
Abstract
Despite the overwhelming success of vaccines in preventing infectious diseases, there remain numerous globally devastating diseases without fully protective vaccines, particularly human immunodeficiency virus (HIV), malaria and tuberculosis. Nanotechnology approaches are being developed both to design new vaccines against these diseases as well as to facilitate their global implementation. The reasons why a given pathogen may present difficulties for vaccine design are unique and tied to the co-evolutionary history of the pathogen and humans, but there are common challenges that nanotechnology is beginning to help address. In each case, a successful vaccine will need to raise immune responses that differ from the immune responses raised by normal infection. Nanomaterials, with their defined compositions, commonly modular construction, and length scales allowing the engagement of key immune pathways, collectively facilitate the iterative design process necessary to identify such protective immune responses and achieve them reliably. Nanomaterials also provide strategies for engineering the trafficking and delivery of vaccine components to key immune cells and lymphoid tissues, and they can be highly multivalent, improving their engagement with the immune system. This Review will discuss these aspects along with recent nanomaterial advances towards vaccines against infectious disease, with a particular emphasis on HIV/AIDS, malaria and tuberculosis.
Similar content being viewed by others

Main
According to the World Health Organization, HIV/AIDS, malaria and tuberculosis (TB) combined caused more than 2.5 million deaths worldwide in 2017, most of them occurring in low-income countries1,2,3,4. In resource-limited countries, these three pathogens are among the top 10 causes of mortality, highlighting the need to develop prophylactic strategies. Although HIV/AIDS, malaria and TB are caused by dissimilar pathogens, the challenges they present for vaccination share commonalities, and nanomaterials are uniquely positioned to address some of these challenges. One common feature is that in each, the immune response to infection does not generally result in protective immunity, in contrast to other vaccine-preventable diseases such as measles or varicella, which do generate protective immunity in most individuals. Because of this, successful vaccine strategies for HIV, malaria or TB must produce immune responses that are uniquely engineered to be distinct from that of natural infection. The synthetic origins of nanomaterials, along with their defined and tailorable structures and increasingly clear engineering design rules offer potential routes to such vaccines. A second commonality is that these three diseases impact tropical and developing locations of the globe most significantly, resulting in challenges of distribution and implementation. The synthetic composition of nanomaterials can also offer advantages in this regard. Third, for each of these diseases, a successful vaccine strategy will likely involve more complex immune responses than simple antibody production, potentially including engagement of a precise combination of humoral and cellular immunity. Here again, the multifunctionality and tailorability of synthetic and nanomaterial-based vaccines offers advantages. Finally, with the global health community currently responding to a pandemic caused by the novel coronavirus SARS-CoV2, the insights gained from vaccines against the infectious diseases described here may also be critical for developing a prophylactic vaccine for this pathogen.
This Review will focus on nanomaterial vaccines under development toward malaria, TB and HIV, emphasizing recent work and an emerging understanding of how such materials engage the immune system. Vaccines based on designed protein, peptide, lipid, polymer and inorganic nanomaterials will be discussed (Fig. 1). We will not address other areas of nanomaterials immune engineering such as anticancer immunotherapies, tolerance induction to treat autoimmunity, or other infectious diseases, despite intense interest in these areas and similar nanomaterials being explored. The reader is instead referred to other recent reviews in these sub-areas5,6,7 as well as recent comprehensive reviews8,9,10,11.

a,b, Ferritin nanoparticle core decorated with gp140 HIV envelope proteins. c,d, Self-assembling protein nanoparticles displaying malaria epitopes. Scale bar, 100 nm. e,f, Self-assembled peptide nanofibres bearing malaria epitopes. Scale bar, 100 nm. g,h, Inter-bilayer crosslinked multilamellar vesicles with surface bound HIV envelope trimers and thiolated polyethylene glycol (PEG-SH). Scale bar, 20 nm. i,j, Fullerenol particles used as nanoadjuvants for a HIV-1 DNA vaccines. Scale bar, 100 nm. k,l, Liposomes incorporating nickel-binding (DGS-NTA(Ni)) and thiol-reactive (MPB) lipids covalently bound to HIV-1 trimers (MD39-HHHHHHC). Scale bar, 100 nm. Panels a and b adapted with permission from ref. 82, AAAS. Panels c and d adapted from ref. 164. Panel f adapted with permission from ref. 156, Elsevier. Panel g adapted with permission from ref. 109, American Chemical Society. Panel h adapted with permission from ref. 165, Springer Nature Ltd. Panels i and j adapted with permission from ref. 153, Wiley. Panels k and l adapted from ref. 148, distributed under a CC BY 4.0 license (http://creativecommons.org/licenses/by/4.0/).
Since the beginning of the HIV-1 pandemic, more than 30 million infected individuals have died. In 2017, 36.9 million people were living with HIV-1, including 1.8 million newly infected individuals, and HIV-1 was responsible for 940,000 deaths with 110,000 deaths occurring among children under 15 years of age4. It is well recognized that a vaccine is critically needed to contain the pandemic, yet despite over 30 years of research, a vaccine has not yet been developed successfully. Several virologic and immunological factors stand in the way of developing an effective prophylactic HIV-1 vaccine. One of these is that the highly dynamic HIV-1 genome, driven by its error-prone reverse transcriptase and recombination between its genome copies, represents a moving target for the design of an optimal vaccine immunogen. Moreover, as HIV-1 infected individuals are not able to eradicate the virus, there are no clear immune correlates of protection in humans. Early studies have demonstrated that the emergence of virus-specific CD8+ T cells in the acute phase of infection was coincident with viral control12 and long term non-progressors have been reported to have potent, polyfunctional cellular immune responses13. However, elicitation of robust cellular responses does not provide the sterilizing immunity needed to prevent infection.
In a subset of individuals, antibodies that target vulnerable sites of the viral envelope (Env) incrementally develop neutralizing potency and evolve into broadly neutralizing antibodies (bnAbs) after several years of untreated infection14. These bnAbs have characteristics such as a high level of somatic hypermutation, long complementary determining regions 3 (CDR3) and poly-reactivity15,16. It has been shown that passive immunization with bnAbs can prevent viral acquisition in non-human primates17,18. While elicitation of broad neutralization is a priority in the HIV-1 vaccine field, designing vaccine immunogens capable of recapitulating the process of bnAb development has proven extremely difficult. This is in part due to the fact that some bnAb epitopes are shielded by the extensive, minimally-immunogenic glycan fence of the Env19. Moreover, the highly variable regions on the HIV-1 Env present as decoys, directing the humoral response away from the relative constant regions that are usually bnAb target sites20. Induction of bnAbs may also require breaking immune tolerance mechanisms21 and may be dependent on follicular helper T cells (TFH)22.
Compared to bnAbs, non-neutralizing antibodies are easier to elicit with vaccination and may provide modest protection against HIV infection, as exemplified in the RV144 vaccine efficacy trial conducted in Thailand23. In this trial, vaccinees were primed with a recombinant canarypox vector expressing HIV-1 Gag, Pro and Env antigens from subtypes B and E, and then boosted with an Env subunit vaccine adjuvanted with Alum23. The immune correlate analysis of this moderately efficacious vaccine trial indicated that IgG antibodies against the envelope variable loops 1 and 2 (V1V2) were associated with reduced risk of HIV-1 acquisition24, whereas high plasma levels of vaccine-elicited IgA were associated with increased HIV-1 acquisition, probably by mitigating the protective effect of IgG against the same epitopes25. Importantly, the mechanism by which V1V2 antibodies mediated protection is not fully understood. As the vaccine-elicited antibodies only neutralized minimally evolved (Tier 1) viruses26, it has been speculated that Fc-mediated, non-neutralizing antibody functions may mediate protection. In fact, monoclonal antibodies isolated from RV144 vaccines are able to mediate antibody-dependent cellular cytotoxicity27, and vaccine-elicited V1V2-specific antibodies can activate the complement cascade28. Nevertheless, the potential for protection of this vaccine approach is now uncertain, as a recent efficacy trial conducted in South Africa attempting to replicate the results of RV144 (HVTN 702) was halted prematurely due to the lack of efficacy29. In this trial, HIV-1 subtype C antigens were included in the vaccine in order to increase coverage against the virus strains circulating in South Africa, which has infection rates significantly higher than in Thailand. Whether differences in vaccine regimens, infection rates, or trial populations contributed to the differences observed between the two trials is still unclear29. Vaccination studies in non-human primates have also supported a protective role for non-neutralizing Fc-mediated effector functions30,31. Yet, studies in non-primates have indicated that passive immunization with non-neutralizing antibodies confer limited or no protection against virus acquisition32,33.
Achieving protective immunity against HIV-1 may require the elicitation of both robust antibody responses with the aim of blocking/reducing infection acquisition and effective cellular responses to control breakthrough infections. Similar to natural infection, immunization with monomeric Env vaccine immunogens can induce robust antibody responses against immuno-dominant epitopes, but these antibodies are unable to neutralize circulating virus strains34. Strategies being pursued to design B cell immunogens with the goal of inducing bnAbs include: immunogens that mimic the native Env structure such as virus-like particles (VLP) bearing only pure native functional Env proteins35, stabilized soluble immunogens that reproduce the trimeric structure of Env36, and bnAb epitope-focused immunogens37. Several approaches are also being pursued for the induction of effective T cell responses including immunogens targeting the most conserved regions of HIV-138 and mosaic immunogens, which are designed using computational approaches to generate combined protein sequences from multiple naturally-occurring proteins39. In addition to challenges in eliciting antibodies of protective specificities, immunization with HIV-1 Env constructs usually induces antibody responses of limited duration40. This issue, along with the need to engage multiple arms of the immune system to achieve protective immunity, makes synthetic and nanomaterial-based approaches attractive.
Malaria, like HIV-1, presents unique challenges for vaccination. While preventive measures have significantly impacted the incidence and severity of malaria infections, continued progress has been difficult to achieve. In 2017, 219 million cases of malaria occurred, 2 million more than in 2016, and the number of malaria-related deaths remained above 400,0003. Children under the age of five have been especially vulnerable, accounting for 61% of malaria-related deaths in 2017. Most are due to Plasmodium falciparum, which has a complex life cycle and exists in several forms in humans: sporozoites, which infect hepatocytes; merozoites, which infect red blood cells; and gametocytes that continue the life cycle in mosquitos, where they undergo sexual replication41. Because of this complexity, vaccines against each stage are currently being pursued42.
