
Abstract
Membrane nanopores are key for molecular transport in biology, portable DNA sequencing1,2,3,4, label-free single-molecule analysis5,6,7,8,9,10,11,12,13,14 and nanomedicine5. Transport traditionally relies on barrel-like channels of a few nanometres width, but there is considerable scientific and technological interest for much wider structures of tunable shape. Yet, these nanopores do not exist in nature and are challenging to build using existing de novo routes for proteins10,15,16,17. Here, we show that rational design with DNA can drastically expand the structural and functional range of membrane nanopores. Our design strategy bundles DNA duplexes into pore subunits that modularly arrange to form tunable pore shapes and lumen widths of up to tens of nanometres. Functional units for recognition or signalling can be optionally attached. By dialling in essential parameters, we demonstrate the utility and potential of the custom-engineered nanopores by electrical direct single-molecule sensing of 10-nm-sized proteins using widely used research and hand-held analysis devices. The designer nanopores illustrate how DNA nanotechnology can deliver functional biomolecular structures to be used in synthetic biology, single-molecule enzymology and biophysical analysis, as well as portable diagnostics and environmental screening.
Similar content being viewed by others

Main
The lumen of membrane nanopores defines their function in biology and technology. In nanopore sensing, channel width controls the entrance and passage of individual molecules and influences the electrical read-out signal caused when the analyte blocks the channel lumen18,19,20. Consequently, biological protein pores of around 1 to 5 nm width enable sensing of equally sized DNA strands2,3,4, organic molecules21,22 and small proteins11,12,13,20. Going beyond the current size remit is scientifically exciting and of relevance in research and sensing. For example, wider nanopores could transform fast and direct sensing and examination of large enzymes, immunoglobulins, protein complexes or even viruses at the single-molecule level. Going beyond the biological remit could also lead to non-cylindrical nanopores better matched to irregularly shaped analytes. As an additional desirable characteristic, wide and shape-defined nanopores should carry molecular receptors to enable highly specific analyte recognition or tethering of analyte molecules for detailed biophysical inspection11,12,23,24. Finally, the next-generation nanopores should be compatible with a wide range of electrical read-outs including the hand-held MinION kit pioneered for portable DNA sequencing2,3,4 but has not been used for protein detection so far. The desirable nanopores also offer applications beyond sensing, such as to form synthetic cells and to puncture biological cells for shuttling bioactive cargo.
Up to now, the next-generation nanopores have not been accessible by engineering proteins25,26 or peptide assemblies, despite considerable progress10,15,16,17. An underpinning challenge is the small size of amino acids and the complex folding of polypeptides into large unique protein structures27. DNA is an alternative material for designing bigger nanoscale architectures28,29,30,31,32 given the larger size of nucleotides27, simple base-pairing rules and the ease of predicting DNA folding33,34. Indeed, DNA membrane nanopores have previously been made with channel diameters up to 10 nm (refs. 35,36,37,38,39,40,41). All existing DNA nanopores use the classical parallel arrangement of DNA duplexes33,34, which puncture the membrane in a perpendicular fashion35,36,37,38,39,40,41.
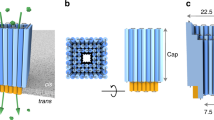
Here we explore how molecular design with DNA can expand the size and shape of membrane nanopores beyond the current biological and engineering remit. Upon initiating the design of the DNA nanopores, we noted that our freedom was unduly restricted by orienting all duplexes in a perpendicular fashion to the membrane. We decided to overcome this limitation with a pore that exploits the architectural freedom offered by building with DNA. In this pore, bundled DNA duplexes form subunits that are modularly arranged parallel to the membrane to form a defined shape (Fig. 1a–c). The use of modular subunits (Fig. 1b) can provide unprecedented design scope over pore shape and size, as demonstrated by a series of pore polygons including triangle, square, pentagon and hexagon (Fig. 1d). In combination with the controllable subunits’ side lengths (10 and 20 nm), the designed channel lumen can be altered from 43 nm2 for a triangle of 10 nm subunit length, to 400 nm2 for a square with 20 nm subunit length (Fig. 1d and Supplementary Table 1). The upper-sized DNA nanopore is up to 260-fold larger when compared with the 1.5 nm2 lumen area of a widely used protein pore (Fig. 1d, α-hemolysin).

a, A DNA nanopore with square-shaped lumen composed of bundled DNA duplexes (cylinders, blue) inserted into a membrane. b, Top-down view of the DNA nanopore in schematic and molecular representations. The pore is composed of four inter-connected subunits. c, The pore in a side view illustrates its outer-membrane cap (light blue) and the membrane-spanning barrel (dark blue). Cholesterol anchors (orange) at the bottom of the cap facilitate the insertion of the pore’s barrel into the membrane. d, In the top-down view, caps of DNA nanopores with different polygonal shapes and sizes cover a lumen area from 43 to 400 nm2. The names of the caps of the DNA nanopores refer to their polygonal shape and the side length of subunits in nanometres, such as Tri-10 for triangle and 10 nm. From left to right, d shows the Tri-10 cap, Sqr-10 cap, Pent-10 cap, Hex-10 cap, Tri-20 cap and Sqr-20 cap. The protein pore α-hemolysin (αHL) is shown for size comparison. ss, single-stranded DNA links; ds, double-stranded stabilisers.
