
Abstract
Large amounts of data are generated in chemistry labs—nearly all instruments record data in a digital form, yet a considerable proportion is also captured non-digitally and reported in ways non-accessible to both humans and their computational agents. Chemical research is still largely centred around paper-based lab notebooks, and the publication of data is often more an afterthought than an integral part of the process. Here we argue that a modular open-science platform for chemistry would be beneficial not only for data-mining studies but also, well beyond that, for the entire chemistry community. Much progress has been made over the past few years in developing technologies such as electronic lab notebooks that aim to address data-management concerns. This will help make chemical data reusable, however it is only one step. We highlight the importance of centring open-science initiatives around open, machine-actionable data and emphasize that most of the required technologies already exist—we only need to connect, polish and embrace them.

Similar content being viewed by others


Main
In the era when scientific results were published only on real paper, the compression of information was of paramount importance. As a consequence of limited page counts, most scientific data were not published. Now, we live in a digital era and a large fraction of our data is captured in digital form. Yet, most scientific data that are collected are still not published1, and the part that is often in a form that makes it difficult for other researchers to build on.
Scientists have also long been concerned about the reproducibility of results2,3. This has lead most funding agencies to insist on a commitment by researchers as to how scientific data are managed (for instance in the form of a data management plan, that is, a clear outline of the types of data generated and used during a study, where and by whom they can be accessed, how and by whom they are protected and how and by whom they can be shared or published) and often to require all data to be made publicly available. Having a data management plan is important but, as we argue here, it does not guarantee that data will be shared in an easily findable, accessible, interoperable and reusable (FAIR) and ultimately machine-actionable, form4.
Additionally, recent advances in machine learning illustrate very clearly why chemistry would benefit from embracing open and reusable data. In chemistry, we have many problems of irreducible complexity5, such as the prediction of synthesizability, where complexity arises from the interaction of many diverse components (such as kinetics of side reactions or impurities) that are often not fully understood. Owing to these unknowns and complex interactions, some problems seem impossible to address with the current theory. Here, data-intensive research might be key. For example, many chemists would welcome a tool that recommends reaction conditions. One can envision building such a recommender system that harvests knowledge from all reactions that have been performed (including the ‘failed’ ones) to recommend conditions for the desired reaction. Building this tool, however, will only be possible if all the data are automatically collected in an interoperable and reusable form, such that machines can read datasets then rather autonomously discover the ones that are most relevant and in turn make decisions. This requires machines to not only parse the data but also understand the data and its context — that is, data must be machine actionable.
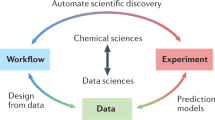
Our key thesis is that, if we want to advance chemistry with data-intensive research and also address reproducibility issues we need to change how experimental data are collected and reported. Structured data alone is not enough; open data alone is also not enough. We need to have both (thesis 1 in Fig. 1), together with additional tools such as semantic web technologies, that allow chemists and their computational agents to understand the meaning and intent of the data objects.

Machine learning has fundamentally changed the way that data can be used in chemistry, which in turn requires a change in how it is reported. In addition, raw data are also needed to verify any conclusions presented in scientific publications—as stated with the ‘Nullius in verba’ principle (take nobody’s word for it)—as the results presented in a paper are always a compression of the original research record6. Only a few groups creating and sharing FAIR data is not sufficient, it needs to be embraced by all chemists. Importantly, this can only happen if there are little or no overheads in publishing all the data in a FAIR, machine-actionable form. For this reason, the most crucial functionality an ELN can provide is to assist chemists in doing so; it is essential to avoid that chemical data become an afterthought in the publication process. Following this logic, developers of ELNs need to work together towards this goal of machine-actionable open science. We can only expect this to be widely adopted if ELNs implement a common standard for data representation and exchange, also with computational tools69, and allow the integration of reusable plugins that can be used to create a custom data management infrastructure that is interoperable with other solutions. Clearly, there will not be one perfect solution that works for all subfields of chemistry. However, we can start by reusing the many existing parts, making them interoperable and ensuring the code is open source, and in this way create a practical solution that works today. This seems more effective than to aim for large-scale, all-encompassing and overcomplicated solutions. Importantly, the development of new data formats (alone) will also not lead us towards the goal of FAIR chemical data.
To make this feasible, we envision a platform that seamlessly integrates the process of data collection, data processing and data publication with minimal overheads for the researcher:
-
(1)
Data collection. A key component of chemistry research is the collection of chemical data (for example, reaction conditions and characterization data). Ideally, the raw6,7,8 (characterization) data are directly captured from the instrument, directly converted into a standard structured form4, in which all the important metadata are systematically added and all the field names, such as ‘adsorption’ or ‘pressure’, are linked to an open vocabulary or ontology (which defines the meaning of the terms and their relation). One should not rely on individual chemists to manually perform such file transfer, annotation or conversion operations. This is not only time consuming and error prone, but also, more importantly, ensuring that all the data are in a form ready for FAIR sharing should not be an afterthought, it should the very first step.
-
(2)
Data processing and collaboration. Once we have converted our data into a standard form, we can apply the same analysis tools to all data types—which makes developments dramatically more efficient. Research groups that use different instruments could compare the data directly, and use the same analysis tools. Also, as soon as all the data are stored in a structured form, an electronic lab notebook (ELN) can make it searchable. For example, if an instrument was incorrectly calibrated, the ELN could allow the users to search for all the spectra that were measured with a specific instrument configuration at a specific range of time (or even automatically apply the correct calibration).
-
(3)
Data publishing. Data that remain locked in an ELN are not useful for the community. As soon as the researcher(s) are ready to publish a project, they could choose the relevant samples from the ELN and export them to a repository from where it can be used by machines, but also reimported by other ELNs.
From this viewpoint, the ELN is the central hub for all chemical research, from which analyses can be requested, analysed, shared, published and integrated with other platforms—and, also, a place to take notes. However, we emphasize that the most important functionality an ELN can provide is to automatically convert the data into an open, standardized and interoperable form (thesis 2 in Fig. 1). Only in this way can we leverage web technologies that allow computational tools to autonomously understand data and hence provide more meaningful (search) results (Box 1). Note that this is quite different from the functionality most current ELNs offer. The majority of current ELNs only store data digitally as an attachment—they do not convert it into such a reusable form (thesis 2 in Fig. 1).
