
Abstract
Over the past decade, CRISPR has become as much a verb as it is an acronym, transforming biomedical research and providing entirely new approaches for dissecting all facets of cell biology. In cancer research, CRISPR and related tools have offered a window into previously intractable problems in our understanding of cancer genetics, the noncoding genome and tumour heterogeneity, and provided new insights into therapeutic vulnerabilities. Here, we review the progress made in the development of CRISPR systems as a tool to study cancer, and the emerging adaptation of these technologies to improve diagnosis and treatment.
Similar content being viewed by others
Introduction
Cancer is a complex and multifaceted disease. Fundamentally, it is a disease of the genome, initiated by mutations in DNA that activate oncogenes and inactivate tumour suppressors, as well as dysregulation of the epigenome, which coordinates normal gene expression. It is also a disease of the cell, feeding off changes in metabolism, cell structure and motility to enable growth in inhospitable environments. Ultimately, it is a disease of the organism, co-opting normal cell types and tissue functions, and circumventing defence systems of the host. Understanding how genomic changes, cellular adaptations and changes to the microenvironment drive the initiation, progression and therapeutic response of individual cancers is crucial for developing more effective treatment options and improving outcomes for the millions diagnosed with cancer each year1. Since its adaptation for mammalian cells, CRISPR has emerged as a powerful and flexible tool for interrogating nearly all aspects of cell function. It has had a major impact on our understanding of cancer biology and continues to drive new discoveries that promise to accelerate the diagnosis and treatment of this deadly disease.
CRISPR and CRISPR-associated (Cas) proteins are key components of an ancient bacterial adaptive immune system2,3,4,5. Over the past three decades, hundreds of scientists have contributed to the understanding of CRISPR biology and development of CRISPR technologies, including landmark papers demonstrating programmable DNA editing in mammalian cells6,7,8,9. Since then, it has been realized as a tool for programmable genome modification in nearly all cell types.
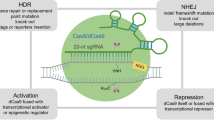
CRISPR systems exist across a wide range of bacterial species, providing a rich source of functional diversity for genome editing in eukaryotic cells10,11,12. The first described, and most commonly used, is the type-II CRISPR–Cas9 system from Streptococcus pyogenes (SpCas9), a DNA endonuclease that is directed to induce double strand breaks (DSBs) at specific genomic loci via a programmable guide RNA (gRNA) molecule that mediates complementary DNA–RNA base pairing. For SpCas9 to efficiently bind and cleave DNA, the target sequence must be flanked on the 3′ side by an ‘NGG’ protospacer adjacent motif (PAM) sequence. The DSB created by Cas9 can be resolved by either precise homology-directed repair (HDR) or, more commonly, by error-prone non-homologous end joining (NHEJ) or microhomology-mediated end joining (MMEJ; also known as alternative NHEJ (Alt-NHEJ))13. HDR enables the introduction of specific changes, while insertions and deletions (indels) from NHEJ can be exploited to disrupt coding and noncoding sequences13.
Since the initial implementation of CRISPR systems in eukaryotic cells there has been a rapid expansion of variant enzymes that broaden the capabilities of CRISPR-based platforms. One source of variants is the diverse set of Cas9 orthologues such as Staphylococcus aureus Cas9 (SaCas9), or other Cas enzymes (for example, Cas12) present in a range of bacterial species11,12. Each has its own set of features and criteria for sequence recognition that provides added flexibility for adaptation as a research or therapeutic tool. For example, SaCas9 recognizes a different PAM sequence from SpCas9, enabling the targeting of alternative genomic loci. Some enzymes (for example, SaCas9, Neisseria meningitidis Cas9 (NmeCas9)14 or Campylobacter jejuni Cas9 (CjCas9))15 are also smaller than SpCas9, allowing easier packaging into size-limited delivery vectors such as adeno-associated virus (AAV)16, while others, such as Cas12a (formerly referred to as Cpf1), can catalyse the maturation of their own gRNAs, simplifying the process of target multiplexing17. Yet another family of Cas enzymes named Cas13 (previously known as C2c2) target RNA instead of DNA, providing an alternative approach to manipulate gene expression18,19.
Where natural variants do not exist, there is no shortage of lab-evolved mutants. Kleinstiver and others20,21,22 have demonstrated the potential for creating Cas9 enzymes with alternative PAM recognition or increased specificity, and subsequent efforts using directed evolution or rational design have resulted in the creation of Cas variants, with broad PAM recognition23,24,25,26 or refined editing activity20,23,25,27,28,29,30,31,32,33.
CRISPR base editing (BE) facilitates the creation of single base edits in target DNA by tethering a nuclease-defective Cas9D10A nickase variant (Cas9n) to a deaminase (for example, apolipoprotein B mRNA-editing enzyme catalytic subunit (APOBEC) or tRNA adenine deaminase (TadA)) to achieve C∙G to T∙A and A∙T to G∙C transition base substitutions without double-stranded DNA cleavage34,35. Alternatively, the fusion of human RNA-modifying adenine deaminase domain of adenosine deaminase (ADARDD) to catalytically inactive (‘dead’) dCas13b can induce precise adenosine deamination in RNA instead of DNA36. Providing further flexibility to engineer changes beyond transition mutations, Liu and colleagues37,38 developed prime editing (PE), a fusion of Cas9H840A to a reverse transcriptase enzyme that promotes genome modification via a sequence template encoded within an extended PE gRNA (pegRNA). PE37 overcomes the restrictions on the types of mutation that can be engineered, but current iterations require substantial optimization to achieve the same levels of editing as is possible with BE37,38. Engineered pegRNAs (epegRNAs) designed to stabilize their structure have improved the efficiency of PE39, and the evolution of new and improved BE and PE enzymes is a very active area in CRISPR technology development. We expect to see more active variants and adaptations appear in the coming years. Until then, the use of BE and PE reporters40,41,42,43 is an effective approach to enrich target editing and streamline the creation of engineered cells and animals for cancer gene discovery.
CRISPR has become a useful tool not only in gene-editing applications, but also for targeting transcriptional and epigenome machinery using dead Cas9D10A/H840A (dCas9), which cannot cleave DNA10,44. Combining the specific DNA recognition of dCas9 with the Krüppel-associated box (KRAB) repressor inhibits the transcription of target genes — so-called CRISPR interference (CRISPRi)44,45. In a similar approach, dCas9 tethered to transcriptional activators such as VP64 and VP64–p65–Rta (VPR) proteins achieves robust gene induction at the target site (CRISPR activation (CRISPRa))46,47,48,49. Finally, multiple groups have described direct epigenetic regulation of DNA and histones by fusing dCas9 to methyltransferases (for example, DNA methyltransferase 3A (DNMT3A) or PR domain-containing protein 9 (PRDM9)), demethylation enzymes (for example, tet methylcytosine dioxygenase 1 (TET1) or lysine-specific histone demethylase 1 (LSD1; also known as KDM1A)) or histone acetyltransferases (for example, p300)50,51,52,53,54,55,56,57,58,59,60. CRISPR can also be used to map subnuclear proteomes onto 3D genome landscapes by tethering engineered ascorbate peroxidase (APEX2) to dCas9 guided to specific regions in the cell with a single guide RNA (sgRNA)61. Common to all CRISPR technologies is the need for potent and specific gRNAs. Many design algorithms have been developed for a wide variety of CRISPR-based platforms, including standard CRISPR nucleases, CRISPRi, BE and PE (Table 1).
The ongoing evolution of CRISPR tools has created a diverse array of opportunities for dissecting cell function (Fig. 1). In this Review, we highlight how this once-in-a-generation technology has transformed our understanding, diagnosis and treatment of cancer. We discuss how CRISPR-based approaches have been a catalyst to reveal new insights into aspects of cancer biology that have otherwise been difficult to probe, such as defining coding and noncoding cancer drivers, understanding the dynamics of tumour heterogeneity and evolution, and improving the diagnosis and treatment of cancer.

Since the initial implementation of CRISPR-associated 9 (Cas9) editing in mammalian cells we have witnessed a fast-moving CRISPR technology boom with variants that can be applied to various specific problems6,7,8,9. Variants developed by rational design and directed evolution have led to increased protospacer adjacent motif (PAM) flexibility and targeting fidelity20,21,22. Naturally occurring variants derived from alternative bacterial species such as Cas12a (also known as Cpf1) and Cas13 have enabled efficient combinatorial knockout (KO) and targeting of RNA, respectively17,18,19. Transcriptional (Trans.) effectors tethered to catalytically dead Cas9 (dCas9) allow for targeting of the transcriptome and epigenome45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60. CRISPR base editing allows the introduction of specific transition mutations with a Cas9 nickase (Cas9n) tethered to an adenine or cytosine deaminase and in the case of cytosine base editing enzymes (BEs), a uracil glycosylase inhibitor (UGI) to limit base excision repair and promote C>T transition mutations34,35. The development of prime editing, whereby a dCas9 is tethered to a reverse transcriptase, enables engineering of many types of mutation such as missense, insertions and deletions guided by a sequence template and extended prime editing guide RNA (pegRNA)37. Engineered ascorbate peroxidase (APEX2) tethered to dCas9 allows for targeted biotinylation at specific genomic loci for unbiased proteome mapping61. gRNA, guide RNA; Me, methylation; nt, nucleotide; sgRNA, single guide RNA.
Separating drivers from passengers
Tumour sequencing over the past two decades has produced an extensive catalogue of genetic alterations from nearly every cancer type. The success of burgeoning precision medicine strategies depends on being able to identify driver mutations that promote cancer growth and to separate them from passenger mutations that do not contribute to tumour progression. Before CRISPR, this relied on comparison of large panels of cancer cell lines that harboured different genetic mutations, small interfering RNA (siRNA) and short hairpin RNA (shRNA) gene silencing, and/or overexpression of mutant cDNAs. CRISPR has complemented and extended these approaches, enabling fast and efficient generation of ‘clean’ genetic knockout (KO), modulation of endogenous gene expression and direct engineering of cancer-associated genomic changes.
Revealing gene function with CRISPR knockouts
A central approach for understanding gene function in tumorigenesis is the generation of cancer models from the bottom up, recreating cancer-linked events to understand their contribution to each stage of the process. In addition to streamlining the process of creating simple gene disruptions in established cancer cell lines, CRISPR enables rapid creation of complex organoid cultures and animal models. Owing to the simplicity and efficiency of CRISPR–Cas technology, the production of KO mice has become routine practice for institutional core facilities and commercial entities (Fig. 2a). Moreover, by eliminating the need for complex vector design and laborious screening of targeted embryonic stem cells (ESCs), it has become feasible to engineer multiple in vivo models in parallel or to derive combinations of genetic alterations in the same mice, in a single step62,63,64. Such efforts are enabled by improved zygote targeting strategies such as CRISPR ribonucleoprotein (RNP) electroporation of zygotes (CRISPR-EZ), CRISPR RNP electroporation and AAV donor infection (CRISPR-READI) and improved genome editing via oviductal delivery of nucleic acids (i-GONAD) that significantly increase throughput and editing efficiency over microinjection methods65,66,67. CRISPR tools have also accelerated the generation of tool strains essential for building mouse cancer models. One recent example described the generation of 70 new tissue-restricted DreER recombinase mice, most facilitated by CRISPR-enhanced HDR targeting68.

a | Transfection of mouse embryonic stem cells with CRISPR-associated 9 (Cas9) and a single guide RNA (sgRNA) (and +/− donor template to promote homology-directed repair (HDR)) enable efficient knockout or knock-in and development of transgenic mouse models62,63,64,65,66,67,68,77,78. b | The development of Cas9 and the inducible Cas9 mouse have made somatic editing in vivo efficient, with various organs as possible targets using either adeno-associated virus (AAV), lentivirus or nanoparticle sgRNA delivery69,70,71,72,74,79,80. c | Genome engineering of cell lines and organoids can lead to a single knockout mutant, base edited mutant and chromosomal rearrangements73,82,83,84,85,86,107,113,115,123,127,128,133,134,135,136,137,138,139. d | Combinatorial knockout of cells and organoids can be achieved with Cas9 and multiple sgRNAs in a pooled or sequential format or with Cas12a and multiple sgRNAs69,83,84,182. e | CRISPR screens have become an impactful tool in many areas of cancer biology. Cas9 and a library of pooled sgRNAs can be infected into cells or organoids, and, after selective pressure is applied by proliferation over time or drug treatments, sgRNA enrichment and depletion are measured to determine targets of interest87,88,89,90,93,94. In addition, infection of a cytosine base editor or adenine base editor and a pooled sgRNA library and addition of drug has led to the identification of missense mutations that confer resistance or sensitivity to poly(ADP-ribose) polymerase (PARP) inhibitors, BH3 mimetics or response to DNA damaging agents124,125,129. BE, base editing; Cas9n, Cas9 nickase; LSL, lox-stop-lox; TRE, tetracycline-responsive.
An alternative strategy to create in vivo KO cancer models is the introduction of all CRISPR components to somatic tissues, either through ex vivo manipulation and transplant of cultured cells or by direct in vivo targeting. For instance, editing of haematopoietic stem cells with a combination of sgRNAs targeting Tet2, Runx1, Dnmt3a, Nf1 (encoding neurofibromin) and structural maintenance of chromosomes 3 (Smc3) induces the development of acute myeloid leukaemia (AML) in engrafted recipients69, while direct in vivo delivery of CRISPR components in the liver, pancreas or lung is a rapid way to derive diverse cancer models with complex cancer genotypes70,71,72,73,74,75,76 (Fig. 2b). Inducible Cas9 mice provide a setting to engineer somatic mutations in a tissue-restricted manner (Fig. 2b). Both Cre- and doxycycline-dependent approaches have been reported77,78, and these, notably, allow the induction of multiple mutations with minimal animal breeding, highlighting a path towards development of more complex autochthonous cancer models with CRISPR. The Winslow lab79,80 has exploited such systems to great effect in lung cancer models, using Cre-controlled Cas9 transgenic mice and lentivirus-delivered panels of cancer gene-focused sgRNAs to classify the impact of specific gene loss on the development of disease in different genetic backgrounds.
