
Abstract
Interleukins and associated cytokines serve as the means of communication for innate and adaptive immune cells as well as non-immune cells and tissues. Thus, interleukins have a critical role in cancer development, progression and control. Interleukins can nurture an environment enabling and favouring cancer growth while simultaneously being essential for a productive tumour-directed immune response. These properties of interleukins can be exploited to improve immunotherapies to promote effectiveness as well as to limit side effects. This Review aims to unravel some of these complex interactions.
Similar content being viewed by others


Introduction
Advances in tumour biology during the past century have demonstrated the tight interplay between the immune system and healthy and malignant cells. These insights laid the foundation for the concept of immunosurveillance: the ability of the immune system to recognize and eliminate transformed cells. In contrast, immunoediting describes the reciprocal interaction and shaping of the immune system and cancer cells, eventually culminating in cancer development and progression1,2. The resistance to immune attack and the presence of protumoural inflammation are two of the major hallmarks of cancer3. Highlighting its importance, the immune milieu at the cancer site on presentation (immune contexture) can define the outcome of patients with colorectal cancer4,5.
Cytokines mediate key interactions between immune and non-immune cells in the tumour microenvironment (TME). It was recently demonstrated how the TME in, for example, lung adenocarcinoma allows malignant cells to co-evolve with immune responses6. Among cytokines, several interleukins are particularly relevant in the development and progression of cancer. The multitude of cellular sources, receptors and signalling pathways and even dose-dependency define the pleiotropic role of interleukins in cancer. Along these lines, interleukin action can be cell specific and spans cancer initiation, tumour progression and tumour control7.
Delineating the exact mechanisms of tumour immune control and evasion enabled the development of novel, tailored and highly effective therapies. The therapeutic potential of interleukins has been of interest in both basic and translational cancer research in recent years. The increasing number of clinical trials currently under way highlights their value as a therapeutic agent and a target. Cytokines have been tested in clinical trials as singular therapeutic agents with limited success rates (extensively reviewed in8) and are now undergoing a revival in combinations with synthetic biology, gene and cellular therapies. In most cases, classification of cytokines relies on structural or receptor homology and gene proximity but not necessarily on their biological role in cancer, which is the purpose of the present work9. In this Review, interleukins will be discussed on the basis of their biological role in cancer rather than family membership. Further information about cytokine classification is presented in Table 1.
This Review will cover the milestones of the latest discoveries of interleukin-related mechanisms in cancer, together with their application in clinical practice. We provide a current overview of clinical trials, newly approved therapeutic agents and breakthrough preclinical concepts. Although this Review focuses on cancer, as many of the same principles apply to a host of other diseases, it may prove useful to readers in a broad range of disciplines.
Carcinogenesis
Chronic inflammation has long been established as one of the drivers for carcinogenesis in many cancer entities, such as lung, skin, oesophageal, gastric, colorectal and pancreatic cancer and hepatocellular carcinoma4. Some interleukins directly induce signalling in non-immune cells and sustain tissue homeostasis. However, after the oncogenic event, interleukin signalling in cancer cells can become a pathological mechanism of tumour growth, metastatic spread and cancer progression (Fig. 1).

Persistent inflammation in response to pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns triggers activation of nuclear factor-κB (NF-κB), which primes pro-IL-1β production, and nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) inflammasome activation, which causes the release of active IL-1β from fibroblasts, epithelial cells and myeloid cells such as dendritic cells (DCs), monocytes and macrophages (MΦ). In turn, IL-33 derived from tumour-initiating cells recruits macrophages, which upon activator protein 1 (AP-1) signalling produce transforming growth factor-β (TGFβ) that suppresses the function of cytotoxic T lymphocytes (CTLs). IL-1β induces production of nitric oxide (NO) and reactive oxygen species (ROS) by epithelial cells, which may cause DNA damage, and promotes the production of IL-6 and IL-11 from epithelial and myeloid cells, and IL-22 from type 3 innate lymphoid cells (ILC3s) and γδ T cells. Under homeostatic conditions, IL-22 facilitates DNA repair caused by bacterial genotoxins, but in transformed cells, IL-6 and IL-11 together with IL-22 rapidly induce phosphorylation (P) of signal transducer and activator of transcription 3 (STAT3). Activation of STAT3 signalling is observed in multiple types of cancer and induces proliferation, survival, stemness, epithelial–mesenchymal transition (EMT) and migration of transformed cells. IL-1β together with TGFβ induces differentiation of T helper 17 (TH17) cells, which upon IL-23 stimulation from DCs secrete IL-17A and IL-17F (IL-17A/F). IL-17, which typically activates NF-κB to mediate wound-healing signalling, and may exacerbate nascent tumour outgrowth.
IL-1 has long been implicated in inflammation-induced carcinogenesis10,11. IL-1α and IL-1β, which have more recently gained attention for their role in tumour biology, and their family members (Table 1) are produced by immune (for example, myeloid) and non-immune (for example, epithelial) cells in the context of chronic inflammation12.
IL-1α and IL-1β are alarm cytokines (also known as alarmins) that both act through IL-1 receptor (IL-1R) to initiate and amplify local inflammation9. The production of the IL-1β precursor pro-IL-1β occurs rapidly in response to danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) by pathogen-recognition receptors, such as Toll-like receptors, C-type lectin receptors or retinoic acid-inducible gene I protein (RIG-I)-like receptors, and requires inflammasome activation following proteolytic cleavage by caspase 1 into its active form13. The sensing of danger-associated molecular patterns by RIG-I-like receptors, nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) and absent in melanoma 2 (AIM2)-like receptors initiates the canonical inflammasome assembly by recruiting and forming procaspase 1 filaments and activating caspase 1, which in turn cleaves pro-IL-1β and pro-IL-18, releasing their active forms14.
Recent work demonstrated that IL-33 can create a self-amplifying tumorigenic niche that furthers the development of a nascent tumour15. As shown in a model of squamous cell carcinoma, once cells have transformed, they acquire tumorigenic capacity (also known as tumour-initiating cells). Tumour-initiating cells can secrete IL-33, which in turn causes infiltration of tumour-associated macrophages and promotes transforming growth factor-β (TGFβ) signalling, which creates a tumorigenic niche15,16.
In the context of chronic inflammation, IL-1α and IL-1β may directly promote the production of carcinogenic mediators, such as nitric oxide and reactive oxygen species12,17. IL-1 initiates the downstream release of proinflammatory cytokines, such as IL-6, and mediates the recruitment of innate immune cells, triggering a cascade of inflammatory mechanisms12. Here, the effects of IL-1 are mediated by phosphorylation of nuclear factor-κB (NF-κB), which in turn inactivates suppressor of cytokine signalling 3 (SOCS3) and potentiates phosphorylation of signal transducer and activator of transcription 3 (STAT3)17,18. Recently, cell type-specific carcinogenic effects of IL-1 were demonstrated in mice harbouring a deletion of the adenomatous polyposis coli (Apc) tumour suppressor gene in colon epithelial cells, where IL-1 secretion was elicited from monocytes, tumour epithelial cells and stromal cells19. Acting through IL-1R on epithelial cells, IL-1 directly promoted the malignant transformation of epithelial cells mediated by nuclear accumulation of NF-κB. In addition, IL-1 signalling in T cells triggered the secretion of protumorigenic IL-17 and IL-22 (ref.19). This observation is in line with the previously reported protumorigenic effects of IL-23 production by myeloid cells and subsequent T helper 17 (TH17) cell responses to microbial products in the same model of colon carcinogenesis20. Similarly, in a mouse model of lung adenocarcinoma, myeloid differentiation primary response gene 88 protein (MyD88)-dependent IL-1 production from myeloid cells acted on γδ T cells to produce an inflammatory loop involving IL-17A and IL-22 secretion that led to the malignant transformation of cells bearing Kras and Trp53 mutations21.
IL-22, which is induced by IL-1 signalling in cancer settings22, has been associated with STAT3 activation and cancer-promoting properties23. IL-22 acts through IL-22R, exclusively expressed on non-haematopoietic cells, to promote wound healing and the production of microbicidal peptides23,24. Recent findings, however, highlight its stage-specific dual properties in carcinogenesis25. Under homeostatic conditions IL-22 is secreted mainly by group 3 innate lymphoid cells (ILC3s) and γδ T cells and can repair genotoxicity-induced DNA damage in the intestinal epithelium to prevent the malignant transformation of cells25. However, when its activity is not controlled by IL-22-binding protein (IL-22BP), its natural inhibitor, IL-22 has a protumorigenic effect in a mouse model of colitis-associated colon cancer26. IL-22-producing T cells were reported to accumulate in nascent lung and colon tumours in mouse and human samples22,27,28. Through STAT3 phosphorylation IL-22 provides proliferation and migration signals to transformed malignant cells and/or cells bearing oncogenic mutations, sustaining their stemness through the induction of SRY (sex-determining region Y)-box 2 (SOX-2) and NANOG as demonstrated in human colon tissues, and similarly in mouse models of lung, pancreatic and breast cancers29,30,31,32. IL-20 has been shown to act similarly to IL-22 in hepatocellular carcinoma and breast, prostate and oral cancer, and also induces immune inhibition through programmed cell death protein 1 (PD1) upregulation in pancreatic cancer33.
Tissue damage and release of alarm cytokines induce IL-6, IL-10, IL-11 and IL-23 expression by sentinel myeloid and tissue cells. Under homeostatic conditions, this results in a self-inhibitory loop to resolve inflammation and promote healing34. In turn, IL-6 and IL-11 are potent orchestrators of innate immune responses and inflammation35,36. Moreover, IL-6 is also a regulator of development and metabolic processes36. These effects are attributed to the ubiquitously expressed common transducing gp130 receptor subunit, which can dimerize with membrane IL-6Rα or IL-11Rα to initiate classical cis signalling or a soluble receptor form resulting in non-classical trans signalling37,38,39,40. gp130 is associated with activation of Janus kinase 1 (JAK1), JAK2 and non-receptor tyrosine-protein kinase 2 (TYK2)38,39,40, as well as tyrosine-protein phosphatase SH-PTP2 (SHP2) and SRC–Yes-associated protein (YAP)–Notch signalling, which activates proliferation and tissue regeneration35,36,41. Furthermore, activation of phosphoinositide 3-kinase (PI3K)–AKT–mechanistic target of rapamycin complex 1 (mTORC1) signalling integrates interleukin signalling and the metabolic cell programme35,36.
Classical IL-6 signalling is considered essential for homeostatic processes, whereas trans signalling was specifically demonstrated to amplify the inflammation and promote inflammation-induced carcinogenesis42,43,44. Excessive activation of STAT3 by the overabundance of IL-6 and IL-11 combined with oncogenic driver mutations licenses the development of malignant tumours such as colon and gastric45,46,47,48, pancreatic49,50 and lung51 cancers. IL-1 and IL-6 released by myeloid cells induce PI3K–AKT–mTOR signalling, which, via activation of hypoxia-inducible factor 1α (HIF1α), shifts their metabolism towards glycolysis and decreases oxidative phosphorylation leading to amplification of IL-1 and IL-6 production and exacerbated inflammation-induced carcinogenesis52,53. Of note, deregulated IL-6 and IL-1 signalling is also a contributor to cancer-induced cachexia (see Box 1).
Importantly, IL-6 also induces angiogenesis and tumour vascularization mediated by vascular endothelial growth factor (VEGF)34. Further, classical IL-6 signalling via IL-6R in lymphocytes promotes proliferation of T cells and lineage commitment to TH17 cells and T follicular helper cells43. IL-6 suppresses forkhead box P3 (FOXP3), limiting the ability of TGFβ to promote regulatory T cell (Treg cell) development, which enables TH17 cell differentiation and amplifies proinflammatory response, as demonstrated in a mouse melanoma model of TH17 cell adoptive cell transfer (ACT)54.
IL-23 is another interleukin produced in response to DAMPs at epithelial barriers20. Typically, it counteracts the antitumour action of IL-12, but it was also demonstrated to directly promote tumour incidence and growth. IL-23 triggers IL-17 production from ILC3s and committed TH17 cells, synergizing with IL-6 in the amplification of inflammation, and prompting epithelial cells to acquire stemness and undergo malignant transformation20,55,56,57. IL-23 together with IL-1, IL-6 and IL-21 is capable of inducing IL-17 production independently of T cell receptor (TCR) signalling12,18,58. During chronic inflammation, an abundance of microbial antigens may drive IL-17 and subsequent aberrant wound healing that results in tumorigenesis, as seen in murine models of skin and colon cancers59,60,61,62. In skin stem cells, IL-17A signalling can recruit and transactivate epidermal growth factor receptor (EGFR), which induces expansion and migration of these cells62,63. Hence, the inflammatory responses initiate cellular programmes in conditions of uncontrolled chronic activation and may provide a direct link to tumorigenesis.
