
Abstract
Nosocomial acquisition and transmission of vancomycin-resistant Enterococcus faecium (VREfm) is the driver for E. faecium carriage in hospitalized patients, which, in turn, is a risk factor for invasive infection in immunocompromised patients. In the present study, we provide a comprehensive picture of E. faecium transmission in an entire sampled patient population using a sequence-driven approach. We prospectively identified and followed 149 haematology patients admitted to a hospital in England for 6 months. Patient stools (n = 376) and environmental swabs (n = 922) were taken at intervals and cultured for E. faecium. We sequenced 1,560 isolates (1,001 stool, 559 environment) and focused our genomic analyses on 1,477 isolates (95%) in the hospital-adapted clade A1. Of 101 patients who provided two or more stool samples, 40 (40%) developed E. faecium carriage after admission based on culture, compared with 64 patients (63%) based on genomic analysis (73% VREfm). Half of 922 environmental swabs (447, 48%) were positive for VREfm. Network analysis showed that, of 111 patients positive for the A1 clade, 67 had strong epidemiological and genomic links with at least one other patient and/or their direct environment, supporting nosocomial transmission. Six patients (3.4%) developed an invasive E. faecium infection from their own gut-colonizing strain, which was preceded by nosocomial acquisition of the infecting isolate in half of these. Two informatics approaches (subtype categorization to define phylogenetic clusters and the development of an SNP cut-off for transmission) were central to our analyses, both of which will inform the future translation of E. faecium sequencing into routine outbreak detection and investigation. In conclusion, we showed that carriage and environmental contamination by the hospital-adapted E. faecium lineage were hyperendemic in our study population and that improved infection control measures will be needed to reduce hospital acquisition rates.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Data availability
The whole-genome sequences from this study have been deposited at the European Nucleotide Archive (www.ebi.ac.uk/ena) under the study nos. PRJEB12937, PRJEB13191 and PRJEB13192. Individual accession nos. and isolate metadata are listed in Supplementary Data 1. Supplementary Data 2 includes the genetic and epidemiological links characterized in this study. Source data are provided with this paper.
References
Arias, C. A. & Murray, B. E. The rise of the Enterococcus: beyond vancomycin resistance. Nat. Rev. Microbiol. 10, 266–278 (2012).
Werner, G. et al. Emergence and spread of vancomycin resistance among enterococci in Europe. Eurosurveillance 13, 19046 (2008).
Prematunge, C. et al. VRE and VSE bacteremia outcomes in the era of effective VRE therapy: a systematic review and meta-analysis. Infect. Control Hosp. Epidemiol. 37, 26–35 (2016).
Tacconelli, E. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327 (2017).
Taur, Y. et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin. Infect. Dis. 55, 905–914 (2012).
Moradigaravand, D. et al. Within-host evolution of Enterococcus faecium during longitudinal carriage and transition to bloodstream infection in immunocompromised patients. Genome Med. 9, 119 (2017).
Falk, P. S., Winnike, J., Woodmansee, C., Desai, M. & Mayhall, C. G. Outbreak of vancomycin-resistant enterococci in a burn unit. Infect. Control Hosp. Epidemiol. 21, 575–582 (2000).
Neely, A. N. & Maley, M. P. Survival of enterococci and staphylococci on hospital fabrics and plastic. J. Clin. Mcrobiol. 38, 724–726 (2000).
Mitchell, B. G., Dancer, S. J., Anderson, M. & Dehn, E. Risk of organism acquisition from prior room occupants: a systematic review and meta-analysis. J. Hosp. Infect. 91, 211–217 (2015).
Turabelidze, D., Kotetishvili, M., Kreger, A., Morris, J. G. & Sulakvelidze, A. Improved pulsed-field gel electrophoresis for typing vancomycin-resistant enterococci. J. Clin. Microbiol. 38, 4242–4245 (2000).
