
Abstract
Placozoa is an enigmatic phylum of simple, microscopic, marine metazoans1,2. Although intracellular bacteria have been found in all members of this phylum, almost nothing is known about their identity, location and interactions with their host3,4,5,6. We used metagenomic and metatranscriptomic sequencing of single host individuals, plus metaproteomic and imaging analyses, to show that the placozoan Trichoplax sp. H2 lives in symbiosis with two intracellular bacteria. One symbiont forms an undescribed genus in the Midichloriaceae (Rickettsiales)7,8 and has a genomic repertoire similar to that of rickettsial parasites9,10, but does not seem to express key genes for energy parasitism. Correlative image analyses and three-dimensional electron tomography revealed that this symbiont resides in the rough endoplasmic reticulum of its host’s internal fibre cells. The second symbiont belongs to the Margulisbacteria, a phylum without cultured representatives and not known to form intracellular associations11,12,13. This symbiont lives in the ventral epithelial cells of Trichoplax, probably metabolizes algal lipids digested by its host and has the capacity to supplement the placozoan’s nutrition. Our study shows that one of the simplest animals has evolved highly specific and intimate associations with symbiotic, intracellular bacteria and highlights that symbioses can provide access to otherwise elusive microbial dark matter.
Similar content being viewed by others

Main
Placozoa are marine invertebrates at the base of the animal tree and are considered among the simplest animals known. These millimetre-sized benthic animals can be easily cultured and are considered key models for understanding metazoan evolution, developmental biology and tissue formation1,14,15,16. Electron microscopy studies as early as in the 1970s revealed the presence of intracellular bacteria in these animals3,4,5,6. Remarkably, nearly five decades later, only very little is known about the biology of these symbionts and their interactions with their hosts.
The phylum Placozoa encompasses 19 cryptic species, on the basis of mitochondrial haplotypes2,6. These benthic animals do not have a mouth or gut and feed on algae and bacterial biofilms by external digestion and subsequent uptake via their ventral epithelium17,18. All placozoans have three cell layers and six morphologically differentiated cell types3,6,19,20. The thick ventral epidermis consists of ciliated epithelial cells in which glandular and lipophilic cells are irregularly interspersed17,18. Ciliated epithelial cells make up the thin dorsal epidermis in which crystal cells occasionally occur. An internal meshwork of fibre cells, sandwiched between the two epidermal layers, connects the ventral and dorsal body walls20. Intracellular symbionts were first described in these fibre cells3,5,20. The bacteria were present in all seven haplotypes examined, independent of sampling site or time, and were hypothesized to reside in the lumen of the rough endoplasmic reticulum (rER)3,5,6,20. Persistent and stable residence of a bacterium in the rER of a host would be remarkable as the vast majority of intracellular symbionts live in the cytoplasm or vacuoles, and the few known exceptions inhabit the nucleus or mitochondria21,22,23.
In this study, we focused on the Trichoplax sp. haplotype H2 (Trichoplax H2), previously reported to host two bacterial morphotypes5. Sequencing of placozoan genomes consistently yielded rickettsial and other bacterial sequences6,24,25. However, as thousands of host individuals were pooled for these analyses, it was neither clear whether these bacterial sequences originated from contaminants or symbionts nor whether they were present in all host individuals. Our recent advances in high-throughput sequencing of single placozoan individuals, together with correlative imaging analyses and three-dimensional (3D) reconstruction, allowed us to explore the patterns, structure and function of the placozoan symbiosis at the individual and cellular level.
The Trichoplax H2 microbiome is dominated by two bacterial symbionts. We isolated a placozoan H2 haplotype lineage from a seawater tank at the Kewalo Marine Laboratory, University of Hawai’i (Supplementary Fig. 1). To characterize the microbiome of this Trichoplax H2, we combined highly sensitive DNA and RNA extraction and library preparation protocols to sequence the metagenomes and metatranscriptomes of microscopic single individuals that have an estimated biovolume of 0.02 µl and from which we could isolate 0.5 to 4 ng of nucleic acids (n = 5). All five individuals had similar microbial communities based on 16S ribosomal RNA (rRNA) gene reads, but only two taxa were consistently dominant in all five host individuals (Supplementary Fig. 2 and Supplementary Table 1).
The first and most abundant 16S rRNA phylotype was an alphaproteobacterium from the family Midichloriaceae (Rickettsiales)7 (Fig. 1a). Midichloriaceae are obligate intracellular, often pathogenic, bacteria found in protists and animals, including humans8. In 16S rRNA analyses, the Trichoplax H2 midichloriacean phylotype formed an unnamed lineage that consisted of sequences recovered from diverse invertebrate hosts and sequences from subsurface sediment samples (98.4–99.4% pairwise identity; Fig. 1a). We recovered a high-quality26 1.26 Mb metagenome-assembled genome that included the midichloriacean 16S rRNA phylotype. Sequences from the Trichoplax adhaerens haplotype H1 genome project15 included a midichloriacean 16S rRNA gene fragment and a partial genome of a rickettsial phylotype (RETA1) was also recovered25. Phylogenetic analyses based on the 16S rRNA gene and phylogenomic analyses based on 43 conserved marker genes placed the Trichoplax H2 phylotype and Trichoplax H1 RETA1 in the Midichloriaceae. The Trichoplax H2 and H1 phylotypes were phylogenetically distinct and, according to amino acid sequence identity, these two symbionts belong to two separate but undescribed genera, with Candidatus ‘Bandiella’27,28 as the closest characterized genus (Fig. 1a,b, Supplementary Note 1). We propose the Candidatus taxon ‘Grellia incantans’ for the midichloriacean phylotype from our haplotype H2 isolate (see Supplementary Note 1 for description and etymology).

Bootstrap support values below 0.5 are not shown. Scale bars indicate substitutions per site. Colours and shades of grey indicate taxonomic groups. a, A 16S rRNA tree of ‘G. incantans’ and related Midichloriaceae. For each sequence, the accession number, the percentage identity to ‘G. incantans’ and the published taxonomic names and hosts (where available) are indicated. b,c, Phylogenomic analyses using 43 conserved marker genes based on metagenome-assembled genomes and reference genomes: ‘G. incantans’ and Midichloriaceae are placed in the Rickettsiales (b) and ‘R. eludens’ in the Margulisbacteria (c). Phylum-level classification follows the Genome Taxonomy Database. Taxon names from the Genome Taxonomy Database are indicated in parentheses where available.
