
Abstract
The most widespread form of malaria is caused by Plasmodium vivax. To replicate, this parasite must invade immature red blood cells through a process requiring interaction of the P. vivax Duffy binding protein (PvDBP) with its human receptor, the Duffy antigen receptor for chemokines. Naturally acquired antibodies that inhibit this interaction associate with clinical immunity, suggesting PvDBP as a leading candidate for inclusion in a vaccine to prevent malaria due to P. vivax. Here, we isolated a panel of monoclonal antibodies from human volunteers immunized in a clinical vaccine trial of PvDBP. We screened their ability to prevent PvDBP from binding to the Duffy antigen receptor for chemokines, and their capacity to block red blood cell invasion by a transgenic Plasmodium knowlesi parasite genetically modified to express PvDBP and to prevent reticulocyte invasion by multiple clinical isolates of P. vivax. This identified a broadly neutralizing human monoclonal antibody that inhibited invasion of all tested strains of P. vivax. Finally, we determined the structure of a complex of this antibody bound to PvDBP, indicating the molecular basis for inhibition. These findings will guide future vaccine design strategies and open up possibilities for testing the prophylactic use of such an antibody.
Similar content being viewed by others
Main
In large parts of the world, Plasmodium vivax is the dominant species causing human malaria1,2. An effective blood-stage vaccine would reduce morbidity, lower blood-stage asexual and sexual parasite densities, and aid progress towards elimination. The symptoms of malaria occur as parasites invade, then replicate within and burst out of red blood cells (RBCs) of infected individuals. P. vivax parasites invade immature RBCs, selecting reticulocytes expressing the transferrin receptor CD71 (refs. 3,4). P. vivax Duffy binding protein (PvDBP) interacts with the Duffy antigen receptor for chemokines (DARC) on the surface of reticulocytes, and is essential for invasion by most strains5,6. Indeed, genetic knockout of the orthologous DBPα gene from the closely related simian malaria Plasmodium knowlesi also prevents invasion of Duffy-positive erythrocytes in vitro7, and antibodies that target PkDBP can block invasion of erythrocytes by P. knowlesi8. Moreover, a human polymorphism that results in Duffy-negative erythrocytes9 is widespread across Africa and is associated with protection from P. vivax infection10, leading to low levels of this form of malaria across much of the continent11. PvDBP is therefore the most promising candidate for inclusion in a vaccine to prevent P. vivax12.
PvDBP has been divided into six distinct regions, with the DARC-binding site mapped to a Duffy binding-like domain of ~350 amino acid residues known as Region II (PvDBPII)6. This domain is split into three regions, known as subdomains 1–3 (ref. 5). PvDBPII binds to the 60-residue extracellular amino (N)-terminal ectodomain of DARC in an interaction that requires DARC to be post-translationally modified by sulfation of 2 tyrosine residues, Tyr30 and Tyr41 (refs. 13,14). Nevertheless, the interaction between PvDBPII and DARC is only partially understood. Structural studies of PvDBPII show it to form dimers in both solution and a crystal13. In addition, structures of PvDBPII crystallized in the presence of the ectodomain of DARC reveal an ordered 12-residue helical peptide (DARC19–30) that lies close to the PvDBPII dimer interface in subdomain 2 (ref. 15). However, the DARC protein used in these studies was not tyrosine-sulfated, and sulfated Tyr41, which could not be seen in this study, was previously shown to be essential for high-affinity PvDBPII binding14. Indeed, mapping residues identified by mutagenesis onto the structure of PvDBPII reveals an additional patch on subdomain 2 proposed to contribute to DARC binding16,17, and it is likely that the DARC-binding site spans much of the surface of subdomain 2, including both this patch and the region that interacts with the helical DARC19–30 peptide.
The development of PvDBPII-based vaccine candidates has progressed through preclinical studies to two recent phase I human clinical trials18,19. Immunization of mice, rabbits and non-human primates using PvDBPII-based vaccines induces inhibitory antibodies that block the binding of recombinant PvDBPII to DARC20,21. Challenges in sustaining P. vivax in long-term culture22 have prevented these antibodies from being robustly tested in standardized functional growth inhibition assays of the type traditionally performed for vaccines targeting Plasmodium falciparum12. Nevertheless, in humans, high titres of naturally acquired antibodies that target PvDBPII and prevent DARC binding in vitro are associated with a reduced risk of P. vivax infection23, lower parasite densities following invasion, and a decreased risk of clinical malaria24,25. Encouragingly, in a recent phase Ia clinical trial, immunization of human volunteers using recombinant viral vectors expressing PvDBPII induced strain-transcending antibodies that prevented recombinant PvDBPII from binding to DARC19, confirming that such antibodies could be raised by human vaccination.
Several studies have investigated the molecular basis for antibody-mediated inhibition of DARC binding by PvDBPII13,15,26,27. Screening a linear peptide array with non-inhibitory and inhibitory human serum identified peptides that recognize antibodies found specifically in inhibitory serum27. These peptides are located within subdomain 2, in regions involved in PvDBPII dimerization and DARC19–30 peptide binding15, suggesting that antibodies that directly block dimerization and DARC binding are desirable. In addition, a study of monoclonal antibodies (mAbs) derived from PvDBPII-immunized mice used structural studies and mass-spectrometry-based mapping approaches to identify three epitopes for antibodies shown to block DARC binding in vitro26. Surprisingly, these epitopes are located on subdomain 3 of PvDBPII, which is distant from the DARC-binding site.
To understand the epitopes for human inhibitory antibodies, and to guide future structure-guided immunogen development, we cloned a panel of antibodies from PvDBPII-immunized human volunteers. We determined their ability to inhibit the binding of recombinant PvDBPII to DARC in vitro, as well as their capacity to neutralize parasite invasion in two distinct parasite-based functional assays. This provides important insight into these different in vitro readouts of antibody function, and led to the structural characterization of an epitope for an antibody that shows broadly neutralizing activity against parasite invasion.
Results
Cloning of a panel of vaccine-induced human mAbs that bind to PvDBPII
A panel of anti-PvDBPII mAbs was isolated from antibody-secreting cells of immunized volunteers enroled in a human phase Ia clinical trial of a PvDBPII-based vaccine delivered using recombinant chimpanzee adenovirus and poxvirus viral vectors19. Reverse-transcription PCR and PCR were used to isolate variable region-coding genes, and these were cloned into a human immunoglobulin G1 (IgG1) scaffold. Cognate heavy-chain and light-chain plasmids were co-expressed in HEK293 cells, and vaccine antigen-specificity was confirmed by enzyme-linked immunosorbent assay (ELISA) reactivity of culture supernatants to recombinant PvDBPII. Ten genetically distinct mAbs were isolated (Supplementary Table 1), which recognized parasite-derived PvDBP from culture supernatant (Supplementary Fig. 1). These were aligned with the most similar germline variable region-coding genes in IgBLAST28, showing them to contain only a small amount of non-germline sequence. Their monovalent binding affinities for PvDBPII, as determined by surface plasmon resonance (SPR; Fig. 1a–d and Supplementary Table 2), reveal low nanomolar to high picomolar affinities. Assessment of their binding to biotinylated PvDBPII by biolayer interferometry (BLI; Fig. 1e,f) defined two distinct epitope bins containing mAbs that bind overlapping epitopes, and three mAbs with distinct non-overlapping epitopes (Fig. 1g). An ELISA-based binding-inhibition assay29 was used to assess their inhibition of the binding of five sequence-divergent PvDBPII proteins19 to DARC (Fig. 2 and Supplementary Fig. 2). Most mAbs showed differential binding inhibition, with only two (DB2 and DB9) inhibiting the DARC binding of all five PvDBPII variant alleles (Fig. 2).

Kinetic rate constants of binding for ten human mAbs (DB1–10) to PvDBPII, as determined by SPR. a, Association rate constant (kon). b, Dissociation rate constant (koff). c, Association constant (1/Kd). d, Iso-affinity plot of kon against koff. e, A ‘relative binding’ matrix showing the fraction of mAb bound to PvDBPII in the presence of a second bound mAb. Assays were conducted in both orientations. Boxes are colour coded with ≥0.75 in blue, 0.75 > X > 0.05 in pink and ≤0.05 in red. Negative values were normalized to 0, and values >1 were normalized to 1. f, A ‘binding profile correlation’ matrix showing the Pearson product-moment correlation values of each mAb pair. The correlation threshold was set to 0.7; values equal to or above this are coloured in red, as the threshold chosen to represent competition. g, Epitope bins determined from e and f.

