
Abstract
Better methods to predict and prevent the emergence of zoonotic viruses could support future efforts to reduce the risk of epidemics. We propose a network science framework for understanding and predicting human and animal susceptibility to viral infections. Related approaches have so far helped to identify basic biological rules that govern cross-species transmission and structure the global virome. We highlight ways to make modelling both accurate and actionable, and discuss the barriers that prevent researchers from translating viral ecology into public health policies that could prevent future pandemics.
Similar content being viewed by others


Main
Most emerging human infectious diseases originate in wild animals1. Of these zoonoses, viruses account for the majority of severe epidemics and pose the greatest pandemic threat due to their transmissibility, evolvability and lack of therapeutic options. Every year, a growing number of viruses are detected in human hosts for the first time2, both because of better surveillance and because the rate of viral emergence is increasing3. Factors that contribute to emergence risk include weak health systems, globalization, inequality, conflict, increasing human–wildlife contact, agricultural intensification, deforestation and climate change4,5,6. Given the urgency of understanding and containing these threats, substantial research effort has been directed towards modelling and predicting host–virus interactions in recent decades.
With the advent of the COVID-19 (coronavirus disease 2019) pandemic, the global scientific community may finally have a broad public and policy audience willing to tackle emerging zoonotic diseases, with the aim of ‘predicting and preventing the next pandemic’7. ‘Prevention’ ultimately falls on healthcare systems and policy interventions, but ‘prediction’—knowing which possible threats should be countered most urgently—is a task that draws on various fields, including microbiology, virology, ecology, evolutionary biology and statistics. Experts in these fields face a massive problem of triage: today’s public health emergencies are caused by only a small subset of the thousands of animal viruses that have zoonotic potential—the ability to infect a human host8,9. Modelling can accelerate the identification of potential future threats if general rules determine which animals and which viruses will pose a future threat to humans with enough specificity to make these predictions actionable. Statistical models have helped to identify reservoir species of novel human pathogens10, map the geographic distribution of risk11, identify seasonal trends in spillover12, estimate transmissibility and virulence post-emergence in humans13, quantify outbreak detectability and under-detection14, and project onward spread15. All of these objectives are part of a prediction pipeline intended to make basic science actionable in public health, but currently these fundamental endeavours rarely reach their full applied potential.
Here we review the subset of modelling studies that predict the potential for specific viruses to infect host species (hereafter host–virus associations). We aim to highlight the main approaches, hypotheses and innovations in this area. These studies have helped to synthesize the basic biological mechanisms that structure the global virome and have shown increasing potential to identify the highest-risk hosts and viruses. However, risk assessment for known viruses is still carried out using a mix of expert opinion and laboratory work16,17, largely in the absence of predictive methods. As such, most modelling work has translated into limited veterinary or public health benefits, leading to concern that predictive approaches may have limited utility for outbreak prevention, especially when compared with direct investments in aid programmes, capacity building, syndromic surveillance or vaccine development18,19. We assess how models may better deliver on their promise and contribute meaningfully to the prediction of future viral threats.
Using patterns for prediction
When the public or policymakers talk about ‘prediction’ in a One Health or pandemic preparedness setting, this idea often refers to anticipating future events. In this view, knowing an outbreak is coming may conceivably allow it to be circumvented or, at least, substantially lessened in scope and duration. However, in computational biology, ‘prediction’ more often refers to the ability of quantitative tools to recapitulate and explain the world as it exists today and has done through history and, by extension, anticipate both unknown contemporary patterns and potential future ones. Conventionally, these approaches fall on a continuum between mechanistic, hypothesis-driven statistics (often associated with the idea of explanatory prediction, based on iterative confirmation of theory) and mechanism-agnostic, exploratory machine learning (used to make predictions over new data, also called anticipatory prediction)20,21. However, the two approaches are synergistic, and the boundary between the approaches is increasingly blurred owing to both an expanding set of tools for interpretable (non-‘black box’) machine learning and a growing set of opportunities (and expectations) to use model–lab–field feedbacks to challenge and improve predictive models.
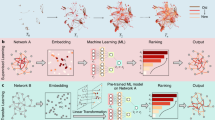
Predictive tools can be used to explain and anticipate many aspects of pathogen transmission. Here we review a subset of those tools, which aim to identify and predict why some host species can be infected with some viruses and others cannot. Models can, with increasing accuracy, predict the zoonotic potential, reservoir hosts or host range of a virus species; the viral diversity of a given host species; and viral sharing among host species. These basic model formulations can all be viewed as subsets of one overarching statistical approach: using statistical models trained on host–virus association data to explain, reproduce and infer the structure of the host–virus network (Fig. 1).

Studies can use data describing known host–virus interactions to predict potential cross-species transmission. Several approaches involve predicting the structure of host–virus association networks (depicted here using real associations known from large datasets in disease ecology), including zoonotic risk prediction (A: can a virus infect humans?) and reservoir identification (B: where does a zoonotic virus come from?); work trying to predict viral host range (C: how many hosts?) and viral diversity (E: how many viruses?); and viral sharing analysis (D: which hosts share viruses?). All of these are subsets of a general problem (F): the prediction of bipartite network links, as a way of representing host–virus associations. Approaching these problems as general link prediction may lead to new insights, especially when considering a more complete network. For comparison, we show the full range of recorded interactions compiled across the HP3 database (G: black lines match the smaller examples, but additional known links are added in grey), for example a recent link prediction study found a high probability that two of these viruses (bovine viral diarrhoea disease virus 1 and bluetongue virus, red links) may have undiscovered zoonotic potential (red lines in ref. 37). Light and dark blue nodes on the right (G) represent the same viruses and hosts, respectively depicted on the left (A–F).
