Institute of Molecular Bioscience, The University of Queensland, Brisbane,, 4072, Queensland, Australia
Matthew R. Robinson, Anna A. E. Vinkhuyzen, Jian Yang & Peter M. Visscher
Department of Research, 23andMe Inc., Mountain View, California, 94041, USA
Aaron Kleinman
Department of Epidemiology, Gillings School of Global Public Health, University of North Carolina, Chapel Hill, 27514, North Carolina, USA
Mariaelisa Graff & Kari E. North
Department of Biostatistics, Gillings School of Global Public Health, University of North Carolina, Chapel Hill, 27514, North Carolina, USA
David Couper
Department of Psychology, University of Minnesota, Minneapolis, Minnesota, USA
Michael B. Miller, William G. Iacono & Matt McGue
Department of Psychiatry, VU University Medical Centre & GGZ inGeest, Amsterdam, The Netherlands
Wouter J. Peyrot
Department of Biological Psychology, VU University Amsterdam, Amsterdam, The Netherlands
Abdel Abdellaoui
School of Psychology, The University of Queensland, Brisbane, , 4072, Queensland, Australia
Brendan P. Zietsch
Department of Epidemiology, University of Groningen, University Medical Center Groningen, Groningen, 9700 RB, The Netherlands
Ilja M. Nolte, Jana V. van Vliet-Ostaptchouk & Harold Snieder
Department of Endocrinology, University of Groningen, University Medical Center Groningen, Groningen, 9700 RB, The Netherlands
Jana V. van Vliet-Ostaptchouk
QIMR Berghofer Medical Research Institute, 300 Herston Road, Herston, 4006, Queensland, Australia
Sarah E. Medland & Nicholas G. Martin
Karolinska Institutet, Stockholm, SE-171 77, Sweden
Patrik K. E. Magnusson
Carolina Centre for Genome Sciences, Gillings School of Global Public Health, University of North Carolina, Chapel Hill, 27514, North Carolina, USA
Kari E. North
The Queensland Brain Institute, The University of Queensland, Brisbane, 4072, Queensland, Australia
Jian Yang & Peter M. Visscher
Department of Epidemiology, University of Groningen, University Medical Center Groningen, The Netherlands
Harold Snieder, Behrooz Z. Alizadeh & H. Marike Boezen
Department of Genetics, University of Groningen, University Medical Center Groningen, The Netherlands
Lude Franke, Morris Swertz & Cisca Wijmenga
Department of Cardiology, University of Groningen, University Medical Center Groningen, The Netherlands
Pim van der Harst
Department of Internal Medicine, Division of Nephrology, University of Groningen, University Medical Center Groningen, The Netherlands
Gerjan Navis
Department of Medical Biology, University of Groningen, University Medical Center Groningen, The Netherlands
Marianne Rots
Department of Endocrinology, University of Groningen, University Medical Center Groningen, The Netherlands
Bruce H. R. Wolffenbuttel
Department of Biostatistics, Center for Statistical Genetics, University of Michigan, Ann Arbor, 48109, Michigan, USA
Goncalo R. Abecasis, Michael Boehnke, Jennifer L. Bragg-Gresham, Anne U. Jackson, Adam E. Locke, Laura J. Scott, Nicole Soranzo, Heather M. Stringham, Tanya M. Teslovich, Ryan Welch, Robert J. Weyant & Cristen J. Willer
Hudson Alpha Institute for Biotechnology, Huntsville, 35806, Alabama, USA
Devin Absher & Lindsay L. Waite
Estonian Genome Center, University of Tartu, Tartu, 50410, Estonia
Helene Alavere, Tõnu Esko, Krista Fischer, Reedik Mägi, Andres Metspalu, Evelin Mihailov, Lili Milani, Mari Nelis, Markus Perola, Mari-Liis Tammesoo & Maris Teder-Laving
Institute of Genetic Epidemiology, Helmholtz Zentrum Mìnchen - German Research Center for Environmental Health, Neuherberg, 85764, Germany
Eva Albrecht, Christian Gieger & Martina Mìller-Nurasyid
Genetics of Complex Traits, Peninsula College of Medicine and Dentistry, University of Exeter, Exeter, EX1 2LU, UK
Hana Lango Allen, Timothy Frayling, Andrew T. Hattersley, John R. B. Perry, Michael N. Weedon & Andrew R. Wood
Department of Clinical Sciences, Lund University Diabetes Centre, Lund University, Malmö, 20502, Sweden
Peter Almgren & Leif C. Groop
Department of Epidemiology, Erasmus MC, Rotterdam, 3015GE, The Netherlands
Najaf Amin, Cornelia M. van Duijn, Karol Estrada, Albert Hofman, Carolina Medina-Gomez, Joyce B. J. van Meurs, Marjolein J. Peters, Fernando Rivadeneira, André G. Uitterlinden, Sophie van Wingerden & Jacqueline C. M. Witteman
Institut Pasteur de Lille, INSERM U744, Université Lille Nord de France, Lille, F-59000, France
Philippe Amouyel
Telethon Institute for Child Health Research, West Perth Western Australia, 6872, Australia
Denise Anderson & Aarno Palotie
Centre for Child Health Research, The University of Western Australia, Australia
Denise Anderson
Departments of Biostatistics, University of Washington, Seattle, 98195, Washington, USA
Alice M. Arnold & Barbara McKnight
Collaborative Health Studies Coordinating Center, Seattle, 98115, Washington, USA
Alice M. Arnold
Department of Epidemiology and Public Health, Faculty of Medicine, Strasbourg, France
Dominique Arveiler & Kaarel Krjutškov
Icelandic Heart Association, Kopavogur, Iceland
Thor Aspelund, Gudny Eiriksdottir, Vilmundur Gudnason & Albert Vernon Smith
University of Iceland, Reykjavik, Iceland
Thor Aspelund, Vilmundur Gudnason & Albert Vernon Smith
Department of Cardiology, Division Heart & Lungs, University Medical Center Utrecht, The Netherlands
Folkert W. Asselbergs
Department of Medicine, Stanford University School of Medicine, Stanford, 94305, California, USA
Themistocles L. Assimes, Joshua W. Knowles & Thomas Quertermous
Institute of Biomedicine/Physiology, University of Eastern Finland, Kuopio Campus, Finland
Mustafa Atalay & Timo A. Lakka
Wellcome Trust Sanger Institute, Hinxton, Cambridge, CB10 1SA, UK
Antony P. Attwood, Inês Barroso, Panos Deloukas, Sarah Edkins, Sarah E. Hunt, Stavroula Kanoni, Willem H. Ouwehand, Leena Peltonen, Aparna Radhakrishnan, Joshua C. Randall, Samuli Ripatti, So-Youn Shin, Kathleen Stirrups & Eleanor Wheeler
Department of Haematology, University of Cambridge, Cambridge, CB2 0PT, UK
Antony P. Attwood, Jennifer Jolley, Willem H. Ouwehand, Aparna Radhakrishnan, Augusto Rendon, Jennifer G. Sambrook & Jonathan C. Stephens
NIHR Cambridge Biomedical Research Centre, Cambridge, UK
Antony P. Attwood, Willem H. Ouwehand & Augusto Rendon
Department of Neurology, Boston University School of Medicine, Boston, 02118, Massachusetts, USA
Larry D. Atwood & Nancy L. Heard-Costa
Department of Internal Medicine, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands
Stephan J. L. Bakker & Gerjan Navis
INSERM CESP Centre for Research in Epidemiology and Public Health U1018, Epidemiology of diabetes, obesity and chronic kidney disease over the lifecourse, Villejuif, 94807, France
Beverley Balkau
University Paris Sud 11, UMRS 1018, Villejuif, 94807, France
Beverley Balkau
