
Abstract
Thrombosis is the most feared complication of cardiovascular diseases and a main cause of death worldwide, making it a major health-care challenge. Platelets and the coagulation cascade are effectively targeted by antithrombotic approaches, which carry an inherent risk of bleeding. Moreover, antithrombotics cannot completely prevent thrombotic events, implicating a therapeutic gap due to a third, not yet adequately addressed mechanism, namely inflammation. In this Review, we discuss how the synergy between inflammation and thrombosis drives thrombotic diseases. We focus on the huge potential of anti-inflammatory strategies to target cardiovascular pathologies. Findings in the past decade have uncovered a sophisticated connection between innate immunity, platelet activation and coagulation, termed immunothrombosis. Immunothrombosis is an important host defence mechanism to limit systemic spreading of pathogens through the bloodstream. However, the aberrant activation of immunothrombosis in cardiovascular diseases causes myocardial infarction, stroke and venous thromboembolism. The clinical relevance of aberrant immunothrombosis, referred to as thromboinflammation, is supported by the increased risk of cardiovascular events in patients with inflammatory diseases but also during infections, including in COVID-19. Clinical trials in the past 4 years have confirmed the anti-ischaemic effects of anti-inflammatory strategies, backing the concept of a prothrombotic function of inflammation. Targeting inflammation to prevent thrombosis leaves haemostasis mainly unaffected, circumventing the risk of bleeding associated with current approaches. Considering the growing number of anti-inflammatory therapies, it is crucial to appreciate their potential in covering therapeutic gaps in cardiovascular diseases.
Key points
-
Inflammation and thrombosis are tightly connected processes that contribute to the containment of pathogen spreading in a host defence effector mechanism termed immunothrombosis.
-
The dysregulated and excessive activation of immunothrombosis results in thromboinflammation, causing tissue ischaemia by microvascular and macrovascular thrombosis.
-
The main factor in immunothrombosis and thromboinflammation is a vicious circle of platelet and innate immune cell activation, unleashing the complement system and coagulation cascade.
-
Inflammatory conditions such as infection, chronic autoimmune diseases and clonal haematopoiesis of indeterminate potential are associated with an increased risk of thrombotic events, providing clinical evidence for the partnership between inflammation and thrombosis.
-
Pulmonary immunothrombosis is a prominent feature of severe COVID-19, aggravating respiratory failure and correlating with a systemic prothrombotic phenotype.
-
The inflammatory component of thrombosis is a therapeutic gap and a promising target for the prevention and treatment of cardiovascular diseases such as myocardial infarction, stroke and venous thromboembolism.
Similar content being viewed by others

Introduction
Thrombosis and inflammation are separate physiological processes yet an intense interdependence between these mechanisms has been recognized over the past decade. Physiologically, inflammation-dependent activation of the coagulation system is part of the host response to pathogens, aiming to limit their systemic spread in the bloodstream. This response is achieved through an interplay between innate immune cells and platelets, triggering the activation of the coagulation system in a process termed immunothrombosis1. This process has received considerable attention in the past year owing to the coronavirus disease 2019 (COVID-19) pandemic. Patients with severe COVID-19 progressing to respiratory failure are characterized by an exceedingly high risk of thrombosis and these processes are linked by excessive pulmonary immunothrombosis2,3,4. Excessive activation of immunothrombosis contributes not only to pulmonary failure but also results in thromboinflammation, characterized by a systemic prothrombotic phenotype that can be only partly prevented or reversed with the use of conventional antithrombotic regimens5,6,7,8,9,10,11,12. However, thromboinflammation — that is, the aberrant and excessive activation of immunothrombosis — not only contributes to the thrombotic complications of acute infectious diseases but is also a crucial trigger of non-infectious cardiovascular diseases. The trigger in this setting can be chronic inflammation (such as in atherosclerosis), atherosclerotic plaque rupture or erosion (such as in acute myocardial infarction (MI) and stroke), or stagnant blood flow (such as in venous thromboembolism)13,14,15. Therefore, targeting inflammation to prevent cardiovascular events is an emerging concept not yet covered by current preventive and therapeutic measures. In this Review, we discuss the interplay between inflammation and thrombosis in cardiovascular diseases with a general focus on the clinical implications for cardiovascular medicine and beyond.
Immunothrombosis: an effector mechanism in host defence
The invasion of pathogens into the bloodstream poses a lethal threat to the host and, therefore, containment of the infection is attempted by all available means. The host defence system does not only involve a pronounced immune response but also comprises the activation of thrombotic mechanisms, resulting in immunothrombotic vessel occlusions. Immunothrombosis has a net benefit in the case of a local barrier breach as seen in skin wounds. However, in systemic infections and ensuing sepsis, immunothrombotic dysregulation results in a systemic coagulopathy and multiple organ failure caused by microvascular obstructions depriving the tissue from the blood supply. The main cellular drivers of this process are platelets and innate immune cells, primarily neutrophils, monocytes and macrophages (Box 1). The activating interplay between platelets and immune cells is flanked by the coagulation and complement system, all of which form an intertwined process connecting inflammation and thrombosis.
Limiting bacterial spreading in the bloodstream
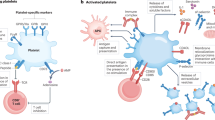
The recognition of bacteria in the bloodstream triggers immunothrombosis through several mechanisms. Neutrophils and platelets detect pathogens through pattern recognition receptors such as Toll-like receptors (TLRs), NOD-like receptors and C-type lectin receptors16,17 (Fig. 1). These cell types have a tightly regulated interplay, which has been uncovered during the past decade. Platelets can migrate within the vasculature — a process distinct from adhesion, aggregation or clot contraction — and function as mechano-scavengers that collect bacteria on their surface and present them to neutrophils18,19. Platelet migration is actomyosin dependent and mediated through the interaction of αIIbβ3 integrin with the surrounding fibrinogen environment. The physical contact between neutrophils and bacteria-loaded migrating platelets triggers neutrophil activation and neutrophil extracellular trap (NET) formation18. NETs are structures consisting of nuclear DNA, histones and neutrophil-derived granule proteins (such as myeloperoxidase and neutrophil elastase) and are expelled from neutrophils to kill bacteria20,21,22,23 (Box 2). NET release is a tightly regulated process involving NADPH oxidases and protein-arginine deiminase type 4 (PAD4). PAD4 converts arginine residues to citrulline, resulting in citrullination of histones, which has been proposed to be crucial for NET formation but its importance seems to depend on the stimulus and the species24,25,26. PAD4-deficient mice are thought to be unable to form NETs, thereby impairing their immune response and protecting them from immunothrombosis26,27. However, citrullinated histone H3 has been used as a marker of NETosis in these studies, an epitope that is absent in PAD4-deficient mice. Therefore, the overall contribution of PAD4 to NETosis might be overestimated; indeed, PAD4-independent pathways of NET release have been described24,28. Moreover, the propensity to form NETs might be an intrinsic property of different neutrophil subpopulations. For example, aged neutrophils have a higher tendency to form NETs and have a higher phagocytic activity than neutrophils just released from the bone marrow29,30,31. Host tools that limit the toxicity of NETosis during immunothrombosis include DNase I and DNase I-like 3, which have protective effects in sepsis by preventing uncontrolled and detrimental microvascular obstruction in animal models32. NETs have also been shown to be degraded within 6 h in vitro by serum from healthy human donors33.

Immunothrombosis is triggered by an invasion of the bloodstream by pathogens. After activation by ADP and thromboxane A2 (TXA2), migrating platelets collect and bundle bacteria and fibrinogen (mediated by αIIbβ3 integrin) on their surface (part a). By actomyosin-dependent migration, platelets present bacteria to neutrophils and boost neutrophil activation. In addition, platelets are activated by Toll-like receptor 4 (TLR4) binding to bacterial products such as lipopolysaccharide (LPS) and pathogen-associated molecular patterns. Together with the complement system, platelets trigger the formation of neutrophil extracellular traps (NETs) through interaction between P-selectin on the platelet surface and P-selectin glycoprotein ligand 1 (PSGL1) on neutrophils. This process depends on NADPH oxidase and protein-arginine deiminase type 4 (PAD4) in neutrophils. NETs trap and kill bacteria but also promote the initiation of the coagulation cascade by activation of the intrinsic pathway (catalysing the activation of factor XII (FXII) to FXIIa) and by degrading the natural anticoagulant tissue factor (TF) pathway inhibitor. The activation of monocytes and macrophages in infection involves the process of pyroptosis (part b). These cells carry on their surface inactive TF, which is a trigger of the extrinsic coagulation pathway. A lytic cell death programme that is dependent on caspase 1, caspase 11, gasdermin D and anoctamin 6 is initiated, resulting in gasdermin pore formation and phosphatidylserine exposure in the plasma membrane. These processes together with the release of protein disulfide-isomerase (PDI) lead to TF activation and the release of TF-rich microvesicles. Finally, activation of the coagulation cascade culminates in thrombin-mediated fibrin generation and formation of an obstructive clot (part c). In parallel, a positive feedback loop is initiated by the cleavage of pro-IL-1α by thrombin into its active form, fostering the activation of innate immune cells. NETosis is counter-regulated by DNase I and DNase I-like 3, which disrupt NETs and prevent the excessive activation of immunothrombosis (part d).
In addition to supporting NETosis, platelets also influence the migratory properties of neutrophils. In inflammation, neutrophils show a distinct polarization with P-selectin glycoprotein ligand 1 (PSGL1) concentrated at the leading edge of the cell, scanning the circulation for activated platelets34. When neutrophils engage in physical interactions with platelets, their migratory capacities and effector functions are boosted34. However, the communication between platelets and neutrophils depends not only on direct cell–cell contacts but also on soluble signals. The detection of bacterial pathogen-associated molecular patterns by platelet TLR4 triggers NET formation and trapping of bacteria in sepsis35. In addition, neutrophil-derived extracellular vesicles are involved in the platelet–neutrophil crosstalk, shuttling the cyclooxygenase 1 substrate arachidonic acid from neutrophils into platelets, thereby fostering thromboxane A2 formation36. Platelet thromboxane A2 release results in endothelial cell activation and neutrophil recruitment in an experimental model of pulmonary infection36.
A major hub in the bidirectional interplay between inflammation and thrombosis is the complement system37. The complement system is activated through several pathways depending on antibodies or pathogen surface molecules and directly lyses or opsonizes invading pathogens. Several complement factors (C1q and C3), anaphylatoxins (C3a and C5a) and the membrane attack complex can activate platelets37,38. Conversely, platelets also provide a surface for complement activation in infectious diseases and platelet-bound complement boosts the inflammatory functions of innate immune cells such as neutrophils39,40. In addition, platelets capture bacteria in the bloodstream in a C3-dependent and platelet glycoprotein Ibα (GPIbα)-dependent manner and shuttle them to antigen-presenting dendritic cells, which prime a T cell-mediated adaptive immune response41. Therefore, multiple reciprocal connections exist between inflammatory pathways and prothrombotic mechanisms that act synergistically to contain bacteria. This defence system limits bacterial spreading beyond the entry site or supports the elimination of bacteria from the circulation in systemic infections by sequestration in the hepatic and pulmonary vasculature18,35,42,43.