Over time, an ‘insane’9 number of different ELNs and laboratory infrastructure management systems (LIMS) have been developed. Many of these different ELNs have been compared in previous works (for example, by the Harvard Medical School, the Library of the University of Cambridge, LIMSWiki or peer-reviewed articles10,11,12,13). In this Perspective, we aim to focus on the ideas and design principles that we think are essential to create a successful open-science infrastructure—for the full lifetime of data from inception, creation and processing to publication. As the infrastructure we propose to embrace is already implemented in parts, we review examples (from Table 1) that we think offer some key aspects of such an infrastructure to support open science. In a similar vein, we highlight examples in which chemical data have already been shared in a reusable form. Taking into account the many attempts to generate new data schema—describing the abstract structure of the data—and file formats for chemical data, we propose that a more efficient route to open science would be for the chemistry community to embrace and connect existing systems instead (thesis 4 in Fig. 1).

Data capture, data processing and data publication
To be practical, the data capture step needs to be both as close as possible to the way chemists work and it should ensure that the chemical data generated can be practically reused by other researchers. We give examples for what ‘machine-actionable data’ means in Box 1.
In chemistry, most samples in the lab are produced with a chemical reaction. Trying to predict the conditions at which a reaction can take place optimally is still one of the major challenges in chemistry. Machine-learning methods are expected to help us in this area14. However, for this to work we need to report data in a format that can be used in machine learning, and also report ‘failed’ experiments15,16. One can easily see the dilemma here; if an experiment—after 99 ‘failed’ attempts—finally works, there is little motivation, if any, for a researcher to spend 1% of their time in reporting the one successful experiment and the remaining 99% of the time on the ‘failed’ ones.
Capturing synthetic data
In chemistry, the number of possible steps and combinations of steps is nearly infinite. For example, the order in which the reagents are added can clearly decide whether a reaction will be successful or not17,18—and any machine-learning efforts will fail if such information is not reported correctly. This is exactly what is missing in many of the existing databases. For example, by mining the patent literature19 one can obtain a wealth of information on which chemicals can be synthesized20. However, the actual procedure of the syntheses cannot be mined systematically: the order of addition, the heating, the stirring and, of course, the workup and purification. And the situation is even more dire for inorganic chemistry21. Similarly, all the databases contain no information about the attempts that did not work and are biased towards certain reaction types22,23,24. This lack of reports on ‘failed’ reactions adds to other factors that lead to certain types of reactions being more prominent than others—for example, looking into the most used reactions in medicinal chemistry, Brown and Boström found that amide formation was mentioned at least once in about half of the selected set of manuscripts published in the Journal of Medicinal Chemistry in 2014 (ref. 25).
Ideally, to capture synthesis information we need to find a balance between the flexibility of a sheet of paper, on which chemists can record anything they want in any format they like26, and imposing a structure such that the captured data can be easily reused for machine-learning applications. The flexibility is key to ensure chemists will widely adopt the tool10,27, whereas from a data-management perspective a highly structured database (for example, filled via a long form) would be much easier to use. In high-throughput experimentation settings the latter might clearly be a natural approach, but for many manually created, small datasets1, this might not be a feasible approach, as to capture all the possible scenarios would result in such a gigantic form that chemists would need special training to navigate it.
Among the different ELNs no consensus has been reached about this design point. Some allow complete flexibility and have the look and feel of a typical note-taking app, whereby one needs natural-language processing to make the information machine-readable which, unavoidably, leads to information loss. At the other end of the spectrum are those that have a lot of structure with the design of a new form for every eventuality, which might be ideal for machine learning but poses a burden to use for non-routine chemistry.
A possible solution to these challenges, which is implemented in the chemotion and the cheminfo ELNs (Table 1), is to stick to the text-based form chemists are used to, but to combine it with templates to structure the text. This hybrid approach is described in Box 2. In practice, we found that some free text fields are always required to give chemists the necessary flexibility to express their motivation, thought process and interpretation). Parts of this can be captured via specific fields, for instance, the related literature, or spectral annotations. For many other parts, the free, potentially unstructured, thought process is exactly what one would like to capture (for example, to annotate when an experiment failed for an unexpected reason, such as a beam drop at the synchrotron).

Characterization data formats and metadata
After a sample has been synthesized, it needs to be characterized. Thereby, we want to ensure that researchers all over the world, as well as their computational agents, can use the data. Clearly, data models, which describe how data are stored in a data format, and metadata, which describe datasets, are not the typical focus of a chemist. However, a lot of chemical data is currently stored in a wide variety of proprietary files (Supplementary Table 2). In the short term, this might not look like a real problem, but in the long term, this is not sustainable. For example, one can lose access to all the files once the software license associated with particular equipment expires; or collaborators in another institute that want to use the data do not have access to the same software. Also, a hodgepodge of inconsistent formats clearly hampers data mining efforts.
Requiring all individual researchers to manually convert all their spectra into a standard format will be a large, potentially insurmountable and non-scalable burden on the researchers. Therefore, an essential step in progressing towards such an open platform is to convert the data into a standardized structured form before it even enters the ELN (thesis 2 in Fig. 1). This is an essential service an ELN must provide to a user. That is, the ELN will take the data as they are provided by the spectrometer, and convert it into a standardized form. The cheminfo implementation, for example, uses JCAMP-DX files (Joint Committee on Atomic and Molecular Physical Data Exchange format; see Extended Data Fig. 1 for an example) as a standard representation for most spectra. This format has been recommended by IUPAC (International Union of Pure and Applied Chemistry) for many spectra together with recommended vocabulary28, and is also recommended by the chemotion ELN, and used in the Open Spectral Database29. However, in principle, any other format (Supplementary Table 4) can be used as long as it is standardized and openly documented. Indeed, some newer formats have a native support for advanced features, such as linking to standardized vocabularies, and might be preferable (see Extended Data Fig. 2 for an example). For example, there were efforts (spearheaded by the pharmaceutical industry) to develop a ‘unified data model’ for compound synthesis and testing, or the ‘Allotrope data format’, which tries to collect the full data life cycle in one file. Some, like the autoprotocol or XDL30, even try to capture the link between hardware (such as reaction vessels) and synthesis steps in a way that can be understood (and executed) by both robots and humans.