Cancer growth and progression
Malignant tumours possess several characteristic traits termed by Hanahan and Weinberg ‘hallmarks of cancer’64. Importantly, several of these traits, namely sustained proliferation, inflammation, angiogenesis, active invasion and migration, are also the hallmarks of wound healing, and therefore may maliciously utilize cytokine signalling aimed at tissue repair65,66 (Fig. 2).

Immune evasion and tumour progression rely on cancer cell-intrinsic and cancer cell-extrinsic cytokine signalling. Several cancer types were demonstrated to overexpress certain cytokines, for example IL-6 or IL-11, which may act in an autocrine manner to activate phosphoinositide 3-kinase (PI3K)–AKT–mTOR signalling to upregulate glycolysis and induce metabolic reprogramming, nuclear factor kappa-κB (NF-κB), rat sarcoma (RAS)–rapidly accelerated fibrosarcoma (RAF)–mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription 3 (STAT3). These pathways in turn can lead to epithelial–mesenchymal transition (EMT), increased proliferation, reduced apoptosis, increased migration and production of cytokines, such as IL-8, metalloproteinases and vascular endothelial growth factor (VEGF), which induces angiogenesis. Other cytokines, such as IL-1β, IL-13, IL-17, IL-22, IL-23 and IL-35 can also induce EMT and, thus, tumour progression. Tumour-secreted IL-8, in turn, induces recruitment of polymorphonuclear leukocytes (PMNs). Together with monocytes, they differentiate into myeloid-derived suppressor cells (MDSCs), which inhibit T helper 1 (TH1) responses. MDSCs, tumour-associated macrophages (TAMs) and M2 macrophages (MΦ), polarized by TH2-type cytokines, contribute to the pool of transforming growth factor-β (TGFβ) to shape an immunosuppressive microenvironment. In turn, TGFβ together with IL-33 promotes differentiation of regulatory T cells (Treg cells), which bear a high-affinity IL-2 receptor (IL-2R) and are a major source of IL-10 that under chronic conditions suppresses antitumour responses. Alternatively, together with IL-6, TGFβ promotes the differentiation of TH17 cells to produce IL-17 and promote further MDSC recruitment and differentiation. pSTAT3, phosphorylated STAT3; ZEB, zinc-finger E-box-binding homeobox.
Thus, it was demonstrated that IL-1 not only promotes inflammation-induced carcinogenesis but also contributes to tumour invasiveness and angiogenesis67. Although activation of, for example, the NLR family pyrin-domain containing 3 (NLRP3) inflammasome is reported ubiquitously in cancer, the consequences of NLRP3 activation and release of IL-1β and its family member IL-18 range from antitumour activity to cancer growth and metastasis68,69. IL-18 was demonstrated to promote angiogenesis through the activation of NF-κB, which triggers the secretion of VEGF from cancer cells, induces their proliferation and invasion and prevents apoptosis, and may also induce PD1-dependent immunosuppression of natural killer (NK) cells70,71,72,73.
In addition to its ability to amplify procarcinogenic chronic inflammation, IL-6 drives tumour-intrinsic mechanisms of cancer progression that recapitulate most of the hallmarks of cancer74. IL-6 induces PI3K–AKT, mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK), NF-κB and STAT3 signalling. These pathways augment the expression of antiapoptotic proteins (B cell lymphoma 2 (BCL-2), B-cell lymphoma, extra large (BCL-xL), induced myeloid leukaemia cell differentiation protein MCL1 and baculoviral inhibitor of apoptosis domain (IAP) repeat-containing protein 5 (BIRC5; also known as survivin)), evade growth control, activate metabolism and induce angiogenesis via VEGF production (reviewed in74). STAT3 phosphorylation in cancer cells is also induced by other cytokines, such as IL-22, exacerbating cancer-promoting pathways22,27,29,75.
Furthermore, chronic STAT3 activation in cancer cells induces proliferation, expression of matrix metalloproteinases, and migration, thereby increasing the aggressiveness of carcinomas34,76,77. These effects are typically associated with epithelial-to-mesenchymal transition (EMT), which is orchestrated by EMT transcription factors, such as zinc-finger E-box-binding homeobox (ZEB), SNAI1, SNAI2, SLUG and TWIST, and enables cell detachment, metastatic spread and invasiveness78,79,80. EMT induced by the integrated network of IL-1β, IL-6, IL-17, IL-22 and IL-23 mediates the invasiveness of many cancer types. For example, STAT3 activation by IL-22 was found to upregulate EMT markers in mouse models of pancreatic ductal adenocarcinoma and breast carcinoma30,32. Moreover, chronic exposure to IL-1β induces MAPK (ERK and JUN amino-terminal kinase (JNK))–activating protein 1 (AP-1) engagement, which results in epigenetic modifications that cause long-lasting EMT phenotypes, including SLUG and ZEB2 transcription in lung cancer81,82. Similarly, EMT is promoted by IL-6-mediated and IL-11-mediated paracrine activation of STAT3 in invasive pancreatic ductal adenocarcinoma, IL-13-mediated activation of STAT6 in colorectal cancer and IL-35 in breast cancer49,83,84,85. IL-23 utilizes the Wnt–β-catenin pathway to induce EMT in oesophageal cancer86, whereas NF-κB activation induced by IL-17 directly promotes ZEB1 in lung cancer87. IL-8 induces EMT through PI3K–AKT and rat sarcoma (RAS)–rapidly accelerated fibrosarcoma (RAF)–mitogen-activated protein kinase kinase (MEK)–ERK in various cancers88. However, permanent activation of ZEB1 by IL-1β in breast cancer cells entraps them in a mesenchymal state and prevents metastatic colonization; hence, cancer cells require mesenchymal–epithelial transition for further progression82. Taken together, these findings provide a rationale for the therapeutic neutralization of these cytokines to reduce EMT-mediated cancer progression.
Cancer immunosurveillance
Innate immunity
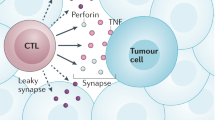
Despite the plethora of oncogenic factors that constantly induce malignant transformation of cells, our immune system recognizes and eliminates most of these transformed cells through immunosurveillance2. Innate immune cells, namely NK cells, natural killer T cells (NKT cells) and γδ T cells, possess a wide array of the evolutionarily conserved ‘altered self’ and activation receptors that can recognize and eliminate transformed cells2. This process depends on production of perforin, granzyme B and interferon-γ (IFNγ) in these cells89. The last of these directly induces apoptosis in tumour cells and triggers a cascade of chemokines to initiate the recruitment of innate immune cells and induce the production of proinflammatory antitumoural cytokines2 (Fig. 3).

Natural killer (NK) cells bear a set of receptors that allow the recognition and elimination of transformed cells. Danger-associated molecular patterns, such as high mobility group protein B1 (HMGB1), which are released from malignant cells, are processed by resident antigen-presenting cells, such as dendritic cells (DCs) and macrophages (MΦ). In turn, these cells produce IL-12 and IL-15 to promote the cytotoxic activity of NK cells and cytotoxic T lymphocytes (CTLs) and induce interferon-γ (IFNγ) release. Resident and recruited monocytes upon priming differentiate into macrophage-like cells and contribute to the IL-12 and IFNγ pool. DCs loaded with tumour antigens migrate into the draining lymph nodes, where they present processed antigens together with major histocompatibility complex class II molecules to naive T cells. Naive T cells originate from the lymphoid progenitors in the bone marrow, where they require an IL-3 proliferation signal, and further IL-7-promoted development in the thymus. IL-12 secreted by DCs in the lymph node triggers expression of T-box transcription factor T-bet (also known as TBX21), which defines T helper 1 (TH1) cell polarization. Upon stimulation, TH1 cells and CTLs migrate to the tumour site and produce IL-2, which leads to rapid lymphocyte proliferation (represented by a circular arrow) and amplification of antigen-specific responses. CTLs and TH1 cells use perforin and granzymes to kill tumour cells and release IFNγ, which can directly induce apoptosis of tumour cells and primes M1 macrophage polarization. DCs together with M1 macrophages produce IL-12 necessary to sustain TH1 cell polarization and amplify IFNγ production. Cytotoxic effects may be also enhanced by IL-10 secreted by M1 macrophages, IL-27 from macrophages and DCs and IL-21 from TH17 cells and T follicular helper (TFH) cells.
Proliferation, maturation and cytotoxic effector functions of innate immune cells are tightly controlled by IL-2 family cytokines, namely IL-2, IL-7, IL-15 and IL-21, which utilize heteromeric receptors and share a common γc receptor subunit8 (Table 1). IL-15 is the primary cytokine to regulate the biology of NK cells, which have particular importance for the control of haematological malignancies90. Unlike other IL-2 family members, IL-15 is typically presented in conjunction with the IL-15Rα subunit on the surface of antigen-presenting cells (APCs)8. To elicit signalling, the IL-15–IL-15Rα complex binds a heterodimeric IL-2Rβ–γc receptor complex shared with IL-2 (refs8,91). Upon binding to its receptor on NK cells, IL-15 triggers a cascade of reactions resulting in phosphorylation of the serine-threonine kinase AKT. Consequently, it causes accumulation and translocation of the transcription factor X-box-binding protein 1 (XBP1) into the nucleus, where T-box protein expressed in T cells (T-bet), the essential transcription factor for cytotoxicity of NK cells and differentiation of TH1 cells, is recruited90,92. T-bet, in turn, induces transcription of granzyme B and genes responsible for proliferation, maturation and survival of NK cells90. Importantly, the activity of AKT downstream of IL-15 induces homeostatic priming in NK cells and cytotoxic T lymphocytes (CTLs)93.
NK cell-secreted IFNγ primes APCs, such as dendritic cells (DCs) and macrophages, to produce IL-12, which shares one of its subunits, p40, with IL-23 (refs8,56). Importantly, APCs serve as a bridge between the innate and adaptive immune responses, and their recruitment, maturation and activation are governed by cytokines94. IL-12 secreted from APCs, in turn, amplifies IFNγ production in NK cells (requires priming by IL-15 and/or IL-18 (refs91,95)) and thereby antitumour responses96.
IL-18 acts through its receptor highly expressed in NK cells and triggers IFNγ production, cytotoxicity and FAS ligand (FASL) expression12. In addition to the reported ability to directly promote the proliferation and migration of cells in certain types of cancer, tumour-infiltrating NK cells and CD8 T cells express high levels of IL-18R73,97. However, IL-18 activity is moderated by IL-18-binding protein (IL-18BP), a soluble receptor that acts as a decoy for IL-18 and inhibits antitumoural and antiviral activities of NK cells and CTLs97,98. Moreover, mouse and human NK cells have high expression of IL-1R8, another member of the IL-1R family, which together with IL-1R5 (also known as IL-18Rα) binds IL-37, which is known for its anti-inflammatory properties12,99. IL-1R8 could inhibit IL-18-dependent mTOR and JNK signalling pathways, which are essential for NK cell metabolism, differentiation and activation, as demonstrated in mouse models of liver carcinogenesis and haematogenous liver and lung metastasis99.
Recently, eosinophils, a pleiotropic innate immune cell type primarily known for antihelminth and allergic responses, have received attention due to their distinct antitumour mechanisms in hepatocellular carcinoma and breast cancer100,101. IL-5, which together with IL-3 and granulocyte–macrophage colony-stimulating factor (GM-CSF) constitutes a family of haematopoietic cytokines, is a key regulator of eosinophil differentiation, maturation, expansion and survival101. Recruited by the tumour-released alarmin IL-33, eosinophils can directly exert cytotoxicity but also aid antitumoural TH1 cell responses100.
IL-28A, IL-28B and IL-29 are cytokines distantly related to the IL-10 family, but are also interferons and therefore are also termed ‘type III interferons’ or ‘λ-interferons’. They typically mediate innate immune antiviral activity but also directly induce apoptosis of malignant cells102,103.
Adaptive immunity
Upon immunogenic cancer cell death, antigens released by tumour cells are taken up by APCs, primarily DCs, and enter draining lymph nodes to initiate the development of antigen-specific adaptive immune responses. There, the cytokine milieu plays a fate-defining role for T cells2,104 (Fig. 3). Proliferation, survival, differentiation and termination of T cell responses are mainly governed by IL-2, IL-7 and IL-15 (ref.8), whereas IL-3 is necessary for survival and proliferation of lymphocyte progenitors105,106. IL-2 binds with intermediate affinity to γc–IL-2Rβ, inducing the clonal expansion of T cells, but also for termination of the T cell responses through FAS/FASL-mediated apoptosis of T cells8. IL-2Rα chain (CD25) expressed mainly in Treg cells increases the affinity of the IL-2R, mediates FOXP3 expression and is essential for self-tolerance8.