Multidisciplinary Cardiovascular Research Centre (MCRC), Leeds Institute of Genetics, Health and Therapeutics (LIGHT), University of Leeds, Leeds, LS2 9JT, UK
Anthony J. Balmforth
Department of Medicine, University of Milan, Surgery and Dentistry, Milano, 20139, Italy
Cristina Barlassina
University of Cambridge Metabolic Research Labs, Institute of Metabolic Science Addenbrooke’s Hospital, Cambridge, CB2 OQQ, UK
Inês Barroso
Department of Vascular Medicine, Academic Medical Center, Amsterdam, The Netherlands
Hanneke Basart, Kees G. Hovingh & Mieke D. Trip
Regensburg University Medical Center, Innere Medizin I, Regensburg, 93053, Germany
Sabrina Bauer, Christa Buechler & Kristina Eisinger
Department of Medical Genetics, University of Lausanne, Lausanne, 1005, Switzerland
Jacques S. Beckmann, Sven Bergmann, Toby Johnson, Karen Kapur, Zoltán Kutalik, Diana Marek & Carlo Rivolta
Service of Medical Genetics, Centre Hospitalier Universitaire Vaudois (CHUV) University Hospital, Lausanne, 1011, Switzerland
Jacques S. Beckmann
Department of Molecular Genetics, PathWest Laboratory of Western Australia, J Block, QEII Medical Centre, Nedlands, 6009, Western Australia, Australia
John P. Beilby & Jennie Hui
Busselton Population Medical Research Foundation Inc., Sir Charles Gairdner Hospital, Nedlands, 6009, Western Australia, Australia
John P. Beilby, Jennie Hui, Joseph Hung, Alan L. James, Arthur W. Musk & Lyle J. Palmer
School of Pathology and Laboratory Medicine, University of Western Australia, Nedlands, 6009, Western Australia, Australia
John P. Beilby & Jennie Hui
Department of Surgery and Pathology, University of Western Australia, Nedlands, 6009, Australia
John P. Beilby
Oxford Centre for Diabetes, Endocrinology and Metabolism, University of Oxford, Oxford, OX3 7LJ, UK
Amanda J. Bennett, Christopher J. Groves, Neelam Hassanali, Fredrik Karpe, Cecilia M. Lindgren, Mark I. McCarthy, Matt J. Neville, Inga Prokopenko, Nigel W. Rayner & Neil R. Robertson
Department of Social Medicine, University of Bristol, Bristol, BS8 2PS, UK
Yoav Ben-Shlomo
Department of Physiology and Biophysics, Keck School of Medicine, University of Southern California, Los Angeles, 90033, California, USA
Richard N. Bergman, Thomas A. Buchanan & Richard M. Watanabe
Swiss Institute of Bioinformatics, Lausanne, 1015, Switzerland
Sven Bergmann, Toby Johnson, Karen Kapur, Zoltán Kutalik & Diana Marek
Division of Cancer Epidemiology and Genetics, Department of Health and Human Services, National Cancer Institute, National Institutes of Health, Bethesda, 20892, Maryland, USA
Sonja I. Berndt, Stephen J. Chanock, Kevin B. Jacobs, Jianxin Shi & Zhaoming Wang
Zentrum fìr Zahn-, Mund- und Kieferheilkunde, Greifswald, 17489, Germany
Reiner Biffar
Molecular Biology Department, Istituto Auxologico Italiano, Milano, Italy
Anna Maria Di Blasio & Davide Gentilini
Division of Endocrinology and Diabetes, Department of Medicine, University Hospital, Ulm, Germany
Bernhard O. Boehm
Department of Epidemiology, German Institute of Human Nutrition Potsdam-Rehbruecke, Nuthetal, 14558, Germany
Heiner Boeing & Eva Fisher
Human Genetics Center and Institute of Molecular Medicine, University of Texas Health Science Center, Houston, 77030, Texas, USA
Eric Boerwinkle
Centre for Population Health Sciences, University of Edinburgh, Teviot Place, Edinburgh, EH8 9AG, Scotland
Jennifer L. Bolton, Harry Campbell, Gerry Fowkes, Ross M. Fraser, Jackie F. Price, Igor Rudan, Sarah H. Wild & James F. Wilson
CNRS UMR8199-IBL-Institut Pasteur de Lille, Lille, F-59000, France
Amélie Bonnefond, Philippe Froguel, Cecile Lecoeur, David Meyre & Boris Skrobek
National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland, 20892, USA
Lori L. Bonnycastle, Peter S. Chines, Francis S. Collins, Michael R. Erdos, Mario A. Morken, Narisu Narisu & Amy J. Swift
Genome Technology Branch, National Human Genome Research Institute, NIH, Bethesda, 20892, MD, USA
Lori L. Bonnycastle, Peter S. Chines, Francis S. Collins, Narisu Narisu & Amy J. Swift
Department of Biological Psychology, VU University Amsterdam, Amsterdam, 1081, BT, The Netherlands
Dorret I. Boomsma, Eco J. C. Geus, Jouke-Jan Hottenga & Gonneke Willemsen
Department of Genetics, Washington University School of Medicine, St Louis, 63110, Missouri, USA
Ingrid B. Borecki, Mary F. Feitosa, Shamika Ketkar, Aldi T. Kraja, Lyle J. Palmer & Michael A. Province
Division of Biostatistics, Washington University School of Medicine, St Louis, 63110, Missouri, USA
Ingrid B. Borecki
Department of Medicine III, University of Dresden, Dresden, 01307, Germany
Stefan R. Bornstein & Barbara Ludwig
Department of Medicine III, University of Dresden, Medical Faculty Carl Gustav Carus, Fetscherstrasse 74, Dresden, 01307, Germany
Stefan R. Bornstein & Peter E. H. Schwarz
CNRS UMR8199-IBL-Institut Pasteur de Lille, Lille, F-59019, France
Nabila Bouatia-Naji, Christine Cavalcanti-Proença, Philippe Froguel, Stefan Gaget, Cecile Lecoeur & Vincent Vatin
University Lille Nord de France, Lille, 59000, France
Nabila Bouatia-Naji, Christine Cavalcanti-Proença, Philippe Froguel, Stefan Gaget, Cecile Lecoeur, David Meyre, Boris Skrobek & Vincent Vatin
Montreal Heart Institute, Montreal, H1T 1C8, Quebec, Canada
Gabrielle Boucher & Guillaume Lettre
Dipartimento di Medicina Sperimentale. Università degli Studi Milano-Bicocca, Monza, Italy
Paolo Brambilla
LifeLines Cohort Study, University Medical Center Groningen, University of Groningen, The Netherlands
Harold Snieder, Marcel Bruinenberg, Lude Franke, Melanie M. Van der Klauw, Ronald P. Stolk, Jana V. Van Vliet-Ostaptchouk & Bruce H. R. Wolffenbuttel
Division of Endocrinology, Keck School of Medicine, University of Southern California, Los Angeles, 90033, California, USA
Thomas A. Buchanan
Genetic Epidemiology and Biostatistics Platform, Ontario Institute for Cancer Research, Toronto, M5G 1L7, Canada
Gemma Cadby
Prosserman Centre for Health Research, Samuel Lunenfeld Research Institute, Toronto, M5G 1X5, Canada
Gemma Cadby & Lyle J. Palmer
Clinical Pharmacology and Barts and The London Genome Centre, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London, EC1M 6BQ, UK
Mark J. Caulfield, Toby Johnson & Patricia B. Munroe
Department of Clinical Medicine, University of Milano-Bicocca, Monza, Italy
Giancarlo Cesana
Harvard Medical School, Boston, 02115, Massachusetts, USA
Daniel I. Chasman, Lee M. Kaplan & Paul M. Ridker
Division of Preventive Medicine, Brigham and Women’s Hospital, Boston, 02215, Massachusetts, USA
Daniel I. Chasman, Paul M. Ridker & Lynda M. Rose
Department of OB/GYN and Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, CA, USA
Yii-Der Ida Chen
Department of Medicine, David Geffen School of Medicine at University of California, Los Angeles, California, USA
Yii-Der Ida Chen
University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, 75390-8854, Texas, USA