Following natural malaria infection, sterilizing immunity does not develop, so the immune response elicited by a successful vaccine must be distinctly different. The most advanced malaria vaccine candidate (RTS,S) contains the central repeat and terminal epitopes of the major surface antigen circumsporozoite protein (CSP) and has exhibited 43.9% efficacy after four immunization doses in children and 27.8% efficacy in infants43. However, efficacy quickly wanes and is variable among the paediatric populations studied44. Thus, there is a need for new strategies to improve efficacy and durability. Another major challenge is the lack of clearly defined correlates of protection, making target selection and formulation optimization challenging. Other hurdles include the complexity of the parasite, with hundreds of potential vaccine targets, the immune evasion inherent in its complex life cycle, the high level of polymorphisms in the most immunogenic antigens45, and poor immunogenicity of conserved epitopes46. Interestingly, in addition to anti-CSP antibodies, polyfunctional T cell responses may also contribute to protection against malaria. In fact, polyfunctional CD4+ T cell responses were associated with protection in individuals primed with the adenoviral vector Ad35-CSP and boosted with RTS,S47. In other studies, vaccine-elicited CD8+ T cell responses have also been associated with protection48. These results demonstrate that several branches of the adaptive immune system may need to be recruited to increase the efficacy of a malaria vaccine. Nanomaterial-based vaccines may thus help overcome some of these challenges by enhancing vaccines targeting conserved epitopes, by providing routes for engaging both humoral and cellular immunity, and by providing defined candidates whose design can be optimized systematically as correlates of protection are increasingly revealed.
TB infected 10 million individuals in 2017 and was responsible for an estimated 1.3 million deaths, making it the leading cause of mortality due to a single infection2. TB cases are complicated by the emergence of multi-drug resistant bacteria and frequent HIV co-infections. In fact, in 2017, 9% of new TB infections occurred in HIV-infected individuals, and TB was responsible for 300,000 deaths among HIV-infected patients2. Mycobacterium tuberculosis (M. tuberculosis) generally infects the lungs, but it can also infect extrapulmonary organs. Following primary infection, TB can evolve towards active disease, latent infection, or be eradicated by the host immune system. Macrophages constitute the first line of defence against TB, but the pathogen can successfully persist in macrophages. Another mechanism of immune evasion includes the transition into a dormant stage that is highly resistant to the host immune system and can persist for years within granulomas49. In young children, immunization with the live-attenuated Bacillus Calmette Guerin (BCG) vaccine can provide protective immunity against severe TB disease50, but the efficacy of this vaccine is low and it poorly protects from TB acquisition. Moreover, the BCG vaccine is less efficacious in adults and variable across different geographic settings51. Interestingly, a recent BCG vaccine study indicated that when immunized intravenously, 90% of macaques were protected against M. tuberculosis challenge, suggesting that it may be possible to broaden the efficacy of BCG vaccination52.
As with HIV and malaria, a major hurdle to the development of an effective TB vaccine is the lack of well-established immune correlates of protection and an incomplete understanding of protective mechanisms53. IFN‐γ producing CD4+ T cells are thought be critical for TB control, but this response may not be sufficient to induce protection. Other T cell subsets including CD8+ T cells and unconventional T cells subsets such as HLA-E restricted CD8+ T cells may also contribute to protection, but their role is not yet clearly established. There are currently multiple ongoing TB vaccine clinical trials. Two subunit vaccines showing promising effects include M72/AS01E and H4:IC3154. M72/AS01E, which consists of a recombinant fusion protein (M72) of two M. tuberculosis antigens with the AS01 liposomal adjuvant system, was shown to protect 54% of adults in a study of individuals with latent TB infection55. H4:IC31 consists of a recombinant fusion protein (H4) of three M. tuberculosis antigens with the IC31 TLR-9 agonist adjuvant system. In a study of adolescents previously vaccinated with BCG, both H4:IC31 or a secondary BCG vaccination were shown to reduce the rates of persistent TB infection56. Most current TB vaccine candidates are designed primarily to induce cellular immune responses. However, whether these vaccines will be able to elicit the sterilizing immunity required to prevent acquisition is not yet completely clear. The development of a TB vaccine is further complicated by the fact that M. tuberculosis expresses about 4,000 proteins at levels that vary with the metabolic stage, complicating antigen selection57. Nanomaterials that can carry multiple antigens are therefore of interest for TB vaccine development, and they are further appealing because they can provide adjuvant effects required to tune the immune response towards potentially important mechanisms such as elicitation of Th1 responses.
Mechanisms by which nanomaterials improve vaccine responses
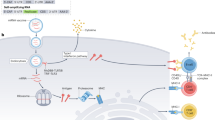
A central aspect in the engineering of nanomaterial vaccines against HIV, malaria and TB involves their delivery to key cells and tissues of the immune system. However, unlike other drug delivery applications that may target a narrowly defined cell type, a productively immunogenic nanomaterial vaccine needs to interact with many different types of cells. These may include various professional antigen presenting cells (APCs), B cells, macrophages, neutrophils and T cells of various types after the vaccine-associated antigens have been processed. These interactions occur across extended time periods, in multiple tissue locations and after considerable processing, making it a complicated endeavour to rationally engineer the trafficking of a nanomaterial vaccine (Fig. 2). Features under control can include the size and shape of the nanomaterial, its durability in vivo, the number of antigen copies on or within the nanomaterial, the specific co-delivery of adjuvants, the physical orientation of antigens, or complement activation. Each of these factors influences how the vaccine traffics to various lymphoid tissues, with considerable impact on the quality and strength of immune responses raised. These aspects are discussed in this section.

Lymph node (LN) trafficking: nanomaterial trafficking to lymph nodes is largely dependent on size but is also influenced by charge, hydrophobicity, flexibility and other physical properties. Mucosal Targeting: to induce mucosal immunity, nanoparticles must be able to penetrate mucosal barriers. Hydrophilic polymeric coatings limit entanglement with mucin fibres, and positively charged nanoparticles are mucoadhesive. Persistence: nanoparticle persistence prolongs the release of antigen, allowing for greater antigen uptake by APCs over time. Examples of nanoparticle persistence include depots present at injection sites or in the draining lymph nodes. Controlled Release: the controlled release of antigens from nanoparticles can take many forms. Endosomal escape is illustrated here with polymeric nanoparticles being endocytosed and then degraded in the endolysosomal pathway. As the polymer degrades, it disrupts the endolysosomal membrane, which allows for release of antigen into the cytoplasm. Cross presentation of the antigen on MHC I ensues. APC Targeting: there are several methods for targeting APCs. DCs can be targeted via receptors such as DEC-205. Macrophages have been shown to preferentially phagocytose anionic nanoparticles. B cell receptors recognize B cell epitopes on the surface of nanomaterials, and uptake is facilitated by multivalency. MØ, macrophage.
Encapsulation or conjugation of antigens within nanomaterials can greatly increase the persistence of antigens at the injection site, in the circulation, in lymphoid tissues, or even within APCs (Fig. 2). It has long been appreciated that extending the persistence of an antigen can enhance its immunogenicity, and a variety of nanotechnological strategies for achieving this have been explored. For example, Moon et al. demonstrated that interbilayer-crosslinked multilamellar vesicles (ICMVs) with both encapsulated and surface-conjugated malaria antigens can improve vaccination compared with the soluble antigen, owing to the prolonged presence of the antigen in the draining lymph nodes (LNs)58. Demento et al. similarly illustrated the effect of prolonged antigen delivery using poly(lactic-co-glycolic acid) (PLGA) nanoparticles encapsulating ovalbumin as a model antigen59. The prolonged release of antigen allowed for improved immunogenicity, and the degradation rates of the PLGA nanoparticles could be manipulated to further extend the ovalbumin release. The slowly degrading nanoparticles served as a durable source of antigen for APCs to acquire and present to follicular helper T cells (Tfh), which play a central role in germinal centre reactions in LNs, where antibody affinity maturation takes place. This process of generating high affinity antibodies to vaccine targets is critical for generating protective immunity against infectious diseases.
Although the creation of depots at injection sites can help prolong antigen exposure and facilitate germinal centre reactions, strong cellular immune responses still require that antigens be internalized, processed and presented efficiently by APCs. In order to mount a specific cytotoxic CD8+ T cell response against HIV, TB, or Malaria, cross-presentation of their exogenous antigens on class-I MHC must first be achieved. For successful cross-presentation to be achieved, antigen fragments must further escape to the cytosol. To accomplish this, nanomaterials sensitive to the environment of the endolysosomal pathway have been explored. This strategy mimics some viral infections and enables enhanced CD8+ T cell responses compared to the delivery of free antigen60. For example, Hirosue et al. demonstrated that when ovalbumin peptide antigens were linked to the surface of pluronic-stabilized poly(propylene sulfide) (PPS) nanoparticles via disulfide bonds that were cleavable in the reductive environment of the endolysosomal pathway, cross presentation was enhanced compared to antigens attached via non-reducible linkages61.
The controlled and predictable delivery of nanoparticle vaccines to the large population of B cells, T cells, follicular dendritic cells, and subcapsular sinus macrophages residing in the LNs is highly sought for vaccine design. While antigens can be delivered to the LNs via APCs that acquire the antigens at the injection site and migrate to the LNs, drainage to the LNs also occurs, largely dependent on the size of the nanoparticles (Fig. 2). With the caveat that multiple physical properties of nanoparticles influence lymphatic drainage, including charge, shape and flexibility, it has been observed that nanoparticles smaller than 100 nm tend to drain to the LNs, with 10–50 nm being near the optimal size62, whereas those close to 6 nm or smaller tend to drain to the vasculature63. The larger the nanoparticles were within this range, however, the better they were retained in the LNs64. Larger nanoparticles that passively drained to the LNs were acquired by subcapsular macrophages, while smaller particles were more likely to enter conduits and travel to B and T cell zones65. Beyond size, characteristics such as flexibility can also influence the drainage of nanoparticles to LNs66, and different nanomaterial platforms may have subtly different optimal sizes for lymphatic drainage. Intranodal injection, though invasive, volume limiting and subject to rapid clearance via afferent lymph, may begin to circumvent such considerations altogether67,68,69.
