
Abstract
Microbial colonization of the human intestine impacts host metabolism and immunity; however, exactly when colonization occurs is unclear. Although many studies have reported bacterial DNA in first-pass meconium samples, these samples are typically collected hours to days after birth. Here, we investigated whether bacteria could be detected in meconium before birth. Fetal meconium (n = 20) was collected by rectal swab during elective breech caesarean deliveries without labour and before antibiotics and compared to technical and procedural controls (n = 5), first-pass meconium (neonatal meconium; n = 14) and infant stool (n = 25). Unlike first-pass meconium, no microbial signal distinct from negative controls was detected in fetal meconium by 16S ribosomal RNA gene sequencing. Additionally, positive aerobic (n = 10 of 20) and anaerobic (n = 12 of 20) clinical cultures of fetal meconium (13 of 20 samples positive in at least one culture) were identified as likely skin contaminants, most frequently Staphylococcus epidermidis, and not detected by sequencing in most samples (same genera detected by culture and sequencing in 2 of 13 samples with positive culture). We conclude that fetal gut colonization of healthy term infants does not occur before birth and that microbial profiles of neonatal meconium reflect populations acquired during and after birth.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

Data availability
All sequencing data associated with this study have been made publicly available in the National Center for Biotechnology Information Sequence Read Archive under project ID PRJNA666699. Source data are provided with this paper.
Code availability
Custom scripts for the microbiome analyses are available from the GitHub repository at https://github.com/kennek6/Kennedyetal2021.
References
Sprockett, D., Fukami, T. & Relman, D. A. Role of priority effects in the early-life assembly of the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 15, 197–205 (2018).
Durack, J. et al. Delayed gut microbiota development in high-risk for asthma infants is temporarily modifiable by Lactobacillus supplementation. Nat. Commun. 9, 707 (2018).
Weström, B., Arévalo Sureda, E., Pierzynowska, K., Pierzynowski, S. G. & Pérez-Cano, F.-J. The immature gut barrier and its importance in establishing immunity in newborn mammals. Front. Immunol. 11, 1153 (2020).
Axelsson, I. et al. Macromolecular absorption in preterm and term infants. Acta Paediatr. Scand. 78, 532–537 (1989).
Nanthakumar, N. et al. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: an immature innate immune response. PLoS ONE 6, e17776 (2011).
Chen, K., Magri, G., Grasset, E. K. & Cerutti, A. Rethinking mucosal antibody responses: IgM, IgG and IgD join IgA. Nat. Rev. Immunol. 20, 427–441 (2020).
Yoshida, M. et al. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity 20, 769–783 (2004).
Hanson, M. A. & Gluckman, P. D. Developmental origins of health and disease: new insights. Basic Clin. Pharmacol. Toxicol. 102, 90–93 (2008).
Fujimura, K. E. et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat. Med. 22, 1187–1191 (2016).
Soderborg, T. K. et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nat. Commun. 9, 4462 (2018).
Aagaard, K. et al. The placenta harbors a unique microbiome. Sci. Transl. Med. 6, 237ra65 (2014).
Antony, K. M. et al. The preterm placental microbiome varies in association with excess maternal gestational weight gain. Am. J. Obstet. Gynecol. 212, 653.e1–653.e16 (2015).
Stinson, L. et al. Comparison of bacterial DNA profiles in mid-trimester amniotic fluid samples from preterm and term deliveries. Front. Microbiol. 11, 415 (2020).
Collado, M. C., Rautava, S., Aakko, J., Isolauri, E. & Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 6, 23129 (2016).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87 (2014).
Lauder, A. P. et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome 4, 29 (2016).
Theis, K. R. et al. Does the human placenta delivered at term have a microbiota? Results of cultivation, quantitative real-time PCR, 16S rRNA gene sequencing, and metagenomics. Am. J. Obstet. Gynecol. 220, 267.e1–267.e39 (2019).
Olomu, I. N. et al. Elimination of ‘kitome’ and ‘splashome’ contamination results in lack of detection of a unique placental microbiome. BMC Microbiol. 20, 157 (2020).
Lim, E. S., Rodriguez, C. & Holtz, L. R. Amniotic fluid from healthy term pregnancies does not harbor a detectable microbial community. Microbiome 6, 87 (2018).