Our pore architecture features an extra-membrane cap, which defines overall pore shape, and a barrel, which punctures the membrane (Fig. 1c). The cap is tunable in height and width by defining the subunits’ number of duplexes (Fig. 1c, Supplementary Figs. 1 and 2 and Supplementary Table 1). The subunits of the cap structure are connected by short, single-stranded DNA links at the subunits' innermost duplex position (Fig. 1d, inset, ss). Furthermore, rigid inter-subunit duplexes prevent the caps from flopping and prevent deviation from the designed shape (Fig. 1d, inset, ds; and Supplementary Figs. 1 and 2); stabilizing duplexes are not required in self-supporting triangles (Fig. 1d). The defined geometric shape of the caps also determines the pore’s transmembrane barrel (Fig. 1c, Supplementary Figs. 3 and 4 and Supplementary Table 2). In our current design, the pore wall is one duplex thick (Fig. 1b), but thicker barrel walls are possible. The barrel punctures the membrane with the aid of cholesterol lipid anchors (Fig. 1a,c, orange) that are positioned at the underside of the subunits’ caps (Fig. 1a,c). This pore design should allow pores to insert into classical lipid bilayers but also into membranes of MinION flow cells from Oxford Nanopore Technologies for direct and portable sensing of immunologically relevant proteins.
We validated the pore design by first fabricating a series of six polygonal caps without a membrane-spanning barrel, that is, Tri-10 cap, Sqr-10 cap, Pent-10 cap, Hex-10 cap with 10 nm subunits, Tri-20 cap and Sqr-20 cap with 20 nm subunits (Figs. 1d and 2a). The simple self-assembly route involves programmed folding of a mix of DNA scaffold and DNA oligonucleotide staples to sequence-specifically anneal the strands into the designed nanostructure28,33,34 (Supplementary Table 1; Supplementary Table 3, sequences of DNA staples; and Supplementary Table 4, restriction enzymes for cutting scaffold). The resulting self-assembled DNA caps were homogeneous as implied by single bands in gel electrophoresis (Fig. 2a, isolated gel lanes; and Supplementary Fig. 5, entire electropherogram).

a, Schematic illustration and gel electrophoretic band (left) and TEM image (right) for DNA nanopore caps Tri-10 cap, Sqr-10 cap and Pent-10 cap (top row), and Hex-10 cap, Tri-20 cap and Sqr-20 cap (bottom row). Scale bar, 25 nm. b, Schematic illustration of membrane-inserted DNA nanopore Sqr-20. c, Gel electrophoretic analysis of Sqr-20 without (left) and with (right) attached cholesterols, leading to a gel upshift typical of cholesterol tagging41. d, TEM analysis of Sqr-20 alone (left), and Sqr-20 bound to a POPC bilayer vesicle (right). Scale bar, 30 nm. e, Fluorescence microscopic images of the binding of Atto647N-tagged pore Sqr-20 (red channel) to the bilayer membrane of a GUV composed of lipid POPC and doped with 0.5% BODIPY (green channel). Scale bar, 20 µm.
The caps’ fine structures were confirmed by negative staining and transmission electron microscopy (TEM) following gel purification. In line with the design, the caps featured the expected polygonal shapes (Fig. 2a and Supplementary Fig. 6) and subunit lengths, as shown for Tri-10 cap, Sqr-10 cap and Sqr-20 cap with lengths of 9.62 ± 0.69 nm, 9.86 ± 0.37 nm and 20.17 ± 1.74 nm, respectively (n = 15, 17 and 16; n = number of analysed caps; Supplementary Tables 1 and 5).
After confirming the validity of the cap design, we tested whether membrane-spanning barrel-cap pores can also be tuned in shape and size. The target pores were Tri-10, Sqr-10, Tri-20 and Sqr-20 (Fig. 2b for Sqr-20; and Supplementary Figs. 3 and 4 and Supplementary Table 2 for other pores). For membrane interaction, the pores carry on the underside of the 5 × 2 duplex caps several cholesterol tags, ten per 10 nm subunit and fifteen per 20 nm subunit (Supplementary Figs. 3 and 4 and Supplementary Table 2). Fabrication was achieved by first assembling pores without cholesterol tags (Supplementary Table 6, sequences of DNA staples). Assembly yielded the anticipated single, defined bands in gel electrophoresis (Fig. 2c, Sqr-20; and Supplementary Fig. 7, other pores) and the expected pore structure in TEM analysis (Supplementary Fig. 8). In the second step, the pores were equipped with cholesterol tags by hybridizing cholesterol-modified DNA oligonucleotides to handles at the underside of caps (Supplementary Figs. 3 and 4; Supplementary Table 6, DNA sequences). The transition to cholesterol-modified pores was complete as inferred from the disappearance of the non-cholesterol pore band and the appearance of the upshifted cholesterol pore band in gel electrophoresis (Fig. 2c, Sqr-20; and Supplementary Fig. 7, other pores).
To verify that the cholesterol tagging enables pores to bind to lipid membranes, TEM analysis was first conducted to directly visualize the cholesterol-tagged Sqr-20 pore on small unilamellar vesicles (SUVs; 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) lipid; Fig. 2d and Supplementary Fig. 9). Efficient membrane binding was further demonstrated with an Atto647N-labelled Sqr-20 pore and its fluorescence microscopic colocalization with the membrane of giant unilamellar vesicles (GUVs) doped with BODIPY Fig. 2e).
