
Abstract
Respiratory failure is associated with increased mortality in COVID-19 patients. There are no validated lower airway biomarkers to predict clinical outcome. We investigated whether bacterial respiratory infections were associated with poor clinical outcome of COVID-19 in a prospective, observational cohort of 589 critically ill adults, all of whom required mechanical ventilation. For a subset of 142 patients who underwent bronchoscopy, we quantified SARS-CoV-2 viral load, analysed the lower respiratory tract microbiome using metagenomics and metatranscriptomics and profiled the host immune response. Acquisition of a hospital-acquired respiratory pathogen was not associated with fatal outcome. Poor clinical outcome was associated with lower airway enrichment with an oral commensal (Mycoplasma salivarium). Increased SARS-CoV-2 abundance, low anti-SARS-CoV-2 antibody response and a distinct host transcriptome profile of the lower airways were most predictive of mortality. Our data provide evidence that secondary respiratory infections do not drive mortality in COVID-19 and clinical management strategies should prioritize reducing viral replication and maximizing host responses to SARS-CoV-2.
Similar content being viewed by others


Main
The earliest known case of SARS-CoV-2 infection causing COVID-19 is thought to have occurred on 17 November 2019 (ref. 1). As of 3 August 2021, 198.7 million confirmed cases of COVID-19 and 4.2 million deaths have been reported worldwide2. As the global scientific community has rallied in a concerted effort to understand SARS-CoV-2 infections, our background knowledge is rooted in previous experience with the related zoonotic betacoronaviruses Middle East Respiratory Syndrome coronavirus (MERS-CoV) and SARS-CoV-1, which have caused severe pneumonia with 34.4% and 9% case fatality, respectively3. As observed for these related coronaviruses, SARS-CoV-2 infection can result in an uncontrolled inflammatory response4 leading to acute respiratory distress syndrome (ARDS) and multi-organ failure, both associated with increased mortality. While a large proportion of the SARS-CoV-2 infected population is asymptomatic or experiences mild illness, a substantial number of individuals will develop severe disease and require hospitalization, with some progressing to respiratory failure and death.
Mortality in other viral pandemics, such as the 1918 H1N1 and 2009 H1N1 influenza pandemics, has been attributed in part to bacterial co-infection or super-infection5,6. To determine whether this is also the case for COVID-19, we can use next generation sequencing (NGS) to probe the complexity of the microbial environment (including RNA and DNA viruses, bacteria and fungi) and how the host (human) responds to infection. Recent studies have used this approach to uncover microbial signatures in patients with ARDS7,8. Increased bacterial burden and the presence of gut-associated bacteria in the lung were shown to worsen outcomes in these critically ill patients7,9, highlighting the potential role of the lung microbiome in predicting outcomes in ARDS. In a recent study using whole genome sequencing to profile the gut microbiome of 69 patients from Hong Kong, investigators identified an increased abundance of opportunistic fungal pathogens among patients with confirmed COVID-19 (ref. 10). While there is emerging interest in understanding the microbial environment in patients with SARS-CoV-2 infections, few studies have attempted to characterize this at the primary site of disease activity: the lower airways11,12.
In this investigation, we characterized the lung microbiome and lower airway markers of host immunity in a cohort of hospitalized COVID-19 patients. While we did not find that isolation of a secondary respiratory pathogen was associated with prolonged mechanical ventilation (MV; >28 days) or fatal outcome, we did identify critical microbial signatures—characterized by enrichment of oral commensals, high SARS-CoV-2 load and decreased anti-SARS-CoV-2 IgG response—associated with fatal outcome, suggesting a need for more targeted antiviral therapeutic approaches for the care of critically ill COVID19 patients.
Results
Cohort description
From 3 March to 18 June 2020, a total of 589 patients with laboratory-confirmed SARS-CoV-2 infection were admitted to the intensive care units of two academic medical centres of NYU Langone Health in New York (Long Island and Manhattan) and required invasive MV (Supplementary Results and Supplementary Tables 1 and 2). This included a subset of 142 patients from the Manhattan campus who underwent bronchoscopy for airway clearance and/or tracheostomy, from whom we collected and processed lower airway (bronchoalveolar lavage, BAL) samples for this investigation (Extended Data Fig. 1). Table 1 shows demographics and clinical characteristics of the 142 patients who underwent bronchoscopy divided into three clinical outcomes: survivors with ≤28 days on MV; survivors with >28 days on MV and deceased. The median post admission follow-up time was 232 days (confidence interval 226–237 days). Patients within the bronchoscopy cohort had higher overall survival than the rest of the NYU COVID-19 cohort since the most critically ill patients were not eligible for bronchoscopy or tracheostomy.
Among the factors associated with clinical outcome within the bronchoscopy cohort, patients who survived were more commonly placed on veno-venous extracorporeal membrane oxygenation (ECMO), whereas patients who died had frequently required dialysis (Table 1). These trends were also observed across the whole NYU cohort. Neither hydroxychloroquine nor azithromycin were significantly associated with clinical outcome. However, patients who survived were more frequently treated with the combination antibiotic piperacillin/tazobactam.
Within the first 48 hours from admission, respiratory bacterial cultures were rarely obtained (n = 70/589, 12%) with very few positive results (n = 12, 17%). Blood cultures were more commonly obtained (n = 353/589, 60%) but the rate of bacterial culture positivity was much lower (n = 5, 1.4%). These data support that community-acquired bacterial co-infection was not a common presentation among critically ill COVID-19 patients.
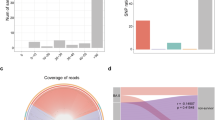
We evaluated whether respiratory or blood culture results obtained as per clinical standard of care were associated with clinical outcome. Risk analyses for the culture results during hospitalization for the whole cohort (n = 589) demonstrated that bacterial culture positivity with suspected pathogens—excluding possible contaminants such as oral microbes in respiratory samples—was not associated with increased odds of dying but was associated with prolonged MV in the surviving patients (Fig. 1). Since length of stay could potentially affect these results (patients who died could have a shorter hospitalization and therefore may have had fewer specimens collected for cultures), we repeated the analysis using culture data obtained during the first two weeks of hospitalization. This analysis showed that bacterial pathogen culture positivity (both respiratory and blood) during the early period of hospitalization was not associated with worse outcome (Fig. 1 and Supplementary Table 3). Interestingly, identification of oral bacteria in respiratory culture, commonly regarded as procedural contaminants, was associated with higher odds of prolonged MV (>28 days) among survivors. Similar trends were noted when analysis was performed on subjects from NYU LI and NYU Manhattan separately or for the bronchoscopy cohort (Supplementary Table 2). Among the bronchoscopy cohort, there was no statistically significant association between culture results and clinical outcome, but there was a trend towards an increased rate of positive respiratory cultures for S. aureus (including MRSA), Staphylococcus epidermidis and K. pneumoniae in the survival groups (Table 1). These data suggest that in critically ill patients with COVID-19 requiring MV, in whom broad-spectrum antimicrobials were frequently used, hospital isolation of a secondary respiratory bacterial pathogen is not associated with worse clinical outcome.

Odds ratios and corresponding 95% confidence intervals for rates of culture positivity for the whole cohort (n = 589) during length of hospitalization (left) and during the first 2 weeks of hospitalization (right).
SARS-CoV-2 load in the lower airways
Using bronchoscopy samples from 142 patients, we evaluated the viral load by real-time reverse transcription PCR (rRT–PCR) for the SARS-CoV-2 N gene, adjusted by levels of human RNase P gene (RP). Of note is that the majority of samples were obtained in the second week of hospitalization (Table 1, median[interquartile range] = 10[6–14], 13[8–16] and 13[8–16] for the ≤28 day MV, >28 day MV and deceased groups, respectively, P = non-significant). Paired analysis of upper airways (UA) and BAL samples revealed that, while there was a positive association between SARS-CoV-2 viral load of the paired samples (ρ = 0.60, P < 0.0001), there was a subset of subjects (21%) for which the viral load was greater in the BAL than in the supraglottic area, indicating topographical differences in SARS-CoV-2 replication (Fig. 2a). Importantly, while the SARS-CoV-2 viral load in the UA samples was not associated with clinical outcome (Supplementary Fig. 1), patients who died had higher viral load in their lower airways than patients who survived (Fig. 2b). Several studies have explored the relationship between SARS-CoV-2 viral load and mortality13,14,15,16,17,18. In a large cohort of 1,145 patients with confirmed SARS-CoV-2, viral load measured in nasopharyngeal swab samples was found to be significantly associated with mortality, even after adjusting for age, sex, race and several comorbidities18. Similar results were found in a cohort of patients in New York City with or without cancer, where in-hospital mortality was significantly associated with a high SARS-CoV-2 viral load in the UA17.