Rehbinder, E. M. et al. Is amniotic fluid of women with uncomplicated term pregnancies free of bacteria? Am. J. Obstet. Gynecol. 219, 289.e1–289.e12 (2018).
de Goffau, M. C. et al. Human placenta has no microbiome but can contain potential pathogens. Nature 572, 329–334 (2019).
Wang, J. et al. Dysbiosis of maternal and neonatal microbiota associated with gestational diabetes mellitus. Gut 67, 1614–1625 (2018).
Hu, J. et al. Diversified microbiota of meconium is affected by maternal diabetes status. PLoS ONE 8, e78257 (2013).
Bittinger, K. et al. Bacterial colonization reprograms the neonatal gut metabolome. Nat. Microbiol. 5, 838–847 (2020).
Hall, I. C. & O’Toole, E. Bacterial flora of first specimens of meconium passed by fifty new-born infants. Am. J. Dis. Child. 47, 1279–1285 (1934).
Rackaityte, E. et al. Viable bacterial colonization is highly limited in the human intestine in utero. Nat. Med. 26, 599–607 (2020).
Chen, C. et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun. 8, 875 (2017).
Moreno-Indias, I., Cardona, F., Tinahones, F. J. & Queipo-Ortuño, M. I. Impact of the gut microbiota on the development of obesity and type 2 diabetes mellitus. Front. Microbiol. 5, 190 (2014).
Gohir, W., Ratcliffe, E. M. & Sloboda, D. M. Of the bugs that shape us: maternal obesity, the gut microbiome, and long-term disease risk. Pediatr. Res. 77, 196–204 (2015).
Singer, J. R. et al. Preventing dysbiosis of the neonatal mouse intestinal microbiome protects against late-onset sepsis. Nat. Med. 25, 1772–1782 (2019).
Galazzo, G. et al. Development of the microbiota and associations with birth mode, diet, and atopic disorders in a longitudinal analysis of stool samples, collected from infancy through early childhood. Gastroenterology 158, 1584–1596 (2020).
Erb-Downward, J. R. et al. Critical relevance of stochastic effects on low-bacterial-biomass 16S rRNA gene analysis. mBio 11, e00258-20 (2020).
Stacy, A. & Belkaid, Y. Microbial guardians of skin health. Science 363, 227–228 (2019).
Weyrich, L. S. et al. Laboratory contamination over time during low-biomass sample analysis. Mol. Ecol. Resour. 19, 982–996 (2019).
Karstens, L. et al. Controlling for contaminants in low-biomass 16S rRNA gene sequencing experiments. mSystems 4, e00290-19 (2019).
de Goffau, M. C., Charnock-Jones, D. S., Smith, G. C. S. & Parkhill, J. Batch effects account for the main findings of an in utero human intestinal bacterial colonization study. Microbiome 9, 6 (2021).
Dominguez-Bello, M. G. et al. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat. Med. 22, 250–253 (2016).
Valdivia-Arenas, M. A. Bloodstream infections due to Micrococcus spp and intravenous epoprostenol. Infect. Control Hosp. Epidemiol. 30, 1237 (2009).
Oudiz, R. J. et al. Micrococcus-associated central venous catheter infection in patients with pulmonary arterial hypertension. Chest 126, 90–94 (2004).
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226 (2018).
Kennedy, K., Hall, M. W., Lynch, M. D. J., Moreno-Hagelsieb, G. & Neufeld, J. D. Evaluating bias of Illumina-based bacterial 16S rRNA gene profiles. Appl. Environ. Microbiol. 80, 5717–5722 (2014).
Sinha, R. et al. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat. Biotechnol. 35, 1077–1086 (2017).
Whelan, F. J. et al. The loss of topography in the microbial communities of the upper respiratory tract in the elderly. Ann. Am. Thorac. Soc. 11, 513–521 (2014).
Bartram, A. K., Lynch, M. D. J., Stearns, J. C., Moreno-Hagelsieb, G. & Neufeld, J. D. Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl. Environ. Microbiol. 77, 3846–3852 (2011).
Sze, M. A. & Schloss, P. D. The impact of DNA polymerase and number of rounds of amplification in PCR on 16S rRNA gene sequence data. mSphere 4, e00163-19 (2019).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 17, 10–12 (2011).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013).