One can argue that some existing formats and data schema are old-fashioned and that we should develop new ones. However, anyone proposing a new format should realize that if a characterization method has N formats provided by the instrument manufacturers and M ‘standard’ formats are invented, we need to write and maintain N × M conversion programs and M2 programs to be able to compare the different ‘standard’ formats. This indicates that it can be more productive to update existing solutions and make them interoperable compared with creating new ones (thesis 5 in Fig. 1).
It is important to note that data become much more useful, and interoperable, if they are linked and described using a controlled, hierarchical vocabulary, that is, an ontology. Using a formal ontology allows us to infer information from the context encoded in the vocabulary. For example, we might have Raman and infrared spectra, as well as the cities of the measurement stored in our database. The ontology will not only remove ambiguities in spelling of the cities, but it will also tell us which cities to include if we search for, say, all organic samples with vibrational spectra measured in a particular country. At the technical level, this is enabled by the fact that the ontology will encode that both infrared and Raman spectroscopy are forms of vibrational spectroscopy and that cities are located in countries. That is, it allows us to go from machine-readable to machine interpretable on a global scale (global because the terms are standardized and shared via uniform resource identifiers (URIs)). In practice, however, ontologies (and related semantic web technologies) remain underused. The main reasons are probably that the diversity of the ontologies is too large and that existing ones are not well integrated31. Clearly, we cannot expect chemists to manually annotate their data using an ontology. This is something an ELN needs to do automatically in the background. However, for this to be practical, ELN developers need to connect with other initiatives to register, standardize, link32 and adopt ontologies.
Let us now assume the ideal situation that most chemists have settled on a standard data reporting form (for the most important characterization techniques in a subdomain, such as gas adsorption isotherms, X-ray adsorption spectroscopy and cyclic voltammetry), and also accept that open science should not be an afterthought. This implies that the ELN must take the file in whatever form it comes from the instrument, convert into a standard form and permanently connect it to the chemical that was characterized (Fig. 2). Such conversion tools (see Supplementary Table 2 for examples) can be developed independently of each other and reused in all ELNs. For instance, the chemotion ELN reuses some of the libraries that we have been developing for the cheminfo ELN (cheminfo.github.io). Having such common conversion tools would also create the incentive to adopt a common schema.

If an instrument is coupled to the network one can, through scanning the barcode on the sample, upload the analysis result directly into a database. Alternatively, one can upload files via drag and drop through a web interface (front end). In both cases, the ELN ensures that the data are converted into a standard form such that anyone with a web browser can visualize and further analyse it. Other parties can access the data, for example, using an access token mechanism70, via a representational state transfer (REST) application programming interface (API) or published on a repository. Importantly, all the steps can take place from a different location, and hence enable collaboration. This data infrastructure is implemented in the open-source cheminfo ELN. Folder icon reproduced from image designed using resources from Flaticon.com; laptop photo by Scott Graham on Unsplash.
Provenance of data
One crucial step in this process is to match the spectrum with the correct sample. A URI system (can be printed as barcodes) can help to avoid mistakes in this step. For instance, in the cheminfo ELN, scanning the barcode will create the upload information for automatic importation from the computers to which the spectrometers are connected. From there, the system can take the file from the computer, convert it into the standard form and store it as an attachment to a sample that has been created in the ELN (for example, as the product of some reaction). This automatic importation not only makes it much easier, and less error-prone, for the chemist to store the data in the ELN, but also it allows us to automatically record a lot of metadata—for example, the importation workflow can fill in information about the instrument (such as the manufacturer, serial number, humidity and temperature of the room) that is not always recorded in the output files of the measurements (see Extended Figs. 1 and 2 for examples).
Data processing
After data have been produced and imported into the ELN, they usually need to be further analysed. At present, chemists have to switch between different, often proprietary, software to carry out this analysis. They might rely on the software provided by the instrument manufacturer to perform peak picking or baseline correction, and then use another plotting tool to overlay the data. In an open-science vision, one would like to ensure that one can not only access data, but, equally important, can also reproduce the subsequent analyses. Likewise, if the chemistry community embraced the view that the ELN converted data into a commonly agreed standard form, the analysis tools become independent of a particular instrument or even characterization technique (Box 3).
If we design the platform with a common interface, ensure a modular architecture and ensure a reusability of the key components, we have the first step towards an ecosystem in which libraries are developed for specific tools that accelerate the workflow of chemists (thesis 4 in Fig. 1 and Box 3). The modular nature would allow that experts in one technique, for example, NMR spectroscopy develop tools that can then be reused by other ELNs. An example for this is the NMRium project33, which is a reusable web component that can, with three lines of code, be plugged into another ELN system. To make this work, it is important that the components can talk with each other via standardized protocols.
In an open-science vision, the code for these components should be open. One of the concerns regarding open-source software is the danger that a project might ‘die out’ if one maintainer leaves the project, whereas a successful commercial software might seem to have the promise of continuity. However, there are many successful examples (such as Linux and Python) of open-source projects are maintained by the community, yet leave many options for commercial initiatives (for instance, support contracts and maintenance of a custom installation). Similarly, at universities a common analytical infrastructure (such as the routine NMR service) is often supported using institutional funding—a similar model might also be appropriate for a digital infrastructure. Importantly, open-source code has the advantage that the underlying assumptions and equations for any analysis are documented and everyone can verify, replicate or even improve the analysis. Also, in contrast to closed-source (commercial) tools that are discontinued because of a change in business interests, the development can be reanimated at any time, as the code is openly accessible and reusable.