In addition to animal models, CRISPR, combined with recent technological advances in 3D culture systems81, has fostered the genesis of tailored and genetically defined human cancer models to interrogate gene function and test new therapies (Fig. 2c). Lo et al.82 recently showcased this experimental paradigm, building early-stage human gastric cancer organoid models with and without AT-rich interactive domain 1A (ARID1A) mutations. They used these engineered organotypic models to clearly define a context-dependent role for ARID1A in early transformation and identify genotype-dependent therapeutic vulnerabilities82. The groups of Sato and Clevers83,84 established even more complex models in human colon organoids, recreating the classic ‘Vogelgram’ sequence with up to five different oncogenic mutations in adenomatous polyposis coli (APC), KRAS, TP53, SMAD4 and PI3K catalytic subunit-α (PIK3CA) (Fig. 2d). Similar studies in breast and lung cancer models have demonstrated that KO mutations in known or suspected drivers can lead to tumours in vivo that can subsequently be used to study drug response85,86.
Screening for drivers
Perhaps where CRISPR has had the biggest impact in cancer research is in pooled genetic screens87,88,89. The ease of design, cloning, efficiency and ongoing development of improved sgRNA libraries89,90 has made CRISPR KO screens the ‘go-to’ method for interrogating gene function in cancer. In cell lines, organoids and animals, positive selection CRISPR screens continue to refine our understanding of how genes and pathways contribute to tumorigenesis (reviewed in91,92; Fig. 2e). There are hundreds of examples of effective screening studies in cell lines, although CRISPR has also enabled pooled genetic screens in more complex settings. Michels et al.75 screened a focused array of tumour suppressors in human colon organoids treated with the transforming growth factor-β receptor (TGFβR) inhibitor A83-01 to pinpoint genes that restrict tumorigenic outgrowth. It is also possible to perform large-scale screens in vivo. Through ex vivo transduction of a genome-wide library and subsequent engraftment in recipient mice, we identified potential regulators of non-small-cell lung cancer (NSCLC) metastasis93 (Fig. 2e). Direct delivery of viral or plasmid-based vectors to organs in situ is challenging, but it is possible to maintain representation of complex libraries in vivo. In one example, Chow et al.94 delivered a genome-wide AAV sgRNA library to the brain of inducible Cas9-expressing mice to reveal a subset of cancer drivers in resultant glioblastomas.
To date, most screens for cancer drivers have been proliferation based, but there are other strategies to identify key cancer regulators. Using cell surface proteins as markers for flow cytometry or magnetic-activated cell sorting (MACS)-based screens offers a direct avenue to isolate cells with specific changes in effector proteins that are not involved in proliferation or cell death. For instance, using functional markers such as programmed cell death protein 1 (PD1), PD1 ligand 1 (PDL1) or major histocompatibility complex (MHC) for cell enrichment in a screening context enables the identification of gene programmes that control antigen presentation or immune activation95,96,97,98. More recent and complex technologies enable ‘marker-free’ pooled CRISPR library screens by directly measuring the transcriptome of single cells following CRISPR-mediated gene disruption99,100,101. Methods such as expanded CRISPR-compatible cellular indexing of transcriptomes and epitopes by sequencing (ECCITE-seq) and Perturb-seq (also known as CROP-seq)99,100,101 integrate pooled gRNA libraries with single-cell RNA sequencing (scRNA-seq) read-outs to provide high-resolution transcriptional information on cellular response to CRISPR perturbations99,101,102,103. Additionally, CRISPR gRNAs paired with protein barcodes (Pro-Code) can be used to interpret the effect of a single genetic disruption on the function of multiple proteins. Pro-Code, paired with a CRISPR gRNA library, has been used to screen breast cancer cells for sensitivity or resistance to antigen-mediated T cell killing104. This kind of fine resolution mapping of cancer phenotypes may reveal subtle changes in cell function that do not alter cell proliferation alone but could suggest alternative treatment strategies. Integrating such high-dimensional data to isolate important changes is a significant and evolving challenge, which includes limitations to scaling up, lentiviral template switching and computational hurdles105.
Making sense of missense mutations
The vast majority of mutations in cancers are single nucleotide variants (SNVs) that may cause hypo, hyper or neomorphic changes in protein function106. CRISPR has had an impact on our ability to engineer and study SNVs in two major ways. First, through the ability to target DNA DSBs, it enhances HDR-based gene targeting, and second, through Cas fusion enzymes, it enables direct DNA modification.
CRISPR-assisted HDR has been used effectively to interrogate the impact of distinct codon 12 and 13 mutations in the Kras oncogene. We and others have used HDR-driven editing to generate an allelic series of Cre-dependent (Lox-stop-Lox (LSL)) Kras mutant mice, which revealed unexpected but profound differences in tumour initiation and progression in the pancreas and colon107,108,109. Moreover, isogenic organoid models derived from these animals highlighted genotype-dependent responses to targeted therapies, underscoring the need to engineer and study individual SNVs in cancer models107. Similar mutation-specific drug responses have been observed in patient-derived KRAS mutant colorectal cancer organoid models110. Winters et al.111 used a sophisticated AAV-based, somatic HDR approach to engineer a wide range of KRAS mutations in a pooled fashion in the lungs and pancreas of otherwise KrasWT mice. Through quantitative analysis of individual barcoded tumours, they provided the first in vivo evidence that particular KRAS variants exhibit distinct oncogenicity, highlighting the importance of modelling the exact mutations seen in human cancers111. The same conceptual approach can be applied broadly in mouse or cell-based models as has been done to reveal the impact of SNVs in the most commonly mutated tumour suppressor gene, TP53. By engineering isogenic cell lines with a CRISPR–HDR approach, Boettcher et al.112 showed that p53 missense mutations (R175H, Y220C, M237I, R248Q, R273H and R282W) can exert a dominant-negative effect on wild-type p53 function. Fraser and colleagues113 described an HDR-driven screening strategy to characterize more than 16,000 defined mutants in yeast. This study took advantage of a strong bias toward HDR-driven DNA repair in yeast that is not seen in mammalian cells, making a similar approach in standard cancer models not yet possible.
CRISPR BE offers an exciting alternative to engineer SNVs without the need for exogenous DNA templates. BE enzymes catalyse C∙G to T∙A (cytosine base editor (CBE)) or A∙T to G∙C (adenine base editor (ABE)) transition mutations with high purity and efficiency and with low off-target effects35,114,115,116,117, while some recently developed enzyme variants also enable C∙G to G∙C transversion events118,119,120,121 (Fig. 2c). BE is becoming an increasingly popular tool for genome engineering, simultaneously enabling the creation of both missense and nonsense mutations122. BE is effective in a wide range of cell types and organisms123, and recent reports describing the first BE screens have demonstrated the unique utility of BE for identifying functional cancer variants that influence drug treatment response124,125. Our lab126 used optimized CBE enzymes to dissect the impact of specific APC disruptions on response to targeted WNT inhibitors, showing that early nonsense mutations drive resistance to tankyrase (TNKS) inhibitors, while late truncating events promote sensitivity127. Correction of mutations in the TERT promoter (seen in 89% of glioblastomas) using ABE is sufficient to inhibit brain tumour growth, defining these mutations as cancer drivers128. The latter example is a clear case in which CRISPR tools have enabled interrogation of direct genotype–phenotype relationships in noncoding genomic elements, which has traditionally been very difficult. We recently described cancer mutation-focused BE ‘sensor’ libraries that enable the simultaneous induction of missense mutations and measurement of BE efficiency in a pooled format129. BE is a powerful tool for engineering cancer-associated mutations but with the current enzymes, it cannot capture all alterations. In theory, PE could enable the creation of almost all known SNVs and small indel variants. Erwood et al.130 recently reported an approach that exploits pooled, saturating PE mutagenesis to interrogate disease variants in two genes. BE and PE libraries such as these provide a means to rapidly assess the impact of cancer-associated point mutations across various experimental settings.
Rearranging the genome
Chromosomal rearrangements are a clinically important subtype of cancer-driving mutations, and their identification has increased exponentially over the past decade131. Yet, the precise functional consequence of many gene rearrangements and fusions remains a mystery as they are often rare and notoriously difficult to recreate in model systems132. Through its ability to catalyse targeted DNA breaks, CRISPR is a powerful tool to engineer large-scale chromosome aberrations. Introduction of paired sgRNAs that target fusion breakpoints with Cas9, can result in multi-megabase deletions133, inversions134,135, duplications136 and translocations137,138 frequently found in patients with cancer. Initial studies showed the feasibility of modelling multiple chromosomal rearrangements in the lung, prostate and colon using cells, organoids and mice, building preclinical models to assess potential drug sensitivities. Still, challenges remain. For instance, two DSBs on the same chromosome can induce inversions, deletions and/or tandem duplications, and it is difficult to bias towards specific outcomes. Furthermore, for studies using mice, synteny is not always maintained, meaning some rearrangements cannot be created. Finally, using CRISPR to engineer targeted gene amplification is theoretically possible, but has yet to be reported in the literature. As an alternative to engineering locus amplification, increased expression can be mimicked using CRISPRa. Dammert et al.139 used CRISPRa to drive expression of MYC paralogues, revealing gene-specific effects on apoptotic priming in small-cell lung cancer. Although driving elevated expression of already well-expressed genes can be a challenge with CRISPRa, it is a powerful approach to drive transcription of endogenous elements, particularly those that contain complex regulatory structures that cannot be mimicked by cDNA approaches.
The noncoding genome
Over the past decade, several mutations in noncoding regions of the human genome have been implicated in cancer risk140. This is unsurprising, as these noncoding regions contain diverse functional elements that regulate the expression of oncogenes, tumour suppressors and related genes140. Pan-cancer genetic association studies have pinpointed prevalent single nucleotide polymorphisms (SNPs) in noncoding regions141,142; however, it remains unclear what role they play in tumorigenesis (reviewed in141). Another factor that influences cancer risk is dysregulation of noncoding RNAs (ncRNAs), which have crucial roles in regulating cellular pathways143,144,145. Several ncRNA-targeting cancer drugs are currently in clinical trials, such as MRX34, a microRNA-34a (miR-34a) mimic, and cobomarsen, a miRNA-155 inhibitor146,147. However, most ncRNAs remain understudied and elucidating their functions could yield novel therapeutic targets144. Recent additions to the CRISPR genome and transcriptome engineering toolbox have enabled a deeper understanding of how cancer phenotypes arise from perturbations to these regulatory elements and ncRNAs.
Targeting noncoding regions using Cas9 knockout
Several groups have used pooled saturation mutagenesis CRISPR nuclease screens to identify essential cis-regulatory elements surrounding a gene or genes148,149 (Fig. 3a,b). Using this approach, we found that cis-regulatory elements of previously established resistance genes of the BRAF inhibitor vemurafenib tend to be 5′ and in close proximity to the gene, in regions of open chromatin148. This study demonstrated how CRISPR mutagenesis can be used to build a high-resolution map of cis-regulatory elements that control a key cancer phenotype (namely, drug resistance).

a | There are multiple CRISPR effectors that can be used to disrupt coding and noncoding regions of DNA, and, more recently, RNA. The CRISPR-associated 9 (Cas9) nuclease cleaves DNA at a target site specified by a guide RNA (gRNA)5,6,7,8,9. Noncoding regions can be repressed (CRISPR interference (CRISPRi)) by targeting promoters and enhancer regions with a catalytically dead Cas9 (dCas9) fused to repressor domains such as methyl-CpG-binding protein 2 (MeCP2) and Krüppel-associated box (KRAB)44,45,46,155. There are several methods to increase gene expression by targeting dCas9 fusion proteins to regions flanking transcription start sites (TSSs). One method is the fusion of dCas9 to the transcriptional activators VP64, p65 and Rta (VPR)152. Another method is the fusion of dCas9 to VP64 along with a modified single gRNA (sgRNA) that recruits the activator fusion complex MS2–p65–HSF1 (collectively known as synergistic activation modulator (SAM))153. The Cas13 nuclease cleaves RNA at a site specified by a gRNA18,19. The fusion of dCas9 to methyltransferases, such as DNA methyltransferase 3A (DNMT3A), or proteins involved with DNA demethylation, such as tet methylcytosine dioxygenase 1 (TET1), enables targeted DNA methylation or demethylation, respectively52,55,56. Furthermore, the fusion of dCas9 to acetyltransferases such as p300 or histone deacetylase proteins such as histone deacetylase 3 (HDAC3) enables targeted histone acetylation or deacetylation, respectively58. b | The design of gRNAs depends on both the CRISPR effector and the intended targets of the CRISPR screen. For screens to target protein-coding genes, gRNAs are designed to target either the exons (CRISPR nuclease) or near the TSS of the gene (CRISPRi or CRISPR activation (CRISPRa)) (gRNA target sites represented by downward arrows). For saturation mutagenesis using nucleases, gRNAs are designed to target many noncoding regions surrounding a gene of interest. For noncoding genome-wide screens using CRISPR nucleases, CRISPRi or CRISPRa, gRNAs are designed to target a specific genomic feature (for example, cis-regulatory elements). For silencing or amplification of noncoding RNAs using CRISPRi and CRISPRa, respectively, sgRNAs are targeted to regions flanking the TSS of a noncoding RNA gene. Ac, acetylation; Me, methylation.
In addition to probing enhancers of genes that impart cancer drug resistance, Cas9 can be used to target transcription factor binding sites genome-wide150. Agami and colleagues150 interrogated binding sites of two transcription factors — p53 and oestrogen receptor-α (ERα) — with known roles in cancer. In two independent CRISPR–Cas9 screens, they found multiple enhancers required for p53-induced senescence and ERα-positive breast cancer cell growth. As these enhancer elements are typically active only in specific cancer types, they may make good therapeutic targets given their greater specificity than protein-coding genes, which may also be expressed in non-malignant cells.