CTLs and effector TH1 cells are the primary mediators of antitumoural adaptive immunity107. IL-12 derived from DCs provides an essential signal that drives expression of T-bet, and thus the differentiation of effector TH1 cells, and CTLs104,107,108. Successful anti-PD1 immunotherapy relies on IL-12-dependent crosstalk between T cells and DCs to promote tumour cell killing109. Similarly to signalling in NK cells, IL-2, IL-15 and IL-18 synergize with IL-12 to trigger IFNγ production and direct cytotoxicity in CTLs and TH1 cells8,107. Thus, soluble IL-15–IL-15Rα complexes produced by myeloid cells were shown to be essential for establishing NK cell and CTL populations in tumours in mouse models of melanoma and colon cancer110. In addition, priming of T cells by IL-18 activates NF-κB, augments STAT4 activation by IL-12 and enables TCR-independent production of IFNγ18. Despite its ability to suppress IL-12 production and resolve inflammation, IL-10 also potently induces CTL cytotoxicity towards tumours with potential for clinical application111.
The plasticity of the interleukin-dependent T cell transcriptional profile is essential for tailoring immune responses and necessary effector mechanisms112. TH9 cells and their prototypical cytokine IL-9, despite being closely related to the TH2 cell lineage, which typically antagonizes TH1-type responses, can serve as persistent antitumour effectors that resist exhaustion and can be exploited in cellular therapy113. TH9 cells are typically induced by IL-4 and TGFβ, and experiments have shown that IL-1β can replace TGFβ to promote the TH9 cell lineage. TH9 cells induced that way mounted a persistent response and possessed potent antitumoural properties, secreting IL-9, IL-21 and granzyme B114,115,116,117. IL-21, besides its typical role in B cell responses, enhances cytotoxicity and increases persistence of NK cells and CTLs in tumours as demonstrated in a melanoma mouse model of ACT118,119. Secretion of IL-21 can also be induced in T cells by IL-27, and limits differentiation of Treg cells, enhances proliferation of NK cells and CD8+ T cells but also triggers the exhaustion programme in these cells as also recently investigated in a mouse model of melamoma120,121. TH2 cells, typically seen as antagonists to antitumoural TH1 cell responses, can cause remodelling of tumour vasculature, thus leading to tumour starvation and regression122.
Cancer immune evasion
Despite the potent action of cancer immunosurveillance and immunoediting, malignant cells may evolve to evade antitumoural responses and exploit extrinsic immunoinhibitory mechanisms for progression6,123. In this way, they may secrete substances that enable domination of cell types that suppress antitumour immunity, such as Treg cells, ILC3s, TH17 cells, TH2 cells, M2 macrophages and myeloid-derived suppressor cells (MDSCs), and induce anergy of antitumour cells through their metabolic reprogramming, tipping the balance towards cancer progression.
Treg cells
High-affinity IL-2 signalling together with TGFβ induces Foxp3. FOXP3 drives Treg cell differentiation and IL-10 production, thereby establishing an immunosuppressive TME4,8,15. Besides its role in creating a macrophage-dependent tumorigenic niche, IL-33 may directly promote TGFβ-elicited Treg cell differentiation, suppress IFNγ and promote Treg cell stability in the tumour124,125.
The regulatory role of Treg cells in the TME is largely mediated by the secretion of IL-10, IL-35 and TGFβ126. IL-10 is anti-inflammatory and relies on its ability to suppress B lymphocyte-induced maturation protein 1 (BLIMP1)-dependent cytokine secretion and regulate cytotoxic effector function, whereas IL-35 induces inhibitory surface receptors (including PD1, lymphocyte-activation gene 3 protein (LAG3), T cell immunoglobulin and mucin domain-containing protein 3 (TIM3), T cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibitory motif (ITIM) domains (TIGIT) and 2B4 (also known as CD244)) and limits the formation of T cell memory as demonstrated in mouse models of melanoma126. Mechanistically, IL-10 may also directly reduce TCR sensitivity upon increased cell membrane glycosylation127. Treg cells further limit the availability of IL-2 to CD4+ T cells in the TME, causing deficiency of granzyme B expression in these cells and uncontrolled tumour outgrowth as demonstrated in a mouse sarcoma model128.
TH17-type responses and myeloid suppressors
In response to IL-1β stimulation, TH17 cells and γδ T cells produce IL-17, which recruits a large number of immunosuppressive granulocytes (reviewed in60,129), which was demonstrated to promote lung metastasis in a murine breast cancer model130. Moreover, in lung cancer, IL-23 converts group 1 innate lymphoid cells into IL-17-producing ILC3s that promote IL-17 mediated tumour cell proliferation131. Additionally, tumour-derived IL-8 (also known as CXCL8) provides a chemotactic signal for myeloid cells in patients with breast cancer and confers resistance to immunotherapy132,133. Similarly, IL-18 produced upon inflammasome activation drives generation of MDSCs in multiple myeloma, conferring a mechanism of immunosuppression134.
TH2-type responses
Tumour and stromal cells may secrete factors to promote TH2 cell and M2 macrophage polarization, which suppress antitumoural TH1 cell polarization and responses135,136. In the same way, IL-33-induced group 2 innate lymphoid cells (ILC2) were demonstrated to antagonize NK cell function in a mouse model of melanoma137. In turn, IL-33, secreted by cancer-associated fibroblasts in breast cancer, is a potent enhancer of ILC2 and TH2-type responses, and may induce TCR-independent secretion of IL-13 and recruitment of immunosuppressive granulocytes58,135. Moreover, IL-33 activates intrinsic signalling in Treg cells necessary for their immunosuppressive action in cancer124.
Immune cell metabolic reprogramming
Cancer cells can induce metabolic reprogramming of immune cells in the TME and systemic changes in metabolism, which can induce the transition from proinflammatory to immunosuppressive responses53. A glycolytic switch in effector T cells dependent on NF-κB-inducing kinase (NIK) signalling was recently demonstrated as pivotal for antitumour immunity in a mouse melanoma model138. The accumulation of glycolysis-derived lactate in the TME can increase oxidative phosphorylation and together with IL-4 induces anti-inflammatory reprogramming (involving M2 polarization of macrophages and secretion of IL-10 and TGFβ)53. In addition, accumulation of lactate and depletion of amino acids and glucose in the TME in combination with TGFβ signalling can induce Treg cell polarization and amplify immunosuppression and T cell exhaustion, thereby rendering tumours resistant to immunotherapy (reviewed in53). In pancreatic ductal adenocarcinoma, it was shown that stromal cells released IL-6, which increased glycolysis and lactate efflux from the tumour cells, leading to M2 polarization of macrophages and reduction of the efficacy of anti-PD1 treatment139.
Hence, the accumulation of tumour-derived substances together with chronic inflammatory responses renders antitumour immunity unable to restrain tumour growth and progression, resulting in an uncontrolled tumour outgrowth and distant metastasis and limiting the efficacy of targeted therapies.
Interleukins in cancer therapy
As some interleukins regulate immunosurveillance and thereby tumour control, clinicians have tried to utilize them for cancer therapy in the past few decades. However, cancer treatment by interleukins proved to be challenging as it has not lived up to its expectations in terms of effectiveness, while also encountering serious toxic effects in some cases. Because interleukins also nurture cancer progression, neutralization of their activity has emerged as a potential avenue.
There are reports suggesting several interleukins as biomarkers74,140,141,142,143, but so far no interleukin has been implemented as a biomarker in the US Food and Drug Administration guidelines for oncologic therapeutics, and their potential overall value has been extensively reviewed in ref.143.
Here, we summarize recent advances in the translation of therapeutic exploitation of interleukins, covering therapeutic neutralization of interleukins as well as the use of their recombinant and engineered forms (Fig. 4).

a | Recombinant and engineered cytokines. Increased persistence: prolonging the half-life and controlling toxic effects by the progressive release of the active drug from conjugated polymers or Fc tags. Targeted toxicity: interleukin–toxin fusion proteins target the toxin to cells bearing the interleukin receptor, leading to cell death. Gene therapy: to avoid systemic toxic effects, the interleukin is expressed directly at the tumour site. Selective receptor affinity: interleukins can be engineered to alter their receptor affinity, thereby increasing efficacy or reducing side effects. Targeted interleukin delivery: by coupling of interleukins to tumour-targeting antibodies (Abs), they reach higher concentrations at the tumour site while decreasing side effects associated with high systemic concentrations. b | Complementing adoptive cell therapy (ACT). T cells redirected for antigen-unrestricted cytokine-initiated killing (TRUCKs): expression of interleukins by ACT cells leads to an accumulation of the interleukin at the tumour site, thereby avoiding systemic toxicity and mounting non-ACT immune responses by the activation of endogenous T cells (tumour-infiltrating T cells (TILs)) for targeted cancer cell killing, as well as macrophages, which can also mount an innate immune response against cancer cells that do not express the antigen. Sensitizing chimeric antigen receptor (CAR) T cells to interleukins: expression of interleukin receptors (for example, IL-7R) on ACT cells increases the likelihood of signalling at low interleukin concentrations. Dendritic cell (DC) vaccine adjuvant: DC vaccination can be accompanied by therapy with interleukins (for example, IL-2), enhancing the activation of DC-primed T cells. ACT adjuvant: increasing persistence, survival, proliferation and activation of ACT by interleukin therapy (for example, IL-2 or IL-15).
Therapeutic neutralization
Interleukin-1
The Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) provided the strongest evidence for the potential of IL-1-neutralization in cancer so far. The study enrolled patients with stable coronary artery disease receiving the standard of care, but with persistent elevation of the level of high-sensitivity C-reactive protein (hsCRP). Patients were treated with the anti-IL-1β antibody canakinumab and did not have a diagnosis of cancer at the time of enrolment. Canakinumab not only reduced hsCRP levels but also significantly lowered overall cancer mortality compared with the placebo group, especially concerning lung cancer144. This finding likely reflected the ability of IL-1β to impede invasion and metastasis of existing subclinical cancers rather than cancer initiation due to the median duration of the trial of 3.7 years145. In support of this, a substantial number of patients with diagnosed lung cancer already had circulating tumour DNA at the time of enrollment146. These findings spawned several clinical trials blocking IL-1β and other inflammatory interleukins in cancer with expected primary completion dates until 2024.
In 2018, a phase III trial of canakinumab as monotherapy compared with placebo started in patients with non-small-cell lung cancer (NCT03447769)147. Similarly, a phase II trial studying canakinumab monotherapy in myelodysplastic syndrome and chronic myelogenous leukaemia was initiated in 2020 (NCT04239157)148. Also, in 2018, a phase III trial using canakinumab in combination with pembrolizumab and chemotherapy in non-small-cell lung cancer (NCT03631199)149, and in 2019 a phase III trial with canakinumab and chemotherapy alone (NCT03626545) were started150. Several additional studies were initiated between 2016 and 2020 (see Supplementary Table 1).
Anakinra is an IL-1R antagonist in clinical trials for cancer therapy. In a phase II trial, 47 patients with smouldering myeloma were treated with anakinra alone for 6 months. If a patient did not achieve a minor response or better, anakinra was combined with dexamethasone. Three of 47 patients achieved a minor response to anakinra alone, highlighting the need for a combinatorial approach. Upon addition of dexamethasone, five partial responses and four minor responses were achieved151,152. Anakinra is also under investigation in several other clinical trials in multiple cancer types as monotherapy and in combination therapy (see Supplementary Table 1).
Interleukin-6
While the anti-IL-6 antibody siltuximab reduced tumour growth in a xenografted cholangiocarcinoma model153, it achieved only small effects in multiple myeloma as monotherapy154, in combination with the proteasome inhibitor bortezomib155 and with bortezomib, the cytostatic agent melphalan and the anti-inflammatory glucocorticoid prednisone156. Siltuximab was also not effective in solid tumours as monotherapy. In prostate cancer, the best outcome achieved was stable disease157, in renal cell carcinoma (RCC) the best outcome was a single partial response158 and no clinical efficacy was observed in a group of mixed solid tumours159. Furthermore, several clinical trials observed treatment-associated side effects, such as higher rates of infections upon siltuximab treatment154,155,156, which overall does not support a benefit of siltuximab therapy.
IL-6 can protect cancer cells from therapy-induced DNA damage, oxidative stress and apoptosis and is known to induce immunosuppression and maintains highly therapy-resistant cancer stem cell populations74. Thus, IL-6 neutralization might reverse resistance to conventional cancer therapy. In combination with chemotherapy, the anti-IL-6R antibody tocilizumab yielded first positive results160. However, combining IL-6 neutralization with immune checkpoint inhibitors (ICIs) has engendered controversy. While IL-6 neutralization might enable patients to continue receiving ICIs by countering cytokine release syndrome (see Box 2), IL-6 inhibition was shown to suppress the expression of PD1 and programmed death ligand 1 (PDL1). This might decrease the effectiveness of ICIs161. As preclinical studies mainly demonstrate asynergy between IL-6 neutralization and ICIs, several clinical trials have been initiated to test the combination in a clinical setting (see Supplementary Table 1).
Publications highlight the potential of blocking IL-6 trans signalling in hepatocellular carcinoma162 and colorectal cancer163 by comparing carcinogenesis in wild-type mice and mice transgenic for soluble gp130Fc, which consists of gp130 fused to a human IgG1 domain164. These data may lead to clinical trials for olamkicept (also known as soluble gp130Fc) in patients with cancer in the future.