Gouliouris, T. et al. Genomic surveillance of Enterococcus faecium reveals limited sharing of strains and resistance genes between livestock and humans in the United Kingdom. mBio 9, e01780-18 (2018).
Arredondo-Alonso, S. et al. Plasmids shaped the recent emergence of the major nosocomial pathogen Enterococcus faecium. mBio 11, e03284-19 (2020).
Lee, T. et al. A three-year whole genome sequencing perspective of Enterococcus faecium sepsis in Australia. PLoS ONE 15, e0228781 (2020).
Raven, K. E. et al. A decade of genomic history for healthcare-associated Enterococcus faecium in the United Kingdom and Ireland. Genome Res. 26, 1388–1396 (2016).
Pinholt, M. et al. WGS of 1058 Enterococcus faecium from Copenhagen, Denmark, reveals rapid clonal expansion of vancomycin-resistant clone ST80 combined with widespread dissemination of a vanA-containing plasmid and acquisition of a heterogeneous accessory genome. J. Antimicrob. Chemother. 74, 1776–1785 (2019).
van Hal, S. J. et al. Evolutionary dynamics of Enterococcus faecium reveals complex genomic relationships between isolates with independent emergence of vancomycin resistance. Microbial. Genom. 2, e000048 (2016).
Wang, G. et al. Evolution and mutations predisposing to daptomycin resistance in vancomycin-resistant Enterococcus faecium ST736 strains. PLoS ONE 13, e0209785 (2018).
Raven, K. E. et al. Complex routes of nosocomial vancomycin-resistant Enterococcus faecium transmission revealed by genome sequencing. Clin. Infect. Dis. 64, 886–893 (2017).
Liese, J. et al. Expansion of vancomycin-resistant Enterococcus faecium in an academic tertiary hospital in southwest germany: a large-scale whole-genome-based outbreak investigation. Antimicrob. Agents Chemother. 63, e01978-18 (2019).
Brown, D. F. J. & Walpole, E. Evaluation of selective and enrichment media for isolation of glycopeptide-resistant enterococci from faecal specimens. J. Antimicrob. Chemother. 51, 289–296 (2003).
Tonkin-Hill, G., Lees, J. A., Bentley, S. D., Frost, S. D. W. & Corander, J. Fast hierarchical Bayesian analysis of population structure. Nucleic Acids Res. 47, 5539–5549 (2019).
Prieto, A. M. G. et al. The two-component system ChtRS contributes to chlorhexidine tolerance in Enterococcus faecium. Antimicrob. Agents Chemother. 61, e02122-16 (2017).
Pidot, S. J. et al. Increasing tolerance of hospital Enterococcus faecium to handwash alcohols. Sci. Transl. Med. 10, eaar6115 (2018).
de Regt, M. J. A. et al. High acquisition and environmental contamination rates of CC17 ampicillin-resistant Enterococcus faecium in a Dutch hospital. J. Antimicrob. Chemother. 62, 1401–1406 (2008).
Ludden, C. et al. A One Health study of the genetic relatedness of Klebsiella pneumoniae and their mobile elements in the east of england. Clin. Infect. Dis. 70, 219–226 (2020).
Passaretti, C. L. et al. An evaluation of environmental decontamination with hydrogen peroxide vapor for reducing the risk of patient acquisition of multidrug-resistant organisms. Clin. Infect. Dis. 56, 27–35 (2013).
Grabsch, E. A. et al. Significant reduction in vancomycin-resistant enterococcus colonization and bacteraemia after introduction of a bleach-based cleaning–disinfection programme. J. Hosp. Infect. 82, 234–242 (2012).
Lebreton, F. et al. Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. mBio 4, e00534-13 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Croucher, N. J. et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43, e15 (2015).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2017).
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P.-L. & Ideker, T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 (2011).
Wiegand, I., Hilpert, K. & Hancock, R. E. W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175 (2008).
