
Abstract
Natural killer (NK) cells comprise a unique population of innate lymphoid cells endowed with intrinsic abilities to identify and eliminate virally infected cells and tumour cells. Possessing multiple cytotoxicity mechanisms and the ability to modulate the immune response through cytokine production, NK cells play a pivotal role in anticancer immunity. This role was elucidated nearly two decades ago, when NK cells, used as immunotherapeutic agents, showed safety and efficacy in the treatment of patients with advanced-stage leukaemia. In recent years, following the paradigm-shifting successes of chimeric antigen receptor (CAR)-engineered adoptive T cell therapy and the advancement in technologies that can turn cells into powerful antitumour weapons, the interest in NK cells as a candidate for immunotherapy has grown exponentially. Strategies for the development of NK cell-based therapies focus on enhancing NK cell potency and persistence through co-stimulatory signalling, checkpoint inhibition and cytokine armouring, and aim to redirect NK cell specificity to the tumour through expression of CAR or the use of engager molecules. In the clinic, the first generation of NK cell therapies have delivered promising results, showing encouraging efficacy and remarkable safety, thus driving great enthusiasm for continued innovation. In this Review, we describe the various approaches to augment NK cell cytotoxicity and longevity, evaluate challenges and opportunities, and reflect on how lessons learned from the clinic will guide the design of next-generation NK cell products that will address the unique complexities of each cancer.
Similar content being viewed by others
Introduction
Adoptive cell therapy using engineered immune effectors is a promising new approach to treat haematological and solid malignancies for which treatment options are limited1,2,3,4,5,6,7,8,9. Autologous chimeric antigen receptor (CAR) T cell therapy, the first to enter clinical translation and commercialization, has led to remarkable improvements in patients with aggressive B cell malignancies, producing long-term sustained remissions in many cases. However, despite these successes, challenges remain. The complex manufacturing process of CAR T cell therapy increases costs and lengthens the vein to vein time, thus representing an obstacle for patients who, owing to rapidly progressing disease, are in critical need of treatment. Furthermore, the requirement for the patient’s own cells as the source material restricts eligibility, as many patients are often heavily pretreated and lymphopenic, and may not have sufficient cells to yield a viable product. In the clinic, CAR T cell-related cytokine-release syndrome (CRS) and neurotoxicities represent an additional concern, requiring inpatient monitoring. Given these limitations, there is growing interest in exploring cell sources that are off the shelf and could be applied universally. Because T cells recognize and mediate a response against non-self via the T cell receptor (TCR), use of CAR T cells in the allogeneic setting requires additional genetic editing steps to remove the TCR to mitigate the risk of graft-versus-host disease (GvHD). Natural killer (NK) cells, on the other hand, recognize their targets in a human leukocyte antigen (HLA)-unrestricted manner and thus do not present these same risks, making them attractive candidates for universal cellular immunotherapy10. NK cell effector function is controlled by a complex array of activating and inhibitory receptors that can distinguish between healthy and ‘stressed’ cells11. The cumulative cues resulting from receptor–ligand engagements determine whether NK cells deliver a ‘kill’ or ‘not kill’ signal. Healthy cells are spared via recognition of self-major histocompatibility complex (MHC) class I molecules which bind inhibitory killer cell immunoglobulin-like receptors (KIRs) that signal to stop NK cell function. By contrast, through the mechanism of missing-self recognition, NK cells attack abnormal self-cells, such as tumour cells, which downregulate expression of MHC class I molecules in an attempt to evade T cell responses, and upregulate activating ligands that are induced by stress such as DNA damage or malignant transformation12,13,14,15,16.
These various attributes provide NK cells with unique advantages for allogeneic therapeutic applications. With the accelerated development of innovative strategies and the emergence of next-generation technologies that allow for deeper biological investigations, various NK cell products can be designed for cancer treatment. In this Review, we describe how these unique properties of NK cells are leveraged for adoptive NK cell immunotherapy, and provide an overview of the evolving engineering strategies to augment NK cell potency and persistence, as well as the efforts to redirect NK cell specificity to tumours through next-generation CAR molecules, engineered TCR and pre-complexing with cell engagers. Lastly, we conclude with a perspective on the challenges and opportunities ahead as we confront solid tumours and safeguard immune effector cells from the suppressive pressures within the tumour microenvironment, while also devising strategies to monitor and mitigate inadvertent safety concerns.
Biological properties of NK cells
Despite the successes of engineered T cell immunotherapies1,2,3,4,5,6,7,8, the clinical benefit has been limited to a fraction of patients and a few indications, thus highlighting the need for new strategies. Leveraging innate immunity to broaden the scope of antitumour responses is an attractive option. Within the innate immune system, NK cells are specialized immune effector cells, and are suspected to have a role in tumour immunosurveillance10, as suggested by the correlation of low NK cell activity with increased cancer susceptibility and higher risk of metastasis observed in both preclinical and clinical studies17,18,19. NK cells develop from CD34+ progenitor cells in the bone marrow, although it is as yet unclear whether they arise from a unique set of precursor cells or from multipotent progenitors that also give rise to T lymphocytes, B lymphocytes and myeloid cells20. Unlike T cells and NKT cells, NK cells lack expression of the clonotypic TCR and the associated CD3 complex responsible for signal transduction. NK cells are generally classified under a dichotomous distribution based on the relative expression of surface proteins CD56 and CD16: CD56brightCD16low/– (immunomodulatory, cytokine-producing) and CD56dimCD16+ (cytotoxic)21. Recent advancements in high-parameter cytometry and single-cell proteo-genomics, however, have led to the understanding that NK cells may, in fact, exhibit greater phenotypic heterogeneity that extends beyond these two subsets, giving rise to diverse cell populations endowed with varying functional properties22.
NK cells possess strong cytotoxicity and, upon forming immunological synapses with targets, elicit a potent response through the release of cytolytic granules and cytotoxic cytokines23. Moreover, they can recognize antibody-coated cells through their Fc\({\rm{\gamma }}{\rm{RIIIA}}\) (CD16) receptor and trigger antibody-dependent cellular cytotoxicity (ADCC) and cytokine production24. NK cells have also been described as ‘immune-regulatory’ because of their ability to produce an array of cytokines and chemokines, through which they help shape B cell and T cell responses, and impact the function of dendritic cells, macrophages and neutrophils11. This broad range of attributes reveals the sophisticated network of biological mechanisms associated with NK cell function and supports the value of NK cells for immunotherapy.
Memory-like function in NK cells
Early studies reported memory-like responses by NK cells in mouse models of cytomegalovirus infection25,26, a behaviour not typically associated with innate immune cells. In these studies26,27, mouse NK cells, when stimulated with a combination of IL-12 and IL-18, acquired a functional phenotype characterized by increased production of IFN\({\rm{\gamma }}\). Interestingly, after a resting phase, these cells were able to reactivate upon cytokine stimulation or engagement of activating receptors and exhibited an enhanced IFN\({\rm{\gamma }}\) response resembling the memory-like properties of adaptive immune cells. Later, Todd Fehniger’s group hypothesized that human NK cells should, likewise, be endowed with memory-like properties. Consistent with this hypothesis, their study demonstrated that human NK cells, preactivated with IL-12, IL-15 and IL-18, followed by 1–3 weeks rest, were able to generate a robust response driven by enhanced IFN\({\rm{\gamma }}\) production upon subsequent exposure to cytokines or to K562 leukaemia cells28. Since then, many more groups have described similar memory-like function in various immunological settings, including observations of such responses in humans29,30.
NK cell source and donor selection
In patients with cancer, NK cells typically display a dysfunctional phenotype marked by altered gene expression profiles and reduced cytotoxic function31,32, thus diminishing the feasibility of autologous NK cell therapy applications. Moreover, autologous manufacturing platforms are cumbersome and may limit accessibility if patients are not able to provide sufficient cells to undergo downstream processing and engineering.
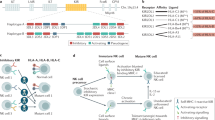
Because allogeneic NK cells do not cause GvHD, current NK cell therapy programmes rely largely on allogeneic sources to avoid the incumbrances associated with autologous approaches. There are various sources from which NK cells can be derived (Fig. 1), namely peripheral blood mononuclear cells28,29,30, cord blood28,29,30,33,34,35,36,37, immortalized cell lines38,39,40, haematopoietic stem and progenitor cells (HSPCs)33,34,41 and induced pluripotent stem cells (iPSCs)42,43. All sources can provide clinically meaningful cell doses, are amenable to CAR receptor engineering and have transitioned into in-human studies. They, nevertheless, come with unique advantages and challenges, and may possess different underlying transcriptional, phenotypic and functional properties44.

Natural killer (NK) cells can be derived from several different sources, each of which presents its own advantages and potential challenges. Chimeric antigen receptor (CAR) NK cells have successfully been engineered from different platforms including cord blood35,37, peripheral blood52,112,264, NK-92 cells39,265,266,267,268,269,270,271,272,273,274 and induced pluripotent stem cell (iPSC)-derived NK cells43,48,49. ADCC, antibody-dependent cellular cytotoxicity; HPSC, haematopoietic stem and progenitor cells; MDACC, University of Texas MD Anderson Cancer Center.
NK-92, the first NK cell-based immunotherapy to receive Investigational New Drug approval by the US Food and Drug Administration (FDA) for clinical testing, is a homogeneous, immortalized NK lymphoma cell line that can be expanded ex vivo to achieve large cell numbers38 (Table 1). NK-92 cells lack expression of most KIRs and are thus less likely to become inhibited, which makes them attractive for cell therapy use38. However, their cancerous origin raises safety concerns, and irradiation of NK-92-derived cell products prior to patient administration is required, which, in turn, can negatively impact their long-term in vivo persistence and overall therapeutic potential37. Another disadvantage is that, owing to a lack of CD16 expression, NK-92 cells are devoid of the ability to mediate cell killing via ADCC.
NK cells can also be derived from CD34+ progenitor cells upon in vitro differentiation33. In a first-in-human clinical trial, Dolstra et al. showed clinical efficacy in older patients with minimal residual disease-positive acute myeloid leukaemia (AML) who received adoptively transferred HSPC-NK cells34. Although preliminary results are encouraging, clinical studies exploring the safety and efficacy of HSPC-NK cells in larger patient populations and various indications will help determine the range of potential clinical applications for these cells. Future work will also need to address whether HSPC-NK cells can be effectively engineered to achieve enhanced tumour specificity.
iPSCs are an appealing source for NK cells given their clonal growth and high expansion capacity, as well as their ability to differentiate in vitro, allowing for the manufacturing of large numbers of homogeneous NK cell products. A potential disadvantage is that iPSC-derived NK cells often express low levels of endogenous CD16, although this can be mitigated through genetic engineering45. Another possible concern is that iPSCs may harbour DNA methylation signatures consistent with their somatic tissue of origin46. This ‘epigenetic memory’ could influence the development of specific cell lineages that differ from the donor cell, and should, therefore, be considered when employing iPSC platforms47. Nonetheless, a growing number of genetically engineered iPSC-NK cell candidates are emerging in preclinical studies, with a few that have transitioned into clinical trials48,49,50,51. In a phase I/II trial, iPSC-NK cells expressing CAR showed encouraging results in patients with relapsed or refractory (R/R) diffuse large B cell lymphoma, both as monotherapy and in combination with CD20-targeting agents43,49.