Publication of reusable and machine-actionable data
The work of a scientist is not completed when all the materials are synthesized and characterized. An essential part of the scientific process is the dissemination of the results to make sure that others can build on top of one’s work. Typically, we are used to thinking of ‘others’ as other scientists in the same field. However, science is increasingly multidisciplinary, and hence non-specialists might also need to understand the data. Additionally, the move towards open science is a logical consequence of the notion that if the taxpayer paid for the research, the ownership of the research data should be the public at large, which can empower citizen (data) science34,35. We have a glimpse of the power of the reuse of data with the discoveries of Don Swanson, an information scientist without formal training in medicine who analysed literature from the Medline database and found previously undiscovered knowledge, such as links between magnesium deficiency and migraine35. Clearly, there is nothing fundamental about chemistry that prohibits us from leveraging such approaches to science.
Usually, however, in contrast to the publication of an article, the publication of all the scientific data on which the article is based is reduced to an afterthought. Most of us have still been educated with the idea that we need to be selective about which data to publish, instead of embracing the idea that all the scientific data we generate is an integral part of the science we do: data are typically only published to fulfil the requirements of journal policy or data management plan—without reuse in mind. This probably explains why many ELNs do not feature an option to export data to a repository.
In the open-science platform we propose, the publication of the scientific data is simply seen as one of the applications of the ELN. The users can select the samples which they want to publish and create an entry on a repository that contains all the relevant raw data (Fig. 3). The application ensures that data are reported in a form that can be easily reused by other researchers as well as by machines. For the chemists writing a publication, this means that they can provide a DOI (digital object identifier) to supplementary material and augment every figure with a link at which readers can interact with the raw data or download it for follow-up studies. Both the chemotion and cheminfo ELNs implement parts of this functionality. The cheminfo ELN exports data to the general-purpose Zenodo36 repository whereas the chemotion ELN can export data to the chemotion repository37, which focuses on chemical synthesis and characterization data).

Once all the chemicals for which the synthesis and characterization data needs to be published are selected, the ELN compiles the data and uploads it to a repository (in this case Zenodo36). These data are not only machine-readable, but also can be accessed through a browser, and a human reader can also use the same visualization tools as the authors of the article71. The implementation sketched in this figure is implemented in the open-source cheminfo ELN. Panel b screenshot reproduced from Zenodo under a Creative Commons license CC BY 4.0.
In a similar vein, an ELN might also allow importing entries from a repository. This means that researchers might import the entire lab notebook used to produce the published results. Importantly, as the characterization data are also provided in the repository, researchers also have access to the original characterization data and might overlay them with their new results. To our knowledge, at the moment no ELN fully implements this automatic reimportation procedure.
Discussion and outlook
The open-science platform we propose in this Perspective provides a central hub for all the synthetic or analytical work of a chemist or materials scientist. Underpinning this platform are two common principles we feel are essential to make it truly open science, such that it can benefit data-intensive research and address reproducibility problems (thesis 1 in Fig. 1). First, FAIR data should be at the core; all data that enter the platform need to be converted into an open, structured and standardized form with the appropriate linked metadata—this is the main functionality that an ELN should provide (thesis 2). Second, open science also implies ensuring that other researchers can reproduce and build on the results. Therefore, the platform should be able to export the data in a form that is machine-readable and interpretable and that can easily be reused by other groups (thesis 3). In addition, in an open-science vision the tools used to analyse the data should be made available to anyone in the world who might be interested in reproducing the results or reinterpreting the data. This leads to the notion that such a platform is ideally developed as a modular open-source infrastructure in which the analysis code can be scrutinized, reused and improved by the community (thesis 4).
If such a platform becomes widely used and supported by the community, the possibilities are unlimited. The way we assess scientific work and credit scientific outputs has the potential to change. Trusted time stamps can provide unique proofs of discovery, going beyond the compressed and delayed priority claim that preprints can provide38, and peers can continuously provide feedback about the raw data, the analyses and the conclusions. An interesting form of making the full research record public, and hence open for feedback, has already been proposed in the context of open notebook science39. If this information is shared with the community, one can build a community-driven version of the Organic Syntheses journal in which the verification of the results is done continuously by the community and not (only) in a lab of one of the members of the editorial board. Importantly, this version would also contain information about the attempts that did not work and in this way document the process, and the learnings, that led to the final result. If data are available in digital form, the peer-review process can be supported with automated checks, for example, to verify the consistency of NMR assignments, and so highlight potential issues for peer reviewers.
The most important reason for embracing the approach described in this Perspective is that it can change the way we do chemistry. Many of us were educated before the digital era, with the idea that if we publish all the data that we generate, any human being will become lost in the sheer volume of data. Data-intensive science, however, fundamentally changed this point of view. With machine learning, we have the tools to analyse orders of magnitude more data than human being can process, discover correlations in millions of data points and build predictive models40. For example, if we aim to synthesize a compound, a simple query in the collective ELN database might show that for one synthesis route there are 100 ‘failed’ reactions and two successful ones, whereas another route shows 90 successful and ten ‘failed’ attempts—which clearly indicates which synthesis route should be tried first. Undoubtedly, a very experienced chemist might have very good intuitions about what works and what does not. However, for a new student in the field, this collective knowledge now becomes accessible. Clearly, we can go beyond this simple search and try to harvest the collective knowledge generated by all chemists, using machine-learning techniques to capture subtle correlations across the chemical space of the millions of reactions that have been carried out in the world. In this respect, machine learning is not different from the experienced chemist; most probably, it can learn even more from ‘failed’ and partially successful experiments as from the successful ones. However, in contrast with the chemist, it typically needs large amounts of structured data—which we could easily generate in chemistry.
Another issue that the chemistry community faces with open data is that everyone agrees that there are benefits in making data reusable and in reporting ‘failed’ experiments, but often there is hesitation from individual researchers to adopt this behaviour until all members of the community do so. The social sciences give us a range of possible solutions to this problem setting35,41. One approach is some kind of compulsion. For example, the fact that the submission of DNA sequences is a condition for publication in the leading scientific journals of the field is seen as one of the reasons for the success of the GenBank database42. This, in turn, opened many doors for bioinformatics research. We also witnessed that for small groups, which include leaders of the field, agreements such as the ‘Bermuda Principles’, which require that DNA sequence data are automatically released in publicly accessible databases directly after the measurement, can be achieved. In chemistry, we have observed similar dynamics in crystallography, in which crystallographic information files must be deposited with the Cambridge Structural Database, where they are made freely accessible (and searchable) on publication. This led the European Commission to conclude that “the requirement from academic journals that authors provide data in support to their papers has proven to be potentially culture-changing, as has been the case in crystallography”31. What we can also learn from crystallography is that once some standards are adopted, automatic checks (such as checkCIF) can be implemented.