Excessive activation of immunothrombosis in sepsis causes thromboinflammation, in which the distinct immunological process of pyroptosis in macrophages causes a pronounced activation of the coagulation system44. Pyroptosis is a caspase 1-dependent cell death programme that involves pore formation in the cell membrane and the release of inflammatory mediators such as IL-1β and IL-18. In addition, inflammasome activation triggers the gasdermin D-dependent lytic pyroptosis of macrophages, leading to the release of tissue factor (TF)-containing microparticles from macrophages44. TF is the main activator of the extrinsic pathway of coagulation and has strong prothrombotic properties14. In general, the activity of TF is influenced by the composition of the plasma membrane and is markedly increased by phosphatidylserine exposure on the cell surface45. TF activation is also controlled by caspase 11-dependent and gasdermin D-dependent processes triggering phosphatidylserine exposure mediated by anoctamin 6 (also known as TMEM16F)46. However, the relevance of this process for the activation of TF on intravascular monocytes in vivo is unclear. In addition, the activity of intravascular TF is regulated by protein disulfide-isomerase (PDI) through the isomerization of a mixed disulfide and a free thiol to an intramolecular disulfide. The release of PDI is tightly regulated to prevent continuous clot formation under physiological conditions. Adherent platelets and damaged vessel wall cells release PDI and facilitate thromboinflammation47.
Not only does inflammation cause thrombosis but thrombosis can in turn directly trigger inflammation. Thrombin cleaves fibrinogen (coagulation) and activates the cytokine IL-1α (inflammation), providing a direct link between coagulation and inflammation. IL-1α is secreted by platelets, macrophages, and keratinocytes and its active form (IL-1α p18) drives innate immunity in response to skin wounds in mice and is also found in the plasma of patients with sepsis48. Furthermore, in a feedback loop, IL-1α p18 boosts thrombopoiesis by inducing the fragmentation of megakaryocytes in the bone marrow48. This increased thrombopoiesis allows the rapid replacement (within hours) of platelets consumed during inflammation. By contrast, thrombopoietin-triggered platelet production requires several days until more platelets are released into the circulation. In general, inflammation causes the activation of haematopoietic stem cells in the bone marrow and their differentiation into the myeloid and megakaryocytic lineages49. During infection-triggered platelet consumption, stem-like megakaryocyte-committed progenitors are activated and rapidly replenish systemic platelet counts50. Therefore, a tight, bidirectional connection exists between inflammation and thrombosis to limit pathogen spreading and causing tissue damage if activation becomes excessive.
Containment of viral infections
As well as being a prominent feature of bacterial infections, immunothrombosis is also involved in host defence in viral diseases, including respiratory infections51. Systemic viral diseases induce the recruitment of platelets and neutrophils to the microvasculature to protect host cells from viral infection52. Platelets can be activated by the binding of immune complexes consisting of H1N1 influenza virus and IgG to the low-affinity FcγRIIA receptor, triggering thrombin generation53. Activated platelets propagate immunothrombosis, for example, by triggering neutrophils to form NETs, a process that involves platelet αIIb integrin and neutrophil αM integrin52,54. Moreover, in influenza A infection, platelets engulf viral particles mediated by TLR7, triggering complement C3-dependent NET formation54. However, influenza A virus and respiratory syncytial virus can also directly trigger NETosis55,56, which has detrimental effects on the course of the disease. Overshooting NETosis causes excessive tissue and endothelial damage by promoting microvascular thrombosis, thereby contributing to the immunopathology of respiratory failure in influenza H1N1 pneumonia in mice57. However, compared with patients with COVID-19 pneumonia, immunothrombotic vessel occlusion in the lungs is a less prominent feature in patients with influenza pneumonia2,58.
Based on the identification of NETs in various viral diseases, the antiviral effects of neutrophil effector proteins have been investigated. These neutrophil effector proteins include defensins, a group of specialized proteins that limit viral infectivity and replication59,60,61. The human cathelicidin antimicrobial peptide LL37 (as well as the murine homologue CRAMP) have been shown to have potent antiviral effects by reducing viral replication in mouse models62.
Thromboinflammation links host defence and cardiovascular diseases
Immunothrombosis is an emergency mechanism of the host to contain infections at the entry site into the bloodstream. However, exaggerated and uncontrolled immunothrombosis generates disastrous collateral damage, compromising organ functions through microvascular thrombosis, which is termed thromboinflammation. This process generates a systemic prothrombotic environment with typical manifestations including venous thromboembolism, MI and stroke. Thromboinflammation is of particular interest in light of the COVID-19 pandemic. In patients with COVID-19, thromboembolic complications are thought to result from aberrant immunothrombosis, as outlined in detail below.
Infectious diseases as a trigger of thrombotic complications
The intimate connection between inflammation and thrombosis is supported by clinical data showing an association between acute systemic infections and thrombotic events. Even in the absence of classic cardiovascular risk factors, respiratory tract infections are associated with an increased risk of MI63. This transiently increased risk of thrombosis induced by viral and bacterial infections is also consistently found for stroke and venous thromboembolism64. Bacteraemia and severe pneumonia are associated with an increased rate of vascular events, suggesting that, rather than infection per se, the ensuing immune reaction of the host is the predominant trigger of macrovascular thrombotic events65,66. This notion is supported by the observation that the increased risk of thrombosis in patients with infection is generally independent of the type of invading pathogen67. In addition, long-term antibiotic therapy has no benefit for the secondary prevention of coronary artery disease68,69. However, some pathogens directly alter both thrombotic and antithrombotic pathways and the exact individual contribution of infection versus the host inflammatory response to thrombotic outcomes is difficult to separate in these scenarios1.
Mechanistically, animal models provide new insights into the pathophysiology of thromboinflammation. NETs are a crucial factor of the coagulopathy seen in experimental sepsis caused by various bacteria35,70. Correspondingly, the disruption of NETs by the administration of DNase or by PAD4 deficiency results in attenuated activation of the coagulation system and platelets, thereby limiting thrombotic vessel occlusions70. In addition, NETs foster monocyte recruitment during endotoxemia, thereby accelerating atherosclerotic plaque formation in a mouse model71. Histones released within NETs provide a molecular link between infection and thrombosis in sepsis. Histones trigger platelet aggregation and endothelial activation and can be deactivated by activated protein C, a natural anticoagulant23,72,73,74. In addition, the activation of the complement system is involved in the hypercoagulable state in thrombosis as shown by bacteria inducing TF-mediated coagulation by complement factor C3 and C5 activation in vitro75,76. This finding is supported by clinical data showing that high C3 levels in the plasma are associated with an increased risk of venous thromboembolism in the general population77. Therefore, infection-induced thrombosis remains an unmet clinical challenge for which potential anti-inflammatory and antithrombotic approaches are very hard to balance with immunosuppression and the risk of bleeding complications.
COVID-19: infection leads to systemic coagulopathy
The crosstalk between inflammation and thrombosis can have severe consequences if one or both systems are thrown out of balance. Currently, the most prominent example is COVID-19, which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. In the ongoing COVID-19 pandemic with millions of people infected worldwide, the most-feared complications are acute respiratory distress syndrome and cardiovascular events such as venous thromboembolism, MI and stroke5,6,7,8,9,10,11,12. Data suggest that the cardiovascular events are caused by a coagulopathy induced by the activation of host defence mechanisms against SARS-CoV-2 (refs3,4,78,79,80). The presence of coagulopathy makes COVID-19 a paradigmatic example of the clinical relevance of immunothrombosis, linking respiratory failure with thrombotic events2,3,4 (Fig. 2).

Immunothrombosis occurring in the lung induced by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection triggers thrombotic events throughout the body. Pulmonary endothelial cells express angiotensin-converting enzyme 2 (ACE2), which is the cell-entry receptor for SARS-CoV-2 (part a). The invasion of endothelial cells by SARS-CoV-2 induces their activation and the exposure of von Willebrand factor (vWF). Platelets and neutrophils are recruited to the site of endothelial activation and engage in a mutually stimulating interplay. In patients with coronavirus disease 2019 (COVID-19), platelets are activated via pathways involving mitogen-activated protein kinase (MAPK) and interferon-induced transmembrane protein 3 (IFITM3). The release of the chemokines CCL5 and CXCL4 by platelets as well as complement activation on platelets triggers neutrophil activation and neutrophil extracellular trap (NET) formation mediated by the binding of the complement factor C5 with its receptor C5aR on neutrophils and the interaction of P-selectin on platelets with the P-selectin glycoprotein ligand 1 (PSGL1) on neutrophils. In addition, neutrophils can be directly infected by SARS-CoV-2 through ACE2 and transmembrane protease serine 2 (TMPRSS2). NETs induce microvascular thrombosis and destroy alveolar epithelial cells, thereby impairing pulmonary gas exchange and aggravating pulmonary failure (part b). Systemically, thromboinflammation is triggered, generating a prothrombotic environment characterized by activated netting neutrophils, stimulated platelets and an activated coagulation system together with elevated fibrinogen and vWF levels and decreased ADAMTS13 levels (part c). The clinical manifestations of this COVID-19-associated coagulopathy are an increased risk of ischaemic stroke, myocardial infarction and venous thromboembolism in patients with severe disease (part d). PAD4, protein-arginine deiminase type 4.
Several clinical findings indicate that immunothrombosis is not just an innocent bystander in COVID-19.
-
The presence of coagulopathy has been reported in patients with COVID-19, characterized by elevated levels of fibrinogen and d-dimer in the plasma, which also correlate with disease severity and prognosis and are indicators of a prothrombotic phenotype80,81,82. Initial studies in Wuhan, China, indicated that most of the patients with COVID-19 who did not survive had a coagulopathy, characterized by a prolonged prothrombin time and activated partial thromboplastin time, mimicking the characteristics observed in disseminated intravascular coagulation80.
-
Both alveolar epithelial cells and vascular endothelial cells express ACE2, which serves as a cell-entry receptor for SARS-CoV-2 (ref.83). Pathological assessment of autopsy specimens indicates that the direct SARS-CoV-2 infection of endothelial cells is associated with necrosis and inflammation, which might foster immune cell and platelet recruitment84. Indeed, patients with severe COVID-19 have elevated levels of von Willebrand factor (vWF) in the blood3,78,85 and a rhesus monkey model of COVID-19 showed increased deposition of vWF on the pulmonary endothelial surface86. In addition, the plasma levels of the metalloproteinase ADAMTS13, which cleaves vWF to regulate vWF multimer size and prothrombotic effects, are decreased in patients with severe COVID-19 compared with those with mild disease and decreased levels of ADAMTS13 are associated with an increased mortality85,87. Therefore, the coagulopathy observed in patients with COVID-19 has similarities to acquired thrombotic microangiopathies.
-
The prothrombotic phenotype in patients with COVID-19 has similarities to the vascular complications seen in patients with antiphospholipid syndrome and, indeed, antiphospholipid antibodies have been detected in patients with COVID-19 (ref.79,88). These antibodies are associated with thrombosis, increased NET formation and more severe respiratory diseases. Therefore, assessment of the presence of these prothrombotic antibodies in convalescent plasma is warranted.
