
Abstract
The evolution of costly traits such as deer antlers and peacock trains, which drove the formation of Darwinian sexual selection theory, has been suggested to both reflect and affect patterns of genetic variance across the genome, but direct tests are missing. Here, we used an evolve and resequence approach to reveal patterns of genome-wide diversity associated with the expression of a sexually selected weapon that is dimorphic among males of the bulb mite, Rhizoglyphus robini. Populations selected for the weapon showed reduced genome-wide diversity compared to populations selected against the weapon, particularly in terms of the number of segregating non-synonymous positions, indicating enhanced purifying selection. This increased purifying selection reduced inbreeding depression, but outbred female fitness did not improve, possibly because any benefits were offset by increased sexual antagonism. Most single nucleotide polymorphisms (SNPs) that consistently diverged in response to selection were initially rare and overrepresented in exons, and enriched in regions under balancing or relaxed selection, suggesting they are probably moderately deleterious variants. These diverged SNPs were scattered across the genome, further demonstrating that selection for or against the weapon and the associated changes to the mating system can both capture and influence genome-wide variation.
Similar content being viewed by others

Main
Sexual selection, arising from reproductive competition1,2,3, has the potential to play a major role in shaping biodiversity, affecting not only phenotypic evolution, but also rates of adaptation, speciation and extinction4,5,6,7,8,9,10,11,12,13. This link, to some degree, is due to the effect sexual selection can have on the amount and nature of genetic variance segregating in populations, which all these evolutionary processes are contingent on. As long as sexual selection aligns with natural selection and causes total selection on males to be stronger than on females14, it should enhance fixation of adaptive variants and selection against deleterious mutations8,15,16,17,18. Sexual selection against mutations detrimental to both sexes can occur because successful competition for mates requires general health and vigour17,19. In addition, purifying selection may also be enhanced by the evolution of costly sexually selected traits (henceforth, SSTs), such as elaborated ornaments or weapons, if their expression captures genetic variation20,21 and reproductive success is skewed in favour of males with the most elaborate SSTs17.
Typically, SSTs act to increase male competitive ability over access to mating and fertilization, but simultaneously impose costs to the bearer due to, for example, their development and maintenance1,2. Male investment in costly SSTs will therefore be limited by the pool of available resources, or an individual’s ‘condition’. In addition to the environment, condition is probably determined by many organismal processes across much of the genome22. SSTs should thus reflect genetic variation at many fitness-related loci, a mechanism known as ‘genic capture’20. Although the genic capture hypothesis was developed to account for the heritability of SSTs (on the order of 20–40%, ref. 23) involved in mate choice, the same mechanism will also apply to other condition-dependent SSTs, such as weapons24. Furthermore, continuous variation in condition can be captured by the discontinuous expression of a SST, as is often the case when SSTs are expressed only above a certain threshold of individual condition, resulting in alternative reproductive phenotypes (ARPs25,26,27,28). ARPs expressing costly SSTs typically will engage in direct competition26, further enhancing selection for general health and vigour within that particular group of males. While potentially beneficial to population level-fitness in the short term, fixation of beneficial alleles and purifying selection deplete genetic variance in populations that may ultimately reduce their evolutionary potential if environmental change renders previously deleterious genetic variation beneficial29.
In opposition, recent theory has highlighted that sexual selection may promote balancing selection30, due to negative pleiotropic effects of SSTs on other components of fitness31,32 or due to sexual antagonism33,34,35 caused by differences between the sexes in the direction of selection on shared traits, a common consequence of sexual selection36. In species with ARPs, the scope for balancing selection may be further increased by alleles being selected in opposing directions between ARPs37,38 or if sexual antagonism is stronger between males expressing SSTs and females39,40,41. Therefore, by increasing the scope for antagonistic selection, sexual selection may augment the amount of genetic variance segregating in natural populations42,43. Thus, understanding how SSTs affect efficacy of selection, and the quantity and quality of genetic variation of a population, is critical to our understanding of the effect of sexual selection in general, and SSTs in particular, in shaping evolutionary processes. Indeed, some studies support the role of sexual selection in removing deleterious load at both phenotypic44,45,46,47,48 and genomic49 levels. However, other studies have not found such support50,51, suggesting that beneficial effects of sexual selection can be negated by sexual conflict52,53,54,55,56. Moreover, recent genomic evidence has supported the role of sexual antagonism in promoting balancing selection57,58. The limited number of studies and mixed findings indicate considerable knowledge gaps still exists, and this is particularly true for the role of costly SSTs, for which we are not aware of any study investigating their effect on the amount and nature of genetic variance segregating in populations. Here, we fill this gap using an evolve and resequence (E&R) approach in the bulb mite, R. robini, which has two male ARPs, each distinct in expression of sexually selected weaponry and associated reproductive strategy (Fig. 1).

a, Fighter male. b, Scrambler male. c, Female. The scale bar in a and b is 300 µm and 400 µm in c. Comparison of legs highlighted in red in a and b shows the dimorphic expression of the sexually selected weapon, a thickened and terminally pointed third pair of legs in fighters. Males can be easily assigned a morph by visual inspection of the thickness of third pair of legs using a standard stereomicroscope (see Extended Data Fig. 2 for the allometric relationship between width of third leg and idiosoma length). Male morph has previously been shown to be heritable66,109 and that the expression of the thickened legs of fighters is to some degree condition dependent65,66. Bearing the weaponry imposes costs90,110, and here we find similar support for such costs by showing that at F20, F-line males have reduced longevity compared to S-line males when the possibility of lethal combat was removed (z = −2.76, d.f. = 1, P < 0.001; Supplementary Fig. 6). In contrast, F- and S-line female longevity did not differ (z = −1.37, d.f. = 1, P = 0.17), indicating it is probably the presence of the weapon that is imposing costs to the bearer. Furthermore, each male morph is associated with different reproductive strategies: fighter males typically engage in aggressive contests with other males and use their thickened third pair of legs to puncture the cuticle of rivals. In contrast, scrambler males will typically avoid direct contests with rival males59. Unlike in some other systems with ARPs111, earlier work on R. robini did not detect negative frequency-dependent selection acting on morphs reproductive success69. Instead, indirect evidence suggests higher mutation load carried by scrambler males67, probably making expression of the aggressive, armoured phenotype too costly for them to benefit from. Additionally, a negative genetic correlation between weapon expression and female fitness39,67, possibly due to correlated gene expression72, may cause balancing selection on morphs and explain the maintenance of both fighters and scramblers in populations112.
Results and discussion
We sequenced and de novo assembled the genome of a single inbred line of R. robini, which resulted in a 307.5 Mb reference genome (1,533 contigs, N50/L50 = 1.59 Mb/43) and putatively identified X-linked contigs. We then investigated the effect of weapon evolution and associated changes to the competitive mating environment on the amount and nature of genetic variation, as well as phenotypic responses, in populations selected for either fighter (F-lines) or scrambler (S-lines) morphs (Fig. 2). Male morphs are characterized by the presence or absence of an enlarged third pair of legs (Fig. 1), which are used in contests over females that can be lethal59. The selection protocol (Extended Data Fig. 1) was facilitated by efficient scoring of discontinuous ARPs (Extended Data Fig. 2) and carried out in large, replicated populations with census size of 1,000 individuals (n = 4 F-lines and n = 4 S-lines). Selection led to substantial responses of morphs in both directions, however, the proportion of the selected morph exceeded 90% faster in F-lines compared to S-lines (χ2 = 39.9, d.f. = 6, P < 0.001; Extended Data Fig. 3).

Shown are the generations when sampling for resequencing was performed (blue boxes), from which Illumina reads were mapped to the reference genome and genomic analysis performed. Also shown are the generations at which morph proportion assays were performed (red boxes) to track morph proportion in experimental evolution populations and when phenotypic assays were performed (purple boxes), including a brief description.
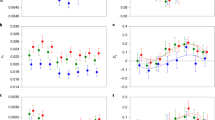
By sampling from each population at different generations (F1, F12, F29) and performing whole-genome resequencing on pools of individuals60 (DNA from 100 males and 100 females per replicate per generation was pooled by sex and sequenced to the mean coverage of 53× range 35−67×; Supplementary Fig. 1 and Supplementary Table 1) we were able to show striking changes and clear differences in genome-wide variation across the selection experiment between F- and S-lines (Fig. 3 and Supplementary Table 2). When analysing variation within autosomal genes (13,389 in total), we found that in F-lines both nucleotide diversity (Tajima’s Pi, π) and the number of segregating sites (Watterson’s theta, ϴ) declined over the duration of the experiment in all three considered classes of SNPs: non-synonymous, synonymous and intronic sites. In S-lines non-synonymous π (πns, Fig. 3a) did not decline, and the decline in all other categories was considerably lower compared to F-lines. Qualitatively similar patterns were also found when performing analyses using 10 kilobase (kb) sliding windows that included intergenic regions (Extended Data Fig. 4 and Supplementary Table 3).

Mean value from lines either selected for fighter (F, green; n = 4) or scrambler males (S, orange; n = 4) shown by large points with error bars (1 s.d.) in each of the three generations in which pool-seq sampling was performed (F1, F12, F29). Each smaller point represents the mean value from an individual experimental evolution population. Estimates are based on 13,389 autosomal genes. The left column (a–c) shows the estimates of nucleotide diversity (π) and the right column (d–f) the number of segregating sites (ϴ) for three classes of SNPs: non-synonymous SNPs (a: πns, d: ϴns), synonymous SNPs (b: πs, e: ϴs) and intronic SNPs (c: πi, f: ϴi). In all cases, there was a significant interaction between morph treatment and generation (P < 0.05) by repeated measures ANOVA analysis demonstrating substantially greater reduction in all measures of genetic diversity in all classes of SNP in F-lines compared to S-lines over the duration of the experiment (see Supplementary Table 2 for test statistics and post hoc comparisons).
In contrast to the differences between F- and S-lines in autosomal regions, there was no apparent consistent difference between F- and S-lines in the genetic diversity of putative X-linked genes (587 in total, Supplementary Fig. 2 and Supplementary Table 4) or 10 kb windows (Supplementary Fig. 3 and Supplementary Table 5), and no diverged SNPs were found on X-linked contigs (below). This suggests sex chromosomes responded differently to selection compared to autosomes, possibly because their effective population size (Ne) is lower61 or due to their hemizygosity in males exposing deleterious recessives to strong selection even at low frequencies62, minimizing differences in morph-specific selection. Further investigation is clearly warranted but would require a chromosome-scale reference genome assembly. At present, assignment of contigs to the X-chromosome is putative making any conclusions with respect to the X-chromosome tentative and we will therefore return our focus to autosomal variation.
Differences between F- and S-lines in autosomal genetic variation, whether estimated from π or ϴ, are the outcome of several processes, most notably, the selection imposed on males (that is, for or against the SST) by experimenters, natural and sexual selection, and drift acting within populations. For example, a decline in genetic diversity (π and ϴ) is expected as a consequence of drift and/or purifying selection. This has led to a common limitation of previous E&R studies of sexual selection (for example, refs. 49,63), in which the limited sizes of evolving populations make it difficult to discern the effects of selection from drift, particularly for variants with low to moderate selection coefficients. We attempted to overcome this limitation by using population sizes of 1,000 individuals, an order of magnitude larger than earlier E&R studies of sexual selection.