In the case of respiratory, sexually and orally transmitted pathogens such as TB and HIV, the mucosal immune response is particularly relevant, so mucosal delivery of nanomaterials has received particular attention (Fig. 2). Mucus is a porous network of mucin glycopoymers70,71, and the ability of nanoparticles to pass through or adhere to it is largely dependent on size and surface characteristics. Pore sizes vary within different mucosal barriers, with cervicovaginal mucus reported to have average pore diameters of 340 nm72 and respiratory mucus up to 200 nm, so nanoparticles smaller than these cut-offs can traverse these barriers more easily73. Hydrophilic polymers such as poly(ethylene glycol) (PEG) have also been found to greatly facilitate transit of particles across the mucus layer (see Box 1 regarding immunological considerations of PEG in nanomaterials)80,83,84. Additionally, cationic nanoparticles are mucoadhesive, allowing them to be retained in the mucus and giving them a greater opportunity to interact with mucosal immune cells. Chitosan, a cationic polysaccharide derived from the shells of crustaceans, has been employed in mucosally delivered vaccine nanoparticles74, including those against TB75.
Beyond targeting LNs or mucosal tissues, considerable work has been undertaken to direct the uptake of nanoparticles to specific subsets of APCs (Fig. 2). In many sites of delivery, nanomaterials are avidly acquired by macrophages owing to their broadly phagocytic nature. Although the design rules for achieving cell-type-specific uptake are multifactorial and interdependent, nanoparticle size, charge and shape can be manipulated to enhance macrophage uptake or to favour uptake by other APCs76,77. For example, it has been reported that macrophages preferentially ingest anionic particles78, whereas both DCs and macrophages can be passively targeted based on nanoparticle size, with particles from 20–200 nm being endocytosed by DCs and particles from 500–5,000 nm being phagocytosed by macrophages in some contexts79. Specific receptors on DCs also provide opportunities for affinity-based targeting strategies of specific DC subsets80. These include C-type lectin receptors such as DEC-205 and DC-SIGN, which are heavily expressed on epithelium-resident Langerhans cells and LN-resident DCs81. Other strategies have targeted follicular dendritic cells (FDCs) in LNs. Multivalent ferritin nanoparticles displaying HIV antigen glycoproteins targeted FDCs and enhanced the development of germinal centres, discussed in greater detail in the section below on multivalency82. B cells, another important APC in the context of nanomaterial based vaccines, can be targeted by repetitive, multivalent antigen structures, which cross-link B-cell receptors and lead to enhanced uptake and activation83, also discussed in greater detail in the next section.
Nanomaterial vaccines also offer routes to improve the function of adjuvants and to minimize negative systemic or local effects such as inflammation or toxicity. Dosing adjuvants so as to achieve strong immune responses without such negative effects is a persistent challenge in vaccine design84,85, and the delivery of adjuvants via nanomaterials can provide routes towards dose reduction by controlling adjuvant release near or inside APCs86. For instance, Moon et al. demonstrated that multilamellar ‘stapled’ lipid vesicles incorporating a malaria antigen and delivered in combination with the TLR4-agonist adjuvant MPLA allowed for a 10-fold dose reduction of the adjuvant when compared to a soluble antigen/MPLA vaccine and induced more diverse humoral responses57. Other adjuvanted lipid nanomaterial formulations have also been shown to raise higher antibody titers than simply mixing antigen with adjuvant87. As another example, Huang et al. demonstrated that His-tagged Pfs25 malaria antigens inserted into the lipid bilayer of adjuvant-encapsulating liposomes generated durable antigen-specific plasma cells88. Furthermore, some nanoparticles have intrinsic adjuvanting properties, even without augmentation by TLR ligands or other adjuvants. Pathways by which this may occur include complement activation, inflammasome signalling or B cell activation8. Such intrinsically adjuvanting nanoparticles may be advantageous because they can limit inflammation and toxicity arising from other adjuvants, and they simplify the formulation and dosing of a vaccine.
The impact of multivalent protein display by nanomaterials
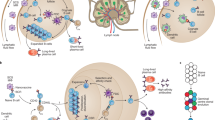
Spatially repetitive structures are intrinsic to viruses, bacteria, and other pathogens, and the immune system has evolved to recognize and respond to them with exquisite sensitivity. Many viruses and bacteria contain repetitive antigens spaced at around 5-10 nm apart89, yet notably some pathogens have evolved to minimize such structures. For example, the envelope spikes of HIV are fewer and more separated than on other spherical viruses, leading to the hypothesis that the virus has evolved specifically to minimize the effects of multivalency, even at a cost of its transmissibility90. Thus, the ability to finely manipulate, enhance and optimize antigen density and orientation is one of the most advantageous features of nanomaterials (Fig. 3), because such control is more difficult to achieve in other forms of vaccines. As evidence of the power of multivalency in nanomaterial vaccines, several highly repetitive structures have been found to elicit strong humoral and cellular immune responses even without supplemental adjuvants or other immunostimulating components. This property is exhibited by supramolecular peptide assemblies91,92,93,94,95, self-assembled polypeptide nanoparticles96,97, and other multivalent particulates62,98, but it is less common for purely soluble proteins. The effect of multivalency is so powerful that even epitopes that would otherwise be tolerized, such as those within autologous cytokines, can be made immunogenic95.

Multivalent displays of proteins, carbohydrates and B cell epitopes activate immune cells through a variety of pathways. a, Several modes of immune cell engagement occur via complement activation through binding of complement protein C3 or its cleavage product C3b to nucleophilic groups on surfaces and polymers, or by binding of ficolin or mannose-binding lectin to carbohydrates. Complement activation leads to association of C3d and C3b with nanomaterials, which bind to complement receptors 1 (CR1) and 2 (CR2), respectively. b, Activation of complement causes nanomaterials to be shuttled to follicular dendritic cells, where they can be displayed for long periods of time82. c, Macrophages in the subcapsular sinus also capture particulate antigens and transfer them to follicular B cells in a complement-dependent manner166. Both b and c are processes that drive affinity maturation. d, Multivalent particles can also activate B cells directly by crosslinking B cell receptors to increase uptake and activation by signalling through immunoreceptor tyrosine-based activation motifs (ITAMs)103.
Defined materials offer the opportunity to investigate the structural features and immunological mechanisms that lead to the immunogenicity-enhancing effects of multivalency. Early work by Howard and Renee Dintzis and co-workers with multivalent polymer-conjugated dinitrophenyl haptens demonstrated that such materials were able to activate T-independent B cell responses99. In a series of landmark studies, it was demonstrated that repetitive displays of polymer-linked haptens were able to activate immune cells in a fashion that was dependent on molecular weight, ligand density and ligand number99,100,101. This phenomenon is quantized, and a critical number of hapten molecules are required to produce a T-independent antibody response100. For example, for polyacrylamide-linked haptens, a sharp threshold of immunogenicity was observed when more than 12–16 copies of the hapten were conjugated to the polymer. Higher molecular weight polymers with a lower density of ligands could also crosslink receptors because of the polymers’ flexible backbones100. Notably, the phenomena described were distinct from the processes by which nanoparticle vaccines typically raise immune responses, as these generally contain T-cell epitopes and raise T-dependent responses, yet multivalency still facilitates T-dependent processes as well102. Though these examples utilize model epitopes, they shed light on the mechanisms by which nanomaterials raise immune responses.
Particulate displays of antigen, such as those created by conjugating antigens to nanoparticles, promote uptake by B cells even if their BCRs have low affinity for the antigen. Work by Batista and Neuberger demonstrated that linking hen egg lysozyme (HEL) to the surface of beads allowed equal levels of uptake and presentation by high- and low-affinity B cells, whereas the uptake of soluble proteins was correlated to the affinity of the BCR for HEL103. The authors speculated that this was due to increased avidity interactions when multiple BCRs were crosslinked, lowering the upper limit for B cell discrimination between high- and low-affinity BCR–antigen interactions103. In agreement with the quantized model proposed by Dintzis, this effect was conserved for multiple levels of antigen density, then dropped off at a lower threshold of antigen density103. Activation of B cells with multivalent antigen displays can be particularly useful for engaging B cells with low affinity BCRs for vaccination against autologous targets95,104, or for the engagement of HIV bnAb precursors with low affinity to the antigen37.
Kiessling and co-workers used highly defined polymeric systems to investigate how multivalent hapten displays influenced multiple steps of B cell internalization and activation to produce functional antibody responses105. Polymers containing 500 hapten molecules per polymer chain induced higher levels of antibodies than polymers with 10 haptens per chain and correlated with increased BCR clustering and intracellular Ca2+ signalling but did not significantly influence polymer internalization105. This work was then extended to incorporate T-cell epitopes, finding that B cells functioned as important APCs for polymer constructs of immunogens106. The ratio of B cell epitope to T cell epitope has also been shown to significantly impact antibody titers against B cell epitopes in self-assembling peptide systems95,107. While the exact mechanism of this phenomenon is still not completely understood, it is becoming clear that B cell epitope density is critical both for the activation of B cells and for their role as APCs in coordinating humoral and cellular immune responses. For example, recent work has shown that B cells are the key APCs for orchestrating immune responses to VLPs and can function independently of dendritic cells to generate germinal centre responses108.
The effects of nanomaterial-multimerized antigens on antibody repertoire have been observed in several infectious disease vaccine design settings. Tethering the Plasmodium vivax circumsporozoite antigen VMP001 to multi-layered liposomes improved the avidity, durability and breadth of antibody responses compared to soluble antigen when adjuvanted with the Toll-like receptor (TLR) agonist MPLA58. HIV trimers tethered to liposomal carriers induced antibodies against more regions on the target antigen109 and more HIV viral proteins110 compared to non-multimeric vaccines. This increase in antibody breadth was hypothesized to be caused by activating a more diverse set of B cells by clustering relatively low-affinity BCRs.
Recently, self-assembling two-component protein nanoparticles have been utilized for the display of viral antigens to elicit immune responses against strategically displayed B cell epitopes111,112. These particles also allow for detailed study of how the number of antigens affects immune response, as varying ratios of assembling components can be expressed without an antigen. Although not directed against TB, HIV or malaria, the work is illustrative, as increasing copies of antigen per particle were shown to improve neutralization of viral strains after immunization with the RSV antigen DS-Cav1. More highly multivalent particles also enhanced the numbers of TFH cells and germinal centre B cells111. Fully decorated particles, bearing 20 copies of DS-Cav1 promoted neutralizing antibody responses 10-fold higher than soluble DS-Cav1111, demonstrating the potential for highly multivalent nanoparticle vaccines to elicit protective immune responses.