These approaches can be identified by the shared data structure they use, which always consists of an edgelist—a set of known host–virus associations, in which most missing or negative records of association represent untested potential interactions—and linked metadata that may include data collection methods, host and virus traits (microbiological, ecological or phylogenetic), and infection characteristics22,23,24,25,26. The quality of these datasets varies in terms of scope, completeness, accuracy and documentation, reflecting the challenges of both wildlife virology and data synthesis. For example, matrix sparsity is a major limitation for computational power: most datasets only record known interactions, with few ‘true negatives’, and many presumed negatives are actually unrecorded associations. Even in long-term ecosystem studies, a third of ‘cryptic’ host–pathogen interactions may go unrecorded27, while at the planetary scale, over 90% of possible mammal–virus associations may never have been observed28. At the same time, reported interactions may also include a mix of data quality (that is, a mix of true and false positives). For example, serological evidence can be confounded by cross-reactivity among closely related viruses, and the process of digitization can also introduce new errors and inconsistencies, particularly in host and virus taxonomy. Every dataset contains unique iterations of these challenges, and discrepancies among them can create significant problems for reproducible hypothesis testing26. Researchers may therefore aim to work from standardized datasets such as The Global Virome in One Network (VIRION)29, which aims to compile every available source of information on vertebrate viruses into one dynamic, open dataset with a reconciled taxonomic backbone and rich metadata on host and virus taxonomy, data provenance and evidence of interactions.
Owing to a common data structure, most studies explore the same basic biological patterns, leading to a broad set of similar findings. For example, exposure and susceptibility within host populations have analogues at eco-evolutionary scales in ‘opportunity’ and ‘compatibility’, captured by geographic and phylogenetic data, respectively4. Host species with broader geographic ranges develop more population genetic structure, encounter more habitats and contact more species—facilitating pathogen exchange at an ecological scale, and viral diversification at an evolutionary scale30,31. Consequently, host geographic range size is a common predictor of viral diversity22,32 and reservoir status11,22, and range overlap is a strong predictor of viral sharing between hosts30. Most studies find a similarly strong phylogenetic effect: some animal clades disproportionately host particular viruses22,33, closely related host species share more viruses30, and zoonoses disproportionately originate in non-human primates13,22. In predictive models, host phylogenies can help to identify and recapitulate a combination of intrinsic autocorrelation (closely related hosts share coevolutionary histories with specific viruses) and latent biological mechanisms (closely related hosts share traits such as metabolic pathways, viral receptors or innate immune mechanisms through identity by descent34). The relative contribution of the two is rarely identifiable, particularly because large databases of within-host traits (for example, receptor chemistry or innate immune responses) are mostly unavailable, thus most studies use host phylogeny as a broad, correlative proxy. Together, phylogeographic predictors are often the strongest across modelling approaches.
Broad similarities such as these point to a set of emerging ‘universal laws’—for example, ‘phylogeographic proximity increases the similarity of host viromes’—that have been repeatedly supported across modelling studies, were often suggested in advance by theory and experimental evidence34,35,36 and predict unknown host–virus associations with surprising accuracy30. However, different study designs can produce very different kinds of ‘predictions’ (Fig. 2), and even though many studies use the same data and statistical methods, the lack of a shared modelling framework has made it difficult to synthesize these findings, for example varying and complex reporting formats prevent researchers from conducting formal meta-analyses. We outline such a framework (Table 1) and the broad patterns that each approach has so far uncovered. To help researchers build on those patterns, we have organized the last decade of this scientific work into our taxonomy in the Host–Virus Model Database (HVMD, available at viralemergence.org/hivemind), an evidence base of predictive studies and their data, methodology and key findings (and one that we hope will, over time, become more comprehensive than the necessarily limited coverage of studies here).

a, A link prediction model inferring the probability of host–parasite interactions using link prediction. Data are from ref. 27. b, A host inference model predicting the richness of probable Nipah virus reservoirs at specific locations. Data are from ref. 41. c, A zoonotic risk model, demonstrating that vector-borne viruses and those able to replicate in the cytoplasm are more likely to be able to infect humans. Individual data points indicate partial residuals, black lines are the mean partial effect and shaded areas indicate the 95% confidence interval. Data are from ref. 22. d, A viral sharing model showing that closely related sympatric mammals are more likely to share viruses. Black lines are the mean partial effects, and shaded areas indicate 95% confidence intervals. Data are from ref. 30.
Model design shapes insights and applications
Predicting species interactions is a fundamental task in ecology, especially with the emergence of ecological network science as a subfield. Here we discuss six approaches researchers can use to understand host–virus interactions as a network science problem. The most general approach, link prediction models, uses known associations in an ecological network to infer the probable association of any two species. For symbiotic interactions, the model is usually structured on the basis of a bipartite network of hosts and symbionts (for example hosts and viruses, or plants and pollinators), with species traits used to predict binary link values that denote the presence or absence of an interaction27,37. Link prediction is a general case of other specialized models: for example, zoonotic risk models calculate the link probability between all virus nodes and one host node (humans) (Fig. 1a), and link prediction models can similarly be used to identify potential zoonoses as a subset of their predictions37. However, different kinds of link prediction may have subtle conceptual differences. For example, a ‘link’ can be hard to define: is the aim to predict all existing hosts of a virus or all potentially compatible hosts? Generic host–virus association data are often mismatched with the study aims; these sources primarily catalogue viruses in natural hosts, but they also contain a mix of experimental infections that may be unrepresentative of infection dynamics in the wild, and serological detections that indicate exposure but not necessarily competence (and that are confounded by cross-reactivity38). Careful training data selection and analytical design can help to predict ‘links’ with more biological meaning39, and reporting the sensitivity of model results to different evidentiary standards can improve transparency22.
Host inference considers a one-sided subset of the link prediction problem, focused on predicting the hosts, reservoirs or sometimes vectors of one specific virus. The approach is most valuable when the definitive reservoir is unknown40, additional reservoirs are suspected41 or intermediate hosts are of interest42. Models can be easily tailored to these circumstances by using training data that reflect host competence (for example viral isolation instead of PCR or serology, or testing tick larvae for pathogens as a proxy of a host’s ability to transmit39) or distinguish reservoirs from incidental hosts43. These approaches have also been widely suggested as a way of triaging viral sampling in wildlife41, but model performance is variable, and this approach has only been tested in limited settings. As genomic data become more integral to the field, these approaches also show tremendous promise to narrow the search for hosts of ‘orphan viruses’ with no known non-human hosts10. Finally, mapping the distribution of predicted hosts can help to reveal the spatial extent of spillover risk and can inform possible futures after climate and land use change4.