Inhibition and activation of noncoding regions
As mentioned above, inhibition or activation of gene promoters and enhancers is possible via dCas9 (refs44,45,151,152,153,154). Although targeting gene regions with dCas9 alone sterically blocks the binding of transcription factors and RNA polymerase44, it is generally more efficient in mammalian cells to use fused transcriptional repressor modules such as KRAB155. Multiple KRAB variants have been developed (Fig. 3a), and a recent study showed that, of these variants, ZIM3 KRAB is the most potent155. Conversely, several gene activation constructs have been developed using similar rational design methods: dCas9–VPR, dCas9–VP64 co-delivered with MS2–p65–HSF1 (collectively known as synergistic activation mediator (SAM)) and dCas9–SunTag–VP64 (refs152,153,154) (Fig. 3a). These new dCas9-based tools are often better suited for cancer biology studies than nuclease-based CRISPR, because, first, CRISPRi overcomes the challenge of CRISPR-mediated apoptosis from accumulation of DSBs that can occur when targeting amplified genes, or the noncoding regions surrounding them156, and second, CRISPRa enables gain-of-function (increased gene expression) studies. Collectively, dCas9-coupled effectors permit more elaborate, bidirectional exploration of pathways that contribute to tumour growth or suppression.
Using CRISPRi and CRISPRa screens, several groups have identified cancer cell type-specific noncoding mutations. Engreitz and colleagues157 targeted regions flanking two genes that encode transcription factors involved in cancer cell proliferation — GATA1 and MYC — identifying nine enhancers that contribute to gene expression and cell proliferation in leukaemia cells. Gersbach and colleagues158 used both CRISPRi and CRISPRa to examine enhancers around β-globin and the oncogene human epidermal growth factor receptor 2 (HER2) in various human cell lines. This strategy enabled a functional distinction between necessary enhancers (CRISPRi) and sufficient enhancers (CRISPRa) for promoting oncogene expression and highlighted cell type-specific enhancer activity. Together, these studies demonstrate that dCas9-based tools can reveal tumour-specific enhancers, which could lead to new therapeutic strategies.
Interrogating chromatin regulation as a cancer driver
Mutations in protein-coding chromatin remodellers are among the most common across all cancer types159,160; however, chromatin regulation may also be perturbed by noncoding mutations that disrupt CCCTC-binding factor (CTCF)-controlled topologically associating domain (TAD) boundaries, driving gene dysregulation linked to cancer161. CRISPR is ideally suited to interrogating such noncoding alterations in mediating tumour growth156,160.
A recent study of ~2,000 genomes across 21 cancer types identified several cancer drivers at insulator sites160. Creating synthetic mutations using CRISPR at the two most commonly mutated CTCF binding sites in cancer cells without these mutations phenocopied the increase in proliferation. In a separate example, disruption of the CTCF binding site near the platelet-derived growth factor receptor-α (PDGFRA) in cells of a gliomasphere also increased proliferation as well as gene expression162. Furthermore, perturbing the PDGFRA insulator using dCas9–DNMT3A and dCas9–KRAB increased methylation and repressive heterochromatin, respectively, at the target site163 (Fig. 3a). Subsequent increased contact between the PDGFRA promoter and a nearby superenhancer element drove increased expression of PDGFRA and downstream platelet-derived growth factor (PDGF) pathway genes.
Although these studies implicate mutations in CTCF binding sites as potential mechanisms underlying gene-regulatory gain of function for nearby oncogenes, a recent study suggests that some effects are likely due to off-target activity164. When CTCF binding sites were perturbed, the authors indeed found a change in cell proliferation and CTCF binding, but expression of genes within the CTCF binding region, as determined by RNA sequencing (RNA-seq) and reverse transcription–quantitative real-time polymerase chain reaction (RT–qPCR), did not change nor did chromatin accessibility, as determined by assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq). Thus, it is important to consider off-target activity for top hits and validate changes in gene expression and chromatin structure near the targeted binding site when targeting genome insulator elements such as CTCF164.
Modulating noncoding RNAs using Cas9 and Cas13
Several classes of noncoding RNA are associated with cancer development and progression144. miRNAs, long noncoding RNAs (lncRNAs), very long intergenic noncoding RNAs (vlincRNAs) and circular RNAs (circRNAs) have all been investigated and perturbed using CRISPR tools. Pooled screens using Cas9-based approaches, CRISPRi and CRISPRa have enabled researchers to probe the role that miRNAs and lncRNAs play in the proliferation and drug resistance of cancer cells. A genome-wide CRISPR nuclease screen of miRNA in leukaemia cells found that miR-150 and miR-155 promote cell growth165. An in vivo genome-wide screen in mice revealed that loss of tumour suppressor miRNAs can act as strong drivers of metastasis93. Pooled nuclease and CRISPRi screens of lncRNAs have identified more than 750 lncRNAs that contribute to cancer cell proliferation, some of which are cancer type specific166,167,168. Lim and colleagues169 used CRISPRi to identify 33 lncRNAs, which, after inhibition, sensitize glioma cells to fractionated radiation therapy. Another team used CRISPRa in melanoma cells to pinpoint 11 lncRNAs that lead to vemurafenib resistance170, while a similar screen of 15,000 lncRNA promoters in leukaemia cells found 10 lncRNAs that contribute to resistance to cytarabine (also known as cytosine arabinoside (ara-C)), a standard treatment for patients with leukaemias and lymphomas171. The latter study identified a novel lncRNA (GAS6-AS2) that hyperactivates the inflammatory growth arrest-specific protein 6 (GAS6)–TYRO3, AXL and MERTK (TAM) resistance pathway.
In addition to blocking or activating transcription with Cas9-based tools (CRISPRi and CRISPRa), Cas13 orthologues can be used to potently knock down RNA species in mammalian cells36,172,173 (Fig. 3a). The RNA-targeting Cas13 enzyme has two key advantages over DNA-targeting (Cas9) methods: first, it directly targets noncoding RNAs without modifying the genome172,173 and second, it does not modulate expression of nearby protein-coding genes, which can be a potential confounder with CRISPRi or CRISPRa screens168,170. A pooled Cas13 screen of 22 vlincRNAs that are upregulated in response to anticancer drugs found that 64% of the vlincRNAs enhanced cell survival174. A Cas13 screen of highly expressed circRNAs in cervical and colon cancer cell lines revealed a family of circRNAs that cause cell type-specific proliferation175. When one oncogenic circRNA identified in this screen — circFAM120A — was knocked down, translation of its parent gene FAM120A, a known oncogene176, was suppressed. Moving forward, Cas13 will be an important tool to complement Cas9-based approaches and uncover noncoding RNA function in tumorigenesis and tumour progression.
Mutational and clonal heterogeneity
Cancer is not a monogenic, monoclonal or static disease. Cancer cells continually acquire alterations that result in complex genetic and epigenetic profiles. Clonal derivatives branch and compete as cancer cell populations evolve into distinct and varied mutational entities, while the intercellular composition of a tumour (cancer cells, stroma and immune cells) is remarkably dynamic. Understanding intratumoural heterogeneity and the emergence and evolution of tumour subclones is important for building a complete picture of tumorigenesis. CRISPR technologies are uniquely suited to tackle these difficult questions, enabling researchers to both engineer complex cancer-associated mutations across cell populations and trace clonal evolution via genetic scars from CRISPR-induced genome repair.
Modelling complex mutational profiles in cancer
The accumulation of diverse combinations of genetic alterations is a hallmark of cancers. As highlighted above, CRISPR has been used to model the stepwise acquisition of cancer-driving changes83,84, but it can also be very effective for exploring the impact of different mutational combinations (Fig. 2). CRISPR editing makes it feasible to create large panels of cells, organoids or animal models each with different mutational patterns. We recently exploited this ability to easily ‘mix and match’ genetic events to reveal a genotype-specific context that leads to acquired drug resistance that is not observed with other combinations of mutations177. Where specific cancer-driving genetic combinations are not obvious, CRISPR approaches can be extensively multiplexed. Rad and colleagues178,179 showcased the potential of this approach, demonstrating that in vivo delivery of small collections of sgRNAs into the pancreas can reveal synergistic genetic interactions that drive tumorigenesis. Given the enormous mutational complexity seen in each human cancer, approaches such as this serve to refine our understanding of how specific mutation patterns influence disease progression and response to therapy. The development of focused combinatorial sgRNA libraries180,181 could provide a way to interrogate the large matrix of possible mutation combinations at a feasible scale182 (see Identifying gene interactions, below).
Clone wars: tracing evolution dynamics in tumours
Multiple CRISPR-based strategies have been devised to delineate distinct clones within a mixed population and enable monitoring of clonal dynamics over time. In addition to inclusion of unique molecular identifiers on sgRNA libraries to tag CRISPR clones183, CRISPR machinery itself can be used to introduce static barcodes through HDR template integration containing unique identifiers184 (Fig. 4a,b), although it is also capable of more dynamic lineage marking (Fig. 4c). By exploiting the heritable ‘semi-random’ indel patterns created by Cas nucleases, CRISPR can be used as a molecular recorder, creating unique and evolving barcodes within the genome185,186,187. As indels accrue over time, the timing, rate and specific combination of indels within a cell population can be deconvoluted to map rate and directionality of clonal evolution of metastatic cells and enable the assembly of lineage or phylogenetic trees188. Initially validated in vivo as a tool to trace cell lineages in the development of zebrafish and mice187,189, the method has been used to reveal metastatic potentials and behaviour of different clones within lung cancer xenografts188. Combining Cas9 molecular recording with regulatable Cas9 expression in a transgenic mouse model known as the CRISPR array repair lineage tracing (CARLIN) mouse enables inducible, Cas9-dependent accumulation of indel barcodes that can be used to lineage trace somatic single-cell events and gene expression profiles in vivo and over time190 (Fig. 4c).

a | Lineage tracing with static barcoding enables the capture of enriched clones over time and after selective pressures (bottlenecks). After initial delivery of a pooled guide RNA (gRNA) library with unique encoded barcodes, and a Cas enzyme, barcodes can be used to delineate clonal outgrowth. b | Retroactive lineage detection is made possible through static barcode lineage tracing as in part a, followed by CRISPR technologies such as CRISPR activation tracing of clones in heterogeneous cell populations (CaTCH)194 and control of lineages by barcode enabled recombinant transcription (COLBERT)195, which employ CRISPR activation (CRISPRa)-mediated activation of GFP in barcode-specific cells. This can be used to identify a clone of interest (for example, a drug-resistant clone) within the originating, naive pooled population retrospectively. c | Genetic scars left by CRISPR machinery can be harnessed as molecular recording events to gather information such as rate and directionality of subclonal events. These molecular barcodes can be deconvoluted into complex phylogenetic trees across tumour evolution and selective pressures185,186,187,188,189. CARLIN, CRISPR array repair lineage tracing; Cas9, CRISPR-associated 9; CROP-seq, CRISPR droplet sequencing; dCas9, dead Cas9; ECCITE-seq, expanded CRISPR-compatible cellular indexing of transcriptomes and epitopes by sequencing; FACS, fluorescence-activated cell sorting; GESTALT, genome editing of synthetic target arrays for lineage tracing; indel, insertion or deletion; MARC1, mouse for actively recording cells; sgRNA, single guide RNA; VPR, VP64–p65–Rta.
Cas9 is not the only tool adaptable for molecular recording; BE offers similar barcoding advantages to Cas9 but avoids DSBs that can result in deletion of previously barcoded events, reducing sequence complexity (Fig. 4c). Hwang et al.191 showed that BE can be used to create barcodes at endogenous repetitive elements instead of using exogenous barcode arrays. The predictable nature of BE outcomes can also be exploited to enable fluorescence in situ hybridization (FISH)-based detection of lineage-traced events192. In addition, Halperin et al.193 devised an alternative strategy of barcode mutagenesis (EvolvR) by tethering DNA polymerase variants to Cas9n, allowing the generation of random mutations within a sequence window that can serve as a lineage barcode (Fig. 4c).
In addition to tracing the forward evolution of cell lineages, retrospectively tracing the origin of some lineages offers key advantages (Fig. 4b), for instance, in defining the cell or clone of origin of drug resistance. Umkehrer et al.194 and Al’Khafaji et al.195 developed an elegant CRISPR-driven system to achieve this. Both approaches use a 20mer DNA barcode that doubles as a sgRNA binding site that can be used to activate GFP expression in the presence of CRISPRa constructs194,195. Identifying cells that contain specific barcodes (that is, those that develop drug resistance) is as simple as introducing dCas9–VP64 with a sgRNA corresponding to the barcode of interest and identifying fluorescent green cells. Using this approach, Umkehrer et al.194 identified rare populations in untreated mouse melanoma YUMM 1.7 cells that have intrinsic resistance to combined RAF and MEK inhibition. Integrating functional barcoding with single-cell transcriptomics provides another layer of detail to understand heterogeneity and cancer cell behaviour under different conditions, as recently showcased by Brock and colleagues196.
Defining vulnerabilities in cancer
Identifying true pan-cancer or cancer type-specific dependencies is a central goal of cancer research. Although conceptually simple, the complex regulatory networks that control cell function in different cell types and cancers makes identifying singular targets a major challenge197,198,199,200. Large-scale pooled genetic screens provide a means to tackle this complexity and allow unbiased classification of essential genes across different cancer types. Both CRISPR and shRNA technologies offer similar targeted loss-of-function outcomes and can complement one another in gene discovery efforts201, although CRISPR is generally favoured owing to its high efficiency and reproducibility202. Moreover, CRISPR systems offer the flexibility of constitutive or inducible induction of either genomic203,204 or transcriptomic (Cas13 (refs175,205), CRISPRi180,206,207,208) perturbations. Furthermore, Cas12 and Cas13 systems are easily adapted to multiplexing173,209, making it possible to investigate complex genetic interactions at scale.