From the Structural Genomics Consortium and related initiatives (for example, Open Source Malaria43 and COVID Moonshot44) we can learn that openness can also be enforced at the level of a consortium, for example, by requesting that members openly publish the protein structures and not file patents for the research outputs. This public–private partnership model seems to be successful because the private sector, which provides the funding and ‘chemical probes’ (potent inhibitors of protein function), can guide the research—that is, prioritize structures that should be solved—without disclosing the companies research and development priorities as the consortium anonymizes the ‘wish lists’45. The utility of such a consortium can best be seen at the precompetitive stage (that is, the early stages of drug discovery) during which it can share risks, enhance collective learning and avoid duplication in new areas of (basic) science46. This is particularly interesting in the case of ‘chemical probes’, which are best produced by experienced industrial medicinal chemists. However, industry would profit enormously if academia could use such probes to validate drug targets47. For this reason, the Structural Genomics Consortium makes them available as ‘open access’ reagents—under the conditions that the research outputs are made available in the public domain. A similar ‘physical open access’ approach is pursued by the Molecule Archive of the Compound Platform at the Karlsruhe Institute of Technology, which acts as a mediator for compound exchange: synthetic chemists can ‘archive’ their compounds (which increases their visibility), which can then be requested for biological screenings48.
Beyond these measures, we need to change incentive structures by creating better ways to give researchers credit for curating data. ELNs could help in this regard by storing the ‘credit’ chain when data are imported and automatically append the citation when datasets are prepared for publication.
Beyond that, the adaption of this data-centric approach to chemistry requires changes in the curriculum at universities to raise the awareness of such new developments, as well as the need for, and the promises of, data curation. Ideally, open-science solutions, such as the infrastructure we describe here, should already be introduced in the undergraduate curriculum. Students can record the results of their labs in ELNs, harvest the data in machine-learning classes, predict the infrared spectrum they just measured in computational chemistry classes49 and use open notebooks to comment on and improve each other’s work. Towards this goal, we define commonly used technical terms in a glossary in the Supplementary Information.
The question that might still be open at this point is how realistic the widespread adoption of such an open-data platform across the chemistry community is. We argue that we have all the basic tools and technology in place. For many of the key design aspects, here we use examples from our own work, which is openly available, can be tried out by the community and can be reused in other implementations. There are also several initiatives (Supplementary Table 3) that work on some of the aspects we emphasize in this Perspective. One example is the German NFDI4Chem consortium50,51, which is embedded in the larger German initiative for the creation of National Research Data Management Infrastructures (which also includes NFDI4Cat52 for catalysis research and NFDI4Ing for the engineering sciences), and aims to ‘FAIRify’ the full data life cycle in chemistry. However, we, as a community, also have to realize that we are in a phase in which there are an insane number of initiatives, proposed data schema and ELNs. The task we as a community face is to embrace and connect the efforts. Only if we succeed in making these tools interoperable we will be able to leverage the full potential of data and the digital age. One promising way forward is the formation of data communities53, in which experimentalists and ELN developers work together to develop a domain-specific (for example, porous materials or batteries) open-science infrastructure by combining, extending and polishing the existing building blocks.
From our perspective, there are a concrete few steps that need to be implemented to reach this goal:
-
The chemistry community should embrace their own existing standards and solutions. We will only be able to make progress as a community if we start to connect and use existing solutions. The feedback can then be used to improve the tools. If we as community do not move beyond the stage of just proposing new formats or implementations—instead of using them in practice—we will not make any progress. Clearly, this also requires that the existing tools are made reusable (that is, packages are extracted from monolithic code bases and augmented with documentation) and shared on platforms such as GitHub.
-
Where community standards exist, journals need to make the deposition of reusable raw data mandatory. This is motivated by the success of the Bermuda agreement and the deposition of crystallographic information files, and is needed to address the collective-action problem. Just using ELNs does not solve the problem. We also need to open our ELNs. Notably, this does not mean that data should be provided as PDFs, but in a standard machine-actionable form. Where community standards exist or are emerging, for example, as is happening in the field of gas adsorption54, journals should start to embrace such formats by requesting the deposition in a community repository53. The same holds for the basic characterization of organic compounds (NMR, infrared and mass spectroscopy), for which the chemotion repository already offers tools and curation that are reminiscent of the Cambridge Structural Database. Importantly, often disconnected pieces of data in different repositories can only practically be used if they are linked. Therefore, for instance, the gas adsorption data in one community repository (such as the NIST/ARPA-E database of Novel and Emerging Adsorbent Materials55) needs to be linked, ideally using hyperlinks, to the crystal structure in the Cambridge Structural Database56.
-
We need to embrace the publication of ‘failed’ experiments. With a digital infrastructure this can be easily done to tell the story of how the final result was reached. It also requires that we as a community realize that the outcome of an experiment is not a binary ‘is this a breakthrough or not’, but simply an observation that is valuable and can be reported. For this to be successful we must take care to properly acknowledge such datasets, for example, when we use them for data-mining exercises or they helped us to avoid some costly experiments.
-
ELNs that do not allow the export of all data into an open machine-actionable form should be avoided. This reflects the core of thesis 2: the most important service an ELN can provide is to remove the hassle from making data FAIR. This is not only to avoid losing access to the data if a licence expires or being unable to build on previous work as it was in the ‘old ELN’ format, but also it is about being able to collaborate and share data with groups independent of the ELN. ELNs that just store data as provided, and might not even allow the export of this data, do not bring us closer to the goal of reusable data in chemistry.