To confirm that DNA nanopores puncture the bilayer membrane via a defined yet tunable channel lumen, single-channel current recordings with a desktop kit were used. The technique measures the flow of electrolyte ions through a channel when a voltage is applied across the membrane. We analysed pores Tri-10, Sqr-10 and Sqr-20 in expectation of stable pore current traces with conductances increasing along the channel size (Fig. 3a(i),b(i),c(i)). Indeed, all pore currents were without fluctuations (Fig. 3a(ii),b(ii),c(ii)) under the standard conditions of 1 M KCl, 10 mM HEPES, pH 7.4 and a transmembrane potential of +50 mV relative to the cis side. Furthermore, larger pores led to higher conductances (Fig. 3a(ii),b(ii),c(ii)) with averages of 2.10 ± 0.21 nS (n = 19), 4.58 ± 0.27 nS (n = 29) and 8.91 ± 0.33 nS (n = 17; Fig. 3a(iv),b(iv),c(iv)). The 2.2-fold conductance step from Tri-10 to Sqr-10 is in good agreement with the 1.8-fold calculated enhancement42. The 1.9-fold step from Sqr-10 to Sqr-20 is lower than the 2.8-fold calculated increase. Possibly, lateral membrane pressure might reduce the effective pore size of the inserted channel, something that can be overcome by increasing rigidity in the pore designs. Any compression did not, however, cause any deviation from the linear current–voltage relationship (Fig. 3a(iii),b(iii),c(iii) and Supplementary Figs. 10 and 11), as is expected for the vertically symmetric DNA nanopores.

a–c, Analysis via single-channel current recordings of the Tri-10 (a), Sqr-10 (b) and Sqr-20 (c) DNA nanopores, illustrated in schematic drawings (i), using representative single-current traces recorded at +50 mV relative to the cis chamber (ii). Average current–voltage graphs (±s.e.m.) for voltages ranging from −100 mV to +100 mV at 20 mV steps (iii), and conductance histograms obtained at +10 mV (iv). For the recordings, the cholesterol-tagged nanopores were mixed with mild detergent octyl polyoxyethylene (OPOE) and then added to the cis chamber for membrane insertion. The recordings were acquired in 1 M KCl, 10 mM HEPES, pH 7.4. Green dots in i represent electrolyte ions which carry ionic current indicated as green arrow. The magnitude of the ionic current increases with a larger pore lumen. Data in iii and iv are from 19, 29 and 17 individual insertions of pores Tri-10, Sqr-10 and Sqr-20, respectively.
We next tested whether DNA nanopores permit label-free, direct and specific detection of IgG antibodies at the single-molecule level. A Sqr-10 pore was chosen to accomodate the 10-nm-sized IgG analyte. The pore was equipped with a biotin tag (Sqr-10-Biot) for binding of the target anti-biotin antibodies (Fig. 4a and Supplementary Fig. 3). Dot blotting confirmed specific binding of the antibody to the biotin tag in Sqr-10-Biot (Supplementary Fig. 12). In single-channel current recordings, the tag did not alter the conductance properties (Fig. 4b), yet antibody addition led to anticipated current blockades representing individual binding events to the biotin tag (Fig. 4b, the downward arrow indicates antibody addition; and Supplementary Fig. 13). The frequency of events increased linearly with the antibody concentration (Supplementary Fig. 13) as summarized in a plot of the antibody concentration versus 1/τon (Fig. 4c), where τon is the inter-event interval (Fig. 4b). Binding is specific because antibody did not elicit blockades in non-biotinylated Sqr-10 (Supplementary Fig. 14) and neither did addition of a non-specific antibody to biotinylated Sqr-10-Biot (Supplementary Fig. 15). Similarly, Sqr-10-Biot did not yield blockades with non-cognate bovine serum albumin (BSA; Supplementary Fig. 16).

a, Anti-biotin antibody (dark pink) binding to Sqr-10-Biot carrying biotin (green). b, Single pore current trace at +50 mV transmembrane potential before and after (arrow) addition of anti-biotin antibody at 9.5 nM to the cis side of the DNA nanopore. A single event is characterized by the event dwell time, τoff, and the event amplitude, A, from the open-channel current, Io. τon is the time between events. The schemes on the right illustrate the DNA nanopore in side-view without (top) and with a bound antibody (bottom). The pore current, represented as green arrow, is smaller when the antibody blocks the channel lumen. c, The frequency of binding events, as described by 1/τon (data from three independent experiments, ±s.d.), increases with antibody concentration, recorded at +50 mV. d, Scatter plot of n = 206 anti-biotin antibody binding events represented by their τoff and relative amplitude, A/Io, at 47 nM concentration of antibody, recorded at +50 mV. e, Antibody binding events in traces occur only at positive transmembrane potential. f, Current power spectral density characterization of Sqr-10-Biot after the addition of 47 nM antibody, comparing the noise analysis at +50 mV and −50 mV. The recordings (b–f) were acquired in 1 M KCl, 10 mM HEPES, pH 7.4. g, Tri-20-Spike pore inserted into schematically drawn membrane arrays of the high-throughput MinION device allows for highly parallel SARS-CoV-2 antibody sensing. The MinION is 10 cm in length. h, Single-channel current traces of Tri-20-Spike at −50 mV before and after (arrow) addition of 44.5 nM SARS-CoV-2 anti-spike antibody. i, Scatter plot of n = 358 antibody binding events of Tri-20-Spike with 44.5 nM SARS-CoV-2 anti-spike antibody, represented in relation to the τoff and relative amplitude, A/Io, at −40 mV.