Deborah J Clegg & Thomas Person
Department of Epidemiology and Biostatistics, School of Public Health, Faculty of Medicine, Imperial College London, London, W2 1PG, UK
Lachlan Coin, Paul Elliott, Marjo-Riitta Jarvelin & Ulla Sovio
British Heart Foundation Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, G12 8TA, UK
John M. Connell
University of Dundee, Ninewells Hospital & Medical School, Dundee, DD1 9SY, UK
John M. Connell
National Heart and Lung Institute, Imperial College London, London, SW3 6LY, UK
William Cookson & Miriam F. Moffatt
Centre for Genetic Epidemiology and Biostatistics, University of Western Australia, Crawley, 6009, Western Australia, Australia
Matthew N. Cooper, Jennie Hui, Robert W. Lawrence & Lyle J. Palmer
Department of Genetics, University of North Carolina, Chapel Hill, 27599, North Carolina, USA
Damien C. Croteau-Chonka, Jennifer R. Kulzer & Karen L. Mohlke
Department of Biostatistics, Boston University School of Public Health, Boston, 02118, Massachusetts, USA
L. Adrienne Cupples, Julius S Ngwa & Charles C. White
Department of Health Sciences, University of Milan, Ospedale San Paolo, Milano, 20139, Italy
Daniele Cusi & Francesca Frau
Fondazione Filarete, Milano, Italy
Daniele Cusi
MRC Epidemiology Unit, Institute of Metabolic Science, Addenbrooke’s Hospital, CB2 0QQ, Cambridge, UK
Felix R. Day, Tuomas O. Kilpeläinen, Claudia Langenberg, Shengxu Li, Ruth J. F. Loos, Jian’an Luan, Ken K. Ong, Nicholas J. Wareham & Jing Hua Zhao
Department of Social Medicine, MRC Centre for Causal Analyses in Translational Epidemiology, Oakfield House, Bristol, BS8 2BN, UK
Ian N. M. Day, Debbie A. Lawlor, George Davey Smith & Nicholas John Timpson
Department of Dietetics-Nutrition, Harokopio University, 70 El. Venizelou Str, Athens, Greece
George V. Dedoussis, Maria Dimitriou & Eirini V. Theodoraki
Istituto di Neurogenetica e Neurofarmacologia del CNR, Monserrato, 09042, Cagliari, Italy
Mariano Dei, Andrea Maschio, Serena Sanna, Manuela Uda & Gianluca Usala
Istituto di Ricerca Genetica e Biomedicadel CNR, Monserrato, 09042, Cagliari, Italy
Mariano Dei & Serena Sanna
Department of Genetic Medicine and Development, University of Geneva Medical School, Geneva, 1211, Switzerland
Emmanouil T. Dermitzakis & Antigone S. Dimas
Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, OX3 7BN, UK
Antigone S. Dimas, Teresa Ferreira, Jon P. Krohn, Cecilia M. Lindgren, Reedik Mägi, Iain Mathieson, Mark I. McCarthy, Josine L. Min, Andrew P. Morris, John F. Peden, Inga Prokopenko, Joshua C. Randall, Nigel W. Rayner & Neil R. Robertson
Biomedical Sciences Research Center Al, 16672, Fleming, Vari, Greece
Antigone S. Dimas
Department of Pharmacy and Pharmacology, University of Bath, Bath, BA1 1RL, UK
Anna L. Dixon
Department of Internal Medicine B, Ernst-Moritz-Arndt University, Greifswald, 17475, Germany
Marcus Dörr
Netherlands Genomics Initiative (NGI)-sponsored Netherlands Consortium for Healthy Aging (NCHA), The Netherlands
Cornelia M. van Duijn, Karol Estrada, Albert Hofman, Carolina Medina-Gomez, Joyce B. J. van Meurs, Ben A. Oostra, Marjolein J. Peters, Fernando Rivadeneira, André G. Uitterlinden, Jacqueline C. M. Witteman & M. Carola Zillikens
Center of Medical Systems Biology, Leiden University Medical Center, Leiden, 2333, ZC, The Netherlands
Cornelia M. van Duijn & Gert-Jan van Ommen
The London School of Hygiene and Tropical Medicine, London, WC1E 7HT, UK
Shah Ebrahim
South Asia Network for Chronic Disease,
Shah Ebrahim
Department of Chronic Disease Prevention, National Institute for Health and Welfare, Unit of Public Health Genomics, Helsinki, 00014, Finland
Niina Eklund, Johannes Kettunen, Kati Kristiansson, Niina Pellikka, Markus Perola, Samuli Ripatti, Kaisa Silander & Ida Surakka
Institute of Molecular and Cell Biology, University of Tartu, Tartu, 51010, Estonia
Niina Eklund, Tõnu Esko, Andres Metspalu, Evelin Mihailov, Mari Nelis & Maris Teder-Laving
MRC-HPA Centre for Environment and Health, London, W2 1PG, UK
Paul Elliott
Clinic of Cardiology, West German Heart Centre, University Hospital of Essen, University Duisburg-Essen, Germany
Raimund Erbel
Nordic Center of Cardiovascular Research (NCCR), Lìbeck, 23538, Germany
Jeanette Erdmann & Heribert Schunkert
Universität zu Lìbeck, Medizinische Klinik II, Lìbeck, 23562, Germany
Jeanette Erdmann, Christina Loley & Heribert Schunkert
Universität zu Lìbeck, Medizinische Klinik II, Lìbeck, 23538, Germany
Jeanette Erdmann, Michael Preuss & Heribert Schunkert
Deutsches Zentrum fìr Herz-Kreislaufforschung e. V. (DZHK), Universität zu Lìbeck, Lìbeck, 23538, Germany
Jeanette Erdmann & Heribert Schunkert
Department of General Practice and Primary health Care, University of Helsinki, Helsinki, Finland
Johan G. Eriksson
National Institute for Health and Welfare, Helsinki, 00271, Finland
Johan G. Eriksson, Eero Kajantie & Leena Peltonen
Helsinki University Central Hospital, Unit of General Practice, Helsinki, 00280, Finland
Johan G. Eriksson
Folkhalsan Research Centre, Helsinki, 00250, Finland
Johan G. Eriksson, Bo Isomaa & Tiinamaija Tuomi
Vasa Central Hospital, Vasa, 65130, Finland
Johan G. Eriksson
Estonian Biocenter, Tartu, 51010, Estonia
Tõnu Esko, Andres Metspalu, Mari Nelis & Maris Teder-Laving
Department of Internal Medicine, Erasmus MC, Rotterdam, 3015GE, The Netherlands
Karol Estrada, Carolina Medina-Gomez, Joyce B. J. van Meurs, Marjolein J. Peters, Fernando Rivadeneira, André G. Uitterlinden & M. Carola Zillikens
Department of Social Medicine, MRC Centre for Causal Analyses in Translational Epidemiology, University of Bristol, Bristol, BS8 2BN, UK
David M Evans & Lavinia Paternoster
Division of Cardiovascular Epidemiology, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden
Ulf de Faire & Bruna Gigante
Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, 171 77, Sweden
Tove Fall, Andrea Ganna, Henrik Grönberg, Stefan Gustafsson, Per Hall, Erik Ingelsson, Yudi Pawitan, Nancy L. Pedersen, Emil Rehnberg & Patrik K. E. Magnusson
Cardiovascular Medicine, University of Oxford, Wellcome Trust Centre for Human Genetics, Oxford, OX3 7BN, UK
Martin Farrall, Anuj Goel & Hugh Watkins
Department of Clinical and Experimental Medicine, Epidemiology and Preventive Medicine Research Center, University of Insubria, Varese, Italy
Marco M. Ferrario
Department of Cardiology, Toulouse University School of Medicine, Rangueil Hospital, Toulouse, France
Jean Ferrières
Division of Intramural Research, National Heart, Lung and Blood Institute, Framingham Heart Study, Framingham, 01702, Massachusetts, USA
Caroline S. Fox
Department of Genetics, University Medical Center Groningen, University of Groningen, The Netherlands
Lude Franke & Pim van der Harst
Department of Clinical Sciences, Genetic and Molecular Epidemiology Unit, Skåne University Hospital Malmö, Lund University, Malmö, Sweden