The second most abundant and consistently present bacterial taxon in the Trichoplax H2 metagenomes belonged to the Margulisbacteria, a phylum without isolated representatives that forms the sister clade to Cyanobacteriota11,12,13,29. No 16S rRNA gene sequences with >90% identity to this bacterium were found in public sequence databases, warranting the establishment of a taxon at the genus or even family level. We therefore propose the Candidatus taxon ‘Ruthmannia eludens’ for this bacterium (see Supplementary Note 2 for a detailed description and etymology). Using metagenomics binning, we recovered a high-quality 1.51 Mb metagenome-assembled genome for ‘R. eludens’. Our phylogenomic analyses confirmed our 16S rRNA gene analysis and placed ‘R. eludens’ in the Marinamargulisbacteria (Margulisbacteria) (Fig. 1c). Marinamargulisbacteria are aquatic bacteria that occur worldwide11,13. ‘R. eludens’ is distantly related to single-cell amplified genomes and metagenome-assembled genomes from marine pelagic samples13 (Fig. 1c). Marinamargulisbacteria are known only from sequence-based studies, with recovered draft genomes of 0.5–2.0 Mb, and all genomes are classified as medium to low quality26. Despite the small genome size, our metagenome-assembled genome was classified as a high-quality draft genome (Supplementary Note 2).
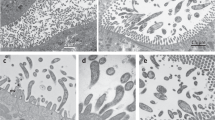
Both symbionts are intracellular, spatially segregated and specific to the host cell type. We used fluorescence in situ hybridization (FISH; Fig. 2a; Supplementary Table 2) to link the bacterial sequences to their morphotypes and visualize the distribution of the two symbionts in Trichoplax. No bacteria except the two symbionts ‘G. incantans’ and ‘R. eludens’ were detected in all the placozoan individuals examined (Fig. 2a; Supplementary Fig. 4). ‘G. incantans’ was thin and rod-shaped (≤1.2 × 0.30 μm2; Fig. 2a and Supplementary Fig. 4). In contrast, ‘R. eludens’ had a wider and stouter rod-shaped morphology (≤1.2 × 0.47 μm2; Fig. 2a and Supplementary Fig. 4; for details see Supplementary Notes 1 and 2). Our correlative FISH and transmission electron microscopy (TEM) analyses of five Trichoplax H2 individuals revealed that the two bacterial symbionts were always intracellular, spatially segregated and specific to one of the six host cell types (Fig. 2b and Supplementary Figs. 5−7). ‘G. incantans’ was observed only in fibre cells and was the only bacterium located in these cells (Fig. 2b and Supplementary Figs. 5 and 6). All ‘G. incantans’ cells were surrounded by a host membrane that was densely covered with ribosomes (Fig. 2c,d and Supplementary Fig. 6; n = 49 symbiont cells in 9 specimens). Similar host structures surrounding the bacteria in other Trichoplax lineages were interpreted as indicating that the bacteria reside inside the host’s rER3. An alternative interpretation for such host membrane structures was shown for the human intracellular pathogens Brucella and Legionella, as well as the amoebal midichloriacean parasite Candidatus ‘Jidaibacter’. These bacteria remodel the phagosome surfaces of their hosts so that they become covered by host ribosomes as an effective strategy for avoiding digestion by their hosts21,30.

a, A false-coloured FISH image using probes specific for ‘G. incantans’ (GRIN-61-2, Atto-647) and ‘R. eludens’ (RUEL-846-22, Atto 594); host nuclei are stained with 4,6-diamidino-2-phenylindole (DAPI). The results are representative of five independent experiments. b, A TEM image of a cross-section of Trichoplax H2 with ‘G. incantans’ and ‘R. eludens’ indicated in false colour (for raw image data see Supplementary Fig. 5). c,d, TEM images of fibre cells. ‘G. incantans’ is indicated with white arrows and the rER with white arrowheads. e,f, TEM images of ventral epithelial cells containing ‘R. eludens’. Outer membrane vesicles are indicated with black arrowheads, fimbriae-like structures with black arrows and internal structures by a white asterisk. Results in b−f are representative of three independent experiments.
To resolve the subcellular architecture of ‘G. incantans’ symbiosis, we used high-resolution 3D TEM tomography to determine whether the structures surrounding the symbiont cells were remodelled phagosomes or rER. Our 3D electron tomographic reconstructions revealed that the ribosome-covered membranes, in which ‘G. incantans’ occurred, formed networks that were connected to the nuclear envelope31. This indicates that the structure in which ‘G. incantans’ is embedded is in fact rER. ‘G. incantans’ symbionts were only observed in the rER, some even within the same rER lumen, and never in other host structures (Fig. 3; Supplementary Fig. 8; Supplementary Video 1). These analyses suggest that ‘G. incantans’ persistently resides in the rER of its host. The second symbiont, ‘R. eludens’, colonized only the ventral epithelial cells. These symbionts were always found within cytoplasmic vacuoles of the host (Fig. 2e,f). The vacuoles contained numerous membrane-bound vesicles, presumably outer membrane vesicles produced by ‘R. eludens’ (Supplementary Fig. 7). Thin, tubular structures that resemble fimbriae appeared to connect the bacterial cells to the host vacuole membrane (Fig. 2f; Supplementary Fig. 7).

All panels are the results from a single experiment. a−c, 3D volume rendering of reconstructed ‘G. incantans’, the rER and the nucleus of a fibre cell, superimposed on a virtual slice of the 3D TEM tomography stack; the rER has been virtually removed to partially (b) and fully (c) show the symbionts within the rER lumen. No scale bar is shown as the scale varies with perspective. d−i, Selected tomography slices (on which the 3D reconstruction is based) at various depths (see z values indicated) show the connection between the nucleus, the rER and the bacteria. For ease of interpretation, d has been false-coloured using the colour key from a. For raw data, see Supplementary Fig. 8.
Bacteria that live inside animal cells are known from only 6 of the 114 recognized bacterial phyla32. The number of bacterial phyla with representatives that can live as intracellular symbionts has not increased since the characterization of Mycoplasmatales in the early 1960s, despite huge advances in the sequencing of animals from a wide range of phyla and environments that have led to the discovery of numerous lineages of microbiota11,32. Marinamargulisbacteria is one of the most phylogenetically remote clades of bacteria, discovered through high-throughput sequencing of environmental samples33. The remote position of the placozoans in the animal tree of life has probably contributed to this late discovery of Margulisbacteria as the seventh bacterial phylum with intracellular symbionts of animals. Our study of the Trichoplax microbiome highlights how bacteria captured by eukaryotes provide a route for studying bacterial groups that are otherwise known only from sequences found in water or sediment samples.
