
Abstract
Dew is a common form of water that deposits from saturated air on colder surfaces. Although presumably common on primordial Earth, its potential involvement in the origin of life in early replication has not been investigated in detail. Here we report that it can drive the first stages of Darwinian evolution for DNA and RNA, first by periodically denaturing their structures at low temperatures and second by promoting the replication of long strands over short, faster replicating ones. Our experiments mimicked a partially water-filled primordial rock pore in the probable CO2 atmosphere of Hadean Earth. Under heat flow, water continuously evaporated and recondensed as acidic dew droplets that created the humidity, salt and pH cycles that match many prebiotic replication chemistries. In low-salt and low-pH regimes, the strands melted at 30 K below the bulk melting temperature, whereas longer sequences preferentially accumulated at the droplet interface. Under an enzymatic replication to mimic a sped-up RNA world, long sequences of more than 1,000 nucleotides emerged. The replication was biased by the melting conditions of the dew and the initial short ATGC strands evolved into long AT-rich sequences with repetitive and structured nucleotide composition.
Similar content being viewed by others
Main
The autonomous replication, mutation and selection of genetic information is central to the origin of life and the onset of Darwinian evolution. It is necessary for RNA or DNA to periodically separate their strands to start a successive replication cycle. Without a denaturation mechanism, the replication encounters a dead-end1. Melting by heat has limitations because high temperatures can induce damaging side reactions and lead to the hydrolysis of chemical reagents2 as well as the RNA3 itself. Moreover, the temperature required to separate RNA strands can become higher than 100 °C and therefore impossible to reach in aqueous solution.
Low salt concentrations can destabilize double strands and facilitate denaturation4. However, many replication reactions and catalytic RNAs require elevated concentrations of ions to function5. For example, Mg2+ strongly stabilizes oligonucleotides in the duplex form. It thus seems that the physico-chemical conditions that favour replication reactions are, at the same time, incompatible with the melting of oligonucleotides. Many studies suggest other denaturation methods, such as Na+ oscillations6, low pH7,8 or chaotropic agents (for example, urea9 or formamide10). However, how any of these methods could be integrated in a primordial autonomous replication remains unclear.
A replicative system must also drive the evolution of the sequence information. It is known that the longer oligonucleotides are quickly lost, because the shorter oligomers are replicated with faster kinetics and quickly outcompete them. This means that, in a pool of sequences, shorter sequences will prevail and longer ones will progressively die out. Mutations in the replication process cause changes in the sequence length and thus start an evolutionary race towards the shortest oligomers, leading to a progressive loss of genetic information. This phenomenon, first demonstrated by Spiegelman11, is known as the ‘tyranny of the shortest’ and is a problem for the onset of Darwinian evolution.
The sequence space of long oligonucleotides is immense. In this vast entropic land, the spontaneous emergence of sequences that contain particular motifs, secondary structures or that are enriched in specific nucleotides becomes rare. Without a selective mechanism that can reduce the sequence space, any sequence bias is likely to remain unexplored and not emerge12. For example, the assembly of short oligomers into active ribozymes requires the presence of particular sequences that can assemble by complementarity to build a ribozyme with catalytic activity13,14.
The composition of Earth’s atmosphere has changed substantially over geological time15. High CO2 pressure (between 0.1 and 10 bar) was likely in the Hadean eon16. Such an atmosphere could have drastically affected the prebiotic chemistry—when gaseous CO2 is dissolved in water, it leads to the formation of carbonic acid, bicarbonate and carbonate, lowering the pH of the water (Supplementary Section 1). The pH is a direct function of the partial pressure of CO2 (\(p_{{\mathrm{CO}}_2}\))17 and the surface waters of early Earth probably had an acidic pH. Condensed water can reach particularly low pH (~4) due to its high purity and thus lack of pH-buffering salt molecules.
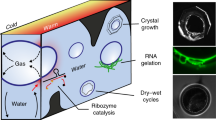
We developed a laboratory model of a heated rock pore18 to cyclically generate dew droplets at the microscale level. This consisted of a thin rock-like pore containing gas and liquid. The gas was enriched with Hadean levels of CO2 (0.1–1 bar) and the pore was heated by a temperature gradient to imitate the heat sources of the Early Earth18. Water evaporated and condensed to form salt-free dew droplets that absorbed CO2 and became acidified (pH of ~4), then induced RNA and DNA melting by means of their acidity and low salt levels. The bulk retained high salt levels and neutral pH, preserving conditions ideal for reannealing and replication. The dew chamber allowed for DNA replication at 30 K below the bulk melting temperature.
Moreover, oligonucleotides accumulated at the gas–liquid boundaries at up to 10,000-fold (where fold indicates the increase in concentration relative to the starting concentration). The accumulation favoured the longer strands, which were then preferentially replicated. The DNA wet–dry cycles induced unspecific annealing of the strands, which recombined into sequences of increasing complexity and length. As the sequences grew longer, their stability against denaturation increased. It is known that the stability of double-stranded DNA (dsDNA) is also determined by the fraction of AT versus GC19. We found that the replication by CO2 fog cycles modulated the selection of oligonucleotides with an ATGC composition compatible with the melting conditions of the dew, drastically reducing the sequence space. At the same time, the length of the DNAs increased by more than 25×, but, to ensure their melting, they were strongly enriched in AT (AT fraction >80%). This showed that fog cycles avoid Spiegelman’s dominance of the shortest sequence, promote the Darwinian evolution of long DNAs and allow low-entropy sequence biases to emerge.