Conversely, zoonotic risk models aim to identify which viruses can infect one specific host (humans), a task that is often framed as the most important for public health applications. Most statistical analyses have focused on the factors that predict innate cross-species transmission potential, with the assumption that humans are ‘just another host’ and that high host plasticity predicts zoonotic potential44,45. More often, the risk factors used in zoonotic risk models are quite coarse and describe thousands of candidate viruses, such as RNA viruses46,47 with broad host range44,45, larger genomes48, vector-borne transmission22, replication in the cytoplasm22,33,49 or lipid envelopes22,33. Interestingly, many of these traits also have contradictory impacts on transmissibility or severity, adding a layer of complexity; some approaches extend this modelling framework to explicitly predict these downstream properties of zoonotic viruses13,50. Some cutting-edge approaches focus more on the specific underpinnings of human–virus compatibility, anticipating structural and biochemical interactions between viruses and cell receptors51 and using genomic features and machine learning to identify human viruses from metagenomic samples52 or to predict zoonotic potential across different influenza or bacterial strains53,54.
Statistical analyses of viral sharing reframe bipartite host–virus networks as unipartite host–host networks and predict whether two hosts share any viruses on the basis of host traits alone. These approaches are limited by viewing viral ecology as an emergent property of hosts, but this reframing reduces network sparsity and opens up an underexplored computational toolkit for unipartite network models55. These models readily identify neutral processes in the ecology of pathogen transmission30, predict cross-species transmission with surprising accuracy56 and can be easily interfaced with models of macroecological change4. However, because they treat viruses as interchangeable, these approaches lose potentially important signals in the data. A handful of studies also model one specific aspect of viral sharing: the probability that an animal host shares any viruses at all with humans (that is, whether the host is a zoonotic reservoir). As with viral sharing generally, sympatry and synanthropy determine opportunities for human–animal contact and predict sharing, both through domestication and through geographic overlap between wildlife ranges and human population centres22,57. Some traits may uniquely predict zoonotic reservoirs, such as a fast life history strategy, in which lifespan is traded off in favour of fertility58, and because they are smaller and more numerous, fast-lived species are more likely to thrive in disturbed ecosystems or alongside human settlements and thus may often be sources of zoonotic outbreaks59,60.
Finally, viral richness models and host range models investigate node degree in the bipartite network, that is, how many hosts a given virus can infect (host range) and how many viruses have infected a given host (viral richness). By collapsing the bipartite network into node-level traits, they provide coarse measures that can be used in species-level analyses (for example, see ref. 22). Identifying viral traits, such as vector transmission, that predict broad host range helps in exploring the evolutionary theory of cross-species transmission events and could inform zoonotic risk models22,45. Conversely, understanding ecological drivers of viral diversity can help to prioritize sampling for viral discovery22,61 and, potentially, to understand the distribution of zoonotic risk if some ‘hyper-reservoirs’ host disproportionately many zoonotic viruses32,49. In this special case, some studies investigate zoonotic viral richness and test whether some animals host a greater number of viruses with observed zoonotic potential and whether this effect differs from overall viral richness62. Increasingly, careful analysis often rejects widely held assumptions (for example, bats or urban-adapted animals host most more zoonotic viruses) in favour of the null hypothesis that zoonotic viral richness is often simply a product of higher total viral diversity49,62.
The limits of prediction
Each of these modelling approaches shows tremendous promise — but each is limited, first and foremost, by the availability of data on the global vertebrate virome. At most, 1% of mammal viruses have been described to date9 and even fewer are known from other vertebrates. At such an early stage in viral discovery, even the most basic statistics, such as host-level viral richness estimates, may say more about sampling effort than the underlying biological reality63,64. When a new zoonotic virus emerges, researchers are disproportionately likely to sample related host species and viral taxa in the vicinity of the spillover event (bottom-up sampling bias). Surveillance also often targets well-studied cosmopolitan species due to availability, and is therefore more likely to discover more viruses, and more zoonotic viruses, in these species (top-down sampling bias49). Similarly, screening efforts have historically focused on hosts and viruses with known relevance to human or domestic animal health; this impact bias may be especially salient in regions with underfunded veterinary and public health surveillance infrastructure. Although high-throughput sequencing and broad-range serological approaches65 can counteract some of these biases, these approaches are not always cost-effective or practical to implement in resource-limited laboratory settings. As a result, targeted screening remains the primary source of host–virus association data, and biases remain pervasive.
Together, the limitations and priorities of these sampling processes heavily shape the observed structure of the host–virus network and are difficult to correct for in modelling efforts30. At present, this is a notable barrier to the advancement of quantitative viral ecology. Most published disease modelling studies use one of only a few small datasets with substantial overlap and similar biases, test the same hypotheses and, unsurprisingly, have generated largely congruent findings (for example, ‘phylogenetic distance structures viral sharing’), most of which are underinformed by microbiology and use phylogeographic or ecological proxies. While independent verification of results is a critical part of the scientific method, especially if data easily facilitate re-analysis or meta-analysis, re-analysing these few datasets so intensely risks pseudo-replication and could entrench spurious findings that are readily explained by sampling bias. For example, a recent study showed that urban-adapted mammals have a higher recorded diversity of zoonotic viruses, but only because they also have a higher total diversity of recorded pathogens, which is probably a clear-cut example of top-down sampling bias62. Cases such as these have engendered scepticism of modelling approaches as a useful tool for applied risk assessment, particularly given the high diversity of wildlife viruses, significant gaps in both host and virus sampling, the spurious patterns generated by sampling bias and even the pace of viral diversification19,63,66. At present, scientists are unlikely to be able to ‘predict and prevent’ outbreaks using these tools. However, models will become more reliable if viral discovery continues at its current pace and, particularly, if data synthesis is a priority for quantitative research. As these datasets grow, they will open doors for more advanced methodologies that have greater impact.