‘R. eludens’ gains nutrition from lipids degraded by its host. We sequenced the metatranscriptomes of the same single placozoan individuals that were used for metagenomic analyses (n = 3) and generated metaproteomes from pooled samples of 10 to 30 individuals (n = 3) to investigate the physiology of ‘R. eludens’. Physiological modelling of these expression data revealed that ‘R. eludens’ is an aerobic chemoorganoheterotroph, with a complete tricarboxylic acid (TCA) cycle that generates energy and biomass from glycerol and the β-oxidation of fatty acids (Fig. 4a; Supplementary Table 3). The source of the glycerol and fatty acids is probably lipids from the algal diet of the host. Our analyses of the host’s transcriptome revealed that Trichoplax H2 expressed several lipases, most probably for the digestion of the algae it feeds on (Supplementary Table 4). These host lipases hydrolyze lipids to glycerol and fatty acids. The genome of ‘R. eludens’ also encodes lipases that would allow ‘R. eludens’ to digest lipids independently of its host. Interestingly, we found neither transcripts nor peptides for these symbiont lipases, suggesting that ‘R. eludens’ relies on the lipases expressed by its host (Supplementary Table 3).

Physiological reconstructions based on RAST annotations and Pathway Tools metabolic modelling. Functions that are discussed in the text and highly expressed are indicated in red. a, ‘R. eludens’. b, ‘G. incantans’. Bold font indicates primary function. ABC, ATP-binding cassette; AdoMet, S-adenosyl-l-methionine; MFS, major facilitator superfamily; nt, nucleotide; nt-ACTUI, the nucleotides a cell can import (all but guanine); P, phosphate; PEP, phosphoenolpyruvate; T4SS, type IV secretion system; TRAP, tripartite ATP-independent periplasmic.
The transfer of glycerol and even-chain fatty acids from the host to ‘R. eludens’ probably occurs passively, as they can easily diffuse through cell membranes. We predict that the fatty acids are taken up and activated by ‘R. eludens’ on the basis of its high expression of a long-chain fatty acid coenzyme A (CoA) ligase (among the top 25% of expressed genes; Fig. 4a; Supplementary Table 3). The fatty acids are then probably catabolized to acetyl-CoA and respired, as indicated by the expression of all the genes needed for β-oxidation and the oxidative TCA cycle. However, the anabolic incorporation of fatty acids is unlikely, as we could not detect the genes for the glyoxylate shunt.
‘R. eludens’ encoded genes for synthesizing all nucleotides and amino acids, including the nine amino acids considered essential for animals. However, we found no genomic or transcriptomic indications that ‘R. eludens’ exports nutrients to its host, for example via amino acid exporters (see Fig. 4a and Supplementary Note 3). Moreover, in our TEM analyses, we found no evidence for the intracellular, lysosomal digestion of ‘R. eludens’, such as lamellar bodies or tertiary lysosomes commonly observed in other nutritional symbioses34,35. Our ultrastructural analyses did, however, reveal large numbers of putative outer membrane vesicles in the host vacuole surrounding ‘R. eludens’ (Fig. 2e,f and Supplementary Fig. 7). It is tempting to speculate that the host takes up outer membrane vesicles produced by ‘R. eludens’ via phagocytosis and thus supplements its diet, as the host lacks synthesis pathways for essential amino acids. However, the beneficial effects of such putative amino acid provisioning by ‘R. eludens’ are not clear, given that the animal’s algal diet may contain sufficient amounts of essential amino acids.
‘G. incantans’ has the genes for energy parasitism but does not express them: it lives in the rER of fibre cells and seems to be a typical Rickettsiales based on genomic features alone, namely a heterotroph that relies on its host for biomass and energy generation (Fig. 4b). The ‘G. incantans’ genome encodes the hallmark feature for intracellular energy parasites that is present in all Rickettsiales genomes: a fully functional ADP/ATP-translocase for importing ATP from its host9. In contrast to all other known energy parasites, we found no transcripts or respective peptides of the ADP/ATP-translocase in ‘G. incantans’ (Supplementary Table 5). Instead, ‘G. incantans’ generated ATP with an ATP synthase, and the subunits a and b were highly expressed in the bacterium’s proteome (Supplementary Table 6). Compared to the typical energy-parasitic lifestyle of cytosolic Rickettsiales that rely on ATP imported from their hosts10, the ability of ‘G. incantans’ to synthesize ATP by itself likely lowers its detrimental impact on its host considerably.
High transcription of key genes of the oxidative TCA cycle and the presence of a complete electron transport chain in the genome, with some of the subunits of the electron transport chain among the most highly transcribed genes, suggests that the proton gradient for ATP synthesis is fuelled by oxidative phosphorylation (Fig. 4b and Supplementary Table 5). An incomplete glycolysis pathway and several importers for α-ketoacids and C4-dicarboxylates suggest that the metabolites respired in the TCA cycle are imported from the host (Fig. 4b).
The genome and transcriptome of ‘G. incantans’ revealed a strong host dependence on both amino acid and nucleotide supply (Fig. 4b; see Supplementary Note 4 for details). In contrast, the transcription profile of ‘G. incantans’ suggested that it could supply its host with riboflavin (vitamin B2), an essential vitamin that cannot be synthesized by most metazoans. Our analyses of the transcriptomic data of Trichoplax H2, as well as the genome and proteome of the closely related haplotype H124,36, revealed that both seem to lack the known genes for synthesizing riboflavin (Supplementary Fig. 9) and rely on an external source of riboflavin (Supplementary Table 4). This suggests that when riboflavin availability is limiting for the host, ‘G. incantans’ could supplement the nutrition of its host.
‘G. incantans’ does not seem to be detrimental to Trichoplax H2, despite the fact that it has to import most of the compounds it needs for generating energy and biomass from its host. Our metagenomic, FISH and TEM data revealed 2−20 symbiont cells per fibre cell, so that the total number of ‘G. incantans’ cells per host individual is roughly the same as the number of host cells (Supplementary Note 5). This indicates closely regulated control of symbiont growth by the symbiont, the host or both partners. Pathogen abundances are typically orders of magnitude higher per host cell and often result in rapid exploitation and destruction of host cells and the impairment of host reproduction37. The relatively low abundance of ‘G. incantans’ in Trichoplax H2 together with the rapid doubling rates of these hosts (2−3 d in our aquaria) are in contrast to virulent pathogenic infections. Unlike all other known energy parasites, ‘G. incantans’ seems to generate its own ATP and might even modulate its host immune response to prevent apoptosis (Supplementary Note 4).