Interleukin-8
HuMax-IL-8 is an antibody neutralizing IL-8. While no objective responses were observed in monotherapy, no dose-limiting toxic effects were observed, and 73% of patients with solid tumours had stable disease165. IL-8 induces resistance to several chemotherapeutic agents and ICIs, as summarized elsewhere133,140,166. Therefore, several clinical trials are exploring the effect of HuMax-IL-8 in combination with nivolumab (NCT03400332 (ref.167), NCT04123379 (ref.168) and NCT04050462 (ref.169)), nivolumab and degarelix (NCT03689699 (ref.170)) and the anti-PD1 antibody nivolumab with stereotactic body radiotherapy (NCT04572451 (ref.171)).
Interleukin-17
Blocking IL-17 in a murine model of gastric cancer led to a reduction of tumour growth and in combination with anti-PD1 therapy to tumour rejections in 80% of the mice172. It also decreased the development of prostate cancer in a preclinical mouse model173. IL-17 can accelerate the progression of multiple myeloma174, and another study showed that IL-17 neutralization indeed can lead to a reduction in multiple myeloma growth175. A clinical trial that started in 2017 (NCT03111992 (ref.176)) is now exploring the efficacy of the anti-IL-17A antibody CJM112 in multiple myeloma.
Interleukin-23
In the context of castration-resistant prostate cancer, MDSC-derived IL-23 was shown to activate the androgen receptor pathway, thereby supporting survival and proliferation of cancer cells under androgen-deprived conditions. Neutralization with an anti-IL-23 antibody significantly reduced tumour growth and extended survival of mice177. This suggests anti-IL-23 therapy might support anti-androgen therapy regimens. To assess the potential of this, the anti-IL-23 antibody tildrakizumab is now being tested in a phase I/II trial in patients with castration-resistant prostate cancer in combination with abiraterone acetate, an inhibitor of androgen synthesis (NCT04458311 (ref.178)).
Interleukin-33
It was shown in a preclinical study that IL-33 may directly act on tumour cells, inducing polyploidy and rapid proliferation, and causing therapy resistance. Use of a specific IL-33-blocking antibody could restore the efficacy of anti-PD1 therapy in an otherwise ICI-resistant subclone of the B16F10 melanoma model179.
Recombinant and engineered interleukins
IL-2 was the first interleukin to be approved for cancer treatment, although its use entails major safety concerns180. Recent advances focusing on modifying interleukins to reduce toxicity and increase the efficacy of treatment with recombinant and/or engineered interleukins might help to overcome this issue.
Interleukin-2
Although high-dose therapy with IL-2 induced durable complete responses in some patients with melanoma and RCC, it comes with potentially life-threatening adverse events. Therefore, low-dose IL-2 schedules were evaluated in several trials. However, in low abundance, IL-2 binds preferentially to its high-affinity receptor expressed on Treg cells, potentially leading to immune evasion. Thus, several IL-2 variants with increased affinity for the IL-2Rβ–γc complex expressed by CTLs are in development.
Bempegaldesleukin, a pegylated IL-2 variant, is a non-α IL-2 variant that is already in clinical trials. In addition to increasing its half-life, the polyethylene glycol (PEG) residues coupled to IL-2 block its binding site for the high-affinity IL-2Rα subunit. Over time, the PEG groups are lost and continually release the active form of bempegaldesleukin, thereby minimizing the risk of overdosing181. Bempegaldesleukin was evaluated as monotherapy in a phase I clinical trial in patients with metastatic solid tumours, predominantly consisting of patients with melanoma or RCC. The best overall responses observed were stable disease182, which is disappointing in comparison with the 7% and 9% durable complete responses achieved by conventional IL-2 therapy in melanoma and RCC, respectively180,182. Nevertheless, bempegaldesleukin showed a favourable safety profile, encouraging combination with ICIs. A phase I study of bempegaldesleukin in combination with nivolumab in metastatic solid tumours achieved a complete response rate of 19%183. Several other clinical trials in combination with ICIs, including phase III clinical trials in patients with bladder cancer (NCT04209114 (ref.184)), RCC (NCT03729245 (ref.185)) or melanoma (NCT03635983 (ref.186)), are currently ongoing. Other non-α IL-2 variants are being tested in clinical trials in combination with ICIs, such as ALKS 4230, a circularly permuted IL-2 and IL-2Rα designed to selectively activate the intermediate-affinity IL-2R. It is tested in four clinical trials in patients with advanced solid tumours or non-cutaneous squamous cell carcinoma of the head and neck alone and in combination with pembrolizumab (see Supplementary Table 1). THOR707 is an engineered non-α IL-2 being tested in patients with solid tumours alone and in combination with an unspecified ICI or with an anti-EGFR antibody (NCT04009681 (ref.187)).
NARA1leukin is a fusion construct that consists of human IL-2 engineered into the anti-IL-2 antibody NARA1. NARA1 thereby permanently masks the CD25-binding site of IL-2 and thus abolishes the CD25-mediated Treg cell development, while mediating potent antitumour responses in a murine melanoma model188.
Another approach is the de novo design of Neo-2/15, a hyperstable interleukin that mimics only the binding site for IL-2Rβ–γc, but otherwise is structurally independent of IL-2 and IL-15. Therefore, it can mediate IL-2 and IL-15 signalling independently of IL-2Rα or IL-15Rα, and showed superior therapeutic activity compared with IL-2 in mouse models of melanoma and colon cancer189.
Several attempts have been made to couple IL-2 to tumour-targeting antibodies to reduce side effects caused by high systemic concentrations while increasing the efficacy by concentrating IL-2 at the tumour. Some of those IL-2-antibody fusion constructs reached the stage of clinical trials190. However, no breakthrough results have emerged so far (see Supplementary Table 1).
Interleukin-10
IL-10 has anti-inflammatory as well as CTL-stimulating properties, which led to its exploration as an anticancer therapeutic. The PEGylated IL-10 variant pegilodecakin can induce systemic immune activation, an increase of the levels of immunostimulatory cytokines and a reduction of TGFβ levels in patients with solid cancer. Additionally, partial responses and prolonged stable disease were observed upon pegilodecakin treatment191. In a follow-up trial, in a mixed group of solid cancers, expansion and activation of intratumoural CTLs in response to treatment were observed192. The first clinical data on the combination of pegilodecakin with ICIs were promising as well193; however, two follow-up trials testing pegilodecakin in combination with nivolumab (NCT03382912 (ref.194)) and the anti-PD1 antibody pembrolizumab (NCT03382899 (ref.195)) were terminated early due to an unfavourable risk–benefit ratio.
CmAb-(IL-10)2, a bispecific fusion protein with one arm derived from the anti-EGFR antibody cetuximab and the other arm containing an IL-10 dimer, is an agent still in preclinical investigation. It has shown superior antitumour activity compared with IL-10 fused to a non-tumour-targeting antibody196.
Interleukin-12
IL-12 is an interleukin that demonstrates striking immune activation and tumour control but causes severe adverse effects with the doses necessary to induce an antitumour effect. However, targeted delivery strategies to achieve high concentrations at the tumour site while avoiding high systemic amounts led to a renaissance of IL-12 cancer therapy197.
Gene therapy allows better control of the localization and amount of interleukins in a therapy regimen. Tavokinogene telseplasmid, an IL-12-encoding plasmid electroporated into melanoma lesions, achieved an overall response rate of 36% and a complete response rate of 18% in a phase II trial. Of note, 46% of patients also showed regression in at least one lesion and 25% had a net regression in all lesions into which tavokinogene telseplasmid had not been injected198, suggesting the induction of systemic antitumour immunity. In combination with pembrolizumab, the overall response rate was increased to 41%, with a complete response rate of 36%199. Several follow-up trials were initiated between 2017 and 2020 in triple-negative breast cancer (NCT03567720 (ref.200)), head and neck squamous cell carcinoma (NCT03823131 (ref.201)) and melanoma (NCT04526730 (ref.202) and NCT03132675 (ref.203)).
Other therapeutic strategies using IL-12 have been extensively discussed elswhere107,197, and recent clinical trials are listed in Supplementary Table 1.
CBD–IL-12 is a fusion protein consisting of IL-12 and a collagen-binding domain. It accumulates in the tumour due to exposed collagen in the disordered tumour vasculature and demonstrates antitumour effects superior to those of IL-12 by a broad activation of the immune system at the tumour site204.
Li et al. designed multifunctional oncolytic nanoparticles with the intent to send several different signals triggering antitumour responses at once. They showed that the composition of nanoparticles containing self-replicating RNA can induce immunogenic cell death of tumour cells, as the contained RNA serves as a signal for pathogen-recognition receptors as well as coding for IL-12 to further enhance the induced immune response. When delivered intratumourally, control of large tumours is achieved in several tumour models205.
Interleukin-15
In contrast to IL-2, IL-15 does not lead to Treg cell expansion, while having similar immunostimulatory properties. However, monotherapy with IL-15 and several engineered variants was ineffective, although there are several promising combination therapies in clinical trials206.
An example of this is ALT-803, consisting of recombinant IL-15 with an N72D mutation linked to the Sushi domain of IL-15Rα fused to an immunoglobulin G (IgG) Fc-fragment. While the N72D mutation increases binding to IL-15Rβ, the Sushi domain provides the IL-15Rα part and the Fc fragment increases its half-life. However, ALT-803 had only a limited effect as monotherapy in patients with solid tumours207 and showed responses in only 19% of patients with haematological malignancies208. Strikingly, in combination with intravesical bacillus Calmette–Guérin (BCG) therapy, complete responses were achieved in all nine patients with bladder cancer209, which led to the expansion of the trial to enrol nearly 600 patients (NCT02138734 (ref.210)).
Other IL-15-based therapeutic strategies include combination with ICIs and tumour-targeting antibodies (Supplementary Table 1); approaches for IL-15 in combination therapy for cancer have been extensively reviewed206,211.
Interleukin-18
IL-18 therapy has shown little preclinical efficacy, potentially due to IL-18BP, a high-affinity IL-18 decoy receptor. Using direct evolution, Zhou et al. developed decoy-resistant IL-18 (DR-18), which maintains signalling potential and unlike IL-18 exerted potent antitumour responses in mice, by supporting effector T cell development, reducing T cell exhaustion and enhancing activity and maturation of NK cells97.
Interleukin-23 and interleukin-36
IL-36γ together with IL-23 can be successfully used in combination with OX40 ligand to promote acute inflammation for efficient tumour control212 and is currently under investigation in a clinical trial (NCT03739931 (ref.213)).
Complementing ACT
Interleukins play a key role in ACT, a breakthrough achievement in cancer therapy in the past decade. Interleukins such as IL-2, IL-7 and IL-15 have been widely used to improve in vitro expansion and differentiation of adoptive cells and to complement ACT therapy in humans by co-administration or by genetically engineering them into the transferred cells.
Interleukin-2
Treatment of patients with IL-2 to support ACT has been studied with variable success in several cancer types. For example, IL-2 was combined with tumour-infiltrating lymphocytes in melanoma214. While some of those trials yielded benefits, for example a correlation of chimeric antigen receptor (CAR) T cell engraftment with IL-2 concentrations215, this approach still faces toxicity problems similar to those of using interleukins alone.
A strategy to overcome the toxic effect of systemic IL-2 treatment while retaining its ability to support ACT therapy is orthogonal IL-2–IL-2R pairs. The designed orthogonal IL-2 cannot bind to the wild-type IL-2R but transfers native IL-2 signalling when binding to the designed orthogonal IL-2Rβ, which was transduced into T cells. This creates a system where only the ACT product reacts to the orthogonal IL-2 treatment and systemic toxicity is avoided. Orthogonal IL-2 showed efficacy in a mouse melanoma model with the enhanced expansion of the orthogonal IL-2Rβ ACT product and negligible toxic effects. These data might indicate a potential advantage for clinical use in ACT216.
Interleukin-8
A quite different approach is to use tumour-associated interleukins, such as IL-8, to improve infiltration of CAR T cells into the tumour by overexpressing their respective receptors. The expression of the IL-8 receptors CXCR1 or CXCR2 in CAR T cells markedly enhanced migration and persistence in the tumour and induced complete regression and long-lasting immunological memory in preclinical models of glioblastoma, ovarian cancer and pancreatic cancer217.
Interleukin-12
Another strategy currently pursued in several trials is the expression of certain interleukins by cellular therapy products. In this manner, the interleukins should be expressed only locally at the tumour site, thereby avoiding systemic toxic effects and recruiting additional immune cells to establish immune responses against CAR antigen-negative tumour cell populations. In the case of T cells, this approach was named ‘T cells redirected for antigen-unrestricted cytokine-initiated killing’ (TRUCKs). The first interleukin used as a proof of concept for TRUCKs was IL-12 (ref.218), and other interleukins, such as IL-7, IL-15 and IL-23, have since been used to engineer T cells as well.