Paul W. Franks & Dmitry Shungin
Department of Nutrition, Harvard School of Public Health, Boston, MA, USA
Paul W. Franks
Department of Public Health & Clinical Medicine, Umeå University, Umeå, Sweden
Paul W. Franks, Göran Hallmans & Dmitry Shungin
Center for Neurobehavioral Genetics, University of California, Los Angeles, 90095, California, USA
Nelson B. Freimer
Department of Genomics of Common Disease, School of Public Health, Imperial College London, London, W12 0NN, UK
Philippe Froguel
Department of Medicine, University of Maryland School of Medicine, Baltimore, 21201, Maryland, USA
Mao Fu, Jeffrey R. O’Connell & Alan R. Shuldiner
University of Chicago, Chicago, IL
Pablo V. Gejman & Alan R. Sanders
Northshore University Healthsystem, Evanston, 60201, Ilinois, USA
Pablo V. Gejman & Alan R. Sanders
Hagedorn Research Institute, Gentofte, 2820, Denmark
Anette P. Gjesing, Torben Hansen, Oluf Pedersen & Camilla Sandholt
Department of Medicine, University of Washington, Seattle, 98101, Washington, USA
Nicole L. Glazer
Cardiovascular Health Research Unit, University of Washington, Seattle, 98101, Washington, USA
Nicole L. Glazer, Guo Li & Bruce M. Psaty
University of Melbourne, Parkville, 3010, Australia
Michael E. Goddard
Department of Primary Industries, Melbourne, 3001, Victoria, Australia
Michael E. Goddard
Institute of Epidemiology, Helmholtz Zentrum Mìnchen - German Research Center for Environmental Health, Neuherberg, 85764, Germany
Harald Grallert, Iris M. Heid, Thomas Illig, Claudia Lamina & H-Erich Wichmann
Unit for Molecular Epidemiology, Helmholtz Zentrum Mìnchen - German Research Center for Environmental Health, Neuherberg, Germany
Harald Grallert & Thomas Illig
Research Unit for Molecular Epidemiology, Helmholtz Zentrum Mìnchen - German Research Center for Environmental Health, Neuherberg, Germany
Harald Grallert, Thomas Illig & Annette Peters
Department of Medicine III, Pathobiochemistry, University of Dresden, Dresden, 01307, Germany
Jìrgen Gräßler
Department of Medicine, University of Iceland, Reykjavik, Iceland
Vilmundur Gudnason & Albert Vernon Smith
Metabolism Initiative and Program in Medical and Population Genetics, Broad Institute, Cambridge, 02142, Massachusetts, USA
Candace Guiducci, Joel N. Hirschhorn, Elizabeth K. Speliotes, Brian Thomson & Sailaja Vedantam
Department of Genetics and Pathology, Rudbeck Laboratory, University of Uppsala, Uppsala, SE-75185, Sweden
Ulf Gyllensten, Wilmar Igl & Åsa Johansson
Department of Immunology, Genetics and Pathology, Uppsala University, Sweden
Ulf Gyllensten & Åsa Johansson
Division of Cardiovascular and Neuronal Remodelling, Multidisciplinary Cardiovascular Research Centre, Leeds Institute of Genetics, Health and Therapeutics, University of Leeds, UK
Alistair S. Hall
Department of Medicine, Atherosclerosis Research Unit, Solna,Karolinska Institutet, Karolinska University Hospital, Stockholm, 171 76, Sweden
Anders Hamsten & Rona J Strawbridge
Faculty of Health Science, University of Southern Denmark, Odense, 5000, Denmark
Torben Hansen
Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, 90048, California, USA
Talin Haritunians
Laboratory of Epidemiology, Demography, Biometry, National Institute on Aging, National Institutes of Health, Bethesda, 20892, Maryland, USA
Tamara B. Harris & Lenore J. Launer
Department of Cardiology, University Medical Center Groningen, University of Groningen, The Netherlands
Pim van der Harst & Irene M. Leach
Department of Clinical Sciences/Obstetrics and Gynecology, University of Oulu, Oulu, 90014, Finland
Anna-Liisa Hartikainen & Anneli Pouta
Department of Chronic Disease Prevention, National Institute for Health and Welfare, Chronic Disease Epidemiology and Prevention Unit, Helsinki, 00014, Finland
Aki S. Havulinna, Pekka Jousilahti, Seppo Koskinen & Veikko Salomaa
MRC Human Genetics Unit, Institute for Genetics and Molecular Medicine, Western General Hospital, Edinburgh, EH4 2XU, Scotland, UK
Caroline Hayward, Jennifer E. Huffman, Veronique Vitart & Alan F. Wright
Department of Psychiatry, Washington University School of Medicine, St Louis, MO 63108, USA
Andrew C Heath & Pamela A. Madden
Department of Child and Adolescent Psychiatry, University of Duisburg-Essen, Essen, 45147, Germany
Johannes Hebebrand & Anke Hinney
Department of Epidemiology and Preventive Medicine, Regensburg University Medical Center, Regensburg, 93053, Germany
Iris M. Heid, Michael F. Leitzmann & Thomas W. Winkler
Public Health and Gender Studies, Institute of Epidemiology and Preventive Medicine, Regensburg University Medical Center, Regensburg, Germany
Iris M. Heid & Thomas W. Winkler
Institute of Epidemiology I, Helmholtz Zentrum Mìnchen - German Research Center for Environmental Health, Neuherberg, Germany
Iris M. Heid & H-Erich Wichmann
Department of Internal Medicine, VU University Medical Centre, Amsterdam, The Netherlands
Martin den Heijer
Klinik und Poliklinik fìr Innere Medizin II, Universität Regensburg, Regensburg, 93053, Germany
Christian Hengstenberg
Regensburg University Medical Center, Innere Medizin II, Regensburg, 93053, Germany
Christian Hengstenberg
Klinik und Poliklinik fìr Innere Medizin II, Universitätklinikum Regensburg, Regensburg, 93053, Germany
Christian Hengstenberg & Klaus Stark
Biocenter Oulu, University of Oulu, Oulu, 90014, Finland
Karl-Heinz Herzig, Marjo-Riitta Jarvelin & Marika Kaakinen
Department of Physiology, Institute of Biomedicine, University of Oulu, Oulu, 90014, Finland
Karl-Heinz Herzig
Department of Psychiatry, Kuopio University Hospital and University of Kuopio, Kuopio, 70210, Finland
Karl-Heinz Herzig
Institute of Genetic Medicine, European Academy Bozen/Bolzano (EURAC), Affiliated Institute of the University of Lìbeck, Lìbeck, Germany, Bolzano/Bozen, 39100, Italy
Andrew A. Hicks, Irene Pichler, Peter P. Pramstaller & Claudia B. Volpato
Center for Biomedicine, European Academy Bozen/Bolzano (EURAC), Affiliated Institute of the University of Lìbeck, Lìbeck, Germany, Bolzano/Bozen, 39100, Italy
Andrew A. Hicks & Peter P. Pramstaller
Center for Biomedicine, European Academy Bozen/Bolzano (EURAC), Affiliated Institute of the University of Lìbeck, Lìbeck, Germany, Bolzano/Bozen, 39100, Italy
Andrew A. Hicks & Peter P. Pramstaller
Department of Epidemiology and Public Health, University College London, 1-19 Torrington Place, London, WC1E 6BT, UK
Aroon Hingorani, Mika Kivimaki, Mika Kivmaki, Meena Kumari & Claudia Langenberg
Divisions of Genetics and Endocrinology and Program in Genomics, Children’s Hospital, Boston, 02115, Massachusetts, USA
Joel N. Hirschhorn & Sailaja Vedantam
Department of Genetics, Harvard Medical School, Boston, 02115, Massachusetts, USA
Joel N. Hirschhorn & Sailaja Vedantam