Identifying essential genes
CRISPR–Cas9 screens allow for unbiased interrogation of large gene sets in various model systems and contexts, including in vivo models. In most cases, essential genes are identified in focused or genome-wide proliferation-based negative selection screens, where the level of sgRNA depletion is used as a surrogate for the essentiality of any given gene. The efficiency and success of CRISPR screens have fostered two independent large-scale efforts to probe pan-cancer dependencies across more than 300 cancer cell lines210,211. The Sanger Institute implemented a pipeline for prioritizing candidates for approved drugs, which culminated in identification of WRN helicase as a top priority cancer dependency in mismatch repair-deficient (dMMR) cancers210. WRN disruption was validated in several independent studies as a synthetic lethality in dMMR or microsatellite instability-high (MSI-H) cancers and has become a potential therapeutic target in this context210,212,213, even for those cancers that are resistant to standard treatments214. Extensive work from the Broad Institute integrated copy number-specific effects in their screening analysis after observing that concurrent induction of multiple DSBs in amplified genomic regions can cause cell death unrelated to gene targeting. This approach removes cell line-specific false positives, thereby refining viable candidate targets resulting from cancer dependency screens211. Although conducted in different continents, the two screening efforts show remarkable agreement in identified cancer dependencies and biomarkers215. The combined effort has led to the cancer dependency map, or DepMap, which serves as a powerful community resource for hypothesis generation or to quickly check how essential your gene of interest is across hundreds of different cancer cell lines and/or cancer types. DepMap can also be used to define genetic associations. For instance, three recent studies described a role for the E3 ligase activating molecule in BECN1-regulated autophagy protein 1 (AMBRA1) in controlling the stability of D-type cyclins216,217,218; in two cases, AMBRA1 was identified by biochemical and/or genetic screens217,218. Loss of AMBRA1 increases cyclin D levels, in part mimicking the downstream consequences of RB1 loss, including increased proliferation and tumorigenesis216,217,218. Remarkably, analysis of DepMap data shows that the top co-dependency for RB1 (that is, the gene with an essentiality profile that most closely matches RB1) is AMBRA1. There are undoubtedly many such discoveries yet to be unearthed in this rich data set. Recently, Stegmaier and colleagues219 described cancer dependencies from 82 paediatric cancer cell lines (PedDep), providing an important data set and highlighting that paediatric cancer dependencies are usually distinct from adult cancer essential genes.
Even with notable progress in identifying cancer dependencies, the vast majority of drug targets entering trials do not translate to clinical responses220,221. Efforts to enrich for meaningful clinical targets including those from CRISPR screens are necessary and will involve fine tuning of CRISPR screen design as well as improvements in the model systems in which they are performed. The Bassik group222 identified crucial differences between genome-wide CRISPR screens performed in cancer cell line monolayers and those performed in cancer cell spheroids, highlighting cancer dependencies specific to 3D growth with more relevance to human cancers. Although in vitro screens are a fast, cost-effective and high-throughput way of screening cancer growth vulnerabilities, in vivo screens provide tissue context and cell–cell interactions that cannot be mimicked outside the organism. Bajaj et al.223 used a genome-wide library in a syngeneic transplant leukaemia model to identify the RNA-binding protein STAU2 as a regulator of chromatin dynamics and a dependency in two different types of myeloid cancer. Lebrun and colleagues224 used a similar strategy with xenografted human triple negative breast cancer cells to reveal mTOR complex (mTORC) and Yes-associated protein 1 (YAP1) as potential therapeutic targets. In vivo screens would be particularly effective in settings where tumours cells cannot be easily cultured ex vivo — for instance, in patient-derived xenograft (PDX) models. To enable efficient editing in PDX lines, Hulton et al.225 built antibody-based selection systems to quickly enrich Cas9-expressing cells from serially transplanted tumours, thereby minimizing the time required in cell culture.
In addition to identifying new gene and protein targets, sgRNA tiling across a gene can be used to probe protein domain function and essentiality226,227 as well as to identify novel protein–drug interactions or mechanisms of action228, or scan large noncoding regions for key regulatory loci149. In this vein, CRISPR can also be employed to test the target specificity of existing small-molecule or biologic therapies229,230. For example, Sheltzer and colleagues231 used a CRISPR KO approach to show that in some cases, drugs entering clinical trials that are presumed to inhibit a single protein can engage more than one target, and this off-target activity is important for drug response. Mattsson et al.230 used an elegant screen-based strategy to define the cell-surface targets of dozens of antibodies. Such approaches streamline the validation of novel antibody-based therapeutics.
Identifying gene interactions
In addition to probing single gene perturbations, CRISPR can be used to identify essential genetic interactions232,233. Targeting multiple genes simultaneously in a single cell can reveal synthetic lethal interactions or potential drug combinations, or define cancer dependencies that are buffered by related, redundant family members (for example, MEK1 and MEK2)234. Multiple large-scale combinatorial CRISPR screens have been described using both Cas9 and CRISPRi systems, revealing novel genetic interactions and identifying possible synergistic anticancer therapeutic strategies180,206,207,208 (Fig. 2d). Owing to the increased complexity of combinatorial screens, most pairwise sgRNA libraries used to date have focused on small gene sets such as ‘cancer genes’235 or ‘druggable genes’180. However, Zhou et al.181 went one step further, and developed a system to clone and co-express three gRNAs simultaneously to identify higher-order genomic interactions and combinatorial targets.
Despite improved pipelines to generate multiplexed libraries208,236, cloning complex pools with multiple sgRNA promoters can be challenging owing to recombination of repetitive elements236,237. CRISPR–Cas12a systems overcome these issues, enabling the production of complex multi-guide libraries with a single cassette, owing to the ability of Cas12a to process its own CRISPR RNA (cRNA) arrays17,182. Dede et al.238 used a pooled gRNA library targeting pairs of gene paralogues and a modified Cas12a enzyme (Fig. 2d) to identify novel synthetic lethal gene interactions that were masked in individual KO studies because of functional buffering between gene paralogues. The Doench group239 found an alternative method of combinatorial screening, exploiting unique features of Cas9 orthologues and creating libraries containing paired guides for SpCas9 and SaCas9. This approach eliminates gRNA competition for enzyme loading during multiplexing by creating distinct gRNA-to-Cas9 orthologue pairs expressed within the same cell with unique targets. They found that this approach increased dual KO efficiencies for robust screening applications239.
Identifying anticancer immune targets
Tumour cell-extrinsic factors such as immune regulation play an essential part in cancer cell behaviour240. In particular, interactions that enable immune evasion or immunotherapy resistance are exciting therapeutic opportunities. CRISPR screens using immune cell–tumour cell co-cultures and in vivo transplants in immune-competent hosts have been used to identify tumour-intrinsic factors that govern tumour–immune interactions (reviewed in241). In one example, a genome-wide screen using cancer cells co-cultured with cytotoxic T cells revealed cancer cell-intrinsic regulators of T cell killing242. The study identified receptor-interacting serine/threonine-protein kinase 1 (RIPK1) as a potential druggable target that upon inhibition sensitizes tumours to antitumour T cell cytotoxicity. Consistent with the genetic screen, pharmacological RIPK1 inhibition sensitized tumours to immunotherapy in a preclinical transplant model242. As a result of this study, a RIPK1 inhibitor proceeded into clinical trials, although ultimately did not achieve its predefined end point243. Towards understanding immune evasion, we and others have examined tumour mutations that drive escape from adoptive T cell therapies244 and immune checkpoint blockade245,246. Using a genome-wide CRISPR KO library with recombinant T cell receptor (TCR)-engineered T cells244, we identified well-known mediators of immune resistance (such as human leukocyte antigen (HLA-A) and β2-microglobulin (β2m), which form the major histocompatibility complex class I (MHC-I)) and previously unknown mechanisms of immune escape, such as loss of the gene encoding the apelin receptor (APLNR), which hinders release of key cytokines such as interferon-γ (IFNγ). Co-culturing during CRISPR screening has now been demonstrated in many immune cell contexts including T cells247,248,249, natural killer (NK) cells249,250,251,252,253,254 and macrophages255, revealing a variety of regulators of immune suppression and tumour evasion. In vivo screening of CRISPR-manipulated cancer cell lines has also identified numerous tumour immune modulators (reviewed in256, and257,258). In a focused screen targeting epigenetic regulators, Li et al.258 identified the histone chaperone ASF1A as a tumour-intrinsic regulator of macrophage differentiation; ASF1A disruption sensitizes tumours to immune checkpoint blockade. Studies such as this identify promising targets to enhance activity of or sensitize resistant tumours to known immunotherapies.
Conversely, modulation of the immune component of the tumour microenvironment can have an equally important impact on tumour progression. Defining new therapeutic targets in immune regulators is an active yet challenging area of research. Although primary immune cells are classically difficult to genetically manipulate, CRISPR has facilitated efficient editing in immune cells and enabled screens for tumour cell-extrinsic regulators of tumour progression and immune evasion. Arrayed CRISPR screening in T cells was demonstrated by Gurusamy et al.259 using electroporated RNP complexes with gRNAs that target 25 kinases known to sustain activation after TCR stimulation. They showed that p38 kinase is linked to immune suppression and its inhibition improves anticancer immunity in mouse models259. A major challenge in engineering primary cells is the efficient delivery of the large Cas enzymes. Electroporation or ‘nucleofection’ of RNP complexes into immune cells including myeloid cells260 has proved a highly efficient method of Cas9 delivery for gene KO (reviewed in261). Schumann et al.262 delivered RNPs in a pooled format to human regulatory T (Treg) cells to perturb 40 transcription factors predicted to regulate Treg cell identity. Validation of a select group of transcription factors using arrayed RNP delivery paired with scRNA-seq read-out produced a comprehensive map of signalling networks governed by proteins encoded by essential regulatory genes in Treg cells (for example, forkhead box protein P3 (FOXP3))262. Studies such as this provide a resource for initiating functional studies in immune populations and identifying potential therapeutic targets to promote antitumour immunity. Effective pooled screening in primary T cells is also possible by combining sgRNA lentiviral transduction with Cas9 protein electroporation (SLICE)263 or through the isolation and interrogation of immune populations from Cas9-expressing transgenic mice264,265,266,267. The use of transgenic mice also offers an opportunity to incorporate other established mouse alleles. For instance, OT-1;Cas9 double transgenic mice, which produce MHC-I-restricted, ovalbumin-specific, CD8+ T cells, have also been used to screen for regulators of T cell infiltration and killing activity265.
CRISPR can also be used to identify the regulatory mechanisms that govern the efficacy of cellular immunotherapies. Legut et al.268 showed that CRISPR KO of the endogenous TCR in primary human T cells enhances the activation of a transgenic TCR and increases cytotoxicity against B cell acute lymphoblastic leukaemia (B-ALL) blasts in co-culture. Guo et al.269 provided preclinical evidence of improved chimeric antigen receptor (CAR) T cell tumour killing after CRISPR–Cas9 deletion of PD1 in those CAR T cells; this strategy is now being evaluated in clinical trials (see section Ex vivo CRISPR therapies below). In addition to modifying known factors that promote immune evasion, a CRISPR screen in CAR T cells identified novel dependencies that upon deletion improve efficacy of T cell killing of glioblastoma stem cells270. In the same study, a reciprocal screen in glioblastoma stem cells identified genes that confer susceptibility to T cell killing and found overlapping gene dependencies between the two screens that inform ways to potentiate CAR T cell efficacy270.
CRISPR has catalysed the identification of cancer vulnerabilities at a rapid rate. Cumulatively, CRISPR tools have defined hundreds of potential tumour cell-intrinsic and tumour cell-extrinsic therapeutic targets. However, the path from putative target to clinical translation is notoriously challenging. It remains to be seen how many CRISPR-validated hits will ultimately become clinically actionable targets. Time (and money) will tell.
CRISPR for clinical cancer care
CRISPR technologies have exciting clinical opportunities in a range of monogenic disorders271,272 but have not been a dominant player in the development of cancer therapeutics. That said, there are tangible applications for CRISPR in clinical cancer management, and it is poised to make an impact in both cancer diagnostics and therapies in the coming years.
CRISPR-driven cancer diagnostics
Targeted enzymatic digestion mediated by CRISPR machinery can be harnessed as a diagnostic tool to identify cancer-specific sequence changes. Microsatellites, a diagnostic marker in cancers273, can be sensitively detected using CRISPR-mediated digestion targeted to short tandem repeats (STRs), which make up microsatellites. Sequencing the resulting DNA fragments (STR-seq) showed greater accuracy and sensitivity for microsatellite detection in a high-throughput manner than fragmentation by sonication274. Bennet-Baker et al.54 demonstrated the targeted release of megabase-sized fragments from genomic DNA through Cas9-mediated digestion. When paired with duplex sequencing that incorporates double-stranded DNA barcodes to prevent errors in sequencing, Cas9-mediated fragmentation allows for targeted sequencing of genomic regions even with very little DNA input (termed CRISPR-DS)275. CRISPR-DS is currently being evaluated in a clinical trial for detection of p53 mutations in ovarian tumours276. In addition, Cas12 and Cas13-mediated detection of nucleic acids via specific high-sensitivity enzymatic reporter unlocking (SHERLOCK) and DNA endonuclease-targeted CRISPR trans reporter (DETECTR)277,278 has been used to identify cancer-associated mutations in tumour biopsy samples from patients279,280. These same platforms have been used to develop affordable, point-of-care diagnostics for SARS-CoV-2 infection281,282. Thus, it is foreseeable that CRISPR technologies could serve as a personalized, sensitive detection and monitoring system for patients with cancer.