-
Platelets from patients with severe COVID-19 have a hyperreactive phenotype and form aggregates with neutrophils, triggering NETosis, which correlates with disease severity3,89. SARS-CoV-2 mRNA has been detected in platelets, although platelets do not express or contain ACE2 (ref.89). Platelets from patients with severe COVID-19 have a prothrombotic phenotype that is at least partially mediated by the increased generation and release of thromboxane A2 and activation of the MAPK signalling pathway. An immunological function of platelets in COVID-19 is also supported by the finding of upregulated interferon-induced transmembrane protein 3 (IFITM3)89, which is a viral restriction factor against influenza A virus90. In addition, genetic variants in IFITM3 are associated with disease severity in both COVID-19 and influenza90,91. This observation is supported by the presence of circulating megakaryocytes showing features of a marked type I interferon response in critically ill patients with COVID-19, characterized by an upregulation of IFITM3 levels and platelet aggregation pathways92. Therefore, this finding suggests that a bone marrow feedback mechanism is operative in patients with COVID-19, linking inflammation and thrombosis92,93.
-
In patients with COVID-19, neutrophils are prone to NET formation94 and the circulating levels of the chemokines CXCL4 and CCL5, which are platelet-derived triggers for NETosis96, are elevated95. Plasma from patients with COVID-19 induces NETosis in vitro3, which can be attenuated by the administration of neonatal NET inhibitory factor, an endogenous NET inhibitor found in umbilical cord blood97. Moreover, SARS-CoV-2 directly enters neutrophils through ACE2 and TMPRSS2, a process that in turn induces NETosis in a PAD4-dependent manner98.
-
A high burden of NETs has been reported in the coronary thrombi of patients with ST-segment elevation MI (STEMI) and COVID-19 compared with patients with STEMI without COVID-19 (ref.99). NETs are also found in pulmonary microthrombi of autopsy specimens from patients with COVID-19 and platelets are colocalized with netting neutrophils3. Within the lung tissue, NETs destroy lung epithelial cells98. Moreover, markers of NET formation correlate with the degree of respiratory failure in patients with COVID-19 (refs3,95).
-
In severe COVID-19, platelets form aggregates with monocytes, inducing monocyte TF expression in a GPIIb/IIIa-dependent and P-selectin-dependent manner100.
-
The complement system is another effector arm of the immune response to viruses and is involved in the immunopathology of COVID-19. Complement and coagulation activation correlates with disease severity101. In addition, complement factors (C5b-9 and C4b) and the enzyme MASP2 are present in the pulmonary microvasculature of patients with severe COVID-19 (ref.102). Moreover, plasma from patients with COVID-19 induces complement activation in vitro, triggering NETosis in a C5a-dependent manner103. In support of an important role for the complement system in COVID-19, C3 deficiency or inhibition of the C5a–C5a receptor axis attenuates pulmonary disease severity and neutrophil influx in mouse models of SARS-CoV infection104,105.
Therefore, in patients with COVID-19, the dysregulation of immunothrombosis through excessive platelet and neutrophil activation results in coagulopathy, triggering respiratory failure and a systemic prothrombotic state. Some of the above responses have also been reported in the context of other viral diseases53,57,60,90,106. However, COVID-19 is associated with an unprecedented prevalence and degree of intravascular thromboinflammatory activation2,58. Targeting thromboinflammation is therefore an attractive option in these patients and eculizumab (an inhibitor of the terminal complement pathway) is currently being tested in patients with COVID-19 in the SOLID-C19 study107.
Thromboinflammation and cardiovascular events
Although immunothrombosis is a host defence mechanism to contain invading pathogens, its aberrant activation in thromboinflammation has a marked effect on the risk of thrombotic events in sterile inflammatory conditions, particularly in cardiovascular diseases, as well as in autoimmune diseases. However, despite the increasing evidence for the relevance of thromboinflammation in the prevention and therapy of thrombotic and atherosclerotic diseases, the targeting of thromboinflammation is currently not covered in clinical practice. Therefore, a better understanding of the pathophysiology of thromboinflammation and the identification of therapeutic targets that leave the host defence mechanisms intact is an unmet clinical need.
Vicious circle of innate immunity, platelet activation and coagulation in sterile thromboinflammation
A crucial event in thromboinflammation is the mutual activation of platelets and neutrophils resulting in clot formation and vessel occlusion. Platelets are involved in neutrophil recruitment and activation through the release of soluble mediators, such as chemokines (including CCL5, CXCL4, CXCL5 and MIF) and serotonin, and through adhesion molecules such as via P-selectin–PSGL1 interactions108,109,110. In addition, this interplay is supported by platelet GPIbα binding to αMβ2 integrin (also known as MAC1) on neutrophils111. The targeting of these adhesive interactions prevents thrombus formation in various mouse and baboon models of thrombosis without impairing haemostasis22,112,113. Elevated levels of soluble P-selectin are found in patients with cardiovascular diseases and are associated with an elevated risk of MI, stroke and cardiovascular death114,115. Such binding between platelets and neutrophils leads to a drastic change in neutrophil function. These activated neutrophils not only arrest at sites of thrombus formation but also contribute to the propagation of thrombus formation through NETosis. NETs are present in both venous and arterial thrombi in humans, supporting the clinical relevance of NETosis116,117. Platelet-derived P-selectin binding to PSGL1 on neutrophils is a major trigger of NETosis118. In addition, the prototypical damage-associated molecular pattern (DAMP) high mobility group protein B1 (HMGB1) is involved in platelet-triggered NETosis119,120. Platelet-derived HMGB1 induces NET formation through the receptor for advanced glycation end products (RAGE) in vitro and in vivo, promoting thrombotic vessel occlusion119,120. The application of DNase I had protective effects in a mouse model of acute lung injury, whereas DNase I-deficient mice had an aggravated course of disease96.
NETs are an essential interface between thrombosis and inflammation. NETs form a scaffold for the activation of platelets and the coagulation system, boosting their prothrombotic properties. The extracellular DNA backbone of NETs binds vWF, providing a substrate for platelet adhesion and thereby fostering their aggregation21,22. Platelet aggregation is stimulated by NET histones, which activate platelets and promote thrombus formation74. Moreover, NETs directly and indirectly activate the coagulation cascade. The granule enzyme neutrophil elastase degrades TF pathway inhibitor, disinhibiting the extrinsic coagulation pathway and augmenting thrombin formation42. In addition, the intrinsic coagulation pathway can also be activated by NETs. The DNA of NETs provides a negatively charged surface that allows the binding and activation of factor XII22. However, how exactly NETs contribute to thrombosis is still controversial. In vitro, isolated NET components, such as purified histones H3 and H4 or DNA, trigger thrombin generation, whereas whole NETs do not121,122. Therefore, the prothrombotic effect of NETs in vivo might be based on the concentration of prothrombotic factors, such as TF, PDI, factor XII, vWF, complement factors and HMGB1, on their surface, which triggers the coagulation system and activates platelets and endothelial cells.
The interplay between platelets and neutrophils involves several reinforcing feedback loops. A prominent example is the release of cathelicidins by neutrophils during thrombosis. These antimicrobial peptides (such as LL37 or the murine homologue CRAMP) are found in human and mouse arterial thrombi and stabilize experimental arterial thrombosis in vivo123. Cathelicidins induce platelet degranulation and the release of pro-inflammatory mediators, such as HMGB1 and IL-1β, without inducing platelet aggregation123. Cathelicidin-primed platelets interact with neutrophils through P-selectin, which fosters neutrophil activation and NETosis123. In addition to this direct role in thrombosis, neutrophil-derived cathelicidins are also involved in the recruitment of classic monocytes to the arterial endothelium through binding to the formyl-peptide receptor 2, promoting atherosclerotic plaque growth124. By contrast, neutrophil-derived cathelicidins limit neointima formation by promoting re-endothelialization125.
In addition to neutrophils, eosinophils have also been found to stabilize developing thrombi. Eosinophils, which are mainly implicated in fighting parasitic infections and driving autoimmune disease, are a main source of activated TF in the bloodstream and support platelet aggregation through the formation of eosinophil extracellular traps (EETs)126,127,128. Although eosinophils constitute a rare immune cell subset, their prothrombotic effect is remarkably high. Several lines of clinical evidence indicate that elevated eosinophil counts are associated with an increased risk of thrombosis in patients with autoimmune diseases or hypereosinophilic syndrome. The incidence of thrombosis is markedly increased in Churg–Strauss syndrome (eosinophilic granulomatosis with polyangiitis)129, which is characterized by eosinophilia. In idiopathic hypereosinophilic syndrome the high risk of thrombosis has been explained by increased TF expression by eosinophils130,131. Indeed, eosinophil deficiency protects against thrombosis induced by endothelial disruption in mice126,128. This finding is explained by the fact that eosinophils are not only a rich source of TF but they also contain a particularly active form of TF. The thrombin-generation potential of eosinophils is very high because of the 12/15-lipoxygenase-mediated enzymatic generation of a procoagulant phospholipid surface128. Pronounced hydroxyeicosatetraenoic acid–phosphatidylethanolamine exposure activates TF on the eosinophil surface128. Apart from this coagulation-activating effect, eosinophils engage in mutual activating interactions with platelets. P-selectin on platelets triggers the release of EETs, which are decorated with the granule protein major basic protein132. The release of EETs in turn fosters platelet aggregation and stabilizes arterial thrombi in animal models in vivo126. Notably, EETs are found not only in experimental models of thrombosis but also in coronary artery thrombi from patients with MI126. Therefore, eosinophils are an interesting new cellular target for the development of anti-inflammatory approaches in the prevention and therapy of thrombotic diseases.
Venous thrombosis as an aberrant activation of immunothrombosis
Venous thrombosis is still considered to be initiated by the classic factors of the Virchow triad (reduced blood flow velocity, hypercoagulability and endothelial activation) and is characterized by a different thromboinflammatory profile to that of arterial thrombosis14,15. Whereas neutrophils are involved in the propagation of venous thrombosis, the absence of eosinophils does not impair stasis-induced venous thrombosis in mice128. In addition, platelet activation is weaker than in arterial thrombosis owing to the preserved endothelial integrity and lack of exposure of subendothelial extracellular matrix in most cases of venous thrombosis, which are triggered by flow reduction due to patient immobility (Fig. 3). Venous thrombosis can cause lethal pulmonary embolism and has a high rate of recurrence133,134. The current cornerstone of prevention and treatment of venous thrombosis is anticoagulant therapy, which is associated with an inherent risk of bleeding135,136.

Venous thrombosis is mainly triggered by a reduction in blood flow velocity. This reduced blood flow results in an aberrant activation of immunothrombosis, in which a sterile inflammatory response sets the coagulation cascade in motion (part a). Reduced blood flow activates mast cells within the venous vessel wall, which release histamine and activate endothelial cells to mobilize the adhesion molecules P-selectin and von Willebrand factor (vWF) to their surface. Innate immune cells and platelets are recruited to the endothelial surface, which is supported by the binding of all-thiol high mobility group protein B1 (HMGB1) released from platelets to the receptor for advanced glycation end products (RAGE) and Toll-like receptor 2 (TLR2) on monocytes and CXCR2 on neutrophils (part b). CXCR2 activation together with oxidized disulfide HMGB1 interacting with RAGE induce the release of neutrophil extracellular traps (NETs), which are formed in a protein-arginine deiminase type 4 (PAD4)-dependent mechanism. Monocytes are activated by platelet-derived oxidized HMGB1 to release pro-inflammatory mediators such as IL-6 and IL-1β, reinforcing innate immune cell activation (part c). In addition, monocytes release tissue factor (TF), which is activated by protein disulfide-isomerase (PDI) and unleashes the extrinsic coagulation cascade. Clot formation is triggered by TF-dependent thrombin generation and supported by the intrinsic coagulation pathway initiated by the activation of factor XII (FXII) on NETs (part d).