a,b, Copies of the SARS-CoV-2 N gene per ml, normalized to the human RNase P gene, comparing paired upper and lower airway samples (a) and levels in BAL comparing clinical outcome groups in 142 subjects (b). LLD, lowest level of detection at 0.3 normalized copies ml–1. Horizontal lines represent the median. *Two-sided Mann–Whitney U P = 0.038. **Two-sided Mann–Whitney U P = 0.001. c, Correlation analysis between levels of sgRNA and viral load as estimated by RT–PCR for the SARS-CoV-2 N gene levels. d,e, Bubble plot showing DESeq results of RNA viruses (d) and expressed DNA phages (e) enriched in each clinical outcome comparison (bubble size based on median relative abundance for those found to be statistically significant; the red dashed line separates positive (to the right) and negative (to the left) fold changes; text in blue indicates statistical significance).
We then evaluated virus replication in BAL samples by measuring levels of subgenomic RNA (sgRNA) targeting the E gene of SARS-CoV-2. This mRNA is only transcribed inside infected cells and is not packaged into virus particles; thus, its presence is indicative of recent virus replication19,20,21. In BAL, levels of sgRNA correlated with viral load as estimated by rRT–PCR for the SARS-CoV-2 N gene (Fig. 2c), and the highest percentage of measurable sgRNA was in the deceased group, followed by the ≤28 day MV group and the >28 day MV group (17.7%, 11.5% and 3.7%, respectively; chi-square P = 0.028 for the comparison deceased versus >28 day MV group). Thus, while in most cases levels of sgRNA were not measurable in BAL, suggesting that no active virus replication was ongoing in the lower airways of COVID-19 patients at the time of bronchoscopy (overall median[interquartile range] = 12[7–16] days from hospitalization), the lower airway viral burden, as estimated by rRT–PCR, is associated with mortality in critically ill COVID-19 patients.
Microbial community structure of the lower and upper airways
Considering the bacterial species and the viral loads identified in the BAL and UA of this cohort and their association with outcomes, we profiled in detail their viral and microbial composition. Microbial communities were evaluated using parallel datasets of RNA and DNA sequencing from 118 COVID-19 patients with BAL samples that passed appropriate quality control and a subset of paired 64 UA samples, along with background bronchoscope controls.
Given the low biomass of BAL samples in the metatranscriptome data, we first identified taxa as probable contaminants by comparing the relative abundance between background bronchoscope and BAL samples (Extended Data Fig. 2a and Supplementary Table 4). However, we did not remove any taxa identified as probable contaminants from subsequent analyses. A comparison of the microbial community complexity captured in these data, determined using the Shannon diversity index, showed there was significantly lower α-diversity in the BAL samples than in the UA and background controls (Extended Data Fig. 3a). Similarly, β-diversity analysis based on the Bray–Curtis dissimilarity index indicated that the microbial composition of the lower airways was distinct from the UA and background controls (Extended Data Fig. 3b, PERMANOVA P < 0.01). Sequence reads indicated a much higher relative abundance of SARS-CoV-2 in the lower airways than in the UA for this cohort (Extended Data Fig. 3c). Comparisons of the most dominant bacterial and fungal taxa that were functionally active showed that S. epidermidis, M. salivarium, S. aureus, Prevotella oris and Candida albicans, many of which are often considered oral commensals, were present in both UA and BAL samples (Extended Data Fig. 3c). Interestingly, the lytic phage Proteus virus Isfahan, known to be active against biofilms of Proteus mirabilis22, was found to be highly transcriptionally active in the BAL.
As with the metatranscriptome data, we first identified taxa as probable contaminants in the metagenome data, but these were not removed for subsequent analyses (Extended Data Fig. 2b). Both α- and β-diversity analyses of the metagenome supported distinct microbial community features in the lower airways as compared with the UA and background controls (Extended Data Fig. 4a,b). Interestingly, S. epidermis ranked as the most highly functional taxon in both BAL and UA, based on RNA-Seq reads (Extended Data Fig. 3c), and was 33rd in relative abundance in the BAL DNAseq data but present at very high relative abundance in the UA (ranked no. 3). These data suggest that microbes that colonize the UA and the skin were common in the lower airways in this cohort of COVID-19 patients requiring invasive MV.
Airway microbiota are associated with clinical outcomes
Consistent with the SARS-CoV-2 viral load assessed by reverse transcriptase PCR (RT–PCR), differential expression analysis (DESeq) of the RNA virome identified SARS-CoV-2 as being enriched in the deceased group, as compared with both ≤28 day and >28 day MV groups (fold change greater than 5, Fig. 2d). Cox proportional hazards modelling supports that enrichment with SARS-CoV-2 was associated with increased risk for death (hazard ratio (HR) 1.33, 95% confidence interval 1.07–1.67, P value = 0.011, false discovery rate (FDR) adjusted P value = 0.06; Supplementary Table 5).
The phage metatranscriptome α- and β-diversity was similar across the clinical outcome groups. There were, however, various taxonomic differences at the RNA level, with enrichment of Staphylococcus phages CNPx in the deceased and >28 day MV groups when compared with the ≤28 day MV group (Fig. 2e). Differential expression from two other Staphylococcus phages was also observed in the >28 day MV group as compared with the ≤28 day MV group (Fig. 2e). None of the described taxa were identified as possible contaminants (Supplementary Table 4).
Oral commensals and poor clinical outcome
We evaluated the overall bacterial load by quantitative PCR, targeting the 16S rRNA gene. As expected, the bacterial load in the BAL was several-fold lower than in the UA but clearly higher than the background bronchoscope control (Supplementary Fig. 2). Patients who died had higher total bacterial load in their BAL than patients who survived (Fig. 3a).

a, Bacterial load measured by Droplet Digital-PCR (ddPCR) targeting 16S rRNA gene in 142 subjects (**two-sided Mann–Whitney U P < 0.01; black horizontal lines represent the median). b,c, PCoA based on Bray–Curtis dissimilarity index of BAL metagenome (b) and metatranscriptome (c) data comparing clinical outcome (single-variable PERMANOVA P value). PC1, principal component 1; PC2, principal component 2. d, Gene set enrichment analysis (GSEA) was used to compare the taxonomic signatures identified in BAL metagenome (diamonds) and metatranscriptome (circles) as distinctly enriched for comparisons between clinical outcome groups (differential enrichment performed based on DESeq2 analysis). e, Bubble plot showing DESeq results of bacteria found concordantly differentially enriched between clinical outcome groups (bubble size based on median relative abundance for those found statistically significant; the red dashed line separates positive (to the right) and negative (to the left) fold change; blue text indicates statistical significance).
While no statistically significant differences were noted in α- or β-diversity across clinical outcome groups (Fig. 3b,c), several differences were noted when differential enrichment was evaluated using DESeq. For the comparisons made across the clinical outcome groups, we focused on consistent signatures identified in the lower airway metagenome and metatranscriptome. Coherence of differentially enriched taxa was determined by GSEA (Fig. 3d) and directionality of enrichment between the two datasets was evaluated (Fig. 3e). Among the most abundant taxa, the oral commensal M. salivarum was enriched in the deceased and >28 day MV groups as compared with the ≤28 day MV group. In contrast, a different oral commensal, P. oris, was enriched in the ≤28 day MV group as compared with the deceased and >28 day MV groups. From previous data published by us, enrichment of the lower airway microbiota with oral commensals was seen to be associated with a pro-inflammatory state in several diseases, including lung cancer23,24 and non-tuberculosis mycobacterium-related bronchiectasis25. The data in this analysis support that oral commensals are frequently found in the lower airways of critically ill COVID-19 patients and that differences between groups could be due to differential microbial pressures related to host factors. Interestingly, most of the statistically significant taxa were identified in the metatranscriptome rather than in the metagenome data, with only P. oris identified in both datasets. None of the described taxa were identified as possible contaminants (Supplementary Table 4).
For the fungal data, there were no statistically significant differences in α- or β-diversity identified between clinical outcome groups in the metagenome or the metatranscriptome data (Extended Data Fig. 5a,c). However, in the metagenome data, we identified Candida glabrata as being enriched in the deceased group as compared with the ≤28 day MV and the >28 day MV groups, but this was not consistent in the metatranscriptome data (Extended Data Fig. 5b,d).