Bacterial phylotypes highly similar or identical to ‘G. incantans’ occur worldwide in aquatic environments. To assess how widespread the two Trichoplax symbionts are in other environments and hosts, we surveyed the ~300,000 publicly available amplicon-based 16S rRNA sequence libraries using the IMNGS pipeline. We did not find any sequences related to ‘R. eludens’, using a cut-off of 99% identity. In contrast, sequences highly similar or identical to ‘G. incantans’ were present in aquatic environments, both marine and limnic, from across the globe (Supplementary Table 7). Of the 8,026 libraries from aquatic environments, we found sequences that were at least 99% identical to ‘G. incantans’ in almost 10% of these libraries (n = 845). One third of the sequences were identical to ‘G. incantans’ and almost all were attributed to the genus Grellia on the basis of evolutionary placement analysis (Supplementary Fig. 10). This is remarkable for Midichloriaceae, because all other genera are much rarer and were found in only 0–55 libraries, depending on the genus (Supplementary Table 7). The presence of Grellia phylotypes in such a wide range of environments, including limnic ones, indicates that these bacteria have host ranges beyond placozoans. Indeed, our phylogenetic 16S rRNA analyses showed that sequences that group with the genus Grellia have been found in marine protists (Eutreptiella), sea cucumbers (Apostichopus) and oysters (Crassostrea), as well as in the limnic cnidarian Hydra oligactis (see Fig. 1a). The Hydra sequences came from specimens collected freshly from their natural environments and animals reared in the laboratory for more than 30 yrs, indicating the stability of this association in these hosts38.
The recent realization that human pathogens such as Chlamydiae, Legionellales and Rickettsiales have close relatives that live in hosts ranging from protists to fish and from aquatic and soil habitats has led to a paradigm shift in our view of the ecology and evolution of intracellular bacteria27,39,40. ‘G. incantans’ extends our conceptual understanding of the pervasiveness of such bacteria and shows that a single environmental rickettsial genus occurs worldwide in marine and limnic habitats. This remarkable distribution raises the question of whether all Grellia are host-associated. If ‘G. incantans’ had a free-living stage, this would be in contrast to all other known Rickettsiales that infect animals27.
Unlike other animals at the base of the animal tree, such as sponges, cnidarians or ctenophores, Placozoa is the only phylum in which intracellular bacteria have been observed in all individuals and haplotypes investigated. Intracellular symbiosis thus seems to be an invariant trait across this phylum. Our study identifies these bacteria in Trichoplax H2, shows that they are found in every specimen examined and defines the specificity and fidelity to the host cell type in which the symbionts reside.
How might the Trichoplax symbionts be transmitted within a growing individual and to its offspring? Within a host individual, the symbiont-containing cells could pass on their bacteria during division, or the symbionts could continuously reinfect host cells derived from aposymbiotic cells. However, little is known about cell turnover and proliferation in placozoans and it remains to be determined whether they even have stem cells. Similarly, we can only speculate on transmission during asexual reproduction (the main mode of reproduction in placozoans). In Trichoplax H2, which has been reproducing asexually in our aquaria for several years, the symbionts are transmitted with high fidelity, as all host individuals had both symbionts. Information on sexual reproduction, which is much rarer and has not been observed in nature, is too limited to allow us to know whether the symbionts are incorporated into resulting embryos. If not, the symbionts must be obtained from the environment. Symbiont uptake from the environment could explain why the midichloriacean symbionts of Trichoplax H1 and H2 do not belong to the same clade, although their hosts are very closely related and separated only a few decades ago24. This split could have been caused by their midichloriacean symbionts, as Rickettsiales are well known to induce reproductive incompatibility in insects41. Future studies of the microbiomes of the large number of extant haplotypes are needed to fully understand the ecology and evolution of symbioses between placozoans and their bacterial symbionts.
Methods
Isolation and cultivation
The placozoans were isolated from a coral tank at the Kewalo Marine Laboratory, University of Hawai’i at Mānoa in October 2015 by placing glass slides, mounted in plastic slide boxes that had the top and bottom cut out, into the tank for 10 d (ref. 17). Placozoans were identified under a dissection microscope, transferred to 400 ml glass beakers with 34.5‰ artificial seawater and fed weekly with 2 × 106 cells ml−1 of Isochrysis galbana from a log-phase culture. Doubling times were 2−3 d at 25 °C in 34.5‰ artificial seawater and with a 16:8 h light:dark regime.
Nucleic acid extractions
DNA was extracted from two single individuals from the Trichoplax H2 cultures using the DNeasy Blood & Tissue Kit (Qiagen) and DNA and RNA from three additional single individuals were extracted using the AllPrep DNA/RNA Micro Kit (Qiagen), according to the manufacturer’s protocols for both kits except for the following modifications. Proteinase K digests were performed overnight. Elution volumes were halved and all samples were eluted twice, reusing the first eluate. Elutions were carried out with a 10-min-long waiting step before centrifugation.
DNA and RNA sequencing
Illumina-library preparation and sequencing were performed by the Max Planck Genome Centre. In brief, DNA/RNA quality was assessed with the Agilent 2100 Bioanalyzer (Agilent) and genomic DNA was fragmented to an average fragment size of 500 base pairs (bp). For the DNA samples, the concentration was increased (MinElute PCR Purification Kit; Qiagen) and an Illumina-compatible library was prepared using the Ovation Ultralow Library Systems Kit (NuGEN) according to the manufacturer’s protocol. For the RNA samples, the Ovation RNA-seq System V2 (NuGen) was used to synthesize complementary DNA and sequencing libraries were then generated with the DNA Library Prep Kit for Illumina (BioLABS). All libraries were size selected by agarose gel electrophoresis and the recovered fragments quality-assessed and quantified by fluorometry. For each DNA library, 14–22 million 150 bp paired-end reads were sequenced on a HiSeq 4000 (Illumina) and, for the RNA libraries, 150 bp single-end reads were sequenced to a depth of 42–44 million.
Host mitochondrial 16S rRNA gene phylogenetic analyses
The metagenomic assembly was screened for the contig containing the host mitochondrial 16S rRNA gene (m16S) using BLAST v2.7.1 as implemented in Geneious R1142. The gene was extracted from the contig and aligned together with a database of publicly available m16S sequences using MAFFT v7.394 in G-Insi mode. The phylogenetic tree was reconstructed using FastTree v2.1.5 (ref. 43) with a GTR model, 20 rate categories and Gamma20 likelihood optimization, generating approximate likelihood-ratio-test values for node support. The tree was drawn with Geneious42. The tree was rooted with clade A placozoans2.
Bacterial diversity 16S rRNA gene phylogenetic analyses
For the 16S rRNA gene database of all phylotypes recovered, the phyloFlash v3.0 beta1 pipeline (https://github.com/HRGV/phyloFlash) assembled full-length SSU genes for all samples. The dataset was aligned and phylogenetic trees were calculated and visualized as for the host m16S dataset above. The tree was rooted with the Eukarya and only the bacterial part of the tree is shown in this Letter.
Genome analyses
Full-length 16S rRNA gene sequences were reconstructed for each metagenomic and metatranscriptomic library using phyloFlash v3.0 beta1 (https://github.com/HRGV/phyloFlash) from raw reads.