Ex vivo CRISPR-based therapies
For a biotechnology that emerged only 8 years ago, it is impressive that the first direct clinical applications of CRISPR are already being realized (Table 2). Multiple groups have shown that ex vivo CRISPR-based targeting of PD1 on T cells can enhance antitumour activity following adoptive transfer269,283,284. This therapeutic pipeline is already in clinical trials285,286. In a pilot clinical study, engineered T cells showed low off-target editing and minimal adverse events285,286. In an independent clinical study, patient T cells were similarly engineered using CRISPR–Cas9 gRNA-mediated KO; however, PD1 was targeted alone or in combination with the endogenous TCR (T cell receptor α-chain constant (TRAC) and T cell receptor-β constant (TRBC)) genes287,288 (Fig. 5). Eyquem et al.289 used Cas9 to enrich the targeting of a CD19 CAR to the TRAC locus in T cells, resulting in uniform CAR expression and increased CAR T cell potency compared with those integrated randomly in the genome (Fig. 5). CRISPR editing in human cells is not without concern. Cas9 induces off-target cutting, and chromosomal rearrangements have been identified in edited T cell populations287 (see section Limitations below). Although, in this particular study, cells with chromosomal rearrangements decreased after transplantation, suggesting some selection against them, there is still potential for alternative CRISPR technologies to address this issue. For instance, Gaudelli and colleagues290 provided a proof-of-concept study that BE can be used to target multiple potential checkpoint regulators in T cells without inducing DSBs. To date, there are several ongoing phase I clinical trials employing ex vivo CRISPR engineering of allogeneic or autologous T cells for the treatment of cancer (Table 2). The strategies designed in these trials involve either gene disruption by CRISPR KO to inactivate immunosuppressive factors and/or integration of a CAR element into the TRAC locus for CAR T cell engineering; both strategies serve to promote the antitumour efficacy of tumour-specific cytotoxic T cells as an immunotherapeutic approach to the treatment of cancer (Fig. 5).

Ongoing clinical trials to evaluate the efficiency and safety of CRISPR engineered T cells through ex vivo manipulation and adoptive transfer aim to improve antitumour activity of healthy donor or patient-derived T cells. Both allogeneic and autologous T cells are being tested as strategies to explore the efficacy of CRISPR engineering in tumour-infiltrating lymphocytes (TILs) and chimeric antigen receptor (CAR) T cells. Deletion of immunosuppressive factors such as the programmed cell death protein 1 (PD1) ligand in human primary T cells has been shown using CRISPR–CRISPR-associated 9 (Cas9) and is being tested therapeutically for adoptive T cell therapy employing both TILs and CAR T cells284,285,286,287,288,332,333,334,335,336,341,344. CRISPR–Cas9 ribonucleoproteins (RNPs) deliver guide RNA (gRNA) and Cas9 to targeted immunosuppressive factors (for example, PD1). Deletion of the endogenous T cell receptor (TCR) is also being explored using CRISPR–Cas9 to prevent TCR priming or immune rejection in the case of allogeneic T cells335,338,339,340,341,342. Replacement of the endogenous TCR with a cancer antigen-specific TCR either with a TCR transgene or with a CAR element has also been shown to improve cancer cell-specific T cell killing. CAR elements delivered using CRISPR–Cas9 homology-directed repair (HDR)-mediated knock-in to the T cell receptor α-chain constant (TRAC) locus are currently being tested in clinical trials339,340,341,342,343. In the same cells CRISPR can be used to delete the endogenous T cell receptor-β constant (TRBC) locus, endogenous major histocompatibility complex class I (MHC-I) to prevent immune rejection after transplant, and immunosuppressive factors with the aim of improving T cell activity, all of which are being tested in ongoing clinical trials using CAR T cells338,339,340,342. Engineered T cells are validated by next-generation sequencing (NGS) to ensure on-target editing with minimal off-target effects and subsequently expanded for transplant into the patient with cancer. Disease progression is monitored to assess the safety and efficacy of the engineered T cells (see Table 2 for details).
Preclinical potential of in vivo CRISPR therapies
As described above, although manipulation of primary patient-derived cells for transplant is a challenging clinical goal, targeting tumours directly with CRISPR is a much harder task. It will require multiple hurdles to be overcome, including achieving efficient and possible tumour-selective delivery, as well as target editing efficiency in a setting where there is strong selection against editing. Martinez-Lage et al.291 describe one clever preclinical example that targets oncogenic gene fusions, providing both tumour cell selectivity owing to the unique fusion and disruption of a tumour-promoting genetic lesion. In another preclinical example, Gao et al.292 exploited nuclear factor-κB (NF-κB) — selectively activated in cancer cells — to drive transcription of CRISPR–Cas13a components and induce cancer cell-restricted oncogene silencing. Delivery of nucleic acids via lipid nanoparticles (LNPs) is an exciting concept that has had enormous success in the delivery of mRNA as a SARS-CoV-2 vaccine293,294,295,296. LNPs encapsulating Cas9 mRNA and gRNAs showed efficacy in a proof-of-concept study targeting the essential gene polo-like kinase 1 (PLK1), achieving efficient gene editing at the target locus in a mouse model of glioblastoma297. Together, these preclinical efforts show promise, but much work needs to be done to make CRISPR itself a viable clinical therapy for cancer.
Limitations
Despite the broad utility of CRISPR in cancer biology, there are still several limitations and concerns for its use that it will be important to address moving forward, particularly in therapeutic settings. The induction of DSBs in nuclease approaches can lead to unintended large deletions298 and in some cases, drive chromothripsis, which could lead to the loss of tumour suppressors and impair otherwise normal cell function299. The downstream effect of DNA damage can also lead to induction of the p53 pathway, resulting in cell death or selection for cells with reduced p53 function300. Although this has raised concerns about CRISPR therapeutics possibly enriching for cancer-prone cells, it is important to note that there are no clinical data suggesting that CRISPR drives or supports cancer growth. In addition to potential issues with on-target activity, CRISPR does show sequence-dependent off-target effects20,301,302,303, which can result in unwanted gene disruption or large-scale chromosome aberrations304. In a research setting, such concerns can be minimized with appropriate controls and multiple sgRNAs. The use of high-fidelity Cas enzymes (HF1, HiFi and HypaCas9 (refs20,29,305)) and improved sgRNA design tools (Table 1) can further reduce the prevalence of off-target events, although we should remain mindful that any genome manipulation may have functional consequences that impact our use and/or interpretation of CRISPR-based strategies. CRISPR therapies that require ongoing Cas9 expression must overcome pre-existing immunity, which is widespread in the population306,307,308. Engineered Cas9 variants to avoid common immunogenic epitopes309 or removal of antigenic regions of CRISPR machinery after editing310 may offer a solution.
Conclusions
As Yogi Berra, an American professional baseball catcher, famously said, “It’s tough to make predictions, especially about the future”. In which case, we will start by mentioning the easy one: CRISPR is here to stay. Although still a relatively young technology, CRISPR has affected nearly all aspects of cancer biology, catalysed the generation of vast amounts of functional data and revealed countless new insights into an already well-studied disease. Yet, there is still more to learn. The integration of CRISPR-based tools with single-cell multiomics approaches offers an enormous array of possible applications for exploring gene function and tumour heterogeneity. Incorporating spatial transcriptomics with pooled CRISPR libraries will be a powerful strategy to interrogate the impact of gene disruptions on tumour microenvironment interactions. We expect that following the recent expansion of BE and PE technologies, our ability to engineer and study disease-specific mutational variants will provide new understanding of how mutational signatures and gene variants drive cancer and hone our collective efforts to develop effective precision medicine-based approaches to treat individual tumours. Over the next 5–10 years, CRISPR will take its first real steps into clinical medicine. How prominently cancer-focused treatments will feature in the CRISPR arsenal is an unanswered question, but promising early work in CAR T cell therapies and other engineered immune cells signals that it will have a part to play. Although the potential for genome damage and off-target editing loom as hurdles for CRISPR, tools to diagnose and minimize such events are already in hand and ultimately they will likely not significantly curtail its use in a clinical setting.
The rapid progress and advances in CRISPR technologies have already begun to address many of the fundamental and puzzling questions we have about cancer. By delineating the role of individual genes in cancer cell behaviour, enabling the creation of next-generation immunotherapies, ascribing the functional effect of recurrent coding variants, and revealing the role of elusive noncoding and regulatory elements in tumorigenesis, CRISPR has been, and will continue to be, a key element in our quest to understand and treat human cancers.
References
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Mojica, F. J., Díez-Villaseñor, C., García-Martínez, J. & Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 60, 174–182 (2005).
Barrangou, R. et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007).
Bolotin, A., Quinquis, B., Sorokin, A. & Ehrlich, S. D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2551–2561 (2005).
Pourcel, C., Salvignol, G. & Vergnaud, G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151, 653–663 (2005).
Cho, S. W., Kim, S., Kim, J. M. & Kim, J.-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230–232 (2013).
Jinek, M. et al. RNA-programmed genome editing in human cells. eLife 2, e00471 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 9, 1911 (2018).
Hendriks, D., Clevers, H. & Artegiani, B. CRISPR-Cas tools and their application in genetic engineering of human stem cells and organoids. Cell Stem Cell 27, 705–731 (2020).
Pickar-Oliver, A. & Gersbach, C. A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 20, 490–507 (2019).
Ventura, A. & Dow, L. E. Modeling cancer in the CRISPR era. Annu. Rev. Cancer Biol. 2, 111–131 (2018).
Zhang, Y. et al. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol. Cell 50, 488–503 (2013).
Kim, E. et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 8, 14500 (2017).
Ran, F. A. et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015).
Zetsche, B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015).
Abudayyeh, O. O. et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573 (2016).
East-Seletsky, A. et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273 (2016).
Kleinstiver, B. P. et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016).
Kleinstiver, B. P. et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485 (2015).
Slaymaker, I. M. et al. Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88 (2016).
Hu, J. H. et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 (2018).
Nishimasu, H. et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, 1259–1262 (2018).
Walton, R. T., Christie, K. A., Whittaker, M. N. & Kleinstiver, B. P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368, 290–296 (2020). This paper describes the development of a ‘PAMless’ CRISPR–Cas9 variant, SpRY, that dramatically broadens the number of possible genomic targets for nuclease- or non-nuclease-mediated editing.
Miller, S. M. et al. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat. Biotechnol. 38, 471–481 (2020).
Richter, M. F. et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 38, 883–891 (2020).
Chen, J. S. et al. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410 (2017).
Vakulskas, C. A. et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 24, 1216–1224 (2018).
Gehrke, J. M. et al. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat. Biotechnol. 36, 977–982 (2018).
Grünewald, J. et al. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 37, 1041–1048 (2019).
Yu, Y. et al. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat. Commun. 11, 2052 (2020).
Rees, H. A., Wilson, C., Doman, J. L. & Liu, D. R. Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci. Adv. 5, eaax5717 (2019).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). This study describes the generation of a cytidine deaminase-tethered dCas9 enzyme to allow targeted C>T missense mutations without DSBs.
Gaudelli, N. M. et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). This study describes the generation of an adenosine deaminase-tethered dCas9 enzyme to allow targeted A>G missense mutations without DSBs.
Cox, D. B. T. et al. RNA editing with CRISPR-Cas13. Science 358, 1019–1027 (2017).
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). This study describes PE technology that enables programmable targeted small insertions, deletions and sequence changes without DSBs by tethering a reverse transcriptase to a Cas enzyme.
Petri, K. et al. CRISPR prime editing with ribonucleoprotein complexes in zebrafish and primary human cells. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-00901-y (2021).
Nelson, J. W. et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-01039-7 (2021).
Brookhouser, N. et al. A Cas9-mediated adenosine transient reporter enables enrichment of ABE-targeted cells. BMC Biol. 18, 193 (2020).
Standage-Beier, K. et al. A transient reporter for editing enrichment (TREE) in human cells. Nucleic Acids Res. 47, e120 (2019).
Martin, A. S. et al. A panel of eGFP reporters for single base editing by APOBEC-Cas9 editosome complexes. Sci. Rep. 9, 497 (2019).
Katti, A. et al. GO: a functional reporter system to identify and enrich base editing activity. Nucleic Acids Res. 48, 2841–2852 (2020).
Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Gilbert, L. A. et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 (2013). Along with Qi et al., this paper describes the use of Cas9 for targeted control of transcriptional activation and repression.
Gilbert, L. A. et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159, 647–661 (2014).
Mali, P. et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 31, 833–838 (2013).
Maeder, M. L. et al. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 10, 977–979 (2013).
Perez-Pinera, P. et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 10, 973–976 (2013).
Liu, X. S. et al. Editing DNA methylation in the mammalian genome. Cell 167, 233–247.e217 (2016).
Amabile, A. et al. Inheritable silencing of endogenous genes by hit-and-run targeted epigenetic editing. Cell 167, 219–232.e214 (2016).
Vojta, A. et al. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 44, 5615–5628 (2016).
Lei, Y. et al. Targeted DNA methylation in vivo using an engineered dCas9-MQ1 fusion protein. Nat. Commun. 8, 16026 (2017).
Bennett-Baker, P. E. & Mueller, J. L. CRISPR-mediated isolation of specific megabase segments of genomic DNA. Nucleic Acids Res. 45, e165 (2017).
Xu, X. et al. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2, 16009 (2016).
Choudhury, S. R., Cui, Y., Lubecka, K., Stefanska, B. & Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 7, 46545–46556 (2016).
Kearns, N. A. et al. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 12, 401–403 (2015).
Hilton, I. B. et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 33, 510–517 (2015).
Cano-Rodriguez, D. et al. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat. Commun. 7, 12284 (2016).
Nuñez, J. K. et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 184, 2503–2519.e2517 (2021).
Gao, X. D. et al. C-BERST: defining subnuclear proteomic landscapes at genomic elements with dCas9-APEX2. Nat. Methods 15, 433–436 (2018).
Wang, H. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 (2013). This study demonstrates the powerful functionality of CRISPR–Cas in rapidly engineering new knockout and ‘floxed’ mouse models.