Recent work with multivalent HIV nanovaccines has shed light on some of the mechanisms by which repetitive antigen presentation enhances the retention of vaccine particles within germinal centres and improves B cell activation compared to soluble proteins (Fig. 3)82,110. Recently Tokatlian, Irvine and co-workers demonstrated that immune responses to multivalent protein nanoparticles were complement-dependent and significantly influenced by the repetitive displays of glycans on the particle surface82. They illustrated that multivalent saccharides promoted the binding of mannose binding lectin (MBL), which subsequently induced complement-dependent trafficking of the particles to the follicular dendritic cell network within LNs. This shuttling led to an increased association of antigen with follicles and induced higher numbers of germinal centre B cells. In other work, conjugation of trimannose groups to polystyrene nanoparticles facilitated shuttling of these materials to follicles82. This articulation of a key mechanism of nanomaterial multivalency now enables the engineering and optimization of this process using carefully controlled patterns of saccharides on the materials’ surfaces.
Despite the marked improvements endowed by nanomaterials in many examples, some authors have reported minimal improvement of immune responses in other situations113,114. Based on the studies described above, it appears likely that fine adjustments of protein density, spacing and orientation coupled with optimized saccharide identity and arrangement will be key aspects determining the level of success of this approach.
Beyond delivery aspects and multivalency, the tethering of an antigen to a nanomaterial surface affords the opportunity to orient the antigen in a strategic way. By burying undesirable epitopes and exposing choice epitopes, the resultant antibody response can be shaped. Being able to influence the epitope specificity of vaccines presenting folded proteins is critical for minimizing responses to immunodominant, non-neutralizing regions of antigens, such as the base and V3 loop tip of HIV trimers115. The use of nanomaterials to alter protein display has been utilized for vaccines against Epstein–Barr virus (EBV)113 and has recently been exploited by screening for protein displays that bind neutralizing antibodies116. By tethering the EBV antigen gp350 to a ferritin or encapsulin core, antibodies were directed to a desired binding site to interfere with the virus’s ability to bind its target, complement receptor 2 on B cells, thus significantly improving neutralization compared to a soluble control.113 To screen for vaccine materials that would induce neutralizing antibodies, Bazzill et al. used a method termed NanoFACS to demonstrate that neutralization capacity of various protein–nanoparticle vaccines could be predicted by their binding to neutralizing antibodies in vitro, which likely varied according to the orientation of the protein on the particle surface116.
The impact of protein orientation was also demonstrated by Martinez-Murillo et al. when HIV trimers were presented in high density on the surface of liposomes117. Similar to other nanoparticle vaccines, vaccines based on liposomes increased the size of germinal centres and the average number of TFH cells contained within each germinal centre117. Interestingly, the antibodies produced by an animal with high Tier 2 neutralization showed a unique mode of binding to the trimer, resembling the binding displayed by bnAb VRC01117. Tier 2 indicates viruses moderately sensitive to neutralization, is typical of most circulating strains, and is a high priority for vaccination. This binding, characterized by horizontal access to the V2 cap, is not typically observed in antibodies elicited by soluble trimer vaccines, and it indicated that presentation of antigen on multivalent surfaces may direct the antibody response towards antigenic regions, which are typically sub-dominant117. Another system in which epitope accessibility can be controlled is the two-component protein nanoparticles described above112. This structural tailoring directly influences the specificity of antibodies toward surface proximal epitopes, as epitopes closer to the base are masked by other components of the nanoparticle112. Similar strategies to orient antigens have been based on multimerizing proteins such as ferritin, virus-like particles, and other de novo proteins118,119,120.
Examples of nanomaterial vaccines for infectious diseases
In this section, we will describe nanomaterials of various classes and provide examples how each are being employed towards vaccines against HIV, TB and malaria. Many of these vaccines make use of the mechanisms and design aspects discussed previously.
To design vaccines capable of maximally engaging the immunological processes sensitive to multivalency, many approaches for arraying antigens on surfaces have received intense interest, with self-assembling protein nanoparticles being among the most promising class. Ferritin, a protein utilized by most living organisms for iron storage, assembles into a 24-member spherical cage structure (Fig. 1a). Antigen–ferritin fusion proteins can form spherical nanoparticles with the antigens displayed on the particle surface in a multivalent fashion121. Multimeric proteins, such as trimeric HIV-1 Env, can be presented on the particles’ surface as well, with eight multimers displayed per particle. Such particles have improved immunogenicity compared to soluble trimers alone122. As discussed in the section ‘The impact of multivalent protein display by nanomaterials’, ferritin nanoparticles were recently employed to demonstrate that multivalency facilitated the trafficking of HIV native-like Env trimers to the follicular dendritic cell regions in draining LNs, likely through the activation of the complement system via the mannose-binding lectin pathway82. Display of HIV Env trimers on ferritin nanoparticles also showed increased immunogenicity in mice and rabbits, inducing higher neutralizing antibody responses including against the autologous Tier-2 virus122. While this strategy has not yet been shown to induce broad neutralization, the results are encouraging and warrant further exploration of similar approaches for elicitation of bnAbs. Recently, Saunders and co-workers used SOSIP trimers displayed on ferritin nanoparticles to select for B cell clones that share BCR mutations with bnAb precursors123. This study demonstrated significant progress in designing immunogens that could be capable of inducing bnAbs when viral antigens are displayed on nanoparticle surfaces.
Other examples of multimerized protein self-assemblies include those employing IMX313, an oligomerizing domain based on chicken complement inhibitor C4b-binding protein124. IMX313 spontaneously forms soluble seven-member assemblies with each subunit carrying a fused antigen. This system has been explored towards the design of vaccines against malaria124,125,126 and TB127,128. For example, the malaria sexual stage antigen Pfs25 was fused to IMX313, forming heptameric protein nanoparticles. Antibodies induced by the Psf25-IMX313 nanoparticles were durable and capable of binding native antigen expressed on malaria ookinetes125. Moreover, in the presence of adjuvants, IgG purified from Psf25-IMX313-immunized mouse sera was more capable of blocking the formation of oocysts in mosquitos compared to IgG from Psf25-immunized mouse sera. Advantages of the IMX313 system are the small size of the oligomerizing domain (55 amino acids) and encouraging safety data in humans128.
Because ferritin can display 8 trimers or 24 subunits, and IMX313 can display seven subunits, larger particles with greater numbers of subunits are also being explored, including dihydrolipoyl acetyltransferase (E2p) and lumazine synthase (LS), both of which form 60-member spherical structures. Towards HIV vaccination, after designing a novel immunogen to induce VRC01-class bnAbs based on a minimal, engineered outer domain, Schief and co-workers fused their designed antigen to lumazine synthase120. The protein was expressed in mammalian cells with good yield and self-assembled into 60-member spherical nanoparticles120. The monomeric form of their designed antigen, eOD-GT6, failed to activate germline or mature B cells bearing VRC01 IgM, even though it bound germline VRC01 with KD below 50 nM, but the LS-based nanoparticles potently activated the VRC01 IgM germline and mature B cells120. The examples of ferritin, IMX313, E2p, and LS indicate that multimerization on a protein particle can have powerful effects on immunogenicity. It remains to be seen what spacing, number of antigens and particle sizes will maximize protective responses in the various contexts under investigation.
VLPs are non-infectious, non-replicating virions that resemble viruses in structure but lack a viral genome129. Over the past few decades they have contributed tremendously to vaccine development and have been reviewed comprehensively130,131, so they will be discussed briefly here in the context of the diseases of interest. A wide variety of virus families have been used to construct VLPs, and like the oligomerizing proteins previously discussed, antigens presented by them can retain their native conformation, allowing for multivalent antigen presentation. Owing to successful clinical translation in the form of licensed vaccines against human papilloma virus, hepatitis B and E, and influenza, VLPs make attractive candidates for further development as vaccines against HIV, TB and malaria. In fact the first licensed malaria vaccine, RTS,S, is VLP-based, employing the hepatitis B surface antigen platform119. In a recent example of VLP design for HIV vaccination, HIV Env proteins displayed on VLPs have been shown to activate B cells producing the bnAb VRC01 in vitro132. McCurley et al. employed a sequential vaccination regimen on macaques with a series of VLPs displaying different HIV Env proteins to mimic the natural developmental pathway of the CH505 bnAbs133. Although this experiment failed to induce bnAbs, half of the vaccinated animals developed antibodies that neutralized the autologous tier 2 virus with a neutralization mechanism similar to CH505. Advantages of VLPs include their long track record of successful development towards many different diseases and well-established expression protocols. Self-assembled protein nanoparticles (Fig. 1c)96,97, which have been designed using predictably oligomerizing coiled coil domains, have also been explored towards vaccines against malaria96,97,134. When self-assembled protein nanoparticles bearing B- and CD8+ T cell epitopes from the P.falciparum circumsporozoite protein and the universal CD4 T helper epitope PADRE were delivered in saline without adjuvant, high-titer, long-lasting protective antibody responses were observed against P. berghei infection in a rodent model134.
Polymeric materials have received interest as non-protein-based vaccine platforms owing to their advantages of being straightforward to synthesize and generally non-immunogenic by themselves (Box 1). Numerous types of polymeric materials have been investigated, including chitosan, polyesters such as PLGA and polyamides such as gamma polyglutamic acid (γ-PGA). In addition to nanoparticles, polyelectrolyte multilayers (PEM) and dendrimers have received interest for vaccine delivery. These have been reviewed elsewhere as well135,136.
Hydrolysable polyesters such as PLGA have been employed within nanomaterials to encapsulate protein antigens along with adjuvants137,138. PLGA encapsulation offers a number of advantages, including the ability to co-deliver multiple antigens or adjuvanting molecules such as TLR ligands in a prolonged fashion. In engineered vaccines requiring shaping of the immune response beyond that which is elicited during natural infection, this property is advantageous. For example, Pulendran and co-workers recently showed that encapsulation of simian immunodeficiency virus (SIV) antigens and multiple agonists for TLR7/8 and TLR4 within PLGA nanoparticles induced robust and durable antigen-specific antibody responses in macaques139. Interestingly, macaques immunized with the PLGA-encapsulated SIV vaccine had higher levels of protection against repeated, low-dose vaginal challenges with a heterologous SIV strain than animals immunized with SIV antigens adjuvanted with Alum. Nevertheless, one challenge with using PLGA as a vaccine carrier is that solvents utilized during the production of particles may potentially denature antigens and diminish their antigenicity. To address this issue, a ‘self-healing encapsulation’ technology was recently developed to allow the antigens to be loaded in a relatively mild, aqueous condition. Bailey and colleagues showed that ovalbumin delivered by this nanoparticle can induce strong CD8+ T cell and antibody responses140,141.