For assembly, adapters and low-quality reads were removed with bbduk v37.9 (https://sourceforge.net/projects/bbmap/) with a minimum quality value of 2 and a minimum length of 36; single reads were excluded from the analysis. Each library was error corrected using BayesHammer v3.6244. A combined assembly of all the libraries was performed using SPAdes 3.62 (ref. 45) with standard parameters and k-mers 21, 33, 55, 77 and 99.
The reads of each library were mapped back to the assembled scaffolds using bbmap v37.9 (https://sourceforge.net/projects/bbmap/) with the option fast = t. Scaffolds were binned on the basis of the mapped read data using MetaBAT v1.046. The binning was refined using Bandage v0.8.147 by collecting all contigs linked to the contig that contained the full-length 16S rRNA gene of the target organism. The bin quality metrics were computed with QUAST v5.0.248 and the completeness for all bins was estimated using checkM v1.07 (ref. 49).
Annotation of the symbiont draft genomes was performed using RAST50 and verified with PSI-BLAST v2.7.151 for selected genes discussed. Average nucleotide and amino acid identities between genomes52,53 were calculated with the ANI/AAI matrix calculator (http://enve-omics.ce.gatech.edu/g-matrix). Comparative analyses were conducted using the PATRIC database and services54. Pathway Tools v22.055, in combination with the BioCyc database56, was used to analyse the metabolic capacities of ‘G. incantans’ and ‘R. eludens’. The genomes were screened for secretion systems and effectors using EffectiveDB57.
Transcriptomic analyses
Adapters and rRNA gene reads were removed from the RNA-seq reads using bbduk v37.9. The gene expression for each symbiont genome bin and of the host (based on the published predicted proteome of T. adhaerens H1) was calculated from RNA-seq libraries using kallisto v0.45.0 with default settings58. Transcription levels were mapped onto metabolic pathways using Pathway Tools v22.055.
Proteomic analyses
Peptide samples for proteomics were prepared and quantified from two samples of 10 Trichoplax each and one sample of 30 Trichoplax specimens, as described by Kleiner et al.59 and according to the filter-aided sample preparation protocol described by Wisniewski et al.60. In addition to minor modifications described in Hamann et al.61, we did not clear the lysate by centrifugation after boiling the sample in lysis buffer. Instead, as the sample size was extremely limited (10 Trichoplax specimens = 0.2 µl), we loaded the whole lysate onto the filter units used for the filter-aided sample preparation procedure. Centrifugation times before column washes with 100 μl UA (8 M urea in 0.1 M Tris/HCl pH 8.5) were halved as compared to Hamann et al.61. Peptides were not desalted. Peptide concentrations were determined with the Pierce Micro BCA assay (Thermo Fisher Scientific) following the manufacturer’s instructions.
All samples were analysed by one-dimensional LC−MS/MS as described in Kleiner et al.59 with the modification that a 75 cm analytical column was used. Briefly, the sample containing 30 specimens was measured in technical replicate, for the others the whole sample was used in one analysis. The peptide (0.8–3 μg) was loaded with an UltiMate 3000 RSLCnano Liquid Chromatograph (Thermo Fisher Scientific) in loading solvent A (2% acetonitrile, 0.05% trifluoroacetic acid) onto a 5 mm × 300 µm ID C18 Acclaim PepMap100 pre-column (Thermo Fisher Scientific). Elution and separation of peptides on the analytical column (75 cm × 75 µm analytical EASY-Spray column packed with PepMap RSLC C18, 2 µm material, Thermo Fisher Scientific; heated to 60 °C) was performed at a flow rate of 225 nl min−1 using a 460 min gradient going from 98% buffer A (0.1% formic acid) to 31% buffer B (0.1% formic acid, 80% acetonitrile) in 363 min, then to 50% B in 70 min, to 99% B in 1 min and ending with 99% B. The analytical column was connected to a Q Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific) via an Easy-Spray source. Eluting peptides were ionized via electrospray ionization. Carry-over was reduced by two wash runs (injection of 20 µl acetonitrile, 99% eluent B) between samples. Data acquisition in the Q Exactive Plus was performed as in Petersen et al.62.
A database containing protein sequences from the Trichoplax host as well as the two symbionts was used. Sequences of common laboratory contaminants were included by appending the cRAP protein sequence database (http://www.thegpm.org/crap/). The final database contained 13,801 protein sequences. Searches of the MS/MS spectra against this database were performed with the Sequest HT node in Proteome Discoverer v2.2.0.388 (Thermo Fisher Scientific) as in Petersen et al.62. For protein quantification, normalized spectral abundance factors63 were calculated per species and multiplied by 100, to give the relative protein abundance as a percentage.
Phylogenetic and phylogenomic analyses
A 16S rRNA gene database for ‘G. incantans’ was constructed using the assembled 16S rRNA gene sequence from each metagenomic library, the 20 best BLAST64 hits in the nr database and all other sequences of described Candidatus taxa in the Midichloriaceae. We added the five type strains with the best BLAST hit score (five species of Rickettsia) as an outgroup. We also screened the trace reads from the Trichoplax H1 genome project for reads containing Midichloriaceae 16S rRNA gene fragments using BLAST v2.7.164, assembled them in Geneious R9 (http://www.geneious.com)42 and added the resulting sequence to the database. A similar search for margulisbacterial 16S rRNA fragments yielded no hits.
The 16S rRNA gene dataset was aligned using MAFFT v7.39465 and the phylogenetic tree was calculated using FastTree v2.1.1043 with a GTR model for nucleotide substitution. The tree was drawn with Geneious R942.
For ‘G. incantans’, the database of genomes for phylogenetic analysis was compiled from all available genomes from the Midichloriaceae as well as representatives for all genera of the Anaplasmataceae and Rickettsiaceae. We also screened the assembly of the Trichoplax H1 genome project for contigs that belong to Midichloriaceae contamination using BLAST v2.7.164 with the ‘G. incantans’ genome as implemented in Geneious R9 (http://www.geneious.com)42. The identified set of contigs corresponded to the set found by Driscoll et al.25 and was added to the database. We similarly searched for sequences related to ‘R. eludens’ in the H1 genome project, but no hits were detected.
For genome-based alignments of the amino acids of 43 conserved phylogenetic marker genes, the tree workflow as implemented in CheckM v1.0.11 was used49. For Ruthmannia, the genome bin data were integrated into a taxonomically selected part of the alignment from Hug et al.11 that covered all Melainabacteria and Cyanobacteria, WOR-1 and RBX-1 (Margulisbacteria), as well as five short branching Firmicutes as an outgroup. The phylogenetic reconstructions of the concatenated alignments were calculated using FastTree v2.1.10 with the WAG model for amino acid substitutions and visualized and analysed using iTOL66.