a, Assessment of the percentage binding of five naturally occurring variants of PvDBPII (SalI, P, O, AH and HMP013) to the DARC ectodomain in vitro in the presence of a 100 µg ml−1 concentration of each mAb (DB1–10). Individual titration curves are shown in Supplementary Fig. 2. ‘Anti-DBP’ is a polyclonal human anti-PvDBPII serum at a 1:5 dilution, while ‘Ebola’ is an anti-Ebolavirus recombinant human IgG1 mAb included as a negative control. Data points represent the means of three technical replicates. Error bars represent s.d. b, Sequence polymorphisms of the PvDBPII variants used in the assay. Numbering is according to the SalI reference sequence. Amino acid polymorphisms are indicated. Amino acids that are the same as the SalI reference sequence are black, while those divergent from SalI are red. Asterisks indicate the absence of a leucine insertion between V429 and P430 in HMP013.
Assessing the neutralization of parasite invasion by the antibody panel
To characterize the ability of the human mAbs to block merozoite entry into RBCs, assays of growth inhibition activity (GIA) were performed. We used a P. knowlesi line that had been adapted to long-term in vitro culture in human RBCs30 and in which the native PkDBPα gene had been replaced by the PvDBP gene31. P. knowlesi is dependent on DARC for RBC invasion, but unlike P. vivax, has three DBP genes: PkDBPα, PkDBPβ and PkDBPγ32. Both PkDBPβ and PkDBPγ are thought to be required for invasion of rhesus RBCs, whereas PkDBPα is essential for invasion of human RBCs7,33,34. The ten mAbs were assayed for growth inhibition against five P. knowlesi lines: the original human RBC-adapted PkA1-H.1 strain30; two transgenic PkA1-H.1 lines (PvDBPOR, in which the PkDBPα gene had been replaced with the PvDBP gene (this transgenic insert was the full-length PvDBP Salvador I (SalI) strain vaccine-homologous sequence), and PvDBPOR/Δβγ, in which the same gene swap had taken place and, additionally, the native PkDBPβ and PkDBPγ genes had been deleted (PvDBPOR/Δβγ was considered the best model for P. vivax, as the only DBP gene present is that for PvDBP); and two transgenic control lines, PkDBPαOR and PkDBPαOR/Δβγ, in which native PkDBP genes had been deleted and replaced with re-codonized versions of themselves.
One anti-PvDBPII mAb, DB7, at high concentration, inhibited the growth of the three P. knowlesi control lines (Fig. 3a), suggesting an epitope cross-reactive with PkDBPα. In contrast, when assayed against the transgenic PvDBP-expressing P. knowlesi lines, three mAbs showed high levels of inhibition of parasite growth, with four showing intermediate levels, and the remaining three showing modest activity. A control human mAb against Ebolavirus showed no detectable GIA (Fig. 3a). The three most neutralizing anti-PvDBPII mAbs (DB1, DB9 and DB10) had half-maximum effective concentration (EC50) values comparable to mouse-derived mAbs, which potently prevent erythrocyte invasion by P. falciparum35,36. A strong correlation (P = 0.002; Spearman’s rank correlation coefficient (ρ) = −0.951) was observed between the association rate (kon) and GIA (Fig. 3b), suggesting that the opportunity for PvDBP blockade in the context of merozoite invasion is time limited36,37. In contrast, no correlation was observed between GIA and either the dissociation rate or affinity (Fig. 3b).

a, Assays of GIA by the ten anti-PvDBPII human mAbs against five different P. knowlesi lines: ‘wild type’ (A1-H.1), PkDBPαOR, PkDBPαOR/Δβγ, PvDBPOR and PvDBPOR/Δβγ. Inhibition was tested in a twofold dilution series starting at 1 mg ml−1. Data points represent the means of three technical replicates. Error bars represent s.d. The EC50 values (interpolated by nonlinear regression) are shown in ascending rank order for the seven mAbs that reached >50% GIA against PvDBPOR/Δβγ at the maximum concentration (1 mg ml−1). conc., concentration. b, kon (on rate), koff (off rate) and Kd (dissociation constant), plotted against GIA EC30 for PvDBPOR/Δβγ. EC30 values were used to include weaker-neutralizing mAbs, and were interpolated from nonlinear regression curves. Kinetic data are as in Fig. 1 and Supplementary Table 2.
To assess the degree of strain transcendence of mAb inhibition across naturally occurring isolates, we next tested the mAbs directly for their capacity to prevent reticulocyte invasion by P. vivax parasites derived from 13 clinical isolates originating from Thai patients, using ex vivo, short-term culture invasion inhibition assays (Fig. 4a,b). Limited availability of these clinical samples prevented us from assessing the inhibitory potential of every antibody against every isolate. We were able to sequence the PvDBPII gene region from eight of these isolates, revealing multiple polymorphisms (Fig. 4c). Some isolates had PvDBPII sequences identical to the SalI reference strain used in the vaccine (isolates 4 and 7), while others were polymorphic, with as many as ten amino acid substitutions in Region II of the DBP gene (isolates 6 and 8).

a, P. vivax ex vivo invasion assays were performed with 13 separate isolates of infected blood from local patients. Each data point represents the percentage inhibition of each antibody against one of the 13 isolates. All antibodies were tested at a final concentration of 1 mg ml−1, except the positive-control camelid anti-DARC VHH (VHH), which was assayed at 25 μg ml−1. The red bars represent the median percentage inhibition for each antibody. A recombinant human IgG1 anti-Ebolavirus mAb (Ebola) was used as a negative control at 1 mg ml−1. b, Percentage invasion inhibition by mAbs tested against isolate 4 (diamonds) and isolate 7 (triangles), which share the PvDBPII gene sequence of SalI. This shows the degree of correlation with the hierarchy of mAb inhibition in the transgenic P. knowlesi GIA assays (Fig. 3a). c, Amino acid polymorphisms found within the PvDBPII gene segment of the eight Thai P. vivax isolates for which we obtained sequence information. The vaccine-homologous SalI reference sequence is shown in the bottom row. Amino acids that are the same as the reference sequence are shown in black, while divergent residues are shown in red. Asterisks represents the absence of a leucine insertion between V429 and P430 in the SalI reference sequence. d, Summary matrix showing the percentage inhibition of invasion by each mAb for each of the sequenced strains. The positive control was VHH at 25 μg ml−1, and the negative control was a recombinant human IgG1 anti-Ebolavirus mAb (Ebola) at a concentration of 1 mg ml−1.
These assays revealed marked strain-dependent differences in the potency of the anti-PvDBPII mAbs. For example, DB1 and DB10—the two most inhibitory mAbs in the transgenic P. knowlesi GIA assay (containing the SalI PvDBP transgene)—inhibited invasion of Thai isolates with matching SalI PvDBPII sequences (isolates 4 and 7), but not those with heterologous PvDBPII sequences (isolates 5 and 8) (Fig. 4b,d). Notably, the hierarchy of mAb potency for the two P. vivax parasite isolates that possessed the homologous SalI PvDBPII sequence mirrored that observed in the transgenic P. knowlesi model (Fig. 4b), suggesting that this model is highly predictive of P. vivax neutralization. DB3, DB35, DB36 and DB37 showed intermediate median levels of inhibition (~40–60%), while DB2, DB34 and DB38 showed low median levels (~10–20%). Only one of the ten mAbs (DB9) potently inhibited the invasion (~65–90%) of 10 out of 11 isolates. The inhibition of invasion by isolate 12 was lower, but a lack of sequence information, due to the challenges of working with small quantities of samples from field isolates, makes it challenging to speculate on the reasons for this. DB9 also showed potent growth inhibition in the transgenic P. knowlesi assays of GIA (Fig. 3a) and inhibited the binding of all five variant alleles of PvDBPII to DARC (Fig. 2), highlighting it as an antibody with broadly neutralizing activity against a wide range of P. vivax variants.
Antagonism of DB9-mediated inhibitory activity
Next, we sought to assess whether the efficacy of DB9 would be enhanced or diminished by the other nine mAbs. A BLI binding-competition assay (Fig. 1e–g) showed that half of the mAbs, including DB9, compete with each other for binding sites on recombinant PvDBPII. To test for evidence of synergy, additivity or antagonism38, we ran GIA assays using the PvDBPOR line of transgenic P. knowlesi in which DB9 was present at 25 μg ml−1, together with a dilution series of a second mAb (Fig. 5). While no synergy was detected, there was antagonism between DB9 and five other mAbs, with the addition of a second mAb leading to growth inhibition lower than that predicted from adding the inhibition due to the individual mAbs at equivalent concentrations. Surprisingly, these were the five mAbs (DB1, DB4, DB5, DB7 and DB10) that did not compete with DB9 for binding to PvDBPII in the BLI competition assay (Fig. 1e–g). The other four mAbs (DB2, DB3, DB6 and DB8) with epitopes overlapping that of DB9 showed an additive growth inhibitory effect with DB9 in this assay. Therefore, antibodies that bind in and around the DB9 epitope function in an independent and additive manner, while antibodies that bind to distinct epitope regions elsewhere on the PvDBPII molecule are mutually antagonistic. Given that DB9 was able to show strain-transcending, high-level neutralizing activity, this raised the imperative to understand the epitope of DB9 and thus enable the future design of immunogens that specifically induce DB9-like antibodies.

