
Abstract
The removal of carbon dioxide from the atmosphere by the marine biological pump is a key regulator of Earth’s climate; however, the ocean also serves as a large source of nitrous oxide, a potent greenhouse gas and ozone-depleting substance. Although biological carbon sequestration and nitrous oxide production have been individually studied in the ocean, their combined impacts on net greenhouse forcing remain uncertain. Here we show that the magnitude of nitrous oxide production in the epipelagic zone of the subtropical ocean covaries with remineralization processes and thus acts antagonistically to weaken the radiative benefit of carbon removal by the marine biological pump. Carbon and nitrogen isotope tracer incubation experiments and nitrogen isotope natural abundance data indicate enhanced biological activity promotes nitrogen recycling, leading to substantial nitrous oxide production via both oxidative and reductive pathways. These shallow-water nitrous oxide sources account for nearly half of the air–sea flux and counteract 6–27% (median 9%) of the greenhouse warming mitigation achieved by carbon export via the biological pump.
Similar content being viewed by others
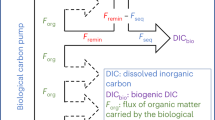
Main
The ocean plays a crucial role in the global climate system through modulating atmospheric greenhouse gases by absorbing nearly 30% of anthropogenic carbon dioxide (CO2) and releasing 20% of total nitrous oxide (N2O) emissions to the atmosphere1,2. The marine biological pump (defined as the biologically driven processes that transfer carbon from the surface ocean to the ocean’s interior) is the dominant mechanism driving long-term CO2 exchange across the air–sea interface and plays a critical role in regulating atmospheric CO2 and climate3,4,5. The efficiency of the marine biological pump, defined by the ratio of carbon export to net primary production at a specific reference depth (for example, the base of the euphotic zone), is often estimated to be ~10% in the global ocean5, and even lower in the subtropical oceans6,7. In the subtropical oceans, a large fraction of newly produced organic material undergoes remineralization in the upper 200 m (the epipelagic zone), resulting in rapid carbon and nitrogen transformations between organic and inorganic forms. Numerous studies on the ocean’s biological pump have focused on the magnitude and controls of carbon removal; however, the potential counter effect of N2O emissions in offsetting the radiative effect of CO2 removal by the biological pump has been largely overlooked.
In the marine nitrogen cycle, N2O is mainly produced as a by-product of nitrification and as an intermediate during denitrification, both of which are largely controlled by organic matter supply and remineralization8. In the oxygenated ocean, nitrification is considered the dominant source of N2O (refs. 8,9,10). However, recent studies on marine N2O suggest the sources are more complex, including both aerobic nitrification and anaerobic nitrate (NO3−) and nitrite (NO2−) reduction in anaerobic micro-niches associated with marine aggregates11 or zooplankton guts12. Ammonia-oxidizing archaea, the dominant ammonia oxidizer in the open oceans13, utilize a hybrid N2O production pathway that is distinct from that of their bacterial counterparts, in which intermediates sourced from ammonium (NH4+) and NO2− co-contribute to N2O formation14. The physiological and enzymatic details of this pathway remain mostly unresolved15,16. Moreover, sources of N2O production to the epipelagic ocean are less well studied than in the mesopelagic waters, despite the fact that remineralization of nitrogen is more intensive in the epipelagic ocean. This lack of investigation stems from the recognition that ammonia oxidation is inhibited by light17 and the abundance of ammonia-oxidizing archaea is relatively low in the upper ocean13. Hence, the pathways, controls, relative contributions from various N2O sources to the N2O pool and linkages between primary productivity and N2O production in the epipelagic ocean remain unclear, hampering our ability to constrain the role of the ocean in the atmospheric N2O budget.
Nitrogen is a primary limiting nutrient to phytoplankton growth over much of the low-latitude oceans18,19. Supply of exogenous sources of nitrogen to the euphotic zone can control net productivity in these waters, with new nitrogen introduced via NO3− supply from subsurface, N2 fixation and nitrogen deposition20. In addition, a large fraction of primary productivity is controlled via nitrogen supplied from remineralization within the epipelagic zone, supporting regenerated production20,21. Under the assumption of steady state, new production should be quantitatively related to export of material out of the epipelagic zone into the interior waters. Thus, regenerated and export production are two competing sides of biological productivity, where regeneration drives recycling (including N2O production), while export determines organic carbon removal to depth. Intensive organic matter remineralization not only reduces CO2 sequestration efficiency, but also contributes to N2O production via nitrogen recycling. The potential counter effect of N2O emission in offsetting the radiative effect of CO2 sequestration via export has been investigated in a few geoengineering and nitrogen deposition experiments22,23,24; however, the source of N2O, and links between biological CO2 sequestration and N2O production, have not been quantified.
We hypothesized that in the vast subtropical oligotrophic oceans, where export production is inefficient3,4,5,6,7, the counter effect of N2O emission on carbon removal by the biological pump could be substantial. We measured rates of N2O production and carbon export in the epipelagic ocean extending from the South China Sea (SCS) into the North Pacific Subtropical Gyre (NPSG) during seven cruises conducted over eight years (Supplementary Fig. 1 and Supplementary Tables 1 and 2). We demonstrate that a large fraction of N2O in the surface ocean is locally produced in the epipelagic waters via both oxidative and reductive pathways. Furthermore, the magnitude of this shallow N2O source appears to covary spatially with biological productivity, offsetting greenhouse warming mitigation achieved by carbon export. Incorporating N2O production that accompanies organic matter remineralization is necessary to quantify net climatic consequences associated with the marine biological pump.
Low export efficiency in the oligotrophic subtropical ocean
The epipelagic waters at all study sites exhibited characteristics typical for the thermally stratified SCS and the NPSG, with high oxygen and low nutrient concentrations throughout the shallow mixed layer (Extended Data Figs. 1 and 2 and Supplementary Table 3). NH4+ concentrations were persistently low (mean ± s.d., 24.4 ± 23.6 nmol l−1 in the SCS and 21.0 ± 24.3 nmol l−1 in the NPSG) with occasional peaks of 50–200 nmol l−1 below the mixed layer. By contrast, NO2− concentrations consistently showed a primary nitrite maximum (PNM) slightly below the deep chlorophyll maximum (DCM). A prominent maximum in the NO3− to Si concentration ratio was also frequently observed in the vicinity of the PNM layer. Particulate organic carbon (POC) and particulate nitrogen (PN) concentrations generally decreased with depth, and concentrations were greater in the shelf region than the open ocean (Extended Data Fig. 3).
Rates of primary production in the nutrient-depleted waters were consistently low (<1 μmol C l−1 d−1) and decreased with depth at both the SCS and the NPSG stations (Extended Data Fig. 4). Depth-integrated (0–125 m) primary production ranged from 10.4 ± 0.4 to 44.1 ± 3.0 mmol C m−2 d−1, similar to long-term observations at the NPSG Station ALOHA and the BATS station in the Atlantic6,7. Carbon export at 200 m in the SCS and NPSG stations ranged from 0.3 ± 0.1 to 2.0 ± 0.4 mmol C m−2 d−1, with low export ratios (averaging 3.9 ± 2.3%; Table 1 and Supplementary Discussion 1). Such results are typical of the subtropical oceans, where most primary production undergoes rapid remineralization, fuelling intensive and rapid nitrogen recycling in the epipelagic zone7,25.