Antibody-binding traces were analysed to reveal the single-molecule binding kinetics for comparison with ensemble measurements. Each binding event was plotted with its relative amplitude, A/Io (Fig. 4b),and duration, τoff (Fig. 4b) as points in a scatter diagram (Fig. 4d). More than 95% of the binding events clustered at a relative amplitude of 48.2 ± 7.7% (±s.d., n = 205; Fig. 4d) and had a mean τoff at 0.66 ± 0.02 s, calculated from the single exponential decay fit to the dwell time frequency histogram (Fig. 4d and Supplementary Fig. 17). Event parameters τoff and τon (Fig. 4c) yielded the kinetic rate constants for dissociation and binding, koff and kon, respectively, whereby koff was obtained from 1/τoff and kon from the gradient of 1/τon plotted against antibody concentration (Fig. 4c). The koff and kon values at 1.79 ± 0.05 s−1 and 1.89 ± 0.38 × 109 M−1 s−1, respectively, are typical for tight antibody binding and slightly higher than the ensemble measurements of 0.056 ± 0.001 s−1 and 1.73 ± 0.05 × 108 M−1 s−1 obtained via switchSENSE technology (Supplementary Fig. 18). The single-molecule equilibrium dissociation constant, Kd = koff/kon, is 9.45 ± 1.92 × 10−10 M (±s.e.m.) is very close to the ensemble-derived value of 3.25 ± 0.11 × 10−10 M (Supplementary Fig. 18), thereby validating the single-molecule binding analysis via the DNA nanopore. Voltage-dependent recordings established that protein transport across the pore is mediated via electrophoresis rather than electro-osmosis43 (Supplementary Fig. 19). In support of electrophoresis, only positive (not negative) potentials led to current blockades for antibody binding (Fig. 4e) as well as a higher power spectrum (Fig. 4f) suggesting antibody movement within the pore. Electrophoresis was also found for the BSA transit through the non-biotinylated DNA nanopore, as inferred from the voltage-dependent power spectrum (Supplementary Fig. 20).
We finally evaluated whether DNA nanopores can insert into membranes of high-throughput MinION flow cells to rapidly sense human SARS-CoV-2 antibodies. To achieve a better geometric match to the Y-shaped analyte, the triangular pore Tri-20 was selected (Fig. 4g). Tri-20 without the antibody receptor inserted successfully into membrane arrays (Fig. 4g) and yielded steady single-channel currents with a mean conductance at 7.83 ± 0.71 nS (±s.e.m., n = 9; Supplementary Fig. 21). The conductance is 1.7-fold higher than for reference Sqr-10 and hence in excellent agreement with the 1.7-fold larger expected conductance increase. Tri-20 was converted into a sensor for SARS-CoV-2 antibodies by specifically attaching the cognate receptor, SARS-CoV-2 spike protein, into the pore lumen (Fig. 4g). The attachment was mediated via an irreversible metal chelate bridge to a linker oligonucleotide (Supplementary Fig. 22). The resulting Tri-20-Spike pore yielded stable read-out traces in highly parallel MinION recordings (Supplementary Fig. 21). The average conductance at 1.8 ± 0.2 nS (±s.e.m., n = 23; Supplementary Fig. 21) is lower than for Tri-20 and confirms the presence of the spike protein in the pore lumen. Addition of human SARS-CoV-2 antibodies to Tri-20-Spike led to current blockades (Fig. 4h and Supplementary Fig. 23), which represent individual binding events to the spike receptor. The binding events had a relative amplitude of 61.7 ± 6.9% (±STD, n = 357) and an average dwell time of 1.7 ± 8.6 s (±STD, n = 357; Fig. 4i), implying that the triangular pores’ shape leads to a high-blockade read-out. Antibody binding is specific as anti-SARS-CoV-2 antibody did not elicit blockades in Tri-20-Link (pore plus linker, without spike protein; Supplementary Fig. 24) and neither did addition of a non-specific antibody (Supplementary Fig. 25).
Reflecting on the importance of membrane nanopores in nature and technology, our study introduces synthetic versions that operate in a size and shape regime not offered by biology. The wide design space is gained by recruiting DNA nanotechnology, and the utility of the approach is demonstrated by directly detecting single protein molecules inside the channel lumen using widely used desktop and hand-held analysis devices. Direct sensing of 10-nm-sized analytes has not yet been possible with membrane-inserted nanopores given the smaller channel width of biological protein pores. Direct placement of protein within the channel is, however, essential to gain the maximum information content about the single molecules’ biophysical, enzymatic or interaction properties13,20,44 as opposed to indirect sensing when analytes do not enter the pore but cause a secondary read-out11,12,23. By custom designing pore shape and size to the analyte, the DNA nanopores also deliver a substantial change to the usual approach of selecting fixed-size biological pores to match a target analyte. The large DNA nanopores may in the future be improved to yield irregular-polygonal pore geometries to match asymmetrically shaped analytes. Irregular geometries are accessible with pore subunits of non-uniform length and changeable inter-subunit angles. Limitations of the DNA nanopores include their propensity to undergo undesirable gating at transmembrane voltages >80 mV; this may be addressed by sensing at lower voltages or by chemically stabilizing the DNA nanostructures45. Another limitation of negatively charged DNA nanopores is the possible electrostatic adsorption of positively charged protein analytes41; a route against adsorption is to sense at pH values that render the protein charge neutral41. Furthermore, the current DNA nanopores show a lower tendency to insert into semifluid membranes of read-out devices than several protein pores, including CsgG, used for DNA sequencing. Higher insertion may be achieved by optimizing the DNA nanopore design including lipid anchor number and position, the membrane composition, the conditions of pore insertion46 or a combination of the above.
Single-molecule analysis with DNA nanopores may be further enhanced by placing any type and number of recognition tags at any lumen position, something that is not possible with any other pore composed of protein25,26, solid-state18,19,20,24,47,48 or hybrid materials15,16, to the best of our knowledge. Attachment of proteins with defined recognition or catalytic activity can also help to add functions not easily accessible with the pore’s construction material of DNA27. One application beyond single-molecule analysis is to use DNA nanopores for transporting bioactive cargo across cells5. Outside of membranes, an equally exciting prospect is to use the highly tunable DNA scaffold to enclose enzymes and design multienzyme assemblies with enhanced catalytic activity27. In conclusion, our route to synthetic pores delivers functional DNA nanostructures for real-world applications, advances synthetic biology with biomolecular structures not accessible in nature and helps transform portable and fast protein sensing for an impact in society.