Assays of GIA were performed to assess the inhibitory activity of DB9 in combination with the other mAbs against the PvDBPOR transgenic P. knowlesi line. DB9 was held at a fixed concentration of 25 μg ml−1, while the other mAbs were in a twofold dilution series starting at 100 μg ml−1. The black line shows the percentage inhibition of each mAb in the absence of DB9. The red line shows the predicted additive inhibition (‘Bliss additivity’, as calculated using the second equation in the Methods section ‘In vitro assay of GIA’) of the indicated mAb plus DB9 at 25 μg ml−1. The blue line shows the actual observed percentage inhibition of the two combined mAbs. The dotted black line gives the percentage inhibition of DB9 alone at 25 μg ml−1. Data points represent the means of three technical replicates. Error bars represent s.d.
Molecular basis for the action of DB9
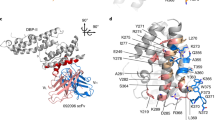
To obtain structural insight, we crystallized a complex of PvDBPII bound to the Fab fragment of DB9. Crystals diffracted to 3.0 Å resolution, allowing structure determination by molecular replacement (Fig. 6 and Supplementary Table 3). Comparison of the structure of PvDBPII in complex with DB9 with that of unliganded PvDBPII (Protein Data Bank (PDB) ID: 4NUV) revealed no change in PvDBPII conformation (with a root-mean-square deviation of 0.523 Å).

a, Structure of PvDBPII (pink) bound to the Fab fragment of DB9 (blue). PvDBPII is shown in two shades of pink, with subdomains 1 and 2 in light pink and subdomain 3 in dark pink. DB9 is shown in two shades of blue, with the light chain in light blue and the heavy chain in dark blue. b, PvDBPII is shown in surface representation in grey, with residues known to be polymorphic highlighted according to their sequence entropy (yellow = 0.15–0.30; orange = 0.30–0.45; red > 0.45). DB9 is shown in blue, and binds to a conserved region of PvDBPII. c, Structure of the PvDBPII–DB9 complex superimposed on the structure of the PvDBPII dimer bound to a peptide from the DARC ectodomain, indicating that DB9 may prevent the binding of PvDBP to DARC in the context of the reticulocyte membrane.
The epitope of DB9 is contained within subdomain 3 of PvDBPII (Fig. 6a). This region consists of two long α-helices (H1 and H2), which form a platform on which three small helices (H3–5) and their intervening loops are arranged (Supplementary Fig. 3). All three complementarity-determining region (CDR) loops of the heavy chain, and CDR1 and CDR2 of the light chain, directly contact PvDBPII (Supplementary Fig. 3 and Supplementary Table 4). The epitope includes a single residue from H1 (K412) of PvDBPII, and residues from each of helices H3, H4 and H5, and the intervening loops. Contacts are predominantly mediated by hydrogen bonds between DB9 and hydrophilic residues from PvDBPII.
Next, we compared the position of the epitope with the locations of known polymorphic residues on PvDBPII, as alignment of globally disparate sequences of PvDBPII revealed 120 polymorphic sites with nucleotide diversity varying between 0.0060 and 0.0109 (ref. 39). We retrieved 383 amino acid sequences of PvDBPII and calculated their sequence entropies. This revealed that the surface of PvDBPII contacted by DB9 is one of the most conserved regions of the domain, with low sequence variation explaining its broadly reactive nature (Fig. 6b and Supplementary Fig. 3).
In the membrane context, PvDBPII is thought to form a dimer that interacts with the extracellular domains of DARC. A crystal structure is available for a dimer of PvDBPII bound to a small helix, consisting of residues 19–30 of the extracellular ectodomain of DARC15. Next, we superimposed our PvDBPII:DB9 structure onto this structure, suggesting a model for how DB9 might inhibit invasion by P. vivax (Fig. 6c). While DB9 binds at a distance from the known region of the DARC-binding site, it protrudes from subdomain 3 in the same direction as the carboxy (C) terminus of the DARC19–30 peptide. This end of the DARC peptide faces the transmembrane region of DARC, located in the reticulocyte membrane. Although the arrangement of DARC31–60 is currently unknown, this model suggests that DB9 can prevent the PvDBPII dimer from approaching the reticulocyte membrane in an orientation that is compatible with DARC binding, thereby preventing initiation of the invasion process.
With the epitope for DB9 contained solely within subdomain 3, we next assessed binding of the other mAbs to this region. Subdomain 3 was produced by refolding material expressed in Escherichia coli. An ELISA-based binding assay showed that 6 of the 10 mAbs bound subdomain 3 (Supplementary Fig. 4). With the exception of DB4, these were the mAbs that competed for binding on PvDBPII (Fig. 1e–g). Indeed, with the exception of DB4, the mAbs that bind to subdomain 3 are not those that antagonize the effect of DB9. Therefore, despite the fact that antibodies that target subdomain 3 differ in their inhibitory potential, these data identify subdomain 3 of PvDBPII as a promising potential target for broadly inhibitory antibodies, and identify the epitope of one such antibody.
Discussion
The Duffy binding protein is the most promising candidate for inclusion in a vaccine to prevent infection by P. vivax, due to its essential interaction with human DARC during reticulocyte invasion. Antibodies that bind to PvDBP are found in individuals from malaria-endemic regions, and their presence associates with protection from clinical vivax malaria23,24,25. However, no human mAb has been reported that targets PvDBP and displays broadly inhibitory potential. We therefore vaccinated human volunteers with PvDBPII to induce a humoral response and isolated a mAb that can inhibit all of the tested isolates of P. vivax from invading blood cells. This opens the possibility for testing the efficacy of this mAb as a prophylactic to prevent vivax malaria, while knowledge of its epitope will guide future vaccine development.
The mAbs generated here have been analysed through a series of complementary approaches, and the differing outcomes of these assays illustrate the need for caution in attributing broadly inhibitory potential (Supplementary Table 5). The most accessible assay involves analysis of purified protein binding in an ELISA-based format, with antibodies assessed for their ability to prevent recombinant PvDBPII from binding to the N-terminal DARC ectodomain, with the assumption that direct prevention of DARC binding is the goal for a protective antibody. This assay allows assessment of a range of PvDBPII variants to analyse the breadth of inhibitory potential, and identified two antibodies that broadly inhibited DARC binding (DB9 and DB2), and three with vaccine-homologous (SalI sequence) binding-inhibition activity (DB5, DB6 and DB7).
With the blood stage of P. vivax currently impossible to sustain in long-term, in vitro culture conditions, we also used a culture-compatible, transgenic, parasite-based assay involving the closely related simian malaria, P. knowlesi. Here, the Duffy binding protein PkDBPα was replaced by PvDBP, generating parasite lines in which human RBC invasion inhibition can be studied in a laboratory setting using long-term culture. This showed a clear hierarchy of mAb potency against transgenic parasites in vitro, whether or not the PkDBPβ and PkDBPγ genes were removed. Most informatively, the outcomes of this study only partially overlapped with those of the protein-based study, with some antibodies (that is, DB1 and DB10) among those with the greatest potency to block parasite invasion but proving ineffective at blocking the PvDBPII–DARC interaction in a protein–protein assay. The failure of the binding-inhibition ELISA, which only uses fragments of DARC and PvDBP, to predict parasite neutralization in both subsequent functional assays calls into question its utility as a method of screening antibodies in this context, suggesting that future studies should instead use parasite-based growth inhibition studies.
Finally, we used the most authentic in vitro assay available, by assessing the invasion of reticulocytes by ex vivo P. vivax taken directly from patient isolates. When studying parasite isolates that expressed the SalI PvDBPII variant incorporated into the transgenic P. knowlesi strain, these studies gave a very similar outcome to the transgenic P. knowlesi assays (Figs. 3a and 4b). However, when studying isolates with heterologous PvDBPII sequences, the outcomes were different. Here, just one antibody, DB9, inhibited reticulocyte invasion by all tested strains. Comparison of the data from these three assays suggests that a protein–protein analysis will miss valuable inhibitory antibodies, and highlights the applicability of a transgenic P. knowlesi-based assay as an accessible in vitro proxy for the ex vivo analysis of P. vivax. In the future, panels of transgenic parasites expressing different PvDBPII variants will allow assessment of the broadly inhibitory potential of therapeutic interventions.