Results and discussion
Figure 1a presents a scheme of the microfluidic CO2 dew chamber, viewed laterally (Supplementary Section 2). The temperature gradient across the thin chamber (250 µm), half-filled with a Mg2+-rich solution and RNA (or DNA), creates a dew cycle at the microscale level. Evaporation at the warm side is balanced by condensation on the cold side, where acidic dew droplets of purified water are formed. Oligomers accumulate at the evaporating gas–liquid interfaces20 (coffee-ring effect) and periodically dry out on the warm wall during the evaporation–condensation–coalescence cycles. Besides condensation, the dew droplets also grow by fusion with other droplets. When the droplet radius exceeds the chamber thickness (250 µm), surface tension rearranges the dew droplets into a capillary bridge between the chamber walls. At this point, the dew redissolves the dry oligomers on the warm wall. The low pH and low salt concentration efficiently denature the oligonucleotides. Figure 1b shows the process via fluorescence, using fluorescently labelled RNA, imaged through the warm side (viewed perpendicular with respect to Fig. 1a; Supplementary Video 1). The dew either dries out and redeposits oligonucleotides on the warm side or grows large by fusion then coalesces with the bulk, where the oligonucleotides can be promptly replicated and elongated (Supplementary Section 3).

The continuous formation of dew in a rock pore at Hadean \(p_{{\mathrm{CO}}_2}\) generates cycles of RNA melting, as well as RNA wet–dry cycles. a, Scheme of the dew cycle at Hadean CO2 levels. The water that evaporates condenses as acidic dew droplets that contain low salt. The dew droplets grow and create a capillary bridge between the pore surfaces. RNA oscillates between the dry and the wet states and denatures in the dew. The salt-rich bulk maintains a neutral pH that allows reannealing and replication of the oligonucleotides. b, Fluorescence images (FAM-labelled (carboxyfluorescein) single-stranded RNA (ssRNA), 51 nt) of the dew cycle, imaged through the z axis.
We expected the dew droplets to have a reduced concentration of buffers and salts and thus be particularly susceptible to the acidification induced by CO2 absorption. On the other hand, the bulk would remain buffer-rich and maintain a quasi-neutral pH. The result would be pH and salt oscillations that lead to periodic melting of RNA in the dew and reannealing in the bulk. The pH and RNA conformation were measured by fluorescence microscopy. pH was measured with the ratiometric dye Lysosensor Yellow/Blue DND-160. RNA conformation was measured by fluorescence resonance energy transfer (FRET), using FRET-pair fluorophores positioned centrally on opposite strands21 (Supplementary Section 4).
The pH of the dew and bulk during the dew cycle was measured at a range of CO2 pressures. The bulk contained 10 mM MgCl2 and 10 mM Tris (initial pH 7.0), to which Lysosensor Yellow/Blue DND-160 was added at 20 µM for pH imaging. A temperature gradient of 5 °C was applied (hot side 27 °C, cold side 22 °C). Fluorescence images are presented in Fig. 2a. At room pCO2 levels (0.4 mbar), the pH of the bulk was neutral (7.0 ± 0.2)(mean ± standard deviation). In the dew droplets we found slightly acidic pH (5.6 ± 0.3), as expected for very low salt water at room CO2. At higher pCO2 (1 bar), the bulk pH dropped to 5.8 (±0.2), even though it contained Tris buffer. The dew acidified down to pH 4.0 (±0.4). The experimental data were compared to a theoretical model based on the rate equations of CO2 absorption into water or into a buffered solution (Fig. 2b). In the dew droplets, the pH drastically dropped with increasing \(p_{{\mathrm{CO}}_2}\). In the bulk, the buffer molecules neutralized the carbonic acid and dampened the pH change.

A synergistic combination of low salts, low pH and temperature is necessary to generate strong denaturing conditions for RNA melting at low temperatures. a, Dew and bulk viewed by RNA fluorescence (top) and corresponding pH (bottom). Dew droplets at a \(p_{{\mathrm{CO}}_2}\) of 1 bar acidify to pH 4, while the bulk remains almost pH-neutral. b, Model (lines) and experimental (points) bulk and dew pH as a function of \(p_{{\mathrm{CO}}_2}\). Error bars represent the standard deviation (n ≈ 3). The dew pH drops with increasing \(p_{{\mathrm{CO}}_2}\), whereas the bulk is buffered by the salts. c, Model (lines) and experiments (bars) of RNA FRET in the dew as a function of \(p_{{\mathrm{CO}}_2}\). Error bars represent the standard deviation (n ≈ 4). RNA melts with increasing \(p_{{\mathrm{CO}}_2}\) inside dew droplets that contain low salts (0.25 mM Mg2+). d, Dew and bulk viewed by RNA fluorescence (top) and corresponding RNA FRET (bottom). RNA melts in the acidic, low-salt dew (RNA FRET = 0.14). In the salt-rich bulk, the RNA remains double-stranded (RNA FRET = 0.86). e, Surface plot (model) showing how pH and Mg2+ cooperatively affect the duplex RNA fraction (RNA FRET). Experimental RNA FRET values for bulk and dew droplets are shown as rectangles.
Prebiotically, the pH of an aqueous solution is probably buffered by the minerals in contact with the liquid by means of mineral dissolution, ion exchange and surface complexation reactions22. Silicate minerals—ubiquitous rock-forming minerals23—can act as a buffer and maintain neutral pH in the long term24. The process of pH buffering by minerals requires several days25, much slower than dew cycles (which require seconds). Accordingly, dew cycles can plausibly create local and temporary acidic spikes on top of a buffered bulk, similar to what we are showing.
We expected the acidity and the low salt content of the dew to be strongly denaturing conditions. The bulk, instead, retained high salt levels and quasi-neutral pH, ideal for stabilizing duplexes. We studied the conformation of a 24-bp dsRNA (33% GC, melting temperature (Tm) = 66 °C) in a solution of 12.5 mM MgCl2 and 10 mM Tris (initial pH of 7.0). The chamber walls were differentially heated at 31 °C and 43 °C (ΔT = 12 °C). The temperature of the chamber was, on average, 29 °C below the RNA Tm. In the dew, the dsRNA fraction decreased proportionally with increasing \(p_{{\mathrm{CO}}_2}\) (Fig. 2c), from 0.78 (±0.05) at ambient CO2 down to 0.17 (±0.08) for \(p_{{\mathrm{CO}}_2}\ge {0.8}\,{\rm{bar}}\). In the bulk, the dsRNA fraction remained close to 100%. Fluorescence images are shown in Fig. 2d and Supplementary Video 2.