A large shallow source of N2O in the epipelagic zone
N2O concentrations in the near-surface waters were consistently near or in excess of the air-saturation state. The mean saturation value of N2O in the coastal and shelf region (121 ± 6%) was slightly higher than the open ocean (110 ± 6%). The corresponding air–sea N2O flux in the shelf and open ocean stations during the observation period was 2.8 ± 0.9 μmol m−2 d−1 and 1.1 ± 0.6 μmol m−2 d−1, respectively, demonstrating that both regions were sources of N2O to the atmosphere (Supplementary Table 4), in agreement with previous observations in the subtropical oceans26,27,28.
Our high-resolution vertical profile sampling of the epipelagic waters provided insights into the vertical variations in N2O concentrations. N2O concentrations and the resulting air-saturation states generally increased with depth. Distinctive N2O concentration peaks, which deviate from simple vertical mixing, were observed at four stations (Fig. 1 and Extended Data Fig. 5). At these sites, peaks in N2O concentrations occurred within narrow depth intervals of 10–20 m and could be easily missed by coarser vertical sampling resolution. The location of these N2O peaks varied in depth, temperature, salinity and density (Extended Data Fig. 6), but consistently overlapped with the PNM and NO3−/Si maximum layers (Fig. 1). The proximity of an N2O peak to the PNM suggests a spatial coupling between N2O accumulation and intensive nitrogen recycling at or around the PNM layer, where high ammonia oxidation rates occur29, leading to enhanced N2O production and accumulation of NO2− (refs. 30,31). The NO3−/Si maximum provides additional evidence of intensive remineralization of organic nitrogen and subsequent nitrification at this depth32,33, reinforcing the contribution of local sources to the N2O accumulation.

a–d, N2O concentration (blue dots), NO2− concentration (green dots) and NO3−/Si (orange dots) at stations A03 (2013; a), B05 (2013; b), the Southeast Asian Time-series Study site (SEATs; 2014; c) and SEATs (2017; d). Blue bars mark the depth range with the distinctive N2O accumulation. Data are presented as mean values ± s.d. of triplicate sample measurement in panels c and d and are smaller than the symbols where not visible.
High-resolution vertical profiles of N2O stable isotopes (δ15N–N2O and δ18O–N2O) at additional stations provided further evidence for near-surface in-situ production of N2O in the subtropical ocean (Fig. 2). The dual isotopes of N2O covaried with depth, decreasing from near equilibrium with the atmosphere in the near-surface waters (average δ15N = 7.4 ± 0.4‰; δ18O = 45.3 ± 0.6‰), to minima in both isotope ratios in the vicinity of the NO3−/Si maximum and the PNM (minima were 0.4 ± 0.2‰ to 4.9 ± 0.3‰ and 0.5 ± 0.4‰ to 4.0 ± 0.4‰ lower than the values in surface water for δ15N and δ18O, respectively). Below the PNM, the δ15N and δ18O values of N2O increased with depth. These results indicate that the prominent dual isotope minima around the PNM probably do not derive from vertical mixing of surface and deep waters. Lateral advection of water with low isotopic signatures is also unlikely because the isotopic minimum layers occurred within waters of varying density and salinity (Extended Data Fig. 7). Hence, the most likely cause for the local δ15N and δ18O minima is in-situ N2O production, as has previously been observed at Station ALOHA in the NPSG10,34. The widespread dual isotope minima in our study area (12 out of 15 stations) further suggest that shallow N2O production is ubiquitous in the subtropical ocean. The shallowness of the feature in the SCS is noteworthy (60–120 m in the epipelagic zone versus ~300 m in the upper mesopelagic zone at ALOHA10,34). This shallow feature points to a potentially sensitive climatic consequence of nitrogen recycling along ocean margins due to the shorter distance from the isotope minimum layer to the air–sea interface and more vigorous physical dynamics in the epipelagic ocean.

a–e, N2O concentration (blue dots), δ15N (green dots) and δ18O (orange dots) at stations D1 (a), X2 (b), X5 (c), J1 (d) and SEATs (e) in the SCS in 2014. f–i, Stations P5 (f), C5 (g), K1 (h) and X1 (i) in the NWP in 2015. j–l, Stations SEATs (j), B2 (k) and SS1 (l) in the SCS in 2017. m–o, Stations A2 (m), A4 (n) and SEATs (o) in the SCS in 2018. The dashed lines mark the depth with NO3−/Si maximum and purple triangles on the right y axis mark the depth of the PNM. Data are presented as mean values ± s.d. of triplicate sample measurement and are smaller than the symbols where not visible. Note that P5 and C5 are shelf stations.
The relative contribution of in-situ N2O production from the isotope minimum layer to air–sea flux can be constrained using an isotope mass balance model under the assumption that N2O in the isotopic minima layer derives from a mixture of N2O diffusing from the concentration maximum layer and locally produced N2O (ref. 10). Applied to our stations, this two-component model reveals that shallow N2O production contributes 41.6 ± 21.0% by using δ15N mass balance and 31.3 ± 11.0% by using δ18O mass balance (Supplementary Discussion 2 and Supplementary Table 5), implying the shallow N2O source is a substantial contributor to air–sea flux in the oligotrophic oceans.
Multiple biological N2O sources in the oxygenated water
The underlying mechanisms that cause the N2O isotope minimum are not fully resolved. Nitrification was previously considered a primary N2O source to the well-oxygenated open ocean8,9,10. However, later observations of δ18O–N2O and isotope labelling incubations suggest part of the N2O in the isotope minima may be produced through nitrifier denitrification or denitrification in particle-associated microenvironments34,35. Because NO2− incorporates oxygen atoms from water30, the δ18O–H2O signal of seawater is incorporated into N2O from NO2−. In addition, NO2− at the PNM layer is usually depleted in 15N, that is, −0.39 ± 3.45‰ (Supplementary Table 6), which is lower than the reported δ15N–NO3− (4.8 ± 0.3‰) and δ15N–PN (4.2 ± 1.0‰) at the base of the euphotic zone in the SCS36. These results indicate NO2− could be an important precursor to N2O, contributing to the dual isotope minima observed in the PNM layer. Nevertheless, because NO2− can be incorporated into N2O via denitrification, nitrifier denitrification or the hybrid pathway, the relative contribution of these potential sources cannot be determined using natural abundance data alone.
We conducted a set of 15N isotope tracer incubations aimed at identifying sources and quantifying their relative contributions to N2O production. Results from these experiments show that multiple precursors contribute to N2O production in the epipelagic ocean (Fig. 3). Notably, N2O production was sometimes detected in the upper mixed layer even though ammonia oxidation rates were below detection limits (Extended Data Fig. 8). Gross N2O production increased with depth to a maximum in the vicinity of the N2O isotopic minima layer, providing additional evidence for active in-situ N2O production. Both NH4+ and NO2− seem to be involved in N2O production, while NO3− reduction to N2O was occasionally detected. Depth-integrated N2O production rates (0–200 m) ranged from 0.10 ± 0.02 to 0.57 ± 0.03 μmol m−2 d−1 (average 0.28 ± 0.04 μmol m−2 d−1) at the open ocean stations. These rates account for 29.5 ± 1.8% to 61.3 ± 6.4% (average 40.0 ± 7.7%) of the air–sea N2O flux in the open ocean stations (Supplementary Table 4), demonstrating that a large proportion of air–sea N2O flux in the subtropical ocean can be produced locally in the epipelagic zone.

a–d, Stations P5 (a), C5 (b), X1 (c) and K1 (d) in the NWP in 2015. e,f, Stations A2 (e) and SEATs (f) in the SCS in 2018. g,h, Stations A32 (g) and Z2 (h) in the NPSG in 2019. The figure shows N2O production rates from NH4+ (green dots), NO2− (orange dots), NO3− (blue dots) and the total N2O production rate (grey dots). Purple triangles on the right y axis mark the depth with lowest δ15N–N2O in the investigated depth profile. The dashed lines in a and b mark the bottom depth of the shelf stations. Data are presented as mean rates ± s.d. of triplicate sample incubation in the SCS and NPSG, and duplicates in the 2015 NWP cruise, and are smaller than the symbols where not visible.