-
Data-intensive research must enter our curricula. ‘Open science’ is gaining momentum in the chemistry community and increasing numbers of researchers are engaging with it (to various extents). We need to raise the awareness of these new developments at the undergraduate level, use ELNs for our lab courses and teach that open science is just science done properly57,58. For example, at the École Polytechnique Fédérale de Lausanne, we teach machine learning and the use of ELNs in the same course, and plan to couple the lab courses with data analysis exercises in the ELN. This also implies that our institutions need to provide faculties with appropriate support, for instance, via the campus library59.
To conclude, we emphasize that the technology is here not only to facilitate the process of publishing data in a FAIR format to satisfy the sponsors, but also to ensure that the combination of chemical data, FAIR principles and openness gives scientists the possibility to harvest all data so that all chemists can have access to the collective knowledge of everybody’s successful, partly successful and even ‘failed’ experiments.
References
Heidorn, P. B. Shedding light on the dark data in the long tail of science. Libr. Trends 57, 280–299 (2008).
Baker, M. 1,500 scientists lift the lid on reproducibility. Nature 533, 452–454 (2016).
Prinz, F., Schlange, T. & Asadullah, K. Believe it or not: how much can we rely on published data on potential drug targets? Nat. Rev. Drug Discov. 10, 712–712 (2011).
Wilkinson, M. D. et al. The FAIR guiding principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016).
Pietsch, W. & Wernecke, J. in Berechenbarkeit der Welt? (eds Pietsch, W., Wernecke, J. Ott, M.) 37–57 (Springer, 2017).
Hunter, M. Establishing the New Science: the Experience of the Early Royal Society (Boydell Press, 1989).
McAlpine, J. B. et al. The value of universally available raw NMR data for transparency, reproducibility, and integrity in natural product research. Nat. Prod. Rep. 36, 35–107 (2019).
Helliwell, J. R., McMahon, B., Guss, J. M. & Kroon-Batenburg, L. M. J. The science is in the data. IUCrJ 4, 714–722 (2017).
Kwok, R. How to pick an electronic laboratory notebook. Nature 560, 269–270 (2018).
Kanza, S. et al. Electronic lab notebooks: can they replace paper? J. Cheminformatics 9, 31 (2017).
Rubacha, M., Rattan, A. K. & Hosselet, S. C. A review of electronic laboratory notebooks available in the market today. J. Lab. Autom. 16, 90–98 (2011).
Guerrero, S. et al. Analysis and implementation of an electronic laboratory notebook in a biomedical research institute. PLoS ONE 11, e0160428 (2016).
Dirnagl, U. & Przesdzing, I. A pocket guide to electronic laboratory notebooks in the academic life sciences. F1000Research 5, 2 (2016).
Coley, C. W. in Artificial Intelligence in Drug Discovery (ed. Brown, N) 327–348 (Royal Society of Chemistry, 2020).
Raccuglia, P. et al. Machine-learning-assisted materials discovery using failed experiments. Nature 533, 73–76 (2016).
Moosavi, S. M. et al. Capturing chemical intuition in synthesis of metal–organic frameworks. Nat. Commun. 10, 539 (2019).
Ojea-Jiménez, I., Bastús, N. G. & Puntes, V. Influence of the sequence of the reagents addition in the citrate-mediated synthesis of gold nanoparticles. J. Phys. Chem. C 115, 15752–15757 (2011).
Huang, Y. et al. Importance of reagent addition order in contaminant degradation in an Fe(II)/PMS system. RSC Adv. 6, 70271–70276 (2016).
Lowe, D. M. Extraction of Chemical Structures and Reactions from the Literature. PhD thesis, Univ. Cambridge (2012).
Jin, W., Coley, C. W., Barzilay, R. & Jaakkola, T. Predicting organic reaction outcomes with Weisfeiler-Lehman network. In Proc. 31st International Conference on Neural Information Processing Systems 2604–2613 (NIPS, 2017).
Kim, E., Huang, K., Kononova, O., Ceder, G. & Olivetti, E. Distilling a materials synthesis ontology. Matter 1, 8–12 (2019).
Roughley, S. D. & Jordan, A. M. The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 54, 3451–3479 (2011).
Schneider, N., Lowe, D. M., Sayle, R. A., Tarselli, M. A. & Landrum, G. A. Big data from pharmaceutical patents: a computational analysis of medicinal chemists’ bread and butter. J. Med. Chem. 59, 4385–4402 (2016).
Brown, D. G., Gagnon, M. M. & Boström, J. Understanding our love affair with p-chlorophenyl: present day implications from historical biases of reagent selection. J. Med. Chem. 58, 2390–2405 (2015).
Brown, D. G. & Boström, J. Analysis of past and present synthetic methodologies on medicinal chemistry: where have all the new reactions gone? J. Med. Chem. 59, 4443–4458 (2015).
L. Bird, C., Willoughby, C. & G. Frey, J. Laboratory notebooks in the digital era: the role of ELNs in record keeping for chemistry and other sciences. Chem. Soc. Rev. 42, 8157–8175 (2013).
Oleksik, G., Milic-Frayling, N. & Jones, R. Study of electronic lab notebook design and practices that emerged in a collaborative scientific environment. In CSCW’14 Proceedings of the 17th ACM Conference on Computer Supported Cooperative Work & Social Computing (ACM Press, 2014).
McDonald, R. S. & Wilks, P. A. Jcamp-dx: a standard form for exchange of infrared spectra in computer readable form. Appl. Spectrosc. 42, 151–162 (1988).
Chalk, S. J. The open spectral database: an open platform for sharing and searching spectral data. J. Cheminformatics 8, 55 (2016).
Mehr, S. H. M., Craven, M., Leonov, A. I., Keenan, G. & Cronin, L. A universal system for digitization and automatic execution of the chemical synthesis literature. Science 370, 101–108 (2020).
Directorate General for Research and Innovation (European Commission) Turning FAIR into Reality: Final Report and Action Plan from the European Commission Expert Group on FAIR Data (Publications Office, 2018).
Harrow, I. et al. Ontology mapping for semantically enabled applications. Drug Discov. Today 24, 2068–2075 (2019).