Primary NK cells can be harvested from peripheral blood (PB-NK cells)28,29,30,36 or from umbilical cord blood (CB-NK cells)33,34,35,37. CB-NK cells are available frozen off the shelf through blood banks, whereas PB-NK cells require apheresis of healthy donors and donor-specific collection. PB-NK cells served as the platform for the very first successful delivery of a CAR construct into NK cells in work led by Dario Campana in 2005 (ref.52) and, today, PB-NK cells provide the basis for various products currently in clinical testing (Table 1). Cord blood NK cells have a proven track record for CAR-redirected antitumour cytotoxicity and clinical activity, as pioneered by our group35,37. In our study, we demonstrated potent clinical efficacy of genetically engineered cord blood-derived CD19-CAR NK cells against R/R CD19+ lymphoid malignancies in an ongoing phase I/II clinical trial37.
NK cells derived from all of these sources have been shown to have advantages and disadvantages for adoptive cell therapy applications. Having various options for source material confers a degree of versatility to the design of therapeutic strategies, allowing platforms to be specifically tailored to address the needs of each patient population and disease indication.
It is important to note that inter-donor variability may influence NK cell profiles and, thus, impact clinical outcomes. Therefore, a thorough understanding of product characteristics is critical to define biomarkers indicative of greater potency and persistence. From an immunological perspective, donor-derived NK cells might prove advantageous when administered across HLA–KIR genotypic boundaries as HLA–KIR ligand disparity might help overcome tumour immune evasion. Early into the journey of NK cell-based immunotherapy, the field recognized a dramatically reduced probability of post-transplant AML relapse in recipients of HLA haplotype-mismatched grafts with KIR ligand incompatibility53. Although KIR–HLA genotype-informed donor selection remains an area of active investigation54,55,56,57, evidence suggests a potential benefit of this approach in the setting of T cell-depleted allogeneic haematopoietic stem cell transplantation (alloHSCT)58,59,60,61,62,63,64,65,66. Further work is needed to determine whether this strategy can be successfully adopted into NK cell therapy pipelines.
Enhancing NK cell fitness and antitumour function
Chimeric antigen receptors
Chimeric antigen receptors (CARs) are synthetic fusion proteins comprising an extracellular antigen-recognition domain and intracellular signalling moieties that trigger cell activation. Most commonly, the single-chain variable fragment (scFv) from a desired antibody is used for the antigen-binding domain, although many CAR modalities comprising the extracellular portion of native cellular receptors have also been constructed, leveraging the natural specificity of receptor–ligand interactions67,68,69. CARs can be expressed on immune effector cells for the purpose of reprogramming their specificity to a particular target (Fig. 2a). CAR-engineered T cell therapeutics were the first to emerge, with various products developed for immuno-oncology applications1,2,3,4,5,6,7,8. The field has since expanded, and NK cells have also been integrated into gene engineering pipelines70,71. CARs conventionally designed for T cells (comprising CD3\({\rm{\zeta }}\) and T cell co-stimulatory molecules) have been used for generation of CAR NK cells, and studies have demonstrated that these cells can target tumours with efficacy and specificity, while maintaining a desirable safety profile37 (Fig. 3a).

Natural killer (NK) cell specificity towards tumour cells can be redirected using different strategies. a,b | Chimeric antigen receptor (CAR) NK cell35,37,43,112,264 (panel a) and T cell receptor (TCR) NK cell94 (panel b) approaches both build upon stable genetic engineering to endow NK cells with synthetic receptors that recognize extracellular and intracellular tumour antigens, respectively. c | Bi-specific and tri-specific engagers96,98,99,100,102,103,105,106,275 deploy two-directional or three-directional antibodies which crosslink NK cells with their respective tumour cell targets and circumvent the need for complex genetic editing. HLA, human leukocyte antigen; MHC, major histocompatibility complex; scFV, single-chain variable fragment.

a,b | Chimeric antigen receptor (CAR) molecules have evolved dramatically over the past two decades, from simple first-generation designs276 (panel a), to second-generation52,277,278 and third-generation39,279 CARs with added co-stimulatory molecules (panel b) and, finally, to current-generation CAR designs resembling a modular system that encompasses optimized extracellular domains for target recognition, intracellular co-stimulatory molecules for effective natural killer (NK) cell activation and added payloads which can enhance NK cell functionality. c–e | Current strategies leverage the core principles of CAR signalling and functionality, and provide innovative methods to improve tumour recognition and enhance cell activation, using sophisticated construct designs to allow targeting of multiple tumour antigens69,280 (panel c), provide cytokine support35,37 (panel e) and activate auxiliary cytotoxicity pathways. Integration of logic-gated circuits to guide selective killing of targeted malignant cells while sparing healthy tissues may lead to improved safety profiles90,92,93 (panel d). aCAR, activating chimeric antigen receptor; AML, acute myeloid leukaemia; DAP12, DNAX-activation protein 12; EMCN, Endomucin; HLA, human leukocyte antigen; HSC, haematopoietic stem cell; iCAR, inhibitory chimeric antigen receptor; scFV, single-chain variable fragment.
Motivated by these encouraging results, there is growing interest in designing CARs based on activating signals associated with NK cell biology (Fig. 3b). Examples of such an approach include DNAX-activation protein 12 (DAP12) and DAP10, which have been used in place of CD3\({\rm{\zeta }}\) in some studies72,73,74,75,76. These adapter proteins are intracellular signalling domains that function via recruitment of PI3K and are associated with activating molecules such as NKp44, activating KIRs (KIR2DS and KIR3DS) and NKG2C (refs.72,76). Similar to CD3\({\rm{\zeta }},\) DAP12 also contains immuno-tyrosine activation motifs (ITAMs) that, once phosphorylated, initiate a cascade of signals that culminate in the release of cytotoxic granules and pro-inflammatory cytokines such as TNFα and IFNγ72. DAP10, although not possessing an ITAM domain, has been shown to induce potent killing activity in NK cells via signalling through the NKG2D–DAP10 axis76.
In the past few years, the behaviour of CARs, once anchored on the cell surface, has garnered considerable interest, especially following observations that, in some cases, CAR T cells demonstrated an elevated basal level of activation independent of antigen engagement, referred to as tonic signalling77,78,79,80,81. Although studies continue to explore potential causes of this phenomenon, it has been reported that CARs may incorporate into the TCR–CD3 complex via the CD3\({\rm{\zeta }}\) domain and may augment T cell activation82,83. More recently, studies have shown that the CD28 transmembrane domain, present in some CAR frameworks, mediates CAR heterodimerization with endogenous CD28 and, thereby, triggers T cell activation84,85. These observations suggest that CARs are not static on the cell membrane but, rather, are prone to interacting with endogenous receptors. Although not much is yet known regarding these types of receptor associations in NK cells, it is conceivable that CARs may form synergistic partnerships with any of the many activating receptors on the membrane of NK cells, resulting in cooperative activation.
Efforts have also focused on refining CAR-based strategies, motivated by learnings from clinical translation of this therapy, which have prompted investigations into approaches to overcome the challenges experienced in the patient. One critical obstacle to CAR-based cell therapy is antigen escape, a process through which tumours, via antigen loss or downregulation, evade the immune response. Various strategies have been employed to address this issue. Targeting multiple antigens is a viable approach to both increase the stringency of tumour detection and extend therapeutic benefit (Fig. 3c). This is often accomplished either by expressing a bi-cistronic construct that encodes for two separate CARs (each specific for a different antigen on the tumour and linked to two separate signalling endodomains) or by expressing a single CAR containing bi-specific recognition domains, each targeting a different antigen in tandem and linked to a single signalling endodomain86. Dual-CAR approaches afford greater design flexibility, accommodating combinations of signalling domains and receptor formats. Each CAR may be formatted to provide both activating and co-stimulatory signals upon target engagement. Alternatively, trans-signalling formats separate these two signals and require engagement to both antigens for full activation. With bi-specific CARs, the antigen-binding moieties are linked to a single receptor, often a second or third-generation CAR87,88. In either approach, considerations regarding antigen density, membrane localization and protein structure are important to ensure both CARs bind their targets efficiently at the immune synapse. Although most of these innovative CAR-based approaches have focused on T cells, it is plausible to anticipate that these systems can also be applied to NK cells.
Strategies for improving the control of CAR-mediated activation have also been explored, especially with a focus on minimizing toxicities resulting from on-target, off-tumour effects. Promoting selective targeting, via the use of logic-gated CARs, has been shown to mitigate some of these problems in the context of CAR T cells89,90,91,92,93 (Fig. 3d). A recent study presented preliminary findings showing the feasibility of controlling CAR NK activity when applying an ‘OR’ and ‘NOT’ logic gated CAR gene circuit approach92,93. In this work, the team targeted FLT3 and/or CD33 on AML blasts via bivalent CARs and employed an inhibitory CAR to bind an antigen on healthy haematopoietic stem cells (HSCs) and elicit a ‘NOT signal’ to prevent cell killing92,93.
One key limitation of CAR-based approaches is that, for the most part, detection capability is limited to surface proteins. Intracellular antigens, which are presented in the form of peptide–HLA complexes, are naturally detected via the TCR. Engineering NK cells to express a TCR could allow detection of such peptides (Fig. 2b). TCR-guided NK-92 cells have recently been shown to mediate successful antitumour responses94. Although additional studies are warranted to validate the clinical applicability of this approach, one potential advantage is that, because NK cells do not possess endogenous TCR, mispairing issues that have been reported in TCR-engineered T cells95 will likely not be a concern. A downside, however, is that NK cells are not equipped with the full signalling machinery found in T cells, thereby potentially compromising the ability to be potently activated via a synthetic TCR.
NK cell engagers
NK cells can also be directed to tumour sites via engagers that elicit a strong NK cell-mediated antitumour response by triggering an activating receptor on the NK cell, while simultaneously binding a target antigen on the tumour cell96,97,98,99,100 (Fig. 2c). Additional NK engager strategies include tri-specific and tetra-specific designs that aim to strengthen the antitumour effect by targeting multiple antigens on the tumour or by cross-linking cytokine moieties to support NK expansion and survival101,102,103,104,105. The use of cell engagers bypasses the need for engineering and does not require vector-mediated gene transfer, therefore representing a simpler and less costly manufacturing process that can deliver a product capable of inducing CAR-like activity.
Various preclinical studies have shown promising results when employing NK cell engagers to target haematological and solid malignancies96,97,98,99,100,101,102,103,104,105,106. Recent work demonstrated a robust NK cell-mediated response against primary patient-derived AML blasts by employing a tri-specific molecule targeting CLEC12A on AML cells and activating NK cells though a humanized anti-CD16 single-domain antibody and IL-15 (ref.105). Moreover, a trifunctional engager targeting two NK activating receptors, CD16 and NKp46, was shown to drive potent antitumour response, culminating in efficient control of tumour growth in vivo in the setting of solid and metastatic malignancies in preclinical models106.