Various properties have been suggested as desirable for inhibitory antibodies that target PvDBP, including the capacity to directly block DARC binding or to inhibit dimerization6,13. In contrast, DB9 binds distantly from either the DARC-binding or dimerization interfaces, on a non-polymorphic site on the distal surface of subdomain 3. Remarkably, it also prevents recombinant PvDBPII from binding to DARC immobilized onto an ELISA plate. A composite model, in which DB9 is docked onto that of a dimer of PvDBPII bound to the 19–30 peptide from DARC, shows that DB9 emerges from the same side of PvDBPII as the C terminus of the DARC peptide. DB9 may therefore sterically prevent dimeric PvDBPII from approaching a DARC-coated surface, and thereby prevent receptor engagement and invasion. A previous study identified antibodies from immunized mice that also bind to subdomain 3 (ref. 26). The inhibitory potential of these antibodies was tested using the protein-based binding assay, which we found to be only partially indicative of efficacy in parasite-based neutralization assays. Nevertheless, one of these antibodies, 2C6, has an epitope close to that of DB9 (Supplementary Fig. 5), and could be operating through the same mechanism.
These findings have significant consequences for vaccine development, identifying subdomain 3 of PvDBPII as an important site of broadly inhibitory epitopes for human antibodies. Not all antibodies that bind to subdomain 3 are inhibitory. However, our discovery of antibodies that bind to PvDBPII and antagonize the function of DB9 also raises the imperative to avoid inducing these deleterious antibodies. While further studies are needed to understand the molecular basis for this antagonism—perhaps involving analysis of their effect on the interaction between full-length PvDBP and DARC in the membrane context—it is encouraging to note that 80% of the antibodies that antagonize the function of DB9 do not recognize subdomain 3. This suggests that future malaria vaccination strategies should specifically present the surface of subdomain 3 containing the DB9 epitope to the immune system, in the form of a vaccine immunogen, to produce a strong and protective immune response.
Methods
Generation of mAbs
Plasmablast isolation and sorting
Volunteers from a phase Ia clinical trial were bled seven days after the second immunization using modified vaccinia virus Ankara encoding PvDBPII19. Blood was collected from volunteers in heparinized tubes and centrifuged in Leucosep tubes (Greiner Bio-One) to separate the peripheral blood mononuclear cells. These cells were enriched for B cells using a Human Pan-B Cell Enrichment Kit (EasySep) and re-suspended in Dulbecco’s modified Eagle medium (DMEM) before staining with a CD19+, CD10−, CD21−, CD27+, CD20−, CD38+ and IgG+ fluorophore-conjugated antibody panel. Plasmablasts were single-cell sorted using a MoFlo cell sorter (DakoCytomation) into 96-well PCR plates containing 10 µl 10 mM Tris-HCl buffer containing 40 U ml−1 RNase inhibitor (Promega). The study received ethical approval from the Oxfordshire Research Ethics Committee A in the UK (Research Ethics Committee reference 13/SC/0001), and was registered with ClinicalTrials.gov (NCT01816113). The volunteers signed consent forms, and consent was verified before each vaccination.
Antibody variable gene amplification
In the wells of a 96-well plate, each of which contained a single antibody-secreting cell, a two-step reverse-transcription PCR was carried out with a first reverse-transcription step using a Sensiscript RT kit (Qiagen) and degenerate primers 1–17 (modified from ref. 40; see Supplementary Table 6). Next, a PCR was performed on 1 µl of the reverse-transcription reaction product using the same set of primers as was used before (1–17), which covered the diversity of all Vγ, Vκ and Vλ sequences using Phusion High-Fidelity PCR Master Mix (New England Biolabs). Following this, a nested PCR was performed with primers 18–51, also using Phusion High-Fidelity PCR Master Mix (Supplementary Table 6) on 1 µl of the previous product diluted 1:100 to amplify inserts that contained plasmid-homologous extensions designed for circular polymerase extension cloning41.
Cloning
The AbVec-hIgG1/AbVec-hIgKappa/AbVec-hIgLambda expression plasmids were a gift from P. C. Wilson (University of Chicago)42. These plasmids were 5′ digested using BshTI and 3′ digested using SalI (AbVec-hIgG1), XhoI (AbVec-hIgLambda) and Pfl23II (AbVec-hIgKappa), to yield linear products. Circular polymerase extension cloning assembly was performed by mixing 100 ng of a 1:1 molar ratio of insert to plasmid in 20 µl containing 1× Phusion High-Fidelity PCR Master Mix, and assembled using an 8-cycle circular polymerase extension cloning protocol (8 cycles of 98 °C for 10 s, slow-ramp anneal from 70–55 °C at 0.1 °C s−1 and 72 °C for 35 s). Full, nicked plasmids were then transformed into Zymo 5α Mix & Go Competent E. coli (Zymo Research) according to the manufacturer’s instructions, streaked on lysogeny broth agar petri dishes containing 100 µg ml−1 carbenicillin, and grown at 37 °C overnight in a static incubator. Colonies were screened by PCR for correctly sized inserts.
Screening
Exponential growth-phase, adherent HEK293 cells were re-suspended in DMEM (Sigma–Aldrich) supplemented with 2 mM l-glutamine, 100 U ml−1 penicillin, 1 mM sodium pyruvate, 0.1 mg ml−1 streptomycin and 10% Ultra-Low IgG Fetal Bovine Serum (Thermo Fisher Scientific), and seeded at 4 × 104 cells well−1 in 100 µl total volume per well, 24 h before transfection in Costar 96-Well Cell Culture Plates (Corning). On the day of transfection, for each well, 50 µl of 60 µg ml−1 linear 25 kDa polyethyleneimine (Alfa Aesar) was mixed with 200 ng of cognate heavy- and light-chain-coding plasmid in a volume of 50 µl, and shaken at 20 °C for 30 min. The DNA–polyethyleneimine complexes were added to the HEK293 cells. The following day, an additional 50 µl of supplemented DMEM (as described above) was added to each well. Supernatants were screened for PvDBPII (produced in S2 cells as described below) binding by indirect ELISA. Over 100 mAb supernatants were screened in this assay; of these, 10 bound PvDBPII.
Recombinant protein constructs, expression and purification
The production of the recombinant PvDBPII used in the IgG screening assay described above, the DARC-binding-inhibition assays (Fig. 2) and the SPR assays (Fig. 1a–d), from Drosophila S2 cells, has been described previously19. In brief, the sequence used was identical to the vaccine sequence: D194–T521 of PvDBPII (SalI), with T257A, S353A and T422A substitutions to remove sites of possible N-linked glycosylation, followed by a shortened Pk/V5 epitope tag (IPNPLLGLD) and a C-tag (EPEA)43 for detection and purification, respectively. Purification was performed on an ÄKTA Pure 25 system (GE Healthcare), consisting of an affinity step with a CaptureSelect C-tag column (Thermo Fisher Scientific) and polishing size exclusion chromatography (SEC) using Superdex 200 16/60 PG (GE Healthcare) in 20 mM Tris-HCl and 150 mM NaCl (pH 7.4). Purified protein was quantified by NanoDrop (Thermo Fisher Scientific) and stored at −80 °C until further use. A gene encoding the PvDBPII HMP013 allele19 (amino acids 194–521) was codon optimized for human expression, and synthesized (Genewiz). This sequence, with a 5′ KpnI site and 3′ XbaI site, was cloned into a mammalian expression plasmid in-frame with an N-terminal mouse IgK light-chain leader sequence and a C-terminal C-tag before the stop codon. Suspension Expi293F cells (Thermo Fisher Scientific) were transiently transfected, and culture supernatants were harvested after four days. Purification was performed on an ÄKTA Pure 25 system (GE Healthcare), consisting of an affinity step with a CaptureSelect C-tag column (Thermo Fisher Scientific) and a polishing SEC step using Superdex 200 Increase 10/300 GL (GE Healthcare) in 20 mM Tris-HCl, 150 mM NaCl and 0.1% Tween 20 (pH 7.4). Purified protein was quantified by NanoDrop (Thermo Fisher Scientific) and stored at −80 °C until further use.
The PvAH, PvO and PvP alleles of PvDBPII44 used in the DARC-binding-inhibition assays (Fig. 2) were a gift from C. Chitnis (Institute Pasteur).
Monobiotinylated PvDBPII supernatant was produced for use in the BLI experiments (Fig. 1e–g) by transient transfection of suspension Expi293F cells, using a plasmid encoding PvDBPII (SalI) with C-terminal rat CD4 domains 3 and 4, followed by a biotin acceptor peptide obtained from Addgene (plasmid number 68529) and courtesy of J. Rayner (Wellcome Sanger Institute)45. This PvDBPII plasmid was co-transfected with another plasmid encoding E. coli biotin ligase (BirA). Supernatant was harvested after four days, clarified, dialysed against phosphate buffered saline (PBS) using snakeskin, and concentrated roughly tenfold using spin columns.