Our estimates of effective population size (Ne) confirm that alleles with relatively low selection coefficients (s = 1/Ne = 0.003; where Ne = lowest estimate of Ne across all lines) should overcome drift and respond to selection64. Furthermore, we find that F-lines had their Ne reduced by approximately 20% compared to S-lines (t = −3.64, d.f. = 6, P = 0.018; mean of F-lines, 369.5 (range 336.9–432.2); S-lines, 459.9 (range 431.7–483.6); Supplementary Table 6). Indeed, sexual selection theory17 predicts that stronger sexual selection (that is, greater variance in male reproductive success) reduces Ne, however, the magnitude of this effect has previously been largely unknown with regard to the expression of a SST. The difference in Ne between morph selection lines is probably driven by greater variance in male reproductive success in F-lines, which is at least partially caused by increased mortality of males due to lethal combat (F = 7.06, d.f. = 1, P = 0.021; Extended Data Fig. 5). In R. robini, the mating system can be described as polyandrous scramble competition, with (F-lines) and without (S-lines) male fights over access to females. We would expect these effects to be further exaggerated in species where males can monopolize mating from multiple females simultaneously, such as those that form harems or leks.
To separate the effects of selection from those of drift on genetic diversity we calculated the relative change in π and ϴ between generations for each autosomal gene at non-synonymous sites (Δπns and Δϴns), as well as in putatively neutral sites (Δπs and Δϴs) that should mostly reflect the effect of drift. The difference between genome-wide averages of Δ at non-synonymous and synonymous sites indicates that at F29 πns had increased in S-lines after controlling for drift, whereas πns in F-lines mostly reflect neutral processes (that is, Δπns − Δπs ≅ 0; Fig. 4a). In contrast, ϴns generally reflects neutral processes in S-lines, whereas in F-lines there is a considerable reduction in ϴns after controlling for drift (Fig. 4b).

a, Estimated using non-synonymous nucleotide diversity πns. b, Estimated using number of non-synonymous segregating sites ϴns after accounting for drift using genome-wide (GW) changes in synonymous π (πs) or ϴ (ϴs), respectively. Mean value from lines either selected for fighter (F, green; n = 4) or scrambler males (S, orange; n = 4) shown by large points with error bars (95% CI), with each smaller point representing the mean value from an individual experimental evolution population. Values were calculated by comparing changes in non-synonymous π or ϴ at F12 and F29 relative to F1 within each gene (n = 13,389), against that of the genome-wide average of presumed neutral sites: log((Δπ(ϴ)ns = π(ϴ)ns at F12 or F29/π(ϴ)ns at F1) + 1) − log((Δ π(ϴ)s = genome-wide average π(ϴ)s at F12 or F29/genome-wide average π(ϴ)s at F1) + 1). Thus, values of 0 (dashed horizontal line) would indicate any change in πns or ϴns mostly reflects drift, positive values that non-synonymous diversity is maintained over the level expected by drift and negative values that non-synonymous values are lost at a greater rate than expected by drift. As calculations are dependent to some degree on starting values, we log transformed Δ values to reduce the impact of outliers on means. Comparison of 95% CIs in a indicate that S-lines differ from drift (that is, no overlap with 0) at the end of the experiment, whereas at F12 in S-lines and all generations of F-lines have some degree of overlap with 0. Indicating that πns mostly reflects genome-wide average in F-lines, whereas in S-lines πns increased (that is, positive values) throughout the experiment when controlling for drift. In contrast to πns, there was considerable reduction in ϴns after controlling for drift in F-lines (that is, negative values and no overlap with 0), whereas, any changes in ϴns within S-lines mostly reflected genome-wide averages (that is, overlapping with 0). For comparisons by repeated measures ANOVA, see Supplementary Table 7.
The decline in autosomal ϴns in F-lines, beyond the expectations of drift, could be due to increased purifying selection compared to S-lines, leading to the enhanced loss of rare deleterious variants. Rare variants, in contrast, have little effect on π, for which no decline was observed in F-lines after controlling for drift. The increased Δπns in S-lines could be a consequence of increased frequencies of initially rare variants due to the selection against fighter morphs favouring males in poor condition, effectively counteracting natural selection and promoting deleterious variants. Overall, these data indicate that selection for male morphs, probably purged fighter populations of, but enriched scrambler populations for rare presumably deleterious variants. This could be due to the previously documented condition-dependence of weapon expression in R. robini65,66 and the indirect evidence that fighter males are probably less burdened with recessive deleterious alleles, as it was shown that inbred lines derived from scrambler males had increased inbreeding depression compared to inbred lines derived from fighter males67. In addition to costs of development and maintenance, evolution of weapons and associated aggressive behaviours59 may impose additional fight-associated energy demands, further enhancing selection against males in F-lines burdened with deleterious mutations. The corresponding reduction of direct male–male competition in S-lines may be another source of weakened purifying selection.
Our phenotypic analyses provide further evidence that our selection protocol affected the load of deleterious mutations differentially between F- and S-lines when estimated at F45. We found that after one generation of full sib × sib mating F-lines suffer reduced inbreeding depression, compared to S-lines (Fig. 5b), indicating that the former has substantially fewer partially recessive deleterious mutations68. This mirrors a set of previous studies: Drosophila melanogaster lines purged from presumed deleterious variants49 had lower levels of inbreeding depression47. However, we only find weak evidence that outbred F-line females have higher fecundity compared to S-line females at F20, whereas, at later time points we find no evidence of higher fecundity in F-lines (Fig. 5).

a, Fecundity assays performed at F20 (n = 233) and F32 (n = 217) using outbred females only. b, Fecundity assay performed at F45 using both inbred and outbred females (n = 210). In a, virgin females were paired with a male from the stock population (both male morphs were used in equal number), in b only fighters from the stock population were used. Fecundity data were collected from days 4–8 after virgin females were initially paired with males. There was a significant two-way interaction between morph selection line (F- or S-lines) and generation (χ2 = 4.10, d.f. = 1, P = 0.043), indicating that in a, F-line female fecundity declined between F20 and F32, whereas the fecundity of S-line females remained similar. There was no effect of the morph of stock population male that a female was paired with (χ2 = 0.44, d.f. = 1, P = 0.509). Inspection of model summaries show that within an individual generation there was, however, no difference between the fecundity of F- and S- line females. Similarly, in b when females were outbred there was no difference in the fecundity of F- and S-line females, however, as indicated by a significant two-way interaction between female status and morph selection line (χ2 = 5.45, d.f. = 1, P = 0.020), when females were inbred, we observed a decline in S-line female fecundity but that the fecundity of inbred F-line females remained similar to that of outbred females. Boxes are composed of the median and hinge values (25th and 75th percentiles), with whiskers ± interquartile range × 1.5. Individual data points from each female are denoted by circles.
That we do not observe an overall increase in female fecundity in F-lines may be reconciled if the benefits of purging deleterious mutations are counteracted by an increase in the strength of intra-locus sexual conflict (IASC) occurring between females and males with greatest expression of SST39,40,41. This increased strength of IASC has previously been described in R. robini inbred lines founded by fighters or scramblers67 and within populations selected for each male morph39, in which females derived from fighter treatments had decreased fitness compared to those from scrambler treatments. This indicates higher fitness of daughters of the less sexually competitive scramblers69, which has been proposed to contribute to their maintenance in populations39. In the case of our populations, these negative effects of IASC in F-lines were probably compensated by the positive effects of purging deleterious mutations. Such purging was minimized in the inbred lines in Łukasiewicz et al.67 and greatly limited in the populations in Plesnar-Bielak et al.39, which were an order of magnitude smaller than ours, thus limiting the effectiveness of selection. This potentially explains the differences in results and is suggestive that the impact of IASC on population fitness may interact with Ne.
Theory30 and empirical evidence57,58 indicates that IASC may increase the scope for balancing selection. Thus, if our selection affected the strength of IASC (intensifying and relaxing it in F- and S-lines, respectively39,67), SNPs consistently differentiated between experimental evolution treatments can be expected to be enriched for variants that were initially under balancing selection due to IASC. Furthermore, because such variants are expected to have more intermediate frequencies, their response to selection should be more easily observable compared to SNPs under purifying selection, which are expected to occur at low frequencies. We therefore identified SNPs that were diverged (across replicates) in frequencies at F29 between F- and S-lines. After filtering steps, we identified roughly 6 million autosomal SNPs at F29, 24,189 of which were found to be consistently diverged between F- and S-lines by using generalized linear models (GLMs) and applying a false discovery rate (FDR) with a q-value of less than 0.05 (ref. 70; henceforth, diverged SNPs). Of these diverged SNPs, around 55% (13,324) were fixed or nearly fixed (frequency of minor alleles <1%) in F-lines, compared to around only 2% (573) in S-lines (Fig. 6a). None of these SNPs were found to be diverged between F- and S-lines at the onset of the experiment (F1) at FDR < 0.05. Most of the diverged SNPs, roughly 65%, were found within protein coding regions, considerably more than expected by chance from randomly drawing SNPs at F29 and determining their locations (46.67%, 46.66–46.67%: 95% confidence interval (CI)). This suggests that mostly variants located in protein coding regions were targets of consistent divergent selection during the experiment.

a, A histogram of the frequency of minor alleles in those SNPs, which diverged between morph selection lines (F-lines, F, green; S-lines, S, orange) at F29; the left panel shows initial frequency of SNPs at F1, middle panel at F12 and right panel at F29. This shows a clear shift to the left in F-lines that is purifying selection and a clear shift to the right in S-lines, indicating that these initially rare alleles substantially increased in frequency. b, A histogram of selection coefficients (s) of diverged SNPs, left panel shows all estimations of s and right panel shows only those that significantly differ from drift (that is, s = 0) after applying FDR at q < 0.05.
In contrast to the expectation that the set of diverged SNPs, including targets of balancing selection, should be enriched for variants at more intermediate frequencies compared to genomic average, we found the opposite. Diverged SNPs had considerably lower initial frequency of minor alleles (median, 0.047), when compared to a distribution of the initial frequencies of all minor alleles (median of 0.160, 0.160–0.161: 95% CI). Similar patterns have previously been described in D. melanogaster, in which, populations with males selected for increased mating success had increased purging of variants that were initially segregating at low frequency when compared to populations selected for lower mating success49, which was interpreted as evidence for the deleterious nature of these SNPs. Consistent with this interpretation was our finding that the distribution of the frequency of minor alleles from diverged SNPs shifted to the left between generations F1 and F29 in F-lines, suggesting increased effectiveness of purifying selection, but substantially shifted to the right in S-lines, as a result of increase in frequency of initially rare alleles (Fig. 6a).
An alternative explanation to the increased purging in F-lines, particularly those SNPs that were initially rare, is that the reduced Ne in F-lines led to stronger effects of drift. To explore if drift alone could cause such patterns, we simulated neutral evolution of SNPs from populations with Ne comparable to our mean estimations for each selection regime (Supplementary Table 6). None of the 1 million simulated SNPs reached significance when applying an FDR with q < 0.05, indicating that divergence of SNPs between F- and S-lines, especially those that were initially rare (Extended Data Fig. 6), is highly unlikely to be due to differences in drift alone, and suggesting that our results are indeed likely to be a consequence of consistent differential selection. This is further corroborated by our estimates of selection coefficients (s) acting on the minor alleles of diverged SNPs (Fig. 6b). In F-lines, the median values were generally negative (s = −0.052, −0.053–−0.050: 95% CI), in contrast to positive values in S-lines (s = 0.099, 0.097–0.100: 95% CI). Also consistent with the scenario of purifying selection is our finding that the mean PN/PS (non-synonymous SNP density/synonymous SNP density) of genes containing diverged SNPs that were fixed or nearly fixed (frequency of minor alleles <1%) within coding regions in F-lines at F29 had a slightly lower median (0.119) at the onset of the experiment (F1) than expected from a random sample of background genes (0.135, 0.134–0.135: 95% CI; Extended Data Fig. 7d). The lower PN/PS suggests that this set of genes was under stronger purifying selection than an average gene from the genome.