Yang, H. et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–1379 (2013).
Huijbers, I. J. et al. Using the GEMM-ESC strategy to study gene function in mouse models. Nat. Protoc. 10, 1755–1785 (2015).
Ohtsuka, M. et al. i-GONAD: a robust method for in situ germline genome engineering using CRISPR nucleases. Genome Biol. 19, 25 (2018).
Chen, S. et al. CRISPR-READI: efficient generation of knockin mice by CRISPR RNP electroporation and AAV donor infection. Cell Rep. 27, 3780–3789.e3784 (2019).
Modzelewski, A. J. et al. Efficient mouse genome engineering by CRISPR-EZ technology. Nat. Protoc. 13, 1253–1274 (2018).
Han, X. et al. A suite of new Dre recombinase drivers markedly expands the ability to perform intersectional genetic targeting. Cell Stem Cell 28, 1160–1176.e1167 (2021).
Heckl, D. et al. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 32, 941–946 (2014).
Xu e, W. et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514, 380–384 (2014).
Chiou, S. H. et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev. 29, 1576–1585 (2015).
Ideno, N. et al. A pipeline for rapidly generating genetically engineered mouse models of pancreatic cancer using in vivo CRISPR-Cas9-mediated somatic recombination. Lab. Invest. 99, 1233–1244 (2019).
Seino, T. et al. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell 22, 454–467.e456 (2018).
Sánchez-Rivera, F. J. et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516, 428–431 (2014). This paper showcases efficient in situ editing by CRISPR–Cas9 in somatic cells of the lung to reveal the impact of specific gene disruption in NSCLC.
Michels, B. E. et al. Pooled in vitro and in vivo CRISPR-Cas9 screening identifies tumor suppressors in human colon organoids. Cell Stem Cell 26, 782–792.e787 (2020).
Xue, W. et al. Response and resistance to NF-κB inhibitors in mouse models of lung adenocarcinoma. Cancer Discov. 1, 236–247 (2011).
Platt, R. J. et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455 (2014).
Dow, L. E. et al. Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol. 33, 390–394 (2015).
Rogers, Z. N. et al. Mapping the in vivo fitness landscape of lung adenocarcinoma tumor suppression in mice. Nat. Genet. 50, 483–486 (2018).
Rogers, Z. N. et al. A quantitative and multiplexed approach to uncover the fitness landscape of tumor suppression in vivo. Nat. Methods 14, 737–742 (2017).
Baliou, S. et al. CRISPR therapeutic tools for complex genetic disorders and cancer (Review). Int. J. Oncol. 53, 443–468 (2018).
Lo, Y.-H. et al. A CRISPR/Cas9-engineered ARID1A-deficient human gastric cancer organoid model reveals essential and non-essential modes of oncogenic transformation. Cancer Discov. 1109, 2020 (2021).
Matano, M. et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 21, 256–262 (2015).
Drost, J. et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 521, 43–47 (2015).
Ng, S. R. et al. CRISPR-mediated modeling and functional validation of candidate tumor suppressor genes in small cell lung cancer. Proc. Natl Acad. Sci. USA 117, 513–521 (2020).
Dekkers, J. F. et al. Modeling breast cancer using CRISPR-Cas9-mediated engineering of human breast organoids. J. Natl Cancer Inst. 112, 540–544 (2020).
Wang, T., Wei, J. J., Sabatini, D. M. & Lander, E. S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80 (2014).
Shalem, O. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84 (2014).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). This study optimizes genome-wide CRISPR screening tools and demonstrates the utility of this approach for identifying gene alterations that drive resistance to cancer therapeutics.
Sanson, K. R. et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 9, 5416 (2018).
Doench, J. G. Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet. 19, 67–80 (2018).
Sánchez-Rivera, F. J. & Jacks, T. Applications of the CRISPR-Cas9 system in cancer biology. Nat. Rev. Cancer 15, 387–395 (2015).
Chen, S. et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160, 1246–1260 (2015).
Chow, R. D. et al. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat. Neurosci. 20, 1329–1341 (2017).
Burr, M. L. et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105 (2017).
Burr, M. L. et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 36, 385–401.e388 (2019).
Okada, M. et al. Blockage of core fucosylation reduces cell-surface expression of PD-1 and promotes anti-tumor immune responses of T cells. Cell Rep. 20, 1017–1028 (2017).
Spel, L. et al. Nedd4-binding protein 1 and TNFAIP3-interacting protein 1 control MHC-1 display in neuroblastoma. Cancer Res. 78, 6621 (2018).
Dixit, A. et al. Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866.e1817 (2016). This work demonstrates that combining CRISPR screening with scRNA-seq provides a means to measure the transcriptional effect of gene KOs in a pooled setting.
Datlinger, P. et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301 (2017).
Mimitou, E. P. et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 16, 409–412 (2019).
Adamson, B. et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867–1882.e1821 (2016).
Replogle, J. M. et al. Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat. Biotechnol. 38, 954–961 (2020).
Wroblewska, A. et al. Protein barcodes enable high-dimensional single-cell CRISPR screens. Cell 175, 1141–1155.e1116 (2018).
Hill, A. J. et al. On the design of CRISPR-based single-cell molecular screens. Nat. Methods 15, 271–274 (2018).
Sharma, Y. et al. A pan-cancer analysis of synonymous mutations. Nat. Commun. 10, 2569 (2019).
Zafra, M. P. et al. An in vivo Kras allelic series reveals distinct phenotypes of common oncogenic variants. Cancer Discov. 10, 1654–1671 (2020).
Poulin, E. J. et al. Tissue-specific oncogenic activity of KRAS(A146T). Cancer Discov. 9, 738–755 (2019).
Johnson, C. W. et al. Isoform-specific destabilization of the active site reveals a molecular mechanism of intrinsic activation of KRas G13D. Cell Rep. 28, 1538–1550.e1537 (2019).
Ponsioen, B. et al. Quantifying single-cell ERK dynamics in colorectal cancer organoids reveals EGFR as an amplifier of oncogenic MAPK pathway signalling. Nat. Cell Biol. 23, 377–390 (2021).
Winters, I. P. et al. Multiplexed in vivo homology-directed repair and tumor barcoding enables parallel quantification of Kras variant oncogenicity. Nat. Commun. 8, 2053 (2017).
Boettcher, S. et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365, 599 (2019).
Sharon, E. et al. Functional genetic variants revealed by massively parallel precise genome editing. Cell 175, 544–557.e516 (2018).
Komor, A. C. et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 3, eaao4774 (2017).
Ma, Y. et al. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat. Methods 13, 1029–1035 (2016).
Nishida, K. et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353, aaf8729 (2016).
Hess, G. T. et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 13, 1036–1042 (2016).
Chen, L. et al. Programmable C:G to G:C genome editing with CRISPR-Cas9-directed base excision repair proteins. Nat. Commun. 12, 1384 (2021).
Koblan, L. W. et al. Efficient C•G-to-G•C base editors developed using CRISPRi screens, target-library analysis, and machine learning. Nat. Biotechnol. 39, 1414–1425 (2021).
Kurt, I. C. et al. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 39, 41–46 (2021).
Zhao, D. et al. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat. Biotechnol. 39, 35–40 (2021).
Billon, P. et al. CRISPR-mediated base editing enables efficient disruption of eukaryotic genes through induction of STOP codons. Mol. Cell 67, 1068–1079.e1064 (2017).
Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020).
Hanna, R. E. et al. Massively parallel assessment of human variants with base editor screens. Cell 184, 1064–1080.e1020 (2021). This study demonstrates the utility of BE screens to characterize mutational variants in cancer susceptibility genes and better define drug–protein interactions.
Cuella-Martin, R. et al. Functional interrogation of DNA damage response variants with base editing screens. Cell 184, 1081–1097.e1019 (2021).
Zafra, M. P. et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 36, 888–893 (2018).
Schatoff, E. M. et al. Distinct colorectal cancer-associated APC mutations dictate response to tankyrase inhibition. Cancer Discov. 9, 1358–1371 (2019).
Li, X. et al. Programmable base editing of mutated TERT promoter inhibits brain tumour growth. Nat. Cell Biol. 22, 282–288 (2020).
Sánchez-Rivera, F. J., Diaz, B. J. & Kastenhuber, E. R. Base editing sensor libraries for high-throughput engineering and functional analysis of cancer-associated single nucleotide variants. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-01172-3 (2022).
Erwood, S. et al. Saturation variant interpretation using CRISPR prime editing. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-01201-1 (2022).
Alonso, S. & Dow, L. E. Engineering chromosome rearrangements in cancer. Dis. Models Mech. 14, dmm049078 (2021).
Maddalo, D. & Ventura, A. Somatic engineering of oncogenic chromosomal rearrangements: a perspective. Cancer Res. 76, 4918–4923 (2016).
Driehuis, E. & Clevers, H. CRISPR-induced TMPRSS2-ERG gene fusions in mouse prostate organoids. JSM Biotechnol. Biomed. Eng. 4, 1076 (2017).
Maddalo, D. et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516, 423–427 (2014).
Blasco, R. B. et al. Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology. Cell Rep. 9, 1219–1227 (2014).
Kraft, K. et al. Deletions, inversions, duplications: engineering of structural variants using CRISPR/Cas in mice. Cell Rep. 10, 833–839 (2015).
Lagutina, I. V. et al. Modeling of the human alveolar rhabdomyosarcoma Pax3-Foxo1 chromosome translocation in mouse myoblasts using CRISPR-Cas9 nuclease. PLoS Genet. 11, e1004951 (2015).
Spraggon, L. et al. Generation of conditional oncogenic chromosomal translocations using CRISPR-Cas9 genomic editing and homology-directed repair. J. Pathol. 242, 102–112 (2017).
Dammert, M. A. et al. MYC paralog-dependent apoptotic priming orchestrates a spectrum of vulnerabilities in small cell lung cancer. Nat. Commun. 10, 3485 (2019).
Zhang, X. & Meyerson, M. Illuminating the noncoding genome in cancer. Nat. Cancer 1, 864–872 (2020).
Sud, A., Kinnersley, B. & Houlston, R. S. Genome-wide association studies of cancer: current insights and future perspectives. Nat. Rev. Cancer 17, 692–704 (2017).
Rheinbay, E. et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 578, 102–111 (2020).
Schmitt, A. M. & Chang, H. Y. Long noncoding RNAs in cancer pathways. Cancer Cell 29, 452–463 (2016).
Slack, F. J. & Chinnaiyan, A. M. The role of non-coding rnas in oncology. Cell 179, 1033–1055 (2019).
Anastasiadou, E., Jacob, L. S. & Slack, F. J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 18, 5 (2018).
Hong, D. S. et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 122, 1630–1637 (2020).
Seto, A. G. et al. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br. J. Haematol. 183, 428–444 (2018).
Sanjana, N. E. et al. High-resolution interrogation of functional elements in the noncoding genome. Science 353, 1545–1549 (2016). One of the first noncoding screens, this study demonstrates how CRISPR screens can be used to identify regulatory elements involved in cancer drug resistance.
Canver, M. C. et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197 (2015).
Korkmaz, G. et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol. 34, 192–198 (2016).
Yeo, N. C. et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 15, 611–616 (2018).
Chavez, A. et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 12, 326–328 (2015).
Konermann, S. et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588 (2015).
Tanenbaum, M. E., Gilbert, L. A., Qi, L. S., Weissman, J. S. & Vale, R. D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159, 635–646 (2014).
Alerasool, N., Segal, D., Lee, H. & Taipale, M. An efficient KRAB domain for CRISPRi applications in human cells. Nat. Methods 17, 1093–1096 (2020).
Montalbano, A., Canver, M. C. & Sanjana, N. E. High-throughput approaches to pinpoint function within the noncoding genome. Mol. Cell 68, 44–59 (2017).
Fulco, C. P. et al. Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science 354, 769–773 (2016).
Klann, T. S. et al. CRISPR–Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 35, 561 (2017). This study demonstrates how dCas9-based screens can be used to identify novel tumour type-specific regulatory elements and how these tools (specifically CRISPRi and CRISPRa) can be combined to further characterize these elements.
Katainen, R. et al. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat. Genet. 47, 818–821 (2015).
Liu, E. M. et al. Identification of cancer drivers at CTCF insulators in 1,962 whole genomes. Cell Syst. 8, 446–455 (2019).
Hnisz, D. et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454–1458 (2016). This study reports the disruption of insulated chromatin neighbourhoods using Cas9, mimicking microdeletions found in T cell ALL, and finds that this perturbation is sufficient to activate proto-oncogenes.
Flavahan, W. A. et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114 (2016).
Tarjan, D. R., Flavahan, W. A. & Bernstein, B. E. Epigenome editing strategies for the functional annotation of CTCF insulators. Nat. Commun. 10, 4258 (2019).
Tycko, J. et al. Mitigation of off-target toxicity in CRISPR-Cas9 screens for essential non-coding elements. Nat. Commun. 10, 4063 (2019).
Wallace, J. et al. Genome-wide CRISPR-Cas9 screen identifies microRNAs that regulate myeloid leukemia cell growth. PLoS ONE 11, e0153689 (2016).
Zhu, S. et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR–Cas9 library. Nat. Biotechnol. 34, 1279–1286 (2016).
Liu, Y. et al. Genome-wide screening for functional long noncoding RNAs in human cells by Cas9 targeting of splice sites. Nat. Biotechnol. 36, 1203–1210 (2018).
Liu, S. J. et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 355, aah7111 (2017).
Liu, S. J. et al. CRISPRi-based radiation modifier screen identifies long non-coding RNA therapeutic targets in glioma. Genome Biol. 21, 83 (2020).
Joung, J. et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature 548, 343–346 (2017).
Bester, A. C. et al. An integrated genome-wide CRISPRa approach to functionalize lncRNAs in drug resistance. Cell 173, 649–664 (2018).
