief report. Arterioscler. Thromb. Vasc. Biol. 37, 1457–1461 (2017).
Mangan, M. S. J. et al. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 17, 588–606 (2018).
Klück, V. et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2, e270–e280 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01768975 (2014).
Wohlford, G. F. et al. Phase 1B, randomized, double-blinded, dose escalation, single-center, repeat dose safety and pharmacodynamics study of the oral NLRP3 indibitor dapansutrile in subjects with NYHA II-III systolic heart failure. J. Cardiovasc. Pharmacol. 77, 49–60 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04540120 (2021).
Monaco, C. et al. Toll-like receptor-2 mediates inflammation and matrix degradation in human atherosclerosis. Circulation 120, 2462–2469 (2009).
Li, Y. et al. VX-765 attenuates atherosclerosis in ApoE deficient mice by modulating VSMCs pyroptosis. Exp. Cell Res. 389, 111847 (2020).
MacKenzie, S. H., Schipper, J. L. & Clark, A. C. The potential for caspases in drug discovery. Curr. Opin. Drug Discov. Dev. 13, 568–576 (2010).
Nilsson, J. & Hansson, G. K. Vaccination strategies and immune modulation of atherosclerosis. Circ. Res. 126, 1281–1296 (2020).
Chyu, K. Y. et al. CD8+ T cells mediate the athero-protective effect of immunization with an ApoB-100 peptide. PLoS ONE 7, e30780 (2012).
Dunér, P. et al. Antibodies against apoB100 peptide 210 inhibit atherosclerosis in apoE−/− mice. Sci. Rep. 11, 9022 (2021).
Herbin, O. et al. Regulatory T-cell response to apolipoprotein B100-derived peptides reduces the development and progression of atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 32, 605–612 (2012).
Pattarabanjird, T., Li, C. & McNamara, C. B cells in atherosclerosis: mechanisms and potential clinical applications. JACC Basic Transl. Sci. 6, 546–563 (2021).
Ait-Oufella, H. et al. B cell depletion reduces the development of atherosclerosis in mice. J. Exp. Med. 207, 1579–1587 (2010).
Kyaw, T. et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J. Immunol. 185, 4410–4419 (2010).
Zhao, T. X. et al. Rituximab in patients with acute ST-elevation myocardial infarction: an experimental medicine safety study. Cardiovasc. Res. https://doi.org/10.1093/cvr/cvab113 (2021).
Porsch, F. & Binder, C. J. Impact of B-cell-targeted therapies on cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 39, 1705–1714 (2019).
Kusters, P. J. H., Lutgens, E. & Seijkens, T. T. P. Exploring immune checkpoints as potential therapeutic targets in atherosclerosis. Cardiovasc. Res. 114, 368–377 (2018).
Foks, A. C. et al. Interruption of the OX40–OX40 ligand pathway in LDL receptor-deficient mice causes regression of atherosclerosis. J. Immunol. 191, 4573–4580 (2013).
Lutgens, E. et al. Requirement for CD154 in the progression of atherosclerosis. Nat. Med. 5, 1313–1316 (1999).
Foks, A. C. et al. Interference of the CD30–CD30L pathway reduces atherosclerosis development. Arterioscler. Thromb. Vasc. Biol. 32, 2862–2868 (2012).
Winkels, H. et al. CD27 co-stimulation increases the abundance of regulatory T cells and reduces atherosclerosis in hyperlipidaemic mice. Eur. Heart J. 38, 3590–3599 (2017).
Kassiteridi, C. et al. CD200 limits monopoiesis and monocyte recruitment in atherosclerosis. Circ. Res. 129, 280–295 (2021).
Poels, K. et al. Antibody-mediated inhibition of CTLA4 aggravates atherosclerotic plaque inflammation and progression in hyperlipidemic mice. Cells 9, 1987 (2020).
Schönbeck, U., Sukhova, G. K., Shimizu, K., Mach, F. & Libby, P. Inhibition of CD40 signaling limits evolution of established atherosclerosis in mice. Proc. Natl Acad. Sci. USA 97, 7458–7463 (2000).
Douna, H. et al. B- and T-lymphocyte attenuator stimulation protects against atherosclerosis by regulating follicular B cells. Cardiovasc. Res. 116, 295–305 (2020).
Waldman, A. D., Fritz, J. M. & Lenardo, M. J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol. 20, 651–668 (2020).
Genovese, M. C. et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor α inhibition. N. Engl. J. Med. 353, 1114–1123 (2005).