Divisions of Genetics and Endocrinology and Centerfor Basic and Translational Obesity Research, Children’s Hospital, Boston, 02115, Massachusetts, USA
Joel N. Hirschhorn & Sailaja Vedantam
MRC Harwell, Harwell Science and Innovation Campus, Oxfordshire, OX11 0RD, UK
Christopher C. Holmes
Department of Statistics, University of Oxford, Oxford, OX1 3TG, UK
Christopher C. Holmes & George Nicholson
Interfaculty Institute for Genetics and Functional Genomics, Ernst-Moritz-Arndt-University Greifswald, Greifswald, 17487, Germany
Georg Homuth & Alexander Teumer
Department of Nutrition, Harvard School of Public Health, Boston, 02115, Massachusetts, USA
Frank B. Hu, David Hunter, Lu Qi & Tsegaselassie Workalemahu
Department of Medicine, Channing Laboratory, Brigham and Women’s Hospital and Harvard Medical School, Boston, 02115, Massachusetts, USA
Frank B. Hu, David Hunter, Lu Qi & Tsegaselassie Workalemahu
Department of Epidemiology, Harvard School of Public Health, Boston, 02115, Massachusetts, USA
Frank B. Hu, David Hunter, Peter Kraft & Liming Liang
Department of Biostatistics andBioinformatics, Emory University, Atlanta, 30322, Georgia, USA
Yi-Juan Hu
School of Population Health, The University of Western Australia, Nedlands, 6009, WA, Australia
Jennie Hui & Arthur W. Musk
Department of Internal Medicine, Institute of Clinical Medicine, University of Oulu, Oulu, 90014, Finland
Heikki Huikuri
Cardiovascular Genetics, British Heart Foundation Laboratories, Rayne Building, University College London, London, United Kingdom
Steve E. Humphries
School of Medicine and Pharmacology, The University of Western Australia, Nedlands, 6009, WA, Australia
Joseph Hung
Department of Public Health and General Practice, HUNT Research Centre, Norwegian University of Science and Technology, Levanger, 7600, Norway
Kristian Hveem, Kirsti Kvaloy, Kristian Midthjell & Carl G. P. Platou
Centre For Paediatric Epidemiolgy and Biostatistics/MRC Centre of Epidemiology for Child Health, University College of London Institute of Child Health, London, UK
Elina Hyppönen & Chris Power
Hannover Unified Biobank, Hannover Medical School, Hannover, 30625, Germany
Thomas Illig
Division of Research, Kaiser Permanente Northern California, Oakland, 94612, California, USA
Carlos Iribarren
Department of Epidemiology and Biostatistics, University of California, San Francisco, San Francisco, 94107, California, USA
Carlos Iribarren
Department of Social Services and Health Care, Jakobstad, 68601, Finland
Bo Isomaa
Core Genotyping Facility, SAIC-Frederick, Inc., NCI-Frederick, Frederick, 21702, Maryland, USA
Kevin B. Jacobs & Zhaoming Wang
School of Medicine and Pharmacology, University of Western Australia, Perth, 6009, Western Australia, Australia
Alan L. James
Department of Physiology, Institute of Neuroscience and Physiology, Sahlgrenska Academy, University of Gothenburg, Gothenburg, 405 30, Sweden
John-Olov Jansson
Institute of Medical Biometry and Epidemiology, University of Marburg, Marburg, 35037, Germany
Ivonne Jarick & Martina Mìller-Nurasyid
Institute of Health Sciences, University of Oulu, Oulu, 90014, Finland
Marjo-Riitta Jarvelin & Marika Kaakinen
National Institute for Health and Welfare, Oulu, 90101, Finland
Marjo-Riitta Jarvelin & Anneli Pouta
Institute for Medical Informatics, Biometry and Epidemiology (IMIBE), University Hospital of Essen, University of Duisburg-Essen, Essen, Germany
Karl-Heinz Jöckel, Susanne Moebus, Sonali Pechlivanis, Carolin Pìtter & Andre Scherag
Department of Cancer Research and Molecular Medicine, Faculty of Medicine, Norwegian University of Science and Technology (NTNU), Trondheim, N-7489, Norway
Åsa Johansson
Uppsala Clinical Research Center, Uppsala university hospital, Sweden
Åsa Johansson
Clinical Pharmacology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary, University of London, London, UK
Toby Johnson
Research Centre for Prevention and Health, Glostrup University Hospital, Glostrup, 2600, Denmark
Torben Jørgensen
Faculty of Health Science, University of Copenhagen, Copenhagen, 2100, Denmark
Torben Jørgensen
Department of Chronic Disease Prevention, National Institute for Health and Welfare, Population Studies Unit, Turku, 20720, Finland
Antti Jula
Department of Epidemiology, School of Public Health, University of North Carolina at Chapel Hill, Chapel Hill, 27514, North Carolina, USA
Anne E. Justice, Keri L. Monda & Kari E. North
Department of Clinical Physiology, University of Tampere and Tampere University Hospital, Tampere, 33520, Finland
Mika Kähönen
Hospital for Children and Adolescents, Helsinki University Central Hospital and University of Helsinki, 00029 HUS, Finland
Eero Kajantie
Department of Epidemiology and Medicine, Johns Hopkins Bloomberg School of Public Health, Baltimore, 21205, Maryland, USA
W. H. Linda Kao
Division of Gastroenterology, Massachusetts General Hospital, Boston, 02114, Massachusetts, USA
Lee M. Kaplan & Elizabeth K. Speliotes
MGH Weight Center, Massachusetts General Hospital, Boston, 02114, Massachusetts, USA
Lee M. Kaplan
Department of Epidemiology and Population Health, Albert Einstein College of Medicine, Bronx, 10461, New York, USA
Robert C. Kaplan
Institute for Molecular Medicine Finland (FIMM), University of Helsinki, Helsinki, 00014, Finland
Jaakko Kaprio, Johannes Kettunen, Kati Kristiansson, Aarno Palotie, Niina Pellikka, Leena Peltonen, Markus Perola, Samuli Ripatti, Kaisa Silander, Ida Surakka & Elisabeth Widen
Department of Public Health, Finnish Twin Cohort Study, University of Helsinki, Helsinki, 00014, Finland
Jaakko Kaprio & Kirsi H. Pietiläinen
Department of Mental Health and Substance Abuse Services, National Institute for Health and Welfare, Unit for Child and Adolescent Mental Health, Helsinki, 00271, Finland
Jaakko Kaprio & Robert N. Luben
National Institute for Health and Welfare, Unit for Child and Adolescent Psychiatry, Helsinki, Finland
Jaakko Kaprio
NIHR Oxford Biomedical Research Centre, Churchill Hospital, Oxford, OX3 7LJ, UK
Fredrik Karpe & Mark I. McCarthy
Oxford National Institute for Health Research Biomedical Research Centre, Churchill Hospital, Old Road Headington, Oxford, OX3 7LJ, UK
Fredrik Karpe & Mark I. McCarthy
Cardiovascular Research Center and Cardiology Division, Massachusetts General Hospital, Boston, 02114, Massachusetts, USA
Sekar Kathiresan
Center for Human Genetic Research, Massachusetts General Hospital, Boston, 02114, Massachusetts, USA
Sekar Kathiresan, Steven A. McCaroll, Shaun Purcell & Benjamin F. Voight
Program in Medical and Population Genetics, Broad Institute of Harvard and Massachusetts Institute of Technology, Cambridge, 02142, Massachusetts, USA
Sekar Kathiresan, Steven A. McCaroll, James Nemesh & Benjamin F. Voight
UKCRC Centre of Excellence for Public Health (NI) Queens University, Belfast