Microbial functional profile and poor outcome
We used the gene annotation of the DNAseq and RNA-Seq data to profile the microbial functional potential of the BAL samples. For the comparisons made across the clinical outcome groups, we focused on consistent functional signatures identified in the lower airway metagenome and metatranscriptome. Coherence of differentially enriched functions was determined using GSEA (Extended Data Fig. 6a) and directionality of enrichment was also evaluated (Extended Data Fig. 6b). Overall, there was coherence of directionality between the metranscriptomics and metagenomics datasets for the comparisons between deceased versus ≤28 day MV and >28 day MV versus ≤28 day MV groups. Interestingly, statistically significant differences were only noted in the metatranscriptome data and not in the metagenome data, suggesting that functional activation of microbes can provide further insights into the lower airway microbial environment of patients with worst outcome. Among the top differentially expressed pathways in the poor outcome groups were glycosylases, oxidoreductase activity, transporters and the two-component system, which is used by bacteria and fungi for signalling. A specific analysis of antibiotic resistance genes showed that there was significant gene enrichment and expression of biocide resistance in the deceased group as compared to the two other MV groups (Extended Data Fig. 7). There was also significant expression of genes resistant to trimethoprim and phenolic compound, as well as multi-drug resistance in the deceased group as compared to the ≤28 day MV group. Presence of the resistance gene against trimethoprim was not significantly associated with prior exposure to the drug. However, only seven patients received this drug before sample collection. These differences may indicate important functional differences leading to a different metabolic environment in the lower airways that could impact host immune responses. It could also be representative of differences in microbial pressure in patients with higher viral loads and different inflammatory environments.
Adaptive and innate immune responses to SARS-CoV-2
To evaluate the host immune response to SARS-CoV-2 infection, we first measured levels of anti-spike and anti-RBD (receptor binding domain) antibodies in BAL samples. For both anti-spike and anti-RBD immunoglobulins, levels of IgG, IgA and IgM were several logs higher than levels found in BAL samples from non-SARS-CoV-2 infected patients. Importantly, IgG levels of anti-spike and anti-RBD were significantly lower in the deceased group as compared to the levels found in patients who survived (Fig. 4a and Extended Data Fig. 8a–c, P < 0.05). Prior investigations have suggested that IgA levels are a key driver of neutralization in the mucosa26,27,28. The differences noted in the current investigation in the IgG pools are intriguing, and future work investigating the antibodies generated during SARS-CoV-2 infections will be essential. Additionally, a neutralization assay performed using BAL fluid showed varying levels of neutralization across all samples (as estimated by EC50) but no statistically significant differences between the clinical outcome groups (Extended Data Fig. 8d).

a, Levels of anti-SARS-CoV-2 spike antibodies in BAL of 142 subjects (*two-sided Mann–Whitney U; deceased versus ≤28 d P =0.0133; deceased vs > 28 d P = 0.0213; horizontal black lines represent the median; mean fluorescence shown as flow cytometry arbitrary units). b, Heat map of canonical pathway analysis based on ingenuity pathway analysis (IPA, RRID:SCR_008653) using the lower airway host transcriptome comparing clinical outcome groups. Orange shows upregulation of pathway, blue shows downregulation of pathway. c, Cell-type abundance quantification plots. Comparison of abundance of mast cells and neutrophils among outcome groups in the BAL fluids of 118 critically ill patients with COVID-19. Cell-type abundance was estimated from the host transcriptome with CIBERSORTx. Each dot denotes the quantification score of a sample, while the box shows the interquartile range with median at the centre, and the whiskers represent the maximum and minimum (*two-sided Mann–Whitney U; macrophages M1, > 28 d versus ≤28 d P = 0.017, deceased versus < 28 d P =0.0014; mast cells, >28 d versus ≤28 d P = 0.0013, deceased versus ≤28 d P = 0.0056; innate T-cells, >28 d versus ≤28 d P = 0.0071, deceased versus ≤28 d P = 0.0068; CCR7+ T-cells, >28 d versus ≤28 d P =0.0192, deceased versus ≤28 d P =0.0068).
Host transcriptome analyses of BAL samples showed significant differences across clinical outcome groups based on β-diversity composition (Extended Data Fig. 9). We identified multiple differentially expressed genes across the clinical outcome groups (Extended Data Fig. 9b–d). First, we noted that the lower airway transcriptomes showed downregulation of heavy constant of IgG (IGHG3) and heavy constant of IgA (IGHA1) genes in those with worse clinical outcome (Supplementary Table 6). We then used IPA to summarize differentially expressed genes across the three clinical outcome groups (Fig. 4b). The sirtuin signalling pathway (a pathway known to be involved in ageing, gluconeogenesis/lipogenesis and host defence against viruses)29 and the ferroptosis pathway (an iron-dependent form of regulated cell death present in bronchial epithelium)30,31 were both upregulated in those with worse outcome. Interestingly, there is evidence to support that STAT3 (ref. 32) and ACSL4 (ref. 33) alleviated ferroptosis-mediated acute lung injury dysregulation, which are both downregulated in COVID-19 patients with worse clinical outcome. While this may reflect the host response to viral infection, other differences in the transcriptomic data showed downregulation of mitochondrial oxidative phosphorylation, HIF1α, STAT3 and phospholipase C signalling. Additional canonical signalling pathways, including insulin secretion, multiple inositol-related pathways, noradrenaline/adrenaline degradation signalling and xenobiotic-related metabolism, were significantly downregulated when comparing the >28 day MV and ≤28 day MV groups. There is evidence that in the neonatal lung, inositol-related components exert an anti-inflammatory effect and can prevent acute lung injury34,35.
To determine if the abundance of immune cells varies between different clinical outcome groups, we estimated cell-type abundance from the host transcriptome with computational cell-type quantification methods, including a deconvolution approach implemented in CIBERSORTx (ref. 36) and a cell-type signature enrichment approach implemented in xCell (ref. 37). As reported recently in other studies38, among the cell types detected in the BAL samples, we observed a consistent enrichment of mast cells and neutrophils in the >28 day MV and deceased groups compared with the ≤28 day MV group (Fig. 4c and Supplementary Table 7). We also identified significantly higher inflammatory macrophages (M1), innate T-cells and memory T-cells (CCR7+) among subjects with worse clinical outcome.
Cross-kingdom network analyses and SARS-CoV-2
To identify potential microbe–microbe and microbe–host interactions that could have an effect on outcome, we used a multiscale network analysis approach (multiscale embedded gene co-expression network analysis, MEGENA)39. We first used the relative abundance from the RNA-Seq data to capture co-expressing taxa in the metatranscriptome network neighbourhood of SARS-CoV-2 (SARS2-NWN). We examined five such network neighbourhoods (constructed by including nodes with increasing distance 1 to 5 from SARS-CoV-2; that is, neighbourhood 1 to neighbourhood 5) that were significantly enriched for taxa functionally active in the deceased group when compared with the ≤28-day MV group. Only the largest cluster, with 504 taxa, had significantly enriched taxa in both the deceased and the ≤28-day MV outcome groups (Extended Data Fig. 10a) (Fisher’s exact test (FET) P value = 4.6 × 10−45, 4.0 fold enrichment (FE)). Many of these taxa are among the top 50 most abundant microbes we had previously identified in the metatranscriptome dataset. Taxa present that are influenced by SARS-CoV-2 and significantly differentially enriched in the deceased group include bacteria such as M. salivarium, Bifidobacterium breve and Lactobacillus rhamnosus (a gut commensal) that we had previously identified by differential expression analysis (Fig. 3e), but also taxa such as S. epidermis, Mycoplasma hominis (urogenital bacteria) and the phage VB_PmiS-Isfahan (also referred to as Proteus virus Isfahan) that we had previously picked up as being highly abundant but not necessarily differentially enriched in the deceased group. Most of the fungi, such as C. albicans, C. glabrata and Candida orthopsilosis were enriched in the ≤28 day MV group. Interestingly, our earlier analysis of the metagenome (Extended Data Fig. 5b) had identified C. glabrata as being enriched in the deceased group with no enrichment in the metatranscriptome. This analysis indicates that some of these abundant taxa could be responding to SARS-CoV-2 disruption in a similar manner or indirectly interacting functionally.
We further investigated the association of the network neighbourhood with host network modules using the host transcriptome data to identify groups of host genes that are co-expressed in response to SARS-CoV-2 disruption. The three host modules with the most significant correlations to SARS2-NWN are M175, M277 and M718. M277 is the parent module of M718, and both are enriched with genes related to respiratory electron transport, while M175 is enriched for IFN-γ signalling (Extended Data Fig. 10b). Module M175 is positively correlated with the SARS2-NWN (ρ = 0.32, P = 2.1 × 10−3). While there was no collective enrichment of the module by differentially expressed genes in the deceased versus ≤28 day MV, there was for >28 day versus ≤28 day MV (FET P = 0.030, 4.5 FE). This module includes well-known antiviral IFN stimulated genes (ISGs), such as IRF7 and OASL.