Production of both 45N2O and 46N2O was detected, with 45N2O frequently comprising a higher fraction of labelled N2O than 46N2O. The fraction of measured 45N2O frequently exceeded that predicted from the binomial distribution. Excess 45N2O has also been observed in several studies conducted in the mid-latitude North Atlantic37, the western North Pacific38 and the eastern tropical South Pacific39,40 and has been interpreted as evidence of hybrid N2O production. However, isotope dilution of the tracer substrates, and ammonia oxidation coupled to NO2− reduction during the incubation, would also cause deviation of the measured 45N2O:46N2O from the predicted ratio, and care should be taken in interpreting the observed excess 45N2O (Supplementary Discussion 3). The presence of 46N2O in both 15NO2− and 15NO3− labelling incubations suggests production of N2O via nitrifier denitrification and/or denitrification in micro-anoxic niches in the oxygenated ocean11,12. As nitrification is widely used as a key component for model parameterization to estimate N2O production in the oxygenated ocean8,9,41, our results strongly support the contribution of multiple precursors and pathways of N2O in the epipelagic ocean that need to be considered in biogeochemical models aiming to estimate marine N2O sources and air–sea flux.
N2O production offsets CO2 removal by the biological pump
The export of organic matter to the ocean’s interior through the marine biological pump is a primary control on the oceanic CO2 sink on long-term timescales3,4,5. However, the magnitude of the marine biological pump depends on complex interactions, including those that alter the vertical length-scale of organic matter remineralization, altering the timescales over which carbon is sequestered42. Rapid (days to weeks) remineralization of organic matter and concomitant nitrogen recycling in the epipelagic zone sustains a large fraction of biological productivity throughout the subtropical oceans43,44,45. Our results highlight that this process also promotes N2O production. We observed that rates of N2O production and ammonia oxidation were significantly positively correlated (Fig. 4a), highlighting the covariance between N2O production and nitrogen regeneration in the epipelagic zone. Moreover, both the integrated ammonia oxidation rates and N2O production rates were also significantly correlated with the POC and PN inventories (Fig. 4b,c and Extended Data Fig. 9), implying the strength of nitrogen recycling and N2O production scales with the availability of organic nitrogen. Particles are known hotspots of microbial metabolism and can provide a source of organic and inorganic substrates to the surrounding seawater. The microenvironment formed in the particle also favours various nitrogen transformation pathways contributing to N2O production11. In our study, larger POC and PN stocks, presumably sustained by efficient epipelagic recycling, seem to promote N2O production.

a, Depth-integrated (0–200 m for the stations in open ocean and surface to bottom for the shelf stations) N2O production rates versus POC inventories. b, Depth-integrated ammonia oxidation rate versus POC inventories. c, Depth-integrated ammonia oxidation rates versus N2O production rates. The blue dots denote the measured rates and POC inventory at stations P5, C5, K1, X1, A2, SEATs (2018), A32 and Z2. Data are presented as the depth-integrated rates ± propagated s.d. of duplicates in the 2015 Northwest Pacific cruise and triplicates in the SCS and NPSG and are smaller than the symbols where not visible. The dashed black lines and grey shadows show linear regressions and the 95% confidence intervals, respectively.
Carbon and nitrogen cycling are intimately coupled to each other because both elements are required by all organisms. Although the regulation of carbon cycling by nitrogen supply has been extensively studied, we provide new perspectives on potential climatic impacts associated with nitrogen recycling and N2O production in the epipelagic ocean. Such shallow N2O production is of particular importance in the vast subtropical oligotrophic oceans, where export efficiencies are low and nutrient recycling is rapid. For example, the average export ratio in our study was 3.9 ± 2.3% (Table 1), suggesting >95% of primary production was remineralized in the epipelagic zone. Our observations indicate that nitrogen recycling promotes production of N2O via multiple pathways in these remineralization-intensive systems. In comparing the potential offset in radiative warming due to N2O production relative to carbon export, we assumed a 100-year time horizon of global warming potential (GWP100) for both processes (where 1 mol N2O would be equivalent to 300 mol CO2 in radiative energy)1. We estimate the integrated N2O production rate associated with nitrogen recycling would be equivalent to offsetting 5.6 ± 0.6% to 27.2 ± 6.7% (median value 8.8%) of the greenhouse gas mitigation capacity supported by carbon export measured at the SCS and the NPSG stations (Fig. 5 and Table 1). However, there are uncertainties associated with this estimate, variation in time and length-scales of particle remineralization and water mass ventilation would alter these radiative warming offset estimates. Therefore, our estimates would probably fall at the lower end of the potential offset attributable to N2O production in this region. For example, ventilation times between 200 and 300 m in this region average 32 ± 5 years, with that age increasing to 50 years at 400 m and 100 years at 500 m46. Assuming the vertical attenuation of sinking particulate matter follows a power-law function47, we estimate ~57 ± 5% of the measured exported carbon would be remineralized above 500 m and could exchange with the atmosphere in <100 years, leading to less CO2 sequestration and a higher N2O offset value (Supplementary Discussion 4). This offset of the effectiveness of the CO2 sink, attributable to a largely overlooked epipelagic N2O source, requires re-examination of the warming mitigation capacity of the marine biological pump.

Carbon (C) and nitrogen (N) undergo rapid cycling in the epipelagic ocean and slow cycling in the ocean’s interior. During the operation of biological pump processes, only a small fraction of newly produced organic matter (in our study, only around 5% of primary production) was exported to depth. This export (EP) drives slow recycling of C and N, during which N2O and CO2 are produced and accumulate at longer timescales (the residence time (τ): centuries to millennia) before exchanging with the atmosphere. In contrast, most of the newly produced organic matter is rapidly remineralized (τ: days to weeks) in the epipelagic ocean to drive intense recycling of C and N, during which N2O is produced through both oxidative and reductive pathways and emitted to the atmosphere. This more rapid recycling in the epipelagic waters can offset a substantial part (6–27%, median 9%) of the decreased radiative forcing (GWP100) due to biological CO2 removal to depth in our study.
Ongoing warming of the ocean and atmosphere may lead to a decline in export efficiency and decreased length-scale of remineralization due to intensified upper-ocean stratification and shifting of phytoplankton communities towards smaller cells48. Together with increased temperatures, these dynamics may enhance organic matter recycling in the epipelagic ocean49,50, with concomitant impacts of N2O production41. Our study suggests enhancement of surface N2O production, through intensified organic matter remineralization, could further exacerbate warming of the climatic system through decreased export and greater N2O production. Our results were derived from a limited number of stations at one time and cannot be directly extrapolated to the large spatial–temporal variation in both carbon export and nitrogen regeneration in the ocean3,6,7. Nevertheless, our findings show that active N2O production, driven by intense organic matter recycling in the epipelagic ocean, can offset a considerable fraction of the benefit of radiative forcing achieved by CO2 sequestration via the marine biological pump. Future work should investigate and compare the N2O/CO2 offset between systems with different export efficiencies. A better integrated assessment should take N2O generation into account for understanding the climatic impact of the marine biological pump in order to devise the best greenhouse gas mitigation strategy.
Methods
Field sampling and on-deck incubations
Samples were collected from high-resolution vertical profiles during four research cruises conducted during 2012 to 2015 to the SCS and the subtropical northwest Pacific aboard the RV Dongfanghong II; additional sampling occurred during three cruises in 2017 to 2019 in the SCS and the NPSG aboard the RV Tan Kah Kee. Stations spanned a wide range of hydrographic conditions and biological activities from the coastal shelf to open ocean (Supplementary Fig. 1 and Supplementary Table 1).