Frank Kee
Faculty of Medicine, Institute of Health Sciences, University of Oulu, Oulu, Finland
Sirkka M. Keinanen-Kiukaanniemi
Unit of General Practice, Oulu University Hospital, Oulu, Finland
Sirkka M. Keinanen-Kiukaanniemi
Department of Public Health and Primary Care, Institute of Public Health, University of Cambridge, Cambridge, CB2 2SR, UK
Kay-Tee Khaw
Department of Epidemiology, Biostatistics and HTA, Radboud University Nijmegen Medical Centre, Nijmegen, 6500, HB, The Netherlands
Lambertus A. Kiemeney, Femmie de Vegt & Sita H. Vermeulen
Department of Urology, Radboud University Nijmegen Medical Centre, Nijmegen, 6500, HB, The Netherlands
Lambertus A. Kiemeney
Comprehensive Cancer Center East, Nijmegen, 6501, BG, The Netherlands
Lambertus A. Kiemeney
National Institute for Health and Welfare, Diabetes Prevention Unit, Helsinki, 00271, Finland
Leena Kinnunen, Jaana Lindström, Jaakko Tuomilehto & Timo T. Valle
Department of Endocrinology, University Medical Center Groningen, University of Groningen, P.O. Box 30001, Groningen, 9700, RB, The Netherlands
Melanie M. Van der Klauw, Jana V. Van Vliet-Ostaptchouk & Bruce H. R. Wolffenbuttel
LURIC Study nonprofit LLC, Freiburg, Germany
Marcus E. Kleber
Mannheim Institute of Public Health, Social and Preventive Medicine, Medical Faculty of Mannheim, University of Heidelberg, Mannheim, Germany
Marcus E. Kleber & Winfried März
Department of Internal Medicine II – Cardiology, University of Ulm Medical Center, Ulm, Germany
Wolfgang Koenig
Andrija Stampar School of Public Health, Medical School, University of Zagreb, Zagreb, 10000, Croatia
Ivana Kolcic, Ozren Polasek & Lina Zgaga
1st Cardiology Department, Onassis Cardiac Surgery Center 356, Sygrou Ave., Athens, Greece
Genovefa Kolovou
Institut fìr Medizinische Biometrie und Statistik, Universität zu Lìbeck, Universitätsklinikum Schleswig-Holstein, Campus Lìbeck, Lìbeck, 23562, Germany
Inke R. König, Christina Loley & Michael Preuss
Interdisciplinary Centre for Clinical Research, University of Leipzig, Leipzig, 04103, Germany
Peter Kovacs
Department of Biostatistics, Harvard School of Public Health, Boston, 02115, Massachusetts, USA
Peter Kraft & Liming Liang
Institut fìr Pharmakologie, Universität Greifswald, Greifswald, 17487, Germany
Heyo K. Kroemer
Croatian Centre for Global Health, School of Medicine, University of Split, Split, 21000, Croatia
Vjekoslav Krzelj & Igor Rudan
MRC Unit for Lifelong Health & Ageing, London, UK
Diana Kuh, Ken K. Ong & Andrew Wong
Department of Chronic Disease Prevention, National Institute for Health and Welfare, Chronic Disease Epidemiology and Prevention Unit, Helsinki, 00271, Finland
Kari Kuulasmaa, Veikko Salomaa & Jarmo Virtamo
Department of Medicine, University of Kuopio and Kuopio University Hospital, Kuopio, 70210, Finland
Johanna Kuusisto & Markku Laakso
Department of Medicine, University of Eastern Finland, Kuopio Campus and Kuopio University Hospital, Kuopio, 70210, Finland
Johanna Kuusisto & Markku Laakso
Finnish Institute of Occupational Health, Oulu, 90220, Finland
Jaana H. Laitinen
Kuopio Research Institute of Exercise Medicine, Kuopio, Finland
Timo A. Lakka & Rainer Rauramaa
Division of Genetic Epidemiology, Department of Medical Genetics, Molecular and Clinical Pharmacology, Innsbruck Medical University, Innsbruck, 6020, Austria
Claudia Lamina
Institut inter-regional pour la sante (IRSA), La Riche, F-37521, France
Olivier Lantieri
Centre National de Genotypage, Evry, Paris, 91057, France
G. Mark Lathrop
The Queensland Brain Institute, The University of Queensland, Brisbane, Queensland, Australia
Sang Hong Lee & Peter M. Visscher
Department of Clinical Chemistry, University of Tampere and Tampere University Hospital, Tampere, 33520, Finland
Terho Lehtimäki
Department of Clinical Chemistry, Fimlab Laboratories, University of Tampere and Tampere University Hospital, Tampere, 33520, Finland
Terho Lehtimäki
Department of Medicine, Université de Montréal, Montreal, H3T 1J4, Quebec, Canada
Guillaume Lettre
Stanford University School of Medicine, Stanford, 93405, California, USA
Douglas F. Levinson
Department of Epidemiology, Tulane School of Public Health and Tropical Medicine, New Orleans, 70112, LA, USA
Shengxu Li
Department of Biostatistics, University of North Carolina, Chapel Hill, 27599, NC, USA
Dan-Yu Lin
Department of Medical Sciences, Uppsala University, Akademiska sjukhuset, Uppsala, 751 85, Sweden
Lars Lind
Human Genetics, Genome Institute of Singapore, Singapore, 138672, Singapore
Jianjun Liu
Department of Internal Medicine, Istituto Auxologico Italiano, Verbania, Italy
Antonio Liuzzi
Transplantation Laboratory, Haartman Institute, University of Helsinki, Helsinki, 00014, Finland
Marja-Liisa Lokki
The Charles Bronfman Institute of Personalized Medicine, Mount Sinai School of Medicine, New York, 10029, NY, USA
Ruth J. F. Loos
Child Health and Development Institute, Mount Sinai School of Medicine, New York, 10029, NY, USA
Ruth J. F. Loos
Department of Preventive Medicine, Mount Sinai School of Medicine, New York, 10029, NY, USA
Ruth J. F. Loos
Department of Internal Medicine, Institute of Medicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, 413 45, Sweden
Mattias Lorentzon, Claes Ohlsson & Liesbeth Vandenput
Department of Twin Research and Genetic Epidemiology, King’s College London, London, SE1 7EH, UK
Massimo Mangino, Nicole Soranzo & Timothy D. Spector
Università Vita-Salute San Raffaele, Chair of Nephrology San Raffaele Scientific Institute, OU Nephrology and Dialysis, Milan, 20132, Italy
Paolo Manunta
Department of Endocrinology, Diabetology and Nutrition, Bichat-Claude Bernard University Hospital, Assistance Publique des Hôpitaux de Paris, Paris, F-75018, France
Michel Marre
Cardiovascular Genetics Research Unit, Université Henri Poincaré-Nancy 1, Nancy, 54000, France
Michel Marre
Genetic Epidemiology Laboratory, Queensland Institute of Medical Research, 4006, Queensland, Australia
Sarah E. Medland & Nicholas G. Martin
Queensland Institute of Medical Research, 4029, Queensland, Australia
Grant W. Montgomery, Sarah E. Medland, Nicholas G. Martin & Jian Yang
Synlab Academy, Mannheim, Germany
Winfried März
Department of Social Medicine, Avon Longitudinal Study of Parents and Children (ALSPAC) Laboratory, University of Bristol, Bristol, BS8 2BN, UK
Wendy L. McArdle
School of Social and Community Medicine, University of Bristol, UK
Wendy L. McArdle
Department of Molecular Biology, Massachusetts General Hospital, Boston, 02114, Massachusetts, USA
Steven A. McCaroll & Benjamin F. Voight
Division of Health, Research Board, An Bord Taighde Sláinte, Dublin, 2, Ireland
Anne McCarthy
Institute of Human Genetics, Klinikum rechts der Isar der Technischen Universität Mìnchen, Munich, 81675, Germany
Thomas Meitinger