Seijkens, T. T. P. et al. Targeting CD40-induced TRAF6 signaling in macrophages reduces atherosclerosis. J. Am. Coll. Cardiol. 71, 527–542 (2018).
Giugliano, G. R., Giugliano, R. P., Gibson, C. M. & Kuntz, R. E. Meta-analysis of corticosteroid treatment in acute myocardial infarction. Am. J. Cardiol. 91, 1055–1059 (2003).
Torii, S. et al. Drug-eluting coronary stents: insights from preclinical and pathology studies. Nat. Rev. Cardiol. 17, 37–51 (2020).
Razavi, M. K., Donohoe, D., D’Agostino, R. B., Jaff, M. R. & Adams, G. Adventitial drug delivery of dexamethasone to improve primary patency in the treatment of superficial femoral and popliteal artery disease: 12-month results from the DANCE clinical trial. JACC Cardiovasc. Interv. 11, 921–931 (2018).
Teunissen, A. J. P. et al. Embracing nanomaterials’ interactions with the innate immune system. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 13, e1719 (2021).
van der Valk, F. M. et al. Prednisolone-containing liposomes accumulate in human atherosclerotic macrophages upon intravenous administration. Nanomed. Nanotechnol. Biol. Med. 11, 1039–1046 (2015).
Fitzgerald, K. et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N. Engl. J. Med. 376, 41–51 (2017).
Flores, A. M. et al. Pro-efferocytic nanoparticles are specifically taken up by lesional macrophages and prevent atherosclerosis. Nat. Nanotechnol. 15, 154–161 (2020).
Tao, W. et al. SiRNA nanoparticles targeting CaMKIIγ in lesional macrophages improve atherosclerotic plaque stability in mice. Sci. Transl. Med. 12, eaay1063 (2020).
Fredman, G. et al. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci. Transl. Med. 7, 275ra20 (2015).
Kamaly, N. et al. Targeted interleukin-10 nanotherapeutics developed with a microfluidic chip enhance resolution of inflammation in advanced atherosclerosis. ACS Nano 10, 5280–5292 (2016).
Tsimikas, S. RNA-targeted therapeutics for lipid disorders. Curr. Opin. Lipidol. 29, 459–466 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02648464 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02874287 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03113773 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04241601 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04762472 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04616872 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04350216 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04148833 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04610892 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03048825 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02898610 (2017).
Jonasson, L., Holm, J., Skalli, O., Gabbiani, G. & Hansson, G. K. Expression of class II transplantation antigen on vascular smooth muscle cells in human atherosclerosis. J. Clin. Invest. 76, 125–131 (1985).
Hansson, G. K., Jonasson, L., Holm, J. & Claesson-Welsh, L. Class II MHC antigen expression in the atherosclerotic plaque: smooth muscle cells express HLA-DR, HLA-DQ and the invariant gamma chain. Clin. Exp. Immunol. 64, 261–268 (1986).
Vedeler, C. A., Nyland, H. & Matre, R. In situ characterization of the foam cells in early human atherosclerotic lesions. Acta Pathol. Microbiol. Immunol. Scand. C. 92, 133–137 (1984).
Aqel, N. M., Ball, R. Y., Waldmann, H. & Mitchinson, M. J. Identification of macrophages and smooth muscle cells in human atherosclerosis using monoclonal antibodies. J. Pathol. 146, 197–204 (1985).
Hansson, G. K., Holm, J. & Jonasson, L. Detection of activated T lymphocytes in the human atherosclerotic plaque. Am. J. Pathol. 135, 169–175 (1989).
Jonasson, L., Holm, J., Skalli, O., Bondjers, G. & Hansson, G. K. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis 6, 131–138 (1986).
Emeson, E. E. & Robertson, A. L. T lymphocytes in aortic and coronary intimas: their potential role in atherogenesis. Am. J. Pathol. 130, 369–376 (1988).
Amento, E. P., Ehsani, N., Palmer, H. & Libby, P. Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 11, 1223–1230 (1991).
Warner, S. J. C. & Libby, P. Human vascular smooth muscle cells. Target for and source of tumor necrosis factor. J. Immunol. 142, 100–109 (1989).
Warner, S. J. C., Auger, K. R. & Libby, P. Human interleukin 1 induces interleukin I gene expression in human vascular smooth muscle cells. J. Exp. Med. 165, 1316–1331 (1987).
Geng, Y. J., Wu, Q., Muszynski, M., Hansson, G. K. & Libby, P. Apoptosis of vascular smooth muscle cells induced by in vitro stimulation with interferon-γ, tumor necrosis factor-α, and interleukin-1β. Arterioscler. Thromb. Vasc. Biol. 16, 19–27 (1996).