To further explore the potential contribution of genomic regions under balancing selection to the divergence between F- and S-lines we focused on windows and genes where high initial Tajima’s D (D) is suggestive of this form of selection. The top 10% of windows with the highest D (mean of F1 samples) were enriched for windows containing diverged SNPs (χ2 = 7.87, d.f. = 1, P = 0.005; Extended Data Fig. 8a), but equivalent analysis of exons showed the top 10% genes had no such enrichment (χ2 = 0.33, d.f. = 1, P = 0.565; Extended Data Fig. 7a). Both exons and windows containing diverged SNPs tended to be more polymorphic (π) than the genomic background (respectively, Wilcoxon signed-rank test; W = 3,125,400, P < 0.001; W = 3,275,300, P < 0.001; Extended Data Figs. 7b and 8c). Finally, there was no difference in PN/PS between genes containing diverged SNPs and background genes (W = 4,576,400, P = 0.332; Extended Data Fig. 7c). Overall, higher polymorphism in windows and genes containing diverged SNPs, coupled with enrichment of the former in high D regions, is consistent with either the action of balancing selection or relaxed evolutionary constraints on these genes/genomic regions. Why diverged SNPs that showed initially low minor allele frequency were enriched in those regions can be explained by their weak deleterious effects, either because of relaxed constrains or due to the sheltering effect of balancing selection71. Indeed, strongly deleterious variants would be unlikely to consistently increase in frequency as we observed in S-lines (Fig. 6a).
As IASC is probably reduced in S-lines39,67,72, we assumed that polymorphisms previously maintained under balancing selection by IASC within the stock population, with mixed male morphs, were more likely to be lost from S-lines compared to F-lines. We consequently predicted that the windows containing diverged SNPs fixed or nearly fixed (frequency of minor alleles <1%) in S-lines would be overrepresented among those with initially high D (top 10%, as described above). However, there was no evidence to support this prediction (χ2 = 1.64, d.f. = 1, P = 0.200; Extended Data Fig. 8b). Next, we investigated the fate of autosomal windows with initially high D (top 10%), predicting that if genetic variation was maintained in these regions by balancing selection driven by fighter-specific IASC, then SNPs would be more often maintained at intermediate frequencies in F-lines and therefore maintain high estimates of D. However, we find no evidence that these windows with initially high D differed between F- and S-lines in estimates of D after 29 generations of selection (and release of S-lines from fighter-specific IASC; t = 0.72, d.f. = 6, P = 0.498; Extended Data Fig. 8d). Thus, overall, we have found no evidence for elevated IASC between fighters and females enhancing balancing selection. We note, however, that this result does not exclude the role of fighter-detrimental female-beneficial alleles in the observed response to selection. Such alleles could have been selected against in our stock population, but sexual antagonism might have weakened selection against them, allowing them to segregate at low frequencies for longer than would be the case with unconditionally deleterious mutations73. Their release from sexual conflict in S-lines would drive their frequency upwards, possibly contributing, alongside an increase in frequency of deleterious alleles, to a mostly positive s of diverged SNPs in S-lines (Fig. 6b). As discussed above, this could explain why fitness of females in S-lines does not decrease despite their higher mutation load.
The model of genic capture20 predicts that SSTs reflects genetic variance across the genome. To explore this, we further analysed the genomic location of the diverged SNPs. We found that diverged SNPs were located in as many as 291 autosomal contigs, including 99 of the 100 longest ones, suggesting that they reflect genome-wide patterns of genetic variance as predicted by the genic capture hypothesis. However, diverged SNPs were found in 4,326, out of a potential 24,290, autosomal windows (expected value 11,958 windows, 11,957–11,959: 95% CI; if drawing random SNPs). This demonstrates that although diverged SNPs are broadly spread across much of the genome, there are many windows/regions containing a high density of diverged SNPs (see Extended Data Fig. 9 for an example from the ten longest contigs). These high-density windows may represent regions of high gene density, as suggested by their exonic enrichment, or a consequence of linkage disequilibrium between SNPs under strong selection and neighbouring regions, resulting in a clustered SNP distribution. Additionally, seven contigs appear to have a disproportionally high number of diverged SNPs (Extended Data Fig. 10), possibly indicative of the existence of linkage groups, for example inversions, associated with morph expression74,75. High-density ‘islands’ of genomic divergence associated with changing strengths of sexual selection have previously been described in D. pseudoobscura, with these regions not associated with inversions but more probably a consequence of selective sweeps63. While it must be acknowledged that the total number of independently selected variants is likely to be less than the total number of diverged SNPs, their location across a large number of contigs is consistent with discontinuous expression of the weapon capturing genetic variance across the genome. It is worth re-emphasizing that SNPs under strong purifying selection, being very rare in our stock population, were unlikely to be repeatedly selected in our replicate populations, and thus be captured in our diverged SNP set. Consequently, this set probably underestimates the impact of purifying selection that was evident from analyses of genomic diversity across the genome (Fig. 3 and Extended Data Fig. 4). That the set is nevertheless enriched for SNPs with some signatures of purifying selection therefore further highlights the purifying role of sexual selection associated with the presence of the weapon.
Concluding remarks
Overall, our E&R experiment demonstrated that selection for a costly SST and associated changes in mating systems lead to a decrease in genetic variation in a population, in line with expectations of the lek paradox76. At the same time, our results support predictions of one of the hypotheses proposed to resolve the lek paradox (that is, genic capture20,22), by demonstrating that selection of SST captured genetic variance across the genome. This allows SSTs to be a large ‘mutational target’77, indicating that genetic variance in SSTs can be maintained indefinitely due to mutation-selection balance, although their evolution may alter this balance genome-wide and reduce genetic variance by enhancing purifying selection, as demonstrated by our results. These genome-wide purifying effects of sexual selection may bolster population resilience to environmental and genetic stress as some empirical work has found45,78,79,80,81,82,83 (also see refs. 50,55,56,84,85,86,87 for counter examples), but our phenotypic data show that despite purging, population fitness does not necessarily improve under benign environmental conditions, possibly due to sexually antagonistic effects associated with SST evolution39,40,41,67. While such effects were proposed to promote balancing selection, thus possibly helping to maintain potentially adaptive genetic variance, we have not found support for signals of balancing selection associated with weapon evolution. However, our results indicated that variants responding to selection on morph can, despite their overall genome-wide distribution, be clustered in polymorphic regions, possibly under relaxed or balancing selection. The reasons for this clustering, also reported in other studies of genome-wide patterns associated with sexual selection63,75, remain to be explained in R. robini.
Methods
For a schematic overview of the experimental design, see Fig. 2.
Experimental evolution
Protocol
The stock population (Stock population below) was allowed to expand for one generation and from this we established eight replicate experimental evolution populations, four selected for fighter morphs (F-lines) and four selected for scrambler morphs (S-lines). Each population was founded by 1,000 recently eclosed adults (500 random females and 500 random males of the desired morph). The classification of the morphs was based on visual inspection using a stereoscopic microscope and was unambiguous due to the discontinuous distribution of the phenotypes (Classifying male morphs below). Adults were allowed to interact freely for 6 days, all surviving adults (with previously laid eggs discarded) were transferred to a new container for 24 h of egg laying, after which adults were removed. The resulting offspring were allowed to mature over 13 days and 1,000 individuals from the newly eclosed adults selected for founding the following generation, again 500 random females and 500 random males of the desired morph, with this protocol repeated every generation (Extended Data Fig. 1). The isolation of nymphs to use virgins was unfeasible with our experimental design and population sizes. However, the period of 6 days after selecting the founders of the next generation and collecting eggs for the next generation was probably enough to displace most sperm stored by females mated with any unselected males due to the high number of remating that will be occurring over this duration (females on average remate after 80 min, ref. 88) and last male sperm precedence89. The timing of generation was chosen to reflect maturation rates from our stock population to avoid indirect selection on this trait. Moreover, a previous study90 showed there was no difference between male morphs in maturation rates and that over similar lengths of time to the protocol here the fertility of both morphs remains similar. Therefore, our protocol was not likely to impose strong differential selection on morph life histories.
Tracking morph proportion
We assayed the proportion of male morph in each population every 6–7 generations, by isolating 200 larvae (ten per vial) from the container, allowing maturation within vials and recording the morph of all males that eclosed (mean n = 86 per population, per generation, range 71–109). Our selection protocol was highly effective in driving an increase in the frequency of the desired male morph to >90% after 20 generations in both treatments, with this effect considerably faster within F-lines indicted by a significant two-way interaction between proportion of the desired morph and generation (χ2 = 39.9, d.f. = 6, P < 0.001; Extended Data Fig. 3). Selection for specific morphs was more effective than a previous experiment by Plesnar-Bielak et al.39, in which it took around 35 generations to drive both the desired morphs above 90%, we note that their lines selected for fighters also responded faster than those selected for scramblers. The difference between experiments in driving the frequency of desired morph >90% is probably a consequence of a longer interaction period (3 versus 6 days) in which the stored sperm of males before selection was able to be displaced and/or because selection was acting more efficiently in our larger populations. The difference between rate of changes in morph proportion between F- and S-lines in the current study, and also found by Plesnar-Bielak et al.39, may be associated with the genetic architecture of morph expression. Alternatively, selection could be less effective in scrambler lines if they are less efficient than fighters in displacing sperm of previous females’ partners, but this is unlikely as R. robini male morphs have previously been demonstrated to not differ in their sperm competiveness89.
Stock population
We established a stock population by mixing three laboratory populations that were collected from three sites in Poland (Krakόw, collected in 1998 and 2008, Kwiejce, collected in 2017 and Mosina, collected in 2017; Extended Data Fig. 1), where the line derived for material used in creating the reference genome (below) was also established from the same collections at Mosina in 2017. All populations were maintained in cultures with several hundred individuals per generation before mixing and establishment of the stock population. The mixing of distinct populations increased the genetic variance in the stock population, which otherwise would probably have been limited due to founder events and the limited population size of each of the contributing populations73, thus decreasing our power to detect the effects of SSTs on genetic variation. The newly mixed stock population was maintained with several hundred individuals per generation for roughly 12 generations before the onset of this experiment. This time period is probably enough to break linkage disequilibria that could have arisen due to mixing (for unlinked loci, linkage disequilibrium should decay by half each generation91).
One generation before establishing experimental evolution populations the proportion of male morphs was determined from 176 random males, indicating a roughly equal morph ratio (95 fighters, 81 scramblers) of the stock population (Extended Data Fig. 3).
General housing and husbandry
The stock population and experimental evolution populations were maintained in plastic containers (approximate length, 9.5 cm; width, 7 cm; height, 4.5 cm), filled with roughly 1 cm of plaster-of-Paris. The same containers were used when sampling mites for sequencing for the reference genome or resequencing from experimental evolution populations, but either replaced the plaster-of-Paris with 5% agarose gel or added a thin layer of 5% agarose gel above the plaster-of-Paris, respectively. The agarose gel was used to reduce the number of contaminates within our samples and on the basis of preliminary extractions that indicated that small pieces of plaster-of-Paris may reduce the quality of DNA during extractions. Individuals, pairs and small groups of ten mites were housed in glass vials (approximate height, 2 cm; diameter, 0.8 cm) and large groups of 60 or 150 mites in plastic containers (approximate height, 1.5 cm; diameter, 2 cm diameter or height, 1.5 cm; diameter, 3.5 cm diameter, respectively) all with an approximate 1 cm base of plaster-of-Paris. All plaster-of-Paris bases were completely soaked in water before mites were transferred into them. All mites were reared at a constant 23 °C, at high humidity (>90%) and were provided an excess of powdered yeast ad libitum.