The production of the PvDBPII and PvDBPII subdomain 3 proteins used in the crystal complex (Fig. 6) and the subdomain 3 binding ELISA (Supplementary Fig. 4) is described in the section ‘Structural methods’ below.
The production of recombinant N-terminal DARC, used in the DARC-binding-inhibition assays, has been described previously19. In brief, a gene encoding the first 60 amino acids of the DARC Fyb allele (GenBank accession: ABA10433.1), followed by a thrombin cleavage site (LVPRGS) and an AviTag (GLNDIFEAQKIEWHE), was codon optimized for E. coli expression, and synthesized (GeneArt; Life Technologies). Cysteines 4, 51 and 54 of DARC were mutated to alanine. This sequence with the 5′ KpnI site and 3′ BamHI site was cloned into a mammalian expression plasmid in-frame with an N-terminal human tissue plasminogen activator (tPA) leader sequence20 and a C-terminal hexa-histidine (His6) tag before the stop codon. Suspension HEK293E cells grown in Expi293 expression medium (Thermo Fisher Scientific) were transiently transfected with plasmid and allowed to grow for three days before the supernatant was harvested, purified using a HisTrap Excel column (GE Healthcare) and buffer exchanged into PBS. Purified protein was quantified by NanoDrop (Thermo Fisher Scientific) and stored at −80 °C until further use.
Recombinant mAbs were transiently expressed in HEK293F cells using the Expi293 Expression System (Thermo Fisher Scientific) according to the manufacturer’s recommendations. Cognate heavy- and light-chain-coding plasmids were co-transfected at a 1:1 ratio. Supernatants were harvested by centrifuging the culture at 2,500g for 15 min and filtering the supernatant with a 0.22 µm vacuum filter. All mAbs were purified using a 5 ml Protein G HP column (GE Healthcare) on an ÄKTA start FPLC system or an ÄKTA Pure FPLC system (both GE Healthcare). Equilibration and wash steps were performed with Dulbecco’s PBS, and mAbs were eluted in 0.1 M glycine (pH 2.7). The eluates were pH equilibrated to 7.4 using 1.0 M Tris-HCl (pH 9.0), then immediately buffer exchanged into Dulbecco’s PBS and concentrated using an Amicon Ultra centrifugal concentrator (Millipore) with a molecular weight cut-off of 30 kDa.
BLI
BLI was carried out on an Octet RED384 (Pall FortéBio) using streptavidin-coated biosensors (Pall FortéBio) to immobilize PvDBPII enzymatically monobiotinylated on a C-terminal AviTag. Assays were carried out in 96-well format in black plates (Greiner). For epitope binning studies (Fig. 1e–g), a six-step sequential assay was performed: baseline (PBS, 30 s); protein immobilization (neat supernatant, 120 s); wash (PBS, 60 s); first mAb (mAb1) binding (300 nM mAb1, 120 s); wash (PBS, 60 s); and second mAb (mAb2) binding (150 nM mAb2, 120 s). ‘Relative binding’ in Fig. 1e shows the ratio (signalmAb2 with mAb1 bound)/(signalmAb2 with no mAb1) where ‘signalmAb2’ was normalized for the amount of PvDBPII bound to the biosensor, such that ‘signalmAb2’ = the raw signal in ‘mAb2 binding’ divided by the raw signal in the ‘protein immobilization’ phase. The resulting ‘binding profile’ for any given mAb corresponds to the column of ‘relative binding values’ under that mAb in the ‘relative binding’ table. To establish the epitope bins, binding profiles between each mAb pair were correlated using a Pearson product-moment correlation coefficient, the values of which are shown in the ‘binding profile correlation’ matrix in Fig. 1f. mAb pairs whose binding profile correlation was >0.7 were grouped into the same epitope bin (Fig. 1g).
Measurement of binding by ELISA
Qualitative mAb binding ELISAs such as those used in Supplementary Fig. 4 were carried out by coating PvDBPII or PvDBPII subdomain 3, produced as described in the section ‘Structural methods’, on MaxiSorp flat-bottom 96-well ELISA plates (Nunc) at 2 µg ml−1 in 50 µl at 4 °C overnight. The plates were then washed twice with PBS and 0.05% Tween 20 (PBS/T) and blocked with 200 µl of Blocker Casein (Thermo Fisher Scientific) for 1 h. Next, the wells were incubated with 10 µg ml−1 of mAb for approximately 45 min at 20 °C, then washed 4 times with PBS/T before the addition of 50 µl of a 1:1,000 dilution of goat anti-human gamma-chain alkaline phosphatase-conjugated secondary antibody (Sigma–Aldrich) for 45 min at 20 °C. Wells were then washed 6 times with PBS/T, and developed with 100 µl of p-nitrophenyl phosphate substrate at 1 mg ml−1 (Sigma–Aldrich) and an optical density read at 405 nm (OD405) using a Model 550 Microplate Reader (Bio-Rad).
Affinity determination by SPR
Data were collected on a Biacore X100 (GE Healthcare). Experiments were performed at 25 °C in Dulbecco’s PBS +0.005% Polysorbate 20 (GE Healthcare). In Fig. 1 and Supplementary Table 2, Sensor Chip Protein A (GE Healthcare) was used to capture 50–100 resonance units of purified mAb diluted in SPR running buffer at a flow rate of 5 µl min−1 on flow cell 2. Next, an appropriate range (typically 20–0.625 nM) of six twofold dilutions, with one replicate, of PvDBPII (expressed in S2 cells as described above) was injected for 90 s at 60 µl min−1, and dissociation was measured for 1,600 s (7,200 s when necessary). Specific binding of the PvDBPII protein to mAb was obtained by reference-subtracting the response of a blank surface from that of the mAb-coated surface. The sensor surface was regenerated with a 60 s pulse of 10 mM glycine-HCl (pH 1.5) (GE Healthcare). Sensorgrams were fitted to a global Langmuir 1:1 interaction model, allowing determination of the kinetic association and dissociation rate constants using Biacore X100 evaluation software.
PvDBPII–DARC-binding-inhibition assays
This assay methodology has been reported previously19,29. In brief, recombinant N-terminal DARC protein was coated onto Nunc-Immuno MaxiSorp plates at 1 μg ml−1. mAb samples were diluted down in a twofold series (starting at 100 µg ml−1) and pre-incubated with PvDBPII protein for 30 min at room temperature. The pre-incubated PvDBPII protein plus mAb mixture was then added to the DARC-coated plates (in duplicate wells). The plates were incubated for 1 h at 37 °C, washed and then incubated with anti-PvDBPII polyclonal rabbit serum20. After a further wash step, the plates were incubated with a 1:1,000 dilution of anti-rabbit IgG alkaline phosphatase (Sigma–Aldrich) and then developed with 1 mg ml−1 p-nitrophenyl phosphate in diethanolamine buffer (Pierce). OD405 was read using a Model 550 Microplate Reader (Bio-Rad) when control wells containing PvDBPII protein and buffer only reached a value of 1.0. In one column of each 96-well plate, wells contained only mAb (100 µg ml−1) and buffer (no PvDBPII protein), and these ‘background’ OD405 values were subtracted from all test values. To calculate the percentage binding inhibition for each mAb sample, we used the following formula:
Five allelic variants of recombinant PvDBPII were used in the assay: SalI, which was made in S2 cells as described above; PvAH, PvO and PvP alleles44, which were a kind gift from C. Chitnis (Institut Pasteur); and PvHMP013, which was sequenced from a vivax-infected patient in Australia46. The expression of recombinant PvDBPII HMP013 protein has been described previously19 and is summarized above. An anti-Ebolavirus glycoprotein-reactive human IgG1 mAb, EBL04047, was used as a negative isotype control for the mAb samples. Polyclonal human anti-PvDBPII serum from the clinical trial19 was used as the positive control.
Dot blot assay
Briefly, 1.5 μl of each of the 10 human anti-PvDBPII mAbs, a human anti-Zaire Ebolavirus glycoprotein IgG1 mAb, recombinant PvDBPII (all at 1 mg ml−1) and PBS were spotted onto 0.2 μm nitrocellulose membrane and air dried for 10 min. The membrane was then blocked in 3% bovine serum albumin plus 3% skimmed milk in PBS for 1 h and washed in PBS. It was then immersed in P. vivax culture supernatant for 1 h and washed again in PBS. This supernatant was taken from a short-term in vitro parasite viability assay using a frozen P. vivax blood-stage inoculum produced for controlled human malaria infection clinical trials (Clinicaltrials.gov NCT03797989). Bound PvDBPII was detected by incubating the membrane in anti-PvDBPII rabbit serum20 diluted 2,000-fold in PBS, followed by an alkaline phosphatase-conjugated anti-rabbit IgG mAb (clone RG-96; Sigma–Aldrich), also diluted 1:2,000, separated by two wash steps in PBS. After a final series of six PBS washes, the dot blot was developed with SIGMAFAST BCIP/NBT Alkaline Phosphatase Substrate at 1 mg ml−1 (Sigma–Aldrich).