Methods
Materials
All DNA oligonucleotides were purchased from Integrated DNA Technologies, except the 5′-cholesterol-labelled DNA oligonucleotide and the 5′-nitrilotriacetate (NTA)-tagged DNA oligonucleotide, which were obtained from Eurogentec and Gene Link, respectively. The M13 and phiX174 DNA scaffolds and all restriction enzymes were ordered from New England Biolabs. The 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC, 207131-40-6) and POPC (26853-31-6) were procured from Avanti Polar Lipids. All other reagents were purchased from Sigma-Aldrich unless stated otherwise.
DNA nanopore design
The DNA origami-based caps and nanopores were designed using CaDNAno software34. For the polygonal caps, the single-stranded M13 DNA was used as the scaffold strand, and for the DNA nanopores, the phiX174 DNA was used as the scaffold. Two-dimensional DNA maps for DNA caps and pores are provided in Supplementary Figs. 1–4. In lipid anchor-modified pores, DNA oligonucleotides carrying a cholesterol tag at the 5′ or 3′ terminus are attached via adaptor oligonucleotides to the pores (Supplementary Figs. 3 and 4). The Sqr-10-Biot pore is modified at the channel entrance (Supplementary Fig. 3) with a 5′-biotin-labelled DNA oligonucleotide. The sequences of staple strands for DNA caps are provided in Supplementary Table 3, and the sequences for the DNA pores’ staple strands, adaptor strands, cholesterol-modified anchor strands, biotin-tagged strand and NTA-modified strand are summarized in Supplementary Table 6.
Preparation of polygonal DNA caps and nanopores
To fold the DNA origami-based polygonal caps and nanopores, the M13 or phiX174 scaffold was mixed at a 1:10 ratio with corresponding staples in 0.5× TAE buffer (20 mM tris(hydroxymethyl)aminomethane (Tris) base, 10 mM acetic acid, 0.5 mM ethylenediaminetetraacetic acid (EDTA), pH 8.3) supplemented with 16 mM MgCl2. The DNA origami structures were folded using a 40 h folding programme: firstly, the solutions were heated at 75 °C for 5 min to denature undesired DNA secondary structures; then, for annealing, the solutions were cooled from 65 °C to 25 °C at a rate of 1 °C per h, followed by cooling to 10 °C at a rate of 1 °C per 5 min, and kept at 4 °C until collection. For the biotin-tagged pore, the 5′-biotinylated strand was used to replace the corresponding non-biotinylated staple strand. For the polygonal caps, the M13 scaffold was first treated using restriction enzymes before assembly. The enzyme and buffers used for the restriction cutting are summarized in Supplementary Table 4. After the folding process, the DNA origami structures were purified by excision from 1% agarose gel in 0.5× TBE buffer (45 mM Tris–borate, 1 mM EDTA, pH 8.3) supplemented with 11 mM MgCl2. The cholesterol-tagged pores were prepared by adding cholesterol-labelled DNA oligonucleotides (Supplementary Table 5) to the purified DNA nanopores at a stoichiometry of 1.5 relative to the total number of DNA cholesterol attachment sites at the DNA nanopores. It is essential that the cholesterol-tagged pores be freshly prepared and used for the membrane binding and current recording experiments on the same day.
To prepare Tri-20-Spike, the NTA-modified DNA oligonucleotide (30 pmol) was mixed with the His-tagged receptor-binding domain (45 pmol) of the SARS-CoV-2 spike 1 protein in HEPES buffer (25 mM, pH 7.6) containing CoCl2 (20 µM), NaCl (400 mM) and Tween 20 (0.02 %v/v) and incubated for 1 h at room temperature. The solution was treated with H2O2 (20 mM) for 1 h to form an irreversible Co3+-mediated chelate bridge between NTA and hexa-His49. The DNA–protein conjugate was then examined by 10% polyacrylamide gel electrophoresis analysis. The freshly prepared DNA–protein conjugate was then mixed with purified Tri-20 bearing the complementary binding strand for the DNA–protein conjugate (Supplementary Fig. 4a) at a ratio of 1.5:1 and incubated for 1 h at room temperature. The Tri-20-Spike pore was aliquoted and stored at –20 °C for further use.
Agarose gel electrophoresis analysis of the DNA caps and nanopores
The folded DNA caps and nanopores were analysed using 1.5% agarose gel electrophoresis in 0.5× TAE buffer (20 mM Tris base, 10 mM acetic acid, 0.5 mM EDTA, pH 8.3), supplemented with 11 mM MgCl2. Gels were run at 70 V in an ice-water bath for 1 h.
TEM characterization of the DNA caps and nanopores
Purified DNA caps and pores without cholesterol tags (6 μl) were added onto TEM grids treated with a glow discharge (Agar Scientific, AGG2050C) and stained with 2% uranyl formate solution. The samples were analysed on a JEM-2100 electron microscope operated at 200 kV, and the images were acquired with an Orius SC200 camera. Pores carrying cholesterol tags were incubated with POPC SUVs in 0.5× TAE supplemented with 500 mM NaCl for 20 min, followed by deposition onto TEM grids, drying, negative staining and TEM analysis. The preparation of SUVs involved drying a POPC solution (20 mg ml–1 in chloroform, 50 µl) in a glass vial (2 ml) by argon air flow, resuspension of the dried film in 1× incubation buffer (0.5× TAE with 500 mM NaCl to 1 ml) and sonication for 30 min.