Metatranscriptome and host transcriptome signatures can predict mortality
We evaluated the strength of the metatranscriptomic, metagenomic and host transcriptomic profiles to predict mortality in this cohort of critically ill COVID-19 patients. To this end, we identified features in each of these datasets and constructed risk scores that best predicted mortality. Figure 5a shows that the metatranscriptome data, alone or combined with the other two datasets, were most predictive of mortality. Importantly, the predictive power (as estimated by the area under the curve) of the metatranscriptome data was improved by excluding probable contaminants and worsened when SARS-CoV-2 was removed from the modelling. The selected features we used to construct the metatranscriptome, metagenome and host transcriptome risk scores are reported in Supplementary Table 8. Using the means of the scores, we classified all subjects into high risk and low risk groups for mortality. Figure 5b shows Kaplan–Meier survival curve comparisons evaluating the predictive power of risk score stratification based on metatranscriptome, metagenome and host transcriptome data. Combining risk scores from different datasets showed an optimal identification of mortality when metatranscriptome and host transcriptome were considered (Fig. 5c). We then used the gene signature found to be the most predictive of mortality to conduct IPA analyses (Supplementary Table 9). Among the upstream regulators, mortality was associated with predicted activation of IFN-α while chemotaxis and infection by RNA virus were predicted as activated in diseases and functions. These observations may be inconsistent with the current suggestion that, based on systemic levels, early interferon responses are associated with poor outcome in COVID19 (refs. 40,41). Others have suggested that a robust interferon response may lead to a hyperinflammatory state that could be detrimental in the disease process, justifying the use of Janus kinase inhibitor inhibitors in patients with COVID-19 (ref. 42). Studies comparing transcriptomic signatures in BAL of patients with severe COVID-19 and controls have shown activation of type 1 interferons43. While further longitudinal data will be needed to clarify the role of interferon signalling on the disease, the data presented here suggest that combining microbial and host signatures could help understand the increased risk for mortality in critically ill COVID-19 patients. Overall, these data highlight the importance of SARS-CoV-2 abundance in the lower airways as a predictor for mortality and the significant contribution of the host cell transcriptome, which reflects the lower airway cell response to infection.

a, Area under the curve median and 95% confidence interval for receiver operating characteristic curve analyses calculated from each sequencing dataset as predictor and mortality (death time from ICU admission) as outcome from 118 subjects. b, Kaplan–Meier survival analyses with 95% confidence interval based on a cut-off value estimated from features selected from each sequencing dataset. The ‘High risk’ and ‘Low risk’ groups are the mean of predicted risk scores in all samples (P value with Mantel–Cox test). c, Scatterplot of risk scores from metatranscriptome, metagenome and host transcriptome. Dotted line denotes the mean of the risk scores across all subjects, which is also the threshold for dividing the samples into ‘High risk’ and ‘Low risk’ groups. d, IPA analyses of host transcriptomic signatures identified as most predictive of mortality.
Discussion
The samples used in this investigation were obtained during the first surge of cases of COVID-19 in New York City, and management reflected clinical practices at that time. Among the differences with current therapeutic approaches in COVID-19 patients, corticosteroids and remdesivir, two medications that likely affect the lower airway microbial landscape, were rarely used during the first surge. Other medications, such as antibiotics and anti-inflammatory drugs, could affect our findings, and we therefore considered them as potential confounders. However, the use of these medications was not found to be associated with clinical outcome. Of note is that although our institutions were responding in ‘surge mode’, both the Long Island and Manhattan campuses did not suffer from shortages in medical staff, supplies or equipment and the decision to start MV did not differ from the standard of care. The cross-sectional study design precluded evaluation of the temporal dynamics of the microbial community or the host immune response in this cohort, which could have provided important insights into the pathogenesis of this disease. Performing repeated bronchoscopies without a clinical indication would be challenging in these patients, and other, less invasive, methods might need to be considered to study the lower airways at earlier time points and serially over time in patients with respiratory failure. It is important to note that there were no statistically significant differences in the timing of sample collection across the three outcome groups. Evaluation of microbial signals at earlier time points in the disease process might also be important to identify changes occurring prior to use of broad-spectrum antimicrobials. Also, the presented data from lower airway samples are restricted to those subjects for whom bronchoscopy was performed as part of their clinical care. Thus, the culture-independent data are biased towards patients who, while critically ill with COVID-19, were not representative of the extremes in the spectrum of disease severity. Investigations focusing on early sample collection time points may be warranted to include subjects on MV with early mortality or early successful discontinuation of MV.
In summary, we present here the first evaluation of the lower airway microbiome using a metagenomic and metatranscriptomic approach, along with host immune profiling, in critically ill patients with COVID-19 requiring invasive MV. The RNA metatranscriptome analysis showed an association between the abundance of SARS-CoV-2 and mortality, consistent with the signal found when viral load was assessed by targeted rRT–PCR. These viral signatures correlated with lower anti-SARS-CoV-2 spike IgG and host transcriptomic signatures in the lower airways associated with poor outcome. Importantly, both through culture and NGS data, we found no evidence for an association between untreated infections with secondary respiratory pathogens and mortality. Together, these data suggest that active lower airway SARS-CoV-2 replication and poor SARS-CoV-2-specific antibody responses are the main drivers of increased mortality in COVID-19 patients requiring MV. The potential role of oral commensals such as M. salivarium needs to be explored further. It is possible that M. salivarium can impact key immune cells, and it has recently been reported at a high prevalence in patients with ventilator-acquired pneumonia44. Critically, our finding that SARS-CoV-2 evades and/or derails effective innate/adaptive immune responses indicates that therapies aiming to control viral replication or induce a targeted antiviral immune response may be the most promising approach for hospitalized patients with SARS-CoV-2 infection requiring invasive MV.
Methods
Subjects
Enrolled subjects were 18 years or older and were admitted to the ICUs at NYU Langone Health from 10 March to 10 May 2020 with a nasal swab confirmed diagnosis of SARS-CoV-2 infection by RT–PCR assay and respiratory failure requiring invasive MV (see Table 1 for subject demographics). Research samples were obtained during clinically indicated bronchoscopies performed for airway clearance or for percutaneous tracheostomy placement, with verbal informed consent from legal authorized representative due to infection control measures that limited the presence of close contacts. All patients or their legal representative agreed to participate via our NYU IRB approved protocol (IRB no. s16-00122/01598). Signed consent was then obtained from patients upon recovery. For those that remained incapacitated, signed consent was obtained from legally authorized representative. All analyses were then performed in de-identified data. Comprehensive demographic and clinical data were collected. We also collected longitudinal data on clinical laboratory culture results and treatment. Extended Data Figure 1 shows the distribution of subjects and sampling strategy used for this study. The study protocol was approved by the Institutional Review Board of New York University.
Lower airway bronchoscopic sampling procedure
Both background and supraglottic (buccal) samples were obtained prior to the procedure, as previously described23. The background samples were obtained by passing sterile saline through the suctioning channel of the bronchoscope prior to the procedure. BAL samples were obtained from one lung segment as per discretion of the treating physician as clinically indicated. Samples were then transferred to a BSL3 laboratory for processing. Once there, 2 ml of whole BAL was stored in a tube prefilled with 2 ml of Zymo Research’s DNA/RNA Shield (catalogue no. R1100-250, https://www.zymoresearch.com/pages/covid-19-efforts) for RNA/DNA preservation and virus inactivation. In addition, background control samples (saline passed through the bronchoscope prior to bronchoscopy) and supraglottic aspirates were stored in the same RNA/DNA shield.
Viral load detection targeting the N gene
SARS-CoV-2 viral load was measured by quantitative rRT–PCR targeting the SARS-CoV-2 nucleocapsid (N) gene and an additional primer/probe set to detect the human RNase P gene (RP). Assays were performed using Thermo Fisher Scientific TaqPath 1-Step RT–qPCR Master Mix, CG (catalogue no. A15299) on the Applied Biosystems 7500 Fast Dx RealTime PCR Instrument. Using the positive controls provided by the CDC, which are normalized to 1,000 copies per ml, we converted the different Ct positive to copies per ml. This was done using the DDCT method, applying the formula Power (2, (CT (sample, N1 gene) − CT (PC, N1 gene)) − (CT (sample, RP gene) − CT (PC, RP gene)) × 1,000.
SARS-CoV-2 viral viability through measurement of subgenomic transcripts
Viral sgRNA is transcribed in infected cells and is not packaged into virions. Thus, presence of sgRNA is indicative of active infection of a mammalian cell in samples. We therefore measured sgRNA in all BAL samples obtained, targeting the E gene as previously described19,20. Briefly, 5 µl RNA was used in a one-step real-time RT–PCR assay to sgRNA (forward primer 5′-CGATCTCTTGTAGATCTGTTCTC-3′, reverse primer 5′-ATATTGCAGCAGTACGCACACA-3′ and probe 5′-FAM-ACACTAGCCATCCTTACTGCGCTTCG-ZEN-IBHQ-3′) and using the Quantifast Probe RT–PCR kit (Qiagen) according to instructions of the manufacturer. In each run, standard dilutions of counted RNA standards were run in parallel to calculate copy numbers in the samples.
Bacterial load assessment
We measured bacterial load in background, BAL and supraglottic samples using a QX200 Droplet Digital PCR System (Bio-Rad). For this, primers were 5′-GCAGGCCTAACACATGCAAGTC-3′ (63F) and 5′-CTGCTGCCTCCCGTAGGAGT-3′ (355R). Cycling conditions included 1 cycle at 95 °C for 5 minutes, 40 cycles at 95 °C for 15 seconds and 60 °C for 1 minute, 1 cycle at 4 °C for 5 minutes and 1 cycle at 90 °C for 5 minutes, all at a ramp rate of 2 °C s−1. PCR cycling was performed on the Bio-Rad C1000 Touch Thermal Cycler and droplets were quantified using the Bio-Rad Quantisoft software. Each sample was run in duplicate.