The RNA FRET experiments were compared to a hybridization model based on the Van ‘t Hoff equation (Supplementary Section 5). The model was created by fitting experimental RNA melting curves measured at different pH values, Mg2+ concentrations and for different RNA sequences. The best agreement between experiments and the model was for a Mg2+ concentration in the dew of between 0.5 and 0.1 mM (Fig. 2c), which corresponds to an ~50–100× reduction compared to the bulk. This concentration agrees well with measurements of the salt content of rainwater26. With the same hybridization model we calculated the duplex RNA fraction (RNA FRET) as a function of pH and Mg2+ concentration (Fig. 2e). The relationship between Mg2+, pH and RNA FRET was nonlinear. We observed a sharp drop in RNA FRET at certain values of pH and Mg2+, suggesting that pH, Mg2+ and temperature are acting synergistically.
It is important to note that, due to the technical limitations of the pH-imaging method, the pH measurements of Fig. 2a were performed at 27 °C (warm side). However, the FRET measurements of Fig. 2d or the later replication experiments of Fig. 4a–d were instead performed at a higher temperature (warm side: 43 °C, 67 °C and 60 °C, respectively). Increasing the temperature can affect the pH of aqueous solutions27,28. The pH of CO2-saturated buffer-free water (that is, the dew) at a \(p_{{\mathrm{CO}}_2}\) of 1 bar undergoes negligible changes with increasing temperature (ΔpH/20 °C = +0.1)29. The dew’s pH is supposed to remain at ~4.0 up to 80 °C. On the other hand, we determined that the bulk could retain annealing conditions (duplex fraction >0.8) down to a pH of ~4.3 (Supplementary Section 5). A pH acidification of this magnitude is, however, not observed for neutral buffered solutions at a \(p_{{\mathrm{CO}}_2}\) of 1 bar, which tend to remain more neutral30,31. Hence, the main features of the water–dew cycles are stable across the different temperatures: acidic low-salt dew for the denaturation of oligonucleotides, and salt-rich and moderate pH in the bulk for annealing.
The dual combination of low salt content and low pH in the dew created a strong denaturing microenvironment that efficiently melted RNA at temperatures 30 K below the bulk Tm. No high-temperature spikes were needed to induce melting, maintaining a low RNA hydrolysis rate and therefore preserving sequence integrity for a longer time. However, selection pressure towards the survival of the longer RNA (or DNA) strands must be present in the system to overcome the ‘tyranny of the shortest’.
By simulation, we studied whether the non-equilibrium conditions of the dew chamber could host selective mechanisms for oligonucleotide length. We performed a three-dimensional (3D) finite-element simulation of an evaporating dew droplet and bulk, using the software COMSOL Multiphysics. The simulation was performed for DNA so as to simulate the DNA accumulation that takes place during the replication reactions that will follow. The diffusion coefficients (D) of DNA and RNA are in fact very similar (DssDNA/DssRNA = 1.15)32. The capillary flow, diffusion, thermophoresis and transport of dissolved DNA in a temperature gradient of 17 °C (hot side 60 °C, cold side 43 °C) were simulated. The size and shape of the simulated dew and bulk were geometrically identical to those in our experiments, as was the shape of the meniscus at the gas–water boundaries (Supplementary Section 6). The thermal gradient induced convection in the bulk and in the dew, with an average speed of 25 µm s−1. The differential evaporation rates at the warm and cold sides created a capillary flow with an average speed of 50 µm s−1 directed towards the hot side (Fig. 3a). The result was a net transport of DNA, driven by a continuous coffee-ring effect, that strongly accumulated DNA (up to 10,000+ fold for a 77-nt DNA) at the gas–liquid interface on the warm side. Our observations by fluorescence microscopy compared qualitatively well with the simulation results (Fig. 3b,c).

The preferential accumulation of longer DNA strands counteracts the tyranny of shorter sequences. a, Lateral distribution of DNA (77 nt) in the dew and bulk at steady state. Surface tension dictates the shape of the dew droplets (capillary bridge) and of the bulk (concave meniscus). Owing to a continuous coffee-ring effect, DNA accumulates at the gas–water boundaries. b, The same simulation viewed frontally (resized). c, Fluorescence image of DNA accumulation. d, Accumulation as a function of DNA length (1–300 nt). Owing to their lower diffusion coefficient, longer DNA strands are favoured and accumulate more strongly. Each data point corresponds to a simulation performed with a given DNA length.
The accumulation favoured longer DNA sequences (Fig. 3d). Because the diffusion coefficient of DNA decreases with length33, longer DNA molecules accumulated more strongly, as they had a slower back-diffusivity. The effect was more pronounced at the gas–water interface of the bulk, where it reached an accumulation factor of 20,000+ for a 300-nt DNA (versus 1,200+ at the dew’s interface).
So far, the CO2 dew chamber has shown characteristics that are essential for a molecular replicator in the RNA world:
-
Melting of oligonucleotides in the dew at 30 K below the bulk Tm. This provides low thermal stress to RNA while allowing long RNA sequences to cyclically melt and reanneal. The bulk also retains the high salt concentrations that are necessary for prebiotic replication chemistries.
-
Preferential accumulation of longer oligonucleotides at the evaporating gas–water interfaces. This feature can promote replication of the longer sequences, avoiding the detrimental kinetic race towards the shortest oligomers, as we have shown in the past for a bulk DNA replication in an open pore under thermal cycling34.