Classifying male morphs
To illustrate the discontinuous distribution of the weapon and to demonstrate that this classification based on visual inspection is non-subjective, we performed phenotypic measurements from male mites from a population collected near Krakόw, Poland, that had previously been fixed onto microscope slides for a separate study66. The measurements taken were idiosoma (body without mouthparts) length and width of third proximal segment of the third right leg (genu). Measurements were preformed using Lecia DM5500B microscope and Lecia Application Suite v.4.6.1. We then performed an analysis to, first, determine whether the allometric relationship between idiosoma length and width of third pair of legs is best described as discontinuous and, second, to verify that classification by simple visual inspection matches the same classification from allometric analysis. One researcher performed all the measurements and classified each male as a fighter (n = 50) or scrambler (n = 50), a separate researcher was then given the measurements but not the classification of the male morph.
Broadly, guidelines for the analysis of non-linear allometries92 were followed. The log–log scatterplots of idiosoma length against leg width were visualized, which showed there was clear evidence for non-linear scaling relationships. Next histograms of idiosoma length, leg width and relative leg width (leg width/idiosoma length) were visualized (Extended Data Fig. 2a–c). Where a normal distribution of idiosoma length, and a binomial distribution in leg width and relative leg width are further indications of a discontinuous relationship. On the basis of the lowest point between the two peaks of the density plot of relative leg width (Extended Data Fig. 2c) males were classified as scramblers (relative leg width <0.125) or fighters (relative leg width >0.125). Replotting the log–log scatterplot of idiosoma length and leg width, and using the classification of morph described above clearly demonstrates the discontinuous allometric relationship of idiosoma length and leg width in R. robini (Extended Data Fig. 2d). Moreover, on the basis of the Akaike information criterion (AIC), the discontinuous model where males were assigned a morph (AIC = 646.5) clearly has a substantially better fit than a simple linear and quadratic models (AIC = 918.5 and 920.2, respectively). Further models were omitted from comparison (for example, breakpoint or sigmoidal) due to the clear discontinuous allometry observed. Finally, all 100 males were assigned the same morph by visual inspection and blind allometric analysis, demonstrating that the former is effective and accurate in classifying male morph.
Phenotypic assays
Fecundity assays were performed using experimental evolution females at F20 and F32. Eggs laid by females between days 4–8 were counted, encompassing the window of time of most evolutionary relevance for female fitness during maintenance of selection lines (that is, egg laying period in selection lines was between days 6–7) and also likely to capture variation in lifetime fecundity that remains largely consistent throughout the first 3 weeks of life93. Nymphs were individually isolated to gain virgin females, which on maturation females from each experimental evolution population (n = 30) were paired with a male from the stock population (15 with fighters and 15 with scramblers). Pairs were transferred to a new vial on day 4, with the pair being removed from the second vial after a further 4 days and all eggs in the second vial counted. If the male had died in the first vial, they were replaced with a stock male of the same morph. Any female deaths in the first or second vials were recorded.
Longevity assays were also performed at F20 and F32. At F20, females used in fecundity assays, including the stock male they were paired with (replaced if dead), were transferred to a new vial at day 8. After this point, vials were then checked every 2 days for female deaths and pairs were moved to new vials every 4 days. Males were replaced with stock males of the same morph if found dead. Similarly, at F20, on maturation males from experimental evolution populations (n = 30) were paired with stock females, vials were checked every 2 days and changed every 4 days, with females being replaced if dead. At F32, only female longevity was determined and was performed in groups; 30 experimental evolution females and 30 stock males (15 of each morph) were placed in plastic containers, two per experimental population. This logistically easier estimate of longevity was done due to local restrictions during the SARS-CoV-2 pandemic and the imposed limitations on people working closely together. Groups were checked for dead females every other day and all remaining live mites transferred to a new container every 4 days. When mites were transferred to a new container the sex and morph ratio were balanced to that of the remaining females, by either removing or adding males of the desired morph from the stock population.
To determine whether the survival of mites differed between F- and S-lines when competition between males was allowed, at F45 we created small colonies from each population and survival of males and females recorded over 6 days, the same period as used between selecting founders of the next generation and subsequent egg laying period. Colonies were at a 50:50 sex ratio, established with 150 newly eclosed mites placed into small plastic containers. This was approximately the same density after selection of the next generations founders during the maintenance of experimental evolution populations (150 mites in roughly 9.5 cm2 = 16 mites per 1 cm2; 1,000 mites in roughly 67 cm2 = 15 mites per 1 cm2). After 3 days, all colonies were checked and any dead mites identified by sex. After another 3 days, again dead mites were recorded and all surviving mites sexed and counted.
Additionally, at F45 we performed further fecundity assays to obtain estimates of inbreeding depression within experimental evolution populations. To establish family groups, larvae were isolated and on maturation F0 males and females (n = 16) from within the same experimental evolution population were paired together. Pairs were allowed to produce eggs for 48 h, after which adults were removed from vials. After hatching from each pair, 12 F1 larvae were isolated into new vials. On their maturation, these F1 mites were either paired with a full sibling, that is, from the same family, or with an individual from a different family but from the same experimental evolution population. When possible, we made two inbred and two outbred pairs with same family lines used. Again, pairs were allowed to produce eggs for 48 h before their removal for the vial. After a further 5 days, vials were checked for larvae, if larvae were present in the first vial six were individually isolated and the second vial discarded, if no larvae were present in the first vial the second vial was checked for larvae and, if present, they were isolated. This protocol therefore produced inbred and outbred individuals from within the same experimental evolution population. Which, as above, on maturation F2 inbred and outbred females were paired with stock males (fighter males only) and number of eggs laid between days 4 and 8 counted. Only a single female from each unique inbred or outbred family was used. Either due to pairs failing to produce offspring or there being no F2 females, samples sizes were not exactly equal. In total, 59 outbred and 55 inbred females from F-lines, and 56 outbred and 54 inbred females from S-lines were paired with stock males.
Phenotypic assay statistical analyses
All phenotypic analysis was conducted using R statistical software94 (v.3.5.2) and data were visualized using ggplot2 (ref. 95).
Analysis of male morph proportion was performed using a generalized linear mixed model with binomial error structure, fitted using lme4 (ref. 96). Where the proportion of desired morph was compared in model with morph selection and generation (as a factor) including their two-way interaction as explanatory variables, and population included as a random effect.
All fecundity data were analysed using generalized linear mixed models with Poisson error structures, fitted using lme4. Due to the differences in stock population males used between F45 and earlier generations, and slightly different rearing conditions between females in the fecundity assays from generations F20 and F32, they were analysed separately from data collected in F45. However, we noted that the fecundity of females in Fig. 5a was comparable to the outbred females in Fig. 5b. Explanatory variables fitted to fecundity data from F20 and F32 were, morph selection treatment, generation, including their two-way interaction term, and stock male morph. The explanatory variables fitted to fecundity data from inbreeding depression data were, morph selection treatment and status of female (that is, inbred or outbred), including their two-way interaction term. In both analyses, we included population as a random effect and an observation level random effect to account for overdispersion, we omitted fitting random slopes due to issues with increasing the complexity of random effects close to reaching a singular fit. Females that died before the end of the fecundity assay and those that laid zero eggs were removed from analysis. This excluded five females from F20 (three F-line and two S-line), 20 from F32 (13 F-line and seven S-line) and 16 from F45 (three inbred and three outbred F-line, and nine inbred and one outbred S-line).
Longevities of females at F20 and F32, and males at F20, were analysed separately using mixed effects Cox models, fitted using coxme97. In all analyses, we included a random effect of population, with morph selection treatment as an explanatory variable and extra variable of male morph included in female longevity analysis at F20. Survival of mites over 6 days at F45 was analysed using a GLM with counts of dead and surviving mites fitted with a quasibinomial error structure, the model included morph selection treatment and sex, including their interaction term, as explanatory variables. If individuals were lost due to handling error (that is, killed or escaped) they were right-censored during analysis.
Genome assembly
Sample origin
A line of R. robini originated from a wild-collected population from the Mosina region (Wielkopolska, Poland). In October 2017, onions were collected from the field and approximately 200 individuals of R. robini were identified under dissecting microscope. The line used for DNA isolation in the genome sequencing project was developed from full sib × sib mating for 14 generations (to maximize homozygosity) following and continuing the protocol described in ref. 67.
DNA extraction
For DNA extraction we used only mite eggs, that were laid by 500 females, collected in a container (see above for a description) Females were kept in this container for 3 days. After that time, they were removed, and eggs were filtered using fine sieves and washed for 1 min in 0.3% sodium hypochlorite solution and in Milli-Q water for 2 × 2 min to remove any potential foreign DNA contamination. These eggs were collected in 1.5 ml Eppendorf tube and after short centrifugation, the remains of the water (supernatant) removed with a pipette. The sample was immediately transferred to ice and prepared for DNA extraction. DNA was extracted using Bionano Prep Animal Tissue DNA Kit for HMW DNA isolation according to the manufacturer’s instructions. Briefly, eggs were smashed with a sterile pellet pestle on ice in 500 μl homogenization buffer; the sample was fixed with 500 μl cold ethanol and incubated 60 min on ice, after that time the sample was centrifuged at 1500g for 5 min at 4 °C and the supernatant was discarded. Next, after resuspension in a homogenization buffer pellet, this was cast in four agarose plugs as described in the original protocol. Agarose plugs were incubated with Proteinase K and Lysis buffer solution for 2 h with intermittent mixing. After that time, the digestion solution was replaced with a freshly made one and incubated overnight with intermittent mixing. According to the original protocol, after RNase A digestion and plug washing, DNA was recovered by incubation of the plugs in TE buffer, followed by plug melting and addition of agarase. Recovered DNA was dialysed and homogenized on a membrane for 45 min at room temperature and transferred to a clean tube with a wide bore tip.
Sequencing
Sequencing was done using Oxford Nanopore Technologies (ONT, MinION). Isolated DNA purified using AMPure XP beads and resuspended in H2O before library preparation. Two separate libraries were prepared using ligation sequencing kit, SQK-LSK109 and Rapid Sequencing Kit SQK-RAD004, respectively, according to the manufacturer’s protocols and were sequenced on a FLO-MIN106 R9.4.1 SpotON flow cell on a MinION Mk 1B sequencer (ONT). The total yields from sequencing were 484,700 reads (2,417,068,187 nt) with a read-N50 of 10,044 nt (ranging from 216,403 to 100). Base calling of the raw reads was done using Guppy (v.3.3) resulting in a total sum of the reads 7,979,616,172, equivalent to 26× coverage aiming for a genome of 300 megabases (Mb). The reads N50/N90 were estimated at 7,958/1,719.