Temperature, salinity, depth and fluorescence concentrations were measured using a Seabird SBE 911 CTD sensor package equipped with fluorometer. Photosynthetically active radiation (PAR) was measured using PAR sensors (LI-COR Biosciences, LI-193 on RV Dongfanghong II and Biospherical QCP-2300L-HP on RV Tan Kah Kee). Discrete seawater samples were collected using 24 12-litre Niskin bottles mounted to the conductivity, temperature and depth (CTD) rosette. The base of the mixed layer was defined as the depth where a difference of 0.8 °C relative to the surface value was observed51. The nitracline was derived as the mid-point (average) of the steepest nitrate concentration gradient with depth51. The depth with 0.1% surface PAR was defined as the base of euphotic zone6.
On-board incubation was conducted at six stations across the shelf to the open ocean of the NPSG; at two of the stations in the SCS basin, experiments were conducted to quantify N2O production rates and nitrification rates in the epipelagic zone (0–200 m) of the open ocean stations and throughout the water column in stations sampled along the shelf.
Samples for chemical, biological and rate measurements were collected from the same casts. Triplicate 150 ml high-density polyethylene Nalgene bottles were used for nutrient collection; 120 ml glass serum bottles were used to collect samples for subsequent N2O concentration measurements in 2012 and 2013; and triplicate 250 ml glass serum bottles (Wheaton) were used for subsequent N2O concentration and isotope measurements from 2014 to 2018. Ammonia oxidation and N2O production rate incubations were conducted in 120 ml glass serum bottles. Seawater for subsequent analyses of POC and PN were collected into 4 l polycarbonate Nalgene bottles. All bottles and equipment were acid washed and rinsed with in-situ seawater at least three times prior to sample collection. During sample collection, glass sample bottles were overfilled two to three times before sealing with 20 mm butyl stopper and aluminium crimp seals (Wheaton). Samples were preserved by adding 0.1 ml to 0.2 ml saturated HgCl2 and were stored at 4 °C. For POC and PN samples, 4–8 l of seawater was gently (<200 mm Hg, 26.6 kPa) filtered through a pre-combusted (450 °C for 4 h) Whatman GF/F filter (25 mm diameter). After filtration, the filters were folded and wrapped in pre-combusted (450 °C for 4 h) aluminium foil and stored at −80 °C.
A comprehensive set of incubations was carried out on-board to determine rates of nitrification and N2O production using 15NH4+, 15NO2− and 15NO3− tracers (Supplementary Table 2). All incubations were conducted in the dark at near in-situ temperatures (±2 °C). On the 2015 cruise, 0.2 ml of tracer was injected into each bottle to obtain final concentrations of 15NH4+ and 15NO2− of 300 nmol l−1. For the 15NO3− tracer, two different stocks were used to obtain final concentrations of 15NO3− of 300 nmol l−1 for samples above the DCM and 1,000 nmol l−1 for samples below the DCM. On the 2018 SCS cruise and 2019 NPSG cruise, 0.2 ml of mixed tracer was added to each bottle to obtain enrichment of 500 nmol l−1 of 15NH4+ + 14NO2− or 14NH4+ + 15NO2−. For 15NO3− tracer, two different stocks were used to get a final enrichment of 500 nmol l−1 of 15NO3− for samples above the DCM and 1,000 nmol l−1 of 15NO3− below the DCM in both the 2018 SCS cruise and 2019 NPSG cruise. The final tracer concentrations were frequently higher than the in-situ substrate concentrations, accounting for 89 ± 12%, 85 ± 15% and 42 ± 30% of the final substrate pool in the 15NH4+, 15NO2− and 15NO3− labelling incubations, respectively. 15NH4+ labelling incubations were conducted for deriving both N2O production and nitrification rates. Immediately after the 15NH4+ tracer injection, 10 ml of sample was pushed out by pure N2 and then filtered through a 0.2 μm syringe filter to represent the initial condition (t0) for the nitrification incubations. The remaining water was preserved with 0.1 ml saturated HgCl2. The remaining bottles were incubated in the dark at near in-situ temperature. At each timepoint, 10 ml of water was sampled and then filtered for subsequent nitrification rate measurements, and the remaining water was preserved using HgCl2 for subsequent determinations of N2O. The filtrate was stored at −20 °C for subsequent analyses. For 15NO2− and 15NO3− incubations, the same procedures were used for on-board incubation, except that the incubation was terminated by adding 0.1 ml HgCl2 without replacing by N2. Primary production rate was also measured in selected stations on 2015, 2018 and 2019 cruises using H13CO3− tracer (99 atom% 13C, Cambridge Isotope Laboratories), and the final concentration of H13CO3− was 100 μmol l−1, accounting for ~5% of the substrate pool. On-deck incubation (duplicates) was performed in 4 l polycarbonate Nalgene bottles for 24 h. Light conditions of the incubators were manipulated by neutral density filter. Seawater was gently (<200 mm Hg, 26.6 kPa) filtered through a pre-combusted (450 °C for 4 h) Whatman GF/F filter (25 mm diameter) and stored at −80 °C.
Nutrient, POC and PN measurements
NH4+ concentrations were measured on-board the research vessels using a fluorometric method with detection limit of 1.2 nmol l−1 and precision of ±3.5%52. Nutrient concentrations below the nitracline were measured using a four-channel Continuous Flow Technicon AA3 Auto-Analyzer. The detection limits for NOx (NO3− + NO2−) and Si(OH)4 were 0.03 μmol l−1 and 0.05 μmol l−1, respectively, with precision better than 1% and 2.8%, respectively53. NO2− and NO3− concentrations above the nitracline were determined using the standard colorimetric method coupled with a Flow Injection Analysis-Liquid Waveguide Capillary Cell system (World Precision Instruments)54; the detection limit was 5 nmol l−1 and precision was better than 3.1%. For POC and PN concentration measurement, the filters were freeze dried and then acidified with 1 ml of 1 N HCl solution to remove carbonates. All filters were dried at 60 °C for 48 h. The decarbonated samples were then analysed for POC and PN using an EA-IRMS (Thermo Finnigan Flash EA 2000 interfaced to a Delta VPLUS isotopic ratio mass spectrometer) system. The precision for both PN and POC concentration is <1% (ref. 55).
N2O concentration measurement
During 2012 to 2013, N2O concentrations were measured using a purge and trap system coupled with a gas chromatograph (Hewlett-Packard model 6890 equipped with a micro-electron capture detector). Calibration of N2O concentrations was determined from peak areas with standard gases of 1.0–5.0 ppmv N2O/N2 (Research Institute of China National Standard Materials), which were run at six-sample intervals. The precision of this method was estimated to be better than ±5% (ref. 56). Beginning in 2014, N2O concentrations were also derived from ion peak area (m/z = 44) during isotope analysis using the gas chromatography-isotope ratio mass spectrometry (GC-IRMS) system (see below). The two methods yielded comparable results; thus, N2O concentrations are shown as the mean value from these independent methods.
234Th measurement
The thorium-deficit method was used to estimate export production. Total 234Th samples were processed using a manganese oxide co-precipitation technique57. Briefly, total 234Th in the seawater was co-precipitated with MnO2 particles and the resulting particles were collected on a 25 mm, 1.0 μm quartz micro-filter (QMA). Suspended particles in the seawater were also analysed for 234Th; for these samples, ~8 l water was filtered onto a QMA filter. All total and particulate 234Th samples were beta counted on a gas flow proportional low-level Risø beta-counter for 16 h until total counts >2,500. A second counting was carried out after >150 days for background correction. The recovery for 234Th was monitored by 230Th spike addition in the seawater and quantified by an alpha-counter with addition of a 228Th internal standard58. The recoveries of 234Th were better than 90%. 238U (dpm l−1) was calculated from the linear relationship of 238U with salinity59.