To mimic the yet-to-be-discovered replication mechanisms of the RNA world, assuming they existed, in the experiment we used a DNA replicating protein. First, we wanted to confirm that our CO2 dew chamber was able to perform DNA replication at temperatures much less than Tm. The Taq DNA polymerase enzyme has been used to amplify a 51-bp DNA (54% GC, Tm 88 °C) by templated polymerization. The mixture (QIAGEN Master Mix) consisted of 10 mM Tris (starting pH 8.2 ± 0.1), KCl and (NH4)2SO4 (concentration not specified), 1.5 mM MgCl2, nucleotides (200 µM each), DNA polymerase (2.5 U µl−1), to which we added the DNA template (0.5 nM) and complementary primers (forward and reverse, each at 0.25 µM). The solution was inserted inside the dew chamber in a temperature gradient of 15 °C (warm side 67 °C, cold side 52 °C). The average chamber temperature was 28 °C lower than the template Tm. After ~5 h of experiment, SYBR Green I fluorescence (a dsDNA fluorescent stain) started increasing in the bulk in the classical exponential manner (Fig. 4a,b and Supplementary Video 3). The replication product was confirmed on a 15% denaturing polyacrylamide gel (Fig. 4c). No replication was observed without CO2 enrichment, because no substantial DNA melting could occur in the dew and the whole replication could not continue further.

The dew chamber enables DNA replication at cold temperatures and drives molecular evolution towards longer strands. a–c, Replication of a single DNA template (51 nt). In a, fluorescence (SYBR Green I) images are shown of the replication reaction at different times. DNA accumulates and replicates at the gas–water interfaces and undergoes repeated wet–dry cycles. Dry replicative spots can be seen above the gas–water interface. In b, SYBR Green I fluorescence over time is shown for the CO2 dew chamber replication, showing classic exponential behaviour. In c, PAGE (15% denaturing, SYBR Gold staining) of the reaction products is shown. DNA replication is only possible at 1-bar pCO2. d, Replication with two DNA templates (47 and 77 nt), shown by PAGE of the reaction. The numbers at the top indicate the lanes. In contrast to the replication in the thermocycler, the CO2 dew chamber avoids the ‘tyranny of the shortest’ sequences and favours longer DNAs.
We studied whether the preferential accumulation of longer DNAs at the evaporating gas–water interfaces could actually prevent the ‘tyranny of the shortest’ sequences during replication. Under standard replication conditions (for example, standard thermocycling protocols), shorter DNA sequences have a kinetic advantage and prevail over the longer ones. This can be seen in the polyacrylamide gel electrophoresis (PAGE) results in Fig. 4d, fourth lane. In this experiment, two DNA templates of different lengths were used—47 nt (53% GC, Tm = 78 °C) and 77 nt (51% GC, Tm = 89 °C)—which shared the same primer binding sites. The replication of the shorter sequence (47 nt) prevailed and completely hindered the longer strand (77 nt).
The replication reaction was then performed in the CO2 dew chamber. We applied a temperature gradient of 17 °C (hot side 60 °C, cold side 43 °C) and enriched the gas with 1 bar of pCO2. After ~12 h, the reaction was stopped and the product was measured on the gel. As shown in Fig. 4d (sixth lane), not only could both DNA templates be replicated with comparable efficiency, but additional longer DNAs unexpectedly emerged in the CO2 dew replication. This indicated that the length-selective features of DNA accumulation at the gas–water interfaces promoted the survival and replication of the longer strands. The longer DNA products were observed only in the dew chamber enriched in pCO2, suggesting that they arose as a combination of enhanced DNA melting + preferential accumulation of long DNAs. In an ambient atmosphere (lane 7), only primer dimers were observed (similar to the negative control experiment without DNA templates, lane 8). The longer DNAs were probably created during the DNA wet–dry cycles, where the extremely high DNA and enzyme concentrations forced unspecific annealing and polymerization, leading to elongation and shortening processes. This created novel and mixed DNA sequences of increasing complexity. Also the formation of hairpins and other forms of self-priming can drive the formation of long replication strands35. But what are those long DNA products?
The products were sequenced using the Nanopore technique MinION (Methods). The sequencing machine yielded 2,856 reads with lengths between 140 and 1,300 nt. Reads shorter than 140 nt were not detected. In the length range of 140–300 nt, the reads contained numerous repetitions of primers and templates, attached one after the other multiple times, in a mixed and repetitive manner (Supplementary Section 7). With increasing lengths (up to 1,300 nt), the nucleotide composition of the reads was revealed to be biased towards AT (AT fraction >80%) and the primer/template repetitions were lost and could only rarely be found. The question is why the nucleotide composition of the sequences is so biased towards AT (>80%). To obtain such a bias in the immense sequence space of long oligonucleotides is entropically unfavourable. The answer lies in the adaptation of the sequences to the melting conditions of the dew, as we will demonstrate in the following sections.
The reads are plotted in Fig. 5a,b according to their AT and CG counts. This was done because the AT:GC ratio is one major factor that determines the stability of DNA duplexes19. The background of Fig. 5a,b corresponds to the duplex DNA fraction (indicated as DNA FRET) computed as a function of the AT and GC counts under the conditions of the CO2 dew (\(p_{{\mathrm{CO}}_2}\) of 1 bar, pH 4, Mg2+ 0.25 mM, temperature range 43–60 °C). The longer AT-rich reads (white circles) lie in a region of low-DNA FRET (0.14 ± 0.12), far away from what is expected from a pool of randomized sequences (black circles). No reads were found in the yellow area to the right or at the left blue extreme of the map. This suggests that the sequences with low stability, which were more prone to undergo multiple cycles of denaturation and reannealing, were the ones preferentially replicated. DNA sequences with a too high or too low melting temperature replicated more slowly, because they struggled to be cyclically melted and to template. In this sense, the DNA FRET landscape of the CO2 dew selected which sequences were replicated and which ones died out.