Recently, our group demonstrated that CB-NK cells, when complexed with AFM13 — a bi-specific engager binding CD16 on NK cells and CD30 on leukaemia or lymphoma targets — exhibited enhanced killing of CD30+ tumour cells, leading to CAR-like responses100. We have since translated this approach to the clinic for treatment of R/R CD30+ Hodgkin lymphoma and non-Hodgkin lymphoma (NHL) (NCT04074746 (ref.107)). As more strategies transition into clinical trials, it will be important to also evaluate the durability of the antitumour effect of engager-loaded NK cells and determine whether multiple treatments are required for sustained therapeutic benefit.
Cytokine armouring
Whereas CAR and TCR engineering technologies seek to enhance NK cell function by genetically redirecting their specificity, there are also initiatives that aim to effectively prime NK cells ex vivo and/or in vivo to sustain optimal antitumour function and persistence. It is well described that freshly isolated NK cells have lower cytolytic capacity compared with NK cells that have been primed108. One approach to address this limitation is cytokine-mediated activation, and various methods are currently under investigation29,100,109,110,111,112,113,114. Ex vivo expansion of NK cells with combinations of IL-2, IL-15 and IL-21 supplementation shows that these cytokines enhance cytotoxic function and support high proliferation rates while maintaining cells in a healthy, non-exhausted state115.
It has now become clear that, when cultured in the presence of IL-12/15/18, PB-NK cells shift to a phenotype referred to as cytokine-induced memory-like NK cells29. Cytokine-induced memory-like NK cells have shown clinical efficacy in patients with R/R relapsed myeloid neoplasias both in the pre-transplant and post-transplant setting29,30. Engineering memory-like NK cells to express CAR augments antitumour responses leading to increased potency against NK-resistant malignancies111,112. Furthermore, in our own clinical trial employing the NK cell engager AFM13, preactivation of NK cells with IL-12/15/18 to induce memory programmes led to enhanced responses against CD30+ lymphomas100 (Fig. 2c) (NCT04074746 (ref.107)).
Although these data strongly point to the advantage of cytokine priming, continuous ex vivo stimulation renders NK cells ‘cytokine-addicted’ and leads to decreased persistence when these cells are infused in the absence of in vivo cytokine support115. To avoid this problem and still leverage the benefits of cytokine activation, the field has turned to genetic engineering, through which NK cells are modified to produce cytokines that will support cell potency, proliferation and persistence (Fig. 3e). This autocrine support has garnered notable interest, and CAR-engineered NK cells are emerging armed with supplemental cytokine signalling35,37,48,49. Cytokine armouring can be programmed such that soluble cytokines are released into the environment or are engineered in membrane-bound form to induce response upon cell to cell interaction116. In soluble form, cytokines that are produced by engineered NK cells can mediate a bystander effect and, thereby, activate other immune effector cells — such as T cells or myeloid cells — that are present in the tumour microenvironment (TME), thus potentially further augmenting the antitumour response117. Our group, as well as others, has demonstrated that IL-15-armoured CAR NK cells exhibit superior persistence in vivo in preclinical models when compared with CAR alone35,48,49. These findings were confirmed in a clinical study of patients with CD19+ lymphoid malignancies, in many of whom we detected CAR NK cells in circulation a year after treatment37. With each cytokine armouring approach, it is important to determine optimal cytokine-dosing strategies that will support increased function and persistence, but will not compromise NK cell functionality by inducing metabolic exhaustion, as has been previously reported118.
Overcoming immunosuppression
The TME consists of a harsh metabolic landscape characterized by a heterogeneous mix of immunosuppressive metabolites, glucose and amino acid deprivation, hypoxia and acidity, which, in concert, prevent effective antitumour immunity. In solid tumours specifically, hypoxia is a common driver of immune cell dysfunction. It has been shown that NK cell function is impaired in the hypoxic TME, in part due to the increased influx of suppressor cells such as myeloid-derived suppressor cells (MDSCs), regulatory T cells (Treg cells) and M2 macrophages in the TME as well as a direct impact of hypoxia on NK cell function119,120,121,122. Inhibition of hypoxia-responsive HIF1a signalling in NK cells has been reported to enhance NK cell potency and unleash NK cell-mediated antitumour function123.
Tumours often display an aberrant metabolic behaviour that results in high levels of lactic acid, depletion of vital nutrients and an increase in concentrations of toxic catabolites, adenosine and reactive oxygen species109. In addition, the uncontrolled proliferation, dysfunctional vasculature and presence of immunosuppressive cell subsets contribute to the dismal fate of immune effector cells that venture into the TME. To overcome the detrimental effects of metabolic immunosuppression, current strategies focus primarily on two areas: altering the metabolic constitution of the tumour or modifying gene expression programmes in immune cells to shield them from the suppressive metabolites in the TME (Fig. 4a). Studies have shown that high levels of lactate dehydrogenase (LDH) in the blood and the TME correlate with poor outcomes for patients with melanoma and low response to checkpoint therapy124. Although still in the preclinical phase, glycolytic inhibitors and LDH blockers may provide an opportunity to favourably modulate the TME. A recent study showed that treatment of patient-derived melanoma cells with a lactate dehydrogenase A (LDHA) inhibitor, GSK2837808A, led to improved T cell antitumour cytotoxicity both in vitro and in vivo125. Lactate levels in the TME can also be decreased by targeting its transporters, MCT1 and MCT4, as has been shown for AstraZeneca’s AZD3965 compound, currently under investigation in the clinic (NCT01791595 (ref.126)).

a–f | Immune cell function is severely compromised by the hostile tumour microenvironment (TME)134. Current strategies leverage engineering tools to disrupt suppressive signals in the TME (panel a) and improve immune cell homing into tumour beds by ectopic expression of chemokine receptors (panel d). A selection of natural killer (NK) cell-relevant pathways that have been targeted through genetic engineering is shown. Genetic engineering strategies that include targeted ablation of inhibitory checkpoints156,164,167,168 (panels b,c) as well as disruption of extracellular receptors which sense inhibitory stimuli including TGFβ140,141 and adenosine135 (panel a) have been shown preclinically to effectively target pathways to enhance metabolic fitness and persistence of NK cells, and efforts are ongoing to advance these findings into the clinic. Ablation of endogenous receptors allows for combinatorial therapeutic approaches, such as by rendering immune cells resistant to corticosteroid-induced immunosuppression (panel e), a principle previously established in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-directed cytotoxic T lymphocytes (CTLs)281. Knockout of CD38 (panel f) renders NK cells resistant to CD38-mediated fratricide, which enables combination strategies of NK cells and anti-CD38-targeting monoclonal antibodies in the context of treating multiple myeloma50. Breg cell, regulatory B cell; MDSC, myeloid-derived suppressor cell; NKG2A, CD94/NK group 2 member A receptor; TIGIT, T cell immunoreceptor with immunoglobulin and ITIM domains; Treg cell, regulatory T cell.
Adenosine, a by-product of ATP metabolism, is generated via the activity of ectonucleotidases CD39 and CD73. Under the hypoxic conditions of the TME, expression of these enzymes is upregulated as part of the hypoxia-driven purinergic signalling pathway, resulting in the accumulation of extracellular adenosine, which in turn acts as a negative regulator of T cell and NK cell function in the TME, suppressing their metabolism and effector function127,128,129,130. Interestingly, adenosine contributes to activation of suppressor cells such as Treg cells, M2 macrophages and MDSCs, all resident populations within the TME. Therapeutic strategies for overcoming adenosine-mediated immunosuppression include blocking CD73 on tumour cells via small-molecule inhibitors or with antagonistic antibodies, both of which are currently under clinical investigation (NCT04148937 (ref.131), NCT03454451 (ref.132) and NCT03616886 (ref.133)). Furthermore, in preclinical studies, genetic editing to delete the adenosine A2A receptor in both CAR T cells and CAR NK cells has shown promise for improving the potency and antitumour efficacy of these cells134,135,136,137 (Fig. 4a).
TGFβ signalling is another mechanism of immunosuppression within the TME that has a deleterious effect on NK cell function. Knockdown of the TGFβ-induced miR-27a-5p led to improved NK cytotoxicity in vitro and in vivo138. Additionally, NK cells equipped with a high-affinity dominant-negative TGFβ receptor can resist the effects of TGFβ and retain their potency139. Similarly, our group successfully rendered CB-NK cells immune to TGFβ signalling by targeted deletion of the TGFβ receptor (TGFBR2)140. We subsequently provided in vivo evidence that disruption of TGFβ signalling safeguards NK cell cytotoxicity in a mouse model of glioblastoma141.
As we seek to modulate immunometabolism in the TME, it is important to achieve a physiologic balance, as some metabolites are an essential component of normal metabolism. Although many strategies are still under development, it is plausible that a combined approach involving TME modulation and NK cell engineering will lead to reduced immunosuppression and will support robust immune cell activity.
Checkpoint disruption
Tumours have evolved sophisticated mechanisms to evade immune surveillance, including the engagement of immune checkpoints, which may restrain NK cells in a similar manner as has been described for T cells. Striving to translate the clinical successes seen with the blockade of T cell-associated immune checkpoints PD1 and CTLA4, several groups have explored the potential of modulating these regulatory circuits in the context of NK cells142,143,144,145,146,147,148,149 (Fig. 4b). Although some studies point to their role as functional suppressors, their overall relevance in NK cell biology is still under debate146,150. With some likelihood, the signalling networks that underpin NK cell activation constitute a more nuanced picture in which these predominantly T cell markers act in concert with various other NK cell regulators151. Other regulators which have been investigated include TIM3 (refs.152,153), T cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT)136,153,154,155,156 and LAG3 (ref.157), and their targeting using monoclonal antibodies has been shown to reverse tumour-induced NK dysfunction in vitro.
Inhibitory KIRs are potent negative regulators of NK cell function and can override any concomitant activating signal when engaged with HLA class I ligands. Given their prominent role in suppressing NK cell function, inhibitory KIRs have attracted considerable interest. Lirilumab, an anti-panKIR2D antibody, functions to skew the diverse and often opposing signalling cues perceived by NK cells towards net activation by blocking these inhibitory KIRs158. In two early-phase clinical trials, however, Lirilumab failed to elicit clinically meaningful responses159,160. One potential reason for these results might stem from the important role of inhibitory KIRs in NK cell education and licensing. Under this assumption, prolonged KIR inhibition due to continuous KIR blockade by Lirilumab might negatively affect NK cell function161,162.
Similar to KIR molecules, CD94/NK group 2 member A receptor (NKG2A) is another prominent negative NK cell regulator, which, when bound to its cognate ligand HLA-E, restrains NK cell cytotoxicity. Monalizumab specifically disrupts this interaction and led to a promising 31% objective response rate in patients with previously treated recurrent or metastatic squamous cell carcinoma of the head and neck when administered in combination with cetuximab163. Monalizumab is currently being tested against other solid tumours including colorectal cancer and non-small-cell lung cancer as well as post alloHSCT.