Polymeric nanoparticles have also been used to deliver DNA vaccines, where delivered genetic material leads to antigen expression in vivo, in strategies to increase immunogenicity and half-life. For example, polyamides such as γ-PGA have been utilized to design a plasmid DNA vaccine encoding the erythrocytic stage malarial antigen merozoite surface protein-1 (MSP-1)142. Chitosan has also been utilized for DNA vaccines, as it can complex with DNA and protect it from nuclease degradation143. DNA coding for T cell epitopes from the TB antigen early secretory antigenic target-6 (ESAT-6) encapsulated in chitosan nanoparticles induced higher T cell functionalities, including cytokine secretion and cytolytic activity, compared to the Bacillus Calmette–Guérin (BCG) vaccine, and offered greater resistance to infectious challenge144. Chitosan nanoparticles have also been used to increase immune responses at mucosal sites, in order to improve pulmonary immunity against TB75. Although linear polymers are commonly used within nanomaterials for vaccine delivery, highly branched structures such as dendrimers have also been explored. Strikingly, a single dose of dendrimer-encapsulated mRNA encoding viral surface antigens conferred protection against influenza, Ebola virus and toxoplasma gondii, an approach that may be applicable to HIV, malaria or TB145.
Other uses of polymers as nanostructured vaccines include polyelectrolyte multilayers (PEM), where layered structures are created with macromolecules of alternating charge. PEM construction occurs in mild aqueous conditions suitable for biomolecular cargos, and depending on the method used, nanoparticles or films can be produced146,147. For example, DeMuth and colleagues reported a microneedle array iteratively coated by layers of SIV gag-expressing plasmids and poly (I:C) as the adjuvant. After administration to mouse skin, the microneedle vaccine induced a potent CD8+ T cell response and antibody responses 10 times higher than injection/electroporation147.
As with polymer-based nanocarriers, liposomal delivery systems allow for the combination and co-delivery of multiple antigens and adjuvant molecules. They also offer opportunities to adjust size, charge, number of antigen copies and other physical parameters that influence trafficking and the immune responses elicited. As emphasized throughout this Review, such properties are especially beneficial for infectious diseases with unclear immune correlate for protection such as HIV, malaria and TB, because they allow iterative engineering of the vaccine. Further, liposomes allow for antigen to be displayed on the surface of a membrane, which for some antigens may be more representative of native conformations on viral particles, and there is sufficient surface area for the display to be highly multivalent, capitalizing on the mechanisms discussed above. Successful adjuvant systems based on liposomes have included AS01, used for example as part of the RTS,S and M72/AS01E malaria vaccines55. AS01 contains in addition to liposomes the saponin QS-21 and the TLR-4 agonist 3-O-desacyl-4′-monophosphoryl lipid A. The ISCOMATRIX system also has similar components (phospholipids, saponins and cholesterol) and forms particulate assemblies on the order of 50 nm114. Designed lipid-based nanomaterials have built on the properties of these previous adjuvants by tailoring the assembly of antigens within them. For example, HIV Env trimers presented on interbilayer-cross-linked multilamellar vesicles (ICMVs, Fig. 1g,h) could induce IgG against a conserved epitope on the trimer close to the HIV envelope membrane termed the membrane-proximal external region (MPER), whereas such IgG was not induced in animals immunized with the soluble trimer109. In a separate study, Env trimers formed a well-ordered high-density array on liposomes, resulting in not only higher levels of B cell activation but the production of neutralizing antibodies against neutralization-resistant HIV strains (tier 2) in rodents110. As mentioned above, ICMVs have also been employed by Moon and co-workers to design a recombinant malaria vaccine using the antigen VMP00158. Liposomal enhancement of humoral responses may result from increased germinal centre formation, because higher numbers of germinal centres and antigen-specific Tfh cells were found in mice immunized with the ICMV vaccines.
Two additional advantages of liposomal nanomaterials include the ability to incorporate multiple antigens and the ability to tune and study the effects of vesicle stability. As an example of the former, Huang, Lovell and co-workers developed a liposomal vaccine platform where cobalt porphyrin-phospholide (CoPoP) was incorporated within the lipid membrane, so antigens expressed with a polyhistidine tag could be coupled to the liposomal surface88. They demonstrated that malaria Psf25 coupled to liposomes containing CoPoP and a TLR4 agonist could induce higher magnitudes of malaria oocyst-blocking antibodies than Psf25 adjuvanted with Alum. This platform then was explored with multiple antigens coupled to the liposomes, raising balanced responses against the antigens Pfg27, Pfs25, AMA-1, Psf30 and NANP, which are expressed in different stages of the malaria life cycle. Immunization with the same combination of antigens using Alum only induced antibodies against Pfs230. The authors surmised that the balanced multi-antigen response may have related to the low antigen dose used in the liposomal formulations or improved antigen uptake by APCs. To study how vesicle stability influenced immunogenicity against stabilized HIV gp140 trimers (BG505 MD39), Tokatlian, Irvine, and co-workers investigated synthetic liposomes variously incorporating sphingomyelin (Fig. 1k,l), which augmented both the stability of the liposomes and germinal centre and antibody responses148.
Gold nanoparticles, with their control over size, shape and surface properties, have received interest as vaccines149. For example, Kumar and colleagues developed malaria transmission-blocking vaccines based on the sexual stage antigen Pfs25 conjugated to gold nanoparticles150. The vaccine constructs were stable at 4°C over an 18-month period, and mice immunized with the nanoparticle-conjugated-Pfs25 vaccines developed robust antibody responses. Interestingly, the immunogenicity and IgG subclass distribution of the vaccine-elicited antibodies varied with the size, shape and other physico-chemical properties of the nanoparticles, with only some nanoparticle formulations able to achieve antibody titers comparable to that of mice immunized with Alum-adjuvanted Pfs25.
Gold nanoparticles modified with viral glycans can also serve as dendritic cell-targeting vaccine carriers, facilitating antigen presentation to T cells and thus inducing more potent cellular antiviral immunity. In the context of HIV, Climent and colleagues engineered gold nanoparticles with high-mannoside derivatives that resemble the N-linked high-mannose glycan clusters on HIV gp120 targeted by DC-SIGN receptors on DCs. They pulsed monocyte-derived dendritic cells isolated from HIV-positive patients with HIV antigen gag p17 conjugated onto the high-mannoside-modified gold nanoparticles, finding that the nanoparticulate formulation induced more potent ex vivo proliferation of the autologous CD8+ T cells than soluble antigen151.
Other inorganic nanoparticles have also been used in TB and HIV vaccine development. Yu and colleagues developed an Ion (III) oxide (Fe2O3)-coated plasmid DNA TB vaccine expressing two immunodominant antigens of M. tuberculosis (Ag85A and ESAT-6) along with IL-21152. Compared to the naked plasmid DNA, the NP-coated vaccine induced greater humoral and cellular responses in mice. Moreover, the bacterial burden in the lungs was significantly lower in animals immunized with the NP-coated vaccine than in animals immunized either with the naked plasmid vaccine or with BCG. The use of fullerenols (Fig. 1i,j) for the delivery of a HIV-1 Env-expressing plasmid DNA vaccine also led to improved T cell responses in mice, but this approach only marginally enhanced the humoral immune response153.
Our group has investigated fibrillar assemblies of short synthetic peptides, and while they have only begun to be explored in the context of the diseases of interest in this Review154,155,156, initial work has indicated that they possess a number of useful properties including considerable multivalency that allows them to raise strong T-cell and B-cell responses without requiring supplemental adjuvants157,158, synthetic definition and the ability to mix multiple epitopes or antigens. Several fibre-forming peptide systems have been reported including those based on β-sheet fibrillizing peptides such as Q11 or KFE891,95,159,160, or α-helical fibrillar assemblies such as Coil29161. In each of these systems, peptide epitopes can be conjugated to fibrillizing assembly domains, which spontaneously self-assemble into defined nanofibres displaying the epitopes on their surfaces. Recent work has explored several facets of these materials, including surface properties necessary for maximizing or minimizing their immunogenicity160, applying them towards intranasal vaccination159, and applying them towards therapeutic vaccines countering inflammation95. To expand the capacity of fibrillar peptide assemblies to incorporate large, folded protein antigens, we have also developed strategies based on the capture enzyme cutinase162 and designed expression tags termed β-tails163.
One of the challenges in developing vaccines towards malaria, HIV and TB is the global distribution of the diseases and their concentration in tropical and developing locations of the world. All current vaccines require uninterrupted maintenance between 4–8 °C, the so-called cold chain, adding additional practical challenges to the already daunting task of designing an effective vaccine. Peptide assemblies carrying peptide epitopes from the M. tuberculosis antigen ESAT-6 were strongly immunogenic even after storage at 45 °C for six months, indicating that this class of materials has advantageous thermal stability154. The repetitive malaria antigen (NANP)3 from the falciparum circumsporozoite protein has also been found to be immunogenic on self-assembled β-sheet nanofibres156, and ESAT-6 has also been explored on nanofibres of the self-assembling peptide KFE8155. Collectively these studies indicate that fibrillar peptide assemblies are attractive platforms not only for enhancing vaccine immunogenicity but also for facilitating vaccine implementation in resource-limited areas.
Conclusion and outlook
There has been an explosion of nanomaterials explored as new vaccines. Coupled with parallel advances in antigen design, it is possible that multiple nanomaterial platforms may ultimately be capable of identifying and reliably raising the defined, engineered immune responses necessary for protection against HIV/AIDS, malaria or TB. Once appropriate immunogens are discovered, more than one platform may likewise offer increased thermal and environmental protection to enable distribution to the most resource-limited locations of the globe. A key question remaining, then, is how to efficiently sort through all of the different platforms and combinations of adjuvants, immunomodulating compounds, delivery considerations, dosing regimens and other factors that have been made available in recent years. In this endeavour, multifactorial Design of Experiment approaches may be particularly powerful. Additionally, benchmarking different platforms against each other during their development will have considerable advantages over more focused and self-referential investigations, as this will help clarify the relative strengths and weaknesses of specific platforms and materials. At present, some of the most valuable research outcomes achieved to date include the consolidation and articulation of concepts by which such materials raise useful immune responses. This basis of knowledge now supports the continued refinement of vaccines towards devastating global diseases. In parallel with advances in antigen design, we are optimistic that vaccines efficacious enough to significantly curtail the global impact of malaria, HIV and TB are within reach.