Acknowledgements
We thank the nurses and healthcare workers on both wards at CUH for assistance with sample collection and the ward matrons, A. Green and C. Cowling, for their support. We thank L. Chaparadza and R. Swayne for assistance with sample and clinical data collection. We thank L. Drumright, A. Chaudhry and the EPIC and Clinical Informatics teams for providing patient movement data. We thank the library construction, sequencing and Pathogen Informatics teams at the Wellcome Trust Sanger Institute for assistance with Illumina sequencing. The flocked swabs were donated by Copan Italia SpA. The present study presents independent research supported by the Health Innovation Challenge Fund (WT098600, HICF-T5-342), a parallel funding partnership between the Department of Health and the Wellcome Trust. The views expressed in this article are those of the author(s) and not necessarily those of the Department of Health or Wellcome Trust. T.G. is a Wellcome Trust Research Training Fellow (103387/Z/13/Z). F.C. is a Wellcome Trust Sir Henry Postdoctoral Fellow (201344/Z/16/Z). C.L. is a Wellcome Trust Sir Henry Postdoctoral Fellow (110243/Z/15/Z). M.E.T. is a Clinician Scientist Fellow supported by the Academy of Medical Sciences, the Health Foundation and the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre. S.J.P. is an NIHR Senior Investigator.
Author information
Authors and Affiliations
Contributions
T.G. and S.J.P. designed the study, wrote the study protocol and case record forms, obtained ethical and research and development approvals for the study, and supervised the data collection. M.E.T. supported ethics approvals. T.G., C.L. and C.C. were responsible for collecting samples, and clinical and epidemiological data. T.G., C.L., B.B. and P.N. isolated and identified E. faecium. B.B. and P.N. undertook susceptibility testing and extracted genomic DNA. N.M.B. and D.A.E. provided access to E. faecium cultures in the routine diagnostic microbiology laboratory and expert opinion relating to infection control. T.G. and F.C. undertook the epidemiological and bioinformatic analyses with contributions from J.P. and K.R. B.B. undertook susceptibility testing to disinfectants with contributions from E.M.H. J.P. supervised the genomic sequencing. F.C. and S.J.P. wrote the first draft of the manuscript. S.J.P. supervised and managed the study. All authors had access to the data, and read, contributed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
N.M.B. is on the advisory board for Discuva Ltd. S.J.P. is a consultant to Specific Technologies. S.J.P., J.P. and F.C. are consultants for Next Gen Diagnostics. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 E. faecium stool culture positivity during study.
Diagram showing E. faecium positivity in patients who provided stool samples within (left branch) or after (right branch) 48 hours from index admission. Subsequent boxes show numbers of patients positive or negative for AREfm and VREfm, and, for patients screened at least twice, whether their positivity status changed, suggestive of E. faecium acquisition. A total of 40 cases acquired E. faecium based on culture, either by acquiring any type of E. faecium after being negative for it (17 and 15 patients in the left and right arms, respectively) or VREfm after being already positive for AREfm (4 and 4 patients in the left and right arms, respectively). Abbreviations: AREfm, ampicillin-resistant E. faecium (which may be vancomycin susceptible or resistant); VREfm, vancomycin-resistant E. faecium.
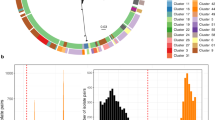
Extended Data Fig. 2 Histogram of pairwise SNP differences between isolates of the same and different subtypes.
Histogram of pairwise SNP differences between 943 clade A1 isolates from stool samples. SNP differences between isolates from the same subtype are shown in dark grey, and between isolates in different subtypes in light grey.
Extended Data Fig. 3 Chlorhexidine and isopropanol susceptibility among selected E. faecium isolates.
Chlorhexidine and isopropanol susceptibility testing results for a subset of phylogenetically representative E. faecium isolates (n=24 biologically independent samples) from the two major subtypes (15A/ST80 (n=3) and 47A/ST78 (n=3)), rest of subtypes in ‘clade A1’ (n=8) and ‘basal’ isolates (n=10) to clade A1. Each dot denotes the median MIC value (panels a and b) or median reduction in colony forming units (CFU) (panel c) across three independent replicates for each isolate tested. In the boxplots, the lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles), and the middle horizontal line to the median. P-values for two-tailed, unpaired Mann-Whitney are showed as NS (non-significant, P > 0.05), * (P < 0.05) or ** (P < 0.01).