Common to these approaches is the reliance on monoclonal antibodies to modulate the patients’ immune cells, a strategy that requires multiple infusions owing to their limited in vivo half-life. With advances in genetic editing capabilities, NK cells can be stably modified to regulate biological mechanisms that augment NK cell effector function. One example is the genetic disruption of the inhibitory receptor NKG2A, which led to superior tumour control in xenograft mouse models inoculated with HLA-E+ tumours164 (Fig. 4b). Moreover, building on seminal work that established CIS (cytokine-inducible SH2-containing protein) as a critical negative regulator of NK cell function165,166, we and others successfully engineered NK cells167,168 lacking this intracellular cytokine checkpoint. The resulting CAR NK cells exhibited enhanced metabolic fitness and increased antitumour activity (Fig. 4c).
These studies foreshadow the potential of targeted genetic perturbations to modulate NK cell biology. Going forward, we anticipate that unbiased high-throughput discovery approaches will elucidate the functional consequences of specific genetic interventions in a more systematic manner to, ultimately, inform the design of the next generation of NK cell immunotherapies.
Enhancing NK cell trafficking to tumours
The ability of NK cells to traffic to and penetrate tumour beds is a critical prerequisite for effective antitumour immunity and has been linked to improved clinical outcomes169,170,171,172,173. NK cells, similar to other immune cells, are guided towards tumour sites by the dynamic interplay of chemokine receptors and their cognate ligands secreted in the TME174,175,176,177. A growing body of work has, in recent years, investigated how modulation of these interactions may be harnessed to augment effective homing to the tumour. Whereas earlier studies focused on the expansion-induced upregulation of chemokine receptors178,179 as well as transient transfection methods180,181, the swift loss of chemokine expression owing to internalization and degradation182 has led to the increasing adoption of genetic engineering strategies to stably equip NK cells with ectopic chemokine receptors182,183 (Fig. 4d). Today, the available studies provide encouraging preclinical evidence to support chemokine receptor modulation to enhance NK cell trafficking into tumour beds across a broad range of hard to treat tumours including multiple myeloma184,185, glioblastoma182, renal cell carcinoma183, pancreatic ductal adenocarcinoma186 and ovarian cancer187.
Despite these advances, three important obstacles remain. First, chemotactic gradients rely on adequate tumour perfusion, and microthrombi-induced circulation deficits may require strategies to increase tumour micro-perfusion182. Second, the release of chemokines from the TME follows distinct tumour-specific kinetics, and intratumoural chemokine levels may need to be artificially boosted to robustly attract engineered NK cells179. Recent work has addressed this concern by locally augmenting chemokine concentrations using a mesothelin-targeting antibody fusion protein loaded with CXCL16, which is cleaved in the TME upon tumour cell engagement186. Furthermore, recent work has highlighted the application of radiation-induced CXCL8 to enhance NK cell migration to the tumour188. Last, chemokine–receptor interactions may be context-specific, either promoting or abrogating NK cell homing depending on the specific tumour type in question, as has been previously observed184,185. Future research will need to address these important questions and, ultimately, validate whether the overall promising findings can prevail in the context of the complex and dynamic interplay of chemokines within the human body.
Clinical lessons learned
CAR T cell therapy has led to remarkable clinical outcomes, with some patients achieving decade-long remissions in the presence of sustained CAR T cell persistence1,2,3,4,5,6,7,8,9. This new form of cellular immunotherapy has also shown potential for treatment of various cancers, driving the development of an ever-expanding number of new strategies to build on these successes189,190,191. The CAR T cell experience has elucidated many requirements for successful development of safe and efficacious cell therapies and has also revealed critical challenges related to mechanisms of resistance and barriers to immune effector cell persistence192,193. Knowledge gained from the clinical journey with CAR T cells has contributed to the emergence of novel cell therapy modalities. Over the past decade, the field has seen NK cells emerge as a strong new therapeutic candidate, possessing biological properties that may help overcome some of the limitations seen with T cell-based approaches. One noteworthy finding is that early clinical data suggest that NK cells are well suited for use in the allogeneic therapy setting as no major adverse events have been reported thus far in ongoing clinical studies194. Although safety results are encouraging, further investigations are required to elucidate whether allogeneic NK cells are capable of evading recipient T cell rejection for long-term persistence. A growing number of clinical studies have shown safety and efficacy of NK cells derived from peripheral blood mononuclear cells, cord blood, iPSCs, HSPCs and cell lines in the treatment of haematological malignancies28,29,30,33,34,37,39,48,49. Moreover, we have learned that, similar to their T cell counterparts, NK cells are quite amenable to genetic modification and numerous clinical candidates have been engineered to target the tumour with greater precision (by expressing CAR) and to increase persistence (through cytokine armouring). In addition, continuous efforts to enhance trafficking to tumour sites (through chemokine receptor editing) (Fig. 4d) and to shield against suppressive factors in the TME (through checkpoint inhibition, metabolic reprogramming (Fig. 4a) and hypoxia tolerance) may lead to enhanced benefits in the clinic. NK cells may also differentiate to a memory-like phenotype that naturally extends the persistence of NK cell function and confers the ability to recall an antitumour response upon antigen re-encounter. Memory NK cells are currently under clinical evaluation in the treatment of leukaemia. Moreover, memory-like NK cells pre-complexed with engager molecules provide an alternative to achieving CAR-like specificity without the need for lengthy manufacture100, and in the clinic, this strategy is driving responses comparable with those seen for CAR T cells and CAR NK cells. Importantly, irrespective of the approach, NK cell therapies have consistently demonstrated a favourable safety profile, and, to date, with no observed CRS or GvHD.
Although tremendous progress has been made, and in a short time NK cells have become an important tool for immuno-oncology, most of the successes reported are limited to haematological malignancies, much like the initial function of CAR T cells. However, there have been challenges. Not all patients respond to NK cell therapy, and some who do, eventually relapse. What are the mechanisms underlying tumour resistance and relapse? Are tumour cells, and, likewise, therapeutic NK cells, evolving in the course of their interactions? Understanding the biological underpinnings that influence patient response is critical to answering these questions. In the current era of single-cell multi-omic capabilities, we are poised to see these challenges as, in fact, opportunities (Box 1). Interrogating therapy evolution through a combination of genomic, proteomic and epigenomic profiling might offer valuable insights into changes that permanently ‘scar’ NK cells into a state of exhaustion and hypo-responsiveness to the tumour, as previously observed in T cells195,196. Indeed, we might also identify epigenetic roots to metabolic reprogramming that results in low bioenergetic reserves and decreased persistence. Once these mechanisms are elucidated, strategizing ways to reverse their effects and rescue cell function, or identifying favourable gene edits that intercept harmful interactions, will play a pivotal role in maximizing the benefit of cellular immunotherapies. Furthermore, implementing combination therapy approaches may provide a strategy for achieving a synergistic effect and increasing the efficacy of engineered adoptive NK cell therapies (Table 2). Integration of antibodies or small-molecule inhibitors to prevent metabolite-driven immunosuppression, or blockade of checkpoint and immunosuppressive mechanisms (Fig. 4b,e), for instance, might lead to a multifaceted outcome: boosting CAR NK cell function, hindering signals from suppressive cells in the TME and supporting the function of other immune effector cells197,198,199. Lastly, engineering NK cells to become resistant to CD38-mediated fratricide will enable NK cell-based treatment approaches to be combined with anti-CD38 monoclonal antibodies and allow NK cell immunotherapy to move into earlier lines of treatment in the setting of multiple myeloma50 (Fig. 4f).
Although many clinical trials are still in the early stages, through an iterative process we will be able to learn directly from patients what the barriers posed by each cancer are and take this information back to the laboratory where therapies can be further refined into applicable solutions to real-life challenges. Knowledge gained from these investigations will serve as the foundation upon which future clinical studies are built and will be critical to achieving success as we move forward and take aim at solid tumours. Through cellular engineering strategies and combination therapy approaches, NK cell adoptive therapies for solid tumours will also need to address and overcome challenges related to poor tumour trafficking and limited persistence and effector function in the immunosuppressive TME.
Ensuring genomic fidelity in NK cell therapies
Given the vast scope and the accelerated pace at which novel insights into fundamental NK cell immunobiology are generated, we anticipate an increase in novel engineered cell products. It is therefore increasingly important to establish platforms for screening and characterization of products to identify unintended genetic alterations resulting from off-target nuclease activity. Concerns that some genomic changes may inadvertently lead to oncogenic mutations continue to reverberate in the background as the field advances. To address this need, the US National Institutes of Health (NIH) have launched the Somatic Cell Genome Engineering (SCGE) programme with two main missions: first to provide financial aid to researchers to facilitate the transfer of genome editing technologies to the clinic, and second, to support the development of more comprehensive assays to investigate potential adverse biological effects that could result from the use of these tools200. In addition, various platforms to investigate genome editing fidelity have been developed, such as GUIDE-Seq201, CIRCLE-Seq202 and rhampSeq203 that rely on sequencing technologies to unbiasedly identify sites in the genome where double-stranded breaks may have occurred. Moreover, unintended genetic rearrangements may be elucidated by assays such as linear amplification-mediated high-throughput genome-wide translocation sequencing (LAM-HTGTS)204. As these tools continue to evolve and newer ones are developed, the implementation of such assays in cell therapy development will be of paramount importance to ensure greater understanding of safety and efficacy of cellular products such as engineered NK cell therapies.
Concluding remarks
Engineered cellular immunotherapies continue to experience tremendous growth, with diverse modalities quickly advancing from preclinical studies into clinical testing. The recent report of in vivo editing of hepatocytes in patients with transthyretin amyloidosis epitomizes the latest frontier in the cell and gene therapy space205. In a related effort, an approach to enable T cell-targeted in vivo CAR transfection using lipid nanoparticles has been developed for the generation of fibroblast activating protein (FAP)-redirected CAR T cells to target cardiac remodelling in the context of myocardial injury206,207. Moreover, recent work has demonstrated that Nipah lentivirus vectors redirected to CD3, CD8 and CD4 could specifically deliver therapeutic genes (such as CAR and TCR) to T cell populations in vivo208. These technical innovations provide a new approach to off-the-shelf personalized therapies, in which targeted viral vectors are readily available to be administered to patients upon need, thereby bypassing the requirement for extensive ex vivo manufacturing, reducing costs and expediting therapy delivery. It is conceivable that targeted in vivo editing may also be applied to tumour cells, driving modifications that will restore their sensitivity to anti-neoplastic agents or eliminate resistance mechanisms. Although current initiatives have mostly focused on CAR T cell therapy, in vivo engineering might also be applicable to NK cell-based therapies, such as to boost endogenous NK cell function and persistence, or to increase tumour sensitivity to NK cell-mediated cytotoxicity.