Institute of Human Genetics, Helmholtz Zentrum Mìnchen - German Research Center for Environmental Health, Neuherberg, 85764, Germany
Thomas Meitinger
Department of Clinical Epidemiology and Biostatistics, McMasterUniversity, Hamilton, L8S 4L8, Ontario, Canada
David Meyre
Human Genetics, Leiden University Medical Center, Leiden, 2333, The Netherlands
Josine L. Min
Merck Research Laboratories, Merck & Co., Inc., Boston, 02115, Massachusetts, USA
Cliona Molony
Center for Observational Research, Amgen, Thousands Oaks, 91320, CA
Keri L. Monda
Molecular Epidemiology Laboratory, Queensland Institute of Medical Research, 4006, Queensland, Australia
Grant W. Montgomery
Genetics Division, GlaxoSmithKline, King of Prussia, Pennsylvania, 19406, USA
Vincent Mooser
Medical Research Institute, University of Dundee, Ninewells Hospital and Medical School, Dundee, DD1 9SY
Andrew D. Morris & Colin N. A. Palmer
Institute of Human Genetics, University of Bonn, Bonn, Germany
Thomas W. Mìhleisen & Markus M. Nöthen
Department of Genomics, Life & Brain Center, University of Bonn, Bonn, Germany
Thomas W. Mìhleisen & Markus M. Nöthen
Department of Medicine I, University Hospital Grosshadern, Ludwig-Maximilians-Universität, Munich, Germany
Martina Mìller-Nurasyid
Institute of Medical Informatics, Biometry and Epidemiology, Chair of Genetic Epidemiology, Ludwig-Maximilians-Universität, Munich, Germany
Martina Mìller-Nurasyid
Department of Respiratory Medicine, Sir Charles Gairdner Hospital, Nedlands, 6009, Australia
Arthur W. Musk
Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, 02114, MA
Benjamin M. Neale
MRC Harwell, Harwell, UK
George Nicholson
Division of Cardiology, Cardiovascular Laboratory, Helsinki University Central Hospital, Helsinki, 00029, Finland
Markku S. Nieminen & Juha Sinisalo
Department of Community Medicine, Faculty of Health Sciences, University of Tromsø, Tromsø, Norway
Inger Njølstad & Tom Wilsgaard
Department of Clinical Medicine, Faculty of Health Sciences, University of Tromsø, Tromsø, Norway
Inger Njølstad
Department of Public Health, Section of Epidemiology, Aarhus University, Denmark
Ellen A. Nohr
Unit of Genetic Epidemiology and Bioinformatics, Dept of Epidemiology, University Medical Center Groningen, University of Groningen, P.O. Box 30001, Groningen, 9700, RB, The Netherlands
Ilja M. Nolte & Harold Snieder
Department of Epidemiology, University of Groningen, University Medical Center Groningen, The Netherlands
Ilja M. Nolte & Ronald P. Stolk
Carolina Center for Genome Sciences, School of Public Health, University of North Carolina Chapel Hill, Chapel Hill, 27514, North Carolina, USA
Kari E. North
Neurogenetics Laboratory, Queensland Institute of Medical Research, 4006, Queensland, Australia
Dale R. Nyholt
Interdisciplinary Center Psychopathology and Emotion Regulation, University of Groningen, University Medical Center Groningen, The Netherlands
Albertine J. Oldehinkel
Department of Human Genetics, Leiden University Medical Center, Leiden, 2333, ZC, The Netherlands
Gert-Jan van Ommen
Department of Clinical Genetics, Erasmus MC, Rotterdam, 3015GE, The Netherlands
Ben A. Oostra
Centre for Medical Systems Biology & Netherlands Consortium on Healthy Aging, Leiden, The Netherlands
Ben A. Oostra
NHS Blood and Transplant, Cambridge Centre, Cambridge, CB2 0PT, UK
Willem H. Ouwehand, Aparna Radhakrishnan, Augusto Rendon, Jennifer G. Sambrook & Jonathan C. Stephens
Department of Pathology and Molecular Medicine, McMaster University, Hamilton, L8N3Z5, Ontario, Canada
Guillaume Paré
Amgen, Cambridge, 02139, Massachusetts, USA
Alex N. Parker
Department of Cardiovascular Medicine, University of Oxford, Level 6 West Wing, John Radcliffe Hospital, Headley Way, Headington, OX3 9DU, Oxford
John F. Peden
Illumina Inc. Cambridge, USA
John F. Peden
Institute of Biomedical Sciences, University of Copenhagen, Copenhagen, 2200, Denmark
Oluf Pedersen
Faculty of Health Science, University of Aarhus, Aarhus, 8000, Denmark
Oluf Pedersen
The Broad Institute of Harvard and MIT, Cambridge, 02142, Massachusetts, USA
Leena Peltonen & Shaun Purcell
Department of Medical Genetics, University of Helsinki, Helsinki, 00014, Finland
Leena Peltonen
Department of Psychiatry/EMGO Institute, VU University Medical Center, Amsterdam, 1081 BT, The Netherlands
Brenda Penninx & Jan H. Smit
Department of Psychiatry, Leiden University Medical Centre, Leiden, 2300 RC, The Netherlands
Brenda Penninx
Department of Psychiatry, University Medical Centre Groningen, Groningen, 9713 GZ, The Netherlands
Brenda Penninx
Institute of Epidemiology II, Helmholtz Zentrum Mìnchen - German Research Center for Environmental Health, Neuherberg, Germany
Annette Peters & Barbara Thorand
Munich Heart Alliance, Munich, Germany
Annette Peters
Department of Psychiatry, Obesity Research unit, Helsinki University Central Hospital, Helsinki, Finland
Kirsi H. Pietiläinen & Aila Rissanen
Department of Medicine, Levanger Hospital, The Nord-Trøndelag Health Trust, Levanger, 7600, Norway
Carl G. P. Platou
Gen-Info Ltd, Zagreb, 10000, Croatia
Ozren Polasek
Faculty of Medicine, University of Split, Croatia
Ozren Polasek
Department of Neurology, General Central Hospital, Bolzano, Italy
Peter P. Pramstaller
Department of Neurology, University of Lìbeck, Lìbeck, Germany
Peter P. Pramstaller
Departments of Epidemiology, Medicine and Health Services, University of Washington, Seattle, 98195, Washington, USA
Bruce M. Psaty
Group Health Research Institute, Group Health, Seattle, 98101, Washington, USA
Bruce M. Psaty
Department of Psychiatry, Harvard Medical School, Boston, 02115, Massachusetts, USA
Shaun Purcell
Research Centre of Applied and Preventive Cardiovascular Medicine, University of Turku, Turku, 20520, Finland
Olli Raitakari
The Department of Clinical Physiology, Turku University Hospital, Turku, 20520, Finland
Olli Raitakari
The Department of Clinical Physiology and Nuclear Medicine, Turku University Hospital, Turku, 20520, Finland
Olli Raitakari
Department of Clinical Physiology and Nuclear Medicine, Kuopio University Hospital, Kuopio, Finland
Rainer Rauramaa
MRC Biostatistics Unit, Institute of Public Health, Cambridge, UK
Augusto Rendon
Department of Clinical Sciences, Lund University, Malmö, 20502, Sweden
Martin Ridderstråle
Finnish Diabetes Association, Kirjoniementie 15, Tampere, 33680, Finland
Timo E. Saaristo
Pirkanmaa Hospital District, Tampere, Finland
Timo E. Saaristo
Medizinische Klinik II, Universität zu Lìbeck Ratzeburger Allee 160, Lìbeck, D-23538, Germany
Hendrik Sager
Department of Cardiovascular Sciences, University of Leicester, Glenfield Hospital, Leicester, LE3 9QP, UK
Nilesh J. Samani
Leicester NIHR Biomedical Research Unit in Cardiovascular Disease, Glenfield Hospital, Leicester, LE3 9QP, UK
Nilesh J. Samani
South Karelia Central Hospital, Lappeenranta, 53130, Finland