De Villiers, W. J. S. et al. Macrophage phenotype in mice deficient in both macrophage-colony- stimulating factor (Op) and apolipoprotein E. Arterioscler. Thromb. Vasc. Biol. 18, 631–640 (1998).
Berk, B. C., Weintraub, W. S. & Alexander, R. W. Elevation of C-reactive protein in ‘active’ coronary artery disease. Am. J. Cardiol. 65, 168–172 (1990).
Rajavashisth, T. et al. Heterozygous osteopetrotic (op) mutation reduces atherosclerosis in LDL receptor-deficient mice. J. Clin. Invest. 101, 2702–2710 (1998).
Smith, J. D. et al. Decreased atherosclerosis in mice deficient in both macrophage colony-stimulating factor (op) and apolipoprotein E. Proc. Natl Acad. Sci. USA 92, 8264–8268 (1995).
Salonen, J. T. et al. Autoantibody against oxidised LDL and progression of carotid atherosclerosis. Lancet 339, 883–887 (1992).
The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007).
Samani, N. J. et al. Genomewide association analysis of coronary artery disease. N. Engl. J. Med 357, 443–453 (2007).
Roman, M. J. et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N. Engl. J. Med. 349, 2399–2406 (2003).
Aviña-Zubieta, J. A. et al. Risk of cardiovascular mortality in patients with rheumatoid arthritis: a meta-analysis of observational studies. Arthritis Care Res. 59, 1690–1697 (2008).
Ha, C., Magowan, S., Accortt, N. A., Chen, J. & Stone, C. D. Risk of arterial thrombotic events in inflammatory bowel disease. Am. J. Gastroenterol. 104, 1445–1451 (2009).
Ridker, P. M., Buring, J. E., Shih, J., Matias, M. & Hennekens, C. H. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation 98, 731–733 (1998).
Ross, R. Inflammation or atherogenesis. N. Engl. J. Med. 340, 115–126 (1999).
Hansson, G. K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 352, 1685–1695 (2005).
Tomita, Y. et al. Acute coronary syndrome as a possible immune-related adverse event in a lung cancer patient achieving a complete response to anti-PD-1 immune checkpoint antibody. Ann. Oncol. 28, 2893–2895 (2017).
Bar, J. et al. Acute vascular events as a possibly related adverse event of immunotherapy: a single-institute retrospective study. Eur. J. Cancer 120, 122–131 (2019).
Hansson, G. K. & Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 12, 204–212 (2011).
Maeda, N. Development of apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 31, 1957–1962 (2011).
Ishibashi, S. et al. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Invest. 92, 883–893 (1993).
Shapiro, M. D., Tavori, H. & Fazio, S. PCSK9 from basic science discoveries to clinical trials. Circ. Res. 122, 1420–1438 (2018).
Maxwell, K. N. & Breslow, J. L. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl Acad. Sci. USA 101, 7100–7105 (2004).
Mestas, J. & Hughes, C. C. W. Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731–2738 (2004).
Shay, T. et al. Conservation and divergence in the transcriptional programs of the human and mouse immune systems. Proc. Natl Acad. Sci. USA 110, 2946–2951 (2013).
Von Herrath, M. G. & Nepom, G. T. Lost in translation: barriers to implementing clinical immunotherapeutics for autoimmunity. J. Exp. Med. 202, 1159–1162 (2005).
Graham, A. L. Naturalizing mouse models for immunology. Nat. Immunol. 22, 111–117 (2021).
Greve, J. M. et al. Allometric scaling of wall shear stress from mice to humans: quantification using cine phase-contrast MRI and computational fluid dynamics. Am. J. Physiol. Hear. Circ. Physiol. 291, 1700–1708 (2006).
Golforoush, P., Yellon, D. M. & Davidson, S. M. Mouse models of atherosclerosis and their suitability for the study of myocardial infarction. Basic Res. Cardiol. 115, 73 (2020).
Schwartz, S. M., Galis, Z. S., Rosenfeld, M. E. & Falk, E. Plaque rupture in humans and mice. Arterioscler. Thromb. Vasc. Biol. 27, 705–713 (2007).
Pasterkamp, G. et al. Human validation of genes associated with a murine atherosclerotic phenotype. Arterioscler. Thromb. Vasc. Biol. 36, 1240–1246 (2016).
Breschi, A., Gingeras, T. R. & Guigó, R. Comparative transcriptomics in human and mouse. Nat. Rev. Genet. 18, 425–440 (2017).