As the field continues to innovate at a rapid rate, it is important to keep abreast of potential safety risks associated with these various gene editing strategies, as concerns associated with possible off-target effects are very relevant. The recent halt of multiple ongoing cell therapy trials owing to cases of chromosomal abnormalities, treatment-associated AML/myelodysplastic syndrome (MDS) or malignant transformation of the infused CAR T cell product209 justify these concerns and serve as reminders of the importance of closely studying the safety of engineered products. Moreover, it is critical that cell therapy programmes implement pipelines for systematic screening of products to assess potential unwanted genetic modifications that may result in deleterious effects. Leveraging resources such as the NIH’s SCGE programme may provide the initial support needed to launch these initiatives.
Although NK cell-based immunotherapy is well positioned as a safe off-the-shelf antitumour therapy, important questions remain open. Elucidating the key parameters that determine NK cell potency and persistence will be important as the field progresses into developing approaches to address challenges specific to each disease indication. Moreover, decisions related to use of co-stimulatory signal, cytokine armouring and combination with other therapeutic modalities will play a role in maximizing the longevity and benefit of adoptively transferred NK cells. Finally, it will be critical to develop and implement optimal methods for expansion and cryopreservation of NK cells to ensure that high product quality is sustained. Logistically, for emerging NK cell therapy programmes to succeed, it will be essential to establish multidisciplinary team structures comprising researchers, clinicians and regulatory officials to jointly sketch a wholistic path to clinical translation.
References
Maude, S. L. et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 378, 439–448 (2018).
Schuster, S. J. et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 380, 45–56 (2019).
Neelapu, S. S. et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017).
Park, J. H. et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 378, 449–459 (2018).
June, C. H., O’Connor, R. S., Kawalekar, O. U., Ghassemi, S. & Milone, M. C. CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018).
Wang, M. et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 382, 1331–1342 (2020).
Munshi, N. C. et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 384, 705–716 (2021).
Raje, N. et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl. J. Med. 380, 1726–1737 (2019).
Melenhorst, J. J. et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 602, 503–509 (2022). This landmark article reports the sustained remissions and in vivo persistence of CD19-CAR T cells for more than 10 years after infusion, and hence highlights the long-term durability of clinical responses achieved using genetically engineered T cells.
Malmberg, K.-J. et al. Natural killer cell-mediated immunosurveillance of human cancer. Semin. Immunol. 31, 20–29 (2017).
Lanier, L. L. Up on the tightrope: natural killer cell activation and inhibition. Nat. Immunol. 9, 495–502 (2008).
Joncker, N. T., Fernandez, N. C., Treiner, E., Vivier, E. & Raulet, D. H. NK cell responsiveness is tuned commensurate with the number of inhibitory receptors for self-MHC class I: the rheostat model. J. Immunol. 182, 4572–4580 (2009). This study elucidates the nature of NK cell responsiveness, which relies on the integration of both inhibitory and activating signalling cues to ensure self-tolerance and immunosurveillance over abnormal cells.
Joncker, N. T., Shifrin, N., Delebecque, F. & Raulet, D. H. Mature natural killer cells reset their responsiveness when exposed to an altered MHC environment. J. Exp. Med. 207, 2065–2072 (2010).
Burshtyn, D. N. et al. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitor receptor. Immunity 4, 77–85 (1996).
Yokoyama, W. M. & Kim, S. How do natural killer cells find self to achieve tolerance? Immunity 24, 249–257 (2006).
Brodin, P., Lakshmikanth, T., Johansson, S., Kärre, K. & Höglund, P. The strength of inhibitory input during education quantitatively tunes the functional responsiveness of individual natural killer cells. Blood 113, 2434–2441 (2009).
Imai, K., Matsuyama, S., Miyake, S., Suga, K. & Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet 356, 1795–1799 (2000).
Guerra, N. et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 28, 571–580 (2008).
López-Soto, A., Gonzalez, S., Smyth, M. J. & Galluzzi, L. Control of metastasis by NK cells. Cancer Cell 32, 135–154 (2017).
Abel, A. M., Yang, C., Thakar, M. S. & Malarkannan, S. Natural killer cells: development, maturation, and clinical utilization. Front. Immunol. 9, 1869 (2018).
Dalle, J.-H. et al. Characterization of cord blood natural killer cells: implications for transplantation and neonatal infections. Pediatr. Res. 57, 649–655 (2005).
Strauss-Albee, D. M. et al. Human NK cell repertoire diversity reflects immune experience and correlates with viral susceptibility. Sci. Transl. Med. 7, 297ra115–297ra115 (2015).
Prager, I. & Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 105, 1319–1329 (2019).
Wang, W., Erbe, A. K., Hank, J. A., Morris, Z. S. & Sondel, P. M. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 6, 368 (2015).
O’Leary, J. G., Goodarzi, M., Drayton, D. L. & von Andrian, U. H. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat. Immunol. 7, 507–516 (2006). This seminal study demonstrates that NK cells can mediate durable recall responses upon antigen re-exposure, establishing the concept of NK cell adaptive memory.
Sun, J. C., Beilke, J. N. & Lanier, L. L. Adaptive immune features of natural killer cells. Nature 457, 557–561 (2009). This important article reveals self-renewing ‘memory’ NK cell subsets that can undergo secondary expansion and elicit strong adaptive immune responses upon viral challenge when transferred to naive animals.
Cooper, M. A. et al. Cytokine-induced memory-like natural killer cells. Proc. Natl Acad. Sci. USA 106, 1915–1919 (2009). This work pioneers the concept of cytokine-induced memory-like NK cells which elicit robust recall responses when transferred to naïve hosts.
Romee, R. et al. Cytokine activation induces human memory-like NK cells. Blood 120, 4751–4760 (2012).
Romee, R. et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 8, 357ra123 (2016).
Shapiro, R. M. et al. Expansion, persistence, and efficacy of donor memory-like NK cells infused for post-transplant relapse. J. Clin. Investig. https://doi.org/10.1172/JCI154334 (2022).
Platonova, S. et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 71, 5412–5422 (2011).
Sun, C. et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology 6, e1264562 (2017).
Spanholtz, J. et al. High log-scale expansion of functional human natural killer cells from umbilical cord blood CD34-positive cells for adoptive cancer immunotherapy. PLoS ONE 5, e9221 (2010).
Dolstra, H. et al. Successful transfer of umbilical cord blood CD34+ hematopoietic stem and progenitor-derived NK cells in older acute myeloid leukemia patients. Clin. Cancer Res. 23, 4107–4118 (2017).
Liu, E. et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 32, 520–531 (2018). This article reports the first successful clinical application of CAR-modified NK immunotherapy in patients with CD19-positive haematologic malignancies.
Berrien-Elliott, M. M. et al. Multidimensional analyses of donor memory-like NK cells reveal new associations with response after adoptive immunotherapy for leukemia. Cancer Discov. 10, 1854–1871 (2020).
Liu, E. et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 382, 545–553 (2020).
Gong, J.-H., Maki, G. & Klingemann, H. G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 8, 652–658 (1994).
Tang, X. et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 8, 1083–1089 (2018).
Zhang, C. et al. Chimeric antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front. Immunol. https://doi.org/10.3389/fimmu.2017.00533 (2017).
Hoogstad-van Evert, J. S. et al. Umbilical cord blood CD34+ progenitor-derived NK cells efficiently kill ovarian cancer spheroids and intraperitoneal tumors in NOD/SCID/IL2Rgnull mice. Oncoimmunology 6, e1320630 (2017).
Knorr, D. A. et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cell Transl. Med. 2, 274–283 (2013).
Li, Y., Hermanson, D. L., Moriarity, B. S. & Kaufman, D. S. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 23, 181–192.e5 (2018). This article demonstrates the first successful generation of iPSC-derived CAR NK cells.
Goldenson, B. H. et al. Umbilical cord blood and iPSC-derived natural killer cells demonstrate key differences in cytotoxic activity and KIR profiles. Front. Immunol. 11, https://doi.org/10.3389/fimmu.2020.561553 (2020).
Zhu, H. et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 135, 399–410 (2020).
Kim, K. et al. Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–290 (2010).
Bar-Nur, O., Russ, H. A., Efrat, S. & Benvenisty, N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet β cells. Cell Stem Cell 9, 17–23 (2011).
Goodridge, J. P. et al. FT596: translation of first-of-kind multi-antigen targeted off-the-shelf CAR-NK cell with engineered persistence for the treatment of B cell malignancies. Blood 134, 301–301 (2019).
Bachanova, V. et al. Safety and efficacy of FT596, a first-in-class, multi-antigen targeted, off-the-shelf, iPSC-derived CD19 CAR NK cell therapy in relapsed/refractory B-cell lymphoma. Blood 138, 823 (2021).
Goodridge, J. P. et al. Abstract 1550: FT576 path to first-of-kind clinical trial: translation of a versatile multi-antigen specific off-the-shelf NK cell for treatment of multiple myeloma. Cancer Res. 81, 1550 (2021).
Strati, P. et al. Preliminary results of a phase I trial of FT516, an off-the-shelf natural killer (NK) cell therapy derived from a clonal master induced pluripotent stem cell (iPSC) line expressing high-affinity, non-cleavable CD16 (hnCD16), in patients (pts) with relapsed/refractory (R/R) B-cell lymphoma (BCL). J. Clin. Oncol. 39, 7541–7541 (2021).
Imai, C., Iwamoto, S. & Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 106, 376–383 (2005). This article describes the first successful generation of CAR NK cells using a 41BB-co-stimulated CD19-directed synthetic CAR.
Ruggeri, L. et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 295, 2097–2100 (2002). This seminal work demonstrates that alloreactive NK cells confer a potent graft-versus-leukaemia effect and protect against GvHD in recipients of T cell-depleted HLA-haploidentical allogeneic transplantation (TCD-haplo-alloHSCT).
Ruggeri, L. et al. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood 94, 333–339 (1999).
Davies, S. M. et al. Evaluation of KIR ligand incompatibility in mismatched unrelated donor hematopoietic transplants. Killer immunoglobulin-like receptor. Blood 100, 3825–3827 (2002).
Ruggeri, L. et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood 110, 433–440 (2007).
Ciurea, S. O. et al. Decrease post-transplant relapse using donor-derived expanded NK-cells. Leukemia 36, 155–164 (2022).
Hsu, K. C. et al. Improved outcome in HLA-identical sibling hematopoietic stem-cell transplantation for acute myelogenous leukemia predicted by KIR and HLA genotypes. Blood 105, 4878–4884 (2005).
Verheyden, S., Schots, R., Duquet, W. & Demanet, C. A defined donor activating natural killer cell receptor genotype protects against leukemic relapse after related HLA-identical hematopoietic stem cell transplantation. Leukemia 19, 1446–1451 (2005).
Venstrom, J. M. et al. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N. Engl. J. Med. 367, 805–816 (2012).
Boudreau, J. E. et al. KIR3DL1/HLA-B subtypes govern acute myelogenous leukemia relapse after hematopoietic cell transplantation. J. Clin. Oncol. 35, 2268 (2017).
Schetelig, J. et al. External validation of models for KIR2DS1/KIR3DL1-informed selection of hematopoietic cell donors fails. Blood 135, 1386–1395 (2020).