Microscopic analysis of DNA nanopore binding to GUVs
GUVs composed of POPC phospholipid were prepared via electroformation. Briefly, two droplets of POPC solution (10 mg ml–1 in chloroform doped with 0.5% β-BODIPY 500/510 C12-HPC, D3793, Thermo Fisher; 3 µl each) were added onto an indium tin oxide (ITO) glass slide. The solvent was left to evaporate in air for 5 min, leading to the formation of a dried lipid film. The glass slide was placed into a Vesicle Prep device (Nanion), and the lipid film patches were confined by two O-rings. Sucrose solution (1 M in water, 600 µl) was added, and another ITO glass slide was placed on top to form a sealed chamber. An alternating electric field of 3 V at 5 Hz was applied across the two slides for 120 min, and the solution of vesicles was collected afterward and stored at 4 °C. For DNA nanopore binding and visualization, DNA pores (20 nM, 20 µl) were incubated with POPC GUVs (500 µl) in 300 mM NaCl, 50 mM HEPES, pH 7.6 for 30 min. GUVs with bound DNA nanopores were imaged with an inverted confocal microscope (SPEinv, Leica).
Dot blot analysis of biotin-tagged DNA nanopores
DNA nanopores Sqr-10-Biot and Sqr-10, with and without biotin modification, respectively, were diluted in 0.5× TAE supplemented with 16 mM MgCl2 and pipetted onto the surface of a positively charged nylon membrane (Sigma-Aldrich, 11209299001) before being left to dry for 2 h. DNA was covalently linked to the membranes using UV illumination for 30 s. Membranes were incubated in blocking buffer TBST (50 mM Tris–Cl, 150 mM NaCl, pH 7.0, 0.1% Tween 20) supplemented with 5% BSA under shaking for 1 h, before incubation with anti-biotin antibody (Sigma-Aldrich, SAB4200680) in TBST with 5% BSA under shaking at 4 °C overnight. The blot was washed three times with TBST before incubation with a fluorescently labelled goat anti-mouse IgG secondary antibody (Thermo Fisher Scientific, A-11001) in TBST with 5% BSA for 1 h with shaking at room temperature. The blot was washed three times with TBST before imaging.
Nanopore recordings with lipid bilayers
Single-channel current recordings were carried out using a parallel recording set-up based on an integrated chip (Orbit Mini and Orbit 16, Nanion Technologies) with multielectrode-cavity-array chips (IONERA)39,41. Bilayers were formed of DPhPC dissolved in octane to a final concentration of 10 mg ml–1. The electrophysiological buffer was composed of 1 M KCl, 10 mM HEPES, pH 7.4. For pore insertions, DNA nanopores were mixed in a 2:1 (v/v) ratio with 0.5% OPOE in 1 M KCl, 10 mM HEPES, pH 7.4. The mixture was applied to the cis chamber and insertions monitored by increases in conductance steps. For protein sensing experiments, anti-biotin antibody (Sigma-Aldrich, SAB4200680) or BSA (Sigma-Aldrich, 9048-46-8) was diluted in electrophysiological buffer to the desired concentration. Upon successful pore insertion, diluted protein was added to the cis chamber. The Orbit 16 was used for protein sensing experiments; the Orbit mini was used in all other electrophysiological experiments. The Orbit 16 and Orbit mini are grounded at the cis and trans, respectively. To aid in comparison, voltages were normalized and are presented as positive in relation to the cis chamber. The current traces were acquired at 10 kHz using Element Data Recorder software (Elements). Traces on protein sensing were Bessel-filtered at 5 kHz before analysis. Single-channel analysis was performed using Clampfit software (Molecular Devices).
Kinetic analysis of antibody binding using switchSENSE technology
Kinetic analysis on ensemble binding of anti-biotin antibody was performed with the switchSENSE technology and heliX biosensor from Dynamic Biosensors. The company’s heliOS software was used to design the experiment, manage workflows and analyse the data. Binding analysis was performed in fluorescence proximity sensing mode with an applied constant voltage of −0.1 V, which renders the surface-tethered DNA at a fixed angle. In fluorescence proximity sensing mode, analyte binding to the DNA target alters the average distance of the fluorescent label from the fluorescence-quenching gold surface. The fluorescence signal may also be influenced by the proximity or direct interaction of the protein to the fluorescent dye. To obtain the kinetics of anti-biotin antibody binding, the protein analyte was diluted to specified concentrations in PE140 buffer (10 mM Na2HPO4/NaH2PO4, 140 mM NaCl, 0.05% Tween 20, 50 µM EDTA, 50 µM egtazic acid) and flushed at a flow rate of 200 μl min–1 over the two electrodes of the biochip; the temperature of the biochip and the sample tray was set at 25 °C. The passing of the analyte over the biotin target molecule in spot 1 led to a decrease in fluorescence signal over seconds, which reflects the binding kinetics. Flushing out the analyte with pure buffer was used to obtain the dissociation kinetics. The association and dissociation rates (kon and koff), dissociation constants (Kd) and respective error values were derived from a global bi-exponential fit model, after correction for blank buffer.
Nanopore recordings using MinION devices
Electrophysiological current recordings were conducted using the MinION analysis device and flow cells (Oxford Nanopore Technologies). The electrolyte solution was 1 M KCl equivalent. Flow cells were made with proprietary membrane materials of Oxford Nanopore Technologies. POPC SUVs were used to facilitate the fusion of the DNA nanopore to the MinION membranes. The vesicles were prepared as described in ‘TEM characterization of DNA caps and nanopores’. For pore insertion, DNA nanopores and vesicles at a ratio of 3 nM:1 mM lipid were incubated at 4 °C overnight. Subsequently, DNA nanopore-containing vesicles were added to the MinION flow cells, 20 µl at a time. To promote fusion with the membranes, a voltage ramp protocol from 50 mV to 300 mV was applied. Upon successful insertions, recordings for conductance and current–voltage curves were acquired. For molecular sensing with the Tri-20-Spike pore, human SARS-CoV-2 antibodies (Antibodies-online, ABIN6952547) were added directly to the flow cell.