Genetic modification of P. knowlesi parasites
The genetic modification of the P. knowlesi A1-H.1 strain30 is described in detail elsewhere31. Briefly, parasites were modified using a two-plasmid CRISPR–Cas9 system comprised of a plasmid (pCas/sg) providing Cas9, single guide RNA (sgRNA) and a hDHFR-yFCU (for positive and negative selection) to create a locus-specific double-strand break and a separate ‘donor’ DNA plasmid (pDonor) to act as the repair template. To create the PkDBPαOR and PvDBPOR lines, the PkDBPα locus (sgRNA: GCTGATCCAGGTTCTCAATC) was targeted using a pDonor plasmid containing full-length re-codonized PkDBPα or PvDBP (SalI) genes, respectively, both flanked by 500-base pair homology regions targeting the 5′ untranslated region (UTR) and 3′ UTR of PkDBPα. To create the PkDBPαOR/Δβγ and PvDBPOR/Δβγ lines, this process was repeated in an A1-H.1 line that had a natural deletion of PkDBPβ (due to a truncation of chromosome 14), and then a subsequent round of gene editing was used to delete the PkDBPγ locus (sgRNA: CATGCAACAATTTACACCCC) using a pDonor plasmid containing a spacer sequence flanked by 500-base pair homology regions targeting the 5′ UTR and 3′ UTR of PkDBPγ.
Assays of GIA with P. knowlesi lines
In vitro parasite culture and synchronization
Human RBC-adapted parasites were maintained in culture as described previously30. Briefly, parasites were grown at 2% haematocrit in O+ human RBCs, which were prepared twice monthly. The culture medium contained 10% heat-inactivated pooled human serum mixed with Roswell Park Memorial Institute 1640 medium supplemented with 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 35 μM hypoxanthine, 2 mM l-glutamine and 20 μg ml−1 gentamycin. Parasite cultures were synchronized at the trophozoite/schizont stage by magnetic separation (MACS LS columns, Miltenyi Biotec).
In vitro assay of GIA
The methodology was adapted from the protocol of the International Growth Inhibition Assay Reference Centre at the National Institutes of Health48. Synchronized trophozoites were adjusted to 1.5% parasitaemia, and 20 μl aliquots were pipetted into 96-well flat/half-area tissue culture cluster plates (Appleton Woods). Test antibody or controls (20 μl) were added in duplicate or triplicate test wells over a concentration range (usually 1, 0.5, 0.25, 0.125, 0.0625, 0.0312, 0.015 and 0.0075 mg ml−1) and incubated for one erythrocytic parasite cycle (26–30 h). Parasitaemia was measured using the lactate dehydrogenase activity assay following standard protocols49. The percentage GIA was calculated as:
An anti-DARC VHH camelid nanobody50—a gift from O. Bertrand (INSERM) was included in the test plate as a positive control in every assay (at a final concentration of 6, 3 or 1.5 μg ml−1), and an anti-Ebolavirus glycoprotein-reactive human IgG1 mAb was used as a negative isotype control for the mAb samples. The assays were performed using five different lines of P. knowlesi parasites: the non-transgenic strain (PkA1-H.1); two transgenic lines containing the PvDBP gene (PvDBPOR and PvDBPOR/Δβγ); and two transgenic control lines (PkDBPαOR and PkDBPαOR/Δβγ)31. In the mAb synergy assays (Fig. 5), the ‘Bliss additivity’ between two mAbs (mAb A and mAb B) was calculated using the following formula:
Ex vivo P. vivax invasion assays
The assay methodology has been described previously51,52. The major steps are summarized below.
Purification of reticulocytes from umbilical cord blood
Aliquots of umbilical cord blood (20 ml) were collected from consenting volunteers (Oxford Tropical Research Ethics Committee at the Centre for Clinical Vaccinology and Tropical Medicine, University of Oxford (027-025) and Mahidol University Faculty of Tropical Medicine Ethics Committee (2008-215)) in lithium heparin tubes, and the white blood cells and platelets were depleted using Nonwoven Filters (Antoshin). The RBCs were then layered onto 70% isotonic Percoll and centrifuged, and the resulting band of enriched reticulocytes was incubated with anti-CD71 MicroBeads (Miltenyi Biotec) before being passed through an LS Column (Miltenyi Biotec) to obtain a CD71-depleted (negative) fraction and a CD71-rich fraction (positive). The purity levels of these CD71+ fractions were assessed with microscopy using New Methylene Blue staining (Sigma–Aldrich).
Purification of schizonts from ex vivo P. vivax-infected blood samples
Samples of whole blood (5 ml) were collected from consenting patients (Oxford Tropical Research Ethics Committee at the Centre for Clinical Vaccinology and Tropical Medicine, University of Oxford (027-025) and Mahidol University Faculty of Tropical Medicine Ethics Committee (2008-215)) diagnosed with P. vivax malaria, and rapidly transported at room temperature back to the laboratory. Samples were not collected from patients who had taken antimalarial or antimicrobial drugs within the previous month, or whose parasitaemia was <0.1% on smear microscopy. The samples were leukodepleted using Nonwoven Filters (Antoshin), and the parasites were cultured to schizont stage. The culture was then treated with trypsin, overlaid on a 45% Percoll (isotonic) cushion and centrifuged to isolate a fine band of concentrated schizonts for use in the assay.
Invasion assay
The concentrated schizont preparation was mixed with the enriched reticulocyte fraction at a ratio of 1:6, giving a starting schizont parasitaemia of ~14%. The mixture was diluted to 1.3% haematocrit in 300 μl of complete McCoy 5A medium and cultured in 96-well cell culture plates under an atmosphere of 5% O2 at 37.5 °C. Test antibodies and controls were buffer exchanged into P. vivax culture medium (McCoy 5A medium (Gibco) supplemented with 2.4 g l−1 d-glucose, 40 mg ml−1 gentamycin sulfate and 20% heat-inactivated human AB serum) and added to the final invasion assay mixture at the desired concentration. In the case of the anti-PvDBPII mAbs, this was 1 mg ml−1. The anti-DARC nanobody, at 25 μg ml−1, and the recombinant IgG1 anti-Ebolavirus mAb, at 1 mg ml−1, were added to all assays as positive and negative controls, respectively. Maturation was obtained after incubation for an average of 24 h. At the end of the incubation period, thin smears (each made with 1 μl packed cells) were made, and stained with Giemsa (Sigma–Aldrich). The numbers of ring stages and trophozoites per 4,000 erythrocytes were determined by examining the Giemsa thin-film smears by light microscopy. For isolates 1–3, the invasion assays were performed in Singapore using cryopreserved Thai P. vivax isolates, and reticulocyte invasion inhibition was quantified by flow cytometry as described previously51.
Sequencing of the PvDBPII gene from Thai isolates
Genomic DNA was extracted either from an aliquot (200 μl) of the initial blood sample or from a dried blood spot, using the DNeasy Blood and Tissue Kit (Qiagen). PCR amplification was performed on 1 μl purified genomic DNA in a total volume of 50 μl, containing 1× PCR buffer, 2.5 mM MgCl2, 0.2 mM of each dNTP, 2 μM of each primer (forward: 5′–GTG ACT GGG CAT GAG GGA AAT TCT CG; reverse: 5′–GCG TAG AAT CTC CTG GAA CCT TCT CC) and 1.25 U of AmpliTaq Gold DNA Polymerase (Applied Biosystems). The cycling conditions were: 95 °C for 5 min, followed by 30 cycles at 94 °C for 1 min, 63 °C for 2 min and 72 °C for 2 min. The product was purified using the QIAquick PCR Purification Kit (Qiagen), then sequenced (GATC Biotech).
Structural methods
Protein cloning, expression and purification
The PvDBPII coding region (residues 211–507; SalI sequence) was cloned into the pET15b vector. This vector allowed expression of the protein with an N-terminal His6 tag and a tobacco etch virus cleavage site in E. coli (BL21-DE3 strain). The transformed bacteria were induced with 1 mM Isopropyl β-d-1-thiogalactopyranoside at an optical density of 0.8 at 600 nm. PvDBPII was expressed exclusively in inclusion bodies and was prepared as described previously15. In short, the inclusion bodies were solubilized in 6 M guanidine hydrochloride, 20 mM Tris (pH 8) and 300 mM NaCl, and refolded by flash-dilution into 400 mM l-arginine, 50 mM Tris (pH 8), 10 mM ethylenediaminetetraacetic acid, 0.1 mM phenylmethylsulfonyl fluoride, 3 mM reduced glutathione and 0.3 mM oxidized glutathione. Refolded proteins were buffer exchanged into 20 mM Tris (pH 8), 150 mM NaCl and 20 mM imidazole, then affinity purified on Ni-NTA resin. The His-tag was removed by tobacco etch virus protease cleavage, and the tagless PvDBPII was separated from the His-tag on Ni-NTA resin and purified by SEC (S75 16/60; GE Healthcare) into 20 mM HEPES (pH 7.5) and 150 mM NaCl.