In preclinical testing, IL-12 expression by CAR T cells led to the recruitment and activation of macrophages critical for the elimination of cancer cells with antigen loss that would normally escape from CAR-induced killing218. The first in-human trial was a study expressing IL-12 under the control of a nuclear factor of activated T cells (NFAT)-inducible promotor in tumour-infiltrating lymphocytes from patients with metastatic melanoma. However, the study had to be terminated due to severe toxic effects likely caused by excessive IL-12 release219. Recently, several clinical trials were started using IL-12-expressing CAR T cells, trying to avoid uncontrolled IL-12 release by the more restricted activation of CARs compared with polyclonal tumour-infiltrating lymphocytes and by using safety switches (see Supplementary Table 1).
Another approach is to induce only transient expression of IL-12 by electroporating IL-12 mRNA into ovalbumin-specific OT-I TCR transgenic CD8+ T cells. Injected intratumorally, but not intravenously, these cells led to complete rejections of lesions into which they had been injected and distant lesions in a B16-ovalbumin tumour model. The study authors claim that the transient expression was likely helpful in restricting IL-12-induced adverse events but do not show any data to prove this assumption220.
A recent clinical trial using intratumourally injected activated DCs showed that successful DC therapy was associated with higher expression of IL-12 subunit p40 and IL-8 (ref.221). This may indicate a favourable interplay between DC therapy and interleukin therapy.
Interleukin-7
Over time, the idea to express interleukins in the ACT product has spread from IL-12 to other interleukins. IL-7 and CC-chemokine ligand 19 (CCL19) are essential for the maintenance of T cell zones in lymphoid organs. In a preclinical setting, CAR T cells engineered to express IL-7 and CCL19 were superior in tumour killing in comparison with conventional CAR T cells. Histopathological analysis showed increased infiltration of DCs and CAR T cells as well as endogenous T cells into the tumour. The depletion of endogenous T cells dampened the therapeutic effect of IL-7 and CCL19-expressing CAR T cells. Together with the observation of abundance of memory CAR T cells and endogenous T cells in the spleen, this suggests co-expression of IL-7 and CCL19 by CAR T cells can initiate a CAR-independent immune response222. Four clinical trials evaluating the effect of CAR T cells overexpressing IL-7 and CCL19 were initiated between 2017 and 2020 (see Supplementary Table 1).
Interleukin-15
Preclinical studies showed that the expression of IL-15 in NKT cells led to a decreased expression of exhaustion markers, enhanced in vivo persistence, increased localization at the tumour site and improved tumour control223. In CAR T cells, membrane-bound IL-15 led to an increase in the abundance of T memory stem cells with long persistence and potent rejection of CD19+ leukaemia224. This led to a collection of ongoing clinical trials engineering IL-15 into CAR T cells and CAR NKT cells (Supplementary Table 1). An interim analysis showed that anti-GD2 CAR NKT cells expressing IL-15 did not cause any dose-limiting toxic effects in three heavily pretreated children with neuroblastoma. CAR NKT cells were shown to be present at the tumour sites, and one patient achieved regression of bone metastatic leasions225.
Recently, a novel approach to reduce the toxicity of IL-15 was reported. Tang et al. designed a cell surface-conjugated nanogel that can be loaded with large quantities of protein drugs226. The nanogel is designed to release its cargo upon an increase in T cell surface reduction potential, which is induced by T cell–antigen encounter. Thereby, the drug release is limited to places of TCR activation, predominantly the tumour site. The study authors also show that the system is translatable to CAR T cells. The potential to expand this method to other cytokines and protein drugs, maybe even combinations thereof, makes this system especially noteworthy, although the approach is inherently self-limiting as the nanogels will be depleted of their drugs over time226.
Interleukin-23
IL-23 is a very recently reported alternative to potentially toxic interleukins such as IL-12 and IL-15. A preclinical study showed that engineering CAR T cells with IL-23 subunit p40 led to autocrine IL-23 signalling promoting the selective proliferation of activated T cells with increased antitumour capacity. Additionally, side effects seemed diminished compared with use of IL-18 or IL-15 (ref.227).
Outlook
Limited understanding of the impact of interleukin therapy on carefully balanced systems in the TME, as well as the Janus-like functions of some interleukins, rendered the therapeutic exploitation of interleukins difficult to predict. This caused much disappointment in the field of interleukin therapy for cancer, as life-threatening side effects occurred and most therapeutic strategies did not live up to the expectations concerning effectiveness.
One example is the targeting of one of the first interleukins to be discovered (IL-1β), in which case neutralization could yield either disease exacerbation or reduction. Recent developments have pointed out that neutralization of this key pathway at a time point where the disease is already present might have therapeutic activity in patients with cancer. Also, IL-2 monotherapy in melanoma and RCC achieved durable complete responses, highlighting the potential of interleukin therapy.
However, in general, interleukin monotherapy still faces major limitations, and new strategies have emerged recently to tackle those problems. Thus, the trend is moving away from applying native forms of interleukins and is going towards sophisticated engineering approaches. Most strategies investigated in the past few years involved altering the receptor affinity for only the target selected cell populations or designing tumour-targeting fusion constructs to increase the efficacy and reduce systemic toxic effects.
Additionally, to improve the application of interleukin therapy, combination approaches are gaining more and more importance. New insights into how interleukins shape tissue repair, cancer progression and immune evasion will greatly help to identify how interleukins are involved in the resistance against established therapies such as chemotherapy or ICI treatment. Thereby, interleukin therapy could unleash the unused potential of other therapeutics and thus help otherwise therapy-resistant patients to benefit therefrom.
Another hypothesis on the low efficacy of interleukin monotherapy is the idea that delivering a single immunostimulatory signal is not sufficient to mount a durable immune response that can eliminate the tumour. Hence, strategies combining several immunostimulatory signals have been fast approaching. A strategy often pursued in this regard is the combination of interleukin therapy to support ACT, either by combining ACT with regular interleukin therapy or by the development of TRUCKs. ACT showed remarkable success in haematological malignancies but lacks efficiency in solid tumours due to poor infiltration and proliferation of the ACT product as well as a highly immunosuppressive TME impairing its function. Interleukins may support ACT by overcoming all of those hurdles, and the number of clinical trials in this area highlights the expectations set by the idea.
Conclusion
In conclusion, interleukins comprise key elements that orchestrate the TME and govern tumour–immune cell crosstalk as they are important players in large cytokine networks. Although interleukin or interleukin-targeted therapy still has significant hurdles to overcome on its path to the clinic, the intensive foundational research conducted on interleukins in cancer biology in the past decade will help us to better understand the mechanisms of interleukin therapy and thereby contribute to the development of new strategies. Also, there is a rich pipeline of exciting new preclinical and clinical studies on interleukin therapy for cancer that will yield interesting results in the coming years.
References
Dunn, G. P., Old, L. J. & Schreiber, R. D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 22, 329–360 (2004).
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J. & Schreiber, R. D. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 3, 991–998 (2002). Dunn and colleagues formulate a theory of individual cancer evolution under immunological pressure.
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Bruni, D., Angell, H. K. & Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer https://doi.org/10.1038/s41568-020-0285-7 (2020).
Pages, F. et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 391, 2128–2139 (2018). This research validates the prognostic value of colon cancer classification according to the immune status of the tumour.
Mascaux, C. et al. Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature 571, 570–575 (2019).
Yuzhalin, A. E. & Kutikhin, A. G. Interleukins in Cancer Biology: their Heterogeneous Role (Academic Press, 2014).
Waldmann, T. A. Cytokines in cancer immunotherapy. Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a028472 (2018).
Dinarello, C. A. The IL-1 family of cytokines and receptors in rheumatic diseases. Nat. Rev. Rheumatol. 15, 612–632 (2019).
Dinarello, C. A. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 29, 317–329 (2010). Here IL-1 blockade is proposed for the first time as the means to tame chronic inflammation and halting tumor progression.
Elinav, E. et al. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 13, 759–771 (2013).
Mantovani, A., Dinarello, C. A., Molgora, M. & Garlanda, C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity 50, 778–795 (2019).
Lamkanfi, M. & Dixit, V. M. Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 (2014).
Zheng, D., Liwinski, T. & Elinav, E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov. 6, 36 (2020).
Taniguchi, S. et al. Tumor-initiating cells establish an IL-33-TGF-beta niche signaling loop to promote cancer progression. Science https://doi.org/10.1126/science.aay1813 (2020). A mechanism by which tumour-initiating cells create a favourable environment that fosters tumour growth is demonstrated here.
Andersson, P. et al. Molecular mechanisms of IL-33-mediated stromal interactions in cancer metastasis. JCI Insight https://doi.org/10.1172/jci.insight.122375 (2018).
Tu, S. et al. Overexpression of interleukin-1 beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 14, 408–419 (2008).
Whitley, S. K. et al. IL-1R signaling promotes STAT3 and NF-kappaB factor recruitment to distal cis-regulatory elements that regulate Il17a/f transcription. J. Biol. Chem. 293, 15790–15800 (2018).
Dmitrieva-Posocco, O. et al. Cell-type-specific responses to interleukin-1 control microbial invasion and tumor-elicited inflammation in colorectal cancer. Immunity 50, 166–180 e167 (2019). Dmitrieva-Posocco and colleagues demonstrate differential cell type-specific immune responses to IL-1 in chronic inflammation and colorectal cancer.
Grivennikov, S. I. et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491, 254–258 (2012).
Jin, C. et al. Commensal microbiota promote lung cancer development via gammadelta T cells. Cell 176, 998–1013 e1016 (2019). This publication delineates a mechanism by which the commensal microbiota may trigger a cascade of protumorigenic interleukin-mediated responses in the lung.
Voigt, C. et al. Cancer cells induce interleukin-22 production from memory CD4+ T cells via interleukin-1 to promote tumor growth. Proc. Natl Acad. Sci. USA 114, 12994–12999 (2017).
Dudakov, J. A., Hanash, A. M. & van den Brink, M. R. Interleukin-22: immunobiology and pathology. Annu. Rev. Immunol. 33, 747–785 (2015).
Ngo, V. L. et al. A cytokine network involving IL-36gamma, IL-23, and IL-22 promotes antimicrobial defense and recovery from intestinal barrier damage. Proc. Natl Acad. Sci. USA 115, E5076–E5085 (2018).
Gronke, K. et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 566, 249–253 (2019). Gronke and colleagues define a mechanism by which IL-22 prevents the malignant transformation of the intestinal epithelium.
Huber, S. et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 491, 259–263 (2012).
Perez, L. G. et al. TGF-beta signaling in Th17 cells promotes IL-22 production and colitis-associated colon cancer. Nat. Commun. 11, 2608 (2020).
Kobold, S. et al. Interleukin-22 is frequently expressed in small- and large-cell lung cancer and promotes growth in chemotherapy-resistant cancer cells. J. Thorac. Oncol. 8, 1032–1042 (2013).
Kryczek, I. et al. IL-22+CD4+ T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 40, 772–784 (2014).
Katara, G. K. et al. Interleukin-22 promotes development of malignant lesions in a mouse model of spontaneous breast cancer. Mol. Oncol. 14, 211–224 (2020).
Khosravi, N. et al. IL-22 promotes Kras-mutant lung cancer by induction of a protumor immune response and protection of stemness properties. Cancer Immunol. Res. 6, 788–797 (2018).
Perusina Lanfranca, M. et al. Interleukin 22 signaling regulates acinar cell plasticity to promote pancreatic tumor development in mice. Gastroenterology 158, 1417–1432 e1411 (2020).
Lu, S. W. et al. IL-20 antagonist suppresses PD-L1 expression and prolongs survival in pancreatic cancer models. Nat. Commun. 11, 4611 (2020).
Huynh, J., Chand, A., Gough, D. & Ernst, M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat. Rev. Cancer 19, 82–96 (2019).
Heinrich, P. C. et al. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374, 1–20 (2003).
Jones, S. A. & Jenkins, B. J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 18, 773–789 (2018).
Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 33, 127–148 (2021).
Putoczki, T. L. et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 24, 257–271 (2013). This work defines a dominant role of IL-11 as a driver of procarcinogenic chronic inflammation in the intestine.
Grivennikov, S. I. IL-11: a prominent pro-tumorigenic member of the IL-6 family. Cancer Cell 24, 145–147 (2013).
Lokau, J. et al. Proteolytic cleavage governs interleukin-11 trans-signaling. Cell Rep. 14, 1761–1773 (2016).
Dong, C., Davis, R. J. & Flavell, R. A. MAP kinases in the immune response. Annu. Rev. Immunol. 20, 55–72 (2002).
Atsumi, T. et al. Inflammation amplifier, a new paradigm in cancer biology. Cancer Res. 74, 8–14 (2014).
Hunter, C. A. & Jones, S. A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 16, 448–457 (2015).
Rose-John, S. Interleukin-6 family cytokines. Cold Spring Harb. Perspect Biol. https://doi.org/10.1101/cshperspect.a028415 (2018).
Ernst, M. et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J. Clin. Invest. 118, 1727–1738 (2008).
Hu, B. et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl Acad. Sci. USA 110, 9862–9867 (2013).