Data availability
Data supporting the results and conclusions are available within this paper and the Supplementary Information. Additional raw data are available at Figshare: https://doi.org/10.6084/m9.figshare.19174400.
References
Clarke, J. et al. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 4, 265–270 (2009).
Quick, J. et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 530, 228–232 (2016).
Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345 (2018).
Van der Verren, S. E. et al. A dual-constriction biological nanopore resolves homonucleotide sequences with high fidelity. Nat. Biotechnol. 38, 1415–1420 (2020).
Majd, S. et al. Applications of biological pores in nanomedicine, sensing, and nanoelectronics. Curr. Opin. Biotechnol. 21, 439–476 (2010).
Howorka, S. Building membrane nanopores. Nat. Nanotechnol. 12, 619–630 (2017).
Huang, G., Voet, A. & Maglia, G. FraC nanopores with adjustable diameter identify the mass of opposite-charge peptides with 44 dalton resolution. Nat. Commun. 10, 835 (2019).
Ouldali, H. et al. Electrical recognition of the twenty proteinogenic amino acids using an aerolysin nanopore. Nat. Nanotechnol. 38, 176–181 (2020).
Cao, C. et al. Discrimination of oligonucleotides of different lengths with a wild-type aerolysin nanopore. Nat. Nanotechnol. 11, 713–718 (2016).
Mahendran, K. R. et al. A monodisperse transmembrane alpha-helical peptide barrel. Nat. Chem. 9, 411–419 (2017).
Rotem, D., Jayasinghe, L., Salichou, M. & Bayley, H. Protein detection by nanopores equipped with aptamers. J. Am. Chem. Soc. 134, 2781–2787 (2012).
Thakur, A. K. & Movileanu, L. Real-time measurement of protein–protein interactions at single-molecule resolution using a biological nanopore. Nat. Biotechnol. 37, 96–101 (2019).
Galenkamp, N. S., Biesemans, A. & Maglia, G. Directional conformer exchange in dihydrofolate reductase revealed by single-molecule nanopore recordings. Nat. Chem. 12, 481–488 (2020).
Brinkerhoff, H., Kang, A. S. W., Liu, J., Aksimentiev, A. & Dekker, C. Multiple rereads of single proteins at single-amino acid resolution using nanopores. Science 374, 1509–1513 (2021).
Spruijt, E., Tusk, S. E. & Bayley, H. DNA scaffolds support stable and uniform peptide nanopores. Nat. Nanotechnol. 13, 739–745 (2018).
Henning-Knechtel, A., Knechtel, J. & Magzoub, M. DNA-assisted oligomerization of pore-forming toxin monomers into precisely-controlled protein channels. Nucleic Acids Res. 45, 12057–12068 (2017).
Xu, C. et al. Computational design of transmembrane pores. Nature 585, 129–134 (2020).
Feng, J. et al. Identification of single nucleotides in MoS2 nanopores. Nat. Nanotechnol. 10, 1070–1076 (2015).
Miles, B. N. et al. Single molecule sensing with solid-state nanopores: novel materials, methods, and applications. Chem. Soc. Rev. 42, 15–28 (2013).
Yusko, E. C. et al. Real-time shape approximation and fingerprinting of single proteins using a nanopore. Nat. Nanotechnol. 12, 360–367 (2017).
Qing, Y., Tamagaki-Asahina, H., Ionescu, S. A., Liu, M. D. & Bayley, H. Catalytic site-selective substrate processing within a tubular nanoreactor. Nat. Nanotechnol. 14, 1135–1142 (2019).
Loudwig, S. & Bayley, H. Photoisomerization of an individual azobenzene molecule in water: an on–off switch triggered by light at a fixed wavelength. J. Am. Chem. Soc. 128, 12404–12405 (2006).
Movileanu, L., Howorka, S., Braha, O. & Bayley, H. Detecting protein analytes that modulate transmembrane movement of a polymer chain within a single protein pore. Nat. Biotechnol. 18, 1091–1095 (2000).
Wei, R. S., Gatterdam, V., Wieneke, R., Tampe, R. & Rant, U. Stochastic sensing of proteins with receptor-modified solid-state nanopores. Nat. Nanotechnol. 7, 257–263 (2012).
Dang, T. X., Hotze, E. M., Rouiller, I., Tweten, R. K. & Wilson-Kubalek, E. M. Prepore to pore transition of a cholesterol-dependent cytolysin visualized by electron microscopy. J. Struct. Biol. 150, 100–108 (2005).
Lukoyanova, N., Hoogenboom, B. W. & Saibil, H. R. The membrane attack complex, perforin and cholesterol-dependent cytolysin superfamily of pore-forming proteins. J. Cell Sci. 129, 2125–2133 (2016).
Pugh, G. C., Burns, J. R. & Howorka, S. Comparing proteins and nucleic acids for next-generation biomolecular engineering. Nat. Rev. Chem. 2, 113–130 (2018).
Rothemund, P. W. Folding DNA to create nanoscale shapes and patterns. Nature 440, 297–302 (2006).
Seeman, N. C. & Sleiman, H. F. DNA nanotechnology. Nat. Rev. Mater. 3, 17068 (2017).
Hong, F., Zhang, F., Liu, Y. & Yan, H. DNA origami: scaffolds for creating higher order structures. Chem. Rev. 117, 12584–12640 (2017).
Praetorius, F. et al. Biotechnological mass production of DNA origami. Nature 552, 84–87 (2017).