The CO2 dew chamber triggers the emergence of long DNA strands, the ATGC compositions of which correlate with the denaturing conditions of the dew. a, Reads (white circles) are plotted according to their AT (y axis) and CG (x axis) count. The background colour corresponds to the simulated duplex fraction (DNA FRET) in the CO2 dew. Denaturing conditions (DNA FRET = 0.14 ± 0.12) correlate with the sequence composition of the reads and drove the evolution of the sequences. The purple colourmap indicates the number of intact templates found in the read. b, Zoomed subsection (dashed box in a) of the surface plot. Starting from the initial templates, the DNA sequences, whether directly evolved towards the long AT-rich strands by means of mutations or into templates-rich strands, are driven by cross-templation. Similar results were observed when starting with a pool of 6–30-nt short templates rich in GC (orange line). c, Shortening, elongation and mutation processes modify the sequences during the replication in the dew cycles. d, Probability of every 4-nt subsequence combination (ordered by AT fraction) that were found in the experimental reads (green), in the model (gold) or in a random pool (black). The replication in the dew drove the evolution of sequences that frequently contain AT-rich motifs. e, DNA replication in the CO2 dew drastically reduces the informational entropy. The model (gold) and the experiment (green) show a similar entropy reduction. f, The average (<>) temperature of the dew can be modulated to drive the final AT:GC composition of the replicated DNA pool, with higher temperatures leading to strands richer in GC. Error bars indicate standard deviation.
To prove the generality of this concept—sequence adaptation to the melting conditions—we emulated an unspecific replication process via a stochastic model (Supplementary Section 8). Starting from an initial pool of DNAs, at every step the model calculated the probability of each sequence to be replicated according to its stability on the DNA FRET landscape of the dew. When a sequence was replicated, its reverse complement was created and a mutation introduced. The mutation consisted of the insertion or deletion of a random subsequence at a random position (Fig. 5c). This was done to mimic the unspecific templation characteristics seen in the CO2 dew replication experiments. Finally, at every step, random single-point mutations in the sequences were introduced with the frequency of the Taq polymerase error rate (1 × 10−4)36. The results are shown in Fig. 5a,b.
The simulation was started from the initial templates or from the sequences rich in template/primer repetitions. In both cases, the sequences replicated and randomly mutated. The DNA FRET landscape preferentially replicated the mutated sequences that best matched the denaturing conditions of the CO2 dew (that is, low FRET). The DNA sequences that were too stable or too unstable could not replicate further and were discarded. As a consequence, the DNA evolved towards longer sequences rich in AT. The ATGC content was thus adapting to the melting conditions of the environment. The simulation also highlighted a progressive loss of the intact templates within the sequence (pink gradient in Fig. 5a,b), similar to what has been observed experimentally, and a consequence of the progressive mutations at every replication step. The dashed lines (in Fig. 5a,b) correspond to other simulations using different parameters.
A similar sequence evolution was also observed for starting templates rich in GC (Fig. 5b, orange line), as is often the case in many non-enzymatic primer extension chemistries37,38.
To understand the nucleotide sequence of these long AT-enriched reads, we counted the frequency of every 4-nt submer combination that could be found within the reads. As shown in Fig. 5d, the most frequent submers were those rich in AT (AT fraction ≥0.75), with some of the most abundant ones being ATAT, AATA (experiment) and AAAA, TTAT (model). Both the experiment and the model showed similar fingerprints, with small differences in the most abundant combinations. The frequency distribution was also very different from a random pool.
The difference from a random pool could also be seen in terms of the relative reduction of Shannon entropy, as shown in Fig. 5e. This indicates the average decrease in the level of information encoded in the replicated sequences, relative to a random pool (the latter contains the maximum possible amount of information)39. Calculated as a function of the submer length, the relative entropy reduction in our experiments and model was <1 overall and reached a minimum of 0.56 (experiment) and 0.66 (model). In other words, the replicative constraint introduced by the DNA FRET landscape drove a selection of sequences that reduced the informational entropy by 44% (experiment) and 34% (model). This drastic reduction in entropy allowed the sequences to replicate and evolve in a subset of the sequence space that would be otherwise too immense to be explored12.
The model reproduced the key features and fingerprints that were measured in the experiments. Owing to its simplicity and purely stochastic nature, the model confirmed the hypothesis of the sequence adaptation to the melting conditions of the environment.
In a molecular replicator where DNAs evolve and adapt to the environment, it should be possible to modulate the final ATGC composition of the sequences by varying the melting conditions of the setting. Higher temperatures facilitate the melting of more stable DNA sequences (those richer in GC), moving the region of low DNA FRET towards higher GC fractions. We varied the average temperature of the dew chamber while maintaining a fixed temperature gradient of 17 °C (Fig. 5f). In the range from 50 to 75 °C, the AT:GC ratio of the replicated sequences changed from 8.0 ± 1.0 to 0.9 ± 0.1, respectively (model). A similar trend was observed in the experiments, where it dropped from 4.7 ± 1.1 to 0.9 ± 0.6 when the average chamber temperature was increased from 51 to 67 °C. The final ATGC composition was driven by the DNA stability landscape of the dew droplets, which was synergistically determined by their Mg2+ concentration (<0.25 mM), their pH (pH 4 at a \(p_{{\mathrm{CO}}_2}\) of 1 bar) and the temperature.
Starting with an out-of-equilibrium condition, we have demonstrated an autonomous molecular replicator of oligonucleotides that is compatible with an RNA world: low-temperature melting minimized the hydrolysis rates, and high salt levels were provided for prebiotic replication chemistries and for the catalytic activity of RNAs.