Sellers, R. S. Translating mouse models: immune variation and efficacy testing. Toxicol. Pathol. 45, 134–145 (2017).
Mair, K. H. et al. The porcine innate immune system: an update. Dev. Comp. Immunol. 45, 321–343 (2014).
Pabst, R. The pig as a model for immunology research. Cell Tissue Res. 380, 287–304 (2020).
Low, L. A., Mummery, C., Berridge, B. R., Austin, C. P. & Tagle, D. A. Organs-on-chips: into the next decade. Nat. Rev. Drug Discov. 20, 345–361 (2021).
Masopust, D., Sivula, C. P. & Jameson, S. C. Of mice, dirty mice, and men: using mice to understand human immunology. J. Immunol. 199, 383–388 (2017).
Abolins, S. et al. The comparative immunology of wild and laboratory mice, Mus musculus domesticus. Nat. Commun. 8, 14811 (2017).
Proto, J. D. et al. Hypercholesterolemia induces T cell expansion in humanized immune mice. J. Clin. Invest. 128, 2370–2375 (2018).
Cai, B. et al. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J. Clin. Invest. 127, 564–568 (2017).
Doran, A. C. et al. CAMKIIγ suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis. J. Clin. Invest. 127, 4075–4089 (2017).
Thorp, E. et al. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cδ, and p38 mitogen-activated protein kinase (MAPK). J. Biol. Chem. 286, 33335–33344 (2011).
Kojima, Y. et al. Cyclin-dependent kinase inhibitor 2B regulates efferocytosis and atherosclerosis. J. Clin. Invest. 124, 1083–1097 (2014).
Kojima, Y. et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536, 86–90 (2016).
Overton, C. D., Yancey, P. G., Major, A. S., Linton, M. F. & Fazio, S. Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circ. Res. 100, 670–677 (2007).
Advani, R. et al. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N. Engl. J. Med. 379, 1711–1721 (2018).
Ansell, S. M. et al. Phase I study of the CD47 blocker TTI-621 in patients with relapsed or refractory hematologic malignancies. Clin. Cancer Res. 27, 2190–2199 (2021).
Brown, E. J. & Frazier, W. A. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 11, 130–135 (2001).
Buatois, V. et al. Preclinical development of a bispecific antibody that safely and effectively targets CD19 and CD47 for the treatment of B-cell lymphoma and leukemia. Mol. Cancer Ther. 17, 1739–1751 (2018).
Moura, R. et al. Thrombospondin-1 deficiency accelerates atherosclerotic plaque maturation in ApoE−/− mice. Circ. Res. 103, 1181–1189 (2008).
Westlake, S. L. et al. Tumour necrosis factor antagonists and the risk of cardiovascular disease in patients with rheumatoid arthritis: a systematic literature review. Rheumatology 50, 518–531 (2011).
Bäck, M., Yurdagul, A., Tabas, I., Öörni, K. & Kovanen, P. T. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat. Rev. Cardiol. 16, 389–406 (2019).
Fredman, G. et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 7, 12859 (2016).
Thul, S., Labat, C., Temmar, M., Benetos, A. & Bäck, M. Low salivary resolvin D1 to leukotriene B4 ratio predicts carotid intima media thickness: a novel biomarker of non-resolving vascular inflammation. Eur. J. Prev. Cardiol. 24, 903–906 (2017).
Laguna-Fernandez, A. et al. ERV1/ChemR23 signaling protects against atherosclerosis by modifying oxidized low-density lipoprotein uptake and phagocytosis in macrophages. Circulation 138, 1693–1705 (2018).
Hasturk, H. et al. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler. Thromb. Vasc. Biol. 35, 1123–1133 (2015).
Salic, K. et al. Resolvin E1 attenuates atherosclerosis in absence of cholesterol-lowering effects and on top of atorvastatin. Atherosclerosis 250, 158–165 (2016).
Petri, M. H. et al. Aspirin-triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E−/− mice. Br. J. Pharmacol. 174, 4043–4054 (2017).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02329743 (2019).
Schrezenmeier, E. & Dörner, T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat. Rev. Rheumatol. 16, 155–166 (2020).
Sharma, T. S. et al. Hydroxychloroquine use is associated with decreased incident cardiovascular events in rheumatoid arthritis patients. J. Am. Heart Assoc. 5, e002867 (2016).
Jung, H. et al. The protective effect of antimalarial drugs on thrombovascular events in systemic lupus erythematosus. Arthritis Rheum. 62, 863–868 (2010).