Assembling reference genome
Reads aligning with the Mitochondrion genome were identified using BLASTN and filtered from the raw reads before assembling the genome. The remaining ONT reads were assembled using the Flye software (v.2.6), with –min-overlap 3,000 to increase stringency at the initial overlay step, and default parameters including five rounds of polishing through consensus, contigs were additionally polished two times with Medaka (v.0.11.2). Illumina paired-end RNA dataset is assembled using CLC Assembler (CLC Assembly Cell). Both RNA assemblies and paired-end 10X genomic dataset (unpublished data) were mapped onto the contigs using minimap2 (v.2.16) and BWA mapper (v.0.7.17), respectively, and the assembly was further polished using PILON (v.1.20) to error correct potential low-quality regions. The resulting assembly yielded a genome of 307 Mb, assembled into 1,533 contigs ranging from 10,840,357 to 100 basepairs (bp) and an assembly-N50 of 1.670 Mb. Moreover, the BUSCO completeness analysis using the Arachnida (odb10) reference set confirmed our assembly represents the complete genome C:94.8%(S:89.1%,D:5.7%),F:0.9%,M:4.3%,n:2934 (=arachnida_odb10), only missing 126 genes from the whole reference set. Knowing that BUSCO only gives a rough estimation, we remain confident that this assembly represents well the bulb mite genome.
Flow-cytometry
Whole individual R. robini were homogenized in 500 μl of ice-cold LB01 detergent buffer along with the head of a male Drosophila melanogaster (1 C = 0.18 pg) as an internal standard. The homogenized tissue was filtered through a 30-μm nylon filter. Then 12 μl of propidium iodide with 2 μl of RNase was added, and stained for 1 h on ice in the dark. All samples were run on an FC500 flow cytometer (Beckman-Coulter) using a 488-nm blue laser, providing output as single-parameter histograms showing relative fluorescence between the standard nuclei and the R. robini nuclei. Six replicate samples were run to account for variation in fluorescence outputs. The genome size of R. robini was estimated at 0.30 pg, or about 293 Mb, and consistent with estimation of size from the genome assembly described above.
Mitochondrial genome
ONT reads aligned with R. robini mitochondrion genome were de novo assembled with Flye (v.2.6) assembler and polished with Racon. Mitochondrion genome is assembled in one single contig with a size of 15,335 bases.
Gene prediction
On the polished final genome, protein coding genes have been predicted. For this, AUGUSTUS was used including hints coming from R. robini RNA-sequencing (RNA-seq) (samples SRR3934324, SRR3934325, SRR3934326, SRR3934327, SRR3934328, SRR3934329, SRR3934330, SRR3934331, SRR3934332, SRR3934333, SRR3934335, SRR3934337, SRR3934338 and SRR3934339 from the PRJNA330592 BioProject deposited at the National Center for Biotechnology Information (NCBI) Short Read Archive) and proteins coming from highly curated Tetranychus urticae (v.2020-03-20) as well as proteins from the previous version of the unpublished, Illumina-sequenced R. robini genome (https://public-docs.crg.eu/rguigo/Data/fcamara/bulbmite.v4a/). The PE RNA-seq reads were mapped on the genome using HISAT2 (-k 1 —no-unal) and further processed with Regtools to extract junction hints and filtered for junctions with a minimum coverage of 10. All the RNA-seq reads were also assembled with CLC Assembly Cell (v.5.2.0) software, setting the word size for the Bruijn graph at 50 and maximum bubble size at 31. The reads were assembled into 689,563 contigs (ranging from 10,675 to 180 bp), which were later mapped on the genome with GenomeTheader to generate complementary DNA hints. Protein hints were generated by using with Exonerate (v.2.2) with Protein2Genome model. To reduce the amount of overprediction due to repeated elements (transposable elements, simple sequence repeats) we de novo predicted high abundant repeats using RepeatModeler. The accompanying parameter file for extrinsic data for AUGUSTUS was adapted to include these hints as well as the softmasking of the genomic sequence. The resulting gene predictions from AUGUSTUS were further curated with EvidenceModeler using the same extrinsic data. The BUSCO analysis confirmed that our gene prediction indeed captured the expected genes well (C:94.6%(S:86.3%,D:8.3%),F:0.4%,M:5.0%,n:2934 (=arachnida_odb10)). The final predicted gene set was subsequently processed to be uploaded into ORCAE (https://bioinformatics.psb.ugent.be/orcae/overview/Rhrob)98.
Resequencing
Genomic sampling and mapping
For genomic analyses we sampled material from each of the morph selection lines (n = 8) at F1, F12 and F29. Following the experimental evolution protocols, after the first 24 h of egg laying all adults were transferred to a new container (described above) for a second 24 h to lay eggs and from these second dishes genomic material was sampled. On maturation, adults were transferred to and kept for 3 days in containers. Adults were then randomly selected and placed into Digestion Solution for MagJET gDNA Kit (F1&12) or ATL buffer (F29) before freezing at −20 °C. From each population two samples were collected consisting of 100 individuals (1 × 100 females and 1 × 100 males of random morph), the two samples separated by sex were used as technical replicates. The tissue from the 100 individuals within each sample was homogenized and DNA was extracted by Proteinase K digestion (24 h) followed by standard procedures using MagJET Genomic DNA Kit (ThermoScientific, F1&12) or DNeasy Blood and Tissue (Qiagen, F29). DNA concentration was controlled with the Qubit double-stranded DNA HS Assay Kit and DNA quality was assessed on agarose gels. The library preparation was performed using NEBNext Ultra II FS DNA Library Prep kit for Illumina.
Whole-genome resequencing was carried out by National Genomics Infrastructure (Uppsala, Sweden) using the Illumina Nova-Seq 6000 platform with S4 flow cell to produce 2 × 150 bp reads (average 160.7 × 106: range 130.7 × 106 − 189.9 × 106). Adaptors were trimmed from reads using Trimmomatic99 software (v.0.39) and unpaired reads discarded. Fastq files were mapped to the assembled genome with bwa mem100 (v.0.7.17-r1188) using default settings. Sam files were converted to bam files, sorted, duplicates marked and ambiguously mapped reads removed using samtools101 (v.1.9). On average, 90% (range, 86–93%) of the reads from each sample were mapped successfully, of which an average of 17% (range, 15–19%) were marked as duplicates. This left us with an average of 117.7 × 106 pair end reads per sample, ranging between 99.6 × 106 and 145.9 × 106 (Supplementary Table 1).
Genomic analysis
File preparation and filtering
Preparation of files used in genomic analysis was done as follows: bam files were converted to a pile-up file using samtools, following which indels and surrounding windows (5 bp either side) were filtered, using identify-genomic-indel-regions.pl and filter-pileup-by-gtf.pl in PoPoolation102 (v.1.2.2) to avoid false SNPs, with the resulting filtered pile-up file converted to a sync file using mpileup2sync.pl in PoPoolation2 (ref. 103) (v.1.201). Using custom python scripts, the distribution of coverage from each sample (single sex) was determined by recording the coverage of positions every 10 kb across the genome from the sync file to give information on expected coverage (Supplementary Fig. 1). On the basis of this, we filtered the sync and pile-up files to contain only regions within a range of informative coverage, where the mean coverage of all samples at every position was between 50% of the expected coverage and 200% of the expected coverage (56×, range 23−112×). The pile-up and sync files containing individual male and female samples (48 in total) were then merged by sex to give files containing allele frequencies from 24 samples (eight populations across three generations), each consisting of allele frequencies of 200 individuals (100 males and 100 females, above) and used in all subsequent analysis (unless stated otherwise). Similarly, we drew coverage of a position every 10 kb from each sample in the sex-merged sync file to determine a distribution from which we decided to subsample to (Supplementary Fig. 1). We putatively identified X-linked contigs (below) and excluded them autosomal analysis. A similar, but, separate analysis on genes and SNPs from X-linked contigs was performed by using different parameters (below).
Estimating nucleotide diversity
Using PoPoolation we determined various estimates of genetic diversity per sample (that is, 24 sex-merged samples). The pile-up file from each sample was subsampled using subsample-pileup.pl to a coverage of 63× (max coverage, 252×) to standardize estimations of genetic diversity across the genome, between populations and across generations. First, nucleotide diversity (Tajima’s Pi, π) and number of segregating sites (Watterson’s theta, ϴ) were estimated within genes. We performed analysis of exons using Syn-nonsyn-at-position.pl, in which genetic diversity of synonymous and non-synonymous positions were determined. Further analysis of overall genetic diversity within exons and introns were performed using Variance-at-position.pl, Tajima’s D (D) also estimated in the former. We used a minimum count of three (equal to a minor allele frequency of roughly 5%) for a SNP to be called, and a phred score >30 and a pool size of 400. Further analysis using 10 kb sliding windows (step size 10 kb) across the genome were performed using Variance-sliding.pl, and also included estimation of D. Estimates of D require the minimum count to be 2, but otherwise all the same parameters were used.
We filtered genes to be included in our analysis (and all subsequent analysis) on the basis of a number of criteria. On the basis of extensive RNA-seq data from both males and females (Plesnar-Bielak, unpublished data with NCBI accession number PRJNA796800), we only included genes in our analyses that were expressed at a mean level of fragments per kilobase of transcript per million mapped reads >1 across 72 samples originated from both sexes and both morphs rearing in three different temperatures (18, 23 and 28 °C). A further filtering step was performed to remove genes with inconsistent mapping between samples, only genes with >60% exons mapped to (calculated from positions used to calculate parameters in the Syn-nonsyn-at-position.pl π outputs), with 63−252× coverage, in all 24 samples were included in the analysis. The final dataset contained 13,389 autosomal genes and subsequently used to filter other datasets to retain this set of genes only (see Supplementary Table 8 for a list of genes). Similarly, windows were discarded from outputs if <60% had been mapped to with 63−252× coverage; when comparing the estimation of genetic diversity between treatments or generations, every window in all 24 samples had to meet these criteria. This means that all comparisons are conservative and based on the same genes or windows, and therefore unlikely to be biased by any differences in mapping between samples.
As our data included estimates of genetic diversity from the same population across multiple generations, we performed analysis by repeated measures analysis of variance (ANOVA), which takes into account the non-independence of samples. Comparisons were made on the mean values of each experimental evolution population.
Estimating X-linked diversity
We then repeated the above analysis on X-linked contigs (below) using the same parameters unless stated otherwise, and also performing the SNP based analysis below. Initially we ran the analysis using 75% of the target coverage and pool size used for autosomes that is a target coverage of 47× and a maximum coverage of 189×, with a pool size of 300. Following filtering steps, it was clear that two samples (PS17 and PS21) with relatively low coverage (Supplementary Fig. 1 and Supplementary Table 1) were having a large effect on filtering steps (that is, >60% of genes being mapped to in all 24 samples) and reducing the final X-linked dataset to contain fewer than 200 genes. We therefore opted to reduce the target coverage further to 40×, in an attempt to retain more genes. This slight reduction of target coverage increased the number of genes in the final dataset substantially to 587 genes. We therefore opted to use a minimum coverage of 40× in all analysis of X-linked SNPs, genes and windows.
Diverging SNPs
To determine divergent SNPs between F- and S-lines, we extracted the allele frequencies of all samples from the sex combined sync file. Samples from F29 were then used to filter the entire dataset to only contain SNPs on the basis of a number of criteria. First, positions within all samples were required to have a coverage >63× and <252×. Second, the frequency of minor alleles (calculated as coverage − major allele count) from all samples combined had to be >5% (that is, the average of all samples but not necessarily above >5% in all samples). Thus, our dataset contained only positions with the target coverage in all F29 samples and in which polymorphisms were unlikely to be a consequence of sequencing errors. After this filtering we were left with roughly 6 million SNPs used in further analysis. We performed a GLM, at each position by comparing the count of the major allele against counts of minor alleles at F29, to determine consistent allele frequency changes between treatments70. If any population had minor or major allele count of 0, +1 was added to minor and major alleles from all samples. To correct for multiple testing, we converted P values to q values using the qvalues R package (v.2.14.1)104 and applied a FDR with a q < 0.05, leaving 24,189 consistently diverged SNPs. Of those SNPs that we classified as diverged at F29, we then performed GLMs at these positions on F1 samples to examine whether they began the experiment diverged.