Subdomain 3 (residues 211–508) was subcloned from the PvDBPII construct in the pET15b vector. Subdomain 3 was also expressed in E. coli BL21 (DE3) cells in the insoluble fraction, and was solubilized in 6 M guanidine hydrochloride, 20 mM Tris (pH 8), 150 mM NaCl and 20 mM imidazole. Subdomain 3 was then refolded by binding to an Ni-NTA column and gradually decreasing the concentration of guanidine hydrochloride until the protein was in 20 mM Tris (pH 8), 150 mM NaCl and 20 mM imidazole. The refolded subdomain 3 protein was then eluted and further purified using SEC (S75 16/60; GE Healthcare) into 20 mM HEPES (pH 7.5) and 150 mM NaCl.
Fab generation and purification
Fab fragments for DB9 were generated from IgG by incubating with Immobilized Papain (Thermo Fisher Scientific) for 16 h at 37 °C. The Fab was separated from the uncleaved DB9 and Fc regions using Protein A resin (Thermo Fisher Scientific). The Fab fragments were then purified by SEC into 20 mM HEPES (pH 7.5) and 150 mM NaCl.
Protein cystallization and data collection
PvDBPII and DB9Fab were mixed in a 1.2:1 molar ratio and incubated at room temperature for 1 h. The complex was purified by SEC (S75; GE Healthcare) in 20 mM HEPES (pH 7.5) and 150 mM NaCl. Broad crystallization trials were set up by sitting-drop vapour diffusion in Swissci 96-well plates by mixing 100 nl protein with 100 nl reservoir solution. Crystals grew in the reservoir solution of 45% v/v polypropylene glycol 400 and 10% v/v ethanol, and were then cryo-cooled in liquid nitrogen. Data were collected on beamline I03 at Diamond Light Source at a wavelength of 0.97625 Å, and were indexed using XDS53 and scaled using Scala54 to a resolution of 3.04 Å.
Structure solution
The PvDBPII:DB9Fab structure was solved by molecular replacement in Phaser55 using the known structures of PvDBPII (PDB ID: 4NUV) and a human mAb Fab fragment (PDB ID: 3DIF), separated into the variable and constant regions, as search models. This identified one copy of the PvDBPII:DB9 complex in the asymmetrical unit. Refinement and rebuilding was completed using Buster56 and Coot57, respectively. The final Ramachandran plot showed 94.9% of residues in the favoured region, 5.1% in the allowed region and none in the disallowed region.
Statistical analyses
Data were analysed using GraphPad Prism version 6.07 for Windows (GraphPad Software). In Fig. 3a, a four-parameter sigmoidal dose–response curve was fitted to the relationship between log10[antibody concentration] and percentage GIA for each dataset and used to interpolate EC30 values. In Fig. 3b, the non-parametric ρ value was used to assess a correlation between the variables kon/koff/Kd and GIA EC30. In all statistical tests, the reported P values are two tailed, with P < 0.05 considered significant.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
Control and Elimination of Plasmodium vivax Malaria: a Technical Brief (WHO, 2015).
Gething, P. W. et al. A long neglected world malaria map: Plasmodium vivax endemicity in 2010. PLoS Negl. Trop. Dis. 6, e1814 (2012).
Gruszczyk, J. et al. Transferrin receptor 1 is a reticulocyte-specific receptor for Plasmodium vivax. Science 359, 48–55 (2018).
Malleret, B., Renia, L. & Russell, B. The unhealthy attraction of Plasmodium vivax to reticulocytes expressing transferrin receptor 1 (CD71). Int. J. Parasitol. 47, 379–383 (2017).
Singh, S. K., Hora, R., Belrhali, H., Chitnis, C. E. & Sharma, A. Structural basis for Duffy recognition by the malaria parasite Duffy-binding-like domain. Nature 439, 741–744 (2006).
Chitnis, C. E. & Sharma, A. Targeting the Plasmodium vivax Duffy-binding protein. Trends Parasitol. 24, 29–34 (2008).
Singh, A. P. et al. Targeted deletion of Plasmodium knowlesi Duffy binding protein confirms its role in junction formation during invasion. Mol. Microbiol. 55, 1925–1934 (2005).
Singh, A. P., Puri, S. K. & Chitnis, C. E. Antibodies raised against receptor-binding domain of Plasmodium knowlesi Duffy binding protein inhibit erythrocyte invasion. Mol. Biochem. Parasitol. 121, 21–31 (2002).
Miller, L. H., Mason, S. J., Clyde, D. F. & McGinniss, M. H. The resistance factor to Plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N. Engl. J. Med. 295, 302–304 (1976).
Howes, R. E. et al. Plasmodium vivax transmission in Africa. PLoS Negl. Trop. Dis. 9, e0004222 (2015).
Culleton, R. L. et al. Failure to detect Plasmodium vivax in West and Central Africa by PCR species typing. Malar. J. 7, 174 (2008).
Draper, S. J. et al. Malaria vaccines: recent advances and new horizons. Cell Host Microbe 24, 43–56 (2018).
Batchelor, J. D., Zahm, J. A. & Tolia, N. H. Dimerization of Plasmodium vivax DBP is induced upon receptor binding and drives recognition of DARC. Nat. Struct. Mol. Biol. 18, 908–914 (2011).
Choe, H. et al. Sulphated tyrosines mediate association of chemokines and Plasmodium vivax Duffy binding protein with the Duffy antigen/receptor for chemokines (DARC). Mol. Microbiol. 55, 1413–1422 (2005).
Batchelor, J. D. et al. Red blood cell invasion by Plasmodium vivax: structural basis for DBP engagement of DARC. PLoS Pathog. 10, e1003869 (2014).
Hans, D. et al. Mapping binding residues in the Plasmodium vivax domain that binds Duffy antigen during red cell invasion. Mol. Microbiol. 55, 1423–1434 (2005).
VanBuskirk, K. M., Sevova, E. & Adams, J. H. Conserved residues in the Plasmodium vivax Duffy-binding protein ligand domain are critical for erythrocyte receptor recognition. Proc. Natl Acad. Sci. USA 101, 15754–15759 (2004).
Singh, K. et al. Malaria vaccine candidate based on Duffy-binding protein elicits strain transcending functional antibodies in a phase I trial. NPJ Vaccines 3, 48 (2018).
Payne, R. O. et al. Human vaccination against Plasmodium vivax Duffy-binding protein induces strain-transcending antibodies. JCI Insight 2, e93683 (2017).
De Cassan, S. C. et al. Preclinical assessment of viral vectored and protein vaccines targeting the Duffy-binding protein Region II of Plasmodium vivax. Front Immunol. 6, 348 (2015).
Moreno, A. et al. Preclinical assessment of the receptor-binding domain of Plasmodium vivax Duffy-binding protein as a vaccine candidate in rhesus macaques. Vaccine 26, 4338–4344 (2008).
Bermudez, M., Moreno-Perez, D. A., Arevalo-Pinzon, G., Curtidor, H. & Patarroyo, M. A. Plasmodium vivax in vitro continuous culture: the spoke in the wheel. Malar. J. 17, 301 (2018).
Cole-Tobian, J. L. et al. Strain-specific Duffy binding protein antibodies correlate with protection against infection with homologous compared to heterologous Plasmodium vivax strains in Papua New Guinean children. Infect. Immun. 77, 4009–4017 (2009).
King, C. L. et al. Naturally acquired Duffy-binding protein-specific binding inhibitory antibodies confer protection from blood-stage Plasmodium vivax infection. Proc. Natl Acad. Sci. USA 105, 8363–8368 (2008).
Nicolete, V. C., Frischmann, S., Barbosa, S., King, C. L. & Ferreira, M. U. Naturally acquired binding-inhibitory antibodies to Plasmodium vivax Duffy binding protein and clinical immunity to malaria in rural Amazonians. J. Infect. Dis. 214, 1539–1546 (2016).
Chen, E. et al. Broadly neutralizing epitopes in the Plasmodium vivax vaccine candidate Duffy binding protein. Proc. Natl Acad. Sci. USA 113, 6277–6282 (2016).