Isotopic analyses of NOx − and N2O
δ15N of NOx− samples for nitrification rate were determined using the denitrifier method60,61. Briefly, NOx− was quantitatively converted to N2O using the bacterial strain Pseudomonas aureofaciens. The evolved N2O was then introduced to the GC-IRMS (Delta VPLUS isotopic ratio mass spectrometer) through an online N2O cryogenic extraction and purification system. δ15N of NOx− values were calibrated against nitrate isotope standards USGS 34, IAEA N3 and USGS 32, which were run before, after and at ten-sample intervals. Accuracy was better than ±0.2‰ according to analyses of these standards at an injection level of 20 nmol N. For samples with NOx− concentrations lower than 0.5 μmol l−1, 1 ml of 5 μmol l−1 of in-house NO3− standard was added as carrier to 9 ml of sample, and the isotopic composition of the sample was then calculated from the measured composition of the mixture and the known in-house standard via mass conservation.
Concentrations and isotopes of N2O were measured using a modified GC-IRMS with large volume purge and trap system62. Briefly, two needles were used for sample transfer and He pressurization, and the sample was transferred into a sparging flask (Pyrex) using ultra-high-purity He (>99.999%) and purged with He. For a 250 ml bottle, the sample was purged for 60 min at a flow rate of 50 ml min−1, and for a 120 ml bottle, the purge time was 30 min. The extracted gases were passed through an ethanol trap with dry ice and a chemical trap filled with magnesium perchlorate and Ascarite to remove H2O and CO2. N2O was trapped by liquid nitrogen twice for purification and concentration and then injected into the GC-IRMS with He as carrier gas. N2O concentrations were determined by ion peak area (m/z = 44), and calibration of N2O concentration was calculated from ion peak areas (m/z = 44) with standard gases of 199.6 and 501.0 ppmv N2O/He, which were run at ten-sample intervals. The serum bottle was weighed before and after transfer to calculate the amount of water transferred. The precision of this method was estimated to be better than ±3% (ref. 62). δ15N and δ18O were calibrated against two reference tanks (R1: 199.6 ppmv N2O/He, δ15N = −3.2 ± 0.1‰ relative to air N2, δ18O = 36.6 ± 0.1‰ relative to Vienna Standard Mean Ocean Water; R2: 501.0 ppmv N2O/He, δ15N = −1.6 ± 0.1‰, δ18O = 36.6 ± 0.3‰), which were measured in the Casciotti lab at Stanford University. The precision of δ15N and δ18O measurements with 2 nmol N2O reference gas was better than 0.3‰ and 0.4‰, respectively (n = 20).
Surface N2O saturation and air–sea flux
Surface N2O saturation was calculated using equation (1):
where R (%) is the saturation of surface N2O; Cobs represents N2O concentration at 5 m depth; Ceq is the expected equilibrium concentration, which is computed using Henry’s law63; and the solubility depends on temperature and salinity64. The air N2O concentration is the average atmospheric N2O concentration at Mauna Loa of the sampling year (NOAA/ESRL programme).
Air–sea N2O flux was computed using equations (2) and (3):
where F (μmol m−2 d−1) is air–sea flux of N2O; k (cm h−1) is the gas transfer velocity depending on wind and water temperature; u is daily mean wind speed at 10 m above sea surface during the cruise, as measured by the on-board meteorological station; and Sc is the Schmidt number calculated from temperature64.
Estimation of the fraction of N2O source derived from the isotope minimum layer
A two-endmember mixing model of isotopically enriched N2O mixing upward from the N2O concentration maximum layer and isotopically depleted N2O produced at the isotope minima layer was used to calculate the fraction of N2O contributed by shallow in-situ production using equation (4)10:
where δtotal is the lowest measured isotopic value of N2O at the isotope minimum. δdeep is the isotopic signature of N2O mixing upward from deep layers; here, we use the N2O concentration maximum layer as an endmember; the measured δ15N was 9.52 ± 0.28‰ and the δ18O was 52.25 ± 0.74‰ in our study sites (Extended Data Fig. 10). δshallow is the isotopic value of the in-situ source in the isotope minimum layer, which is unknown. f is the fraction of N2O contributed from the shallow source to the isotope minimum layer, with the remainder equal to that diffusing upward from the [N2O] maximum. The lower limit of f could be constrained by assuming δshallow was represented by the lowest value in an existing database from the North Pacific, and the δ15N and δ18O was 1.8‰ and 24.5‰, respectively65.
Nitrification and N2O production rate calculation
Rates of nitrification were determined based on the accumulation of 15N in the product pool relative to the initial 15N signature of that pool. Rates were computed based on equation (5):
where RNR is the bulk nitrification rate for all substrates following 15NH4+ enrichment (nmol N l−1 d−1). \(C_{\mathrm{NO}_{x}- }\) is the product concentration at the beginning of incubation (nmol N l−1), f15 is the atom% 15N of the NH4+ pool at the beginning of incubation (the fraction of 15N–NH4+ in the gross NH4+ pool after tracer enrichment), and nt and n0 are the atom% 15N of the product pool (NO2− + NO3−) at the ending and beginning of incubation (%), respectively. t is the duration of incubation (h). This equation quantifies the transformation rate including the concentration due to the tracer addition (that is, ambient substrate + tracer) and thus represents a potential reaction rate.
Rates of N2O production from a particular labelled substrate (for example, 15NH4+) were quantified as the increase in mass 44, 45 and 46 from NH4+ during an incubation. In our calculation, 15N–N2O production during 15NH4+ incubation was obtained from the increase of 45 and 46, and the 14N–N2O production from 14NH4+ was then derived based on the atom fraction of 14N and 15N of the substrate pool. During the incubations, the tracer substrate is enriched in 15N; thus, we assume the accumulation of 45N2O is mainly contributed by 15N14N16O and 14N15N16O (single labelled N2O) and production of 14N14N17O during the incubation is negligible. Similarly, the accumulation of 46N2O is thus mainly contributed by 15N15N16O (double labelled N2O), and the production of 14N15N17O and 14N14N18O are negligible compared with the double labelled N2O. Therefore, the rates of 45N2O production and 46N2O production can be derived using equations (6) and (7):
where R45measure (pmol N2O l−1 d−1) is measured production rate of 45N2O according to the increase of measured R45N2O/44N2O; R46measure (pmol N2O l−1 d−1) is measured production rate of 46N2O according to the increase of measured R46N2O/44N2O; and CN2O is N2O concentration (pmol N2O l−1) at the beginning of the incubation. nt45, n045, nt46 and n046 are the 45N2O% and 46N2O% based on R45N2O/44N2O and R46N2O/44N2O at the ending and beginning of incubation (%), respectively. t is the duration of incubation (h), which is converted to units of days (d) by multiplying by 24 h d−1.