The incidence of venous thrombosis has been increasing over the past decades, although this condition is considered preventable137,138. Therefore, venous thromboembolism is a key health-care challenge and a major cause of death despite established preventive measures139. One reason for the high incidence might be that a crucial feature of the pathophysiology of venous thrombosis, namely inflammation, is not addressed by current therapeutic approaches. In venous thrombosis, an immune reaction is triggered by reduced blood flow velocity in the vein15,21,22,120. Reduced shear stress results in the upregulation of inflammatory NF-κB pathways and increased exposure of adhesion molecules, triggering leukocyte recruitment140,141. However, how a reduction in venous blood flow translates into a full-fledged innate immune response remains unclear. One hint could be derived from the finding of increased venous thrombosis at high altitudes and in the setting of experimental systemic hypoxia142. Under these conditions, hypoxia-inducible factor 1α induces the expression of the NLRP3 inflammasome in endothelial cells, leading to the secretion of IL-1β142. Slowed venous blood flow or stasis might also result in endothelial hypoxia and could trigger similar events. Mast cells have been identified as an additional factor in the venous vessel wall initiating venous thrombosis143. Mast cell mediator release (including histamine) in response to blood flow reduction activates endothelial cells and results in the release of Weibel–Palade bodies containing vWF and P-selectin from endothelial cells143. These adhesion molecules trigger the ensuing immune response, characterized by a complex interplay between platelets, neutrophils and monocytes22. Platelets are crucial for leukocyte recruitment to the vessel wall and activation of innate immune cells, which deliver procoagulant factors, setting in motion the coagulation cascade. In contrast to arterial thrombosis, the activation of the coagulation system in venous thrombosis depends on blood-derived TF, mainly released by monocytes, which is locally activated by PDI22,144. In rolling neutrophils, a cooperative engagement of PSGL1 and the chemokine receptor CXCR2 induces β2 integrin-dependent arrest under low-flow conditions and stimulates the release of NETs145.
The pharmacological disruption of NETs by DNase I administration protects mice from venous thrombosis, providing a proof-of-concept for NETs as therapeutic targets in thrombosis22,146. Another anti-inflammatory approach for the prevention of venous thrombosis is the targeting of HMGB1 (ref.147). The oxidized disulfide form of platelet-derived HMGB1 promotes the release of NETs as well as the expression of TF by monocytes120. HMGB1 acts through several pattern-recognition receptors, including RAGE, TLR2 and TLR4, but only the combined inhibition of these receptors prevents thrombus formation in mice120. Together, this immune response triggers the activation of the extrinsic and intrinsic coagulation pathways, resulting in obstructive clot formation through a dense fibrin network22. Therefore, inflammation is an integral part of the pathophysiology of venous thrombosis that is not yet therapeutically addressed.
Autoimmune diseases as risk factors for thrombotic diseases
Chronic inflammation as in autoimmune diseases is associated with an increased risk of thrombotic events even in the absence of infection. For example, systemic lupus erythematosus (SLE) is associated with a high risk of thrombotic events148. In patients with severe SLE, the degradation of NETs is impaired and NETs therefore permanently activate the complement system149. Conversely, the complement factor C1q present on NETs inhibits NET degradation by interfering with DNase I activity149, creating a vicious circle of NETosis and complement activation. Activated complement factors are also found on platelets in patients with SLE, especially in those with a history of venous thrombosis, suggesting that complement activation is involved in platelet activation and thrombosis in SLE150. Similarly, activation of the complement system is a hallmark of antiphospholipid syndrome, a condition characterized by a high risk of thrombosis and the presence of anti-cardiolipin or anti-β2GPI antibodies151. In addition, in patients with antiphospholipid syndrome, thrombosis and fetal loss are dependent on complement activation, which can be prevented by treatment with heparin, and on neutrophils, highlighting the conserved links between autoimmune diseases and thromboinflammation152,153,154. Moreover, in SLE, NETs can indirectly boost the immune response through the release of DAMPs, such as HMGB1 and histones, which in turn activate other innate and adaptive immune cells155.
The risk of MI is drastically increased in patients with rheumatoid arthritis156. In addition to accelerated atheroprogression157,158, the activation and dysregulation of thromboinflammation is likely to contribute to this observation. Neutrophils from patients with rheumatoid arthritis have a high propensity to form NETs, which in turn support thrombosis (see previous sections)159. At the same time, increased NETosis combined with the inability to degrade NETs becomes a perpetuating factor of autoimmune disease in which NET components, such as DNA and histones, function as autoantigens and induce an antibody response. In particular, protein citrullination during NETosis seems to be an essential modification step triggering the pathognomonic autoantibodies against citrullinated molecules in rheumatoid arthritis, causing an aberrant adaptive and innate immune response159. The studies mentioned in this section indicate that chronic sterile inflammation increases the risk of both venous and arterial thrombotic events.
Crosstalk between platelets and innate immunity promotes atherothrombosis
Arterial thrombosis is primarily triggered by atherosclerotic plaque rupture, resulting in the exposure of the subendothelial extracellular matrix and extensive release of TF from damaged tissue13. The interplay between platelets and innate immune cells is a crucial driver of atherosclerotic plaque initiation and progression as well as of thrombotic vessel occlusion. Platelets adhere to the endothelium during plaque formation through the adhesion molecules GPIbα and αIIbβ3 integrin and support leukocyte recruitment into the growing plaque160,161. The platelet CD40 ligand is involved in inducing both leukocyte and endothelial cell activation162. The arrest of monocytes and neutrophils on the endothelium is mediated by platelet-derived chemokines, among which CCL5–CXCL4 heteromers have a major role163,164. In addition, platelets recruit bone marrow-derived progenitor cells to the atherosclerotic plaque by releasing CXCL12, which potentially contributes to vascular repair165. The destruction of vascular smooth muscle cells by the cytotoxic effects of histone H4 from NETs is involved in the destabilization of the plaque166. Eosinophils are also implicated in atherosclerosis. The plasma levels of eosinophil cationic protein are positively correlated with the extent of atherosclerosis in patients with coronary artery disease167,168. The eosinophil chemokine eotaxin (also known as CCL11) is highly expressed in human atherosclerotic plaques169,170,171. In addition, eosinophil-deficient mice have decreased atherosclerotic plaque formation and attenuated platelet adhesion to the endothelium compared with mice with normal eosinophil levels126. Eosinophils are markedly enriched in coronary artery and ischaemic stroke thrombi from patients116,172, and a genome-wide association study for sequence variants affecting systemic eosinophil counts reported an association with the risk of MI173.
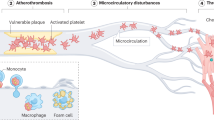
The interplay between platelets and myeloid cells also has a pivotal role during arterial thrombosis after plaque rupture, which causes MI and stroke174. Following plaque rupture and rapid platelet recruitment mediated by GPIbα–vWF and GPVI–collagen interactions, a mutually activating interplay between platelets, neutrophils and eosinophils evolves, mediated by several adhesion molecules, including P-selectin110,111. The targeting of P-selectin has already been tested in clinical trials in patients with non-STEMI175. Infusion of the recombinant monoclonal P-selectin-blocking antibody inclacumab before percutaneous coronary intervention reduced myocardial damage compared with placebo175. A role of the neutrophil α9β1 integrin in arterial thrombosis has been identified in mice176. A deficiency of α9β1 integrin in myeloid leukocytes reduced arterial thrombosis by attenuating platelet-induced NETosis, neutrophil-mediated platelet aggregation and cathepsin G release176 (Fig. 4). Cathepsin G supports platelet activation and myeloid cell recruitment to the arterial vessel wall in mice177,178. In addition, the neutrophil-derived antimicrobial cathelicidin LL37/CRAMP induces platelet activation involving GPVI and downstream signalling via the tyrosine-protein kinases SRC and SYK and phospholipase C123. In a feedback loop, activated platelets stimulate NETosis, mediated by the binding of platelet-derived HMGB1 to RAGE on neutrophils119,120. HMGB1 also activates platelets through a TLR4–MyD88 pathway and the inhibition of HMGB1 attenuates arterial thrombus formation179. Although the contribution of PAD4-dependent NETosis to atherosclerotic plaque formation is controversial, this process is involved in the destabilization and erosion of plaques180,181. Accordingly, PAD4 deficiency or pharmacological inhibition of NETosis impairs arterial thrombosis in several mouse models180,182. In addition to their prothrombotic effects, NETs can activate the complement system, which aggravates endothelial damage and boosts platelet activation180,183,184. Accordingly, the deficiency of complement factor C3 attenuates arterial thrombosis in mice owing to the disruption of the C3a–C3a receptor axis in platelets, an axis that induces platelet activation via RAS-related protein RAP1B185,186. In summary, several inflammatory pathways contribute to arterial vessel occlusions, which are not targeted by current therapeutic approaches.

Arterial thrombosis is induced by endothelial disruption caused by atherosclerotic plaque rupture or erosion. Platelets are recruited to the exposed subendothelial matrix and aggregate (part a). Neutrophils are activated by the binding of platelet-derived high mobility group protein B1 (HMGB1) to the receptor for advanced glycation end products (RAGE), by platelet P-selectin interacting with neutrophil P-selectin glycoprotein ligand 1 (PSGL1) and by α9β1 integrin ligation (part b). In parallel, platelets mediate the recruitment and activation of eosinophils in an integrin-dependent manner. Platelet-induced activation of neutrophils results in the formation of neutrophil extracellular traps (NETs), which promote the activation of the coagulation system (by degrading tissue factor (TF) pathway inhibitor (TFPI) and activating the coagulation factor XII (FXII)) (part c). In addition, platelet activation is reinforced by the release of cathelicidin antimicrobial peptides (LL37 in humans and CRAMP in mice) as well as by HMGB1 binding to platelet Toll-like receptor 4 (TLR4). Activated complement factor C3a binds to its receptor C3aR on platelets, promoting platelet activation. Activated platelets through P-selectin–PSGL1 interactions induce the formation of eosinophil extracellular traps (EETs) containing the granule protein major basic protein (MBP), which fosters platelet aggregation. Together, these processes result in the excessive activation of platelets and the coagulation system, leading to arterial occlusions in myocardial infarction and stroke. vWF, von Willebrand factor.