Beelen, D. W. et al. Genotypic inhibitory killer immunoglobulin-like receptor ligand incompatibility enhances the long-term antileukemic effect of unmodified allogeneic hematopoietic stem cell transplantation in patients with myeloid leukemias. Blood 105, 2594–2600 (2005).
Bishara, A. et al. The beneficial role of inhibitory KIR genes of HLA class I NK epitopes in haploidentically mismatched stem cell allografts may be masked by residual donor-alloreactive T cells causing GVHD. Tissue Antigens 63, 204–211 (2004).
Schaffer, M., Malmberg, K. J., Ringdén, O., Ljunggren, H. G. & Remberger, M. Increased infection-related mortality in KIR-ligand-mismatched unrelated allogeneic hematopoietic stem-cell transplantation. Transplantation 78, 1081–1085 (2004).
Mehta, R. S. & Rezvani, K. Can we make a better match or mismatch with KIR genotyping? Hematol. Am. Soc. Hematol. Educ. Program. 2016, 106–118 (2016).
Schmidts, A. et al. Rational design of a trimeric APRIL-based CAR-binding domain enables efficient targeting of multiple myeloma. Blood Adv. 3, 3248–3260 (2019).
Leivas, A. et al. NKG2D-CAR-transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J. 11, 146 (2021).
Chang, Y. H. et al. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 73, 1777–1786 (2013).
Biederstädt, A. & Rezvani, K. Engineering the next generation of CAR-NK immunotherapies. Int. J. Hematol. 114, 554–571 (2021).
Daher, M. & Rezvani, K. Outlook for new CAR-based therapies with a focus on CAR NK cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Disco 11, 45–58 (2021).
Lanier, L. L., Corliss, B. C., Wu, J., Leong, C. & Phillips, J. H. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature 391, 703–707 (1998). This seminal article elucidates the role of DAP12 as an activating NK cell immunoreceptor through cross-linking with killer cell immunoglobin-like receptor (KIR) family molecules.
Zhao, R. et al. DNAX-activating protein 10 co-stimulation enhances the anti-tumor efficacy of chimeric antigen receptor T cells. Oncoimmunology 8, e1509173 (2018).
Ng, Y. Y. et al. T cells expressing NKG2D CAR with a DAP12 signaling domain stimulate lower cytokine production while effective in tumor eradication. Mol. Ther. 29, 75–85 (2021).
Töpfer, K. et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J. Immunol. 194, 3201 (2015).
Billadeau, D. D., Upshaw, J. L., Schoon, R. A., Dick, C. J. & Leibson, P. J. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 4, 557–564 (2003). This article uncovers the role of the activating NKG2D–DAP10 immune receptor recognition complex which can induce NK cell-mediated killing in a SYK-independent manner.
Frigault, M. J. et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res. 3, 356–367 (2015).
Long, A. H. et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 21, 581–590 (2015). This important article lays out the principle of antigen-independent tonic CAR signalling which can induce T cell exhaustion impairing antitumour efficacy.
Watanabe, N. et al. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology 5, e1253656 (2016).
Mamonkin, M. et al. Tonic 4-1BB signaling from chimeric antigen receptors (CARs) impairs expansion of T cells due to Fas-mediated apoptosis. J. Immunol. 196 (Suppl 1), 143.7 (2016).
Feucht, J. et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 25, 82–88 (2019).
Bridgeman, J. S. et al. CD3ζ-based chimeric antigen receptors mediate T cell activation via cis- and trans-signalling mechanisms: implications for optimization of receptor structure for adoptive cell therapy. Clin. Exp. Immunol. 175, 258–267 (2014).
Bridgeman, J. S. et al. The optimal antigen response of chimeric antigen receptors harboring the CD3ζ transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol. 184, 6938–6949 (2010).
Muller, Y. D. et al. The CD28-transmembrane domain mediates chimeric antigen receptor heterodimerization with CD28. Front. Immunol. 12, 639818 (2021).
Savoldo, B. et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Invest. 121, 1822–1826 (2011).
Cronk, R. J., Zurko, J. & Shah, N. N. Bispecific chimeric antigen receptor T cell therapy for B cell malignancies and multiple myeloma. Cancers 12, 2523 (2020).
Zah, E. et al. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat. Commun. 11, 2283 (2020).
Shah, N. N. et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat. Med. 26, 1569–1575 (2020).
Wallstabe, L. et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight https://doi.org/10.1172/jci.insight.126345 (2019).
Srivastava, S. et al. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell 35, 489–503.e8 (2019).
Cho, J. H. et al. Engineering advanced logic and distributed computing in human CAR immune cells. Nat. Commun. 12, 792 (2021).
Garrison, B. S. et al. FLT3 OR CD33 NOT EMCN logic gated CAR-NK cell therapy (SENTI-202) for precise targeting of AML. Blood 138 (suppl. 1), 2799 (2021).
Gonzalez, A. et al. Abstract LB028: Development of logic-gated CAR-NK cells to reduce target-mediated healthy tissue toxicities. Cancer Res. 81, LB028 (2021).
Mensali, N. et al. NK cells specifically TCR-dressed to kill cancer cells. EBioMedicine 40, 106–117 (2019).
Shao, H. et al. TCR mispairing in genetically modified T cells was detected by fluorescence resonance energy transfer. Mol. Biol. Rep. 37, 3951–3956 (2010).
Wiernik, A. et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16×33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 19, 3844–3855 (2013).
Vallera, D. A. et al. Heterodimeric bispecific single-chain variable-fragment antibodies against EpCAM and CD16 induce effective antibody-dependent cellular cytotoxicity against human carcinoma cells. Cancer Biother. Radiopharm. 28, 274–282 (2013).
Gleason, M. K. et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 123, 3016–3026 (2014).
Schmohl, J., Gleason, M., Dougherty, P., Miller, J. S. & Vallera, D. A. Heterodimeric bispecific single chain variable fragments (scFv) killer engagers (BiKEs) enhance NK-cell activity against CD133+ colorectal cancer cells. Target. Oncol. 11, 353–361 (2016).
Kerbauy, L. N. et al. Combining AFM13, a bispecific CD30/CD16 antibody, with cytokine-activated blood and cord blood-derived NK cells facilitates CAR-like responses against CD30+ malignancies. Clin. Cancer Res. 27, 3744–3756 (2021).
Reusch, U. et al. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs 6, 727–738 (2014).
Schmohl, J. U. et al. Tetraspecific scFv construct provides NK cell mediated ADCC and self-sustaining stimuli via insertion of IL-15 as a cross-linker. Oncotarget 7, 73830–73844 (2016).
Vallera, D. A. et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin. Cancer Res. 22, 3440–3450 (2016).
Schmohl, J. U. et al. Engineering of anti-CD133 trispecific molecule capable of inducing NK expansion and driving antibody-dependent cell-mediated cytotoxicity. Cancer Res. Treat. 49, 1140 (2017).
Arvindam, U. S. et al. A trispecific killer engager molecule against CLEC12A effectively induces NK-cell mediated killing of AML cells. Leukemia 35, 1586–1596 (2021).
Gauthier, L. et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell 177, 1701–1713.e16 (2019). This report outlines a novel tri-specific NK cell engager molecule that cross-links the two NK cell activating receptors CD16 and NKp46 with a specific tumour antigen.
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04074746 (2020).
Bryceson, Y. T., March, M. E., Ljunggren, H. G. & Long, E. O. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 107, 159–166 (2006).
Rosario, M. et al. The IL-15-based ALT-803 complex enhances FcγRIIIa-triggered NK cell responses and in vivo clearance of B cell lymphomas. Clin. Cancer Res. 22, 596–608 (2016).
de Rham, C. et al. The proinflammatory cytokines IL-2, IL-15 and IL-21 modulate the repertoire of mature human natural killer cell receptors. Arthritis Res. Ther. 9, R125 (2007).
Gang, M. et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood 136, 2308–2318 (2020).
Dong, H. et al. Engineered memory-like NK cars targeting a neoepitope derived from intracellular NPM1c exhibit potent activity and specificity against acute myeloid leukemia. Blood 136, 3–4 (2020).
Lasek, W., Zagożdżon, R. & Jakobisiak, M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 63, 419–435 (2014).
McMichael, E. L. et al. IL-21 enhances natural killer cell response to cetuximab-coated pancreatic tumor cells. Clin. Cancer Res. 23, 489–502 (2017).
Miller, J. S. Therapeutic applications: natural killer cells in the clinic. Hematology 2013, 247–253 (2013).
Anton, O. M. et al. Trans-endocytosis of intact IL-15Rα–IL-15 complex from presenting cells into NK cells favors signaling for proliferation. Proc. Natl Acad. Sci. USA 117, 522–531 (2020).
Tarannum, M. & Romee, R. Cytokine-induced memory-like natural killer cells for cancer immunotherapy. Stem Cell Res. Ther. 12, 592 (2021).
Felices, M. et al. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight https://doi.org/10.1172/jci.insight.96219 (2018).
Sarhan, D. et al. Adaptive NK cells resist regulatory T-cell suppression driven by IL37. Cancer Immunol. Res. 6, 766–775 (2018).
Nuñez, S. Y. et al. Human M2 macrophages limit NK cell effector functions through secretion of TGF-β and engagement of CD85j. J. Immunol. 200, 1008–1015 (2018).
Tumino, N. et al. Interaction between MDSC and NK cells in solid and hematological malignancies: impact on HSCT. Front. Immunol. https://doi.org/10.3389/fimmu.2021.638841 (2021).
Zalfa, C. & Paust, S. Natural killer cell interactions with myeloid derived suppressor cells in the tumor microenvironment and implications for cancer immunotherapy. Front. Immunol. https://doi.org/10.3389/fimmu.2021.633205 (2021).
Ni, J. et al. Single-cell RNA sequencing of tumor-infiltrating NK cells reveals that inhibition of transcription factor HIF-1α unleashes NK cell activity. Immunity 52, 1075–1087.e8 (2020).
Van Wilpe, S. et al. Lactate dehydrogenase: a marker of diminished antitumor immunity. Oncoimmunology 9, 1731942 (2020).
Cascone, T. et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 27, 977–-987.e4 (2018).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT01791595 (2013).
Jin, D. et al. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. 70, 2245–2255 (2010).
Stagg, J. et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl Acad. Sci. USA 107, 1547–1552 (2010).
Allard, B., Longhi, M. S., Robson, S. C. & Stagg, J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol. Rev. 276, 121–144 (2017).
Perrot, I. et al. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep. 27, 2411–2425.e9 (2019).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04148937 (2020).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03454451 (2018).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03616886 (2018).
Young, A. et al. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell 30, 391–403 (2016).
Young, A. et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. 78, 1003 (2018).
Lupo, K. & Matosevic, S. 123 Natural killer cells engineered with an inducible, responsive genetic construct targeting TIGIT and CD73 to relieve immunosuppression within the GBM microenvironment. J. Immunother. Cancer 8, A74–A75 (2020).
Giuffrida, L. et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat. Commun. 12, 3236 (2021).