DNA/RNA isolation, library preparation and sequencing
DNA and RNA were isolated in parallel using zymoBIOMIC DNA/RNA Miniprep Kit (catalogue no. R2002) as per manufacturer’s instructions. DNA was then used for whole genome shotgun (WGS) sequencing, using it as input into the NexteraXT library preparation kit following the manufacturer’s protocol. Libraries were purified using the Agencourt AMPure XP beads (Beckman Coulter) to remove fragments below 200 base pairs. The purified libraries were quantified using the Qubit dsDNA High Sensitivity Assay kit (Invitrogen) and the average fragment length for each library was determined using a High Sensitivity D1000 ScreenTape Assay (Agilent). Samples were added in an equimolar manner to form two sequencing pools. The sequencing pools were quantified using the KAPA Library Quantification Kit for Illumina platforms. The pools were then sequenced on the Illumina Novaseq 6000 in one single run. For RNA sequencing, RNA quantity and integrity were tested with a BioAnalyzer 2100 (Agilent). Among bronchoscope control (BKG) samples, only five yielded RNA with sufficient quality and quantity to undergo library preparation and sequencing. Further, in order to ensure sufficient depth on these background samples, we used an equimolar strategy to pool the background samples based on the concentrations of each individual library. Of note is that the same five BKG samples were selected to undergo WGS sequencing, and we used the same pooling strategy. The automated Nugen Ovation Trio Low Input RNA method was used for library prep with 3 ng total RNA input of each sample. After six amplification cycles, samples were sequenced using 2x Novaseq 6000 S4 200 cycle Flowcells using PE100 sequencing.
Microbial community characterization using WGS sequencing and RNA metatranscriptome
For all metagenomic and metatranscriptomic reads, Trimmomatic v.0.36 (ref. 45), with leading and trailing values set to 3 and minimum length set to 36, was used to remove adaptor sequences. All rRNA reads were then removed from the metatranscriptomic reads using SortMeRNA v.4.2.0 (ref. 46) with default settings. Metagenomic and filtered metatranscriptomic reads were mapped to the human genome using Bowtie2 v.2.3.4.1 (ref. 47) with default settings, and all mapping reads were excluded from subsequent microbiome, mycobiome and virome metagenomic and metatranscriptomic analysis. Technical replicates for each biological sample were pooled together for subsequent analyses. Taxonomic profiles for all metagenomic and metatranscriptomic samples were generated using Kraken v.2.0.7 (ref. 48) and Bracken v.2.5 (https://doi.org/10.7717/peerj-cs.104) run with default settings. The database used for quantifying taxonomic profiles was generated using a combined database containing human, bacterial, fungal, archaeal and viral genomes downloaded from NCBI RefSeq on 8 January 2021. Additionally, genomes for Candida auris (Genbank accession nos. GCA_003013715.2, GCA_008275145.1) and Pneumocystic jirovecii (Genbank accession no. GCA_001477535.1) were manually added to the database. Supplementary Table 10 shows sequence depth and taxonomic richness per sample within sample types. Differentially abundant bacterial and viral taxa were identified for the BAL and UA samples groups individually using DESeq2 v.1.28.1 (ref. 49) with the three group clinical outcome metadata readouts set as the sample groupings. Significantly differentially abundant taxa contained at a minimum an aggregate of five reads across samples and had an FDR < 0.2 (refs. 50,51). The specificity of the top hits identified as being enriched by DESeq analysis was confirmed by mapping metagenomic and metatranscriptomics reads against whole genome references. In this manner, we confirmed that sequence reads that were annotated to M. salivarium, B. breve, L. rhamnosus, M. hominis and Streptococcus oralis (Fig. 3e) mapped along the length of the genomes and were not spurious matches (Supplementary Fig. 3).
For functional microbial profiling, processed sequencing reads were further depleted of human-mapping reads by removing all reads classified as human by Kraken v.2.0.7 (ref. 48) using KrakenTools v0.1-alpha (https://github.com/jenniferlu717/KrakenTools). FMAP v.0.15 (ref. 52) was run on both the metagenomic and metatranscriptomic reads to profile the metabolic pathways present in each sample. FMAP_mapping.pl paired with diamond v.0.9.24 (ref. 53) and FMAP_quantification.pl were used with default settings to identify and quantify proteins in the Uniref90 database. Using DESeq2 v.1.28.1 (ref. 49), differentially expressed genes were identified for the BAL samples individually using the three group clinical outcome metadata readouts for all genes that had an aggregate five reads across all samples.
Antibiotic resistance genes were quantified in all metagenome and metatranscriptome samples using Salmon v.1.3.0 (refs. 54) run with flags --keepDuplicates for indexing and --libtype A --allowDovetail --meta for quantification. Genes were filtered such that only genes that actively conferred antibiotic resistance were kept. To assess differentially expressed classes of antibiotic resistance genes, gene counts for individual antibiotic resistance genes were collapsed by their conferred antibiotic resistance.
Extended Data Figure 1 shows a summary of depth achieved with the parallel WGS and metatranscriptome approach across sample types and the number of reads assigned to different microbial subfractions (bacteria, fungi, DNA viruses, RNA viruses and phages). Further analysis was also done to identify possible contaminants in the metatranscriptome and metagenome datasets. To this end, we compared the relative abundance of taxa between background bronchoscope control and BAL samples. Taxa with median relative abundance greater in background than in BAL were identified as probable contaminants and listed in Supplementary Table 4. None of the taxa identified as possible contaminants were removed from the analysed data, but the data are shown for comparison with signatures identified in the rest of the analyses.
Anti-spike SARS-CoV-2 antibody profiling in BAL
BAL samples were heat-treated at 56 °C for one hour and centrifuged at 14,000 g for 5 min. The supernatant was collected and diluted 50-fold in PBST containing 1% skim milk. The diluted samples were incubated at room temperature for 30 min with QBeads DevScreen: SAv (Streptavidin) (Sartorius catalogue no. 90792) that had been loaded with biotinylated spike, biotinylated RBD or biotin (negative control) in wells of a 96-well HTS filter plate (MSHVN4550). As positive controls, we used CR3022 antibody, which recognizes SARS-CoV-2 spike and RBD, in human IgG, IgA and IgM formats (Absolute Antibody; dilutions 1:1,120, 1:1,300 and 1:258, respectively). After washing the beads, bound antibodies were labelled with anti IgG-DyLight488, anti IgA-PE and anti IgM-PECy7, and the fluorescence intensities were measured in Intellicyt IQue3 (Sartorius). The acquired data (median fluorescence intensity (MFI)) were normalized using the MFI values of the CR3022 antibodies to compensate for variations across plates. Extended Data Figure 8 shows that the levels of these antibodies were higher in BAL samples of patients with SARS-CoV-2 than in BAL samples from ten uninfected healthy smokers recruited for research bronchoscopy. Details of method development and validation will be described elsewhere (Koide et al., in preparation).
SARS-CoV-2 preparation and neutralization assay
icSARS-CoV-2-mNG (isolate USA/WA/1/2020, obtained from the UTMB World Reference Center for Emerging Viruses and Arboviruses) was amplified once in Vero E6 cells (P1 from the original stock). Briefly, 90–95% confluent T175 flask (Thomas Scientific) of Vero E6 (1 × 107 cells) was inoculated with 50 μl of icSARS-CoV-2-mNG in 5 ml of infection media (DMEM, 2% FBS, 1% NEAA and 10 mM HEPES) for 1 hour. After 1 hour, 20 ml of infection media were added to the inoculum, and cells were incubated 72 hours at 37 °C and 5% CO2. After 72 hours, the supernatant was collected, and the monolayer was frozen and thawed once. Both supernatant and cellular fractions were combined, centrifuged for 5 min at 500 g and filtered using a 0.22 μm Steriflip (Millipore). Viral titres were determined by plaque assay in Vero E6 cells. In brief, 220,000 Vero E6 cells per well were seeded in a 24-well plate, 24 hours before inoculation. Tenfold dilutions of the virus in DMEM (Corning) were added to the Vero E6 monolayers for 1 hour at 37 °C. Following incubation, cells were overlaid with 0.8% agarose in DMEM containing 2% FBS (Atlanta Biologicals) and incubated at 37 °C for 72 h. The cells were fixed with 10% formalin, the agarose plug removed and plaques visualized by crystal violet staining. All procedures including icSARS-CoV-2-mNG virus were performed using Biosafety Level 3 laboratory conditions.