Bollrath, J. et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 15, 91–102 (2009).
Phesse, T. J. et al. Partial inhibition of gp130-Jak-Stat3 signaling prevents Wnt-beta-catenin-mediated intestinal tumor growth and regeneration. Sci. Signal. 7, ra92 (2014).
Wu, Y. S. et al. Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim. Biophys. Acta Gen. Subj. 1861, 296–306 (2017).
van Duijneveldt, G., Griffin, M. D. W. & Putoczki, T. L. Emerging roles for the IL-6 family of cytokines in pancreatic cancer. Clin. Sci. 134, 2091–2115 (2020).
Caetano, M. S. et al. IL-6 blockade reprograms the lung tumor microenvironment to limit the development and progression of Kras-mutant lung cancer. Cancer Res. 76, 3189–3199 (2016).
Tannahill, G. M. et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496, 238–242 (2013).
Biswas, S. K. Metabolic reprogramming of immune cells in cancer progression. Immunity 43, 435–449 (2015).
Knochelmann, H. M. et al. IL-6 fuels durable memory for Th17 cell-mediated responses to tumors. Cancer Res. 80, 3920–3932 (2020).
Liu, Y. et al. NCR− group 3 innate lymphoid cells orchestrate IL-23/IL-17 axis to promote hepatocellular carcinoma development. EBioMedicine 41, 333–344 (2019).
Teng, M. W. et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 21, 719–729 (2015).
Langowski, J. L. et al. IL-23 promotes tumour incidence and growth. Nature 442, 461–465 (2006).
Guo, L. et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc. Natl Acad. Sci. USA 106, 13463–13468 (2009).
Wu, L. et al. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4-ERK5 axis. J. Exp. Med. 212, 1571–1587 (2015).
Zhao, J., Chen, X., Herjan, T. & Li, X. The role of interleukin-17 in tumor development and progression. J. Exp. Med. https://doi.org/10.1084/jem.20190297 (2020).
Zepp, J. A. et al. IL-17A-induced PLET1 expression contributes to tissue repair and colon tumorigenesis. J. Immunol. 199, 3849–3857 (2017).
Chen, X. et al. IL-17R-EGFR axis links wound healing to tumorigenesis in Lrig1+ stem cells. J. Exp. Med. 216, 195–214 (2019).
Merrouche, Y. et al. IL-17E synergizes with EGF and confers in vitro resistance to EGFR-targeted therapies in TNBC cells. Oncotarget 7, 53350–53361 (2016).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Kitamura, T., Qian, B. Z. & Pollard, J. W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 15, 73–86 (2015).
MacCarthy-Morrogh, L. & Martin, P. The hallmarks of cancer are also the hallmarks of wound healing. Sci. Signal. https://doi.org/10.1126/scisignal.aay8690 (2020).
Voronov, E. et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl Acad. Sci. USA 100, 2645–2650 (2003).
Moossavi, M., Parsamanesh, N., Bahrami, A., Atkin, S. L. & Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 17, 158 (2018).
Lee, H. E. et al. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci. Rep. 9, 12277 (2019).
Deswaerte, V. et al. Inflammasome adaptor ASC suppresses apoptosis of gastric cancer cells by an IL-18-mediated inflammation-independent mechanism. Cancer Res. 78, 1293–1307 (2018).
Park, S., Cheon, S. & Cho, D. The dual effects of interleukin-18 in tumor progression. Cell Mol. Immunol. 4, 329–335 (2007).
Terme, M. et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res. 71, 5393–5399 (2011).
Guo, X. et al. Blocking NF-kappaB is essential for the immunotherapeutic effect of recombinant IL-18 in pancreatic cancer. Clin. Cancer Res. 22, 5939–5950 (2016).
Kumari, N., Dwarakanath, B. S., Das, A. & Bhatt, A. N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 37, 11553–11572 (2016).
De Simone, V. et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 34, 3493–3503 (2015).
Lim, C. & Savan, R. The role of the IL-22/IL-22R1 axis in cancer. Cytokine Growth Factor. Rev. 25, 257–271 (2014).
Markota, A., Endres, S. & Kobold, S. Targeting interleukin-22 for cancer therapy. Hum. Vaccin. Immunother. 14, 2012–2015 (2018).
Brabletz, T., Kalluri, R., Nieto, M. A. & Weinberg, R. A. EMT in cancer. Nat. Rev. Cancer 18, 128–134 (2018).
Liu, X. et al. Epithelial-type systemic breast carcinoma cells with a restricted mesenchymal transition are a major source of metastasis. Sci. Adv. 5, eaav4275 (2019).
Dongre, A. & Weinberg, R. A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 20, 69–84 (2019).
Li, R. et al. Chronic IL-1beta-induced inflammation regulates epithelial-to-mesenchymal transition memory phenotypes via epigenetic modifications in non-small cell lung cancer. Sci. Rep. 10, 377 (2020).
Castano, Z. et al. IL-1beta inflammatory response driven by primary breast cancer prevents metastasis-initiating cell colonization. Nat. Cell Biol. 20, 1084–1097 (2018).
Lee, C. C. et al. Macrophage-secreted interleukin-35 regulates cancer cell plasticity to facilitate metastatic colonization. Nat. Commun. 9, 3763 (2018).
Cao, H. et al. IL-13/STAT6 signaling plays a critical role in the epithelial-mesenchymal transition of colorectal cancer cells. Oncotarget 7, 61183–61198 (2016).
Al-Ismaeel, Q. et al. ZEB1 and IL-6/11-STAT3 signalling cooperate to define invasive potential of pancreatic cancer cells via differential regulation of the expression of S100 proteins. Br. J. Cancer 121, 65–75 (2019).
Chen, D. et al. Interleukin-23 promotes the epithelial-mesenchymal transition of oesophageal carcinoma cells via the Wnt/beta-catenin pathway. Sci. Rep. 5, 8604 (2015).
Gu, K. et al. Interleukin-17-induced EMT promotes lung cancer cell migration and invasion via NF-kappaB/ZEB1 signal pathway. Am. J. Cancer Res. 5, 1169–1179 (2015).
Long, X. et al. IL-8, a novel messenger to cross-link inflammation and tumor EMT via autocrine and paracrine pathways (Review). Int. J. Oncol. 48, 5–12 (2016).
Shankaran, V. et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410, 1107–1111 (2001).
Wang, Y. et al. The IL-15-AKT-XBP1s signaling pathway contributes to effector functions and survival in human NK cells. Nat. Immunol. 20, 10–17 (2019).
Wu, Y., Tian, Z. & Wei, H. Developmental and functional control of natural killer cells by cytokines. Front. Immunol. 8, 930 (2017).
Lazarevic, V., Glimcher, L. H. & Lord, G. M. T-bet: a bridge between innate and adaptive immunity. Nat. Rev. Immunol. 13, 777–789 (2013).
Zhou, X. et al. The deubiquitinase Otub1 controls the activation of CD8+ T cells and NK cells by regulating IL-15-mediated priming. Nat. Immunol. 20, 879–889 (2019).
Vesely, M. D., Kershaw, M. H., Schreiber, R. D. & Smyth, M. J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 29, 235–271 (2011).
Fehniger, T. A. et al. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: implications for the innate immune response. J. Immunol. 162, 4511–4520 (1999).
Gerosa, F. et al. Reciprocal activating interaction between natural killer cells and dendritic cells. J. Exp. Med. 195, 327–333 (2002).
Zhou, T. et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature 583, 609–614 (2020). This publication suggests a decoy-resistant cytokine variant as a way to enable effective IL-18-based cancer immunotherapy.
Dixon, K. O. & Kuchroo, V. K. IL-18: throwing off the shackles to boost anti-tumor immunity. Cell Res. 30, 831–832 (2020).
Molgora, M. et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 551, 110–114 (2017).
Hollande, C. et al. Inhibition of the dipeptidyl peptidase DPP4 (CD26) reveals IL-33-dependent eosinophil-mediated control of tumor growth. Nat. Immunol. 20, 257–264 (2019).
Grisaru-Tal, S., Itan, M., Klion, A. D. & Munitz, A. A new dawn for eosinophils in the tumour microenvironment. Nat. Rev. Cancer 20, 594–607 (2020).
Sheppard, P. et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 4, 63–68 (2003).
Lazear, H. M., Schoggins, J. W. & Diamond, M. S. Shared and distinct functions of type i and type III interferons. Immunity 50, 907–923 (2019).
Zhu, J., Yamane, H. & Paul, W. E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 28, 445–489 (2010).
Testa, U., Pelosi, E. & Castelli, G. CD123 as a therapeutic target in the treatment of hematological malignancies. Cancers https://doi.org/10.3390/cancers11091358 (2019).
Dolgin, E. First CD123-targeted drug approved after wowing in rare cancer. Nat. Biotechnol. 37, 202–203 (2019).
Tugues, S. et al. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 22, 237–246 (2015).
Tait Wojno, E. D., Hunter, C. A. & Stumhofer, J. S. The immunobiology of the interleukin-12 family: room for discovery. Immunity 50, 851–870 (2019).
Garris, C. S. et al. Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-gamma and IL-12. Immunity 49, 1148–1161 e1147 (2018).
Santana Carrero, R. M. et al. IL-15 is a component of the inflammatory milieu in the tumor microenvironment promoting antitumor responses. Proc. Natl Acad. Sci. USA 116, 599–608 (2019).
Mumm, J. B. et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell 20, 781–796 (2011).
DuPage, M. & Bluestone, J. A. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat. Rev. Immunol. 16, 149–163 (2016).
Lu, Y. et al. Th9 cells represent a unique subset of CD4+ T cells endowed with the ability to eradicate advanced tumors. Cancer Cell 33, 1048–1060 e1047 (2018). This publication highlights the potential of TH9 polarized cells for the improvement of immunotherapy for solid tumours.
Angkasekwinai, P. & Dong, C. IL-9-producing T cells: potential players in allergy and cancer. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-020-0396-0 (2020).
Kim, I. K. et al. Glucocorticoid-induced tumor necrosis factor receptor-related protein co-stimulation facilitates tumor regression by inducing IL-9-producing helper T cells. Nat. Med. 21, 1010–1017 (2015).
Vegran, F. et al. The transcription factor IRF1 dictates the IL-21-dependent anticancer functions of TH9 cells. Nat. Immunol. 15, 758–766 (2014).
Xue, G., Jin, G., Fang, J. & Lu, Y. IL-4 together with IL-1beta induces antitumor Th9 cell differentiation in the absence of TGF-beta signaling. Nat. Commun. 10, 1376 (2019).
Spolski, R. & Leonard, W. J. Interleukin-21: a double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 13, 379–395 (2014).
Zander, R. et al. CD4+ T cell help is required for the formation of a cytolytic CD8+ T cell subset that protects against chronic infection and cancer. Immunity 51, 1028–1042 e1024 (2019).
Kourko, O., Seaver, K., Odoardi, N., Basta, S. & Gee, K. IL-27, IL-30, and IL-35: a cytokine triumvirate in cancer. Front. Oncol. 9, 969 (2019).
Chihara, N. et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 558, 454–459 (2018).
Li, S. et al. Cancer immunotherapy via targeted TGF-beta signalling blockade in Th cells. Nature https://doi.org/10.1038/s41586-020-2850-3 (2020).
Zitvogel, L., Tesniere, A. & Kroemer, G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat. Rev. Immunol. 6, 715–727 (2006).
Hatzioannou, A. et al. An intrinsic role of IL-33 in Treg cell-mediated tumor immunoevasion. Nat. Immunol. 21, 75–85 (2020).
Schiering, C. et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 513, 564–568 (2014).
Sawant, D. V. et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 20, 724–735 (2019). This work demonstrates a pathway by which Treg cell-derived IL-10 and IL-35 cooperatively exhaust CD8+ T cells in tumours.
Smith, L. K. et al. Interleukin-10 directly inhibits CD8+ T cell function by enhancing N-glycan branching to decrease antigen sensitivity. Immunity 48, 299–312 e295 (2018).
Sledzinska, A. et al. Regulatory T cells restrain interleukin-2- and Blimp-1-dependent acquisition of cytotoxic function by CD4+ T cells. Immunity 52, 151–166 e156 (2020).
Vitiello, G. A. & Miller, G. Targeting the interleukin-17 immune axis for cancer immunotherapy. J. Exp. Med. https://doi.org/10.1084/jem.20190456 (2020).
Coffelt, S. B. et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522, 345–348 (2015).
Koh, J. et al. IL23-producing human lung cancer cells promote tumor growth via conversion of innate lymphoid cell 1 (ILC1) into ILC3. Clin. Cancer Res. 25, 4026–4037 (2019).
Bakouny, Z. & Choueiri, T. K. IL-8 and cancer prognosis on immunotherapy. Nat. Med. 26, 650–651 (2020).
Yuen, K. C. et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat. Med. 26, 693–698 (2020).
Nakamura, K. et al. Dysregulated IL-18 is a key driver of immunosuppression and a possible therapeutic target in the multiple myeloma microenvironment. Cancer Cell 33, 634–648 e635 (2018).