Oksanen, J. et al. Vegan: Community ecology package. R package version 2.0-10 https://cran.r-project.org/web/packages/vegan/index.html (2013).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
Wright, E. S. RNAconTest: comparing tools for noncoding RNA multiple sequence alignment based on structural consistency. RNA 26, 531–540 (2020).
Schliep, K. P. phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593 (2011).
Acknowledgements
We thank all the participants that were recruited in this study. We thank H. Brinkmann, L. Pasura, L. Maschirow and A. Schwickert for assisting with patient recruitment, L. Ehrlich with sample preparation and K. von Weizsaecker and W. Henrich for their external review of the microbiology protocol and advice on protocol improvements. We thank M. Shah for performing the genomic DNA extractions. T.B. and M.M.H. are supported by the Deutsche Forschungsgemeinschaft (German Research Foundation). K.M.K. is supported by a Farncombe Digestive Health Research Institute Student Fellowship. M.G.S. and D.M.S. are supported by the Canada Research Chairs Program. The laboratory analyses including sequencing were supported by funds from the Canadian Institute for Health Research Team Grant no. MWB 141879.
Author information
Authors and Affiliations
Contributions
K.M.K. analysed the sequencing data and wrote the manuscript. M.J.G. contributed to sample collection and wrote the ‘Study design and sample collection’ section of the Methods. T.A. supervised the culture-based analyses. M.M.H. assisted with study design. L.R. assisted with study design and performed the V3–V4 amplifications and processing of raw sequencing data. M.G.S. assisted with study design and analysis of the sequencing data. D.M.S. contributed to data analysis and manuscript development. T.B. designed the study and contributed to sample collection. All authors discussed the analyses and results and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Microbiology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
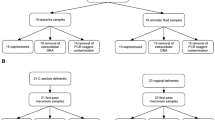
Extended Data Fig. 1 Strobe flowchart.
The study flowchart in line with the STROBE (Strengthening the Reporting of Observational Studies in Epidemiology) statement (http://www.strobestatement.org). Pilot cohort 1A and pilot cohort 1B were used to optimize sample collection. The principal study cohort was analysed by culture and sequencing.
Extended Data Fig. 2 Optimization of collection to reduce contamination.
We considered potential problems related to contamination (birth mode, sampling, sample preparation, general) and sensitivity of culture methods and present our solutions to these problems (see Methods).
Extended Data Fig. 3 Detection of genera across technical replicates.
The abundance (read count) is shown for each sample by sequencing run (run 1 and run 2) for 30 cycles of PCR amplification. Genera are only shown if they were also detected within the same sample’s sequencing data from 40 cycles of PCR amplification.
Supplementary information
Supplementary Tables
Supplementary Table 1. Pilot cohort culture results. Supplementary Table 2. ASV table of negative controls, fetal meconium, neonatal meconium and infant stool samples (listed in ST2). Supplementary Table 3. Sample data for ASV table (ST2). Supplementary Table 4. Agreement of sequencing runs. Supplementary Table 5. Use of negative controls in previous studies.
Source data
Source Data Fig. 2
a, Alpha diversity values for each sample. b, Axes 1 and 2 values for each sample. c, Bray–Curtis dissimilarity values for each sample pair.
Source Data Extended Data Fig. 2
Genus abundance data for each sample and each sequencing run.
Rights and permissions
About this article

Cite this article
Kennedy, K.M., Gerlach, M.J., Adam, T. et al. Fetal meconium does not have a detectable microbiota before birth. Nat Microbiol 6, 865–873 (2021). https://doi.org/10.1038/s41564-021-00904-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-021-00904-0
This article is cited by
-
Investigating prenatal and perinatal factors on meconium microbiota: a systematic review and cohort study
Pediatric Research (2024)
-
Microbiota–gut–brain axis and its therapeutic applications in neurodegenerative diseases
Signal Transduction and Targeted Therapy (2024)
-
Transmission and dynamics of mother-infant gut viruses during pregnancy and early life
Nature Communications (2024)
-
Is there a placental microbiota? A critical review and re-analysis of published placental microbiota datasets
BMC Microbiology (2023)
-
Bacterial extracellular vesicles in the microbiome of first-pass meconium in newborn infants
Pediatric Research (2023)