References
The top 10 causes of death. WHO https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (2018).
Global Tuberculosis Report. WHO https://www.who.int/tb/publications/global_report/en/ (2018).
World Malaria Report (WHO, 2018); https://apps.who.int/iris/bitstream/handle/10665/275867/9789241565653-eng.pdf
Global HIV & AIDS statistics — 2018 fact sheet. UNAIDS http://www.unaids.org/en/resources/fact-sheet (2018).
Gammon, J. M. & Jewell, C. M. Engineering immune tolerance with biomaterials. Adv. Healthc. Mater. 18, 1801419–19 (2019).
Pati, R., Shevtsov, M. & Sonawane, A. Nanoparticle vaccines against infectious diseases. Front. Immunol. 9, 1–16 (2018).
Kim, J., Manspeaker, M. P. & Thomas, S. N. Augmenting the synergies of chemotherapy and immunotherapy through drug delivery. Acta Biomaterialia 88, 1–14 (2019).
Irvine, D. J., Hanson, M. C., Rakhra, K. & Tokatlian, T. Synthetic nanoparticles for vaccines and immunotherapy. Chem. Rev. 115, 11109–11146 (2015).
Scott, E. A., Karabin, N. B. & Augsornworawat, P. Overcoming immune dysregulation with immunoengineered nanobiomaterials. Annu. Rev. Biomed. Eng. 19, 57–84 (2017).
Tostanoski, L. H. & Jewell, C. M. Engineering self-assembled materials to study and direct immune function. Adv. Drug Deliv. Rev. 114, 60–78 (2017).
Kelly, S. H., Shores, L. S., Votaw, N. L. & Collier, J. H. Biomaterial strategies for generating therapeutic immune responses. Adv. Drug Deliv. Rev. 114, 3–18 (2017).
Koup, R. A. et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68, 4650–4655 (1994).
Betts, M. R. et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107, 4781–4789 (2006).
Hraber, P. et al. Prevalence of broadly neutralizing antibody responses during chronic HIV-1 infection. AIDS 28, 163–169 (2014).
Finney, J. & Kelsoe, G. Poly- and autoreactivity of HIV-1 bNAbs: implications for vaccine design. Retrovirology 15, 53 (2018).
Mascola, J. R. & Haynes, B. F. HIV-1 neutralizing antibodies: understanding nature’s pathways. Immunological Rev. 254, 225–244 (2013).
Hessell, A. J. et al. Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog. 5, e1000433 (2009).
Mascola, J. R. et al. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat. Med. 6, 207–210 (2000).
Julien, J. P. et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 342, 1477–1483 (2013).
Williams, W. B. et al. Diversion of HIV-1 vaccine-induced immunity by gp41-microbiota cross-reactive antibodies. Science 349, aab1253 (2015).
Kelsoe, G. & Haynes, B. F. Host controls of HIV broadly neutralizing antibody development. Immunol. Rev. 275, 79–88 (2017).
Borrow, P. & Moody, M. A. Immunologic characteristics of HIV-infected individuals who make broadly neutralizing antibodies. Immunol. Rev. 275, 62–78 (2017).
Rerks-Ngarm, S. et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 361, 2209–2220 (2009).
Haynes, B. F. et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 366, 1275–1286 (2012).
Tomaras, G. D. et al. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc. Natl Acad. Sci. USA 110, 9019–9024 (2013).
Montefiori, D. C. et al. Magnitude and breadth of the neutralizing antibody response in the RV144 and Vax003 HIV-1 vaccine efficacy trials. J. Infect. Dis. 206, 431–441 (2012).
Bonsignori, M. et al. Antibody-dependent cellular cytotoxicity-mediating antibodies from an HIV-1 vaccine efficacy trial target multiple epitopes and preferentially use the VH1 gene family. J. Virol. 86, 11521–11532 (2012).
Perez, L. G. et al. V1V2-specific complement activating serum IgG as a correlate of reduced HIV-1 infection risk in RV144. PLoS ONE 12, e0180720 (2017).
Cohen, J. Combo of two HIV vaccines fails its big test. Science 367, 611–612 (2020).
Barouch, D. H. et al. Protective efficacy of adenovirus/protein vaccines against SIV challenges in rhesus monkeys. Science 349, 320–324 (2015).
Bradley, T. et al. Pentavalent HIV-1 vaccine protects against simian-human immunodeficiency virus challenge. Nat. Commun. 8, 15711 (2017).
Burton, D. R. et al. Limited or no protection by weakly or nonneutralizing antibodies against vaginal SHIV challenge of macaques compared with a strongly neutralizing antibody. Proc. Natl Acad. Sci. USA 108, 11181–11186 (2011).
Santra, S. et al. Human Non-neutralizing HIV-1 envelope monoclonal antibodies limit the number of founder viruses during SHIV mucosal infection in rhesus macaques. PLoS Pathog. 11, e1005042 (2015).
Moore, P. L. et al. Nature of nonfunctional envelope proteins on the surface of human immunodeficiency virus type 1. J. Virol. 80, 2515–2528 (2006).
Crooks, E. T., Tong, T., Osawa, K. & Binley, J. M. Enzyme digests eliminate nonfunctional Env from HIV-1 particle surfaces, leaving native Env trimers intact and viral infectivity unaffected. J. Virol. 85, 5825–5839 (2011).
Sanders, R. W. et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 9, e1003618 (2013).
Jardine, J. et al. Rational HIV immunogen design to target specific germline B cell receptors. Science 340, 711–716 (2013).
Rolland, M., Nickle, D. C. & Mullins, J. I. HIV-1 group M conserved elements vaccine. PLoS Pathog. 3, e157 (2007).
Korber, B. T., Letvin, N. L. & Haynes, B. F. T-cell vaccine strategies for human immunodeficiency virus, the virus with a thousand faces. J. Virol. 83, 8300–8314 (2009).
Sundling, C. et al. Immunization of macaques with soluble HIV type 1 and influenza virus envelope glycoproteins results in a similarly rapid contraction of peripheral B-cell responses after boosting. J. Infect. Dis. 207, 426–431 (2013).
Itsara, L. S. et al. The Development of Whole Sporozoite Vaccines for Plasmodium falciparum Malaria. Front. Immunol. 9, 2748 (2018).
Ouattara, A. & Laurens, M. B. Vaccines against malaria. Clin. Infect. Dis. 60, 930–936 (2015).
Rts, S. C. T. P. Efficacy and safety of RTS, S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet 386, 31–45 (2015).
Rts, S. C. T. P. Efficacy and safety of the RTS, S/AS01 malaria vaccine during 18 months after vaccination: a phase 3 randomized, controlled trial in children and young infants at 11 African sites. PLos Med. 11, e1001685 (2014).
Ouattara, A. et al. Designing malaria vaccines to circumvent antigen variability. Vaccine 33, 7506–7512 (2015).
Dutta, S. et al. Overcoming antigenic diversity by enhancing the immunogenicity of conserved epitopes on the malaria vaccine candidate apical membrane antigen-1. PLoS Pathog. 9, e1003840 (2013).
Kazmin, D. et al. Systems analysis of protective immune responses to RTS, S malaria vaccination in humans. Proc. Natl Acad. Sci. USA 114, 2425–2430 (2017).
Schmidt, N. W. et al. Memory CD8 T cell responses exceeding a large but definable threshold provide long-term immunity to malaria. Proc. Natl Acad. Sci. USA 105, 14017–14022 (2008).
Gengenbacher, M. & Kaufmann, S. H. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol. Rev. 36, 514–532 (2012).
Orme, I. M. The Achilles heel of BCG. Tuberculosis 90, 329–332 (2010).
Dockrell, H. M. & Smith, S. G. What have we learnt about BCG vaccination in the last 20 years? Front. Immunol. 8, 1134 (2017).
Darrah, P. A. et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature 577, 95–102 (2020).
Lienhardt, C. et al. Translational research for tuberculosis elimination: Priorities, challenges, and actions. PLos Med. 13, e1001965 (2016).
Sable, S. B., Posey, J. E. & Scriba, T. J. Tuberculosis vaccine development: Progress in clinical evaluation. Clin. Microbiol. Rev. 33, 16076–30 (2019).
Van Der Meeren, O. et al. Phase 2b Controlled Trial of M72/AS01 EVaccine to Prevent Tuberculosis. N. Engl. J. Med. 379, 1621–1634 (2018).
Nemes, E. et al. Prevention of M. tuberculosisInfection with H4:IC31 Vaccine or BCG Revaccination. N. Engl. J. Med. 379, 138–149 (2018).
Schrager, L. K., Izzo, A. & Velmurugan, K. Immunopathogenesis of tuberculosis and novel mechanisms of vaccine activity. Tuberculosis 99, S3–7 (2016).
Moon, J. J. et al. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc. Natl Acad. Sci. USA 109, 1080–1085 (2012).
Demento, S. L. et al. Role of sustained antigen release from nanoparticle vaccines in shaping the T cell memory phenotype. Biomaterials 33, 4957–4964 (2012).
Swartz, M. A., Hirosue, S. & Hubbell, J. A. Engineering approaches to immunotherapy. Sci. Transl. Med. 4, 148rv9–148rv9 (2012).
Hirosue, S., Kourtis, I. C., van der Vlies, A. J., Hubbell, J. A. & Swartz, M. A. Antigen delivery to dendritic cells by poly(propylene sulfide) nanoparticles with disulfide conjugated peptides: Cross-presentation and T cell activation. Vaccine 28, 7897–7906 (2010).
Reddy, S. T. et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 25, 1159–1164 (2007).
Kaminskas, L. M. & Porter, C. J. H. Targeting the lymphatics using dendritic polymers (dendrimers). Adv. Drug Deliv. Rev. 63, 890–900 (2011).
Kourtis, I. C. et al. Peripherally administered nanoparticles target monocytic myeloid cells, secondary lymphoid organs and tumors in mice. PLoS ONE 8, e61646 (2013).
Sixt, M. et al. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity 22, 19–29 (2005).
Rohner, N. A. & Thomas, S. N. Flexible macromolecule versus rigid particle retention in the injected skin and accumulation in draining lymph nodes are differentially influenced by hydrodynamic size. ACS Biomater. Sci. Eng. 3, 153–159 (2017).
Pape, K. A., Catron, D. M., Itano, A. A. & Jenkins, M. K. The humoral immune response is initiated in lymph nodes by B cells that acquire soluble antigen directly in the follicles. Immunity 26, 491–502 (2007).