Emerging directions for powerful inference
As this subfield advances, the microbiology underpinning models is becoming more detailed, leading to insights that better bridge virology, ecology and computational biology. Across the global virome, an intangible but finite set of host–virus associations are possible, while each impossible pair is prevented by at least one (identifiable and, ideally, predictable) incompatibility between viral and host microbiology. In this lock-and-key framework, a virus’s ability to infect a novel host species depends on the features that allow it to enter cells, hijack cellular machinery, replicate its genome, evade both the innate and adaptive immune response, produce infectious virions, optimize transmission and cause disease. While the ‘phylogenetic distance effect’ has been used as a broadly supported and convenient (but black box) proxy for these mechanisms, researchers are increasingly turning to data that explicitly characterize these processes instead. For example, host cell receptors and viral envelope proteins act as one kind of lock-and-key, which determine a virus’s potential for cell entry36; data on the angiotensin converting enzyme 2 (ACE2) receptor of mammalian host cells have been used to predict the broad host range of SARS-CoV (severe acute respiratory syndrome coronavirus) and SARS-CoV-2 (refs. 67,68). Compatibility is further altered by biochemical modifications of host and viral proteins, such as glycans (the sugars on the outside of host and virus proteins)69; viral proteins inherit host glycosylation, and their cross-species transmission potential may be enhanced or hindered by glycosylation by the source host70. The fractal geometry of these molecules could be represented as quantitative features, and glycan similarity may be predictive of viral sharing. Eventually, it may also be possible to represent more complicated immunology in this framework, for example broadly reactive innate antiviral factors, such as TRIM5α, act as barriers to different groups of viruses to varying degrees71, and while few models currently capture these pathways, this may be an important research horizon in the coming decade.
Increasingly, modellers have also harnessed the genomic revolution to make better predictions in the absence of detailed information on microbiological mechanisms. Genomes are inherently high-dimensional data that encode both meaningful phenotypes and residual signals of coevolution, and they can be used as features for both host and virus nodes in a network. Usually, genomes are analysed by quantifying the usage of dinucleotides, codons and codon pairs; in more advanced cases, these can be augmented with data on amino acid biochemistry, protein–protein interactions53 or longer k-mers72. In the near-term future, these datasets may increasingly be supported by machine learning tools that predict protein folding structures73,74. A number of studies have begun using these genomic features in various forms of link prediction, including predicting reservoir taxonomic orders10, characterizing the broad host and vector associations of flaviviruses75, and predicting the zoonotic potential of circulating strains of avian influenza53,54 (Box 1) and animal viruses more broadly76. Researchers have particularly advanced these methods while studying host–bacteriophage networks77, integrating genomic data into network-based frameworks with other predictors10,77 and exploring the potential for deep learning to identify genes or genomic features that control host specificity or virulence78. These approaches can even be useful in practical outbreak investigation, for example one recent study predicted the reservoirs of three dozen ‘orphan viruses’ with murky origins (for example, the Bas-Congo virus is predicted to be a virus of even-toed ungulates10).
In combination with these growing sources of data on host–virus interactions, researchers have increasingly started using network science to make more complex and more powerful predictive models. The structure of the host–virus network is determined by unobserved biological processes with identifiable signals; tools from graph theory and network science can recover this hidden information and leverage it for better prediction. Often, these recommendations rely on pairwise dissimilarity of virus communities among hosts or vice versa27, or on the degree distributions of viruses and hosts37. These approaches can be supplemented with phylogenetic or ecological traits fairly easily27,37, or even with genomic data77. More sophisticated ways of leveraging network structure have been developed in computer science, but they remain largely untested on viral networks; in particular, as network data expand — in both the number of associations and the dimensionality of predictors — the door for deep learning methods, such as collaborative filtering79 and neural networks, will also open80. The surprising strength of these methods for other link prediction tasks — from protein–protein interaction networks to online social network or shopping algorithms — makes this avenue particularly promising. Many of these approaches rely on graph embedding, a set of methods that use matrix algebra to generate a small number of feature vectors, which encode information about relationships between nodes or the graph as a whole81; these features can be used to improve link prediction or to add a network component to other kinds of models. For example, one recent study imputed missing links in the mammal–virus network using machine learning, generated graph embeddings of the derived network and used these features to substantially improve the performance of a genomics-based classifier of viral zoonotic potential28. By using these kinds of computational tools to characterize the structure of the global virome, scientists may be able to translate a broader understanding of the rules of cross-species transmission into applied problems such as zoonotic risk prediction.
From models to actionable science
Opportunities to apply these models to high-impact problems are abundant, albeit mostly unexplored. For example, host inference models can help target fieldwork during the early stages of zoonotic outbreaks, when origins are unclear (for example, the SARS-CoV-2 pandemic56) or when a familiar virus emerges in an unusual location (for example, Nipah virus in Kerala, India41). These models can also be used to target wildlife sampling more efficiently. Viral discovery is still expensive at scale: the USAID PREDICT programme spent over US$200 million to discover roughly 1,000 novel wildlife viruses in 10 years18, and the proposed Global Virome Project would aim to spend US$1 billion over the next decade discovering a million more8. Future programmes such as these present an opportunity to test model-guided approaches as both a cost-saving measure and shortcut to accelerate scientific progress. Once wildlife viruses are discovered and characterized, their zoonotic potential can be predicted as part of the first scientific report describing their existence82, helping virologists triage laboratory characterization; these tools may increasingly be paired with models that aim to predict dimensions of epidemic potential, such as human-to-human transmissibility45,50 and pathogen virulence13, which often use the same core datasets and machine learning approaches that are used to predict zoonotic potential. Once priority risks have been identified, managers can implement longitudinal, multi-site sampling programmes that can inform (and support other models that predict) where and when people are at risk of zoonotic spillover. Similarly, modelling approaches that integrate data on surveillance and health systems can help understand where those spillovers are most likely to go undetected14 and spread quickly15. When integrated into one pipeline, these different approaches capture all three components of risk: hazard (what the threat is), exposure (where and when it occurs) and vulnerability (what the potential disease burden is, and for whom).
Building predictive models into this pipeline requires that researchers, practitioners and stakeholders have confidence in these approaches. To refine existing models, formalize best practices and convince sceptics (including both colleagues and stakeholders) of the value of this work, modellers need to measure and report model performance in a way that is open, transparent and accountable. Developing standardized meta-datasets26 and forming collaborative teams (for example, the Verena Consortium; see viralemergence.org) can facilitate multi-model study designs that are commonplace in statistical research, such as ensemble models or ‘bake-offs’ testing predictive accuracy. However, these are only a step in the required direction. Actionable forecasting is an iterative process83, and adding feedback loops to the modelling process would help researchers to measure the accuracy of specific approaches, validate or falsify model-generated hypotheses and, ultimately, make more sound, actionable inference about the global virome. A lack of feedback among field, experimental and modelling approaches currently precludes that process of refinement; when predictions are tested, it has mostly been ad hoc. For example, one recent field study84 confirmed model predictions of bat filovirus hosts11, while another found no support85; a recent experimental study86 more definitively refuted another prediction about bat reservoirs of Nipah virus41. These kinds of data are rarely fed back into modelling efforts and are almost never pursued prospectively. In a unique counterexample, we recently generated eight predictive models of undiscovered bat hosts of betacoronaviruses and tracked their performance over more than a year as new viral discoveries were reported56. We found that biology-agnostic network models performed no better than random predictions, while machine learning and network models that also leveraged data on bat biology made strong, accurate predictions. Using measures of model performance, we were able to weight a predictive ensemble to make more accurate predictions, and the updated list of potential undiscovered hosts can now be confidently used to target the screening of samples from field surveys and biological collections. This example highlights several best practices for actionable prediction: making predictions public and interpretable, tracking predictive accuracy over time, and incorporating new data into dynamic predictions that keep pace with changing scientific knowledge.