Initial SNP frequencies
We then compared the initial frequency of alleles (that is, at F1) of diverged SNPs to the genomic average (autosomes only). From the F1 samples, we determined the average allele frequency of all populations (that is, treating all F1 samples as a panmictic population), we then randomly sampled 24,189 positions and recorded the median frequency of minor alleles; this was repeated 10,000 times to draw a distribution. From this distribution, we then determined CIs to examine whether the median frequency of minor alleles of diverged SNPs differed from the genomic average.
Position of diverged SNPs
Using bedtools105 (v.2.27.1) we determined which genes (exons) contained diverged SNPs. Next also using bedtools, the genome was split into 10 kb windows and we determined windows that contained at least one significantly diverged SNP. By then drawing 24,189 random positions from all possible SNPs and counting the number of autosomal windows at least one SNP was within, and repeating this 10,000 times, we were able to draw a distribution and determine CIs of the number of windows containing random SNPs. This was used to examine whether diverged SNPs were distributed randomly across the genome. Position of diverged SNPs were visualized by Manhattan plots using the R package qqman106.
Regions or genes containing diverged SNPs
The ratio of non-synonymous (PN) to synonymous (PS) segregating sites (PN/PS) was compared between exons that contained diverged SNPs against those that did not by Wilcoxon signed-rank test on the basis of the average across replicates of F1 samples. Additionally, to test a specific hypothesis that genes containing diverged SNPs that were fixed in F-lines are under stronger purifying selection than genomic average, PN/PS of exons containing them were compared to those that did not contain diverged SNPs fixed in F-lines. Due to the relatively low numbers in the former set of genes, we compared these two sets using a random sampling approach, in which we randomly sampled 78 times from the set of genes that did not contain diverged SNPs, and the median was calculated. This was repeated 10,000 times to draw a distribution and calculate 95% CIs for the median.
Furthermore, genetic diversity was compared between genes (exons) and 10 kb windows that contained diverged SNPs against those that did not. For this purpose, we calculated the mean π of F1 samples for both exonic diversity and that within 10 kb windows, and compared the two groups using Wilcoxon signed-rank tests due to non-normal distributions.
Finally, we explored the top 10% genes and 10 kb windows with highest values of D (that is, those with signatures of balancing selection) calculated as an average across F1 samples. We investigated whether the top 10% genes and windows were enriched for those containing diverged SNPs using chi-square test analyses. We similarly tested whether the top 10% windows were enriched for those containing SNPs fixed in S-lines, to test the specific hypothesis that these regions were initially under balancing selection due to sexual antagonism and the SNPs were subsequently lost in those lines when sexual antagonism was relaxed. Finally, to test whether the top 10% D set at F1 was more likely to maintain high values of D across the experimental evolution in F-lines compared to S-lines, we compared mean D values of this set of 10 kb windows between treatments at F29 using a simple t-test.
Identifying X-linked contigs
As with most other Acarid mites107, R. robini has a XO sex determination system, with males being the heterogametic sex (Supplementary Fig. 4). On the basis of predicted differences in read coverages of contigs between male and female samples we identified putative X-linked contigs using the Illumina reads from all experimental evolution populations. We calculated the mean coverage of each contig for individual male and female samples, with contig coverage standardized by the mean overall sample coverage. Autosomal contigs are expected to have a ratio between female and male samples of 1:1, whereas X-linked contigs are expected to have a ratio of 1:0.5. These expected differences in the latter are due to females having two copies of the X chromosome but males only having a single copy. A similar approach was used in Callosobruchus maculatus57 to putatively assign contigs as either autosomal or as a sex chromosome (X- and Y-linked). Inspection of Supplementary Fig. 5 shows that few contigs conform to the expectation of X-linked contigs, but that a number of contigs do have relatively low male coverage. It appears that on average male samples have slightly higher than expected coverage; this is probably a consequence of males only having a single sex chromosome and therefore an excess of autosomes and X-chromosome reads are found in male libraries compared to female libraries. With this in mind, we drew an arbitrary cut-off in the ratio of coverage between female and male contigs of 1:0.75, with contigs with a ratio above this being assigned as autosomal, and those with a ratio below this assigned as X-linked contigs (Supplementary Fig. 5).
Estimating N e, selection coefficients and simulating the impact of drift with different N e
Drift under different N e
An estimation of Ne for each population was determined by the changes in allele frequency of all autosomal SNPs between F1 and F29 using the R package poolSeq108 (v.0.3.5) and the estimateNe() function. As these results showed a difference in Ne between morph selection treatments, we performed simulations to determine whether this differences in Ne and consequently changes in drift could explain our patterns of diverged SNPs. Using poolSeq and the wf.traj() function we simulated two populations with Ne the same as our mean estimation of Ne of the F-lines (Ne = 370) and S-lines (Ne = 460), with generation of sampling (excluding F12), census size and sample size identical to those used in the experiment. To save computing time, we ran 20 identical simulations and combined output files. Each simulated population consisted of 1.5 million independent SNPs evolving in a Wright–Fisher population, the starting frequencies of minor alleles were randomly drawn between the range of 0.01 and 0.5. From these two simulated populations we sampled 50,000 times to determine whether allele frequencies consistently diverged. We approximated initial frequency of SNPs in the following way: by binning initial allele frequency from genetic F1 SNP data (<1, 1–5, 5–10% and so on in steps of 5%) we calculated a proportion of total SNPs within each bin. Using this proportion, we sampled from each simulated bin from both simulated populations in proportions matching those from genetic data (that is, 50,000 × proportion SNPs in bin). Each sample consisted of drawing four positions (without replacement) from each of the simulated populations from the same F1 bin, thus, adding a small amount of noise to the initial allele frequency of each sample. To add some further noise associated with differences in coverage, for each sample we drew a random number from a Poisson distribution resembling our actual target coverage (lambda, 125), by then drawing eight random numbers between −20 and +20 (approximate variation estimated from genetic data) and adding these to the random number from the Poisson distribution. Each of the simulated proportions of allele frequencies at F29 in the sample were then multiplied by one of these eight numbers at random, and rounded to the nearest integer, to gain counts of minor and major alleles and introduce some variation in coverage comparable to our molecular data. As with molecular data, we discarded simulated positions that did not meet our criteria of having an average proportion of minor alleles from all simulated populations >5% and if the coverage from any simulated position was <63× and >252×. From those samples that remained (>900,000), GLMs were performed (identical to above) on the simulated major and minor allele counts. Using a FDR with a q < 0.05 no simulated positions reached significance.
Selection coefficients
Next using the estimateSH() function in poolSeq we estimated selection coefficients (s) within each morph selection treatment on the set of diverged SNPs. Initially, we tried to allow dominance to be accounted for at each position using method = ’automatic’, but due to low number of time points at which sampling was performed this was not possible and all outputs reverted to using codominance. We therefore opted to use fixed codominance (h = 0.5), which also enabled us to calculate a P value by running 1,000 simulations for each position to estimate if s differed significantly from drift (that is, s = 0) within each morph selection treatment. Reliable estimates of s are not feasible if allele frequencies are too low, therefore, we were unable to determine s for every position in each morph selection treatments. To account for multiple testing, we applied a FDR with a q < 0.05.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. Raw Illumina reads have been deposited on NCBI and can be found in links on bioproject accession number PRJNA837268. Reference genome and raw ONT reads have been deposited on NCBI and can found in links on bioproject accession number PRJNA771500, the gff annotation file can be found at https://bioinformatics.psb.ugent.be/orcae/overview/Rhrob. Phenotypic data have been deposited on Dryad at https://doi.org/10.5061/dryad.ncjsxksxg.
Code availability
Instructional code for genomic analysis using PoPoolation and PoPoolation2 can be found on Dryad at https://doi.org/10.5061/dryad.ncjsxksxg.
References
Darwin, C. The Descent of Man and Selection in Relation to Sex (Murray, 1871).
Andersson, M. Sexual Selection (Princeton Univ. Press, 1994).
Shuker, D. M. & Kvarnemo, C. The definition of sexual selection. Behav. Ecol. 32, 781–794 (2021).
Martínez-Ruiz, C. & Knell, R. J. Sexual selection can both increase and decrease extinction probability: reconciling demographic and evolutionary factors. J. Anim. Ecol. 86, 117–127 (2016).
Kokko, H. & Brooks, R. Sexy to die for? Sexual selection and the risk of extinction. Ann. Zool. Fennici 40, 207–219 (2003).
van Doorn, G. S., Edelaar, P. & Weissing, F. J. On the origin of species by natural and sexual selection. Science 326, 1704–1707 (2009).
Ritchie, M. G. Sexual selection and speciation. Annu. Rev. Ecol. Evol. Syst. 38, 79–102 (2007).
Lorch, P. D., Proulx, S., Rowe, L. & Day, T. Condition dependent sexual selection can accelerate adaptation. Evol. Ecol. Res. 5, 867–881 (2003).
Rowe, L. & Rundle, H. D. The alignment of natural and sexual selection. Annu. Rev. Ecol. Evol. Syst. 52, 499–517 (2021).
Candolin, U. & Heuschele, J. Is sexual selection beneficial during adaptation to environmental change? Trends Ecol. Evol. 23, 446–452 (2008).
Holman, L. & Kokko, H. The consequences of polyandry for population viability, extinction risk and conservation. Philos. Trans. R. Soc. B. Biol. Sci. 368, 20120053 (2013).
Cally, J. G., Stuart-Fox, D. & Holman, L. Meta-analytic evidence that sexual selection improves population fitness. Nat. Commun. 10, 2017 (2019).
Tanaka, Y. Sexual selection enhances population extinction in a changing environment. J. Theor. Biol. 180, 197–206 (1996).
Winkler, L., Moiron, M., Morrow, E. H. & Janicke, T. Stronger net selection on males across animals. eLife 10, e68316 (2021).
Agrawal, A. F. Sexual selection and the maintenance of sexual reproduction. Nature 411, 692–695 (2001).
Siller, S. Sexual selection and the maintenance of sex. Nature 411, 689–692 (2001).
Whitlock, M. C. & Agrawal, A. F. Purging the genome with sexual selection: reducing mutation load through selection on males. Evolution 63, 569–582 (2009).
Grieshop, K., Maurizio, P. L., Arnqvist, G. & Berger, D. Selection in males purges the mutation load on female fitness. Evol. Lett. 5, 328–343 (2021).
Darwin, C. The Origin of Species (Oxford World’s Classics, 1859).
Rowe, L. & Houle, D. The lek paradox and the capture of genetic variance by condition dependent traits. Proc. R. Soc. B. Biol. Sci. 263, 1415–1421 (1996).
Tomkins, J. L., Radwan, J., Kotiaho, J. S. & Tregenza, T. Genic capture and resolving the lek paradox. Trends Ecol. Evol. 19, 323–328 (2004).
Andersson, M. Evolution of condition-dependent sex ornaments and mating preferences: sexual selection based on viability differences. Evolution 40, 804–816 (1986).
Prokuda, A. Y. & Roff, D. A. The quantitative genetics of sexually selected traits, preferred traits and preference: a review and analysis of the data. J. Evol. Biol. 27, 2283–2296 (2014).