Tag-sequence data analysis
The 16S rRNA gene sequences from ‘G. incantans’, as well as representative sequences from all characterized midichloriacean Candidatus taxa were used as query sequences to search the global collection of the microbial tag-sequencing library. The search was carried out using the IMNGS service67 with a minimal alignment length of 200 bp and a minimal identity of 99%. Identified amplicon libraries were grouped according to their deposited metadata. For the top 10% of libraries with the highest number of sequences from ‘G. incantans’, the habitat type (limnic or marine) and geolocation were manually collected from the deposited metadata and related publications. The detected 16S rRNA reads were aligned to the Rickettsiales dataset using MAFFT—addfragments and the evolutionary placements in the tree were performed using raxml v8.2.1268.
TEM
Live specimens were high-pressure frozen with a HPM 100 (Leica Microsystem) in 3 mm aluminium sample holders, using hexane as filler as needed. The samples were transferred onto frozen acetone containing 1% osmium tetroxide and processed using a very quick freeze-substitution method69. After reaching room temperature, the samples were washed three times with acetone and infiltrated using centrifugation, modified after McDonald70, in 2 ml tubes sequentially with 25%, 50%, 75% and 2 × 100% Agar Low Viscosity Resin (Agar Scientific). For this process, the samples were placed on top of the resin and centrifuged for 30 s with a benchtop centrifuge (Heathrow Scientific) at 2,000g for each step. After the second pure resin step, they were transferred into fresh resin in embedding moulds and polymerized at 60 °C for 12 h.
Ultrathin (70 nm) sections were cut with an Ultracut UC7 (Leica Microsystem) and mounted on formvar-coated slot grids (Agar Scientific). They were contrasted with 0.5% aqueous uranyl acetate (Science Services) for 20 min and with 2% Reynold’s lead citrate for 6 min before imaging them at 20–30 kV with a Quanta FEG 250 transmission electron microscope (FEI Company) equipped with a scanning TEM detector using the xT microscope control software v6.2.6.3123.
For electron tomography, 300 nm serial sections were placed on formvar-coated 2 × 1 mm2 slot grids and stained with uranyl acetate and lead citrate. 30 nm gold fiducials were applied on both sides of the slot grid. Dual-axis tilt series (±60°, step size 1°) were acquired with a FEI Tecnai F30 300 kV electron microscope equipped with an Axial Gatan US1000 CCD camera. SerialEM software was used for the automated acquisition of tomographic tilt series71. Alignment and reconstruction of the tilt series were carried out with IMOD v4.972. The serial tomograms were aligned with TrakEM2 v1.0i73 in Fiji74 and visualization and segmentation were carried out using the software Amira 3D v6.5.
FISH
We used ARB−SILVA database 128 (ref. 75) and the ARB PROBE_DESIGN tool (the ARB software package v6.0.6)76 to design two FISH probes for each symbiont that were specific to their 16S rRNA sequences (Supplementary Table 2). We confirmed the specificity of the probes by comparing their sequences to all available sequences in the ARB−SILVA 128 database and Ribosomal Database Project release 11.5 (ref. 77). The most specific probe for ‘R. eludens’ had two mismatches to first non-target hit sequences; the most specific probe for G. incantans also matched the six most closely related Grellia sequences; detailed results are presented in Supplementary Table 2.
Specimens were fixed on coverslips with 2% formaldehyde and 0.1% glutaraldehyde in 1.5× PIPES, HEPES, EGTA and MgCl2 (PHEM) buffers modified from Montanaro et al.78 at 4 °C for 12 h. After three washing steps in 1.5× PHEM buffer, the samples were stored in 70% ethanol until use. Samples were rehydrated in PBS and hybridization was performed according to Manz et al.79. Mono-labelled-, DOPE-80 or MIL-81 probes (Supplementary Table 2) at a concentration of 8.4 pmol µl−1 were diluted with hybridization buffer containing 35% formamide, 900 mM NaCl, 20 mM Tris/HCl and 0.01% SDS at a ratio of 15:1. Whole animals were incubated in 30 µl of the probe/hybridization buffer mix at 46 °C in 250 µl PCR tubes for 3−4 h, followed by a 30-min-long washing step in washing buffer containing 700 mM NaCl, 20 mM Tris/HCl, 5 mM EDTA and 0.1% SDS. After a 10-min-long washing step in PBS, the animals were stained with DAPI for 30 min, washed twice again in PBS and mounted on glass slides in Vectashield mounting medium.
To test the probes designed for this study, 30 clonal individuals of Trichoplax H2 were pooled, fixed as described above, homogenized by sonication and applied to a filter. The parts of the filter were then tested with different formamide concentrations and the optimal formamide concentration was determined.
Fluorescence images were taken with a Zeiss LSM 780 equipped with a GaAsP detector or an Airyscan detector and a Plan-Apochromat 63×/1.4 and a Plan-Apochromat 100×/1.46 oil immersion objective using the ZEN software (black edition, 64bits, v14.0.1.201; Carl Zeiss Microscopy GmbH).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The metagenomic and metatranscriptomic raw reads and assembled symbiont genomes are available in the European Nucleotide Archive under Study Accession Number PRJEB30343. The mass spectrometry metaproteomics data and protein sequence databases were deposited in the ProteomeXchange Consortium82 via the PRIDE partner repository with the dataset PXD012106. The TEM 3D reconstruction data were deposited in figshare; the aligned tomography slices used for the reconstruction shown in Fig. 4 are available at https://figshare.com/s/886b869a9ada0264ffb2 (ref. 31).
Code availability
The script used for the assembly graph-based binning is available at https://github.com/HRGV/tools_and_scripts.
References
Laumer, C. E. et al. Support for a clade of Placozoa and Cnidaria in genes with minimal compositional bias. eLife 7, e36278 (2018).
Eitel, M., Osigus, H. J., DeSalle, R. & Schierwater, B. Global diversity of the Placozoa. PloS ONE 8, e57131 (2013).
Grell, K. G. & Benwitz, G. Die Ultrastruktur von Trichoplax adhaerens F.E. Schulze. Cytobiologie 4, 216–240 (1971).
Eitel, M., Guidi, L., Hadrys, H., Balsamo, M. & Schierwater, B. New insights into placozoan sexual reproduction and development. PloS ONE 6, e19639 (2011).
Guidi, L., Eitel, M., Cesarini, E., Schierwater, B. & Balsamo, M. Ultrastructural analyses support different morphological lineages in the phylum Placozoa Grell, 1971. J. Morphol. 272, 371–378 (2011).
Eitel, M. et al. Comparative genomics and the nature of placozoan species. PLoS Biol. 16, e2005359 (2018).