Mohanan, D. et al. Administration routes affect the quality of immune responses: A cross-sectional evaluation of particulate antigen-delivery systems. J. Control. Release 147, 342–349 (2010).
Jewell, C. M., López, S. C. B. & Irvine, D. J. In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc. Natl Acad. Sci. USA 108, 15745–15750 (2011).
Lai, S. K., Wang, Y. Y., Wirtz, D. & Hanes, J. Micro- and macrorheology of mucus. Adv. Drug Deliv. Rev. 61, 86–100 (2009).
Lai, S. K., Wang, Y. Y. & Hanes, J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv. Drug Deliv. Rev. 61, 158–171 (2009).
Lai, S. K., Wang, Y. Y., Hida, K., Cone, R. & Hanes, J. Nanoparticles reveal that human cervicovaginal mucus is riddled with pores larger than viruses. Proc. Natl Acad. Sci. USA 107, 598–603 (2010).
Schuster, B. S., Suk, J. S., Woodworth, G. F. & Hanes, J. Nanoparticle diffusion in respiratory mucus from humans without lung disease. Biomaterials 34, 3439–3446 (2013).
Singh, B. et al. Chitosan-based particulate systems for the delivery of mucosal vaccines against infectious diseases. Int. J. Biol. Macromol. 110, 54–64 (2018).
Bivas-Benita, M. et al. Pulmonary delivery of chitosan-DNA nanoparticles enhances the immunogenicity of a DNA vaccine encoding HLA-A*0201-restricted T-cell epitopes of Mycobacterium tuberculosis. Vaccine 22, 1609–1615 (2004).
Champion, J. A. & Mitragotri, S. Role of target geometry in phagocytosis. Proc. Natl Acad. Sci. USA 103, 4930–4934 (2006).
Yu, S. S. et al. Size-and charge-dependent non-specific uptake of PEGylated nanoparticles by macrophages. IJN 7, 799–813 (2012).
Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. IJN 7, 5577–5591 (2012).
Xiang, S. D. et al. Pathogen recognition and development of particulate vaccines: Does size matter? Methods 40, 1–9 (2006).
Leleux, J., Atalis, A. & Roy, K. Engineering immunity: Modulating dendritic cell subsets and lymph node response to direct immune-polarization and vaccine efficacy. J. Control. Release 219, 610–621 (2015).
Figdor, C. G., van Kooyk, Y. & Adema, G. J. C-type lectin receptors on dendritic cells and langerhans cells. Nat. Rev. Immunol. 2, 77–84 (2002).
Tokatlian, T. et al. Innate immune recognition of glycans targets HIV nanoparticle immunogens to germinal centers. Science 363, 649–654 (2018).
Kim, Y. M. et al. Monovalent ligation of the B cell receptor induces receptor activation but fails to promote antigen presentation. Proc. Natl Acad. Sci. USA 103, 3327–3332 (2006).
Smirnov, D., Schmidt, J. J., Capecchi, J. T. & Wightman, P. D. Vaccine adjuvant activity of 3M-052: An imidazoquinoline designed for local activity without systemic cytokine induction. Vaccine 29, 5434–5442 (2011).
Ilyinskii, P. O. et al. Adjuvant-carrying synthetic vaccine particles augment the immune response to encapsulated antigen and exhibit strong local immune activation without inducing systemic cytokine release. Vaccine 32, 2882–2895 (2014).
Hanson, M. C. et al. Nanoparticulate STING agonists are potent lymph node–targeted vaccine adjuvants. J. Clin. Invest. 125, 2532–2546 (2015).
Friede, M., Muller, S., Briand, J.-P., Van Regenmortel, M. H. V. & Schuber, F. Induction of immune response against a short synthetic peptide antigen coupled to small neutral liposomes containing monophosphoryl lipid a. Mol. Immun. 30, 539–547 (1993).
Huang, W. C. et al. A malaria vaccine adjuvant based on recombinant antigen binding to liposomes. Nat. Nanotechnol. 13, 1174–1181 (2018).
Bachmann, M. F. & Zinkernagel, R. M. The Influence of virus structure on antibody responses and virus serotype formation. Immunol. Today 17, 553–558 (1996).
schiller, j & chackerian, b why hiv virions have low numbers of envelope spikes: Implications for vaccine development. PLoS Pathog. 10, e1004254 (2014).
Chen, J. et al. The use of self-adjuvanting nanofiber vaccines to elicit high-affinity B cell responses to peptide antigens without inflammation. Biomaterials 34, 8776–8785 (2013).
Chesson, C. B. et al. Antigenic peptide nanofibers elicit adjuvant-free CD8+ T cell responses. Vaccine 32, 1174–1180 (2014).
Black, M. et al. Self-assembled peptide amphiphile micelles containing a cytotoxic T-cell epitope promote a protective immune response in vivo. Adv. Mater. 24, 3845–3849 (2012).
Barrett, J. C. et al. Modular Peptide Amphiphile Micelles Improving an Antibody-Mediated Immune Response to Group A Streptococcus. ACS Biomater. Sci. Eng. 3, 144–152 (2016).
Mora-Solano, C. et al. Active immunotherapy for TNF-mediated inflammation using self-assembled peptide nanofibers. Biomaterials 149, 1–11 (2017).
Babapoor, S. et al. A Novel vaccine using nanoparticle platform to present immunogenic M2e against avian influenza infection. Influenza Res. Trea. 2011, 1–12 (2011).
Kaba, S. A. et al. ProtectiveaAntibody and CD8+ T-cell responses to the plasmodium falciparum circumsporozoite protein induced by a nanoparticle vaccine. PLoS ONE 7, e48304 (2012).
Skwarczynski, M. et al. Polyacrylate dendrimer nanoparticles: A self-adjuvanting vaccine delivery system. Angew. Chem. Int. Ed. 49, 5742–5745 (2010).
Dintzis, H. M., Dintzis, R. Z. & Vogelstein, B. Molecular determinants of immunogenicity: The immunon model of immune response. Proc. Natl Acad. Sci. USA 73, 3671–3675 (1976).
Dintzis, R. Z., Vogelstein, B. & Dintzis, H. M. Specific cellular stimulation in the primary immune response: Experimental test of a quantized model. Proc. Natl Acad. Sci. USA 79, 884–888 (1982).
Dintzis, R. Z., Okajima, M., Middleton, M. H., Greene, G. & Dintzis, H. M. The immunogenicity of soluble haptenated polymers is determined by molecular mass and hapten valence. J. Immunol. 143, 1239–1244 (1989).
Rudra, J. S. et al. Modulating adaptive immune responses to peptide self-assemblies. ACS Nano 6, 1557–1564 (2012).
Batista, F. D. & Neuberger, M. S. B cells extract and present immobilized antigen: implications for affinity discrimination. EMBO J. 19, 513–520 (2000).
Chackerian, B., Lenz, P., Lowy, D. R. & Schiller, J. T. Determinants of Autoantibody Induction by Conjugated Papillomavirus Virus-Like Particles. J. Immunol. 169, 6120–6126 (2002).
Puffer, E. B., Pontrello, J. K., Hollenbeck, J. J., Kink, J. A. & Kiessling, L. L. Activating B cell signaling with defined multivalent ligands. ACS Chem. Biol. 2, 252–262 (2007).
Bennett, N. R., Zwick, D. B., Courtney, A. H. & Kiessling, L. L. Multivalent antigens for promoting B and T cell activation. ACS Chem. Biol. 10, 1817–1824 (2015).
Pompano, R. R. et al. Titrating T-cell epitopes within self-assembled vaccines optimizes CD4+ helper T cell and antibody outputs. Adv. Healthc. Mater. 3, 1898–1908 (2014).
Hong, S. et al. B cells are the dominant antigen-presenting cells that activate naive CD4+ T cells upon immunization with a virus-derived nanoparticle antigen. Immunity 49, 695–708e4 (2018).
Pejawar-Gaddy, S., Kovacs, J. M., Barouch, D. H., Chen, B. & Irvine, D. J. Design of Lipid Nanocapsule Delivery vehicles for multivalent display of recombinant Env trimers in HIV vaccination. Bioconjugate Chem. 25, 1470–1478 (2014).
Ingale, J. et al. High-density array of well-ordered HIV-1 spikes on synthetic liposomal nanoparticles efficiently activate B cells. CellReports 15, 1986–1999 (2016).
Marcandalli, J. et al. Induction of potent neutralizing antibody responses by a designed protein nanoparticle vaccine for respiratory syncytial virus. Cell 176, 1420–1431e17 (2019).
Brouwer, P. J. M. et al. Enhancing and shaping the immunogenicity of native-like HIV-1 envelope trimers with a two-component protein nanoparticle. Nat. Commun. 10, 4247 (2019).
Kanekiyo, M. et al. Rational design of an Epstein-Barr Virus vaccine targeting the receptor-binding site. Cell 162, 1090–1100 (2015).
Pauthner, M. et al. Elicitation of robust tier 2 neutralizing antibody responses in nonhuman primates by HIV envelope trimer immunization using optimized approaches. Immunity 46, 1073–1088e6 (2017).
Kulp, D. W. et al. Structure-based design of native-like HIV-1 envelope trimers to silence non-neutralizing epitopes and eliminate CD4 binding. Nat. Commun. 8, 1655 (2017).
Bazzill, J. D. et al. Interrogation of antigen display on individual vaccine nanoparticles for achieving neutralizing antibody responses against hepatitis C virus. Nano Lett. 18, 7832–7838 (2018).
Martinez-Murillo, P. et al. Particulate array of well-ordered HIV clade C Env trimers elicits neutralizing antibodies that display a unique V2 cap approach. Immunity 46, 804–817e7 (2017).
Kanekiyo, M. et al. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature 499, 102–106 (2013).
Gordon, D. M. et al. Safety, immunogenicity, and efficacy of a recombinantly produced plasmodium falciparum circumsporozite protein-hepatitis B surface antigen subunit vaccine. J. Infect. Dis. 171, 1576–1585 (1995).
Nieusma, T. et al. Rational HIV immunogen design to target specific germline B cell receptors. Science 340, 711–716 (2013).
He, D. & Marles-Wright, J. Ferritin family proteins and their use in bionanotechnology. N. Biotechnol. 32, 651–657 (2015).
Sliepen, K. et al. Presenting native-like HIV-1 envelope trimers on ferritin nanoparticles improves their immunogenicity. Retrovirology 12, 82 (2015).