Berglund, A., Bisazza, A. & Pilastro, A. Armaments and ornaments: an evolutionary explanation of traits of dual utility. Biol. J. Linn. Soc. 58, 385–399 (1996).
Tomkins, J. L. & Hazel, W. The status of the conditional evolutionarily stable strategy. Trends Ecol. Evol. 22, 522–528 (2007).
Gross, M. R. Alternative reproductive strategies and tactics: diversity within sexes. Trends Ecol. Evol. 11, 92–98 (1996).
Gross, M. R. & Repka, J. Stability with inheritance in the conditional strategy. J. Theor. Biol. 192, 445–453 (1998).
Taborsky, M., Oliveira, R. & Brockmann, H. in Alternative Reproductive Tactics: An Integrative Approach (eds Oliveira, R. et al.) 1–22 (Cambridge Univ. Press, 2008).
Jensen, J. D. On the unfounded enthusiasm for soft selective sweeps. Nat. Commun. 5, 527 (2014).
Connallon, T. & Clark, A. G. Balancing selection in species with separate sexes: insights from fisher’s geometric model. Genetics 197, 991–1006 (2014).
Johnston, S. E. et al. Life history trade-offs at a single locus maintain sexually selected genetic variation. Nature 502, 93–95 (2013).
Mérot, C., Llaurens, V., Normandeau, E., Bernatchez, L. & Wellenreuther, M. Balancing selection via life-history trade-offs maintains an inversion polymorphism in a seaweed fly. Nat. Commun. 11, 670 (2020).
Chippindale, A. K., Gibson, J. R. & Rice, W. R. Negative genetic correlation for adult fitness between sexes reveals ontogenetic conflict in Drosophila. Proc. Natl Acad. Sci. USA. 98, 1671–1675 (2001).
Bonduriansky, R. & Chenoweth, S. F. Intralocus sexual conflict. Trends Ecol. Evol. 24, 280–288 (2009).
Foerster, K. et al. Sexually antagonistic genetic variation for fitness in red deer. Nature 447, 1107–1110 (2007).
Cox, R. M. & Calsbeek, R. Sexually antagonistic selection, sexual dimorphism, and the resolution of intralocus sexual conflict. Am. Nat. 173, 176–187 (2009).
Pike, K. N., Tomkins, J. L. & Buzatto, B. A. Mixed evidence for the erosion of intertactical genetic correlations through intralocus tactical conflict. J. Evol. Biol. 30, 1195–1204 (2017).
Morris, M. R., Goedert, D., Abbott, J. K., Robinson, D. M. & Rios-Cardenas, O. in Advances in the Study of Behavior (eds Jane Brockmann, H. et al.) 45 (Elsevier Inc., 2013).
Plesnar-Bielak, A., Skrzynecka, A. M., Miler, K. & Radwan, J. Selection for alternative male reproductive tactics alters intralocus sexual conflict. Evolution 68, 2137–2144 (2014).
Harano, T., Okada, K., Nakayama, S., Miyatake, T. & Hosken, D. J. Intralocus sexual conflict unresolved by sex-limited trait expression. Curr. Biol. 20, 2036–2039 (2010).
Okada, K. et al. Natural selection increases female fitness by reversing the exaggeration of a male sexually selected trait. Nat. Commun. 12, 3420 (2021).
Radwan, J., Engqvist, L. & Reinhold, K. A paradox of genetic variance in epigamic traits: beyond ‘good genes’ view of sexual selection. Evol. Biol. 43, 267–275 (2016).
Zajitschek, F. & Connallon, T. Antagonistic pleiotropy in species with separate sexes, and the maintenance of genetic variation in life-history traits and fitness. Evolution. 72, 1306–1316 (2018).
Radwan, J. Effectiveness of sexual selection in removing mutations induced with ionizing radiation. Ecol. Lett. 7, 1149–1154 (2004).
Lumley, A. J. et al. Sexual selection protects against extinction. Nature 522, 470–473 (2015).
Almbro, M. & Simmons, L. W. Sexual selection can remove an experimentally induced mutation load. Evolution. 68, 295–300 (2014).
Dugand, R. J., Jason Kennington, W. & Tomkins, J. L. Evolutionary divergence in competitive mating success through female mating bias for good genes. Sci. Adv. 4, eaaq0369 (2018).
Hollis, B., Fierst, J. L. & Houle, D. Sexual selection accelerates the elimination of a deleterious mutant in Drosophila melanogaster. Evolution. 63, 324–333 (2009).
Dugand, R. J., Tomkins, J. L. & Kennington, W. J. Molecular evidence supports a genic capture resolution of the lek paradox. Nat. Commun. 10, 1359 (2019).
Parrett, J. M., Ghobert, V., Cullen, F. S. & Knell, R. J. Strong sexual selection fails to protect against inbreeding-driven extinction in a moth. Behav. Ecol. 32, 875–882 (2021).
Arbuthnott, D. & Rundle, H. D. Sexual selection is ineffectual or inhibits the purging of deleterious mutations in Drosophila melanogaster. Evolution. 66, 2127–2137 (2012).
Holland, B. & Rice, W. R. Experimental removal of sexual selection reverses intersexual antagonistic coevolution and removes a reproductive load. Proc. Natl Acad. Sci. USA. 96, 5083–5088 (1999).
Rundle, H. D., Chenoweth, S. F. & Blows, M. W. The roles of natural and sexual selection during adaptation to a novel environment. Evolution 60, 2218–2225 (2006).
Chenoweth, S. F., Appleton, N. C., Allen, S. L. & Rundle, H. D. Genomic evidence that sexual selection impedes adaptation to a novel environment. Curr. Biol. 25, 1860–1866 (2015).
Holland, B. Sexual selection fails to promote adaptation to a new environment. Evolution. 56, 721–730 (2002).
Berger, D. et al. Intralocus sexual conflict and the tragedy of the commons in seed beetles. Am. Nat. 188, E98–E112 (2016).
Sayadi, A. et al. The genomic footprint of sexual conflict. Nat. Ecol. Evol. 3, 1725–1730 (2019).
Ruzicka, F. et al. Genome-wide sexually antagonistic variants reveal long-standing constraints on sexual dimorphism in fruit flies. PLoS Biol. 17, e3000244 (2019).
Radwan, J., Czyz, M., Konior, M. & Kołodziejczyk, M. Aggressiveness in two male morphs of the bulb mite Rhizoglyphus robini. Ethology 106, 53–62 (2000).
Schlötterer, C., Tobler, R., Kofler, R. & Nolte, V. Sequencing pools of individuals-mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 15, 749–763 (2014).
Ellegren, H. The different levels of genetic diversity in sex chromosomes and autosomes. Trends Genet. 25, 278–284 (2009).
Charlesworth, B., Coyne, J. A. & Barton, N. H. The relative rates of evolution of sex chromosomes and autosomes. Am. Nat. 130, 113–146 (1987).
Wiberg, R. A. W., Veltsos, P., Snook, R. R. & Ritchie, M. G. Experimental evolution supports signatures of sexual selection in genomic divergence. Evol. Lett. 5, 214–229 (2021).
Wright, S. Evolution in mendelian populations. Genetics 16, 97–159 (1931).
Smallegange, I. M. Complex environmental effects on the expression of alternative reproductive phenotypes in the bulb mite. Evol. Ecol. 25, 857–873 (2011).
Radwan, J. Male morph determination in two species of acarid mites. Heredity. 74, 669–673 (1995).
Łukasiewicz, A., Niśkiewicz, M. & Radwan, J. Sexually selected male weapon is associated with lower inbreeding load but higher sex load in the bulb mite. Evolution. 74, 1851–1855 (2020).
Charlesworth, D. & Willis, J. H. The genetics of inbreeding depression. Nat. Rev. Genet. 10, 783–796 (2009).
Radwan, J. & Klimas, M. Male dimorphism in the bulb mite, Rhizoglyphus robini: fighters survive better. Ethol. Ecol. Evol. 13, 69–79 (2001).
Wiberg, R. A. W., Gaggiotti, O. E., Morrissey, M. B. & Ritchie, M. G. Identifying consistent allele frequency differences in studies of stratified populations. Methods Ecol. Evol. 8, 1899–1909 (2017).
Llaurens, V., Whibley, A. & Joron, M. Genetic architecture and balancing selection: the life and death of differentiated variants. Mol. Ecol. 26, 2430–2448 (2017).
Joag, R. et al. Transcriptomics of intralocus sexual conflict: Gene expression patterns in females change in response to selection on a male secondary sexual trait in the bulb mite. Genome Biol. Evol. 8, 2351–2357 (2016).
Connallon, T. & Clark, A. G. A general population genetic framework for antagonistic selection that accounts for demography and recurrent mutation. Genetics 190, 1477–1489 (2012).
Küpper, C. et al. A supergene determines highly divergent male reproductive morphs in the ruff. Nat. Genet. 48, 79–83 (2015).
Hendrickx, F. et al. A masculinizing supergene underlies an exaggerated male reproductive morph in a spider. Nat. Ecol. Evol. 6, 195–206 (2022).
Kirkpatrick, M. & Ryan, M. J. The evolution of mating preferences and the paradox of the lek. Nature 350, 33–38 (1991).
Houle, D. How should we explain variation in the genetic variance of traits? Genetica 102–103, 241–253 (1998).
Parrett, J. M. & Knell, R. J. The effect of sexual selection on adaptation and extinction under increasing temperatures. Proc. R. Soc. B. Biol. Sci. 285, 20180303 (2018).
Parrett, J. M., Mann, D. J., Chung, A. Y. C., Slade, E. M. & Knell, R. J. Sexual selection predicts the persistence of populations within altered environments. Ecol. Lett. 22, 1629–1637 (2019).
Plesnar-Bielak, A., Skrzynecka, A. M., Prokop, Z. M. & Radwan, J. Mating system affects population performance and extinction risk under environmental challenge. Proc. R. Soc. B. Biol. Sci. 279, 4661–4667 (2012).
Jarzebowska, M. & Radwan, J. Sexual selection counteracts extinction of small populations of the bulb mites. Evolution. 64, 1283–1289 (2010).
Godwin, J. L., Lumley, A. J., Michalczyk, Ł., Martin, O. Y. & Gage, M. J. G. Mating patterns influence vulnerability to the extinction vortex. Glob. Chang. Biol. 26, 4226–4239 (2020).
Yun, L. et al. Competition for mates and the improvement of nonsexual fitness. Proc. Natl Acad. Sci. USA 115, 6762–6767 (2018).
Martins, M. J. F., Puckett, T. M., Lockwood, R., Swaddle, J. P. & Hunt, G. High male sexual investment as a driver of extinction in fossil ostracods. Nature 556, 366–369 (2018).
Doherty, P. F. et al. Sexual selection affects local extinction and turnover in bird communities. Proc. Natl Acad. Sci. USA 100, 5858–5862 (2003).
Sorci, G., Møller, A. P. & Clobert, J. Plumage dichromatism of birds predicts introduction success in New Zealand. J. Anim. Ecol. 67, 263–269 (1998).
Grieshop, K., Berger, D. & Arnqvist, G. Male-benefit sexually antagonistic genotypes show elevated vulnerability to inbreeding. BMC Evol. Biol. 17, 134 (2017).
Radwan, J. & Siva-Jothy, M. T. The function of post-insemination mate association in the bulb mite, Rhizoglyphus robini. Anim. Behav. 52, 651–657 (1996).
Radwan, J. Sperm precedence in the bulb mite, Rhiziglyphus robini: context-dependent variation. Ethol. Ecol. Evol. 9, 373–383 (1997).