Jouko Saramies
Pacific Biosciences, Menlo Park, California, 94025, USA
Eric E. Schadt
Sage Bionetworks, Seattle, 98109, Washington, USA
Eric E. Schadt
Department of Genetics and Genomic Sciences, Mount Sinai School of Medicine, One Gustave L. Levy Place, Box 1498, New York, 10029-6574, NY, USA
Eric E. Schadt
Institute of Genomics and Multiscale Biology, Mount Sinai School of Medicine, One Gustave L. Levy Place, Box 1498, New York, 10029-6574, NY, USA
Eric E. Schadt
Institute for Community Medicine, University Medicine Greifswald, Greifswald, Germany
Sabine Schipf
Laboratory of Genetics, National Institute on Aging, Baltimore, 21224, Maryland, USA
David Schlessinger
Institut fìr Klinische Molekularbiologie, Christian-Albrechts Universität, Kiel, Germany
Stefan Schreiber
Department of Medicine III, Prevention and Care of Diabetes, University of Dresden, Dresden, 01307, Germany
Peter E. H. Schwarz
Geriatrics Research and Education Clinical Center, Baltimore Veterans Administration Medical Center, Baltimore, 21201, Maryland, USA
Alan R. Shuldiner
Department of Odontology, Umeå University, Sweden
Dmitry Shungin
Azienda ospedaliera di Desio e Vimercate, Milano, Italy
Stefano Signorini
Institute of Preventive Medicine, Bispebjerg University Hospital, Copenhagen, and Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen, Denmark
Thorkild I. A. Sørensen
Center for Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan, USA
Elizabeth K. Speliotes
Department of Internal Medicine, Division of Gastroenterology, University of Michigan, Ann Arbor, Michigan, USA
Elizabeth K. Speliotes
University of Eastern Finland and Kuopio University Hospital, Kuopio, 70210, Finland
Alena Stančáková
Regensburg University Medical Center, Clinic and Policlinic for Internal Medicine II, Regensburg, 93053, Germany
Klaus Stark
deCODE Genetics, Reykjavik, 101, Iceland
Kari Stefansson, Valgerdur Steinthorsdottir, Gudmar Thorleifsson, Unnur Thorsteinsdottir & G. Bragi Walters
Faculty of Medicine, University of Iceland, Reykjavík, 101, Iceland
Kari Stefansson & Unnur Thorsteinsdottir
Division of Community Health Sciences, St George’s, University of London, London, SW17 0RE, UK
David P Strachan
Division of Population Health Sciences and Education, St George’s, University of London, London, SW17 0RE, UK
David P Strachan
Department of Medicine, University of Leipzig, Leipzig, 04103, Germany
Michael Stumvoll & Anke Tönjes
LIFE Study Centre, University of Leipzig, Leipzig, Germany
Michael Stumvoll
University of Leipzig, IFB Adiposity Diseases, Leipzig, Germany
Michael Stumvoll & Anke Tönjes
Uppsala University / Dept. of Medical Sciences, Molecular Medicine, Uppsala, 751 85, Sweden
Ann-Christine Syvanen
Coordination Centre for Clinical Trials, University of Leipzig, Härtelstr. 16-18, Leipzig, 04103, Germany
Anke Tönjes
INSERM UMR_S 937, ICAN Institute, Pierre et Marie Curie Medical School, Paris, 75013, France
David-Alexandre Tregouet
Department of Pharmacological Sciences, University of Milan, Monzino Cardiology Center, IRCCS, Milan, Italy
Elena Tremoli
Department of Clinical and Experimental Cardiology, Heart Failure Research Centre, Academic Medical Center, Amsterdam, The Netherlands
Mieke D. Trip
Department of Medicine, Helsinki University Central Hospital, Helsinki, 00290, Finland
Tiinamaija Tuomi
Research Program of Molecular Medicine, University of Helsinki, Helsinki, 00014, Finland
Tiinamaija Tuomi
Department of Public Health, Hjelt Institute, University of Helsinki, Helsinki, 00014, Finland
Jaakko Tuomilehto
South Ostrobothnia Central Hospital, Seinajoki, 60220, Finland
Jaakko Tuomilehto
Red RECAVA Grupo RD06/0014/0015, Hospital Universitario La Paz, Madrid, 28046, Spain
Jaakko Tuomilehto
Centre for Vascular Prevention, Danube-University Krems, Krems, 3500, Austria
Jaakko Tuomilehto
Department of Oncology, University of Cambridge, Cambridge, CB1 8RN, UK
Jonathan Tyrer
Department of Public Health and Clinical Nutrition, University of Eastern Finland, Finland
Matti Uusitupa
Research Unit, Kuopio University Hospital, Kuopio, Finland
Matti Uusitupa
Department of Human Genetics, Radboud University Nijmegen Medical Centre, PO Box 9101, Nijmegen, 6500, HB, The Netherlands
Sita H. Vermeulen
Department of Medicine, University of Turku and Turku University Hospital, Turku, 20520, Finland
Jorma Viikari
Queensland Statistical Genetics Laboratory, Queensland Institute of Medical Research, 4006, Queensland, Australia
Jian Yang & Peter M. Visscher
Department of Internal Medicine, Centre Hospitalier Universitaire Vaudois (CHUV) University Hospital, Lausanne, 1011, Switzerland
Peter Vollenweider & Gérard Waeber
Institut fìr Community Medicine, Greifswald, 17489, Germany
Henry Völzke
Institute for Community Medicine, Ernst-Moritz-Arndt-University Greifswald, Greifswald, Germany
Henry Völzke
Institut fìr Klinische Chemie und Laboratoriumsmedizin, Universität Greifswald, Greifswald, 17475, Germany
Henri Wallaschofski
Institute of Clinical Chemistry and Laboratory Medicine, University Medicine Greifswald, Greifswald, 17475, Germany
Henri Wallaschofski
Department of Preventive Medicine, Keck School of Medicine, University of Southern California, Los Angeles, 90089, California, USA
Richard M. Watanabe
Klinikum Grosshadern, Munich, 81377, Germany
H-Erich Wichmann
Ludwig-Maximilians-Universität, Institute of Medical Informatics, Biometry and Epidemiology, Chair of Epidemiology, Munich, 81377, Germany
H-Erich Wichmann
Institute of Medical Informatics, Biometry and Epidemiology, Chair of Epidemiology, Ludwig-Maximilians-Universität, and Klinikum Grosshadern, Munich, Germany
H-Erich Wichmann
Klinikum Grosshadern, Munich, Germany
H-Erich Wichmann
Cardiology Group, Frankfurt-Sachsenhausen, Germany
Bernhard R. Winkelmann
Steno Diabetes Center, Gentofte, 2820, Denmark
Daniel R. Witte
Centre for Public Health, Queen’s University, Belfast, UK
John W. G. Yarnell
Department of Physiatrics, Lapland Central Hospital, Rovaniemi, 96101, Finland
Paavo Zitting
Genetic and Genomic Epidemiology Unit, Wellcome Trust Centre for Human Genetics, Oxford., OX3 7BN
Krina T. Zondervan
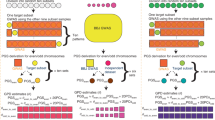
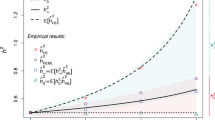
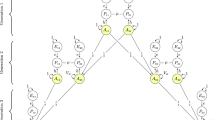
M.R.R., J.Y. and P.M.V. conceived and designed the study. M.R.R., A.K. and M.G. analysed the data. M.R.R. devised and performed the simulations. A.A.E.V., W.J.P., A.A., B.Z., S.M. provided statistical support. 23andMe Inc., The LifeLines cohort, GIANT consortium, G.W.M., N.G.M., M.L., P.L., D.C., J.V.V.O., M.B.M., H.S., W.G.I., P.K.E.M, N.L.P, M.McG. and K.E.N. provided study oversight, sample collection and management. M.R.R. and P.M.V. derived the theory and wrote the manuscript. All collaborators reviewed and approved the final manuscript.