Graßhoff, H. et al. Low-dose IL-2 therapy in autoimmune and rheumatic diseases. Front. Immunol. 12, 902 (2021).
Von Spee-Mayer, C. et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 75, 1407–1415 (2016).
Zhao, T. X., Newland, S. A. & Mallat, Z. 2019 ATVB plenary lecture: Interleukin-2 therapy in cardiovascular disease: the potential to regulate innate and adaptive immunity. Arterioscler. Thromb. Vasc. Biol. 40, 853–864 (2020).
Zhao, T. X. et al. Low dose interleukin-2 in patients with stable ischaemic heart disease and acute coronary syndrome (LILACS). Eur. Heart J. 41, e022452 (2020).
Cole, J. E. et al. Unexpected protective role for Toll-like receptor 3 in the arterial wall. Proc. Natl Acad. Sci. USA 108, 2372–2377 (2011).
Salagianni, M. et al. Toll-like receptor 7 protects from atherosclerosis by constraining inflammatory macrophage activation. Circulation 126, 952–962 (2012).
Cole, J. E., Kassiteridi, C. & Monaco, C. Toll-like receptors in atherosclerosis: a ‘Pandora’s box’ of advances and controversies. Trends Pharmacol. Sci. 34, 629–636 (2013).
Dinarello, C. A. Interleukin-1β and the autoinflammatory diseases. N. Engl. J. Med. 360, 2467–2470 (2009).
Edfeldt, K., Swedenborg, J., Hansson, G. K. & Yan, Z. Q. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation 105, 1158–1161 (2002).
Methe, H. et al. Expansion of circulating Toll-like receptor 4-positive monocytes in patients with acute coronary syndrome. Circulation 111, 2654–2661 (2005).
Mullick, A. E. et al. Increased endothelial expression of Toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J. Exp. Med. 205, 373–383 (2008).
Michelsen, K. S. et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl Acad. Sci. USA 101, 10679–10684 (2004).
Liu, X. et al. Toll-like receptor 2 plays a critical role in the progression of atherosclerosis that is independent of dietary lipids. Atherosclerosis 196, 146–154 (2008).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010).
Hornung, V. et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 (2008).
Lüsebrink, E. et al. AIM2 stimulation impairs reendothelialization and promotes the development of atherosclerosis in mice. Front. Cardiovasc. Med. 7, 223 (2020).
Paulin, N. et al. Double-strand DNA sensing Aim2 inflammasome regulates atherosclerotic plaque vulnerability. Circulation 138, 321–323 (2018).
Bauernfeind, F. G. et al. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791 (2009).
Py, B. F., Kim, M. S., Vakifahmetoglu-Norberg, H. & Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 49, 331–338 (2013).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015).
Acknowledgements
This article is dedicated to Prof. Attilio Maseri (1935–2021), who indicated the way for many of us to follow. The authors received funding from the British Heart Foundation (PG/18/1/33430 and PG/19/41/344), the European Commission under the Seventh Framework Programme (FP7/2007-2013, grant agreement number HEALTH-F2-2013-602114 (Athero-B-Cell), HEALTH-F2-2013-602222 (Athero-Flux), HEALTH.2012-1.2-1, contract number 305739 RiskyCAD, and (TAXINOMISIS) grant agreement H2020-SC1-2016-2017, 797788 STRIKING STREAKS), The Kennedy Trustees and the Novo Nordisk Foundation (NNF15CC0018346 and NNF0064142).
Author information
Authors and Affiliations
Contributions
All the authors contributed substantially to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Cardiology thanks C.J. Binder, who co-reviewed with F. Porsch; A. Abbate; K. Ley; and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Engelen, S.E., Robinson, A.J.B., Zurke, YX. et al. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed?. Nat Rev Cardiol 19, 522–542 (2022). https://doi.org/10.1038/s41569-021-00668-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-021-00668-4
This article is cited by
-
Cardiovascular disease and cancer: shared risk factors and mechanisms
Nature Reviews Cardiology (2024)
-
Canonical and non-canonical roles of complement in atherosclerosis
Nature Reviews Cardiology (2024)
-
Effects of lifestyle factors on leukocytes in cardiovascular health and disease
Nature Reviews Cardiology (2024)
-
Synergistic effects of mesenchymal stem cell-derived extracellular vesicles and dexamethasone on macrophage polarization under inflammatory conditions
Inflammopharmacology (2024)
-
Selenopeptide nanomedicine ameliorates atherosclerosis by reducing monocyte adhesions and inflammations
Nano Research (2024)