Shani, O. et al. Fibroblast-derived IL-33 facilitates breast cancer metastasis by modifying the immune microenvironment and driving type 2 immunity. Cancer Res. 80, 5317–5329 (2020).
Coussens, L. M., Zitvogel, L. & Palucka, A. K. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science 339, 286–291 (2013).
Schuijs, M. J. et al. ILC2-driven innate immune checkpoint mechanism antagonizes NK cell antimetastatic function in the lung. Nat. Immunol. 21, 998–1009 (2020).
Gu, M. et al. NF-kappaB-inducing kinase maintains T cell metabolic fitness in antitumor immunity. Nat. Immunol. 22, 193–204 (2021).
Kesh, K. et al. Stroma secreted IL-6 selects for “stem-like” population and alters pancreatic tumor microenvironment by reprogramming metabolic pathways. Cell Death Dis. 11, 967 (2020).
Fousek, K., Horn, L. A. & Palena, C. Interleukin-8: A chemokine at the intersection of cancer plasticity, angiogenesis, and immune suppression. Pharmacol. Therap. https://doi.org/10.1016/j.pharmthera.2020.107692 (2020).
Ngiow, S. F., Teng, M. W. L. & Smyth, M. J. A balance of interleukin-12 and -23 in cancer. Trends Immunol. 34, 548–555 (2013).
Feng, L. et al. Serum levels of IL-6, IL-8, and IL-10 are indicators of prognosis in pancreatic cancer. J. Int. Med. Res. 46, 5228–5236 (2018).
Lippitz, B. E. Cytokine patterns in patients with cancer: a systematic review. Lancet Oncol. 14, e218–e228 (2013).
Ridker, P. M. et al. Effect of interleukin-1beta; inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842 (2017).
Libby, P. Interleukin-1 beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J. Am. Coll. Cardiol. 70, 2278–2289 (2017).
Wong, C. C. et al. Inhibition of IL-1β by canakinumab may be effective against diverse molecular subtypes of lung cancer: an exploratory analysis of the CANTOS trial. Cancer Res. https://doi.org/10.1158/0008-5472.Can-19-3176 (2020). This work impressively demonstrates how patients with cancer could benefit from IL-1β neutralization.
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03447769 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04239157 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03631199 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03626545 (2018).
Lust, J. A. et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1beta-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin. Proc. 84, 114–122 (2009).
Lust, J. A. et al. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am. J. Hematol. 91, 571–574 (2016).
Nguyen, M. L. T. et al. Targeting interleukin 6 signaling by monoclonal antibody siltuximab on cholangiocarcinoma. J. Gastroenterol. Hepatol. https://doi.org/10.1111/jgh.15307 (2020).
Brighton, T. A. et al. Randomized, double-blind, placebo-controlled, multicenter study of siltuximab in high-risk smoldering multiple myeloma. Clin. Cancer Res. 25, 3772–3775 (2019).
Orlowski, R. Z. et al. A phase 2, randomized, double-blind, placebo-controlled study of siltuximab (anti-IL-6 mAb) and bortezomib versus bortezomib alone in patients with relapsed or refractory multiple myeloma. Am. J. Hematol. 90, 42–49 (2015).
San-Miguel, J. et al. Phase 2 randomized study of bortezomib-melphalan-prednisone with or without siltuximab (anti-IL-6) in multiple myeloma. Blood 123, 4136–4142 (2014).
Dorff, T. B. et al. Clinical and correlative results of SWOG S0354: a phase II trial of CNTO328 (siltuximab), a monoclonal antibody against interleukin-6, in chemotherapy-pretreated patients with castration-resistant prostate cancer. Clin. Cancer Res. 16, 3028–3034 (2010).
Rossi, J. F. et al. A phase I/II study of siltuximab (CNTO 328), an anti-interleukin-6 monoclonal antibody, in metastatic renal cell cancer. Br. J. Cancer 103, 1154–1162 (2010).
Angevin, E. et al. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 20, 2192–2204 (2014).
Dijkgraaf, E. M. et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-alpha2b in patients with recurrent epithelial ovarian cancer. Ann. Oncol. 26, 2141–2149 (2015).
Johnson, D. E., O’Keefe, R. A. & Grandis, J. R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 15, 234–248 (2018).
Bergmann, J. et al. IL-6 trans-signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology 65, 89–103 (2017).
Schmidt, S. et al. ADAM17 is required for EGF-R-induced intestinal tumors via IL-6 trans-signaling. J. Exp. Med. 215, 1205–1225 (2018).
Jostock, T. et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 268, 160–167 (2001).
Bilusic, M. et al. Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J. Immunother. Cancer 7, 240–240 (2019).
Schalper, K. A. et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat. Med. 26, 688–692 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03400332 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04123379 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04050462 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03689699 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04572451 (2020).
Nagaoka, K. et al. Deep immunophenotyping at the single-cell level identifies a combination of anti-IL-17 and checkpoint blockade as an effective treatment in a preclinical model of data-guided personalized immunotherapy. J. Immunother. Cancer 8, e001358 (2020).
Zhang, Q. et al. Targeting Th17-IL-17 pathway in prevention of micro-invasive prostate cancer in a mouse model. Prostate 77, 888–899 (2017).
Calcinotto, A. et al. Microbiota-driven interleukin-17-producing cells and eosinophils synergize to accelerate multiple myeloma progression. Nat. Commun. 9, 4832–4832 (2018).
Prabhala, R. H. et al. Targeting IL-17A in multiple myeloma: a potential novel therapeutic approach in myeloma. Leukemia 30, 379–389 (2016).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03111992 (2017).
Calcinotto, A. et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 559, 363–369 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04458311 (2020).
Kudo-Saito, C. et al. IL-33 is a key driver of treatment resistance of cancer. Cancer Res. 80, 1981–1990 (2020).
Rosenberg, S. A. IL-2: the first effective immunotherapy for human cancer. J. Immunol. 192, 5451–5458 (2014). This work provides an overview of the treatment of patients with cancer with recombinant IL-2, the first interleukin approved for cancer therapy.
Charych, D. H. et al. NKTR-214, an engineered cytokine with biased IL-2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin. Cancer Res. 22, 680–690 (2016).
Bentebibel, S.-E. et al. A first-in-human study and biomarker analysis of NKTR-214, a novel IL2Rβγ-biased cytokine, in patients with advanced or metastatic solid tumors. Cancer Discov. 9, 711–721 (2019).
Diab, A. et al. Bempegaldesleukin (NKTR-214) plus nivolumab in patients with advanced solid tumors: phase I dose-escalation study of safety, efficacy, and immune activation (PIVOT-02). Cancer Discov. 10, 1158–1173 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04209114 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03729245 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03635983 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04009681 (2019).
Sahin, D. et al. An IL-2-grafted antibody immunotherapy with potent efficacy against metastatic cancer. Nat. Commun. 11, 6440–6440 (2020).
Silva, D.-A. et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature 565, 186–191 (2019).
Mortara, L. et al. Anti-cancer therapies employing IL-2 cytokine tumor targeting: contribution of innate, adaptive and immunosuppressive cells in the anti-tumor efficacy. Front. Immunol. https://doi.org/10.3389/fimmu.2018.02905 (2018).
Naing, A. et al. Safety, antitumor activity, and immune activation of pegylated recombinant human interleukin-10 (AM0010) in patients with advanced solid tumors. J. Clin. Oncol. 34, 3562–3569 (2016).
Naing, A. et al. PEGylated IL-10 (pegilodecakin) induces systemic immune activation, CD8(+) T cell invigoration and polyclonal T cell expansion in cancer patients. Cancer Cell 34, 775–791.e773 (2018).
Naing, A. et al. Pegilodecakin combined with pembrolizumab or nivolumab for patients with advanced solid tumours (IVY): a multicentre, multicohort, open-label, phase 1b trial. Lancet Oncol. 20, 1544–1555 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03382912 (2017).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03382899 (2017).
Qiao, J. et al. Targeting tumors with IL-10 prevents dendritic cell-mediated CD8(+) T Cell Apoptosis. Cancer Cell 35, 901–915.e904 (2019).
Nguyen, K. G. et al. Localized interleukin-12 for cancer immunotherapy. Front. Immunol. https://doi.org/10.3389/fimmu.2020.575597 (2020).
Algazi, A. et al. Intratumoral delivery of tavokinogene telseplasmid yields systemic immune responses in metastatic melanoma patients. Ann. Oncol. 31, 532–540 (2020).
Algazi, A. P. et al. Phase II trial of IL-12 plasmid transfection and PD-1 blockade in immunologically quiescent melanoma. Clin. Cancer Res. 26, 2827–2837 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03567720 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03823131 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04526730 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03132675 (2017).
Mansurov, A. et al. Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nat. Biomed. Eng. 4, 531–543 (2020).
Li, Y. et al. Multifunctional oncolytic nanoparticles deliver self-replicating IL-12 RNA to eliminate established tumors and prime systemic immunity. Nat. Cancer 1, 882–893 (2020).
Waldmann, T. A., Dubois, S., Miljkovic, M. D. & Conlon, K. C. IL-15 in the combination immunotherapy of cancer. Front. Immunol. 11, 868–868 (2020).
Margolin, K. et al. Phase I Trial of ALT-803, A novel recombinant IL-15 complex, in patients with advanced solid tumors. Clin. Cancer Res. 24, 5552–5561 (2018).
Romee, R. et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood 131, 2515–2527 (2018).
Rosser, C. J., Nix, J., Ferguson, L., Hernandez, L. & Wong, H. C. Phase Ib trial of ALT-803, an IL-15 superagonist, plus BCG for the treatment of BCG-naïve patients with non-muscle-invasive bladder cancer. J. Clin. Oncol. 36, 510–510 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02138734 (2014).
Waldmann, T. A., Miljkovic, M. D. & Conlon, K. C. Interleukin-15 (dys)regulation of lymphoid homeostasis: Implications for therapy of autoimmunity and cancer. J. Exp. Med. 217, e20191062 (2020).
Hewitt, S. L. et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36gamma, and OX40L mRNAs. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aat9143 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03739931 (2018).
Nguyen, L. T. et al. Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol. Immunother. 68, 773–785 (2019).
Junghans, R. P. et al. Phase I trial of anti-PSMA designer CAR-T cells in prostate cancer: possible role for interacting interleukin-2-T cell pharmacodynamics as a determinant of clinical response. Prostate 76, 1257–1270 (2016).
Sockolosky, J. T. et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science 359, 1037–1042 (2018).
Jin, L. et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun 10, 4016 (2019).
Chmielewski, M., Kopecky, C., Hombach, A. A. & Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 71, 5697–5706 (2011). This study provides a proof of concept for the expression of interleukins by CAR T cells to recruit innate immune cells and mount an antigen-independent immune response.
Zhang, L. et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res. 21, 2278–2288 (2015). This is the first report of interleukin-expressing tumour-infiltrating lymphocytes.
Etxeberria, I. et al. Intratumor adoptive transfer of IL-12 mRNA transiently engineered antitumor CD8+ T cells. Cancer Cell 36, 613–629.e617 (2019).
Subbiah, V. et al. Cytokines produced by dendritic cells administered intratumorally correlate with clinical outcome in patients with diverse cancers. Clin. Cancer Res. 24, 3845–3856 (2018).
Adachi, K. et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 36, 346–351 (2018).
Xu, X. et al. NKT cells coexpressing a GD2-specific chimeric antigen receptor and IL-15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clin. Cancer Res. 25, 7126–7138 (2019).
Hurton, L. V. et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 113, E7788–E7797 (2016).
Heczey, A. et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat. Med. 26, 1686–1690 (2020).
Tang, L. et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol. 36, 707–716 (2018).
Ma, X. et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. biotechnol. 38, 448–459 (2020).
Espinosa-Cotton, M. et al. Interleukin-1 alpha increases anti-tumor efficacy of cetuximab in head and neck squamous cell carcinoma. J. Immunother. Cancer 7, 79 (2019).
Gottschlich, A., Endres, S. & Kobold, S. Therapeutic strategies for targeting IL-1 in cancer. Cancers 13, 477 (2021).
Li, S. et al. Role for nuclear interleukin-37 in the suppression of innate immunity. Proc. Natl Acad. Sci. USA 116, 4456–4461 (2019).
Weinstein, A. M. et al. Association of IL-36gamma with tertiary lymphoid structures and inflammatory immune infiltrates in human colorectal cancer. Cancer Immunol. Immunother. 68, 109–120 (2019).
van de Veerdonk, F. L., de Graaf, D. M., Joosten, L. A. & Dinarello, C. A. Biology of IL-38 and its role in disease. Immunol. Rev. 281, 191–196 (2018).
Kinoshita, F. et al. Interleukin-38 promotes tumor growth through regulation of CD8+tumor-infiltrating lymphocytes in lung cancer tumor microenvironment. Cancer Immunol. Immunother. 70, 123–135 (2021).