Conclusions
A CO2 dew chamber could accommodate the essential features of a molecular replicator in the context of the origin of life on Earth and the RNA world. It mimics the ubiquitous setting of a heated rock pore in a CO2-rich Hadean atmosphere. The acidic dew leads to strand displacement of oligonucleotides at 30 K below the melting temperature. At the same time, the closed rock pore maintains a pH-neutral bulk solution rich in salts that promotes strand reannealing and replication. The low temperatures offer a moderate hydrolysis rate for RNA, allowing for extended survival of the genetic information. As a consequence of the DNA wet–dry cycles and the DNA length-selective accumulation at the gas–water interfaces, the sequences recombine with increasing length and complexity, overcoming the ‘tyranny of the shortest’ dilemma and building new genetic information. In this primordial molecular replicator, the new DNA sequences adapt to the melting conditions of the environment and evolve towards a biased ATGC composition of reduced entropy.
Hadean water cycles in a CO2 atmosphere present adequate physico-chemical conditions to host the first reactions of molecular evolution during the origin of life. The non-equilibrium properties of the dew droplets boost chemical reactions that have limited efficiency under standard conditions. They provide a strong increase in concentrations at gas–water interfaces and during wet–dry cycles, fluctuations between low and high salt concentration and a periodic change between acidic and neutral pH. As shown here, the combination of the above processes enhances the replication of oligonucleotides with increasing lengths, opening the door to open-ended Darwinian evolution.
Methods
The experiments were carried out in a microfluidic chamber (250 µm × 30 mm × 14 mm) made of Teflon, placed on a silicon wafer and then between a transparent sapphire and a copper back plate. The silicon wafer was covered with a thin (50 µm) Teflon foil to increase the hydrophobicity. The liquid volume of every experiment was ~20 µl, corresponding to one-third of the chamber volume. The remaining two-thirds of the volume was left for the gas. A temperature gradient was produced by differentially heating the sapphire with rod resistors and the silicon back plate with a Peltier element. The temperature gradients applied in the experiments ranged from 5 to 17 °C. The software LabVIEW was used to maintain a constant target temperature with an accuracy of ±1 °C. Microfluidic tubings were connected to the chamber for the inflows and outflows of liquid, to control the flux of CO2 and to measure the barometric pressure within the chamber. The fluorescent microscope pointed towards the transparent sapphire and was focused on the cold wall.
The set-up for FRET and pH imaging consisted of a custom-built fluorescence microscope (with the body of a Zeiss Axiotech Vario microscope) equipped with four excitation light-emitting diodes (M340L4, M385L2, M470L2, M590L2; Thorlabs), excitation filters (BP 340/26, BP 379/34, BP 482/35, BP 588/20), beamsplitters (DC 475/40, DC 425 LP, DC 365 LP), a triple-edge dichroic mirror (DC 395/495/610), an Optosplit II with a ratiometric filter set (DC 600 LP, BP 536/40, BP 630/50), a Stingray-F145B ASG camera (ALLIED Vision Technologies) and a ×1 objective (AC254 100-A-ML Achromatic Doublet). The specifications for the light-emitting diodes, filters, beamsplitters and dichroic mirrors are given in nanometres (average/full-width at half-maximum). A schematic of the set-up is provided in Supplementary Section 4.
RNA oligonucleotides were purchased from biomers.net, with high-performance liquid chromatography purification. The sequences used for the FRET experiments were as follows (5′–3′)—24-bp RNA 33% GC: strand forward CGU AGU AAA UAT *FAM* CUA GCU AAA GUG, strand reverse CAC UUU AGC UAG AT*ROX*A UUU ACU ACG; 24-bp RNA 0% GC: strand forward AAA UAA AUA AUA T*FAM*AA AAU AAU AAA, strand reverse UUU AUU AUU UUA T*ROX*AU UAU UUA UUU. The two labelled complementary strands were diluted from stock solution (100 µM in nuclease-free water) and mixed together to a final concentration of 5 µM in salt-rich buffer (Tris 10 mM, MgCl2 10–12.5 mM, pH 7.0). To favour annealing of the two complementary strands, before every experiment the solution was heated and slowly cooled from 80 °C to 4 °C (ramp rate of −1 °C per 5 s) in a standard thermocycler (Bio-Rad CFX96 Real-Time System). Lysosensor Yellow/Blue DND-160 (stock 1 mM in anhydrous dimethyl sulfoxide) at a final concentration of 20 µM in aqueous solution was eventually added to the solution.
The replication reaction was performed using the Taq PCR Master Mix (QIAGEN) at 1× final concentration, primers forward and reverse 0.25 µM, SYBR Green I 2×, BSA 0.1%, DNA template 0.5 nM. The QIAGEN Master Mix 2× contains 3 mM MgCl2, 20 mM Tris, KCl (concentration unspecified), (NH4)2SO4 (concentration not specified), 400 µM of each dNTP and Taq DNA polymerase (5 U µl−1) and has a pH of 8.2 ± 0.1. For the reaction in the thermocycler, the protocol was as follows: initial heat activation at 95 °C for 3 min, 25 cycles of 90 °C for 5 s, 40 °C for 15 s and 60 °C for 15 s, then 4 °C for storage. For the reaction in the dew chamber, the same mixture was inserted after an initial activation step at 95 °C for 3 min. The temperature gradient was then applied and the gas phase was eventually enriched with 1 bar of CO2. For replication of the 51-bp DNA, we used the following sequences (5′–3′): template DNA 51 bp, TTA GCA GAG CGA GGT ATG TAG GCG GGA CGC TCA GTG GAA CGA AAA CTC ACG, primer forward TTA GCA GAG CGA GGT ATG TAG GCG G, primer reverse CGT GAG TTT TCG TTC CAC TGA GCG T. For replication of the 47-bp and 77-bp DNA, we used the following sequences (5′–3′): template DNA 47 bp AGG AGG CGG ATA AAG TAG GAC CAC GGG AGT CAG GCA ACT ATG GAT GA, template DNA 77 bp TCA TCC ATA GTT GCG AGT GTA GAT ACG AGG GCT TAC CAG CGA GAC CCC AGT AAC CAA GTC TAC TTT ATC CGC CTC CT, primer forward TCA TCC ATA GTT GC, primer reverse AGG AGG CGG ATA AAG T.