Abudayyeh, O. O. et al. RNA targeting with CRISPR–Cas13. Nature 550, 280–284 (2017).
Konermann, S. et al. Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell 173, 665–676 (2018). Along with Abudayyeh et al. and Cox et al., this study shows that Cas13 can be used to robustly knock down RNA in mammalian cells with superior specificity compared with RNA interference (RNAi).
Xu, D. et al. A CRISPR/Cas13-based approach demonstrates biological relevance of vlinc class of long non-coding RNAs in anticancer drug response. Sci. Rep. 10, 1794 (2020).
Li, S. et al. Screening for functional circular RNAs using the CRISPR–Cas13 system. Nat. Methods 18, 51–59 (2021).
Bartolomé, R. A. et al. IL13 receptor α2 signaling requires a scaffold protein, FAM120A, to activate the FAK and PI3K pathways in colon cancer metastasis. Cancer Res. 75, 2434–2444 (2015).
Han, T. et al. Lineage reversion drives WNT independence in intestinal cancer. Cancer Discov. 10, 1590 (2020).
Weber, J. et al. CRISPR/Cas9 somatic multiplex-mutagenesis for high-throughput functional cancer genomics in mice. Proc. Natl Acad. Sci. USA 112, 13982 (2015).
Maresch, R. et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Nat. Commun. 7, 10770 (2016). This article demonstrates rapid in vivo multiplexed editing to study gene interactions and their role in tumorigenesis.
Han, K. et al. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol. 35, 463–474 (2017). This study reports a CRISPR-based double KO screening strategy that minimizes recombination and improves ease of cloning for this type of library.
Zhou, P. et al. A three-way combinatorial CRISPR screen for analyzing interactions among druggable targets. Cell Rep. 32, 108020 (2020).
Gier, R. A. et al. High-performance CRISPR-Cas12a genome editing for combinatorial genetic screening. Nat. Commun. 11, 3455 (2020). This study applies modified Cas12a protein and its CRISPR RNA (crRNA) to enable high-efficiency combinatorial screening in cancer cells.
Michlits, G. et al. CRISPR-UMI: single-cell lineage tracing of pooled CRISPR–Cas9 screens. Nat. Methods 14, 1191–1197 (2017).
Guernet, A. et al. CRISPR-barcoding for intratumor genetic heterogeneity modeling and functional analysis of oncogenic driver mutations. Mol. Cell 63, 526–538 (2016).
Frieda, K. L. et al. Synthetic recording and in situ readout of lineage information in single cells. Nature 541, 107–111 (2017).
Kalhor, R., Mali, P. & Church, G. M. Rapidly evolving homing CRISPR barcodes. Nat. Methods 14, 195–200 (2017).
McKenna, A. et al. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907 (2016).
Quinn, J. J. et al. Single-cell lineages reveal the rates, routes, and drivers of metastasis in cancer xenografts. Science 371, eabc1944 (2021). This article describes subclonal tumour dynamics during metastasis through a single-cell Cas9 lineage tracing system.
Kalhor, R. et al. Developmental barcoding of whole mouse via homing CRISPR. Science 361, eaat9804 (2018).
Bowling, S. et al. An engineered CRISPR-Cas9 mouse line for simultaneous readout of lineage histories and gene expression profiles in single cells. Cell 181, 1410–1422.e1427 (2020).
Hwang, B. et al. Lineage tracing using a Cas9-deaminase barcoding system targeting endogenous L1 elements. Nat. Commun. 10, 1234 (2019).
Askary, A. et al. In situ readout of DNA barcodes and single base edits facilitated by in vitro transcription. Nat. Biotechnol. 38, 66–75 (2020).
Halperin, S. O. et al. CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window. Nature 560, 248–252 (2018).
Umkehrer, C. et al. Isolating live cell clones from barcoded populations using CRISPRa-inducible reporters. Nat. Biotechnol. 39, 174–178 (2021). This study introduces a functional lineage tracing tool called CaTCH that can isolate founding rare clones and study their mechanism of emergence during drug resistance.
Al’Khafaji, A. M., Deatherage, D. & Brock, A. Control of lineage-specific gene expression by functionalized gRNA barcodes. ACS Synth. Biol. 7, 2468–2474 (2018).
Gutierrez, C. et al. Multifunctional barcoding with ClonMapper enables high-resolution study of clonal dynamics during tumor evolution and treatment. Nat. Cancer 2, 758–772 (2021).
Hart, T. et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163, 1515–1526 (2015).
Kim, E. et al. A network of human functional gene interactions from knockout fitness screens in cancer cells. Life Sci. Alliance 2, e201800278 (2019).
Lopes, R. et al. Systematic dissection of transcriptional regulatory networks by genome-scale and single-cell CRISPR screens. Sci. Adv. 7, eabf5733 (2021).
Wan, C. et al. Genome-scale CRISPR-Cas9 screen of Wnt/β-catenin signaling identifies therapeutic targets for colorectal cancer. Sci. Adv. 7, eabf2567 (2021).
Morgens, D. W., Deans, R. M., Li, A. & Bassik, M. C. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol. 34, 634–636 (2016). This comparative study identifies the behaviour of shRNA and CRISPR–Cas9 screens in cancer cells and highlights that combining results from the two systems improves performance.
Evers, B. et al. CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat. Biotechnol. 34, 631–633 (2016).
Cao, J. et al. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. 44, e149 (2016).
Lundin, A. et al. Development of an ObLiGaRe doxycycline inducible Cas9 system for pre-clinical cancer drug discovery. Nat. Commun. 11, 4903 (2020).
Wessels, H.-H. et al. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat. Biotechnol. 38, 722–727 (2020).
Horlbeck, M. A. et al. Mapping the genetic landscape of human cells. Cell 174, 953–967.e922 (2018). This work outlines the use of a paired CRISPRi-based approach for interrogating a large set of gene pairs in cancer cell lines.
Thompson, N. A. et al. Combinatorial CRISPR screen identifies fitness effects of gene paralogues. Nat. Commun. 12, 1302 (2021).
Diehl, V. et al. Minimized combinatorial CRISPR screens identify genetic interactions in autophagy. Nucleic Acids Res. 49, 5684–5704 (2021).
Zetsche, B. et al. Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nat. Biotechnol. 35, 31–34 (2017).
Behan, F. M. et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 568, 511–516 (2019). This paper describes the Sanger Institute’s large-scale effort to probe cancer dependencies across 324 human cancer cell lines contributing to DepMap.
Meyers, R. M. et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784 (2017). This paper describes the Broad Institute’s large-scale effort to identify cancer dependencies in 342 cancer cell lines that account for copy number-specific effects through a novel computational method called CERES.
Chan, E. M. et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 568, 551–556 (2019).
Lieb, S. et al. Werner syndrome helicase is a selective vulnerability of microsatellite instability-high tumor cells. eLife 8, e43333 (2019).
Picco, G. et al. Werner helicase is a synthetic-lethal vulnerability in mismatch repair–deficient colorectal cancer refractory to targeted therapies, chemotherapy, and immunotherapy. Cancer Discov. 11, 1923–1937 (2021).
Dempster, J. M. et al. Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat. Commun. 10, 5817 (2019).
Maiani, E. et al. AMBRA1 regulates cyclin D to guard S-phase entry and genomic integrity. Nature 592, 799–803 (2021).
Simoneschi, D. et al. CRL4AMBRA1 is a master regulator of D-type cyclins. Nature 592, 789–793 (2021).
Chaikovsky, A. C. et al. The AMBRA1 E3 ligase adaptor regulates the stability of cyclin D. Nature 592, 794–798 (2021).
Dharia, N. V. et al. A first-generation pediatric cancer dependency map. Nat. Genet. 53, 529–538 (2021).
Wong, C. H., Siah, K. W. & Lo, A. W. Estimation of clinical trial success rates and related parameters. Biostatistics 20, 273–286 (2019).
Scannell, J. W., Blanckley, A., Boldon, H. & Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 11, 191–200 (2012).
Han, K. et al. CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities. Nature 580, 136–141 (2020).
Bajaj, J. et al. An in vivo genome-wide CRISPR screen identifies the RNA-binding protein Staufen2 as a key regulator of myeloid leukemia. Nat. Cancer 1, 410–422 (2020).
Dai, M. et al. In vivo genome-wide CRISPR screen reveals breast cancer vulnerabilities and synergistic mTOR/Hippo targeted combination therapy. Nat. Commun. 12, 3055 (2021).
Hulton, C. H. et al. Direct genome editing of patient-derived xenografts using CRISPR-Cas9 enables rapid in vivo functional genomics. Nat. Cancer 1, 359–369 (2020).
He, W. et al. De novo identification of essential protein domains from CRISPR-Cas9 tiling-sgRNA knockout screens. Nat. Commun. 10, 4541 (2019).
Shi, J. et al. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 33, 661–667 (2015).
Vinyard, M. E. et al. CRISPR-suppressor scanning reveals a nonenzymatic role of LSD1 in AML. Nat. Chem. Biol. 15, 529–539 (2019).
Lin, A., Giuliano, C. J., Sayles, N. M. & Sheltzer, J. M. CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials. eLife 6, e24179 (2017).
Mattsson, J. et al. Accelerating target deconvolution for therapeutic antibody candidates using highly parallelized genome editing. Nat. Commun. 12, 1277 (2021).
Lin, A. et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 11, eaaw8412 (2019).
Ramkumar, P. & Kampmann, M. CRISPR-based genetic interaction maps inform therapeutic strategies in cancer. Transl. Cancer Res. 7, S61–S67 (2018).
Mair, B., Moffat, J., Boone, C. & Andrews, B. J. Genetic interaction networks in cancer cells. Curr. Opin. Genet. Dev. 54, 64–72 (2019).
Aoidi, R., Maltais, A. & Charron, J. Functional redundancy of the kinases MEK1 and MEK2: rescue of the Mek1 mutant phenotype by Mek2 knock-in reveals a protein threshold effect. Sci. Signal. 9, ra9 (2016).
Shen, J. P. et al. Combinatorial CRISPR–Cas9 screens for de novo mapping of genetic interactions. Nat. Methods 14, 573–576 (2017).
Vidigal, J. A. & Ventura, A. Rapid and efficient one-step generation of paired gRNA CRISPR-Cas9 libraries. Nat. Commun. 6, 8083 (2015).
Sack, L. M., Davoli, T., Xu, Q., Li, M. Z. & Elledge, S. J. Sources of error in mammalian genetic screens. G3 6, 2781–2790 (2016).
Dede, M., McLaughlin, M., Kim, E. & Hart, T. Multiplex enCas12a screens detect functional buffering among paralogs otherwise masked in monogenic Cas9 knockout screens. Genome Biol. 21, 262 (2020).
Najm, F. J. et al. Orthologous CRISPR–Cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol. 36, 179–189 (2018).
Vinay, D. S. et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 35, S185–S198 (2015).
Potts, M. A., McDonald, J. A., Sutherland, K. D. & Herold, M. J. Critical cancer vulnerabilities identified by unbiased CRISPR/Cas9 screens inform on efficient cancer Immunotherapy. Eur. J. Immunol. 50, 1871–1884 (2020).
Hou, J. et al. Integrating genome-wide CRISPR immune screen with multi-omic clinical data reveals distinct classes of tumor intrinsic immune regulators. J. Immunother. Cancer 9, e001819 (2021). This preclinical study identifies RIPK1 as a potential therapeutic target to sensitize tumours to immunotherapies.
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT03681951 (2020).
Patel, S. J. et al. Identification of essential genes for cancer immunotherapy. Nature 548, 537–542 (2017).
Manguso, R. T. et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418 (2017).
Pan, D. et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775 (2018).
Frangieh, C. J. et al. Multimodal pooled Perturb-CITE-seq screens in patient models define mechanisms of cancer immune evasion. Nat. Genet. 53, 332–341 (2021).
Gee, S. et al. Developing an arrayed CRISPR-Cas9 co-culture screen for immuno-oncology target ID. SLAS Discov. 25, 581–590 (2020).
Kearney, C. J. et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci. Immunol. 3, eaar3451 (2018).
Zhuang, X., Veltri, D. P. & Long, E. O. Genome-wide CRISPR screen reveals cancer cell resistance to NK cells induced by NK-derived IFN-γ. Front. Immunol. 10, 2879 (2019).
Pech, M. F. et al. Systematic identification of cancer cell vulnerabilities to natural killer cell-mediated immune surveillance. eLife 8, e47362 (2019).
Freeman, A. J. et al. Natural killer cells suppress T cell-associated tumor immune evasion. Cell Rep. 28, 2784–2794.e2785 (2019).
Sheffer, M. et al. Genome-scale screens identify factors regulating tumor cell responses to natural killer cells. Nat. Genet. 53, 1196–1206 (2021).
Dufva, O. et al. CRISPR screens identify mechanisms of natural killer cell evasion across blood cancers. Blood 134, 3597 (2019).
Kamber, R. A. et al. Inter-cellular CRISPR screens reveal regulators of cancer cell phagocytosis. Nature 597, 549–554 (2021).
Chow, R. D. & Chen, S. Cancer CRISPR screens in vivo. Trends Cancer 4, 349–358 (2018).
Wang, X. et al. In vivo CRISPR screens identify the E3 ligase Cop1 as a modulator of macrophage infiltration and cancer immunotherapy target. Cell 184, 5357–5374.e22 (2021).
Li, F. et al. In vivo epigenetic CRISPR screen identifies Asf1a as an immunotherapeutic target in Kras-mutant lung adenocarcinoma. Cancer Discov. 10, 270–287 (2020).
Gurusamy, D. et al. Multi-phenotype CRISPR-Cas9 screen identifies p38 kinase as a target for adoptive Immunotherapies. Cancer Cell 37, 818–833.e819 (2020).