Extended Data Fig. 4 Exemplars of E. faecium transmission clusters.
Each row represents the hospital admission period(s) of patients with the exception of the top four rows, which show different environmental sources. Ward of admission is denoted as A or B, and the room numbered and color-coded. Visits to other hospital wards or areas are colored in grey. Positivity results for stool and environmental samples are shown as circles and squares, respectively. Blunt lines and arrowed lines are drawn to point to the putative sources of index and acquired subtypes respectively, the numbers adjacent to these lines indicating the minimum genetic distance observed between connected samples, which ranged from 0 to 6 SNPs. Solid and dotted lines denote strong and weak epidemiological links, respectively. (a) Exemplar of transmission cluster in the same ward (subtype 49A – ST1454). Strong genetic and epidemiological links point to transmission of this subtype in different rooms of ward B among patients D040, D037, D036, D044 and D041. Strong links to the hospital environment, including communal bathrooms and medical devices, suggest their involvement as reservoirs for onward transmission to patients. (b) Exemplar of transmission cluster spanning both hematology wards and involving 7 patients (subtype 26B – ST80). Strong genetic and epidemiological links point to transmission of this subtype in room A3 among patients C015, C023, C009 and D021, followed by spread in different rooms of ward B among patients D021, D022, D010 and D045.
Extended Data Fig. 5 Midpoint rooted maximum likelihood tree based on SNPs in the core genes of 1,560 E. faecium isolates.
E. faecium genomes (1,001 stool, 559 environmental) labeled by clade (B, A2, and A1), commonest sequence types (STs) (only those with more than 10 isolates shown), van genotype, source, ward of origin and month of isolation. Scale bar, ~10,000 SNPs.
Supplementary information
Supplementary information
Supplementary Methods and Tables 1–5.
Supplementary Data 1
Isolate metadata, including European Nucleotide Archive accessions.
Supplementary Data 2
Details on transmission of subtypes and epidemiological links.
Source data
Source Data Fig. 2
Genetic diversity captured within stool samples and subtypes in the same patient.
Source Data Fig. 3
Subtype assignation and source for all clade A1 isolates.
Source Data Fig. 4
Cytoscape-compliant network files.
Source Data Extended Data Fig. 2
Pairwise SNP distances between clade A1 stool isolates.
Source Data Extended Data Fig. 3
Isopropanol and chlorhexidine MICs, and isopropanol tolerance measurements, for selected E. faecium isolates.
Source Data Extended Data Fig. 5
Maximum likelihood phylogenetic tree including all 1,560 E. faecium isolates.
Rights and permissions
About this article

Cite this article
Gouliouris, T., Coll, F., Ludden, C. et al. Quantifying acquisition and transmission of Enterococcus faecium using genomic surveillance. Nat Microbiol 6, 103–111 (2021). https://doi.org/10.1038/s41564-020-00806-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-020-00806-7
This article is cited by
-
Promiscuous, persistent and problematic: insights into current enterococcal genomics to guide therapeutic strategy
BMC Microbiology (2024)
-
GraphSNP: an interactive distance viewer for investigating outbreaks and transmission networks using a graph approach
BMC Bioinformatics (2023)
-
Structural and functional features of a broad-spectrum prophage-encoded enzybiotic from Enterococcus faecium
Scientific Reports (2023)
-
Antimicrobial resistance and whole genome sequencing of novel sequence types of Enterococcus faecalis, Enterococcus faecium, and Enterococcus durans isolated from livestock
Scientific Reports (2023)
-
PowerBacGWAS: a computational pipeline to perform power calculations for bacterial genome-wide association studies
Communications Biology (2022)