Damoiseaux, J. The IL-2 - IL-2 receptor pathway in health and disease: The role of the soluble IL-2 receptor. Clin. Immunol. 218, 108515 (2020).
Suzuki, A., Leland, P., Joshi, B. H. & Puri, R. K. Targeting of IL-4 and IL-13 receptors for cancer therapy. Cytokine 75, 79–88 (2015).
Lin, J. et al. The role of IL-7 in immunity and cancer. Anticancer Res. 37, 963–967 (2017).
Ferretti, E., Corcione, A. & Pistoia, V. The IL-31/IL-31 receptor axis: general features and role in tumor microenvironment. J. Leukoc. Biol. 102, 711–717 (2017).
Ouyang, W. & O’Garra, A. IL-10 family cytokines IL-10 and IL-22: from basic science to clinical translation. Immunity 50, 871–891 (2019).
Chen, Y. Y., Li, C. F., Yeh, C. H., Chang, M. S. & Hsing, C. H. Interleukin-19 in breast cancer. Clin. Dev. Immunol. 2013, 294320 (2013).
Hsu, Y. H. et al. Anti-IL-20 monoclonal antibody suppresses breast cancer progression and bone osteolysis in murine models. J. Immunol. 188, 1981–1991 (2012).
Menezes, M. E. et al. MDA-7/IL-24: multifunctional cancer killing cytokine. Adv. Exp. Med. Biol. 818, 127–153 (2014).
Hu, H. J. et al. The armed oncolytic adenovirus ZD55-IL-24 eradicates melanoma by turning the tumor cells from the self-state into the nonself-state besides direct killing. Cell Death Dis. 11, 1022 (2020).
Trotter, T. N. et al. IL-26, a noncanonical mediator of DNA inflammatory stimulation, promotes TNBC engraftment and progression in association with neutrophils. Cancer Res 80, 3088–3100 (2020).
Larochette, V. et al. IL-26, a cytokine with roles in extracellular DNA-induced inflammation and microbial defense. Front. Immunol. 10, 204 (2019).
Yan, J., Smyth, M. J. & Teng, M. W. L. Interleukin (IL)-12 and IL-23 and their conflicting roles in cancer. Cold Spring Harb. Perspect Biol. https://doi.org/10.1101/cshperspect.a028530 (2018).
Park, Y. J. et al. IL-27 confers a protumorigenic activity of regulatory T cells via CD39. Proc. Natl Acad. Sci. USA 116, 3106–3111 (2019).
Figueiredo, M. L. et al. Ligand-mediated targeting of cytokine interleukin-27 enhances its bioactivity in vivo. Mol. Ther. Methods Clin. Dev. 17, 739–751 (2020).
Bie, Q., Jin, C., Zhang, B. & Dong, H. IL-17B: A new area of study in the IL-17 family. Mol. Immunol. 90, 50–56 (2017).
Jungnickel, C. et al. IL-17C mediates the recruitment of tumor-associated neutrophils and lung tumor growth. Oncogene 36, 4182–4190 (2017).
Liu, X., Sun, S. & Liu, D. IL-17D: a less studied cytokine of IL-17 family. Int. Arch. Allergy Immunol. 181, 618–623 (2020).
Hasegawa, K. et al. Anti-tumor immunity elicited by direct intratumoral administration of a recombinant adenovirus expressing either IL-28A/IFN-lambda2 or IL-29/IFN-lambda1. Cancer Gene Ther. 23, 266–277 (2016).
Ford, R. et al. Identification of B-cell growth factors (interleukin-14; high molecular weight-B-cell growth factors) in effusion fluids from patients with aggressive B-cell lymphomas. Blood 86, 283–293 (1995).
Richmond, J., Tuzova, M., Cruikshank, W. & Center, D. Regulation of cellular processes by interleukin-16 in homeostasis and cancer. J. Cell Physiol. 229, 139–147 (2014).
Sloot, Y. J. E., Smit, J. W., Joosten, L. A. B. & Netea-Maier, R. T. Insights into the role of IL-32 in cancer. Semin. Immunol. 38, 24–32 (2018).
Baghdadi, M. et al. Interleukin-34, a comprehensive review. J. Leukoc. Biol. 104, 931–951 (2018).
Franzè, E., Stolfi, C., Troncone, E., Scarozza, P. & Monteleone, G. Role of interleukin-34 in cancer. Cancers 12, 252 (2020).
Grupp, S. A. et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 368, 1509–1518 (2013).
Davila, M. L. et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 6, 224ra225 (2014).
Norelli, M. et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 24, 739–748 (2018).
Giavridis, T. et al. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 24, 731–738 (2018).
Ando, K. et al. Tocilizumab, a proposed therapy for the cachexia of Interleukin-6-expressing lung cancer. PloS ONE 9, e102436 (2014).
Rigas, J. R. et al. Efect of ALD518, a humanized anti-IL-6 antibody, on lean body mass loss and symptoms in patients with advanced non-small cell lung cancer (NSCLC): results of a phase II randomized, double-blind safety and efficacy trial. J. Clin. Oncol. 28, 7622–7622 (2010).
Flint, T. R. et al. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab. 24, 672–684 (2016).
Pettersen, K. et al. Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci. Rep. 7, 2046 (2017).
McDonald, J. J., McMillan, D. C. & Laird, B. J. A. Targeting IL-1alpha in cancer cachexia: a narrative review. Curr. Opin. Support Palliat. Care 12, 453–459 (2018).
Hickish, T. et al. MABp1 as a novel antibody treatment for advanced colorectal cancer: a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 18, 192–201 (2017).
Hong, D. S. et al. Xilonix, a novel true human antibody targeting the inflammatory cytokine interleukin-1 alpha, in non-small cell lung cancer. Invest. New Drugs 33, 621–631 (2015).
Callaway, C. S. et al. IL-8 released from human pancreatic cancer and tumor-associated stromal cells signals through a CXCR2-ERK1/2 axis to induce muscle atrophy. Cancers 11, 1863 (2019).
Costamagna, D. et al. Interleukin-4 administration improves muscle function, adult myogenesis, and lifespan of colon carcinoma-bearing mice. J. Cachexia Sarcopenia Muscle 11, 783–801 (2020).
Acknowledgements
This study was supported by the Marie Skłodowska-Curie Programme Training Network for the Immunotherapy of Cancer and Training Network for Optimizing Adoptive T Cell Therapy of Cancer funded by the Horizon 2020 programme of the European Union (grant 641549 to S.E. and S.K. and grant 955575 to S.K), the Hector Foundation, the international doctoral programme i Target: Immunotargeting of Cancer funded by the Elite Network of Bavaria (S.K. and S.E.), Melanoma Research Alliance grant 409510 (to S.K.), the Else Kröner-Fresenius-Stiftung (S.K.), the German Cancer Aid (S.K.), the Ernst-Jung-Stiftung (S.K.), LMU Munich’s institutional strategy LMUexcellent within the framework of the German Excellence Initiative (S.E. and S.K.), the Bundesministerium für Bildung und Forschung project Oncoattract (S.E. and S.K.), European Research Council grant 756017, ARMOR-T (to S.K.), the German Research Foundation (S.K.), the Fritz Bender Foundation (S.K.), the Bavarian Ministry of Economical Affairs (m4 award to S.K. and S.E.) and the José Carreras Foundation (to S.K.). The authors apologize to any colleagues whose important work could not be cited or commented on due to space restrictions.
Author information
Authors and Affiliations
Contributions
S.K., D.B. and J.D. contributed equally to all aspects of the article. S.E., P.L. and C.A.D. contributed to researching literature for the article and discussion of the content, and reviewed or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
J.D. has received remuneration from Novartis for work unrelated to this Review. P.L. is an unpaid consultant to, or involved in clinical trials for, Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Norvo Nordisk, Novartis, Pfizer and Sanofi-Regeneron. P.L. is a member of scientific advisory board for Amgen, Caristo, Cartesian, Corvidia Therapeutics, CSL Behring, DalCor Pharmaceuticals, Dewpoint, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, PlaqueTec and XBiotech, Inc. P.L.’s laboratory has received research funding in the past 2 years from Novartis. P.L. is on the Board of Directors of XBiotech, and has a financial interest in Xbiotech, a company developing therapeutic human antibodies. P.L.’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. S.K. and S.E. are inventors named on several patents in the field of immuno-oncology unrelated to the present work. S.K. and S.E. received research support from TCR2 Inc. and Arcus Bioscience for work unrelated to this Review. S.K. and S.E. have licensed intellectual property to TCR2 Inc. S.K. has received honoraria from GlaxoSmithKline and Novartis. D.B. and C.A.D. declare no competing interests.
Additional information
Peer review information
Nature Reviews Cancer thanks L. Apetoh, who co-reviewed with F. Vegran; and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- Danger-associated molecular patterns
-
(DAMPs). Danger-associated molecular patterns and alarmins are endogenous, constitutively expressed, chemotactic and immune-activating proteins and peptides that are released upon degranulation, cell injury or death or in response to immune induction.
- Pathogen-associated molecular patterns
-
(PAMPs). Exogenous conserved microbial structures recognized by the innate immune receptors.
- Tumour-associated macrophages
-
Macrophage subtype that is highly abundant in tumours and associated with immunosuppression, tumour progression and metastasis.
- γδ T cells
-
A subset of T cells differing from conventional T cells by expressing a distinct γδ T cell receptor in contrast to the usual αβ T cell receptor. They do not depend on histocompatibility complex presentation and possess properties of both innate and adaptive immunity
- Group 3 innate lymphoid cells
-
(ILC3s). Cells with characteristics of lymphoid cells but without rearranged antigen-specific surface receptors that are crucial for maintaining homeostasis in the gut microbiota.
- Regulatory T cell
-
(Treg cell). A subtype of T cells maintaining self-tolerance and preventing excessive immune responses, mainly by the inhibition of proliferation and activation of cytotoxic T cells.
- Adoptive cell transfer
-
(ACT). Treatment of patients with ex vivo expanded or modified immune cells usually derived from the patient.
- Natural killer (NK) cells
-
Cytotoxic lymphocytes that can eliminate virus-infected cells or tumour cells by the recognition of non-self antigens presented on major histocompatibility complex class I molecules of the target cell.
- Natural killer T cells
-
(NKT cells). A subset of T cells exhibiting antigen-specific immunity via a restricted T cell receptor recognizing CD1-presented lipid antigens, but also sharing natural killer cell functions in terms of their independence from major histocompatibility complex.
- Antigen-presenting cells
-
(APCs). Dendritic cells, macrophages and B cells that present non-self peptide antigens to T cells via major histocompatibility complex class II molecules.
- M1 macrophage
-
A macrophage subtype associated with inflammation that is characterized by the secretion of the proinflammatory cytokines IL-1, IL-6 and TNF.
- Immunotherapy
-
Treatment of patients with factors shaping an immune response to enable the patient’s immune system to react to the disease.
- TH1-type responses
-
Proinflammatory responses inducing killing of intracellular pathogens and tumour cells mediated mainly by interferon-γ.
- M2 macrophages
-
An inhibitory macrophage subtype associated with tissue remodelling and repair that secrete anti-inflammatory cytokines such as IL-10 and transforming growth factor-β (TGFβ).
- Myeloid-derived suppressor cells
-
(MDSCs). A heterogeneous group of myeloid cells often recruited by tumour cells that lead to pronounced immunosuppression by inhibiting a wide range of inflammatory immune cells.
- Group 2 innate lymphoid cells
-
(ILC2). Cells with characteristics of lymphoid cells but without rearranged antigen-specific surface receptors. Similarly to CD4+ T cells, they produce type 2 cytokines and are also referred to as natural or innate helper cells.
- TH2-type responses
-
Immune responses against mainly extracellular antigens mediated by IL-4, IL-5 and IL-13.
- Immune checkpoint inhibitors
-
(ICIs). Drugs used for therapeutic neutralization of signalling pathways that activate immune checkpoints, which normally lead to the inhibition of immune cell function to keep immune responses under control.
- Tumour-infiltrating lymphocytes
-
Lymphocytes that migrated into the tumour and can be isolated, expanded and re-infused into the patient as therapy.
Rights and permissions
About this article

Cite this article
Briukhovetska, D., Dörr, J., Endres, S. et al. Interleukins in cancer: from biology to therapy. Nat Rev Cancer 21, 481–499 (2021). https://doi.org/10.1038/s41568-021-00363-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41568-021-00363-z
This article is cited by
-
Targeting inflammation as cancer therapy
Journal of Hematology & Oncology (2024)
-
Lyophilized lymph nodes for improved delivery of chimeric antigen receptor T cells
Nature Materials (2024)
-
Biomaterials to enhance adoptive cell therapy
Nature Reviews Bioengineering (2024)
-
Dictionary of immune responses to cytokines at single-cell resolution
Nature (2024)
-
LAPTM4B-mediated hepatocellular carcinoma stem cell proliferation and MDSC migration: implications for HCC progression and sensitivity to PD-L1 monoclonal antibody therapy
Cell Death & Disease (2024)