We suggest that future sampling efforts would best complement modelling efforts by following up on actionable (high zoonotic risk) leads for public health priorities, as suggested by both expert knowledge and predictive models. If model-generated hypotheses turn out to be largely incorrect, this can help to identify spurious assumptions about a virus’s ecology or identify modelling approaches unsuited for future use; on the other hand, if accurate and effective, these integrated approaches will save time and resources during outbreaks. This will require researchers to match the scope of predictions to the nature of an intended outcome, for example host inference models are used to suggest gaps in known reservoirs11,41,56, and sampling these hosts first can reduce the cost of viral discovery. Similarly, models that predict viral zoonotic potential can identify threats to human health before the first case of infection28,76; in the near-term future, these tools could be used to identify which wildlife viruses should be the focus of testing for new therapeutics and candidate universal vaccines. Matching predictions to purpose will also help to identify potential barriers to implementation; these are discussed more extensively elsewhere87.
Conclusions
The promise of host–virus network prediction should be met with cautious enthusiasm, particularly with regard to zoonotic risk. These models still face many challenges in practice, and a well-trained scientist may be able to identify many of the same patterns or risks as the most advanced predictive models would. For example, a betacoronavirus pandemic was almost inevitable, not just because the zoonotic potential of bat viruses, which had been confirmed experimentally, but also because there had been two previous outbreaks of zoonotic betacoronaviruses and insufficiently responsive policy and planning (SARS and MERS (Middle East respiratory syndrome); Box 2 and Fig. 3).

Even within the different formulations of predicting animal–virus interactions, models can answer a wide range of questions and can be useful at a wide range of points in the history of an outbreak. Leading up to the COVID-19 pandemic, we highlight here where the modelling frameworks (A–G) available today offered useful insights — or may have been able to make a difference if appropriate data, infrastructure and model technology had been available at the time90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105. Reservoir models from sequence data can help to trace orphan viruses to broad host groups10 (A). Some groups of viruses have higher host range and zoonotic potential than others22 (B). Knowing a subset of virus reservoirs allows predictive models to identify potential undiscovered reservoirs11 (C). Models that predict zoonotic potential from sequence data could help to generalize the work of gain-of-function studies with much lower investment and risk76 (D). Viral sharing models can identify hosts with similar viruses, targeting sampling around a given lead30 (E). Link prediction and reservoir inference models can suggest which wildlife hosts may be able to carry a virus in the future37,67 (F). Models can help to guide sampling to trace the origins of SARS-CoV-2 (ref. 56) (G). Meanwhile, hundreds of known wildlife coronaviruses still require risk assessment for zoonotic potential, transmissibility and pandemic risk. CDC, Centers for Disease Control and Prevention.
Just as ‘virus hunting’ has been insufficient to stem the emergence, re-emergence or global spread of several major viral threats18, there are obstacles to turning model-based predictions into disease prevention. Even with massive efforts to mitigate upstream drivers of disease emergence (and quantitative modelling to target those interventions), spillover risk will never be reduced to zero—especially for unknown threats—and after the first human case, the actual levers of pandemic prevention will always lie in diagnostic and surveillance capacity, healthcare access, social safety nets and health system investment—not the tools we discuss here.
However, as future threats emerge, modelling will be a key tool for rapid scientific inquiry, particularly given how much still remains unknown about the global virome. Although scientists may never be able to ‘predict and prevent the next pandemic’, a renewed vision of this work — ‘prediction’ as the development of quantitative tools that can learn the rules of life underpinning host–virus interactions and apply them to information-limited problems to benefit human health and the environment — could be an invaluable step towards true preparedness.
These approaches will help virologists to explore the ecology and evolution of coronaviruses and to build a data-driven risk assessment infrastructure along the lines of the global influenza monitoring system. But there is still no guarantee that the next SARS-like pandemic could be ‘predicted and prevented’, particularly given that the risk of a pandemic such as COVID-19 was ‘predicted’ for two decades by virologists on the basis of other kinds of scientific evidence88,89. Downstream problems preventing the translation of scientific knowledge to public health responses cannot be entirely solved through actionable science; no amount of viral discovery, laboratory characterization, modelling and risk assessment can solve vulnerability due to weak healthcare infrastructure and insufficient funding continuity and support for pandemic preparedness18. Knowing where SARS-CoV-2 came from may help us to target surveillance and slow the emergence of similar viruses, but another highly transmissible coronavirus will inevitably emerge in humans someday. Developing a universal vaccine that protects against bat coronaviruses with predicted zoonotic potential, building pandemic preparedness frameworks that include international governance of vaccine sharing and production, and developing responsive health systems with better syndromic detection of early outbreaks could be enough to achieve a future that never sees another coronavirus pandemic.
References
Jones, K. E. et al. Global trends in emerging infectious diseases. Nature 451, 990–993 (2008).
Woolhouse, M. E. et al. Temporal trends in the discovery of human viruses. Proc. R. Soc. B 275, 2111–2115 (2008).
Smith, K. F. et al. Global rise in human infectious disease outbreaks. J. R. Soc. Interface 11, 20140950 (2014).
Carlson, C. J. et al. Climate change will drive novel cross-species viral transmission. Preprint at bioRxiv https://doi.org/10.1101/2020.01.24.918755 (2020).
Swei, A., Couper, L. I., Coffey, L. L., Kapan, D. & Bennett, S. Patterns, drivers, and challenges of vector-borne disease emergence. Vector Borne Zoonotic Dis. 20, 159–170 (2020).