The samples were run on a 12.5% denaturing (50% urea) polyacrylamide gel, with an acrylamide:bis acrylamide ratio of 29:1, solidified with TEMED (tetramethylethylenediamine) and ammonium persulfate. The replicated DNA samples were mixed with the gel loading dye blue (BioLabs, final concentration 2×) and loaded onto the gel wells. The samples were run in an electrophoretic chamber at 50 V for 5 min (alignment step) followed by 300 V for 15–20 min. The gel was then taken out and stained with SYBR Gold in TBE (Tris/Borate/EDTA) 1× for 5 min, and then imaged at the ChemiDOC MP (Bio-Rad) imaging station.
To prepare the replicated DNA for sequencing with Nanopore we used the following protocol. The DNA was end-repaired and 3′ A-tailed with NEBnext Ultra II repair and an A-tailing module (purchased from New England Biolabs) by incubation at 20 °C for 10 min and subsequent enzyme inactivation at 65 °C for 5 min. The DNA was then purified using magnetic beads (Beckman Coulter AMPure XP). A barcode adapter from the Oxford Nanopore barcoding kit (kit NBD104) was then ligated using the NEBnext Quick ligation module by incubation at 20 °C for 15 min. Barcode-ligated DNA was purified with Beckman Coulter AMPure XP magnetic beads. The barcoded samples were then pooled and ligated to the Oxford Nanopore sequencing adapter preloaded with a stalled motor protein. Ligation was performed with an NEBnext Quick ligation module by incubation at 20 °C for 15 min. The pooled sequencing library was purified with Beckman Coulter AMPure XP magnetic beads and eluted in elution buffer (EB) provided with the Oxford Nanopore sequencing kit. The eluted library was mixed with loading beads and sequencing fuel buffer (ONT sequencing kit LSK109) and loaded to a flowcell on a MinION sequencer. The flowcell was run for 24 h. The raw signals were converted to FASTQ sequence reads by ONT basecaller guppy v3.6.
Data availability
The data used in this paper are available in a public repository at https://doi.org/10.5282/ubm/data.258. Source data are provided with this paper.
Code availability
The analysis scripts used to analyse the data are available in a public repository at https://doi.org/10.5282/ubm/data.258.
References
Szostak, J. W. The eightfold path to non-enzymatic RNA replication. J. Syst. Chem. 3, 2 (2012).
Taran, O., Thoennessen, O., Achilles, K. & von Kiedrowski, G. Synthesis of information-carrying polymers of mixed sequences from double stranded short deoxynucleotides. J. Syst. Chem. 1, 9 (2010).
AbouHaidar, M. G. & Ivanov, I. G. Non-enzymatic RNA hydrolysis promoted by the combined catalytic activity of buffers and magnesium ions. Z. Naturforsch. C. 54, 542–548 (1999).
Schildkraut, C. & Lifson, S. Dependence of the melting temperature of DNA on salt concentration. Biopolymers 3, 195–208 (1965).
Hampel, A. & Cowan, J. A. A unique mechanism for RNA catalysis: the role of metal cofactors in hairpin ribozyme cleavage. Chem. Biol. 4, 513–517 (1997).
Ianeselli, A., Mast, C. B. & Braun, D. Periodic melting of oligonucleotides by oscillating salt concentrations triggered by microscale water cycles inside heated rock pores. Angew. Chem. Int. Ed. 58, 13155–13160 (2019).
Bunville, L. G., Geiduschek, E. P., Rawitscher, M. A. & Sturtevant, J. M. Kinetics and equilibria in the acid denaturation of deoxyribonucleic acids from various sources. Biopolymers 3, 213–240 (1965).
Mariani, A., Bonfio, C., Johnson, C. M. & Sutherland, J. D. pH-driven RNA strand separation under prebiotically plausible conditions. Biochemistry 57, 6382–6386 (2018).
Yoon, J., Thirumalai, D. & Hyeon, C. Urea-induced denaturation of PreQ1-riboswitch. J. Am. Chem. Soc. 135, 12112–12121 (2013).
Blake, R. D. & Delcourt, S. G. Thermodynamic effects of formamide on DNA stability. Nucleic Acids Res. 24, 2095–2103 (1996).
Mills, D. R., Peterson, R. L. & Spiegelman, S. An extracellular Darwinian experiment with a self-duplicating nucleic acid molecule. Proc. Natl Acad. Sci. USA 58, 217–224 (1967).
De Duve, C. The onset of selection. Nature 433, 581–582 (2005).
Wachowius, F. & Holliger, P. Non-enzymatic assembly of a minimized RNA polymerase ribozyme. ChemSystemsChem 1, 1–4 (2019).
Zhou, L., O’Flaherty, D. K. & Szostak, J. W. Assembly of a ribozyme ligase from short oligomers by nonenzymatic ligation. J. Am. Chem. Soc. 142, 15961–15965 (2020).
Kasting, J. F. Earth’s early atmosphere. Science 259, 920–926 (1993).
Walker, J. C. G. Carbon dioxide on the early earth. Orig. Life Evol. Biosph. 16, 117–127 (1985).
Byck, H. T. Effect of dissolved CO2 on the pH of water. Science 75, 224 (1932).
Agerschou, E. D., Mast, C. B. & Braun, D. Emergence of life from trapped nucleotides? Non-equilibrium behavior of oligonucleotides in thermal gradients. Synlett 28, 56–63 (2017).
Wada, A. & Suyama, A. Local stability of DNA and RNA secondary structure and its relation to biological functions. Prog. Biophys. Mol. Biol. 47, 113–157 (1986).
Morasch, M. et al. Heated gas bubbles enrich, crystallize, dry, phosphorylate and encapsulate prebiotic molecules. Nat. Chem 11, 779–788 (2019).