For SARS-CoV-2 neutralization assays, Vero E6 cells (30,000 cells per well) were seeded in a 96-well plate 24 h before infection. Two-fold serial dilutions of BAL lysates were mixed with mixed 1:1 (vol/vol) with SARS-CoV-2 mNG virus (multiplicity of infection (MOI) 0.5) and incubated for 1 h at 37 °C. After incubation, 100 μl of the mixtures of the antibody and SARS-CoV-2 mNG were added to the Vero E6 monolayers, and cells were incubated at 37 °C. After 20 h, cells were fixed with 4% formaldehyde (Electron Microscopy Sciences) at room temperature for 1 h. After fixation, cells were washed twice with PBS and permeabilized with 0.25% triton-100, stained with DAPI (Thermo) and quantified on a CellInsight CX7 High Content microscope (Thermo) using a cut-off for three standard deviations from negative to be scored as an infected cell.
Transcriptome of BAL samples
Using data from the Hi-seq/Illumina55,56,57 platform at the NYU Langone Genomic Technology Center (data available at Sequence Read Archive accession no. PRJNA592149) KEGG (refs. 58,59) annotation was summarized at levels 1 to 3. Genes with an FDR-corrected adjusted P value <0.25 were considered significantly differentiated unless otherwise specified. Pathway analysis using differentially regulated genes (FDR < 0.25) was done using Ingenuity Pathway Analysis, RRID: SCR_008653 (Qiagen)60. GSEA was performed with differential genes (FDR < 0,25) for dataset comparison using R package fgsea v1.4.1 (ref. 61). Normalized host transcriptome data as transcript per million (TPM) from the BAL metatranscriptome was subjected to digital cell-type quantification with CIBERSORTx36 and xCell37 separately. Genes with at least 1 count per million in at least two samples were retained. For digital cytometry with CIBERSORTx, a signature matrix derived from single-cell transcriptome of BAL cells collected from patients with COVID-19 (ref. 38) was first generated with the ‘Create Signature Matrix’ module in the CIBERSORTx online tool. A maximum of 10 cells per cell type per patient were initially sampled from the original data and 20 cells per cell type were then used to build the single-cell reference with the default parameters. Then, the ‘Impute Cell Fractions’ module was used to estimate the absolute cell fraction score of different cell types in bulk transcriptomes using the single-cell signatures with ‘S-mode’ batch correction and 100 permutations in the absolute mode. Bulk transcriptomes with a significant deconvolution P value (≤0.05) were retained. For xCell cell-type signature enrichment analysis, the enrichment scores were inferred with built-in signature of cell types detected in the BAL samples as reported previously38. The two-tailed Wilcoxon rank sum test with Benjamini–Hochberg correction was computed between groups of samples for comparison.
Microbial and host predictive modelling
Cox proportional hazards model was used for investigating the association between the time to death and the relative abundance of each taxon quantified using metatranscriptomic and metagenomic data separately. We first performed the univariate screening test to identify significant features associated with the time to death using the Cox proportion hazards regression model for the relative abundance of taxa from the RNA and DNA data and log-transformed count of host transcriptome data, respectively. Within each type of data, given the P value cut-off, the features with a P value less than the cut-off were selected and integrated as a sub-community. For the RNA and DNA data, the α-diversity (Shannon index) was calculated for each sample on the selected sub-community and the negative of the value was defined as the microbial risk score, because high α-diversity indicates low risk of death. For the host transcriptome data, the log-transformed total count of all selected candidate transcriptome for each sample was defined as the risk score, since most selected candidate transcriptomes increased the risk of death. The leave-one-out cross-validation was used for the predictions. The P value cut-off was set at the value that produces the largest area under the receiver operating characteristic curve in predicting the death/survival status using the risk score we constructed over these features. The additive model was used to integrate when more than one score is used for the prediction.
Multiscale and co-expression network analyses
Raw counts from the human transcriptome were normalized and converted to log2 counts per million using the R packages limma (ref. 60) and voom (ref. 61) (v.3.44.1 with R v.4.0.0) with standard parameters. Microbiome abundance information was converted to relative abundance. Low abundance taxa were removed based on average abundance across all samples to yield a minimum of 1,000 taxa for each metatranscriptome dataset. All datasets were batch adjusted. Differentially expressed genes and differentially abundant taxa were called using the DESeq2 package49 (v.1.28.1), based on the negative binomial (that is, Gamma–Poisson) distribution. According to the recommendation by the authors of the package, we used non-normalized data (that is, raw gene counts and abundance data), as DESeq2 internally corrects data and performs normalization steps. For this purpose, raw microbiome abundance data were converted to DESeq2 dds objects using the phyloseq R library (v.1.32.0). Contrasts are based on outcome groups (≤28 day MV, >28 day MV or death). Differentially expressed genes and differentially abundant tax with FDR of 0.2 or less are considered significant.
MEGENA39 was performed to identify host modules of highly co-expressed genes in SARS-CoV-2 infection. The MEGENA workflow comprises four major steps: (1) fast planar filtered network construction (FPFNC), (2) multiscale clustering analysis (MCA), (3) multiscale hub analysis (MHA) and (4) cluster–trait association analysis (CTA). The total relevance of each module to SARS-CoV-2 infection was calculated by using the rank product method with the combined enrichment of the differentially expressed gene signatures as implemented: \(G_j = \mathop {\prod }\limits_i g_{ji}\), where gji is the relevance of a consensus j to a signature i and gji is defined as \(({\max}_{j} (r_{ji}) + 1 - r_{ji})/\mathop {\sum }\limits_j r_{ji}\), where rji is the ranking order of the significance level of the overlap between the module j and the signature.
To functionally annotate gene signatures and gene modules derived from the host transcriptome data, we performed an enrichment analysis of the established pathways and signatures, including the gene ontology categories and MSigDB. The hub genes in each subnetwork were identified using the adopted Fisher’s inverse chi-square approach in MEGENA; Bonferroni-corrected P values smaller than 0.05 were set as the threshold to identify significant hubs. The calculation of correlations between modules, between modules and clinical traits as well as between modules and individual taxa was performed using Spearman correlation. Other correlation measures, such as Pearson correlation or the maximal information coefficient (MIC)62 proved to be inferior for this task. Categorical trait data were converted to numerical values as suitable.
Statistics and reproducibility
Specific statistical analysis is described in detail for each subsection of the methods above. For association with discrete factors, we used non-parametric tests (Mann–Whitney or Kruskal–Wallis analysis of variance (ANOVA)). We used the ade4 package in R to construct PCoA based on Bray–Curtis distances63,64,65,66. No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All sequencing data used for this analysis are available in the NCBI Sequence Read Archive under project numbers PRJNA688510 and PRJNA687506 (RNA and DNA sequencing, respectively).
Code availability
Code used for the analyses presented in the current manuscript is available at https://github.com/segalmicrobiomelab/SARS_CoV2.
References
Emerging understandings of 2019-nCoV. Lancet 395, 311 (2020).
WHO Coronavirus (COVID-19) Dashboard. WHO Health Emergency Dashboard https://covid19.who.int/ (2020).
Rabaan, A. A. et al. SARS-CoV-2, SARS-CoV, and MERS-COV: a comparative overview. Infez. Med. 28, 174–184 (2020).
Cao, X. COVID-19: immunopathology and its implications for therapy. Nat. Rev. Immunol. 20, 269–270 (2020).
Morens, D. M. & Fauci, A. S. The 1918 influenza pandemic: insights for the 21st century. J. Infect. Dis. 195, 1018–1028 (2007).
Shieh, W. J. et al. 2009 pandemic influenza A (H1N1): pathology and pathogenesis of 100 fatal cases in the United States. Am. J. Pathol. 177, 166–175 (2010).
Dickson, R. P. et al. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat. Microbiol. 1, 16113 (2016).
Kitsios, G. D. et al. Respiratory tract dysbiosis is associated with worse outcomes in mechanically ventilated patients. Am. J. Respir. Crit. Care Med. 202, 1666–1677 (2020).
Dickson, R. P. et al. Lung microbiota predict clinical outcomes in critically ill patients. Am. J. Respir. Crit. Care Med. 201, 555–563 (2020).
Zuo, T. et al. Alterations in fecal fungal microbiome of patients with COVID-19 during time of hospitalization until discharge. Gastroenterology 159, 1302–1310.e5 (2020).
Chen, L. et al. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerg. Microbes Infect. 9, 313–319 (2020).
Shen, Z. et al. Genomic diversity of severe acute respiratory syndrome-coronavirus 2 in patients with coronavirus disease 2019. Clin. Infect. Dis. 71, 713–720 (2020).
Kelleni, M. T. SARS CoV-2 viral load might not be the right predictor of COVID-19 mortality. J. Infect. 82, e35 (2020).
Fajnzylber, J. et al. SARS-CoV-2 viral load is associated with increased disease severity and mortality. Nat. Commun. 11, 5493 (2020).
Bitker, L. et al. Protracted viral shedding and viral load are associated with ICU mortality in COVID-19 patients with acute respiratory failure. Ann. Intensive Care 10, 167 (2020).