Kim, T.-D. et al. Human microRNA-27a* targets Prf1 and GzmB expression to regulate NK-cell cytotoxicity. Blood, J. Am. Soc. Hematol. 118, 5476–5486 (2011).
Yvon, E. S. et al. Cord blood natural killer cells expressing a dominant negative TGF-β receptor: implications for adoptive immunotherapy for glioblastoma. Cytotherapy 19, 408–418 (2017).
Daher, M. et al. The TGF-β/SMAD signaling pathway as a mediator of NK cell dysfunction and immune evasion in myelodysplastic syndrome. Blood 130, 53–53 (2017).
Shaim, H. et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. https://doi.org/10.1172/JCI142116 (2021).
Stojanovic, A., Fiegler, N., Brunner-Weinzierl, M. & Cerwenka, A. CTLA-4 Is expressed by activated mouse NK cells and inhibits NK cell IFN-γ production in response to mature dendritic cells. J. Immunol. 192, 4184–4191 (2014).
Russick, J. et al. Natural killer cells in the human lung tumor microenvironment display immune inhibitory functions. J. Immunother. Cancer 8, e001054 (2020).
Sanseviero, E. et al. Anti-CTLA-4 activates intratumoral NK cells and combined with IL15/IL15Rα complexes enhances tumor control. Cancer Immunol. Res. 7, 1371–1380 (2019).
Judge, S. J. et al. Minimal PD-1 expression in mouse and human NK cells under diverse conditions. J. Clin. Investig. 130, 3051–3068 (2020).
Judge, S. J., Murphy, W. J. & Canter, R. J. Characterizing the dysfunctional NK cell: assessing the clinical relevance of exhaustion, anergy, and senescence. Front. Cell Infect. Microbiol. 10, 49–49 (2020).
Davis, Z. et al. Low-density PD-1 expression on resting human natural killer cells is functional and upregulated after transplantation. Blood Adv. 5, 1069–1080 (2021).
Hsu, J. et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 128, 4654–4668 (2018).
Dong, W. et al. The mechanism of anti-PD-L1 antibody efficacy against PD-L1-negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discov. 9, 1422–1437 (2019).
Newman, J. & Horowitz, A. NK cells seize PD1 from leukaemia cells. Nat. Rev. Immunol. 21, 345–345 (2021).
Wilk, A. J. et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 26, 1070–1076 (2020).
da Silva, I. P. et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol. Res. 2, 410–422 (2014).
Farkas, A. M. et al. Tim-3 and TIGIT mark natural killer cells susceptible to effector dysfunction in human bladder cancer. J. Immunol. 200, 124.114 (2018).
Chauvin, J.-M. et al. IL15 stimulation with TIGIT blockade reverses CD155-mediated NK-cell dysfunction in melanoma. Clin. Cancer Res. 26, 5520–5533 (2020).
Sarhan, D. et al. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res. 76, 5696–5706 (2016).
Zhang, Q. et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 19, 723–732 (2018).
Ali, A. et al. LAG-3 modulation of natural killer cell immunoregulatory function. J. Immunol. 202, 76.77 (2019).
Kohrt, H. E. et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood 123, 678–686 (2014).
Vey, N. et al. Randomized phase 2 trial of Lirilumab (anti-KIR monoclonal antibody, mAb) as maintenance treatment in elderly patients (pts) with acute myeloid leukemia (AML): results of the Effikir trial. Blood 130, 889–889 (2017).
Vey, N. et al. A phase 1 study of Lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget 9, 17675–17688 (2018).
Kim, S. et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 436, 709–713 (2005). This seminal work lays out the concept of NK cell ‘licensing’ through which NK cells acquire functional competence and self-tolerance by interaction of inhibitory receptors and self-MHC molecules.
Kim, S. et al. HLA alleles determine differences in human natural killer cell responsiveness and potency. Proc. Natl Acad. Sci. USA 105, 3053–3058 (2008).
André, P. et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 175, 1731–1743.e13 (2018).
Kamiya, T., Seow, S. V., Wong, D., Robinson, M. & Campana, D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J. Clin. Investig. 129, 2094–2106 (2019).
Delconte, R. B. et al. CIS is a potent checkpoint in NK cell–mediated tumor immunity. Nat. Immunol. 17, 816–824 (2016). This important article identifies the intracellular NK cell checkpoint CIS, which negatively regulates IL-15 signalling.
Delconte, R. B. et al. NK cell priming from endogenous homeostatic signals is modulated by CIS. Front. Immunol. https://doi.org/10.3389/fimmu.2020.00075 (2020).
Daher, M. et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood 137, 624–636 (2021).
Zhu, H. et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell 27, 224–237.e6 (2020).
Coca, S. et al. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 79, 2320–2328 (1997).
Ishigami, S. et al. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer 88, 577–583 (2000).
Villegas, F. R. et al. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer 35, 23–28 (2002).
Donskov, F. & von der Maase, H. Impact of immune parameters on long-term survival in metastatic renal cell carcinoma. J. Clin. Oncol. 24, 1997–2005 (2006).
Geissler, K. et al. Immune signature of tumor infiltrating immune cells in renal cancer. Oncoimmunology 4, e985082 (2015).
Wendel, M., Galani, I. E., Suri-Payer, E. & Cerwenka, A. Natural killer cell accumulation in tumors is dependent on IFN-γ and CXCR3 ligands. Cancer Res. 68, 8437–8445 (2008).
Mlecnik, B. et al. Biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology 138, 1429–1440 (2010).
Park, M. H., Lee, J. S. & Yoon, J. H. High expression of CX3CL1 by tumor cells correlates with a good prognosis and increased tumor-infiltrating CD8+ T cells, natural killer cells, and dendritic cells in breast carcinoma. J. Surg. Oncol. 106, 386–392 (2012).
Castriconi, R. et al. Molecular mechanisms directing migration and retention of natural killer cells in human tissues. Front. Immunol. 9, 2324 (2018).
Levy, E. R., Clara, J. A., Reger, R. N., Allan, D. S. J. & Childs, R. W. RNA-seq analysis reveals CCR5 as a key target for CRISPR gene editing to regulate in vivo NK cell trafficking. Cancers https://doi.org/10.3390/cancers13040872 (2021).
Wennerberg, E., Kremer, V., Childs, R. & Lundqvist, A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol. Immunother. 64, 225–235 (2015).
Somanchi, S. S., Somanchi, A., Cooper, L. J. N. & Lee, D. A. Engineering lymph node homing of ex vivo–expanded human natural killer cells via trogocytosis of the chemokine receptor CCR7. Blood 119, 5164–5172 (2012).
Carlsten, M. et al. Efficient mRNA-based genetic engineering of human NK cells with high-affinity CD16 and CCR7 augments rituximab-induced ADCC against lymphoma and targets NK cell migration toward the lymph node-associated chemokine CCL19. Front. immunol. 7, 105 (2016).
Müller, N. et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1α-secreting glioblastoma. J. Immunother. 38, 197 (2015).
Kremer, V. et al. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J. ImmunoTher. Cancer 5, 73 (2017).
Ponzetta, A. et al. Multiple myeloma impairs bone marrow localization of effector natural killer cells by altering the chemokine microenvironment. Cancer Res. 75, 4766–4777 (2015).
Bonanni, V., Antonangeli, F., Santoni, A. & Bernardini, G. Targeting of CXCR3 improves anti-myeloma efficacy of adoptively transferred activated natural killer cells. J. ImmunoTher. Cancer 7, 290 (2019).
Lee, J. et al. An antibody designed to improve adoptive NK-cell therapy inhibits pancreatic cancer progression in a murine model. Cancer Immunol. Res. 7, 219 (2019).
Ng, Y. Y., Tay, J. C. & Wang, S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol. Ther. Oncolytics 16, 75–85 (2020).
Walle, T. et al. Radiotherapy orchestrates natural killer cell dependent antitumor immune responses through CXCL8. Sci. Adv. 8, eabh4050 (2022).
Larson, R. C. & Maus, M. V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 21, 145–161 (2021).
Sterner, R. C. & Sterner, R. M. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 11, 69 (2021).
Gill, S. & Brudno, J. N. CAR T-cell therapy in hematologic malignancies: clinical role, toxicity, and unanswered questions. Am. Soc. Clin. Oncol. Educ. Book https://doi.org/10.1200/EDBK_320085 (2021).
Shah, N. N. & Fry, T. J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 16, 372–385 (2019).
Strati, P. & Neelapu, S. S. CAR-T failure: beyond antigen loss and T cells. Blood 137, 2567–2568 (2021).
Biederstädt, A. & Rezvani, K. How I treat high-risk acute myeloid leukemia using pre-emptive adoptive cellular immunotherapy. Blood https://doi.org/10.1182/blood.2021012411 (2022).
Abdel-Hakeem, M. S. et al. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat. Immunol. 22, 1008–1019 (2021). This article elucidates how exhausted T cell subsets fail to restore their full functional capacity upon elimination of antigenic stimulation due to persistent epigenetic scarring.
Yates, K. B. et al. Epigenetic scars of CD8+ T cell exhaustion persist after cure of chronic infection in humans. Nat. Immunol. 22, 1020–1029 (2021). This report demonstrates the fixed nature of exhausted T cell epigenetic signatures following resolution of chronic antigenic stimulation.
Biederstädt, A. et al. SUMO pathway inhibition targets an aggressive pancreatic cancer subtype. Gut 69, 1472–1482 (2020).
Kumar, S. et al. Targeting pancreatic cancer by TAK-981: a SUMOylation inhibitor that activates the immune system and blocks cancer cell cycle progression in a preclinical model. Gut https://doi.org/10.1136/gutjnl-2021-324834 (2022).
Demel, U. M. et al. Activated SUMOylation restricts MHC class I antigen presentation to confer immune evasion in cancer. J. Clin. Investig. https://doi.org/10.1172/JCI152383 (2022).
Saha, K. et al. The NIH Somatic Cell Genome Editing program. Nature 592, 195–204 (2021). This report lays out the aims and scope of the NIH SCGE Consortium.
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR–Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015).
Tsai, S. Q. et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat. Methods 14, 607–614 (2017).
Dobosy, J. R. et al. RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 11, 80 (2011).
Hu, J. et al. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat. Protoc. 11, 853–871 (2016).
Gillmore, J. D. et al. CRISPR–Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385, 493–502 (2021). This seminal work demonstrates the first successful clinical application of in vivo CRISPR–Cas9 gene editing.
Aghajanian, H. et al. Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433 (2019).
Rurik, J. G. et al. CAR T cells produced in vivo to treat cardiac injury. Science 375, 91–96 (2022). This study demonstrates the first successful in vivo engineering of CAR T cells using a CD5-directed lipid nanoparticle containing CAR-encoding mRNA.
Bender, R. R., Muth, A., Schneider, I. C., Maisner, A. & Buchholz, C. J. Developing an engineered nipah virus glycoprotein based lentiviral vector system retargeted to cell surface receptors of choice. Mol. Ther. 23, S2 (2015).