Belay, E. D. et al. Zoonotic disease programs for enhancing global health security. Emerg. Infect. Dis. 23, S65 (2017).
Morse, S. S. et al. Prediction and prevention of the next pandemic zoonosis. Lancet 380, 1956–1965 (2012).
Carroll, D. et al. The global virome project. Science 359, 872–874 (2018).
Carlson, C. J., Zipfel, C. M., Garnier, R. & Bansal, S. Global estimates of mammalian viral diversity accounting for host sharing. Nat. Ecol. Evol. 3, 1070–1075 (2019).
Babayan, S. A., Orton, R. J. & Streicker, D. G. Predicting reservoir hosts and arthropod vectors from evolutionary signatures in RNA virus genomes. Science 362, 577–580 (2018).
Han, B. A. et al. Undiscovered bat hosts of filoviruses. PLoS Negl. Trop. Dis. 10, e0004815 (2016).
Schmidt, J. P. et al. Spatiotemporal fluctuations and triggers of Ebola virus spillover. Emerg. Infect. Dis. 23, 415 (2017).
Guth, S., Visher, E., Boots, M. & Brook, C. E. Host phylogenetic distance drives trends in virus virulence and transmissibility across the animal–human interface. Phil. Trans. R. Soc. Biol. Sci. 374, 20190296 (2019).
Glennon, E. E. et al. Syndromic detectability of haemorrhagic fever outbreaks. Preprint at medRxiv https://doi.org/10.1101/2020.03.28.20019463 (2020).
Pigott, D. M. et al. Local, national, and regional viral haemorrhagic fever pandemic potential in Africa: a multistage analysis. Lancet 390, 2662–2672 (2017).
Palmer, S., Brown, D. & Morgan, D. Early qualitative risk assessment of the emerging zoonotic potential of animal diseases. BMJ 331, 1256–1260 (2005).
Grange, Z. L. et al. Ranking the risk of animal-to-human spillover for newly discovered viruses. Proc. Natl Acad. Sci. USA 118, e2002324118 (2021).
Carlson, C. J. From PREDICT to prevention, one pandemic later. Lancet Microbe 1, e6–e7 (2020).
Holmes, E., Rambaut, A. & Andersen, K. Pandemics: spend on surveillance, not prediction. Nature 558, 180–182 (2018).
Breiman, L. Statistical modeling: the two cultures (with comments and a rejoinder by the author). Stat. Sci. 16, 199–231 (2001).
Mouquet, N. et al. Predictive ecology in a changing world. J. Appl. Ecol. 52, 1293–1310 (2015).
Olival, K. J. et al. Host and viral traits predict zoonotic spillover from mammals. Nature 546, 646–650 (2017).
Stephens, P. R. et al. Global mammal parasite database version 2.0. Ecology 98, 1476 (2017).
Wardeh, M., Risley, C., McIntyre, M. K., Setzkorn, C. & Baylis, M. Database of host–pathogen and related species interactions, and their global distribution. Sci. Data 2, 150049 (2015).
Shaw, L. P. et al. The phylogenetic range of bacterial and viral pathogens of vertebrates. Mol. Ecol. 29, 3361–3379 (2020).
Gibb, R. et al. Data proliferation, reconciliation, and synthesis in viral ecology. BioScience https://doi.org/10.1093/biosci/biab080 (2021).
Dallas, T., Park, A. W. & Drake, J. M. Predicting cryptic links in host–parasite networks. PLoS Comput. Biol. 13, e1005557 (2017).
Poisot, T. et al. Imputing the mammalian virome with linear filtering and singular value decomposition. Preprint at https://arxiv.org/abs/2105.14973 (2021).
Carlson, C. J. et al. The Global Virome in One Network (VIRION): an atlas of vertebrate–virus associations. Preprint at bioRxiv https://doi.org/10.1101/2021.08.06.455442 (2021).
Albery, G. F., Eskew, E. A., Ross, N. & Olival, K. J. Predicting the global mammalian viral sharing network using phylogeography. Nat. Commun. 11, 2260 (2020).
Davies, T. J. & Pedersen, A. B. Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proc. R. Soc. B Biol. Sci. 275, 1695–1701 (2008).
Guy, C., Thiagavel, J., Mideo, N. & Ratcliffe, J. M. Phylogeny matters: revisiting ‘a comparison of bats and rodents as reservoirs of zoonotic viruses’. R. Soc. Open Sci. 6, 181182 (2019).
Washburne, A. D. et al. Taxonomic patterns in the zoonotic potential of mammalian viruses. PeerJ 6, e5979 (2018).
Plowright, R. K. et al. Pathways to zoonotic spillover. Nat. Rev. Microbiol. 15, 502 (2017).
Stephens, P. R. et al. The macroecology of infectious diseases: a new perspective on global-scale drivers of pathogen distributions and impacts. Ecol. Lett. 19, 1159–1171 (2016).
Longdon, B., Brockhurst, M. A., Russell, C. A., Welch, J. J. & Jiggins, F. M. The evolution and genetics of virus host shifts. PLoS Pathog. 10, e1004395 (2014).
Farrell, M. J., Elmasri, M., Stephens, D. A. & Davies, T. J. Predicting missing links in global host–parasite networks. bioRxiv https://doi.org/10.1101/2020.02.25.965046 (2020).
Gilbert, A. T. et al. Deciphering serology to understand the ecology of infectious diseases in wildlife. EcoHealth 10, 298–313 (2013).
Becker, D. J., Seifert, S. N. & Carlson, C. J. Beyond infection: integrating competence into reservoir host prediction. Trends Ecol. Evol. 35, 1062–1065 (2020).
Walsh, M. G., Mor, S. M., Maity, H. & Hossain, S. A preliminary ecological profile of Kyasanur Forest disease virus hosts among the mammalian wildlife of the Western Ghats, India. Ticks Tick Borne Dis. 11, 101419 (2020).
Plowright, R. K. et al. Prioritizing surveillance of Nipah virus in India. PLoS Negl. Trop. Dis. 13, e0007393 (2019).
Schmidt, J. P. et al. Ecological indicators of mammal exposure to Ebolavirus. Philos. Trans. R. Soc. B Biol. Sci. 374, 20180337 (2019).