Magleby, R. et al. Impact of SARS-CoV-2 viral load on risk of intubation and mortality among hospitalized patients with coronavirus disease 2019. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa851 (2020).
Westblade, L. F. et al. SARS-CoV-2 viral load predicts mortality in patients with and without cancer who are hospitalized with COVID-19. Cancer Cell 38, 661–671.e2 (2020).
Pujadas, E. et al. SARS-CoV-2 viral load predicts COVID-19 mortality. Lancet Respir. Med. 8, e70 (2020).
Wolfel, R. et al. Virological assessment of hospitalized patients with COVID-2019. Nature 581, 465–469 (2020).
Kim, D. et al. The architecture of SARS-CoV-2 transcriptome. Cell 181, 914–921.e10 (2020).
Speranza, E. et al. Single-cell RNA sequencing reveals SARS-CoV-2 infection dynamics in lungs of African green monkeys. Sci. Transl. Med. 13, eabe8146 (2021).
Yazdi, M., Bouzari, M. & Ghaemi, E. A. Genomic analyses of a novel bacteriophage (VB_PmiS-Isfahan) within Siphoviridae family infecting Proteus mirabilis. Genomics 111, 1283–1291 (2019).
Tsay, J. J. et al. Airway microbiota is associated with upregulation of the PI3K pathway in lung cancer. Am. J. Respir. Crit. Care Med. 198, 1188–1198 (2018).
Tsay, J. J. et al. Lower airway dysbiosis affects lung cancer progression. Cancer Discov. 11, 293–307 (2020).
Sulaiman, I. et al. Evaluation of the airway microbiome in nontuberculous mycobacteria disease. Eur. Respir. J. 52, 1800810 (2018).
Sterlin, D. et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci. Transl. Med. 13, eabd2223 (2021).
Wang, Z. et al. Enhanced SARS-CoV-2 neutralization by dimeric IgA. Science Transl. Med. 11, eabf1555 (2021).
Klingler, J. et al. Role of IgM and IgA antibodies in the neutralization of SARS-CoV-2. Preprint at medRxiv https://doi.org/10.1101/2020.08.18.20177303 (2020).
Budayeva, H. G., Rowland, E. A. & Cristea, I. M. Intricate roles of mammalian sirtuins in defense against viral pathogens. J. Virol. 90, 5–8 (2016).
Dar, H. H. et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J. Clin. Invest. 128, 4639–4653 (2018).
Stoyanovsky, D. A. et al. Iron catalysis of lipid peroxidation in ferroptosis: regulated enzymatic or random free radical reaction? Free Radic. Biol. Med. 133, 153–161 (2019).
Qiang, Z. et al. Nrf2 and STAT3 alleviates ferroptosis-mediated IIR-ALI by regulating SLC7A11. Oxid. Med. Cell Longev. 2020, 5146982 (2020).
Xu, Y., Li, X., Cheng, Y., Yang, M. & Wang, R. Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J. 34, 16262–16275 (2020).
Hallman, M., Bry, K., Hoppu, K., Lappi, M. & Pohjavuori, M. Inositol supplementation in premature infants with respiratory distress syndrome. N. Engl. J. Med. 326, 1233–1239 (1992).
Preuss, S. et al. Inositol-trisphosphate reduces alveolar apoptosis and pulmonary edema in neonatal lung injury. Am. J. Respir. Cell Mol. Biol. 47, 158–169 (2012).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019).
Aran, D., Hu, Z. & Butte, A. J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220 (2017).
Liao, M. et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 26, 842–844 (2020).
Song, W. M. & Zhang, B. Multiscale embedded gene co-expression network analysis. PLoS Comput. Biol. 11, e1004574 (2015).
Bastard, P. et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370, eabd4585 (2020).
Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370, eabd4570 (2020).
Kalil, A. C. et al. Baricitinib plus remdesivir for hospitalized adults with COVID-19. N. Engl. J. Med. 384, 795–807 (2020).
Zhou, Z. et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 27, 883–890.e2 (2020).
Nolan, T. J. et al. Low-pathogenicity Mycoplasma spp. alter human monocyte and macrophage function and are highly prevalent among patients with ventilator-acquired pneumonia. Thorax 71, 594–600 (2016).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kopylova, E., Noe, L. & Touzet, H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Pavel, A. B. et al. Alterations in bronchial airway miRNA expression for lung cancer detection. Cancer Prev. Res. 10, 651–659 (2017).
Seumois, G. et al. Transcriptional profiling of Th2 cells identifies pathogenic features associated with asthma. J. Immunol. 197, 655–664 (2016).
Kim, J., Kim, M. S., Koh, A. Y., Xie, Y. & Zhan, X. FMAP: functional mapping and analysis pipeline for metagenomics and metatranscriptomics studies. BMC Bioinf. 17, 420 (2016).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Wilhelm, B. T. et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 453, 1239–1243 (2008).
Sultan, M. et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321, 956–960 (2008).
Tanabe, M. & Kanehisa, M. Using the KEGG database resource. Curr. Protoc. Bioinformatics https://doi.org/10.1002/0471250953.bi0112s38 (2012).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114 (2012).
Law, C. W. et al. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Res. 5, 1408 (2016).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Reshef, D. N. et al. Detecting novel associations in large data sets. Science 334, 1518–1524 (2011).
Dray, S. & Dufour, A.-B. The ade4 package: implementing the duality diagram for ecologists. J. Stat. Softw. https://doi.org/10.18637/jss.v022.i04 (2007).
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J. & Knight, R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 5, 169–172 (2011).
Holmes, I., Harris, K. & Quince, C. Dirichlet multinomial mixtures: generative models for microbial metagenomics. PLoS ONE 7, e30126 (2012).
Morgan, M. DirichletMultinomial: Dirichlet-Multinomial Mixture Model Machine Learning for Microbiome Data. R package v.1.34.0 (Bioconductor, 2021).
Acknowledgements
We thank the Genome Technology Center (GTC) for expert library preparation and sequencing, the Applied Bioinformatics Laboratories (ABL) for providing bioinformatics support and helping with the analysis and interpretation of the data and the Experimental Pathology Research Laboratory for histopathology services and imaging. GTC and ABL are shared resources partially supported by the Cancer Center Support Grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center. This work has used computing resources at the NYU School of Medicine High Performance Computing Facility (HPCF) and computational resources of the NIH High Performance Computing (HPC) Biowulf cluster (http://hpc.nih.gov). We thank M. Dittmann at the NYU Grossman School of Medicine and the NYU Langone Microscopy Laboratory for the use of the CX7 high content microscope. Financial support for the PACT project is provided to the FNIH by AbbVie Inc., Amgen Inc., Boehringer-Ingelheim Pharma GmbH & Co. KG, Bristol-Myers Squibb, Celgene Corporation, Genentech Inc., Gilead, GlaxoSmithKline plc, Janssen Pharmaceutical Companies of Johnson & Johnson, Novartis Institutes for Biomedical Research, Pfizer Inc. and Sanofi. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. R37 CA244775 (L.N.S., NCI/NIH); R01 HL125816 (L.N.S., NHLBI/NIH); Colton Pilot Project Grant (L.N.S., L.A., S.B.L. and K.K.); UWSC1085.1 (L.N.S., R.P. and L.E., CDC Foundation); PACT grant (L.N.S., FNIH); R21 AI158997 (S.K.); R01 AI143861 (K.M.K., NIAID/NIH); R01 AI143861-02S1 (K.M.K., NIAID/NIH); R01 DK110014 (H.L. and C.W., NIDDK/NIH); P20 CA252728 (C.W. and H.L., NCI/NIH) American Association for Cancer Research Grant (H.P./L.N.S.); The Genome Technology Center is partially supported by the Cancer Center Support Grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center (A.H. and A.T.); FAMRI Young Clinical Scientist Award (B.G.W.); and Stony Wold-Herbert Fund Grant-in-Aid/Fellowship (I.S. and C.B.). This work was supported in part by the Division of Intramural Research (DIR) of the NIAID/NIH (E.G. and E.D.W.).
Author information
Authors and Affiliations
Contributions
This study was conceived and designed by I.S., L.A., L.D., S.K., K.A.S., K.K., E.G. and L.N.S. Data were obtained by I.S., L.A., S.T.Y., K.K., Y.L., R.D., R.S., S.A.T., A.K., S.R., C.B., R.P., G.J., G.S., P.M., J.C., X.L., K.J., D.C., A.L., N.A., S.B., V.M., D.K., J.B., A.L., D.P., L.P.-P., E.d.W., D.H.S., A.H. and L.N.S. Data were analysed by I.S., M.C., J.-C.J.T., B.G.W., S.T.Y., K.K., R.D., Chang Wang, J.C.C., Chan Wang, H.L., C.V.F., B.Z., S.K., K.A.S., E.G. and L.N.S. The first draft of the manuscript was written by I.S., M.C., L.A., E.G. and L.N.S. All authors read, critically revised and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Microbiology thanks Weizhong Li and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Description of patient cohort, samples obtained, analyses performed and sequencing depth.