Freund, E. C. et al. Efficient gene knockout in primary human and murine myeloid cells by non-viral delivery of CRISPR-Cas9. J. Exp. Med. 217, e20191692 (2020).
Simeonov, D. R. & Marson, A. CRISPR-based tools in immunity. Annu. Rev. Immunol. 37, 571–597 (2019).
Schumann, K. et al. Functional CRISPR dissection of gene networks controlling human regulatory T cell identity. Nat. Immunol. 21, 1456–1466 (2020).
Shifrut, E. et al. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell 175, 1958–1971.e1915 (2018).
Henriksson, J. et al. Genome-wide CRISPR screens in T helper cells reveal pervasive crosstalk between activation and differentiation. Cell 176, 882–896.e818 (2019).
Dong, M. B. et al. Systematic immunotherapy target discovery using genome-scale in vivo CRISPR screens in CD8 T cells. Cell 178, 1189–1204.e1123 (2019).
Parnas, O. et al. A genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell 162, 675–686 (2015).
LaFleur, M. W. et al. A CRISPR-Cas9 delivery system for in vivo screening of genes in the immune system. Nat. Commun. 10, 1668 (2019).
Legut, M., Dolton, G., Mian, A. A., Ottmann, O. G. & Sewell, A. K. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood 131, 311–322 (2018).
Guo, X. et al. Disruption of PD-1 enhanced the anti-tumor activity of chimeric antigen receptor T cells against hepatocellular carcinoma. Front. Pharmacol. 9, 1118 (2018). This preclinical study demonstrates that CRISPR–Cas9 disruption of PD1 in CAR T cells enhances their cytotoxicity — an approach that is being investigated currently in clinical trials.
Wang, D. et al. CRISPR screening of CAR T cells and cancer stem cells reveals critical dependencies for cell-based therapies. Cancer Discov. 11, 1192–1211 (2021).
Uddin, F., Rudin, C. M. & Sen, T. CRISPR gene therapy: applications, limitations, and implications for the future. Front. Oncol. 10, 1387 (2020).
Konishi, C. T. & Long, C. Progress and challenges in CRISPR-mediated therapeutic genome editing for monogenic diseases. J. Biomed. Res. 35, 148–162 (2020).
Li, K., Luo, H., Huang, L., Luo, H. & Zhu, X. Microsatellite instability: a review of what the oncologist should know. Cancer Cell Int. 20, 16 (2020).
Shin, G. et al. CRISPR–Cas9-targeted fragmentation and selective sequencing enable massively parallel microsatellite analysis. Nat. Commun. 8, 14291 (2017).
Nachmanson, D. et al. Targeted genome fragmentation with CRISPR/Cas9 enables fast and efficient enrichment of small genomic regions and ultra-accurate sequencing with low DNA input (CRISPR-DS). Genome Res. 28, 1589–1599 (2018).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT03606486 (2021).
Kellner, M. J., Koob, J. G., Gootenberg, J. S., Abudayyeh, O. O. & Zhang, F. SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 14, 2986–3012 (2019).
Chen, J. S. et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436 (2018).
Gootenberg, J. S. et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438 (2017).
Gootenberg, J. S. et al. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444 (2018).
Joung, J. et al. Detection of SARS-CoV-2 with SHERLOCK one-pot testing. N. Engl. J. Med. 383, 1492–1494 (2020).
Broughton, J. P. et al. CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 38, 870–874 (2020).
Choi, B. D. et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 7, 304 (2019).
Zhao, Z. et al. CRISPR knock out of programmed cell death protein 1 enhances anti-tumor activity of cytotoxic T lymphocytes. Oncotarget 9, 5208–5215 (2017).
Lu, Y. et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 26, 732–740 (2020).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT02793856 (2021).
Stadtmauer, E. A. et al. CRISPR-engineered T cells in patients with refractory cancer. Science 367, eaba7365 (2020). The first clinical trial to publish testing of the safety of ex vivo CRISPR–Cas9 deletion of TRAC, TRBC and PDCD1 genes in autologous TILs transplanted back into patients.
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT03399448 (2021).
Eyquem, J. et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117 (2017).
Gaudelli, N. M. et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 38, 892–900 (2020).
Martinez-Lage, M. et al. In vivo CRISPR/Cas9 targeting of fusion oncogenes for selective elimination of cancer cells. Nat. Commun. 11, 5060 (2020).
Gao, J., Luo, T., Lin, N., Zhang, S. & Wang, J. A new tool for CRISPR-Cas13a-based cancer gene therapy. Mol. Ther. Oncolytics 19, 79–92 (2020).
Thi, T. T. H. et al. Lipid-based nanoparticles in the clinic and clinical trials: from cancer nanomedicine to COVID-19 vaccines. Vaccines (Basel) 9, 359 (2021).
Milane, L. & Amiji, M. Clinical approval of nanotechnology-based SARS-CoV-2 mRNA vaccines: impact on translational nanomedicine. Drug Deliv. Transl. Res. 11, 1309–1315 (2021).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403–416 (2020).
Rosenblum, D. et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 6, eabc9450 (2020).
Kosicki, M., Tomberg, K. & Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771 (2018). This study reports unwanted effects of on-target CRISPR–Cas9 gene editing that may have consequences in applying CRISPR–Cas9 as a gene editing tool.
& Liebowitz, M. L. et al. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat. Genet. 53, 895–905 (2021).
Enache, O. M. et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 52, 662–668 (2020).
Tsai, S. Q. et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat. Methods 14, 607–614 (2017).
Wienert, B., Wyman, S. K., Yeh, C. D., Conklin, B. R. & Corn, J. E. CRISPR off-target detection with DISCOVER-seq. Nat. Protoc. 15, 1775–1799 (2020).
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015).
Perez, A. R. et al. GuideScan software for improved single and paired CRISPR guide RNA design. Nat. Biotechnol. 35, 347–349 (2017).
Ikeda, A., Fujii, W., Sugiura, K. & Naito, K. High-fidelity endonuclease variant HypaCas9 facilitates accurate allele-specific gene modification in mouse zygotes. Commun. Biol. 2, 371 (2019).
Charlesworth, C. T. et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 25, 249–254 (2019). This study reveals a large proportion of people with pre-existing immunity to Cas9, which has implications for gene and cell therapies that express Cas9.
Wagner, D. L. et al. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 25, 242–248 (2019).
Simhadri, V. L. et al. Prevalence of pre-existing antibodies to CRISPR-associated nuclease Cas9 in the USA population. Mol. Ther. Methods Clin. Dev. 10, 105–112 (2018).
Ferdosi, S. R. et al. Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes. Nat. Commun. 10, 1842 (2019).
Dubrot, J. et al. In vivo screens using a selective CRISPR antigen removal lentiviral vector system reveal immune dependencies in renal cell carcinoma. Immunity 54, 571–585.e576 (2021).
Doench, J. G. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016).
Doench, J. G. et al. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267 (2014).
Hsu, P. D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013).
Labun, K. et al. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–W174 (2019).
Xu, H. et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 25, 1147–1157 (2015).
Chari, R., Mali, P., Moosburner, M. & Church, G. M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat. Methods 12, 823–826 (2015).
Moreno-Mateos, M. A. et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 12, 982–988 (2015).
Kim, H. K. et al. Deep learning improves prediction of CRISPR–Cpf1 guide RNA activity. Nat. Biotechnol. 36, 239–241 (2018).
Lorenz, R. et al. ViennaRNA Package 2.0. Algorithms Mol. Biol. 6, 26 (2011).
DeWeirdt, P. C. et al. Optimization of AsCas12a for combinatorial genetic screens in human cells. Nat. Biotechnol. 39, 94–104 (2021).
Listgarten, J. et al. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat. Biomed. Eng. 2, 38–47 (2018).
Concordet, J. P. & Haeussler, M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245 (2018).
Tycko, J. et al. Pairwise library screen systematically interrogates Staphylococcus aureus Cas9 specificity in human cells. Nat. Commun. 9, 2962 (2018).
Heigwer, F., Kerr, G. & Boutros, M. E-CRISP: fast CRISPR target site identification. Nat. Methods 11, 122–123 (2014).
McKenna, A. & Shendure, J. FlashFry: a fast and flexible tool for large-scale CRISPR target design. BMC Biol. 16, 74 (2018).
Jost, M. et al. Titrating gene expression using libraries of systematically attenuated CRISPR guide RNAs. Nat. Biotechnol. 38, 355–364 (2020).
Meier, J. A., Zhang, F. & Sanjana, N. E. GUIDES: sgRNA design for loss-of-function screens. Nat. Methods 14, 831–832 (2017).
Park, J., Bae, S. & Kim, J. S. Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics 31, 4014–4016 (2015).
Bae, S., Kweon, J., Kim, H. S. & Kim, J.-S. Microhomology-based choice of Cas9 nuclease target sites. Nat. Methods 11, 705–706 (2014).
Michlits, G. et al. Multilayered VBC score predicts sgRNAs that efficiently generate loss-of-function alleles. Nat. Methods 17, 708–716 (2020).
Arbab, M. et al. Determinants of base editing outcomes from target library analysis and machine learning. Cell 182, 463–480.e430 (2020).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT03081715 (2019).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT03044743 (2017).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT04417764 (2020).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT03545815 (2020).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT04037566 (2019).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT04767308 (2021).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT03166878 (2017).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT04502446 (2021).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT04244656 (2021).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT04637763 (2021).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT04438083 (2021).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT04557436 (2020).
US National Library of Medicine. ClinicalTrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT04426669 (2020).
Acknowledgements
This work was supported by F31 Ruth L. Kirschstein Predoctoral Individual National Research Service Awards for A.K. (F31-CA247351-02) and B.J.D. (F31-CA261061-01).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
L.E.D. is a consultant and holds equity in Mirimus, Inc. L.E.D. has served as a consultant for Volastra Therapeutics and Frazier Healthcare. N.E.S. is an adviser to Vertex and Qiagen. A.K., B.J.D. and C.M.C declare no competing interests.
Peer review
Peer review information
Nature Reviews Cancer thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
BE-Hive: https://www.crisprbehive.design/
Benchling: https://www.benchling.com/crispr/
cas13design: https://cas13design.nygenome.org
CHOPCHOP: http://chopchop.cbu.uib.no
CRISPick: https://portals.broadinstitute.org/gppx/crispick/public
CRISPOR: http://crispor.tefor.net
E-CRISP: http://www.e-crisp.org/E-CRISP/
FlashFry: https://github.com/mckennalab/FlashFry
GUIDES: http://guides.sanjanalab.org
GuideScan: http://www.guidescan.com
RGEN Cas-Designer: http://www.rgenome.net/cas-designer/
Vienna Bioactivity CRISPR (VBC) score: https://www.vbc-score.org
Glossary
- Homology-directed repair
-
(HDR). An error-free DNA damage repair mechanism that uses an existing DNA template.
- Non-homologous end joining
-
(NHEJ). An error-prone DNA repair system that directly ligates broken DNA strands without a homologous template.
- Directed evolution
-
A method that uses the process of natural selection to steer enzymes towards a defined variant.
- Transition base substitutions
-
Single base pair change from purine to purine or pyrimidine to pyrimidine.
- Zygote targeting
-
A genetic engineering method to introduce knockout and knock-in mutations into mouse lines.
- Template switching
-
The unique barcode is uncoupled from its correct element in pooled screening assays by reverse transcriptase or PCR polymerase shuffling.
- Neomorphic changes
-
A type of mutation in which the change results in novel gene function.
- Missense mutations
-
A single nucleotide change that results in alteration of the amino acid sequence.
- Transversion events
-
Single base pair change from a purine to a pyrimidine or vice versa.
- Nonsense mutations
-
Single base pair change that generates a premature termination (stop) codon.
- Synteny
-
Conservation of co-localization of genes on chromosomes across species.
- Single nucleotide polymorphisms
-
(SNPs). Genetic variation in the form of a single base pair change found within a population.
- Saturation mutagenesis
-
The generation of a large number of mutations across a genomic region of interest.
- Topologically associating domain
-
(TAD). Highly interacting genomic region demarcated by CCCTC-binding factor (CTCF) binding sites.
- Insulator sites
-
Genomic regions that are enriched in CCCTC-binding factor (CTCF) or cohesin.
- Gliomasphere
-
The in vitro spheroid culture of patient-derived glioblastoma tumour cells.
- Heterochromatin
-
Transcriptionally repressed, gene-poor, chromatin regions that typically contain repetitive sequences.
- Very long intergenic noncoding RNAs
-
(vlincRNAs). Noncoding RNA molecules >50 kb in size that are transcribed from intergenic regions.
- Circular RNAs
-
(circRNAs). Single-stranded closed RNA molecules.
- Barcoding
-
The addition or creation of identifying sequences to monitor cell lineage.
- Fluorescence in situ hybridization
-
(FISH). A cytogenetic approach using fluorescently labelled probes to detect specific nucleic acid sequences.
- Microsatellites
-
Short tandem repeats in noncoding DNA often used as a genetic marker.
Rights and permissions
About this article

Cite this article
Katti, A., Diaz, B.J., Caragine, C.M. et al. CRISPR in cancer biology and therapy. Nat Rev Cancer 22, 259–279 (2022). https://doi.org/10.1038/s41568-022-00441-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41568-022-00441-w
This article is cited by
-
Functional CRISPR screens in T cells reveal new opportunities for cancer immunotherapies
Molecular Cancer (2024)
-
Comprehensive review of CRISPR-based gene editing: mechanisms, challenges, and applications in cancer therapy
Molecular Cancer (2024)
-
Finely tuned ionizable lipid nanoparticles for CRISPR/Cas9 ribonucleoprotein delivery and gene editing
Journal of Nanobiotechnology (2024)
-
Tafazzin mediates tamoxifen resistance by regulating cellular phospholipid composition in ER-positive breast cancer
Cancer Gene Therapy (2024)
-
Translating p53-based therapies for cancer into the clinic
Nature Reviews Cancer (2024)