Worsley-Tonks, K. E. et al. Using host traits to predict reservoir host species of rabies virus. PLoS Negl. Trop. Dis. 14, e0008940 (2020).
Woolhouse, M. E. & Gowtage-Sequeria, S. Host range and emerging and reemerging pathogens. Emerg. Infect. Dis. 11, 1842 (2005).
Johnson, C. K. et al. Spillover and pandemic properties of zoonotic viruses with high host plasticity. Sci. Rep. 5, 14830 (2015).
Elena, S. F. & Sanjuán, R. Adaptive value of high mutation rates of RNA viruses: separating causes from consequences. J. Virol. 79, 11555–11558 (2005).
Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 16, e3000003 (2018).
Grewelle, R. E. Larger viral genome size facilitates emergence of zoonotic diseases. Preprint at bioRxiv https://doi.org/10.1101/2020.03.10.986109 (2020).
Mollentze, N. & Streicker, D. G. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. Proc. Natl Acad. Sci. USA 117, 9423–9430 (2020).
Walker, J. W., Han, B. A., Ott, I. M. & Drake, J. M. Transmissibility of emerging viral zoonoses. PLoS ONE 13, e0206926 (2018).
Damas, J. et al. Broad host range of SARS-CoV-2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc. Natl Acad. Sci. USA https://doi.org/10.1073/pnas.2010146117 (2020).
Zhang, Z. et al. Rapid identification of human-infecting viruses. Transbound. Emerg. Dis. 66, 2517–2522 (2019).
Eng, C. L., Tong, J. C. & Tan, T. W. Predicting zoonotic risk of influenza A viruses from host tropism protein signature using random forest. Int. J. Mol. Sci. 18, 1135 (2017).
Li, J. et al. Machine learning methods for predicting human-adaptive influenza A viruses based on viral nucleotide compositions. Mol. Biol. Evol. 37, 1224–1236 (2020).
Kim, B., Niu, X., Hunter, D. R. & Cao, X. A dynamic additive and multiplicative effects model with application to the United Nations voting behaviors. Preprint at https://arxiv.org/abs/1803.06711 (2018).
Becker, D. et al. Optimizing predictive models to prioritize viral discovery in zoonotic reservoirs. Lancet Microbe (in the press).
Han, B. A., Schmidt, J. P., Bowden, S. E. & Drake, J. M. Rodent reservoirs of future zoonotic diseases. Proc. Natl Acad. Sci. USA 112, 7039–7044 (2015).
Plourde, B. T. et al. Are disease reservoirs special? Taxonomic and life history characteristics. PLoS ONE 12, e0180716 (2017).
Keesing, F. et al. Impacts of biodiversity on the emergence and transmission of infectious diseases. Nature 468, 647–652 (2010).
Albery, G. F. & Becker, D. J. Fast-lived hosts and zoonotic risk. Trends Parasitol. 37, 117–129 (2021).
Young, C. C. & Olival, K. J. Optimizing viral discovery in bats. PLoS ONE 11, e0149237 (2016).
Albery, G. F. et al. Urban-adapted mammal species have more known pathogens. Preprint at bioRxiv https://doi.org/10.1101/2021.01.02.425084 (2021).
Wille, M., Geoghegan, J. L. & Holmes, E. C. How accurately can we assess zoonotic risk? PLoS Biol. 19, e3001135 (2021).
Gibb, R. et al. Mammal virus diversity estimates are unstable due to accelerating discovery effort. Preprint at bioRxiv https://doi.org/10.1101/2021.08.10.455791 (2021).
Xu, G. J. et al. Comprehensive serological profiling of human populations using a synthetic human virome. Science 348, aaa0698 (2015).
Geoghegan, J. L. & Holmes, E. C. Predicting virus emergence amid evolutionary noise. Open Biol. 7, 170189 (2017).
Fischhoff, I. R., Castellanos, A. A., Rodrigues, J. P., Varsani, A. & Han, B. A. Predicting the zoonotic capacity of mammals to transmit SARS-CoV-2. Proc. R. Soc. B Biol. Sci. https://doi.org/10.1098/rspb.2021.1651 (2021).
Hou, Y. et al. Angiotensin-converting enzyme 2 (ACE2) proteins of different bat species confer variable susceptibility to SARS-CoV entry. Arch. Virol. 155, 1563–1569 (2010).
Thompson, A. J., de Vries, R. P. & Paulson, J. C. Virus recognition of glycan receptors. Curr. Opin. Virol. 34, 117–129 (2019).
Kocher, J. F. et al. Bat caliciviruses and human noroviruses are antigenically similar and have overlapping histo-blood group antigen binding profiles. Mbio 9, e00869-18 (2018).
Chiramel, A. I. et al. TRIM5α restricts flavivirus replication by targeting the viral protease for proteasomal degradation. Cell Rep. 27, 3269–3283 (2019).
Young, F., Rogers, S. & Robertson, D. L. Predicting host taxonomic information from viral genomes: a comparison of feature representations. PLoS Comput. Biol. 16, e1007894 (2020).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Truong, P., Garcia-Vallve, S. & Puigbo, P. An unsupervised algorithm for host identification in flaviviruses. Life https://doi.org/10.3390/life11050442 (2021).
Mollentze, N., Babayan, S. & Streicker, D. Identifying and prioritizing potential human-infecting viruses from their genome sequences. PLoS Biol. 19, e3001390 (2021).
Wang, W. et al. A network-based integrated framework for predicting virus–prokaryote interactions. NAR Genom. Bioinform. 2, lqaa044 (2020).
Bartoszewicz, J. M., Seidel, A. & Renard, B. Y. Interpretable detection of novel human viruses from genome sequencing data. NAR Genom. Bioinform. 3, lqab004 (2021).
He, X. et al. Neural collaborative filtering. In Proc. 26th International Conference on World Wide Web 26, 173–182 (Republic and Canton of Geneva, Switzerland, 2017).
Fout, A., Byrd, J., Shariat, B. & Ben-Hur, A. Protein interface prediction using graph convolutional networks. NIPS’17: Proc. 31st International Conference on Neural Information Processing Systems 31, 6533–6542 (2017).
Hamilton, W. L., Ying, R. & Leskovec, J. Representation learning on graphs: methods and applications. IEEE Data Eng. Bull. 40, 52–74 (2017).