Davies, A. & Patiny, L. NMRium browser-based nuclear magnetic resonance data processing. Spectrosc. Eur. https://doi.org/10.1255/sew.2021.a18 (2021).
Bonney, R. et al. Next steps for citizen science. Science 343, 1436–1437 (2014).
Nielsen, M. Reinventing Discovery: the New Era of Networked Science (Princeton Univ. Press, 2012).
European Organization For Nuclear Research & OpenAIRE Zenodo https://www.zenodo.org/ (2013).
Tremouilhac, P. et al. Chemotion repository, a curated repository for reaction information and analytical data. Chem. Methods 1, 8–11 (2020).
Coudert, F.-X. The rise of preprints in chemistry. Nat. Chem. 12, 499–502 (2020).
Bradley, J.-C. Open notebook science using blogs and wikis. Nat. Prec. https://doi.org/10.1038/npre.2007.39.1 (2007).
Jablonka, K. M., Ongari, D., Moosavi, S. M. & Smit, B. Big-data science in porous materials: materials genomics and machine learning. Chem. Rev. 120, 8066–8129 (2020).
Olson, M. The Logic of Collective Action; Public Goods and the Theory of Groups (Schocken Books, 1971).
Strasser, B. GENETICS: genbank—natural history in the 21st century? Science 322, 537–538 (2008).
Williamson, A. E. et al. Open source drug discovery: highly potent antimalarial compounds derived from the Tres Cantos arylpyrroles. ACS Centr. Sci. 2, 687–701 (2016).
Chodera, J., Lee, A. A., London, N. & von Delft, F. Crowdsourcing drug discovery for pandemics. Nat. Chem. 12, 581–581 (2020).
Perkmann, M. & Schildt, H. Open data partnerships between firms and universities: the role of boundary organizations. Res. Policy 44, 1133–1143 (2015).
Jones, M. M. & Chataway, J. The structural genomics consortium: successful organisational technology experiment or new institutional infrastructure for health research? Technol. Anal. Strategic Manage. 33, 296–306 (2021).
Edwards, A. M., Bountra, C., Kerr, D. J. & Willson, T. M. Open access chemical and clinical probes to support drug discovery. Nat. Chem. Biol. 5, 436–440 (2009).
Jung, N., Deckers, A. & Bräse, S. Ein molekülarchiv als akademisch integrierte service-einrichtung. Biospektrum 23, 212–214 (2017).
Jablonka, K. M., Patiny, L. & Smit, B. Making molecules vibrate: Interactive web environment for the teaching of infrared spectroscopy. J. Chem. Educ. https://doi.org/10.1021/acs.jchemed.1c01101 (2022).
Herres-Pawlis, S., Koepler, O. & Steinbeck, C. NFDI4chem: shaping a digital and cultural change in chemistry. Angew. Chem. Int. Ed. 58, 10766–10768 (2019).
Steinbeck, C. et al. NFDI4chem—towards a national research data infrastructure for chemistry in Germany. Res. Ideas Outcomes 6, e55852 (2020).
Wulf, C. et al. A unified research data infrastructure for catalysis research—challenges and concepts. ChemCatChem 13, 3223–3236 (2021).
Cooper, D. & Springer, R. Data Communities: A New Model for Supporting STEM Data Sharing Technical Report (Univ. Nebraska-Lincoln, 2019).
Evans, J. D., Bon, V., Senkovska, I. & Kaskel, S. A universal standard archive file for adsorption data. Langmuir 37, 4222–4226 (2021).
Siderius, D. NIST/ARPA-E Database of Novel and Emerging Adsorbent Materials (NIST, accessed 29 June 2020); https://doi.org/10.18434/T43882
Ongari, D., Talirz, L., Jablonka, K. M., Siderius, D. W. & Smit, B. Data-driven matching of experimental crystal structures and gas adsorption isotherms of Metal–Organic frameworks. J. Chem. Eng. Data https://doi.org/10.1021/acs.jced.1c00958 (2022).
Watson, M. When will ‘open science’ become simply ‘science’? Genome Biol. 16, 101 (2015).
Tennant, J. Open science: Just science done right? https://figshare.com/articles/Open_Science_Just_science_done_right_/9759353/1 (2019).
Long, M. & Schonfeld, R. Supporting the Changing Research Practices of Chemists Technical Report (Ithaca, 2013).
Tremouilhac, P. et al. Chemotion ELN: an open source electronic lab notebook for chemists in academia. J. Cheminformatics 9, 54 (2017).
Huang, Y.-C., Tremouilhac, P., Nguyen, A., Jung, N. & Bräse, S. ChemSpectra: a web-based spectra editor for analytical data. J. Cheminformatics 13, 8 (2021).
Barillari, C. et al. openBIS ELN-LIMS: an open-source database for academic laboratories. Bioinformatics 32, 638–640 (2016).
Patiny, L. et al. The c6h6 NMR repository: an integral solution to control the flow of your data from the magnet to the public. Magn. Reson. Chem. 56, 520–528 (2017).
A. Badiola, K. et al. Experiences with a researcher-centric ELN. Chem. Sci. 6, 1614–1629 (2015).
Woelfle, M., Olliaro, P. & Todd, M. H. Open science is a research accelerator. Nat. Chem. 3, 745–748 (2011).
Carpi, N., Minges, A. & Piel, M. eLabFTW: an open source laboratory notebook for research labs. J. Open Source Softw. 2, 146 (2017).
Rudolphi, F. Ein elektronisches laborjournal als open-source-software. Nachr. Chem. 58, 548–550 (2010).
Brandt, N. et al. Kadi4mat: a research data infrastructure for materials science. Data Sci. J. 20, 8 (2021).
Jablonka, K. M. et al. Connecting lab experiments with computer experiments: making ‘routine’ simulations routine. Preprint at ChemRxiv https://doi.org/10.26434/chemrxiv-2021-h3381-v2 (2021).
Gray, A. J., Goble, C. A., Jimenez, R. et al. Bioschemas: from potato salad to protein annotation. In 16th International Semantic Web Conference (2017).