The production of 15N–N2O and 14N–N2O from a single substrate (for example, NH4+) can be then derived using equations (8) and (9):
where \(R_{^1{^5}\mathrm{N} - \mathrm{N}{_2}\mathrm{O}}\) (pmol N l−1 d−1) is the measured production of 15N–N2O from the labelled substrate (note, one 15N atom in the 45N2O and two 15N atoms in the 46N2O), and \(R_{^1{^4}\mathrm{N} - \mathrm{N}{_2}\mathrm{{O}}}\) (pmol N l−1 d−1) is the calculated production of 14N–N2O based on the atom fractions of 14N (f14) and 15N (f15) of the substrate pool. The production of N2O from one substrate is defined as the sum of \(R_{^1{^5}\mathrm{N} - \mathrm{N}{_2}\mathrm{{O}}}\) and \(R_{^1{^4}\mathrm{N} - \mathrm{N}{_2}\mathrm{{O}}}\). The same equations were used to calculate the rates of N2O production (\(R_{\mathrm{NH}_4}, R_{\mathrm{NO}_2}, R_{\mathrm{NO}_3}\)) from each of the individual tracers (15NH4+, 15NO2−, 15NO3−).
The gross N2O production rate was derived from the sum of NH4+ sourced, NO2− sourced and NO3− sourced N2O. Therefore, the rate of gross N2O production was calculated using equation (10):
where Rgross is the total N2O production rate during our incubation (pmol N l−1 d−1). The errors of the NH4+ sourced, NO2− sourced and NO3− sourced N2O rate are based on the increase of N2O of our incubation in the 2015 cruise (duplicates), 2018 cruise (triplicates) and 2019 cruise (triplicates), and propagation of the errors during the calculation using the equations listed above.
Detection limits of rate measurements
For nitrification rate measurements, the detection limits depend on the concentration of the product pool and the fraction of 15N in the substrate pool during the incubation66,67. As mentioned, the precision of δ15N–NOx− was better than ±0.2‰, and we here use three times the standard deviation as a reliable enrichment of 15N in the product pool. Therefore, we calculated a detection limit of 0.04–0.16 nmol N l−1 d−1 for nitrification. Similarly, for N2O production rate, the precision of δ15N–N2O and δ18O–N2O was better than ±0.3‰ and ±0.4‰, respectively, and we here use three times the standard deviation as a reliable enrichment of 45N2O and 46N2O in the product pool. Therefore, we calculated a detection limit of 0.1–0.3 pmol N l−1 d−1 for 45N2O production rate and 0.2–0.6 pmol N l−1 d−1 for 46N2O production rate in 15NH4+ tracer incubation; 0.1–0.7 pmol N l−1 d−1 for 45N2O production rate and 0.2–1.0 pmol N l−1 d−1 for 46N2O production rate in 15NO2− tracer incubation; and 0.1–3.0 pmol N l−1 d−1 for 45N2O production rate and 0.2–5.0 pmol N l−1 d−1 for 46N2O production rate in 15NO3− tracer incubation.
Data availability
All data needed to evaluate the conclusions in the paper are deposited in the Zenodo database and can be accessed through https://doi.org/10.5281/zenodo.6867932.
References
Canadell, J. G. et al. in Climate Change 2021: The Physical Science Basis (eds Masson-Delmotte, V. et al.) Ch. 5 (IPCC, Cambridge Univ. Press, 2021).
Tian, H. et al. A comprehensive quantification of global nitrous oxide sources and sinks. Nature 586, 248–256 (2020).
Buesseler, K. O., Boyd, P. W., Black, E. E. & Siegel, D. A. Metrics that matter for assessing the ocean biological carbon pump. Proc. Natl Acad. Sci. USA 117, 9679–9687 (2020).
Ito, T. & Follows, M. J. Preformed phosphate, soft tissue pump and atmospheric CO2. J. Mar. Sci. 4, 813–839 (2005).
Honjo, S. et al. Understanding the role of the biological pump in the global carbon cycle: an imperative for ocean science. Oceanography 27, 10–16 (2014).
Karl, D. M. & Church, M. J. Microbial oceanography and the Hawaii Ocean Time-series programme. Nat. Rev. Microbiol. 12, 699–713 (2014).
Lomas, M. W. et al. Two decades and counting: 24-years of sustained open ocean biogeochemical measurements in the Sargasso Sea. Deep Sea Res. II 93, 16–32 (2013).
Freing, A., Wallace, D. W. R. & Bange, H. W. Global oceanic production of nitrous oxide. Phil. Trans. R. Soc. B 367, 1245–1255 (2012).
Buitenhuis, E. T., Suntharalingam, P. & Le Quéré, C. Constraints on global oceanic emissions of N2O from observations and models. Biogeosciences 15, 2161–2175 (2018).
Dore, J. E., Popp, B. N., Karl, D. M. & Sansone, F. J. A large source of atmospheric nitrous oxide from subtropical North Pacific surface waters. Nature 396, 63–66 (1998).
Klawonn, I., Bonaglia, S., Bruchert, V. & Ploug, H. Aerobic and anaerobic nitrogen transformation processes in N2-fixing cyanobacterial aggregates. ISME J. 9, 1456–1466 (2015).
Graf, J. S. et al. Anaerobic endosymbiont generates energy for ciliate host by denitrification. Nature 591, 445–450 (2021).
Santoro, A. E., Richter, R. A. & Dupont, C. L. Planktonic marine archaea. Annu. Rev. Mar. Sci. 11, 131–158 (2019).
Kozlowski, J. A., Stieglmeier, M., Schleper, C., Klotz, M. G. & Stein, L. Y. Pathways and key intermediates required for obligate aerobic ammonia-dependent chemolithotrophy in bacteria and Thaumarchaeota. ISME J. 10, 1836–1845 (2016).
Lancaster, K. M., Caranto, J. D., Majer, S. H. & Smith, M. A. Alternative bioenergy: updates to and challenges in nitrification metalloenzymology. Joule 2, 421–441 (2018).
Stein, L. Y. Insights into the physiology of ammonia-oxidizing microorganisms. Curr. Opin. Chem. Biol. 49, 9–15 (2019).
Horrigan, S. G., Carlucci, A. F. & Williams, P. M. Light inhibition of nitrification in sea-surface films [California]. J. Mar. Res. 39, 557–565 (1981).
Moore, C. M. et al. Processes and patterns of oceanic nutrient limitation. Nat. Geosci. 6, 701–710 (2013).
Ustick, L. J. et al. Metagenomic analysis reveals global-scale patterns of ocean nutrient limitation. Science 372, 287–291 (2021).
Dugdale, R. C. & Goering, J. J. Uptake of new and regenerated forms of nitrogen in primary production. Limnol. Oceanogr. 12, 196–206 (1967).
Karl, et al. in Nitrogen in the Marine Environment 2nd edn (eds Capone, D. G. et al.) Ch. 16 (Academic Press, 2008).
Duce, R. A. et al. Impacts of atmospheric anthropogenic nitrogen on the open ocean. Science 320, 893–897 (2008).
Jin, X. & Gruber, N. Offsetting the radiative benefit of ocean iron fertilization by enhancing N2O emissions. Geophys. Res. Lett. https://doi.org/10.1029/2003GL018458 (2003).
Dutreuil, S., Bopp, L. & Tagliabue, A. Impact of enhanced vertical mixing on marine biogeochemistry: lessons for geo-engineering and natural variability. Biogeosciences 6, 901–912 (2009).
Martínez‐García, S. & Karl, D. Microbial respiration in the euphotic zone at Station ALOHA. Limnol. Oceanogr. 60, 1039–1050 (2015).
Yang, S. et al. Global reconstruction reduces the uncertainty of oceanic nitrous oxide emissions and reveals a vigorous seasonal cycle. Proc. Natl Acad. Sci. USA 117, 11954–11960 (2020).
Forster, G. et al. Nitrous oxide and methane in the Atlantic Ocean between 50° N and 52° S: latitudinal distribution and sea-to-air flux. Deep Sea Res. II 56, 964–976 (2009).