Saunders, K. O. et al. Targeted selection of HIV-specific antibody mutations by engineering B cell maturation. Science 366, eaay7199–19 (2019).
Ogun, S. A., Dumon-Seignovert, L., Marchand, J. B., Holder, A. A. & Hill, F. The oligomerization domain of C4-binding protein (C4bp) acts as an adjuvant, and the fusion protein comprised of the 19-kilodalton merozoite surface protein 1 fused with the murine C4bp domain protects mice against malaria. Infect. Immun. 76, 3817–3823 (2008).
Li, Y. et al. Enhancing immunogenicity and transmission-blocking activity of malaria vaccines by fusing Pfs25 to IMX313 multimerization technology. Sci. Rep. 6, 1–13 (2016).
Brod, F. et al. Combination of RTS, S and Pfs25-IMX313 induces a functional antibody response against malaria infection and transmission in mice. Front. Immunol. 9, e474 (2018).
Spencer, A. J. et al. Fusion of the Mycobacterium tuberculosis Antigen 85A to an Oligomerization Domain Enhances Its Immunogenicity in Both Mice and Non-Human Primates. PLoS ONE 7, e33555 (2012).
Minhinnick, A. et al. A first-in-human phase 1 trial to evaluate the safety and immunogenicity of the candidate tuberculosis vaccine MVA85A-IMX313, administered to BCG-vaccinated adults. Vaccine 34, 1412–1421 (2016).
Gao, Y., Wijewardhana, C. & Mann, J. F. S. Virus-like particle, liposome, and polymeric particle-based vaccines against HIV-1. Front. Immunol. 9, 1–18 (2018).
Mohsen, M. O., Zha, L., Cabral-Miranda, G. & Bachmann, M. F. Major findings and recent advances in virus–like particle (VLP)-based vaccines. Semin. Immunol. 34, 123–132 (2017).
Zeltins, A. Construction and characterization of virus-like particles: A review. Mol. Biotechnol. 53, 92–107 (2012).
Zwick, M. B. et al. Dense array of spikes on HIV-1 Virion particles. J. Virol. 91, 1–19 (2017).
McCurley, N. P. et al. HIV transmitted/founder vaccines elicit autologous tier 2 neutralizing antibodies for the CD4 binding site. PLoS ONE 12, e0177863 (2017).
Kaba, S. A. et al. A nonadjuvanted polypeptide nanoparticle vaccine confers long-lasting protection against rodent malaria. J. Immunol. 183, 7268–7277 (2009).
Zhao, L. et al. Nanoparticle vaccines. Vaccine 32, 327–337 (2014).
Peek, L. J., Middaugh, C. R. & Berkland, C. Nanotechnology in vaccine delivery. Adv. Drug Deliv. Rev. 60, 915–928 (2008).
Cleland, J. L. et al. Development of a single-shot subunit vaccine for HIV-1. AIDS Res. Hum. Retrovir. 10, S21–6 (1994).
Rostami, H., Ebtekar, M., Ardestani, M. S., Yazdi, M. H. & Mahdavi, M. Co-utilization of a TLR5 agonist and nano-formulation of HIV-1 vaccine candidate leads to increased vaccine immunogenicity and decreased immunogenic dose: A preliminary study. Immunol. Lett. 187, 19–26 (2017).
Kasturi, S. P. et al. Programming the magnitude and persistence of antibody responses with innate immunity. Nature 470, 543–547
Bailey, B. A. et al. Self-encapsulating Poly(lactic- co-glycolic acid) (PLGA) microspheres for intranasal vaccine delivery. Mol. Pharmaceutics 14, 3228–3237 (2017).
Bailey, B. A., Ochyl, L. J., Schwendeman, S. P. & Moon, J. J. Toward a single-dose vaccination strategy with self-encapsulating PLGA microspheres. Adv. Funct. Mater. 6, 1601418 (2017).
Cherif, M. S. et al. Immunogenicity of novel nanoparticle-coated MSP-1 C-terminus malaria DNA vaccine using different routes of administration. Vaccine 29, 9038–9050 (2011).
Cui, Z. & Mumper, R. J. Chitosan-based nanoparticles for topical genetic immunization. J. Control. Release 75, 409–419 (2001).
Feng, G. et al. Enhanced immune response and protective effects of nano-chitosan-based DNA vaccine encoding T Cell epitopes of Esat-6 and FL against Mycobacterium Tuberculosis infection. PLoS ONE 8, e61135 (2013).
Chahal, J. S. et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl Acad. Sci. USA 113, E4133–E4142 (2016).
Chong, S. F. et al. A paradigm for peptide vaccine delivery using viral epitopes encapsulated in degradable polymer hydrogel capsules. Biomaterials 30, 5178–5186 (2009).
DeMuth, P. C. et al. Polymer multilayer tattooing for enhanced DNA vaccination. Nat. Mater. 12, 367–375 (2013).
Tokatlian, T. et al. Enhancing humoral responses against HIV envelope trimers via nanoparticle delivery with stabilized synthetic liposomes. Sci. Rep. 8, 16527 (2018).
Gregory, A. E., Titball, R. & Williamson, D. Vaccine delivery using nanoparticles. Front. Cell. Infect. Microbiol. 3, 1–13 (2013).
Kumar, R. et al. Nanovaccines for malaria using Plasmodium falciparum antigen Pfs25 attached gold nanoparticles. Vaccine 33, 5064–5071 (2015).
Climent, N. et al. Loading dendritic cells with gold nanoparticles (GNPs) bearing HIV-peptides and mannosides enhance HIV-specific T cell responses. Nanomedicine 14, 339–351 (2017).
Yu, F. et al. Nanoparticle-based adjuvant for enhanced protective efficacy of DNA vaccine Ag85A-ESAT-6-IL-21 against Mycobacterium tuberculosis infection. Nanomedicine 8, 1337–1344 (2012).
Xu, L. et al. Morphologically virus-like fullerenol nanoparticles act as the dual-functional nanoadjuvant for HIV-1 vaccine. Adv. Mater. 25, 5928–5936 (2013).
Sun, T. et al. Thermal stability of self-assembled peptide vaccine materials. Acta Biomaterialia 30, 62–71 (2016).
Chesson, C. B. et al. Nanoscale peptide self-assemblies boost BCG-primed cellular immunity against mycobacterium tuberculosis. Sci. Rep. 8, 12519 (2018).
Rudra, J. S. et al. Self-assembled peptide nanofibers raising durable antibody responses against a malaria epitope. Biomaterials 33, 6476–6484 (2012).
Rudra, J. S., Tian, Y. F., Jung, J. P. & Collier, J. H. A self-assembling peptide acting as an immune adjuvant. Proc. Natl Acad. Sci. USA 107, 622–627 (2010).
Solano, C. M., Wen, Y., Han, H. & Collier, J. H. in Peptide Self-Assembly: Methods and Protocols (eds. Nilsson, B. L. & Doran, T. M.) 233–248 (Springer New York, 2018).
Si, Y., Wen, Y., Kelly, S. H., Chong, A. S. & Collier, J. H. Intranasal delivery of adjuvant-free peptide nanofibers elicits resident CD8+ T cell responses. J. Control. Release 282, 120–130 (2018).
Wen, Y., Waltman, A., Han, H. & Collier, J. H. Switching the immunogenicity of peptide assemblies using surface properties. ACS Nano 10, 9274–9286 (2016).
Wu, Y. et al. A Supramolecular vaccine platform based on α-helical peptide nanofibers. ACS Biomater. Sci. Eng. 3, 3128–3132 (2017).
Hudalla, G. A. et al. A Self-adjuvanting supramolecular vaccine carrying a folded protein antigen. 2, 1114–1119 (2014).
Hudalla, G. A. et al. Gradated assembly of multiple proteins into supramolecular nanomaterials. Nat. Mater. 13, 829–836 (2014).
Kaba, S. A. et al. Protective Antibody and CD8+ T-Cell responses to the plasmodium falciparum circumsporozoite protein induced by a nanoparticle vaccine. PLoS ONE 7, e48304–11 (2012).
Moon, J. J. et al. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat. Mater. 10, 243–251 (2011).
Phan, T. G., Grigorova, I., Okada, T. & Cyster, J. G. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat. Immunol. 8, 992–1000 (2007).
Yang, Q. et al. Analysis of pre-existing IgG and IgM antibodies against polyethylene glycol (PEG) in the general population. Anal. Chem. 88, 11804–11812 (2016).
Schellekens, H., Hennink, W. E. & Brinks, V. The immunogenicity of polyethylene glycol: Facts and fiction. Pharm. Res. 30, 1729–1734 (2013).
Zhuang, Y. et al. PEGylated cationic liposomes robustly augment vaccine-induced immune responses: Role of lymphatic trafficking and biodistribution. J. Control. Release 159, 135–142 (2012).
Kumar, R. et al. Nanovaccines for malaria using Plasmodium falciparum antigen Pfs25 attached gold nanoparticles. Vaccine 33, 5064–5071 (2015).
Acknowledgements
Related research in our laboratories is currently supported by the NIH (NIBIB 5R01EB009701; NIAID 5R01AI145016), Duke MEDx, and the Duke Center for AIDS Research (CFAR). C.N.F. and E.J.C are supported by the National Science Foundation Graduate Research Fellowship Program (DGE-1644868). The content of this Review is solely the responsibility of the authors and do not necessarily represent the official views of these funding agencies.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
J.H.C., S.R.P., G.G.F. and C.N.F. are inventors on a patent application associated with the technology areas described.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fries, C.N., Curvino, E.J., Chen, JL. et al. Advances in nanomaterial vaccine strategies to address infectious diseases impacting global health. Nat. Nanotechnol. 16, 1–14 (2021). https://doi.org/10.1038/s41565-020-0739-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41565-020-0739-9
This article is cited by
-
Nanovaccines to combat drug resistance: the next-generation immunisation
Future Journal of Pharmaceutical Sciences (2023)
-
Nanomedicine and nanoparticle-based delivery systems in plastic and reconstructive surgery
Maxillofacial Plastic and Reconstructive Surgery (2023)
-
A nanoadjuvant that dynamically coordinates innate immune stimuli activation enhances cancer immunotherapy and reduces immune cell exhaustion
Nature Nanotechnology (2023)
-
Engineering nanomaterial physical characteristics for cancer immunotherapy
Nature Reviews Bioengineering (2023)
-
Accelerating the prediction and discovery of peptide hydrogels with human-in-the-loop
Nature Communications (2023)