Jablonka, K. M. et al. A data-driven perspective on the colours of metal–organic frameworks. Chem. Sci. 12, 3587–3598 (2021).
Kratsios, M., Kent, S. & Rinat. O. Connecting Americans to coronavirus information online. Trump White House Archives https://trumpwhitehouse.archives.gov/articles/connecting-americans-coronavirus-information-online/ (2020).
COVID-19 Announcements Structured Data (Google Search Central, 2021); https://developers.google.com/search/docs/advanced/structured-data/special-announcements
Fletcher, G., Groth, P. & Sequeda, J. Knowledge scientists: unlocking the data-driven organization. Preprint at https://arxiv.org/abs/2004.07917 (2020).
Kellogg, G., Champin, P.-A. & Longley, D. JSON-LD 1.1—A JSON-based Serialization for Linked Data. (W3C, 2020).
Tennison, J. CSV on the Web: A Primer (W3C, 2016).
Coles, S. J., Frey, J. G., Bird, C. L., Whitby, R. J. & Day, A. E. First steps towards semantic descriptions of electronic laboratory notebook records. J. Cheminformatics 5, 52 (2013).
Lütjohann, D. S., Jung, N. & Bräse, S. Open source life science automation: design of experiments and data acquisition via ‘dial-a-device’. Chemometr. Intell. Lab. Syst. 144, 100–107 (2015).
Chung, Y. G. et al. Advances, updates, and analytics for the computation-ready, experimental metal–organic framework database: CoRE MOF 2019. J. Chem. Eng. Data 64, 5985–5998 (2019).
Gražulis, S. et al. Crystallography Open Database—an open-access collection of crystal structures. J. Appl. Crystallogr. 42, 726–729 (2009).
Gražulis, S. et al. Crystallography Open Database (COD): an open-access collection of crystal structures and platform for world-wide collaboration. Nucleic Acids Res. 40, D420–D427 (2012).
Chalk, S. J. SciData: a data model and ontology for semantic representation of scientific data. J. Cheminformatics 8, 54 (2016).
Acknowledgements
This work was partially supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 666983, MaGic) and the Swiss National Science Foundation (SNSF) through the National Center of Competence in Research (NCCR) and Materials’ Revolution: Computational Design and Discovery of Novel Materials (MARVEL). We thank M. Evans, L. Talirz, M. Moosavi, M. Asgari, N. Marzari, G. Pizzi and fellow EPFL Data Champions for discussion and inputs and thank the cheminfo and Zakodium developers (among others, M. Zasso, D. Kostro, J. Wiest, A. M. Castillo, A. Bolaños, J. Osorio and N. Pellet; also see https://cheminfo.github.io/team for a list of contributors) for their invaluable contributions (conceiving and implementing many of the examples discussed in this perspective). Of course, we also thank the chemists whose feedback about our ELN implementation shaped our Perspective.
Author information
Authors and Affiliations
Contributions
K.M.J. and B.S. wrote the manuscript with inputs from L.P. All the authors contributed to discussions.
Corresponding authors
Ethics declarations
Competing interests
L.P. is chief scientific officer of Zakodium Sàrl, a company dedicated to the development of tools for storing, processing and analysis of scientific information. All the authors are contributors to the cheminfo ecoystem.
Peer review
Peer review information
Nature Chemistry thanks Samantha Kanza, Matthew Todd and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Fragment of a NMR spectrum serialized to a classic standard format.
This is an example of a JCAMP-DX file. This format is a widely used IUPAC-recommended format for spectra that is, for example, supported by the cheminfo and chemotion ELNs. Also, spectra in many databases such as the NIST webbook or the Infrared & Raman Users Group (IRUG) Spectral Database can be downloaded in JCAMP-DX format. A JCAMP-DX file can contain multiple blocks of labelled data records (LDR). That is, one can store multiple related spectra (such as repeated measurements) in the same file. All data blocks must contain a CORE header with basic metadata such as OWNER, DATATYPE. The IUPAC working group also provides a vocabulary of further global labels such as for the temperature/pressure/CAS-number. Data can also be compressed using various compression schemes. Note that the JCAMP-DX format is only one, old standard, and many others have been proposed. The JCAMP-DX format, however, does allow for the addition of an unlimited number of private labels by using the ##$ prefix, which allows every system to tailor the format to its own needs. Drawbacks of this format are, however, that it does not come with native, standardised, support for semantic web features (such as linking to a vocabulary) and, in contrast to formats like xml, csv, or json, that it is not natively supported by many general purpose tools.
Extended Data Fig. 2 Fragment of a NMR spectrum serialized to a modern standard format.
We show another NMR dataset (taken from the SciData website from the Chalk Group at the University of North Florida) serialized to JSON-LD using the SciData data model82. One important part on the JSON-LD file is the @context field. The values in this field links to the vocabularies that are used for naming things in this datafile. For instance, for units, the vocabularies provided by qudt are used, whereas the method is described using the chemical methods ontology (from which it is clear that, for instance, NMR spectroscopy is—similar to electron spin resonance spectroscopy–a magnetic resonance method). Importantly, almost all modern programming languages provide support for reading such json files. The @type field can describe the format of the data, for instance, to let a computer now that it can expect a list of doubles. Different parts of the file (such as methodology, the dataset) can be access by their own address.
Supplementary information
Supplementary Information
Supplementary Note 1 and glossary, Tables 1–4 and Fig. 1.
Rights and permissions
About this article

Cite this article
Jablonka, K.M., Patiny, L. & Smit, B. Making the collective knowledge of chemistry open and machine actionable. Nat. Chem. 14, 365–376 (2022). https://doi.org/10.1038/s41557-022-00910-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41557-022-00910-7
This article is cited by
-
Leveraging large language models for predictive chemistry
Nature Machine Intelligence (2024)
-
LinChemIn: SynGraph—a data model and a toolkit to analyze and compare synthetic routes
Journal of Cheminformatics (2023)
-
Artificial intelligence driven design of catalysts and materials for ring opening polymerization using a domain-specific language
Nature Communications (2023)
-
Using genetic algorithms to systematically improve the synthesis conditions of Al-PMOF
Communications Chemistry (2022)