Montagna, M. et al. “Candidatus Midichloriaceae” fam. nov. (Rickettsiales), an ecologically widespread clade of intracellular alphaproteobacteria. Appl. Environ. Microbiol. 79, 3241–3248 (2013).
Castelli, M., McCarthy, U., Petroni, G. & Bazzocchi, C. in Rickettsiales: Biology, Molecular Biology, Epidemiology, and Vaccine Development (ed. Thomas, S.) 283−292 (Springer, 2016).
Schmitz-Esser, S. et al. ATP/ADP translocases: a common feature of obligate intracellular amoebal symbionts related to Chlamydiae and Rickettsiae. J. Bacteriol. 186, 683–691 (2004).
Driscoll, T. P. et al. Wholly Rickettsia! Reconstructed metabolic profile of the quintessential bacterial parasite of eukaryotic cells. mBio 8, e00859-17 (2017).
Hug, L. A. et al. A new view of the tree of life. Nat. Microbiol. 1, 16048 (2016).
Anantharaman, K. et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 7, 13219 (2016).
Matheus Carnevali, P. B. et al. Hydrogen-based metabolism as an ancestral trait in lineages sibling to the Cyanobacteria. Nat. Commun. 10, 463 (2019).
Simion, P. et al. A large and consistent phylogenomic dataset supports sponges as the sister group to all other animals. Curr. Biol. 27, 958–967 (2017).
Srivastava, M. et al. The Trichoplax genome and the nature of placozoans. Nature 454, 955–960 (2008).
Sebé-Pedrós, A. et al. Early metazoan cell type diversity and the evolution of multicellular gene regulation. Nat. Ecol. Evol. 2, 1176–1188 (2018).
Pearse, V. B. & Voigt, O. Field biology of placozoans (Trichoplax): distribution, diversity, biotic interactions. Integr. Comp. Biol. 47, 677–692 (2007).
Smith, C. L., Pivovarova, N. & Reese, T. S. Coordinated feeding behavior in Trichoplax, an animal without synapses. PloS ONE 10, e0136098 (2015).
Grell, K. G. & Benwitz, G. Ergänzende Untersuchungen zur Ultrastruktur von Trichoplax adhaerens F.E. Schulze (Placozoa). Zoomorphology 98, 47–67 (1981).
Smith, C. L. et al. Novel cell types, neurosecretory cells, and body plan of the early-diverging metazoan Trichoplax adhaerens. Curr. Biol. 24, 1565–1572 (2014).
Martinez, E., Siadous, F. A. & Bonazzi, M. Tiny architects: biogenesis of intracellular replicative niches by bacterial pathogens. FEMS Microbiol. Rev. 42, 425–447 (2018).
Schulz, F. & Horn, M. Intranuclear bacteria: inside the cellular control center of eukaryotes. Trends Cell Biol. 25, 339–346 (2015).
Sassera, D. et al. ‘Candidatus Midichloria mitochondrii’, an endosymbiont of the tick Ixodes ricinus with a unique intramitochondrial lifestyle. Int. J. Syst. Evol. Bacteriol. 56, 2535–2540 (2006).
Kamm, K., Osigus, H. J., Stadler, P. F., DeSalle, R. & Schierwater, B. Trichoplax genomes reveal profound admixture and suggest stable wild populations without bisexual reproduction. Sci. Rep. 8, 11168 (2018).
Driscoll, T., Gillespie, J. J., Nordberg, E. K., Azad, A. F. & Sobral, B. W. Bacterial DNA sifted from the Trichoplax adhaerens (Animalia: Placozoa) genome project reveals a putative rickettsial endosymbiont. Genome Biol. Evol. 5, 621–645 (2013).
Bowers, R. M. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731 (2017).
Castelli, M., Sassera, D. & Petroni, G. In Rickettsiales: Biology, Molecular Biology, Epidemiology, and Vaccine Development (ed. Thomas, S.) 59−91 (Springer, 2016).
Senra, M. V. et al. A house for two—double bacterial infection in Euplotes woodruffi Sq1 (Ciliophora, Euplotia) sampled in southeastern Brazil. Microb. Ecol. 71, 505–517 (2016).
Soo, R. M., Hemp, J., Parks, D. H., Fischer, W. W. & Hugenholtz, P. On the origins of oxygenic photosynthesis and aerobic respiration in Cyanobacteria. Science 355, 1436–1440 (2017).
Schulz, F. et al. A Rickettsiales symbiont of amoebae with ancient features. Environ. Microbiol. 18, 2326–2342 (2016).
Leisch, N. Electron tomography dataset of a serial sectioned Trichoplax haplotype H2 fiber cell. Figshare https://figshare.com/s/886b869a9ada0264ffb2 (2018).
Parks, D. H. et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004 (2018).
Parks, D. H. et al. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542 (2017).
Bright, M. & Sorgo, A. Ultrastructural reinvestigation of the trophosome in adults of Riftia pachyptila (Annelida, Siboglinidae). Invertebr. Biol. 122, 347–368 (2003).
Fiala-Médioni, A., Michalski, J.-C., Jollès, J., Alonso, C. & Montreuil, J. Lysosomic and lysozyme activities in the gill of bivalves from deep hydrothermal vents. C. R. Acad. Sci. III 317, 239–244 (1994).
Ringrose, J. H. et al. Deep proteome profiling of Trichoplax adhaerens reveals remarkable features at the origin of metazoan multicellularity. Nat. Commun. 4, 1408 (2013).
Sedzicki, J. et al. 3D correlative electron microscopy reveals continuity of Brucella-containing vacuoles with the endoplasmic reticulum. J. Cell Sci. 131, jcs210799 (2018).
Fraune, S. & Bosch, T. C. G. Long-term maintenance of species-specific bacterial microbiota in the basal metazoan Hydra. Proc. Natl Acad. Sci. USA 104, 13146–13151 (2007).
Horn, M. et al. Illuminating the evolutionary history of Chlamydiae. Science 304, 728–730 (2004).
Duron, O., Doublet, P., Vavre, F. & Bouchon, D. The importance of revisiting Legionellales diversity. Trends Parasitol. 34, 1027–1037 (2018).
Werren, J. H., Baldo, L. & Clark, M. E. Wolbachia: master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751 (2008).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—approximately maximum-likelihood trees for large alignments. PloS ONE 5, e9490 (2010).
Nikolenko, S. I., Korobeynikov, A. I. & Alekseyev, M. A. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genom. 14, S7 (2013).
Bankevich, A. et al. SPAdes: a new genome assembler and its applications to single cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Kang, D. D., Froula, J., Egan, R. & Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3, e1165 (2015).
Wick, R. R., Schultz, M. B., Zobel, J. & Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352 (2015).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Aziz, R. K. et al. The RAST server: rapid annotations using subsystems technology. BMC Genom. 9, 75 (2008).