The connection between clonal haematopoiesis, inflammation and cardiovascular diseases
Over the past 4 years, another non-infectious aspect of leukocyte biology has been added to the traditional risk factors of cardiovascular diseases, namely clonal haematopoiesis of indeterminate potential (CHIP). This entity is characterized by the accumulation of somatic mutations in haematopoietic stem cells that provide a competitive advantage. These haematopoietic stem cell clones disproportionally contribute to the pool of peripheral leukocytes, which increases with age and has the potential to transform into haematological malignancies. However, even in the absence of the haematological malignancies, individuals with CHIP have an increased mortality compared with matched individuals without CHIP owing to an increased rate of cardiovascular events187,188. Variants in a few genes account for most cases of CHIP: TET2, DNMT3A and ASXL1 (which encode proteins involved in the epigenetic regulation of haematopoiesis) and JAK2 (which encodes a protein involved in growth factor signalling in haematopoietic cells)187,188,189. CHIP-related variants in these genes potentially contribute to increasing the risk of thrombotic events by promoting atherosclerotic plaque formation and by generating a prothrombotic environment caused by a pro-inflammatory effect. The JAK2V617F variant is the most common cause of myeloproliferative neoplasms, but, even in the absence of this malignancy, the presence of this variant is associated with an increased risk of thrombotic events owing to functional changes in neutrophils190,191. This increased risk of thrombosis involves an increased propensity of JAK2V617F neutrophils to form NETs in a PAD4-dependent manner191. Irradiated Ldlr–/– mice receiving transplantation with Jak2V617F bone marrow had increased atherosclerotic plaque formation compared with mice receiving wild-type bone marrow192. In addition, in mice, the absence of Tet2 in macrophages results in a pro-inflammatory phenotype characterized by high levels of NLRP3 inflammasome-dependent IL-1β secretion and by increased release of chemokines such as CXCL1 and CXCL3 and the cytokine IL-6 (refs189,193,194). However, in contrast to the mounting evidence supporting a link between JAK2V617F and thromboinflammation, the link is less clear for TET2. Despite the implication of JAK2 and DNMT3A variants in quantitative and qualitative changes of platelets, the overall importance of the direct effects of CHIP on the megakaryocyte lineage and platelet production are less defined195. However, these interesting findings have not been translated into clinical practice. The screening for the presence of CHIP is still expensive and relies on sequencing, which is not universally available. In addition, the clinical consequences of the presence of CHIP for the prevention of cardiovascular events needs to be clarified.
Thromboinflammation as a therapeutic target
Based on findings showing an intimate connection between inflammation and thrombosis, targeting the interface between these processes to prevent thrombosis seems promising. Increasing evidence shows that current therapies targeting thrombosis also modulate inflammatory processes196,197,198,199,200,201, which contributes to the beneficial effects of these therapies. Conversely, anti-inflammatory approaches can prevent thrombotic events202,203,204, indicating that these strategies might not only affect atherosclerotic plaque formation. However, these additional antithrombotic or anti-inflammatory effects are unselective and a more tailored approach specifically targeting thromboinflammation is an unmet clinical need.
Effects of current antithrombotic therapeutics on inflammation
Although current antithrombotic approaches are designed to target platelets and the coagulation system, these therapies also have distinct effects on inflammation, demonstrating the tight interaction between these processes (Table 1). Findings from the COMPASS198 and VOYAGER PAD196 trials highlight the potential anti-inflammatory effects of anticoagulants that potentially affect plaque progression. The administration of the direct factor Xa inhibitor rivaroxaban prevents ischaemic events in patients with cardiovascular disease but might potentially also attenuate the progression of atherosclerosis as has been shown in mice196,198,200. In experimental models of myocardial ischaemia–reperfusion injury, the inflammatory response mediated by NF-κB and inflammasome activation was attenuated by inhibition of factor Xa, characterized by the decreased expression of pro-inflammatory mediators199. In patients with atrial fibrillation, oral anticoagulation with rivaroxaban or a vitamin K antagonist decreased the levels of IL-6 and C-reactive protein in the plasma205. The molecular link for this bidirectional interplay of thrombosis and inflammation might be proteinase-activated receptors (PARs), which self-activate after proteolytic cleavage by serine proteases such as factor X and thrombin201. PARs are found on many vascular cell types, including platelets, myeloid leukocytes, endothelial cells and smooth muscle cells, in which PAR activation has pro-inflammatory and pro-atherogenic effects197. For example, PAR1 and PAR2 activation induces the production of cytokines and chemokines and the exposure of adhesion molecules in endothelial cells206,207,208. Moreover, factor Xa-dependent PAR1 activation elicits the expression of CCL2, IL-6 and IL-8 in endothelial cells and mononuclear leukocytes206. Rivaroxaban treatment limits arterial hypertension induced by renal damage in mice through the inhibition of the pro-inflammatory effects of PAR2 (ref.209). In addition, heparin has established anti-inflammatory effects, which are at least partly mediated by the disruption of NETs21,22. Heparin also neutralizes the detrimental effect of histones independently of its effects on coagulation210.
Antiplatelet therapies do not only modulate the thrombotic function of platelets but also have effects on inflammation211. ADP binding to platelets, which strongly induces platelet aggregation and the release of pro-inflammatory mediators, is blocked by P2Y purinoceptor 12 inhibitors such as clopidogrel and prasugrel212. Interestingly, treatment with ticagrelor but not with prasugrel reduces IL-6 and TNF levels in the plasma of patients with diabetes mellitus after non-STEMI, potentially by inhibiting adenosine uptake through the equilibrative nucleoside transporter 1 (ref.213). The anti-inflammatory effects of aspirin are mainly dose dependent, mediated by cyclooxygenase inhibition and decreased synthesis of pro-inflammatory prostaglandins and thromboxane A2. The low dose of aspirin used for the prevention of cardiovascular events is considered to have mainly antiplatelet effects. However, even low-dose aspirin attenuates interstitial leukocyte accumulation in experimental inflammation, driven by the increased synthesis of the pro-resolution mediator 15-epi-lipoxin A4 and upregulation of its receptor formyl peptide receptor-like 1 (refs214,215). These findings demonstrate that inflammation is partially and unspecifically targeted by current antithrombotic regimens.
New approaches targeting inflammation to prevent thrombotic cardiovascular events
Several clinical studies have provided proof-of-concept data showing that the specific inhibition of inflammation can provide additional benefits for preventing ischaemic events202,203,204 (Table 2); some of these studies specifically evaluated the period after MI. This period is characterized by marked activation of the innate immune system and data from studies in animals indicate that this detrimental immune response to myocardial injury systemically aggravates atherosclerosis216. MI induces the proliferation and mobilization of bone marrow haematopoietic stem and progenitor cells, which engraft in the spleen, resulting in sustained monocyte production216,217. This increased haematopoietic stem cell proliferation in the bone marrow depends on IL-1β216,217. In accordance with this finding, the CANTOS trial203 showed that, in patients with previous MI and elevated levels of high-sensitivity C-reactive protein in the plasma, treatment with the anti-IL-1β monoclonal antibody canakinumab in addition to state-of-the-art secondary prophylaxis provided an additional benefit in reducing the rate of recurrent MI or coronary revascularization. However, all-cause mortality was not affected, mainly owing to the increased risk of fatal infections203. The risk of infection is not the only concern with this expensive therapy. In experimental models of advanced atherosclerosis, IL-1β promotes plaque stabilization by the formation of a fibrous cap rich in smooth muscle cells and collagen218,219. Other less expensive but less selective anti-inflammatory agents than canakinumab have been tested. The addition of low-dose methotrexate to standard therapy had no benefit in reducing ischaemic events in patients with previous MI or multivessel coronary artery disease in the CIRT trial220. By contrast, in the COLCOT trial204, treatment with the established anti-inflammatory drug colchicine induced a significant reduction in the risk of MI and stroke when given to patients early after MI. Colchicine has profound effects on the activation of neutrophils because this drug inhibits NET formation and attenuates NLRP3 inflammasome activation221,222, which decreases caspase 1 activation and the processing and release of IL-1β and IL-18 (ref.222). Interestingly, in the LoDoCo2 trial223, the administration of colchicine to patients with a history of MI resulted in a marked reduction in inflammatory mediators within 30 days. NLRP3 inflammasome-associated molecules, such as IL-18, and inflammatory mediators released during neutrophil degranulation, such as myeloperoxidase, were markedly decreased. Moreover, levels of platelet GPVI (which mediates collagen-induced platelet activation and aggregation) were also reduced with colchicine treatment223. However, in the COLCOT trial204, colchicine therapy also had an immunosuppressive effect, with an increased incidence of pneumonia. Therefore, the CANTOS and COLCOT trials provide a proof-of-concept that targeting inflammation can prevent ischaemic events in the inflammatory environment of the period after MI. However, a better understanding of the systemic immune response to MI as well as therapeutic approaches specifically addressing thromboinflammation are necessary to target the detrimental pathways boosting atherosclerosis and thrombosis while leaving protective immunity against pathogens unaffected.
Deep vein thrombosis and arterial thrombosis, both prototypical examples of thromboinflammation, share many aspects with sterile inflammation. Therefore, in the context of deep vein thrombosis, DAMPs such as HMGB1 are promising therapeutic targets because they do not affect host defence. In the setting of arterial thrombosis, a potential option is to target eosinophils. Several established therapies deplete eosinophils, which does not result in an increased risk of infection224,225,226. In addition, a growing number of complement therapeutics are available, which might provide attractive approaches for targeting thromboinflammation227. However, targeting the detrimental aspect of immunothrombosis is still challenging and has not been translated into clinical practice.
Conclusions
The link between inflammation and thrombosis is an emerging experimental field in which a lot of mechanistic insight is derived from mouse models, which might not completely reflect human disease pathophysiology and immunity. However, accumulating evidence supports the relevance of this process in cardiovascular diseases in humans. Intriguingly, dysregulated immunothrombosis is involved in the whole spectrum of cardiovascular pathology. Consequently, inflammation is now seen as a promising therapeutic target in addition to optimizing risk factors and targeting platelets and the coagulation system. However, balancing the potential immunosuppressive adverse effects and the impairment of haemostasis make this combined approach challenging. Human data providing specific molecular targets of immunothrombosis are still insufficient and warrant further study. Identifying the potential therapeutic targets of immunothrombosis holds great promise of covering therapeutic gaps in cardiovascular medicine with anti-inflammatory approaches and might also help to fight thrombotic complications in infectious diseases, including COVID-19.
References
Engelmann, B. & Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 13, 34–45 (2013).
Ackermann, M. et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N. Engl. J. Med. 383, 120–128 (2020).
Nicolai, L. et al. Immunothrombotic dysregulation in COVID-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation 142, 1176–1189 (2020).
Wichmann, D. et al. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann. Intern. Med. 173, 268–277 (2020).
Creel-Bulos, C. et al. Acute cor pulmonale in critically ill patients with Covid-19. N. Engl. J. Med. 382, e70 (2020).
Cui, S., Chen, S., Li, X., Liu, S. & Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 18, 1421–1424 (2020).
Klok, F. A. et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 191, 148–150 (2020).
Klok, F. A. et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 191, 145–147 (2020).
Poissy, J. et al. Pulmonary embolism in COVID-19 patients: awareness of an increased prevalence. Circulation 142, 184–186 (2020).
Oxley, T. J. et al. Large-vessel stroke as a presenting feature of Covid-19 in the young. N. Engl. J. Med. 382, e60 (2020).
Bangalore, S. et al. ST-segment elevation in patients with Covid-19 – a case series. N. Engl. J. Med. 382, 2478–2480 (2020).
Stefanini, G. G. et al. ST-elevation myocardial infarction in patients with COVID-19: clinical and angiographic outcomes. Circulation 141, 2113–2116 (2020).