Didenko, V. V. DNA probes using fluorescence resonance energy transfer (FRET): designs and applications. Biotechniques 31, 1106–1116 (2001).
Seibert, S. et al. Identification and quantification of redox and pH buffering processes in a heterogeneous, low carbonate aquifer during managed aquifer recharge. Water Resour. Res. 52, 4003–4025 (2016).
Appelo, C. & Postma, D. Geochemistry, Groundwater and Pollution (CRC Press, 2004).
Lacroix, E., Brovelli, A., Holliger, C. & Barry, D. A. Evaluation of silicate minerals for pH control during bioremediation: application to chlorinated solvents. Water Air Soil Pollut. 223, 2663–2684 (2012).
Lacroix, E., Brovelli, A., Holliger, C. & Barry, D. A. Control of groundwater pH during bioremediation: improvement and validation of a geochemical model to assess the buffering potential of ground silicate minerals. J. Contam. Hydrol. 160, 21–29 (2014).
Carroll, D., Udall, S. L. & Nolan, T. B. Rainwater as a Chemical Agent of Geologic Processes: A Review. Chemical Composition of Rainwater and Its Probable Relation to Soil Water and Exchangeable Cations in Weathering Processes (US Department of the Interior, 1962).
Langelier, W. F. Effect of temperature on the pH of natural waters. J. Am. Water Works Assoc. 38, 179–185 (1946).
Gieskes, J. M. Effect of temperature on the pH of seawater. Limnol. Oceanogr. 15, 329–329 (1970).
Peng, C., Crawshaw, J. P., Maitland, G. C., Martin Trusler, J. P. & Vega-Maza, D. The pH of CO2-saturated water at temperatures between 308 K and 423 K at pressures up to 15 MPa. J. Supercrit. Fluids 82, 129–137 (2013).
Liu, Q. & Maroto-Valer, M. M. Investigation of the effect of brine composition and pH buffer on CO2-brine sequestration. Energy Procedia 4, 4503–4507 (2011).
Holmes, J. D. et al. Buffering the aqueous phase pH in water-in-CO2 microemulsions. J. Phys. Chem. B 103, 5703–5711 (1999).
Baaske, P. et al. Extreme accumulation of nucleotides in simulated hydrothermal pore systems. Proc. Natl Acad. Sci. USA 104, 9346–9351 (2007).
Mast, C. B., Schink, S., Gerland, U. & Braun, D. Escalation of polymerization in a thermal gradient. Proc. Natl Acad. Sci. USA 110, 8030–8035 (2013).
Kreysing, M., Keil, L., Lanzmich, S. & Braun, D. Heat flux across an open pore enables the continuous replication and selection of oligonucleotides towards increasing length. Nat. Chem. 7, 203–208 (2015).
Park, D., Ellington, A. D. & Jung, C. Selection of self-priming molecular replicators. Nucleic Acids Res. 47, 2169–2176 (2019).
Potapov, V. & Ong, J. L. Examining sources of error in PCR by single-molecule sequencing. PLoS ONE 12, e0169774 (2017).
Duzdevich, D. et al. Competition between bridged dinucleotides and activated mononucleotides determines the error frequency of nonenzymatic RNA primer extension. Nucleic Acids Res. 49, 3681–3691 (2021).
Blain, J. C., Ricardo, A. & Szostak, J. W. Synthesis and nonenzymatic template-directed polymerization of 2'-amino-2'-deoxythreose nucleotides. J. Am. Chem. Soc. 136, 2033–2039 (2014).
Shannon, C. E. A mathematical theory of communication. Bell Syst. Tech. J. 27, 623–656 (1948).
Acknowledgements
We thank S. Krebs and colleagues from the Gene Center München for the preparation and sequencing of the DNA samples and L. Keil for sharing his expertise in the preparation of the set-up for FRET measurements and for programming support. We thank L. Karr for extensive support during the preparation of the experiments, J. Stein for useful discussions and A. Serrao and N. Yeh Martinez for proofreading the manuscript. Financial support was provided by the European Research Council (ERC Evotrap, grant no. 787356, D.B.), the Simons Foundation (grant no. 327125, D.B.), the Graduate School of Quantitative Biosciences Munich (QBM, A.I.), the CRC 235 Emergence of Life (Project-ID 364653263, D.B.), the Excellence Cluster ORIGINS (D.B.) funded by the Germany’s Excellence Strategy EXC-2094-390783311, and the Center for NanoScience (D.B.).
Author information
Authors and Affiliations
Contributions
A.I. performed the experiments, A.I. and M.A. performed the simulations and analysed the data. A.I. and P.W.K. analysed the sequencing data with Nanopore. A.I., M.A., U.G., C.B.M. and D.B. conceived the study, designed the experiments and the simulations and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Physics thanks Alexey Tkachenko, Elisa Biondi and Robert Endres for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary figures, tables and equations (1)–(26).
Supplementary Data
Supplementary Videos 1–3 and simulation files.
Source data
Source Data
Statistical source data for Figs. 2–5.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ianeselli, A., Atienza, M., Kudella, P.W. et al. Water cycles in a Hadean CO2 atmosphere drive the evolution of long DNA. Nat. Phys. 18, 579–585 (2022). https://doi.org/10.1038/s41567-022-01516-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41567-022-01516-z
This article is cited by
-
Formation mechanism of thermally controlled pH gradients
Communications Physics (2023)
-
Aqueous microdroplets promote C–C bond formation and sequences in the reverse tricarboxylic acid cycle
Nature Ecology & Evolution (2023)
-
Ribozyme-mediated RNA synthesis and replication in a model Hadean microenvironment
Nature Communications (2023)
-
Physical non-equilibria for prebiotic nucleic acid chemistry
Nature Reviews Physics (2023)
-
Mist and replication
Nature Physics (2022)