Bergner, L. M. et al. Characterizing and evaluating the zoonotic potential of novel viruses discovered in vampire bats. Viruses 13, 252 (2021).
Dietze, M. C. et al. Iterative near-term ecological forecasting: needs, opportunities, and challenges. Proc. Natl Acad. Sci. USA 115, 1424–1432 (2018).
Schulz, J. E. et al. Serological evidence for henipa-like and filo-like viruses in Trinidad bats. J. Infect. Dis. 221, S375–S382 (2020).
Brook, C. E. et al. Disentangling serology to elucidate henipa- and filovirus transmission in Madagascar fruit bats. J. Anim. Ecol. 88, 1001–1016 (2019).
Seifert, S. N. et al. Rousettus aegyptiacus bats do not support productive Nipah virus replication. J. Infect. Dis. 221, S407–S413 (2020).
Carlson, C. J. et al. The future of zoonotic risk prediction. Phil. Trans. R. Soc. B Biol. Sci. 376, 20200358 (2021).
Ge, X.-Y. et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503, 535–538 (2013).
Menachery, V. D. et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 21, 1508–1513 (2015).
Guan, Y. et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302, 276–278 (2003).
Woo, P. C. Y. et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 79, 884–895 (2005).
Li, W. et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 310, 676–679 (2005).
Wang, M. et al. SARS-CoV infection in a restaurant from palm civet. Emerg. Infect. Dis. 11, 1860–1865 (2005).
Hu, B. et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 13, e1006698 (2017).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Xiao, K. et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 583, 286–289 (2020).
Lam, T.-Y. et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 583, 282–285 (2020).
Wacharapluesadee, S. et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat. Commun. 12, 972 (2021).
Holmes, E. C. et al. The origins of SARS-CoV-2: a critical review. Cell 184, 4848–4856 (2021).
Oude Munnink, B. B. et al. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 371, 172–177 (2021).
Chandler, J. C. et al. SARS-CoV-2 exposure in wild white-tailed deer (Odocoileus virginianus). Proc. Natl Acad. Sci. USA 118, e2114828118 (2021).
Jia, P., Dai, S., Wu, T. & Yang, S. New approaches to anticipate the risk of reverse zoonosis. Trends Ecol. Evol. 36, 580–590 (2021).
Lednicky, J. A. et al. Isolation of a novel recombinant canine coronavirus from a visitor to Haiti: further evidence of transmission of coronaviruses of zoonotic origin to humans. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciab924 (2021).
Vlasova, A. N. et al. Novel canine coronavirus isolated from a hospitalized pneumonia patient, East Malaysia. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciab456 (2021).
Lednicky, J. A. et al. Emergence of porcine delta-coronavirus pathogenic infections among children in Haiti through independent zoonoses and convergent evolution. Preprint at medRxiv https://doi.org/10.1101/2021.03.19.21253391 (2021).
Hay, A. J. & McCauley, J. W. The WHO global influenza surveillance and response system (GISRS)—a future perspective. Influenza Other Respir. Viruses 12, 551–557 (2018).
Subbarao, K. et al. Characterization of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science 279, 393–396 (1998).
Kandeel, A. et al. Zoonotic transmission of avian influenza virus (H5N1), Egypt, 2006–2009. Emerg. Infect. Dis. 16, 1101 (2010).
Ke, C. et al. Human infection with highly pathogenic avian influenza A (H7N9) virus, China. Emerg. Infect. Dis. 23, 1332 (2017).
Gaidet, N. et al. Evidence of infection by H5N2 highly pathogenic avian influenza viruses in healthy wild waterfowl. PLoS Pathog. 4, e1000127 (2008).
Webster, R. G., Bean, W. J., Gorman, O. T., Chambers, T. M. & Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Mol. Biol. Rev. 56, 152–179 (1992).
Pawar, S. D. et al. Avian influenza surveillance reveals presence of low pathogenic avian influenza viruses in poultry during 2009–2011 in the West Bengal State, India. Virol. J. 9, 151 (2012).
Parry, R., Wille, M., Turnbull, O. M., Geoghegan, J. L. & Holmes, E. C. Divergent influenza-like viruses of amphibians and fish support an ancient evolutionary association. Viruses 12, 1042 (2020).
Campbell, P. J. et al. The M segment of the 2009 pandemic influenza virus confers increased neuraminidase activity, filamentous morphology, and efficient contact transmissibility to A/Puerto Rico/8/1934-based reassortant viruses. J. Virol. 88, 3802–3814 (2014).
Carlson, C. Evolutionary surprise, artificial intelligence, and H5N8. The Verena Blog https://www.viralemergence.org/blog/evolutionary-surprise-artificial-intelligence-and-h5n8 (2021).
Wardeh, M., Baylis, M. & Blagrove, M. S. Predicting mammalian hosts in which novel coronaviruses can be generated. Nat. Commun. 12, 780 (2021).
Crossman, L. C. Leveraging deep learning to simulate coronavirus spike proteins has the potential to predict future zoonotic sequences. Preprint at bioRxiv https://doi.org/10.1101/2020.04.20.046920 (2020).
Acknowledgements
The Viral Emergence Research Initiative (VERENA) consortium is supported by NSF BII 2021909. For more information, see viralemergence.org.
Author information
Authors and Affiliations
Contributions
C.J.C. and G.F.A. conceived the study and drafted the manuscript, G.F.A. and C.J.C. produced visualizations, and all authors contributed to the writing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Microbiology thanks Jonathan Dushoff, Vincent Munster and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Albery, G.F., Becker, D.J., Brierley, L. et al. The science of the host–virus network. Nat Microbiol 6, 1483–1492 (2021). https://doi.org/10.1038/s41564-021-00999-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-021-00999-5
This article is cited by
-
HostNet: improved sequence representation in deep neural networks for virus-host prediction
BMC Bioinformatics (2023)
-
Using drivers and transmission pathways to identify SARS-like coronavirus spillover risk hotspots
Nature Communications (2023)
-
Worldwide transmission and infection risk of mosquito vectors of West Nile, St. Louis encephalitis, Usutu and Japanese encephalitis viruses: a systematic review
Scientific Reports (2023)
-
Global patterns of phylogenetic diversity and transmission of bat coronavirus
Science China Life Sciences (2023)
-
Climate change increases cross-species viral transmission risk
Nature (2022)