Jackson, S. P. Arterial thrombosis–insidious, unpredictable and deadly. Nat. Med. 17, 1423–1436 (2011).
Mackman, N. Triggers, targets and treatments for thrombosis. Nature 451, 914–918 (2008).
Mackman, N. New insights into the mechanisms of venous thrombosis. J. Clin. Invest. 122, 2331–2336 (2012).
Hottz, E. D. et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood 122, 3405–3414 (2013).
Semple, J. W., Italiano, J. E. Jr. & Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 11, 264–274 (2011).
Gaertner, F. et al. Migrating platelets are mechano-scavengers that collect and bundle bacteria. Cell 171, 1368–1382 (2017).
Nicolai, L. et al. Vascular surveillance by haptotactic blood platelets in inflammation and infection. Nat. Commun. 11, 5778 (2020).
Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535 (2004).
Fuchs, T. A. et al. Extracellular DNA traps promote thrombosis. Proc. Natl Acad. Sci. USA 107, 15880–15885 (2010).
von Bruhl, M. L. et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 209, 819–835 (2012).
Xu, J. et al. Extracellular histones are major mediators of death in sepsis. Nat. Med. 15, 1318–1321 (2009).
Kenny, E. F. et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife 6, e24437 (2017).
Lewis, H. D. et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 11, 189–191 (2015).
Martinod, K. et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl Acad. Sci. USA 110, 8674–8679 (2013).
Li, P. et al. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 207, 1853–1862 (2010).
Sollberger, G., Tilley, D. O. & Zychlinsky, A. Neutrophil extracellular traps: the biology of chromatin externalization. Dev. Cell 44, 542–553 (2018).
Silvestre-Roig, C., Hidalgo, A. & Soehnlein, O. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood 127, 2173–2181 (2016).
Uhl, B. et al. Aged neutrophils contribute to the first line of defense in the acute inflammatory response. Blood 128, 2327–2337 (2016).
Zhang, D. et al. Neutrophil ageing is regulated by the microbiome. Nature 525, 528–532 (2015).
Jimenez-Alcazar, M. et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 358, 1202–1206 (2017).
Hakkim, A. et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl Acad. Sci. USA 107, 9813–9818 (2010).
Sreeramkumar, V. et al. Neutrophils scan for activated platelets to initiate inflammation. Science 346, 1234–1238 (2014).
Clark, S. R. et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 13, 463–469 (2007).
Rossaint, J. et al. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 7, 13464 (2016).
Verschoor, A. & Langer, H. F. Crosstalk between platelets and the complement system in immune protection and disease. Thromb. Haemost. 110, 910–919 (2013).
Schmidt, C. Q. & Verschoor, A. Complement and coagulation: so close, yet so far. Blood 130, 2581–2582 (2017).
Peerschke, E. I., Yin, W. & Ghebrehiwet, B. Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol. Immunol. 47, 2170–2175 (2010).
Polley, M. J. & Nachman, R. The human complement system in thrombin-mediated platelet function. J. Exp. Med. 147, 1713–1726 (1978).
Verschoor, A. et al. A platelet-mediated system for shuttling blood-borne bacteria to CD8α+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat. Immunol. 12, 1194–1201 (2011).
Massberg, S. et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 16, 887–896 (2010).
Yipp, B. G. et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 18, 1386–1393 (2012).
Wu, C. et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity 50, 1401–1411 (2019).
Bach, R., Gentry, R. & Nemerson, Y. Factor VII binding to tissue factor in reconstituted phospholipid vesicles: induction of cooperativity by phosphatidylserine. Biochemistry 25, 4007–4020 (1986).
Yang, X. et al. Bacterial endotoxin activates the coagulation cascade through gasdermin D-dependent phosphatidylserine exposure. Immunity 51, 983–996 (2019).
Reinhardt, C. et al. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J. Clin. Invest. 118, 1110–1122 (2008).
Burzynski, L. C. et al. The coagulation and immune systems are directly linked through the activation of interleukin-1α by thrombin. Immunity 50, 1033–1042 (2019).
Pietras, E. M. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood 130, 1693–1698 (2017).
Haas, S. et al. Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell 17, 422–434 (2015).
Schönrich, G. & Raftery, M. J. Neutrophil extracellular traps go viral. Front. Immunol. 7, 366 (2016).
Jenne, C. N. et al. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 13, 169–180 (2013).
Boilard, E. et al. Influenza virus H1N1 activates platelets through FcγRIIA signaling and thrombin generation. Blood 123, 2854–2863 (2014).
Koupenova, M. et al. The role of platelets in mediating a response to human influenza infection. Nat. Commun. 10, 1780 (2019).
Funchal, G. A. et al. Respiratory syncytial virus fusion protein promotes TLR-4-dependent neutrophil extracellular trap formation by human neutrophils. PLoS ONE 10, e0124082 (2015).
Tripathi, S., Verma, A., Kim, E. J., White, M. R. & Hartshorn, K. L. LL-37 modulates human neutrophil responses to influenza A virus. J. Leukoc. Biol. 96, 931–938 (2014).
Narasaraju, T. et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 179, 199–210 (2011).
Nicolai, L. et al. Vascular neutrophilic inflammation and immunothrombosis distinguish severe COVID-19 from influenza pneumonia. J. Thromb. Haemost. 19, 574–581 (2020).
Doss, M. et al. Interactions of α-, β-, and θ-defensins with influenza A virus and surfactant protein D. J. Immunol. 182, 7878–7887 (2009).
Hartshorn, K. L., White, M. R., Tecle, T., Holmskov, U. & Crouch, E. C. Innate defense against influenza A virus: activity of human neutrophil defensins and interactions of defensins with surfactant protein D. J. Immunol. 176, 6962–6972 (2006).
Salvatore, M. et al. α-Defensin inhibits influenza virus replication by cell-mediated mechanism(s). J. Infect. Dis. 196, 835–843 (2007).
Barlow, P. G. et al. Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS ONE 6, e25333 (2011).
Meier, C. R., Jick, S. S., Derby, L. E., Vasilakis, C. & Jick, H. Acute respiratory-tract infections and risk of first-time acute myocardial infarction. Lancet 351, 1467–1471 (1998).
Smeeth, L. et al. Risk of myocardial infarction and stroke after acute infection or vaccination. N. Engl. J. Med. 351, 2611–2618 (2004).
Corrales-Medina, V. F. et al. Cardiac complications in patients with community-acquired pneumonia: incidence, timing, risk factors, and association with short-term mortality. Circulation 125, 773–781 (2012).
Dalager-Pedersen, M., Sogaard, M., Schonheyder, H. C., Nielsen, H. & Thomsen, R. W. Risk for myocardial infarction and stroke after community-acquired bacteremia: a 20-year population-based cohort study. Circulation 129, 1387–1396 (2014).
Beristain-Covarrubias, N. et al. Understanding infection-induced thrombosis: lessons learned from animal models. Front. Immunol. 10, 2569 (2019).
Grayston, J. T. et al. Azithromycin for the secondary prevention of coronary events. N. Engl. J. Med. 352, 1637–1645 (2005).
O’Connor, C. M. et al. Azithromycin for the secondary prevention of coronary heart disease events: the WIZARD study: a randomized controlled trial. JAMA 290, 1459–1466 (2003).
McDonald, B. et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 129, 1357–1367 (2017).
Schumski, A. et al. Endotoxinemia accelerates atherosclerosis via electrostatic charge-mediated monocyte adhesion. Circulation 143, 254–266 (2020).
Ammollo, C. T., Semeraro, F., Xu, J., Esmon, N. L. & Esmon, C. T. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb. Haemost. 9, 1795–1803 (2011).
Semeraro, F., Ammollo, C. T., Esmon, N. L. & Esmon, C. T. Histones induce phosphatidylserine exposure and a procoagulant phenotype in human red blood cells. J. Thromb. Haemost. 12, 1697–1702 (2014).
Semeraro, F. et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 118, 1952–1961 (2011).
Landsem, A. et al. The key roles of complement and tissue factor in Escherichia coli-induced coagulation in human whole blood. Clin. Exp. Immunol. 182, 81–89 (2015).
Ovstebo, R. et al. Microparticle-associated tissue factor activity is reduced by inhibition of the complement protein 5 in Neisseria meningitidis-exposed whole blood. Innate Immun. 20, 552–560 (2014).
Norgaard, I., Nielsen, S. F. & Nordestgaard, B. G. Complement C3 and high risk of venous thromboembolism: 80517 individuals from the Copenhagen general population study. Clin. Chem. 62, 525–534 (2016).
Goshua, G. et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 7, e575–e582 (2020).
Zhang, Y. et al. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N. Engl. J. Med. 382, e38 (2020).
Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062 (2020).
Tang, N., Li, D., Wang, X. & Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 18, 844–847 (2020).
Chen, G. et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 130, 2620–2629 (2020).
Hamming, I. et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203, 631–637 (2004).
Varga, Z. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 395, 1417–1418 (2020).
Mancini, I. et al. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J. Thromb. Haemost. 19, 513–521 (2020).
Aid, M. et al. Vascular disease and thrombosis in SARS-CoV-2-infected rhesus macaques. Cell 183, 1354–1366 (2020).
Bazzan, M. et al. Low ADAMTS 13 plasma levels are predictors of mortality in COVID-19 patients. Intern. Emerg. Med. 15, 861–863 (2020).
Zuo, Y. et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl Med. 12, eabd3876 (2020).
Manne, B. K. et al. Platelet gene expression and function in COVID-19 patients. Blood 136, 1317–1329 (2020).
Everitt, A. R. et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484, 519–523 (2012).
Zhang, Y. et al. Interferon-induced transmembrane protein 3 genetic variant rs12252-C associated with disease severity in coronavirus disease 2019. J. Infect. Dis. 222, 34–37 (2020).
Bernardes, J. P. et al. Longitudinal multi-omics analyses identify responses of megakaryocytes, erythroid cells, and plasmablasts as hallmarks of severe COVID-19. Immunity 53, 1296–1314 (2020).
Campbell, R. A. et al. Human megakaryocytes possess intrinsic antiviral immunity through regulated induction of IFITM3. Blood 133, 2013–2026 (2019).
Zuo, Y. et al. Neutrophil extracellular traps in COVID-19. JCI Insight 5, e138999 (2020).
Middleton, E. A. et al. Neutrophil extracellular traps (NETs) contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 136, 1169–1179 (2020).
Rossaint, J. et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood 123, 2573–2584 (2014).
Yost, C. C. et al. Neonatal NET-inhibitory factor and related peptides inhibit neutrophil extracellular trap formation. J. Clin. Invest. 126, 3783–3798 (2016).
Veras, F. P. et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 217, e20201129 (2020).
Blasco, A. et al. Assessment of neutrophil extracellular traps in coronary thrombus of a case series of patients with COVID-19 and myocardial infarction. JAMA Cardiol. 6, 469–474 (2021).
Hottz, E. D. et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 136, 1330–1341 (2020).
Ramlall, V. et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat. Med. 26, 1609–1615 (2020).
Magro, C. et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 220, 1–13 (2020).
Skendros, P. et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Invest. 130, 6151–6157 (2020).
Gralinski, L. E. et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio 9, e01753-18 (2018).