Micklethwaite, K. P. et al. Investigation of product-derived lymphoma following infusion of piggyBac-modified CD19 chimeric antigen receptor T cells. Blood 138, 1391–1405 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT02742727 (2016).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT00995137 (2009).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT01974479 (2013).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT02892695 (2016).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03056339 (2017).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03824951 (2019).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03690310 (2019).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT05020678 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04245722 (2020).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04639739 (2020).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04887012 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04796675 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04796688 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT05379647 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT05336409 (2022).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04023071 (2019).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03692767 (2019).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03824964 (2019).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT02944162 (2016).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT05008575 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT05215015 (2020).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT05092451 (2022).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03559764 (2018).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03940833 (2019).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT05008536 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT05182073 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04614636 (2020).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04623944 (2020).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03415100 (2018).
Xiao, L. et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol. Ther. 27, 1114–1125 (2019).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT05213195 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT05247957 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT02839954 (2016).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03383978 (2017).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03692663 (2018).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03692637 (2019).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04630769 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03940820 (2019).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03931720 (2019).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03941457 (2019).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04551885 (2020).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04847466 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT05194709 (2021).
US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04324996 (2020).
Wu, L. et al. lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin. Cancer Res. 14, 4650–4657 (2008).
Tai, Y. T. et al. Immunomodulatory drug lenalidomide (CC-5013, IMiD3) augments anti-CD40 SGN-40-induced cytotoxicity in human multiple myeloma: clinical implications. Cancer Res. 65, 11712–11720 (2005).
Nahas, M. R. et al. Hypomethylating agent alters the immune microenvironment in acute myeloid leukaemia (AML) and enhances the immunogenicity of a dendritic cell/AML vaccine. Br. J. Haematol. 185, 679–690 (2019).
Hicks, K. C. et al. Epigenetic priming of both tumor and NK cells augments antibody-dependent cellular cytotoxicity elicited by the anti-PD-L1 antibody avelumab against multiple carcinoma cell types. OncoImmunology 7, e1466018 (2018).
Leung, E. Y. L. et al. NK cells augment oncolytic adenovirus cytotoxicity in ovarian cancer. Mol. Ther. Oncolytics 16, 289–301 (2020).
Marotel, M., Hasim, M. S., Hagerman, A. & Ardolino, M. The two-faces of NK cells in oncolytic virotherapy. Cytokine Growth Factor. Rev. 56, 59–68 (2020).
Cichocki, F. et al. GSK3 inhibition drives maturation of NK cells and enhances their antitumor activity. Cancer Res. 77, 5664–5675 (2017).
Parameswaran, R. et al. Repression of GSK3 restores NK cell cytotoxicity in AML patients. Nat. Commun. 7, 11154 (2016).
van Hall, T. et al. Monalizumab: inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 7, 263 (2019).
Yalniz, F. F. et al. A pilot trial of Lirilumab with or without azacitidine for patients with myelodysplastic syndrome. Clin. Lymphoma Myeloma Leuk. 18, 658–663.e2 (2018).
Bachier, C. et al. A phase 1 study of NKX101, an allogeneic CAR natural killer (NK) cell therapy, in subjects with relapsed/refractory (R/R) acute myeloid leukemia (AML) or higher-risk myelodysplastic syndrome (MDS). Blood 136, 42–43 (2020).
Pinz, K. G. et al. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. Oncotarget 8, 112783–112796 (2017).
Romanski, A. et al. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J. Cell Mol. Med. 20, 1287–1294 (2016).
You, F. et al. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am. J. Cancer Res. 9, 64–78 (2019).
Xu, Y. et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J. Hematol. Oncol. 12, 49 (2019).
Martín, E. M. et al. Exploring NKG2D and BCMA-CAR NK-92 for adoptive cellular therapy to multiple myeloma. Clin. Lymphoma, Myeloma Leuk. 19, e24–e25 (2019).
Jiang, H. et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol. Oncol. 8, 297–310 (2014).
Han, J. et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 5, 11483 (2015).
Chu, J. et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 28, 917–927 (2014).
Chen, K. H. et al. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia 31, 2151–2160 (2017).
Chen, K. H. et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget 7, 56219–56232 (2016).
Vallera, D. A. et al. NK-cell-mediated targeting of various solid tumors using a B7-H3 tri-specific killer engager in vitro and in vivo. Cancers https://doi.org/10.3390/cancers12092659 (2020).
Cooper, L. J. et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood 101, 1637–1644 (2003).
Imai, C. et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 18, 676–684 (2004).
Maher, J., Brentjens, R. J., Gunset, G., Rivière, I. & Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat. Biotechnol. 20, 70–75 (2002).
Schubert, M.-L. et al. Third-generation CAR T cells targeting CD19 are associated with an excellent safety profile and might improve persistence of CAR T cells in treated patients. Blood 134, 51–51 (2019).
Bjordahl, R. et al. Abstract 1539: development of off-the-shelf B7H3 chimeric antigen receptor NK cell therapeutic with broad applicability across many solid tumors. Cancer Res. 81, 1539 (2021).
Basar, R. et al. Generation of glucocorticoid-resistant SARS-CoV-2 T cells for adoptive cell therapy. Cell Rep. https://doi.org/10.1016/j.celrep.2021.109432 (2021).
Shalem, O. et al. Genome-scale CRISPR–Cas9 knockout screening in human cells. Science 343, 84–87 (2014).
Wang, T., Wei, J. J., Sabatini, D. M. & Lander, E. S. Genetic screens in human cells using the CRISPR–Cas9 system. Science 343, 80–84 (2014).
Manguso, R. T. et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418 (2017).
Patel, S. J. et al. Identification of essential genes for cancer immunotherapy. Nature 548, 537–542 (2017).
Pan, D. et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775 (2018).
Shang, W. et al. Genome-wide CRISPR screen identifies FAM49B as a key regulator of actin dynamics and T cell activation. Proc. Natl Acad. Sci. 115, E4051 (2018).
Ishizuka, J. J. et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48 (2019).
Lawson, K. A. et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 586, 120–126 (2020).
Sheffer, M. et al. Genome-scale screens identify factors regulating tumor cell responses to natural killer cells. Nat. Genet. 53, 1196–1206 (2021). This report uses an orthogonal approach combining genome-wide CRISPR screening with multiplexed cancer cell line screening to decipher cancer type-specific vulnerabilities towards NK cells.
Ting, P. Y. et al. Guide Swap enables genome-scale pooled CRISPR–Cas9 screening in human primary cells. Nat. Methods 15, 941–946 (2018).
Shifrut, E. et al. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell 175, 1958–1971.e15 (2018).
Wang, D. et al. CRISPR screening of CAR T cells and cancer stem cells reveals critical dependencies for cell-based therapies. Cancer Discov. 11, 1192–1211 (2021).
Roth, T. L. et al. Pooled knockin targeting for genome engineering of cellular immunotherapies. Cell 181, 728–744.e21 (2020).
Legut, M. et al. A genome-scale screen for synthetic drivers of T cell proliferation. Nature 603, 728–735 (2022).
Acknowledgements
The authors thank J. S. Moyes for providing editing support during the preparation of this manuscript. A.B. received support from the German Research Foundation as Walter Benjamin Postdoctoral Fellow (464778766). This work was supported, in part, by generous philanthropic contributions to The University of Texas MD Anderson Cancer Center Moon Shots Program, The Sally Cooper Murray endowment, generous support of Ann and Clarence Cazalot, and Lyda Hill Philanthropies; and by grants from CPRIT (RP160693), a Stand Up To Cancer Dream Team Research Grant (Grant number: SU2C-AACR-DT-29-19), grants from the National Institutes of Health (NIH) (1 R01 CA211044-01, 5 P01CA148600-03, and P50CA100632-16), the Specialized Program of Research Excellence (SPORE) in Brain Cancer Grant (P50CA127001) and a grant to MD Anderson Cancer Center from the NIH (CA016672). The SU2C research grant is administered by the American Association for Cancer Research, the scientific partner of SU2C.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
K.R. and The University of Texas MD Anderson Cancer Center have an institutional financial conflict of interest with Takeda Pharmaceutical and Affimed GmbH. K.R. participates on the Scientific Advisory Board for GemoAb, AvengeBio, Virogin Biotech, GSK, Bayer, Navan Technologies and Caribou Biosciences. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Cancer thanks E. Vivier, who co-reviewed with P. Andre, and A. Cerwenka and S. Gill for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Autologous chimeric antigen receptor (CAR) T cell therapy
-
A patient-specific cellular therapy in which the patient’s own T cells are genetically modified to express a chimeric antigen receptor.
- Lymphopenic
-
A lower than normal number of lymphocytes in the blood.
- Allogeneic setting
-
The therapy setting in which adoptive cell therapies are generated from material obtained from a different individual of the same species.
- Graft-versus-host disease
-
(GvHD). A potentially fatal condition that results from the response of allogeneic T cells against the host tissues of recipients who are immunosuppressed, which can occur after adoptive cell therapy or allogeneic haematopoietic stem cell transplantation.
- Killer cell immunoglobulin-like receptors
-
(KIRs). A family of polymorphic activating and inhibitory transmembrane proteins that regulate natural killer cell development and function through interactions with major histocompatibility complex class I molecules.
- Induced pluripotent stem cells
-
(iPSCs). Cells that result from the reprogramming of adult somatic cells into an embryonic-like pluripotent stem cell state, capable of self-renewal and differentiation into tissues originated from the three germ layers (endoderm, mesoderm and ectoderm).
- Apheresis
-
The process that allows for collection of specific components from blood, such as white blood cells, by separating the cellular and soluble fractions, retaining what is of interest and returning the remainder to circulation.
- Chimeric antigen receptors
-
(CARs). Genetically engineered cell surface receptors designed to recognize specific proteins on tumour cells.
- Tonic signalling
-
The constitutive signalling mediated by a chimeric antigen receptor in a ligand-independent manner.
- Logic gated
-
A term derived from electronics that refers to implementation of a Boolean strategy to execute a logical function on one or more input signals to generate a single output signal.
- Licensing
-
A process of education in maturing natural killer (NK) cells that is driven by the interaction of inhibitory receptors and self-major histocompatibility complex class I molecules, which potentiates NK cell responses to activating signals.
Rights and permissions
About this article

Cite this article
Laskowski, T.J., Biederstädt, A. & Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat Rev Cancer 22, 557–575 (2022). https://doi.org/10.1038/s41568-022-00491-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41568-022-00491-0
This article is cited by
-
Advances in metabolic reprogramming of NK cells in the tumor microenvironment on the impact of NK therapy
Journal of Translational Medicine (2024)
-
Natural killer cell therapies
Nature (2024)
-
Multi-omic analyses of m5C readers reveal their characteristics and immunotherapeutic proficiency
Scientific Reports (2024)
-
Pan-cancer analysis reveals PDK family as potential indicators related to prognosis and immune infiltration
Scientific Reports (2024)
-
Advances in Engineered Macrophages: A New Frontier in Cancer Immunotherapy
Cell Death & Disease (2024)