Johnson, M. et al. NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9 (2008).
Richter, M. & Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl Acad. Sci. USA 106, 19126–19131 (2009).
Goris, J. et al. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Bacteriol. 57, 81–91 (2007).
Wattam, A. R. et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 45, D535–D542 (2017).
Karp, P. D. et al. Pathway Tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief. Bioinform. 11, 40–79 (2010).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 44, D471–D480 (2016).
Eichinger, V. et al. EffectiveDB—updates and novel features for a better annotation of bacterial secreted proteins and Type III, IV, VI secretion systems. Nucleic Acids Res. 44, D669–D674 (2016).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Kleiner, M. et al. Assessing species biomass contributions in microbial communities via metaproteomics. Nat. Commun. 8, 1558 (2017).
Wiśniewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009).
Hamann, E. et al. Environmental Breviatea harbour mutualistic Arcobacter epibionts. Nature 534, 254–258 (2016).
Petersen, J. M. et al. Chemosynthetic symbionts of marine invertebrate animals are capable of nitrogen fixation. Nat. Microbiol. 2, 16195 (2016).
Zybailov, B. et al. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5, 2339–2347 (2006).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016).
Lagkouvardos, I. et al. IMNGS: a comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep. 6, 33721 (2016).
Berger, S. A., Krompass, D. & Stamatakis, A. Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum likelihood. Syst. Biol. 60, 291–302 (2011).
McDonald, K. L. & Webb, R. I. Freeze substitution in 3 hours or less. J. Microsc. 243, 227–233 (2011).
McDonald, K. L. Rapid embedding methods into epoxy and LR white resins for morphological and immunological analysis of cryofixed biological specimens. Microsc. Microanal. 20, 152–163 (2014).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Kremer, J. R., Mastronarde, D. N. & McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996).
Cardona, A. et al. TrakEM2 software for neural circuit reconstruction. PloS ONE 7, e38011 (2012).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Ludwig, W. et al. ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371 (2004).
Cole, J. R. et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642 (2014).
Montanaro, J., Gruber, D. & Leisch, N. Improved ultrastructure of marine invertebrates using non-toxic buffers. PeerJ 4, e1860 (2016).
Manz, W., Amann, R., Ludwig, W., Wagner, M. & Schleifer, K.-H. Phylogenetic oligodeoxynucleotide probes for the major subclasses of Proteobacteria: problems and solutions. Syst. Appl. Microbiol. 15, 593–600 (1992).
Stoecker, K., Dorninger, C., Daims, H. & Wagner, M. Double labeling of oligonucleotide probes for fluorescence in situ hybridization (DOPE−FISH) improves signal intensity and increases rRNA accessibility. Appl. Environ. Microbiol. 76, 922–926 (2010).
Schimak, M. P. et al. MiL−FISH: multilabeled oligonucleotides for fluorescence in situ hybridization improve visualization of bacterial cells. Appl. Environ. Microbiol. 82, 62–70 (2015).
Vizcaíno, J. A. et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 (2016).
Acknowledgements
This study was funded by the Max Planck Society, a Gordon and Betty Moore Foundation Marine Microbial Initiative Investigator Award (grant no. GBMF3811 to N.D.), grants to M.G.H. from the Gordon and Betty Moore Foundation (no. 5009) and the US Office of Naval Research (no. N00014-15-1-2658), the German Academic Exchange Service DAAD (T.H.) and the NC State Chancellor’s Faculty Excellence Program Cluster on Microbiomes and Complex Microbial Communities (M.K.). We thank M. Strous for access to proteomic equipment and A. Kouris for LC−MS/MS operation. The purchase of the proteomics equipment was supported by a grant from the Canadian Foundation for Innovation to M. Strous. We thank the Electron Microscopy Facility of the MPI-CBG, the Max Planck Genome Centre and the Electron Microscopy Facility at the MPI-PZ Cologne and the Cell Imaging and Ultrastructure Research Core Facility of the University of Vienna for technical support. We thank C. Peters for nucleic acid extractions, C. Peters, M. Meyer and W. Ruschmeier for support with Trichoplax cultivation, B. Nedved for support in the field and G. Bennett, T. Erb, L. Schada von Borzyskowski and P. A. Chakkiath for discussions on symbiont physiology.
Author information
Authors and Affiliations
Contributions
M.G.H., N.D., M.M.-N. and H.R.G.-V. conceived the study. H.R.G.-V. sampled and cultivated the organisms and performed the assemblies, genome and transcriptome analyses, tag-sequencing analyses and phylogenetic analyses. H.R.G.-V. reconstructed the symbiont physiology with the help of M.L. and M.K. M.K. and T.H. generated the proteomic data and H.R.G.-V. and M.K. analysed the proteomic data. N.L. performed the fluorescence microscopy, electron microscopy and electron tomography, subsequent data analysis and 3D reconstruction. H.R.G.-V. and N.L. wrote the manuscript with support from N.D., M.M.-N. and M.G.H. All authors revised the manuscript and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Notes 1−6, Supplementary Figs. 1−10, legend for Supplementary Video 1, Supplementary Tables 1 and 2, Supplementary Dataset 1 and Supplementary References.
Supplementary Table 3
‘R. eludens’ transcriptome.
Supplementary Table 4
Trichoplax H2 transcriptome analysis.
Supplementary Table 5
‘G. incantans’ transcriptome.
Supplementary Table 6
‘G. incantans’ proteome.
Supplementary Table 7
Grellia is the most widely distributed midichloracean genus. This table shows IMNGS results for the characterized genera of Midichloriacea. Environmental categories were filtered for categories with at least ten cumulative hits accross all taxa.
Supplementary Video 1
3D rendering of reconstructed ‘G. incantans’ in Trichoplax H2 fibre cell rER.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gruber-Vodicka, H.R., Leisch, N., Kleiner, M. et al. Two intracellular and cell type-specific bacterial symbionts in the placozoan Trichoplax H2. Nat Microbiol 4, 1465–1474 (2019). https://doi.org/10.1038/s41564-019-0475-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-019-0475-9
This article is cited by
-
Host association and intracellularity evolved multiple times independently in the Rickettsiales
Nature Communications (2024)
-
Single-cell Microbiomics Unveils Distribution and Patterns of Microbial Symbioses in the Natural Environment
Microbial Ecology (2023)
-
Microbiomes of microscopic marine invertebrates do not reveal signatures of phylosymbiosis
Nature Microbiology (2022)
-
Mixotrophic chemosynthesis in a deep-sea anemone from hydrothermal vents in the Pescadero Basin, Gulf of California
BMC Biology (2021)
-
Placozoan fiber cells: mediators of innate immunity and participants in wound healing
Scientific Reports (2021)