Jiang, Y. et al. Blockade of the C5a-C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg. Microbes Infect. 7, 77 (2018).
Le, V. B. et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am. J. Respir. Crit. Care Med. 191, 804–819 (2015).
Risitano, A. M. et al. Complement as a target in COVID-19? Nat. Rev. Immunol. 20, 343–344 (2020).
Duerschmied, D. et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood 121, 1008–1015 (2013).
Karshovska, E., Weber, C. & von Hundelshausen, P. Platelet chemokines in health and disease. Thromb. Haemost. 110, 894–902 (2013).
Palabrica, T. et al. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature 359, 848–851 (1992).
Wang, Y. et al. Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbα. Nat. Commun. 8, 15559 (2017).
Downing, L. J. et al. Anti-P-selectin antibody decreases inflammation and thrombus formation in venous thrombosis. J. Vasc. Surg. 25, 816–827 (1997).
Myers, D. D. Jr. et al. Decreased venous thrombosis with an oral inhibitor of P selectin. J. Vasc. Surg. 42, 329–336 (2005).
Blann, A. D., Nadar, S. K. & Lip, G. Y. The adhesion molecule P-selectin and cardiovascular disease. Eur. Heart J. 24, 2166–2179 (2003).
Ridker, P. M., Buring, J. E. & Rifai, N. Soluble P-selectin and the risk of future cardiovascular events. Circulation 103, 491–495 (2001).
Riegger, J. et al. Histopathological evaluation of thrombus in patients presenting with stent thrombosis. A multicenter European study: a report of the prevention of late stent thrombosis by an interdisciplinary global European effort consortium. Eur. Heart J. 37, 1538–1549 (2016).
Savchenko, A. S. et al. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J. Thromb. Haemost. 12, 860–870 (2014).
Etulain, J. et al. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 126, 242–246 (2015).
Maugeri, N. et al. Activated platelets present high mobility group Box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 12, 2074–2088 (2014).
Stark, K. et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 128, 2435–2449 (2016).
Noubouossie, D. F., Reeves, B. N., Strahl, B. D. & Key, N. S. Neutrophils: back in the thrombosis spotlight. Blood 133, 2186–2197 (2019).
Noubouossie, D. F. et al. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 129, 1021–1029 (2017).
Pircher, J. et al. Cathelicidins prime platelets to mediate arterial thrombosis and tissue inflammation. Nat. Commun. 9, 1523 (2018).
Doring, Y. et al. Lack of neutrophil-derived CRAMP reduces atherosclerosis in mice. Circ. Res. 110, 1052–1056 (2012).
Soehnlein, O. et al. Neutrophil-derived cathelicidin protects from neointimal hyperplasia. Sci. Transl Med. 3, 103ra198 (2011).
Marx, C. et al. Eosinophil-platelet interactions promote atherosclerosis and stabilize thrombosis with eosinophil extracellular traps. Blood 134, 1859–1872 (2019).
Moosbauer, C. et al. Eosinophils are a major intravascular location for tissue factor storage and exposure. Blood 109, 995–1002 (2007).
Uderhardt, S. et al. Enzymatic lipid oxidation by eosinophils propagates coagulation, hemostasis, and thrombotic disease. J. Exp. Med. 214, 2121–2138 (2017).
Ames, P. R., Margaglione, M., Mackie, S. & Alves, J. D. Eosinophilia and thrombophilia in churg strauss syndrome: a clinical and pathogenetic overview. Clin. Appl. Thromb. Hemost. 16, 628–636 (2010).
Cugno, M., Marzano, A. V., Lorini, M., Carbonelli, V. & Tedeschi, A. Enhanced tissue factor expression by blood eosinophils from patients with hypereosinophilia: a possible link with thrombosis. PLoS ONE 9, e111862 (2014).
Gao, S. J. et al. Hypereosinophilic syndrome presenting with multiple organ infiltration and deep venous thrombosis: a case report and literature review. Medicine 95, e4658 (2016).
Yousefi, S. et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 14, 949–953 (2008).
Heit, J. A. et al. Predictors of recurrence after deep vein thrombosis and pulmonary embolism: a population-based cohort study. Arch. Intern. Med. 160, 761–768 (2000).
Prandoni, P. et al. The risk of recurrent venous thromboembolism after discontinuing anticoagulation in patients with acute proximal deep vein thrombosis or pulmonary embolism. A prospective cohort study in 1,626 patients. Haematologica 92, 199–205 (2007).
Goldhaber, S. Z. & Bounameaux, H. Pulmonary embolism and deep vein thrombosis. Lancet 379, 1835–1846 (2012).
Nieto, J. A. et al. Fatal bleeding in patients receiving anticoagulant therapy for venous thromboembolism: findings from the RIETE registry. J. Thromb. Haemost. 8, 1216–1222 (2010).
Heit, J. A. Epidemiology of venous thromboembolism. Nat. Rev. Cardiol. 12, 464–474 (2015).
Heit, J. A. et al. The epidemiology of venous thromboembolism in the community. Thromb. Haemost. 86, 452–463 (2001).
Cohen, A. T. et al. Venous thromboembolism (VTE) in Europe. The number of VTE events and associated morbidity and mortality. Thromb. Haemost. 98, 756–764 (2007).
Chiu, J. J. & Chien, S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol. Rev. 91, 327–387 (2011).
Mohan, S., Mohan, N., Valente, A. J. & Sprague, E. A. Regulation of low shear flow-induced HAEC VCAM-1 expression and monocyte adhesion. Am. J. Physiol. 276, C1100–C1107 (1999).
Gupta, N. et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc. Natl Acad. Sci. USA 114, 4763–4768 (2017).
Ponomaryov, T., Payne, H., Fabritz, L., Wagner, D. D. & Brill, A. Mast cells granular contents are crucial for deep vein thrombosis in mice. Circ. Res. 121, 941–950 (2017).
Subramaniam, S. et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 129, 2291–2302 (2017).
Yago, T., Liu, Z., Ahamed, J. & McEver, R. P. Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes deep vein thrombosis in mice. Blood 132, 1426–1437 (2018).
Brill, A. et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 10, 136–144 (2012).
Venereau, E., Schiraldi, M., Uguccioni, M. & Bianchi, M. E. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol. Immunol. 55, 76–82 (2013).
Magder, L. S. & Petri, M. Incidence of and risk factors for adverse cardiovascular events among patients with systemic lupus erythematosus. Am. J. Epidemiol. 176, 708–719 (2012).
Leffler, J. et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 188, 3522–3531 (2012).
Lood, C. et al. Increased C1q, C4 and C3 deposition on platelets in patients with systemic lupus erythematosus–a possible link to venous thrombosis? Lupus 21, 1423–1432 (2012).
Espinosa, G. & Cervera, R. Antiphospholipid syndrome: frequency, main causes and risk factors of mortality. Nat. Rev. Rheumatol. 6, 296–300 (2010).
Fischetti, F. et al. Thrombus formation induced by antibodies to β2-glycoprotein I is complement dependent and requires a priming factor. Blood 106, 2340–2346 (2005).
Girardi, G. et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J. Clin. Invest. 112, 1644–1654 (2003).
Girardi, G., Redecha, P. & Salmon, J. E. Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nat. Med. 10, 1222–1226 (2004).
Garcia-Romo, G. S. et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl Med. 3, 73ra20 (2011).
Solomon, D. H. et al. Cardiovascular morbidity and mortality in women diagnosed with rheumatoid arthritis. Circulation 107, 1303–1307 (2003).
Hannawi, S., Haluska, B., Marwick, T. H. & Thomas, R. Atherosclerotic disease is increased in recent-onset rheumatoid arthritis: a critical role for inflammation. Arthritis Res. Ther. 9, R116 (2007).
Maradit-Kremers, H. et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum. 52, 402–411 (2005).
Khandpur, R. et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl Med. 5, 178ra140 (2013).
Massberg, S. et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J. Exp. Med. 196, 887–896 (2002).
Massberg, S. et al. Platelet adhesion via glycoprotein IIb integrin is critical for atheroprogression and focal cerebral ischemia: an in vivo study in mice lacking glycoprotein IIb. Circulation 112, 1180–1188 (2005).
Gerdes, N. et al. Platelet CD40 exacerbates atherosclerosis by transcellular activation of endothelial cells and leukocytes. Arterioscler. Thromb. Vasc. Biol. 36, 482–490 (2016).
Drechsler, M., Megens, R. T., van Zandvoort, M., Weber, C. & Soehnlein, O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 122, 1837–1845 (2010).
Koenen, R. R. et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat. Med. 15, 97–103 (2009).
Massberg, S. et al. Platelets secrete stromal cell-derived factor 1α and recruit bone marrow-derived progenitor cells to arterial thrombi in vivo. J. Exp. Med. 203, 1221–1233 (2006).
Silvestre-Roig, C. et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 569, 236–240 (2019).
Xia, G. L., Wang, Y. K. & Huang, Z. Q. The correlation of serum myeloid-related protein-8/14 and eosinophil cationic protein in patients with coronary artery disease. BioMed. Res. Int. 2016, 4980251 (2016).
Niccoli, G. et al. Eosinophil cationic protein: a new biomarker of coronary atherosclerosis. Atherosclerosis 211, 606–611 (2010).
Haley, K. J. et al. Overexpression of eotaxin and the CCR3 receptor in human atherosclerosis: using genomic technology to identify a potential novel pathway of vascular inflammation. Circulation 102, 2185–2189 (2000).
Pertiwi, K. R. et al. Extracellular traps derived from macrophages, mast cells, eosinophils and neutrophils are generated in a time-dependent manner during atherothrombosis. J. Pathol. 247, 505–512 (2018).
van Dijk, R. A. et al. Systematic evaluation of the cellular innate immune response during the process of human atherosclerosis. J. Am. Heart Assoc. 5, e002860 (2016).
Novotny, J. et al. Thrombus NET content is associated with clinical outcome in stroke and myocardial infarction. Neurology 94, e2346–e2360 (2020).
Gudbjartsson, D. F. et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat. Genet. 41, 342–347 (2009).
Pircher, J., Engelmann, B., Massberg, S. & Schulz, C. Platelet-neutrophil crosstalk in atherothrombosis. Thromb. Haemost. 119, 1274–1282 (2019).
Tardif, J. C. et al. Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention for non-ST-segment elevation myocardial infarction: results of the SELECT-ACS trial. J. Am. Coll. Cardiol. 61, 2048–2055 (2013).
Dhanesha, N. et al. Targeting myeloid-cell specific integrin α9β1 inhibits arterial thrombosis in mice. Blood 135, 857–861 (2020).
Faraday, N. et al. Cathepsin G-dependent modulation of platelet thrombus formation in vivo by blood neutrophils. PLoS ONE 8, e71447 (2013).
Ortega-Gomez, A. et al. Cathepsin G controls arterial but not venular myeloid cell recruitment. Circulation 134, 1176–1188 (2016).
Vogel, S. et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Invest. 125, 4638–4654 (2015).
Franck, G. et al. Roles of PAD4 and NETosis in experimental atherosclerosis and arterial injury: implications for superficial erosion. Circ. Res. 123, 33–42 (2018).