Sacca, B. & Niemeyer, C. M. DNA origami: the art of folding DNA. Angew. Chem. Int. Ed. 51, 58–66 (2012).
Douglas, S. M. et al. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 459, 414–418 (2009).
Douglas, S. M. et al. Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. 37, 5001–5006 (2009).
Langecker, M. et al. Synthetic lipid membrane channels formed by designed DNA nanostructures. Science 338, 932–936 (2012).
Burns, J., Stulz, E. & Howorka, S. Self-assembled DNA nanopores that span lipid bilayers. Nano Lett. 13, 2351–2356 (2013).
Gopfrich, K. et al. DNA-tile structures induce ionic currents through lipid membranes. Nano Lett. 15, 3134–3138 (2015).
Krishnan, S. et al. Molecular transport through large-diameter DNA nanopores. Nat. Commun. 7, 12787 (2016).
Burns, J. R., Seifert, A., Fertig, N. & Howorka, S. A biomimetic DNA-based channel for the ligand-controlled transport of charged molecular cargo across a biological membrane. Nat. Nanotechnol. 11, 152–156 (2016).
Thomsen, R. P. et al. A large size-selective DNA nanopore with sensing applications. Nat. Commun. 10, 5655 (2019).
Diederichs, T. et al. Synthetic protein-conductive membrane nanopores built with DNA. Nat. Commun. 10, 5018 (2019).
Kowalczyk, S. W., Grosberg, A. Y., Rabin, Y. & Dekker, C. Modeling the conductance and DNA blockade of solid-state nanopores. Nanotechnology 22, 315101 (2011).
Firnkes, M., Pedone, D., Knezevic, J., Döblinger, M. & Rant, U. Electrically facilitated translocations of proteins through silicon nitride nanopores: conjoint and competitive action of diffusion, electrophoresis, and electroosmosis. Nano Lett. 10, 2162–2167 (2010).
Schmid, S., Stommer, P., Dietz, H. & Dekker, C. Nanopore electro-osmotic trap for the label-free study of single proteins and their conformations. Nat. Nanotechnol. 16, 1244–1250 (2021).
Stephanopoulos, N. Strategies for stabilizing DNA nanostructures to biological conditions. ChemBioChem 20, 2191–2197 (2019).
Burns, J. R. & Howorka, S. Defined bilayer interactions of DNA nanopores revealed with a nuclease-based nanoprobe strategy. ACS Nano 12, 3263–3271 (2018).
Dekker, C. Solid-state nanopores. Nat. Nanotechnol. 2, 209–215 (2007).
Tunuguntla, R. H. et al. Enhanced water permeability and tunable ion selectivity in subnanometer carbon nanotube porins. Science 357, 792–796 (2017).
Wegner, S. V. & Spatz, J. P. Cobalt(III) as a stable and inert mediator ion between NTA and His6-tagged proteins. Angew. Chem. Int. Ed. 52, 7593–7596 (2013).
Acknowledgements
We thank Z. Cicova, A. Solda and J. Faherty from Dynamic Biosensors for acquiring the ensemble binding kinetics of the anti-biotin antibody, J. Ciccone and R. Gutierrez for helping to optimize conditions for DNA nanopore insertions into MinION chips and J. R. Burns for preparing Figs. 1–4. The Howorka Group receives funding from the Engineering and Physical Sciences Research Council (EP/N009282/1, EP/V02874X/1), the Wellcome Institutional Strategic Support Fund, Moorfields Biomedical Research Centre and Oxford Nanopore Technologies.
Author information
Authors and Affiliations
Contributions
Y.X. conceived the idea of the modular configurations of polygonal DNA origami structures and applied it to forming nanopores based on S.H.’s research project of antibody-sensing ring and simple polygonal DNA nanopores composed of few strands. Y.X. designed all DNA origami nanostructures, prepared DNA origami samples and DNA–protein conjugates and performed gel electrophoresis, TEM imaging and confocal microscopy experiments. A.D. conducted the Western-dot blot assay and all electrophysiological measurements and antibody sensing including data analysis and interpretation. The SARS-CoV-2 conjugate pore was jointly conceived by Y.X., A.D. and S.H.; L.J. coordinated resources, MinION recordings and all other experiments at Oxford Nanopore Technologies. S.H. supervised the project and wrote the manuscript with input from Y.X. and A.D.
Corresponding author
Ethics declarations
Competing interests
Y.X. and S.H. are named inventors on patents claiming for DNA origami membrane pores held by UCL Business Ltd (UCLB) deriving priority from UK patent application nos. 1612458.8 and 1812615.1 and filed 14.07.2016 and 02.08.2018 respectively (published as WO2018011603 and WO2020025974). Oxford Nanopore Technologies plc (ONT) has licensed the patents from UCLB. UCL has a research collaboration agreement with ONT related to origami-derived membrane nanopores. L.J. is an employee of ONT and owns company shares and options.
Peer review
Peer review information
Nature Nanotechnology thanks Yi-Tao Long, Amit Meller and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–25 and Tables 1–6.
Rights and permissions
About this article

Cite this article
Xing, Y., Dorey, A., Jayasinghe, L. et al. Highly shape- and size-tunable membrane nanopores made with DNA. Nat. Nanotechnol. 17, 708–713 (2022). https://doi.org/10.1038/s41565-022-01116-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41565-022-01116-1
This article is cited by
-
Nanopore DNA sequencing technologies and their applications towards single-molecule proteomics
Nature Chemistry (2024)
-
Fabricating higher-order functional DNA origami structures to reveal biological processes at multiple scales
NPG Asia Materials (2023)
-
A generalizable nanopore sensor for highly specific protein detection at single-molecule precision
Nature Communications (2023)
-
De novo design of transmembrane nanopores
Science China Chemistry (2022)