Chootong, P. et al. Mapping epitopes of the Plasmodium vivax Duffy binding protein with naturally acquired inhibitory antibodies. Infect. Immun. 78, 1089–1095 (2010).
Ye, J., Ma, N., Madden, T. L. & Ostell, J. M. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 41, W34–W40 (2013).
Shakri, A. R., Rizvi, M. M. & Chitnis, C. E. Development of quantitative receptor–ligand binding assay for use as a tool to estimate immune responses against Plasmodium vivax Duffy binding protein Region II. J. Immunoass. Immunochem. 33, 403–413 (2012).
Moon, R. W. et al. Adaptation of the genetically tractable malaria pathogen Plasmodium knowlesi to continuous culture in human erythrocytes. Proc. Natl Acad. Sci. USA 110, 531–536 (2013).
Mohring, F. et al. Rapid and iterative genome editing in the zoonotic malaria parasite Plasmodium falciparum: new tools for P. vivax research. Preprint at https://www.biorxiv.org/content/10.1101/590976v1.full (2019).
Adams, J. H. et al. The Duffy receptor family of Plasmodium knowlesi is located within the micronemes of invasive malaria merozoites. Cell 63, 141–153 (1990).
Gruring, C. et al. Human red blood cell-adapted Plasmodium knowlesi parasites: a new model system for malaria research. Cell Microbiol. 16, 612–620 (2014).
Pain, A. et al. The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature 455, 799–803 (2008).
Ord, R. L. et al. A malaria vaccine candidate based on an epitope of the Plasmodium falciparum RH5 protein. Malar. J. 13, 326 (2014).
Douglas, A. D. et al. Neutralization of Plasmodium falciparum merozoites by antibodies against PfRH5. J. Immunol. 192, 245–258 (2014).
Saul, A. Kinetic constraints on the development of a malaria vaccine. Parasite Immunol. 9, 1–9 (1987).
Williams, A. R. et al. Enhancing blockade of Plasmodium falciparum erythrocyte invasion: assessing combinations of antibodies against PfRH5 and other merozoite antigens. PLoS Pathog. 8, e1002991 (2012).
Nobrega de Sousa, T., Carvalho, L. H. & Alves de Brito, C. F. Worldwide genetic variability of the Duffy binding protein: insights into Plasmodium vivax vaccine development. PLoS ONE 6, e22944 (2011).
Tiller, T. et al. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J. Immunol. Methods 329, 112–124 (2008).
Quan, J. & Tian, J. Circular polymerase extension cloning of complex gene libraries and pathways. PLoS ONE 4, e6441 (2009).
Wrammert, J. et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671 (2008).
Jin, J. et al. Accelerating the clinical development of protein-based vaccines for malaria by efficient purification using a four amino acid C-terminal ‘C-tag’. Int. J. Parasitol. 47, 435–446 (2017).
Ntumngia, F. B. et al. Conserved and variant epitopes of Plasmodium vivax Duffy binding protein as targets of inhibitory monoclonal antibodies. Infect. Immun. 80, 1203–1208 (2012).
Hostetler, J. B. et al. A library of Plasmodium vivax recombinant merozoite proteins reveals new vaccine candidates and protein–protein interactions. PLoS Negl. Trop. Dis. 9, e0004264 (2015).
Payne, R. O., Griffin, P. M., McCarthy, J. S. & Draper, S. J. Plasmodium vivax controlled human malaria infection—progress and prospects. Trends Parasitol. 33, 141–150 (2017).
Rijal, P. et al. Therapeutic monoclonal antibodies for Ebola virus infection derived from vaccinated humans. Cell Rep. 27, 172–186.e7 (2019).
Miura, K. et al. Anti-apical-membrane-antigen-1 antibody is more effective than anti-42-kilodalton-merozoite-surface-protein-1 antibody in inhibiting Plasmodium falciparum growth, as determined by the in vitro growth inhibition assay. Clin. Vaccin. Immunol. 16, 963–968 (2009).
Kennedy, M. C. et al. In vitro studies with recombinant Plasmodium falciparum apical membrane antigen 1 (AMA1): production and activity of an AMA1 vaccine and generation of a multiallelic response. Infect. Immun. 70, 6948–6960 (2002).
Smolarek, D. et al. A recombinant dromedary antibody fragment (VHH or nanobody) directed against human Duffy antigen receptor for chemokines. Cell Mol. Life Sci. 67, 3371–3387 (2010).
Cho, J. S. et al. Unambiguous determination of Plasmodium vivax reticulocyte invasion by flow cytometry. Int. J. Parasitol. 46, 31–39 (2016).
Russell, B. et al. A reliable ex vivo invasion assay of human reticulocytes by Plasmodium vivax. Blood 118, e74–e81 (2011).
Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 (2006).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Bricogne, G. et al. Buster version 2.10.2 (Global Phasing Limited, 2017).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Acknowledgements
The authors are grateful for the assistance of J. Furze, D. Llewellyn, D. Worth, K. Johnson, J. Barrett and S. Biswas (Jenner Institute, University of Oxford), P. C. Wilson (University of Chicago) for the expression plasmids, O. Fedorov (Structural Genomics Consortium, University of Oxford) for use of the Octet RED384, A. Duckett and C. Banner (University of Oxford) for arranging contracts, Y. Durocher for the HEK293E cells (CNRC-NRC, Canada), A. Rushdi Shakri (ICGEB, India) and C. E. Chitnis (Institute Pasteur, France) for the PvDBPII variant proteins, J. McCarthy (QIMR, Australia) for the HMP013 P. vivax strain PvDBPII sequence, O. Bertrand (INSERM, Paris) for the VHH camelid anti-DARC nanobody, and the VAC051 PvDBPII vaccine clinical trial study volunteers and staff and patients associated with the Shoklo Malaria Research Unit for the P. vivax sample donation (SMRU, Thailand). This work was supported in part by funding from the European Union’s Horizon 2020 Research and Innovation programme under a grant agreement for MultiViVax (number 733073). T.A.R. held a Wellcome Trust Research Training Fellowship (108734/Z/15/Z), N.M.B. is funded by the Wellcome Trust DPhil programme in structural cell biology, J.S.C. held a Singapore International Graduate Award (SINGA), D.G.W.A. held a UK Medical Research Council (MRC) iCASE PhD Studentship (MR/K017632/1), M.K.H. is a Wellcome Trust investigator (101020/Z/13/Z), and S.J.D. is a Jenner investigator, a Lister Institute Research Prize fellow and a Wellcome Trust senior fellow (106917/Z/15/Z). SMRU is part of the Mahidol Oxford Tropical Medicine Research Unit supported by the Wellcome Trust. R.W.M. and F.M. are supported by a UK MRC Career Development Award (MR/M021157/1) jointly funded by the UK MRC and Department for International Development.
Author information
Authors and Affiliations
Contributions
T.A.R., N.M.B., M.K.H. and S.J.D. conceived the study and wrote the manuscript. T.A.R., N.M.B., F.M., J.S.C., V.K., S.F.G., D.G.W.A., G.M.L., S.C.E., S.E.S., D.Q., J.J., J.M.M., R.W.M. and M.K.H. performed the experiments. T.A.R., N.M.B., F.M., J.S.C., D.G.W.A., M.K.H. and S.J.D. performed the data analysis and interpretation. R.O.P., A.M.M., J.J., R.W.M., B.R., L.R. and F.H.N. contributed reagents or materials and facilities.
Corresponding authors
Ethics declarations
Competing interests
D.G.W.A., M.K.H. and S.J.D. are named inventors on patent applications relating to malaria vaccines, mAbs and/or immunization regimens.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figures 1–5 and Supplementary Tables 1–6.
Rights and permissions
About this article

Cite this article
Rawlinson, T.A., Barber, N.M., Mohring, F. et al. Structural basis for inhibition of Plasmodium vivax invasion by a broadly neutralizing vaccine-induced human antibody. Nat Microbiol 4, 1497–1507 (2019). https://doi.org/10.1038/s41564-019-0462-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-019-0462-1
This article is cited by
-
Human monoclonal antibodies inhibit invasion of transgenic Plasmodium knowlesi expressing Plasmodium vivax Duffy binding protein
Malaria Journal (2023)
-
Elicitation of T-cell-derived IFN-γ-dependent immunity by highly conserved Plasmodium ovale curtisi Duffy binding protein domain region II (PocDBP-RII)
Parasites & Vectors (2023)
-
Development of a Plasmodium vivax biobank for functional ex vivo assays
Malaria Journal (2023)
-
Sequential roles for red blood cell binding proteins enable phased commitment to invasion for malaria parasites
Nature Communications (2023)
-
Structural basis for DARC binding in reticulocyte invasion by Plasmodium vivax
Nature Communications (2023)