Liu, Y. et al. Myeloid-specific deletion of peptidylarginine deiminase 4 mitigates atherosclerosis. Front. Immunol. 9, 1680 (2018).
Novotny, J. et al. Histological comparison of arterial thrombi in mice and men and the influence of Cl-amidine on thrombus formation. PLoS ONE 13, e0190728 (2018).
Schreiber, A. et al. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc. Natl Acad. Sci. USA 114, E9618–E9625 (2017).
Wang, H., Wang, C., Zhao, M. H. & Chen, M. Neutrophil extracellular traps can activate alternative complement pathways. Clin. Exp. Immunol. 181, 518–527 (2015).
Gushiken, F. C., Han, H., Li, J., Rumbaut, R. E. & Afshar-Kharghan, V. Abnormal platelet function in C3-deficient mice. J. Thromb. Haemost. 7, 865–870 (2009).
Sauter, R. J. et al. Functional relevance of the anaphylatoxin receptor C3aR for platelet function and arterial thrombus formation marks an intersection point between innate immunity and thrombosis. Circulation 138, 1720–1735 (2018).
Jaiswal, S. et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498 (2014).
Jaiswal, S. et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121 (2017).
Fuster, J. J. et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 (2017).
James, C. et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434, 1144–1148 (2005).
Wolach, O. et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl Med. 10, eaan8292 (2018).
Wang, W. et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in Jak2 (V617F) Mice. Circ. Res. 123, e35–e47 (2018).
Cull, A. H., Snetsinger, B., Buckstein, R., Wells, R. A. & Rauh, M. J. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 55, 56–70 (2017).
Zhang, Q. et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393 (2015).
Veninga, A., De Simone, I., Heemskerk, J. W. M., Cate, H. T. & van der Meijden, P. E. J. Clonal hematopoietic mutations linked to platelet traits and the risk of thrombosis or bleeding. Haematologica 105, 2020–2031 (2020).
Bonaca, M. P. et al. Rivaroxaban in peripheral artery disease after revascularization. N. Engl. J. Med. 382, 1994–2004 (2020).
Borissoff, J. I., Spronk, H. M. & ten Cate, H. The hemostatic system as a modulator of atherosclerosis. N. Engl. J. Med. 364, 1746–1760 (2011).
Eikelboom, J. W. et al. Rivaroxaban with or without aspirin in stable cardiovascular disease. N. Engl. J. Med. 377, 1319–1330 (2017).
Gadi, I. et al. Different DOACs control inflammation in cardiac ischemia-reperfusion differently. Circ. Res. 128, 513–529 (2020).
Hara, T. et al. Rivaroxaban, a novel oral anticoagulant, attenuates atherosclerotic plaque progression and destabilization in ApoE-deficient mice. Atherosclerosis 242, 639–646 (2015).
Vu, T. K., Hung, D. T., Wheaton, V. I. & Coughlin, S. R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64, 1057–1068 (1991).
Nidorf, S. M. et al. Colchicine in patients with chronic coronary disease. N. Engl. J. Med. 383, 1838–1847 (2020).
Ridker, P. M. et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131 (2017).
Tardif, J. C. et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 381, 2497–2505 (2019).
Kirchhof, P. et al. Effects of rivaroxaban on biomarkers of coagulation and inflammation: a post hoc analysis of the X-VeRT TRIAL. TH Open 4, e20–e32 (2020).
Busch, G. et al. Coagulation factor Xa stimulates interleukin-8 release in endothelial cells and mononuclear leukocytes: implications in acute myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 25, 461–466 (2005).
Daubie, V. et al. Factor Xa and thrombin evoke additive calcium and proinflammatory responses in endothelial cells subjected to coagulation. Biochim. Biophys. Acta 1763, 860–869 (2006).
Senden, N. H. et al. Factor Xa induces cytokine production and expression of adhesion molecules by human umbilical vein endothelial cells. J. Immunol. 161, 4318–4324 (1998).
Ichikawa, H. et al. Rivaroxaban, a direct factor Xa inhibitor, ameliorates hypertensive renal damage through inhibition of the inflammatory response mediated by protease-activated receptor pathway. J. Am. Heart Assoc. 8, e012195 (2019).
Wildhagen, K. C. et al. Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood 123, 1098–1101 (2014).
Mansour, A., Bachelot-Loza, C., Nesseler, N., Gaussem, P. & Gouin-Thibault, I. P2Y12 inhibition beyond thrombosis: effects on inflammation. Int. J. Mol. Sci. 21, 1391 (2020).
Mackman, N., Bergmeier, W., Stouffer, G. A. & Weitz, J. I. Therapeutic strategies for thrombosis: new targets and approaches. Nat. Rev. Drug Discov. 19, 333–352 (2020).
Jeong, H. S. et al. Comparison of ticagrelor versus prasugrel for inflammation, vascular function, and circulating endothelial progenitor cells in diabetic patients with non-ST-segment elevation acute coronary syndrome requiring coronary stenting: a prospective, randomized, crossover trial. JACC Cardiovasc. Interv. 10, 1646–1658 (2017).
Morris, T. et al. Effects of low-dose aspirin on acute inflammatory responses in humans. J. Immunol. 183, 2089–2096 (2009).
Paul-Clark, M. J., Van Cao, T., Moradi-Bidhendi, N., Cooper, D. & Gilroy, D. W. 15-epi-lipoxin A4-mediated induction of nitric oxide explains how aspirin inhibits acute inflammation. J. Exp. Med. 200, 69–78 (2004).
Dutta, P. et al. Myocardial infarction accelerates atherosclerosis. Nature 487, 325–329 (2012).
Sager, H. B. et al. Targeting interleukin-1β reduces leukocyte production after acute myocardial infarction. Circulation 132, 1880–1890 (2015).
Gomez, D. et al. Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 24, 1418–1429 (2018).
Alexander, M. R. et al. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J. Clin. Invest. 122, 70–79 (2012).
Ridker, P. M. et al. Low-dose methotrexate for the prevention of atherosclerotic events. N. Engl. J. Med. 380, 752–762 (2019).
Munoz, L. E. et al. Neutrophil extracellular traps initiate gallstone formation. Immunity 51, 443–450 (2019).
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A. & Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006).
Opstal, T. S. J. et al. Colchicine attenuates inflammation beyond the inflammasome in chronic coronary artery disease: a LoDoCo2 proteomic substudy. Circulation 142, 1996–1998 (2020).
Castro, M. et al. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir. Med. 3, 355–366 (2015).
FitzGerald, J. M. et al. Benralizumab, an anti-interleukin-5 receptor α monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 388, 2128–2141 (2016).
Ortega, H. G. et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N. Engl. J. Med. 371, 1198–1207 (2014).
Mastellos, D. C., Ricklin, D. & Lambris, J. D. Clinical promise of next-generation complement therapeutics. Nat. Rev. Drug Discov. 18, 707–729 (2019).
Liverani, E., Rico, M. C., Tsygankov, A. Y., Kilpatrick, L. E. & Kunapuli, S. P. P2Y12 receptor modulates sepsis-induced inflammation. Arterioscler. Thromb. Vasc. Biol. 36, 961–971 (2016).
Sexton, T. R. et al. Ticagrelor reduces thromboinflammatory markers in patients with pneumonia. JACC Basic Transl. Sci. 3, 435–449 (2018).
Hillmen, P. et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood 110, 4123–4128 (2007).
Dong, Y. et al. TLR4 regulates ROS and autophagy to control neutrophil extracellular traps formation against Streptococcus pneumoniae in acute otitis media. Pediatr. Res. https://doi.org/10.1038/s41390-020-0964-9 (2020).
Guglietta, S. et al. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat. Commun. 7, 11037 (2016).
Metzler, K. D., Goosmann, C., Lubojemska, A., Zychlinsky, A. & Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 8, 883–896 (2014).
Papayannopoulos, V., Metzler, K. D., Hakkim, A. & Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 191, 677–691 (2010).
Vu, T. T. et al. Histidine-rich glycoprotein binds DNA and RNA and attenuates their capacity to activate the intrinsic coagulation pathway. Thromb. Haemost. 115, 89–98 (2016).
Fu, H. et al. Flow-induced elongation of von willebrand factor precedes tension-dependent activation. Nat. Commun. 8, 324 (2017).
Jiang, Y., Fu, H., Springer, T. A. & Wong, W. P. Electrostatic steering enables flow-activated von willebrand factor to bind platelet glycoprotein, revealed by single-molecule stretching and imaging. J. Mol. Biol. 431, 1380–1396 (2019).
Chonn, A., Cullis, P. R. & Devine, D. V. The role of surface charge in the activation of the classical and alternative pathways of complement by liposomes. J. Immunol. 146, 4234–4241 (1991).
Acknowledgements
The authors receive support from the Deutsche Forschungsgemeinschaft (DFG) SFB 914 (S.M. [B02 and Z01], K.S. [B02]), the DFG SFB 1123 (S.M. [B06], K.S. [A07]), the DFG FOR 2033 (S.M.), the German Centre for Cardiovascular Research (DZHK) (MHA 1.4VD [S.M.]), the European Research Council (ERC-2018-ADG “IMMUNOTHROMBOSIS” [S.M.]), the FP7 programme (project 260309, PRESTIGE [S.M.]) and the European Research Council under the European Union Horizon 2020 research and innovation programme (T-MEMORE, grant agreement No. 947611 [K.S.]).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Cardiology thanks A. Assinger, A. Greinacher, A. Hidalgo and O. Soehlein for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Immunothrombosis
-
Protective host defence mechanism to limit the systemic spread of pathogens in the bloodstream, in which inflammation triggers thrombosis.
- Thromboinflammation
-
Dysregulated and excessive form of immunothrombosis causing collateral tissue injury by inducing ischaemia and excessive inflammation.
- Coagulopathy
-
Dysregulation of haemostasis or fibrinolysis caused by an underlying systemic condition such as infection, trauma or cancer as well as hereditary disorders. Coagulopathy can result in an increased risk of thrombosis and/or bleeding.
- Disseminated intravascular coagulation
-
Systemic and excessive activation of the coagulation system resulting in generalized microvascular thrombosis. Platelets and coagulation factors are consumed. This condition is mainly triggered by severe sepsis and trauma.
Rights and permissions
About this article

Cite this article
Stark, K., Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol 18, 666–682 (2021). https://doi.org/10.1038/s41569-021-00552-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-021-00552-1
This article is cited by
-
Levels of peripheral blood routine, biochemical and coagulation parameters in patients with hemorrhagic fever with renal syndrome and their relationship with prognosis: an observational cohort study
BMC Infectious Diseases (2024)
-
Nonalcoholic fatty liver disease is an independent risk factor for ischemic stroke after revascularization in patients with Moyamoya disease: a prospective cohort study
Lipids in Health and Disease (2024)
-
Tea polyphenol-derived nanomedicine for targeted photothermal thrombolysis and inflammation suppression
Journal of Nanobiotechnology (2024)
-
Heparin-network-mediated long-lasting coatings on intravascular catheters for adaptive antithrombosis and antibacterial infection
Nature Communications (2024)
-
Phytochemical profiling, antioxidant, cytotoxic, and anti-inflammatory activities of Plectranthus rugosus extract and fractions: in vitro, in vivo, and in silico approaches
Inflammopharmacology (2024)