Wilson, S. T., Ferrón, S. & Karl, D. M. Interannual variability of methane and nitrous oxide in the North Pacific Subtropical Gyre. Geophys. Res. Lett. 44, 9885–9892 (2017).
Wan, X. S. et al. Ambient nitrate switches the ammonium consumption pathway in the euphotic ocean. Nat. Commun. https://doi.org/10.1038/s41467-018-03363-0 (2018).
Frame, C. H. & Casciotti, K. L. Biogeochemical controls and isotopic signatures of nitrous oxide production by a marine ammonia-oxidizing bacterium. Biogeosciences 7, 2695–2709 (2010).
Santoro, A. E., Buchwald, C., McIlvin, M. R. & Casciotti, K. L. Isotopic signature of N2O produced by marine ammonia-oxidizing archaea. Science 333, 1282–1285 (2011).
Santoro, A. E. et al. Thaumarchaeal ecotype distributions across the equatorial Pacific Ocean and their potential roles in nitrification and sinking flux attenuation. Limnol. Oceanogr. 62, 1984–2003 (2017).
Smith, J. M., Damashek, J., Chavez, F. P. & Francis, C. A. Factors influencing nitrification rates and the abundance and transcriptional activity of ammonia-oxidizing microorganisms in the dark northeast Pacific Ocean. Limnol. Oceanogr. 61, 596–609 (2016).
Popp, B. N. et al. Nitrogen and oxygen isotopomeric constraints on the origins and sea-to-air flux of N2O in the oligotrophic subtropical North Pacific Gyre. Glob. Biogeochem. Cycles 16, 12-1–12-10 (2002).
Wilson, S. T., del Valle, D. A., Segura-Noguera, M. & Karl, D. M. A role for nitrite in the production of nitrous oxide in the lower euphotic zone of the oligotrophic North Pacific Ocean. Deep Sea Res. I 85, 47–55 (2014).
Yang, J. Y. T., Kao, S. J., Dai, M., Yan, X. & Lin, H. L. Examining N cycling in the northern South China Sea from N isotopic signals in nitrate and particulate phases. J. Geophys. Res. Oceans 122, 2118–2136 (2017).
Ji, Q. & Ward, B. B. Nitrous oxide production in surface waters of the mid-latitude North Atlantic Ocean. J. Geophys. Res. Oceans 122, 2612–2621 (2017).
Breider, F. et al. Response of N2O production rate to ocean acidification in the western North Pacific. Nat. Clim. Change 9, 954–958 (2019).
Frey, C. et al. Regulation of nitrous oxide production in low-oxygen waters off the coast of Peru. Biogeosciences 17, 2262–2287 (2020).
Santoro, A. E. et al. Nitrification and nitrous oxide production in the offshore waters of the eastern tropical South Pacific. Glob. Biogeochem. Cycles https://doi.org/10.1029/2020GB006716 (2020).
Landolfi, A., Somes, C. J., Koeve, W., Zamora, L. M. & Oschlies, A. Oceanic nitrogen cycling and N2O flux perturbations in the Anthropocene. Glob. Biogeochem. Cycles 31, 1236–1255 (2017).
Marsay, C. M. et al. Attenuation of sinking particulate organic carbon flux through the mesopelagic ocean. Proc. Natl Acad. Sci. USA 112, 1089–1094 (2015).
Eppley, R. W., Renger, E. H. & Betzer, P. R. The residence time of particulate organic carbon in the surface layer of the ocean. Deep Sea Res. A 30, 311–323 (1983).
Poulton, A. J. et al. Phytoplankton mineralization in the tropical and subtropical Atlantic Ocean. Glob. Biogeochem. Cycles https://doi.org/10.1029/2006GB002712 (2006).
Wilson, S. T. et al. Short-term variability in euphotic zone biogeochemistry and primary productivity at Station ALOHA: a case study of summer 2012. Glob. Biogeochem. Cycles 29, 1145–1164 (2015).
Wang, W. et al. Transit time distributions and apparent oxygen utilization rates in northern South China Sea using chlorofluorocarbons and sulfur hexafluoride data. J. Geophys. Res. Oceans 126, e2021JC017535 (2021).
Martin, J. H., Knauer, G. A., Karl, D. M. & Broenkow, W. W. VERTEX: carbon cycling in the northeast Pacific. Deep Sea Res. A 34, 267–285 (1987).
Flombaum, P. et al. Present and future global distributions of the marine cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl Acad. Sci. USA 110, 9824–9829 (2013).
Garcia-Corral, L. S. et al. Temperature dependence of plankton community metabolism in the subtropical and tropical oceans. Glob. Biogeochem. Cycles 31, 1141–1154 (2017).
Wohlers, J. et al. Changes in biogenic carbon flow in response to sea surface warming. Proc. Natl Acad. Sci. USA 106, 7067–7072 (2009).
Kara, A. B., Rochford, P. A. & Hurlburt, H. E. An optimal definition for ocean mixed layer depth. J. Geophys. Res. Oceans 105, 16803–16821 (2000).
Zhu, Y., Yuan, D., Huang, Y., Ma, J. & Feng, S. A sensitive flow-batch system for on board determination of ultra-trace ammonium in seawater: method development and shipboard application. Anal. Chim. Acta 794, 47–54 (2013).
Han, A. et al. Nutrient dynamics and biological consumption in a large continental shelf system under the influence of both a river plume and coastal upwelling. Limnol. Oceanogr. 57, 486–502 (2012).
Zhang, J. Shipboard automated determination of trace concentrations of nitrite and nitrate in oligotrophic water by gas-segmented continuous flow analysis with a liquid waveguide capillary flow cell. Deep Sea Res. I 47, 1157–1171 (2000).
Kao, S. J. et al. Isotope constraints on particulate nitrogen source and dynamics in the upper water column of the oligotrophic South China Sea. Glob. Biogeochem. Cycles https://doi.org/10.1029/2011GB004091 (2012).
Lin, H. et al. Spatiotemporal variability of nitrous oxide in a large eutrophic estuarine system: the Pearl River estuary, China. Mar. Chem. 182, 14–24 (2016).
Zhou, K. et al. Apparent enhancement of 234Th-based particle export associated with anticyclonic eddies. Earth Planet. Sci. Lett. 381, 198–209 (2013).
Cai, P., Dai, M., Lv, D. & Chen, W. An improvement in the small-volume technique for determining thorium-234 in seawater. Mar. Chem. 100, 282–288 (2006).
Chen, J., Edwards, R. L. & Wasserburg, G. J. 238U, 234U and 232Th in seawater. Earth Planet. Sci. Lett. 80, 241–251 (1986).
Casciotti, K. L., Sigman, D. M., Hastings, M. G., Böhlke, J. K. & Hilkert, A. Measurement of the oxygen isotopic composition of nitrate in seawater and freshwater using the denitrifier method. Anal. Chem. 74, 4905–4912 (2002).
Sigman, D. M. et al. A bacterial method for the nitrogen isotopic analysis of nitrate in seawater and freshwater. Anal. Chem. 73, 4145–4153 (2001).
McIlvin, M. R. & Casciotti, K. L. Fully automated system for stable isotopic analyses of dissolved nitrous oxide at natural abundance levels. Limnol. Oceanogr. Methods 8, 54–66 (2010).
Weiss, R. F. & Price, B. A. Nitrous oxide solubility in water and seawater. Mar. Chem. 8, 347–359 (1980).
Wanninkhof, R. Relationship between wind speed and gas exchange over the ocean revisited. Limnol. Oceanogr. Methods 12, 351–362 (2014).
Snider, D. M., Venkiteswaran, J. J., Schiff, S. L. & Spoelstra, J. From the ground up: global nitrous oxide sources are constrained by stable isotope values. PLoS ONE 10, e0118954 (2015).