Schematic representation of the measurements obtained in this cohort of 589 critically ill COVID-19 patients. Top barplots summarize median number of reads obtained per sample of 142 independent subjects prior to filtering (Pre) and after filtering (Post). Bottom barplots show the median number of reads annotated to different microbial kingdoms in each sample. *SARS-CoV-2, Human with Lungs, RNA and DNA images were created with BioRender.com. **NYU Langone Health is the official brand from NYU Langone Health.
Extended Data Fig. 2 Identification of top taxa found in background samples as compared with BAL and upper airway samples.
Boxplots showing the relative abundance values in log10 relative abundance of taxa ranked ordered based on dominance of Background bronchoscope control samples and compared to abundances in BAL and Upper Airway samples within metatranscriptome (a) and metagenome (b) data. Red labels indicate taxa where relative abundance is higher in background samples than in BAL and therefore considered possible contaminant.
Extended Data Fig. 3 Topographical analyses of metatranscriptome data.
a, Comparison of alpha diversity (Shannon Index, each dot denotes the Shannon diversity of a sample while box inter-quartile range with median at the center and whiskers represent maximum and minimum value) and b) beta diversity (Bray Curtis Dissimilarity index, across 5 background negative controls (bronchoscope), 118 bronchoalveolar lavage (BAL) and 64 upper airway (UA) samples (Kruskal-Wallis p value =0.0006 and PERMANOVA p-values = 0.001, without multiple comparisons, respectively). c, Boxplots showing the relative abundance values in log10 across all metatranscriptome samples for the 118 BAL and 64 Upper Airway samples. The 50 taxa with the highest relative abundance values in the BAL metatranscriptome data are displayed; the top 10 in the BAL are highlighted in bold. Each column consists of four plots displaying in decreasing order of abundance the top RNA vertebrate viruses, DNA phages, bacteria, and fungi identified (from top to bottom). Numbers in parentheses next to the taxa labels display the ranking in relative abundance for either the BAL or UA metatranscriptome samples, respectively. Each dot denotes the relative abundance of a taxa per sample while the box inter-quartile range with median at the center and whiskers represent maximum and minimum value.
Extended Data Fig. 4 Topographical analyses of metagenome data.
Comparison of a) alpha diversity (Shannon Index, each dot denotes the Shannon diversity of a sample while the box inter-quartile range with median at the center and whiskers represent maximum and minimum value) and b) beta diversity (Bray Curtis Dissimilarity index, across 5 background negative controls (bronchoscope), 118 bronchoalveolar lavage (BAL) and 64 upper airway (UA) samples (Kruskal-Wallis p-value = 0.00000000000000022 and PERMANOVA p-value= 0.001, without multiple comparisons, respectively). (c) Boxplots showing the relative abundance values in log10 across all metagenome samples for the 118 BAL and 64 Upper Airway samples. The 50 taxa with the highest relative abundance values in the BAL metagenome are displayed; the top 10 in the BAL are highlighted in bold. Each column consists of two plots displaying the most abundant bacteria and fungi identified. Numbers in parentheses next to the taxa labels displays its ranking in relative abundance for either the BAL or UA metagenome samples, respectively. Each dot denotes the relative abundance of a taxa per sample while the box inter-quartile range with median at the center and whiskers represent maximum and minimum value.
Extended Data Fig. 5 Evaluation of associations between the lower airway mycobiome and clinical outcome.
Fungal taxonomic data was subtracted from metagenome and metatranscriptome data from 5 background negative controls (bronchoscope), 118 bronchoalveolar lavage (BAL) and 64 upper airway (UA) samples. a, Comparisons between the three clinical outcome groups was performed for α diversity (Shannon Index, each dot denotes the Shannon diversity of a sample while the box inter-quartile range with median at the center and whiskers represent maximum and minimum value, left panel), β diversity (based on Bray Curtis Dissimilarity Index, right panel); Kruskal-Wallis p-value and PERMANOVA p-value respectively; on metagenome data. b, Bubble plot showing DESeq results of fungi enriched in each clinical outcome comparisons based on metagenome data (bubble size based on median relative abundance for those found to be statistically significant). c, Comparisons between the three clinical outcome groups was performed for α diversity (Shannon Index, each dot denotes the Shannon diversity of a sample while the box inter-quartile range with median at the center and whiskers represent maximum and minimum value, left panel), β diversity (based on Bray Curtis Dissimilarity Index, right panel); Kruskal-Wallis p-value and PERMANOVA p-value respectively; on metatranscriptome data. d, Bubble plot showing DESeq results of fungi enriched in each clinical outcome comparisons based on metatranscriptome data (bubble size based on median relative abundance for those found to be statistically significant).
Extended Data Fig. 6 Functional microbial compositional analyses.
KOs were summarized to associated pathways and differential expression was calculated based on DESeq2 analysis. a, Gene Set Enrichment Analysis (GSEA) was used to compare the functional signatures identified in BAL metagenome and metatranscriptome as distinctly enriched for comparisons between clinical outcome groups. b, Bubble plot showing DESeq results of microbial functions found concordantly differentially enriched between clinical outcome groups (bubble size based on median relative abundance for those found statistically significant).
Extended Data Fig. 7 Evaluation of associations between the lower airway antibiotic resistance genes and clinical outcome.
Bubble plot showing DESeq results of summarized categories of antibiotic resistant microbial genes taken from MEGARes for the metagenome (top) and metatranscriptome (bottom) data sets for each clinical outcome comparison (bubble size based on median relative abundance for those found to be statistically significant). Colored bubbles indicate significantly enriched antibiotic resistance groups.
Extended Data Fig. 8 Measurement of anti-SARS-CoV-2 Immunoglobulin levels and neutralization activity.
Levels of anti-SARS-CoV-2 Spike (a) and anti-SARS-CoV-2 receptor binding domain (RBD, b) antibodies in from 20 non-SARS-CoV-2 infected smoker controls and 142 severely ill COVID-19 intubated patients. Note that the signals for different isotypes cannot be compared because they are detected with different reagents. c, Comparisons of anti-SARS-CoV-2 RBD antibody levels in 142 BAL samples across subjects in different clinical outcome groups (*= Two-sided Mann–Whitney U p < 0.05). d, Neutralizing activity in BAL samples across subjects in different clinical outcome groups.
Extended Data Fig. 9 Evaluation for associations between the lower airway host transcriptome and clinical outcome.
a, PCoA (based on Bray Curtis Dissimilarity Index, PERMANOVA p-value) comparing the three clinical outcome groups. b, c, d, Volcano plots comparing lower airway host transcriptome between the three clinical outcome groups.
Extended Data Fig. 10 Multi-scale cross-kingdom and co-expression networks.
The neighborhood 5 cross-kingdom metatranscriptome network centered around SARS-CoV-2 is shown. Nodes refer to taxa, edges denote co-abundance after MEGENA. The size of the nodes indicates abundance. Taxa with large nodes are highly abundant. Node shapes are according to the legend and refer to different microbial kingdoms. The differential abundance of taxa in log2(fold change) between the deceased group and the ≤28-day MV groups is shown by node color - red nodes are taxa abundant in the deceased group compared to the ≤28-day MV group, blue colored nodes denote the opposite. b, Modules M175 and M718 of the host transcriptome are shown. The node size refers to the absolute gene expression value. Nodes with wide node border refer to key regulators/hub genes (see Methods). The differential gene expression of taxa in log2(fold change) between the deceased group and the ≤28-day MV groups is shown by node color - red nodes are up-regulated in the deceased group compared to the ≤28-day MV group, blue colored nodes denote the opposite.
Supplementary information
Supplementary Information
Supplementary Figs. 1–4 and Results.
Supplementary Tables
Supplementary Tables 1–14.
Rights and permissions
About this article

Cite this article
Sulaiman, I., Chung, M., Angel, L. et al. Microbial signatures in the lower airways of mechanically ventilated COVID-19 patients associated with poor clinical outcome. Nat Microbiol 6, 1245–1258 (2021). https://doi.org/10.1038/s41564-021-00961-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-021-00961-5
This article is cited by
-
Comprehensive bronchoalveolar lavage characterization in COVID-19 associated acute respiratory distress syndrome patients: a prospective cohort study
Respiratory Research (2023)
-
Pleural fluid microbiota as a biomarker for malignancy and prognosis
Scientific Reports (2023)
-
The dynamic lung microbiome in health and disease
Nature Reviews Microbiology (2023)
-
Characterization of the pig lower respiratory tract antibiotic resistome
Nature Communications (2023)
-
Metatranscriptome of human lung microbial communities in a cohort of mechanically ventilated COVID-19 Omicron patients
Signal Transduction and Targeted Therapy (2023)