Radwan, J. & Bogacz, I. Comparison of life-history traits of the two male morphs of the bulb mite, Rhizoglyphus robini. Exp. Appl. Acarol. 24, 115–121 (2000).
Roff, D. A. Evolutionary Quantitative Genetics (Chapman and Hall, 1997).
Knell, R. J. On the analysis of non-linear allometries. Ecol. Entomol. 34, 1–11 (2009).
Tilszer, M., Antoszczyk, K., Sałek, N., Zajac, E. & Radwan, J. Evolution under relaxed sexual conflict in the bulb mite Rhizoglyphus robini. Evolution 60, 1868–1873 (2006).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2020); https://www.R-project.org/
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (2016).
Bates, D., Mächler, M., Bolker, B. M. & Walker, S. C. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48 (2015).
Therneau, T. M. coxme: mixed effects Cox models (2020); https://CRAN.R-project.org/package=coxme
Sterck, L., Billiau, K., Abeel, T., Rouzé, P. & Van De Peer, Y. ORCAE: online resource for community annotation of eukaryotes. Nat. Methods 9, 1041 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at arXiv https://doi.org/10.48550/arXiv.1303.3997 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Kofler, R. et al. Popoolation: a toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS ONE 6, e15925 (2011).
Kofler, R., Pandey, R. V. & Schlötterer, C. PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 27, 3435–3436 (2011).
Storey, J. D. A direct approach to false discovery rates. J. R. Stat. Soc. Ser.B. Stat. Methodol. 64, 479–498 (2002).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
D. Turner, S. qqman: an R package for visualizing GWAS results using Q-Q and Manhattan plots. J. Open Source Softw. 3, 731 (2018).
Oliver, J. H. Cytogenetics of mites and ticks. Annu. Rev. Entomol. 22, 407–429 (1977).
Taus, T., Futschik, A. & Schlötterer, C. Quantifying selection with pool-seq time series data. Mol. Biol. Evol. 34, 3023–3034 (2017).
Smallegange, I. M. & Coulson, T. The stochastic demography of two coexisting male morphs. Ecology 92, 755–764 (2011).
Plesnar-Bielak, A., Skwierzyńska, A. M., Hlebowicz, K. & Radwan, J. Relative costs and benefits of alternative reproductive phenotypes at different temperatures—genotype-by-environment interactions in a sexually selected trait. BMC Evol. Biol. 18, 109 (2018).
Bleay, C., Comendant, T. & Sinervo, B. An experimental test of frequency-dependent selection on male mating strategy in the field. Proc. R. Soc. B Biol. Sci. 274, 2019–2025 (2007).
Skrzynecka, A. M. & Radwan, J. Experimental evolution reveals balancing selection underlying coexistence of alternative male reproductive phenotypes. Evolution 70, 2611–2615 (2016).
Acknowledgements
We thank A. Plesnar-Bielak for sharing gene expression data; K. Dudek who performed library preparation; N. Jeffery who carried out flow-cytometry; A. Maryańska-Nadachowska who performed karyotyping; M. Łukasiewicz, S. Jedut and I. Bolitho for laboratory assistance; K. Burda for help with schematics; M. Nowicki and J. Dabert for assistance and use of equipment used in scanning electron microscopy and mite measurement photography, respectively; members of the National Genomics Infrastructure in performing sequencing; M. Osowiecki for technical IT support; and F. De Temmerman, M. Langhendries, B. Rombaut and M. De Nolf for their analyses of the reference genome annotation. Finally, we thank R. Knell, A. Plesnar-Bielak and J. Tomkins for useful comments and discussion on an earlier version of this manuscript. This work was supported by National Science Centre grant no. 2017/27/B/NZ8/00077 awarded to J.R.
Author information
Authors and Affiliations
Contributions
J.R. and W.B. conceived the study. J.M.P. and S.C. performed the experiment, with assistance from A.Ł. and A.S.-K. A.Ł. and A.S.-K. prepared mites for the reference genome. E.A. and S.R. performed ONT sequencing and de novo assembly, gene prediction and analyses of gene data, with additional Illumina sequencing provided by M.K. for further improvement of the reference genome. A.S.-K. performed photography. J.M.P. performed the analysis. M.K., W.B. and J.R. provided input. J.M.P. and J.R. wrote the initial manuscript with input from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Ecology & Evolution thanks Locke Rowe and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Schematic representation of experimental evolution protocol.
Three populations were mixed to establish the stock population, which after ca. 12 generations of mixing were divided into replicates selected either for fighter males (F-lines: green; n = 4) and scrambler males (S-lines: orange; n = 4) according the protocol shown. This protocol was the repeated each generation of the experimental evolution.
Extended Data Fig. 2 Allometric relationship between idiosoma length and width of the third pair of legs of R. robini fighter and scrambler males.
Frequency distributions of a) idiosoma length, b) width of the third proximal segment of the right third leg (genu) and c) relative leg width (leg width / idiosoma length). In each the line represents the kernel density and in c) the vertical dashed line (at 0.125) where males with a relative leg width greater or less than 0.125 are classified as fighters and scramblers, respectively. d) shows a log-log scatterplot of idiosoma length against leg width, were the classification of males as fighters or scramblers (based upon c) are shown in green and orange, respectively. Lines of best fit are based on simple linear models and shaded area denote 95% confidence intervals.
Extended Data Fig. 3 Tracking male morph proportion.
Proportion of fighters in fighter (F: green) and scrambler (S: orange) morph selection lines every 6–7 generations of experimental evolution, the stock population started at approximately 50:50 morph ratio. Large points and lines indicate average across each treatment and small points and lines individual replicate populations. The purple point indicates the starting morph proportion of the stock population one generation prior to starting the experiment (F0). Points and error bars indicate means and 95% confidence intervals.
Extended Data Fig. 4 Analysis of the nature of autosomal genetic diversity between morph selection lines within 10 kb windows.
Mean value from lines either selected for fighter (F: green; n = 4) or scrambler males (S: orange; n = 4) shown by large points with error bars (1 s.d.) in each of the three generations in which pool-seq sampling was performed (F1, F12, F29). Each smaller point represents the mean value from an individual experimental evolution population. Estimates are based upon 10 kb windows across autosomal contigs (n = 7067). a) the estimates of nucleotide diversity (π) and b) the number of segregating sites (ϴ). In both cases there was a significant interaction between morph treatment and generation (p < 0.05) by repeated measures ANOVA analysis demonstrating substantially greater reduction in both measures of genetic diversity in F-lines compared to S-lines over the duration of the experiment (see Supplementary Table 3 for test statistics and post-hoc comparisons).
Extended Data Fig. 5 Survival of males and females from F-lines and S-lines.
Proportion of dead males and females within small colonies derived from each experimental evolution line (F45) after 6 days being housed at a 50:50 sex ratio with roughly the same density used during experimental procedures. Mean values are shown by large points with error bars (1 s.d.) and each small point shows values from each individual population. More fighter (F: green) males died compared to scrambler (S: orange) males, whereas, female mortality remained similar between morph selection lines as indicated by a significant two-way interaction between morph selection line and sex (F = 7.06, df = 1, p = 0.021).
Extended Data Fig. 6 Heatmaps of diverged and simulated SNPs.
Representing counts of SNPs and the frequency of minor alleles (MAF) at F1 and F29 of a) 24,189 diverged SNPs at F29 (that is the same data from Fig. 6a&c) in F-lines (left panel) and S-lines (right panel) and b) simulated SNPs used to examine divergence as a consequence of differences in Ne (left panel: Ne = 370; right panel: Ne = 460), displaying SNPs with p-values < 0.025 from GLMs (totaling 41,116), however, none of which reached significance after applying a false discovery rate (FDR) with q-value < 0.05. MAFs were placed into bins at F1 and F29, with the total number of SNPs in each category indicated by the darkness of colour. Note that the scale differs between figures a) and b).
Extended Data Fig. 7 Comparison of genes that contain diverged SNPs (blue) against background genes (red), estimates taken from average values of F1 samples.
a) density plot where genes to the right of the vertical dashed line represent the top 10% of genes (n = 13,389 total genes) with initially high estimates of Tajima’s D (D). b) nucleotide diversity (π) in genes containing diverged SNPs and background genes. c) and d) show values of PN/PS (non-synonymous SNP density / synonymous SNP density), comparing genes containing c) diverged SNPs and d) diverged SNPs that were fixed in F-lines. In b-d boxes are composed of the median and hinge values (25th and 75th percentiles), with whiskers ± interquartile range * 1.5, points outside this range are denoted by individual points.
Extended Data Fig. 8 Comparison of 10 kb windows that contain diverged SNPs against background windows that do not contain diverged SNPs at F29.
a) density plot comparing distribution of Tajima’s D (D) in windows (n = 7067) containing diverged SNPs (blue) and background windows (red); b) As above, but only including a set of diverged SNPs that were fixed in S-lines (blue). In both a) and b) windows to the right of the vertical dashed line represent the top 10% windows with highest D (average of F1 samples). c) nucleotide diversity (π) in 10 kb windows that contain diverged SNPs (blue) and background windows (red), boxes are composed of the median and hinge values (25th and 75th percentiles), with whiskers ± interquartile range * 1.5, points outside this range are denoted by individual points. d) D from 10 kb windows with initially high D (top 10%, average of F1 samples), showing mean value (large points) from lines either selected for fighter (F-line: green) or scrambler males (S-line: orange); error bars represent 1 s.d., with each smaller point representing the mean value from an individual experimental evolution population.
Extended Data Fig. 9 Manhattan plots of SNPs across the 10 longest autosomal contigs.
Left to right from top to bottom: sq0001, sq0002, sq0003, sq0004, sq0005, sq0006, sq0007,sq0009, sq0010, sq0011. X-axis indicates position along the contig and y-axis –log10(p-value), points above the horizontal line indicate diverged SNPs after applying a false discovery rate (FDR) with q-value <0.05. Green and orange points highlight SNPs fixed or nearly fixed (frequency of minor alleles <1%) in fighter and scrambler lines, respectively.
Extended Data Fig. 10 Manhattan plots of seven contigs with a high density of diverged and fixed SNPs.
Left to right from top to bottom: sq0064, sq0091, sq0092, sq0114, sq0124, sq0128, sq0254. X-axis indicates position along the contig and y-axis –log10(p-value), points above the horizontal line indicate diverged SNPs after applying a false discovery rate (FDR) with q-value <0.05. Green and orange points highlight SNPs fixed or nearly fixed (frequency of minor alleles <1%) in fighter and scrambler lines, respectively.
Supplementary information
Supplementary Information
Supplementary Tables 1–7 and Figs. 1–6.
Supplementary Table
List of 13,389 autosomal genes.
Source data
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Rights and permissions
About this article

Cite this article
Parrett, J.M., Chmielewski, S., Aydogdu, E. et al. Genomic evidence that a sexually selected trait captures genome-wide variation and facilitates the purging of genetic load. Nat Ecol Evol 6, 1330–1342 (2022). https://doi.org/10.1038/s41559-022-01816-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41559-022-01816-w
This article is cited by
-
6Pgdh polymorphism in wild bulb mite populations: prevalence, environmental correlates and life history trade-offs
Experimental and Applied Acarology (2024)
-
The roles of sexual selection and sexual conflict in shaping patterns of genome and transcriptome variation
Nature Ecology & Evolution (2023)
-
Small-scale genetic structure of populations of the bulb mite Rhizoglyphus robini
Experimental and Applied Acarology (2023)