Santoro, A. E. et al. Measurements of nitrite production in and around the primary nitrite maximum in the central California Current. Biogeosciences 10, 7395–7410 (2013).
Peng, X. et al. Ammonia and nitrite oxidation in the eastern tropical North Pacific. Glob. Biogeochem. Cycles 29, 2034–2049 (2015).
Acknowledgements
We greatly appreciate S. S. Hsiao, Y. Wu, M. Xu, M. He, X. Zhang, G. Shao, W. Zhang and Q. Wu’s inputs during on-board sampling and incubation in the research cruises. We also thank T. Huang and Y. Zhu for the on-board measurement of NH4+, Y. Wu and L. Wang for NO3−, NO2− and PO43− measurements and Q. Hong, Y. Ma and W. Chen for measuring the 234Th. We are also grateful for the crew of the RV Dongfanghong II and RV Tan Kah Kee for the on-board assistance and providing the CTD data. Comments from T. W. Trull and H. M. Nelson improved earlier versions of the manuscript. This work was supported by the National Natural Science Foundation of China through grants 92058204, 41890802, 922583024, 1721005, 41730533 and 41906040. M.J.C. acknowledges funding from the Simons Foundation via SCOPE (grant 721221).
Author information
Authors and Affiliations
Contributions
X.S.W., M.D. and S.-J.K. conceived the study and designed the experiment. X.S.W., H.-X.S., K.L.C., W.Z., L.L., H.S. and K.Z. performed the experiment and measured the samples. X.S.W., H.-X.S., M.D., K.L.C., M.J.C., B.B.W. and S.-J.K. analysed the results and structured the manuscript. All authors contributed to the discussion of the results and editing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Geoscience thanks Christopher Somes and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Tom Richardson, in collaboration with the Nature Geoscience team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Hydrological and biological characteristics at sampling stations.
a1-g1, Potential temperature. a2−g2, Salinity. a3−g3, Potential density anomaly. a4-g4, Fluorescence. a5-g5, DO at the South China Sea (SCS); Northwest Pacific (NWP) and North Pacific Subtropical Gyre (NPSG). Each row of plots depicts hydrological and biological parameters from a single cruise.
Extended Data Fig. 2 Depth profiles of concentrations of nutrients and N2O at sampling stations.
a1-g1, NH4+. a2-g2, NO2-. a3−g3, NO3-. a4-g4, NO3-/Si. a5-g5, N2O at the South China Sea (SCS); Northwest Pacific (NWP) and North Pacific Subtropical Gyre (NPSG).. Each row depicts nutrient and N2O concentrations from a single cruise.
Extended Data Fig. 3 Depth profiles of POC and PN concentrations.
a-d, Station P5, C5, K1 and X1 in the subtropical Northwest Pacific in 2015. e-f, Station A2 and SEATs in the South China Sea in 2018. g-h, A32 and Z2 in the North Pacific Subtropical Gyre in 2019. POC concentrations are depicted with orange circles and PN concentrations are blue circles.
Extended Data Fig. 4 Depth profiles of primary production (PP) rate.
a-b, Station K1 and X1 in the subtropical Northwest Pacific in 2015. c-d, Station SEATs and A2 in the South China Sea in 2018. e, Station Z2 in the North Pacific Subtropical Gyre in 2019. Data are presented as mean rates ± standard deviation of triplicate sample incubation in the SCS and NPSG, and duplicates in the 2015 NWP cruise, are smaller than the symbols where not visible.
Extended Data Fig. 5 Depth profiles of N2O saturation, NO2− and NO3−/Si at stations with distinctive shallow N2O accumulation in the epipelagic zone.
N2O saturation (orange dots); NO2− concentration (blue dots); NO3−/Si (green dots). a1-a7, 2012 cruise. b1-b4, 2013 cruise. c1-c5, 2014 cruise. d1-d4, 2015 cruise. e1-e3, 2017 cruise. f1-f3, 2018 cruise. g1-g2, 2019 cruise. Data are presented as mean values ± standard deviation of triplicate sample measurement, and are smaller than the symbols where not visible.
Extended Data Fig. 6 Depth profiles of physical parameters and N2O saturation at four stations with distinctive shallow N2O accumulation in the SCS.
a-d, The potential temperature (blue lines); salinity (orange lines); fluorescence (green lines) and potential density anomaly (gray lines). e-h, N2O saturation (orange dots); NO2- concentration (blue dots); NO3−/Si (green dots) a, e, Station A03; b, f, Station B05; c, g, Station SEATs (2014); d, h, Station SEATs (2017). Gray bars mark the depth range with the distinctive N2O accumulation. Data are presented as mean values ± standard deviation of triplicate sample measurement, and are smaller than the symbols where not visible in panels g and h.
Extended Data Fig. 7 Concentration and dual isotopes of N2O plotted against potential density anomaly.
a-e, Station D1, X2, X5, J1, SEATs in the South China Sea (SCS) in 2014. f-i, Station P5, C5, K1, X1 in the Northwest Pacific in 2015. j-l, Station SEATs, B2, SS1 in the SCS in 2017. m-o, Station A2, A4, SEATs in the SCS in 2018. The dashed lines mark the depth with δ15N-N2O isotope minima. Data are presented as mean values ± standard deviation of triplicate sample measurement, and are smaller than the symbols where not visible.
Extended Data Fig. 8 Depth profiles of ammonia oxidation rates.
a-d, Station P5, C5, K1 and X1 in the Northwest Pacific (NWP) in 2015. e-f, Station SEATs and A2 in the South China Sea (SCS) in 2018. g-h, Station A32 and Z2 in the North Pacific Subtropical Gyre (NPSG). Data are presented as mean rates ± standard deviation of triplicate sample incubation in the SCS and NPSG, and duplicates in the 2015 NWP cruise, are smaller than the symbols where not visible.
Extended Data Fig. 9 Relationships between the depth-integrated N2O production rates, ammonia oxidation rates and PN inventories.
a, Depth-integrated (0–200 m for the stations in open ocean and surface to bottom for the shelf stations) N2O production rate vs. particulate nitrogen (PN) inventory. b, Depth-integrated ammonia oxidation rates vs. PN inventories. The blue dots denote the measured rates and PN inventory at station P5, C5, K1, X1, A2, SEATs (2018), A32 and Z2. Data are presented as the depth-integrated rates ± propagated standard deviation of duplicates in the 2015 Northwest Pacific cruise and triplicates in the South China Sea and North Pacific Subtropical Gyre, and are smaller than the symbols where not visible. The black lines and gray shadows show linear regressions and the 95% confidence intervals, respectively.
Extended Data Fig. 10 Depth profiles of concentrations and dual isotopes of N2O for sampling sites.
a-c, Station SEATs, SS1 and B2 in the South China Sea in 2017. Data are presented as mean values ± standard deviation of triplicate sample measurement, and are smaller than the symbols where not visible.
Supplementary information
Supplementary Information
Supplementary Discussions 1–4, Tables 1–7, Figs. 1 and 2 and References.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wan, X.S., Sheng, HX., Dai, M. et al. Epipelagic nitrous oxide production offsets carbon sequestration by the biological pump. Nat. Geosci. 16, 29–36 (2023). https://doi.org/10.1038/s41561-022-01090-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41561-022-01090-2
This article is cited by
-
Particle-associated denitrification is the primary source of N2O in oxic coastal waters
Nature Communications (2023)
-
Tracing surface seawater mixing and nutrient transport by 222Rn on the northern coast of Beibu Gulf, China
Acta Oceanologica Sinica (2023)
