
Abstract
Biomacromolecules are highly promising therapeutic modalities to treat various diseases. However, they suffer from poor cellular membrane permeability, limiting their access to intracellular targets. Strategies to overcome this challenge often employ nanoscale carriers that can get trapped in endosomal compartments. Here we report conjugated peptides that form pH- and redox-responsive coacervate microdroplets by liquid–liquid phase separation that readily cross the cell membrane. A wide range of macromolecules can be quickly recruited within the microdroplets, including small peptides, enzymes as large as 430 kDa and messenger RNAs (mRNAs). The therapeutic-loaded coacervates bypass classical endocytic pathways to enter the cytosol, where they undergo glutathione-mediated release of payload, the bioactivity of which is retained in the cell, while mRNAs exhibit a high transfection efficiency. These peptide coacervates represent a promising platform for the intracellular delivery of a large palette of macromolecular therapeutics that have potential for treating various pathologies (for example, cancers and metabolic diseases) or as carriers for mRNA-based vaccines.

Similar content being viewed by others


Main
Biomacromolecules, including peptides1, proteins2,3 and RNAs4,5,6, are promising therapeutic modalities for the treatment of various diseases owing to key advantages such as high potency, specificity and safety7. However, their therapeutic potential has not yet been fully realized due to their poor cell membrane permeability and/or endosomal entrapment that limits their intracellular exposure8. Hence, there is substantial interest in developing safe vehicles that can deliver therapeutic cargo to the cytosol. Ideally, endosomal escape can be chemically encoded in the carrier to facilitate the release of the therapeutic payload9,10. Alternatively, approaches to use non-endocytic entry mechanisms could also enhance delivery efficiency7,8. In addition, it is important that the encapsulation method does not affect cargo bioactivity and that the carriers exhibit negligible cytotoxicity.
Current strategies to tackle these issues rely on nanoscale carriers such as inorganic nanoparticles11, synthetic polymers12 or nanoscale hybrid assemblies that can mediate cell membrane fusion13,14. In alternative approaches, the macromolecular drugs are conjugated or complexed with cell-penetrating peptides9,15 to enhance endosomal escape. Although these methods are promising and increasingly considered for clinical translation, they also have pitfalls7. Specifically, fabrication methods can be complex and often use organic solvents that can decrease cargo bioactivity16,17. Some carriers are limited to a specific type of biomacromolecule, whereas others are restricted to the release of payloads with relatively small molecular weights18,19. Safety concerns have also been raised for some carriers, such as inorganic and lipid nanoparticles17,20,21. Whether the carriers are inorganic- or organic-based (polymers, lipids, peptides or fusions thereof), it is generally considered that they must have dimensions below ~200 nm to cross the cell membrane8,18. Recent studies in our laboratory have challenged this notion. Specifically, we have found that micrometre-sized peptide coacervates obtained by liquid–liquid phase separation (LLPS), within which both proteins22 and low-molecular-weight compounds23 can be recruited, are also capable of crossing the cell membrane through an endocytosis-independent pathway23, potentially opening new avenues for intracellular delivery of therapeutics. Peptide coacervates inspired by the self-coacervating histidine (His)-rich beak proteins (HBPs)22 exhibit several advantages over traditional nanoscale delivery vehicles24,25, including (1) fast (within seconds) and efficient recruitment of therapeutic cargo within the coacervate microdroplets; (2) bioactivity-preserving aqueous-based recruitment conditions and (3) negligible cytotoxicity of the peptide building blocks. Furthermore, the physicochemical conditions to induce coacervation can be precisely tuned by single amino acid mutations26,27.
Based on these benefits, we hypothesized that peptide coacervates could be used for the intracellular delivery of a broad palette of macromolecular therapeutics featuring a wide range of molecular weights and isoelectric points (IEPs). To achieve this, we developed short His-rich, pH-responsive beak peptide (HBpep) coacervates conjugated with disulfide bond-containing self-immolative moieties that trigger disassembly of the droplets, facilitating payload delivery within the intracellular reducing environment. We show that these coacervate microdroplets bypass classical endocytic pathways and are capable of direct and efficient cytosolic delivery of a wide range of macromolecules, from therapeutic peptides as small as 726 Da to large enzymes as large as 430 kDa. They can also deliver messenger RNAs (mRNAs) with a high transfection efficiency while also preventing their premature degradation by RNase. Overall, these robust conjugated peptide coacervates can be used as general intracellular delivery vehicles for a broad range of macromolecular therapeutics.
Results and discussion
Preparation and characterization of redox-responsive peptide coacervates
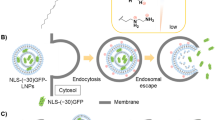
As an initial self-coacervating peptide we selected the histidine-rich beak peptide HBpep, because of advantages including its biological origin, its ability to recruit client molecules with high efficiency (above 95%) and its low toxicity22. Notably, HBpep coacervates can cross the cell membrane via an endocytosis-free pathway23, suggesting their potential for the intracellular delivery of therapeutics. HBpep is characterized by low sequence complexity consisting of five copies of the tandem repeat sequence GHGXY (where X could be leucine (L), proline (P) or valine (V) amino acids) and a single C-terminus tryptophan (W) residue, providing opportunities to further tune its phase separation behaviour. A key feature of the HBpep are the five His residues that confer pH-responsive LLPS behaviour26. Specifically, HBpep adopts a monomeric state at low pH, but then quickly phase-separates into coacervate microdroplets at neutral pH, concomitantly recruiting various macromolecules from the solution in the process (Fig. 1). Preliminary attempts to use HBpep coacervates to recruit and deliver enhanced green fluorescence protein (EGFP) resulted in successful cellular uptake in various cell lines (Supplementary Fig. 1 and Supplementary Videos 1 and 2). However, the EGFP-containing microdroplets formed organelle-like structures within the cells that remained stable for at least three days in HepG2 cells and up to seven days in a T22 cell line without releasing their cargo (Supplementary Figs. 2 and 3).

HBpep-K (top left) remains in solution at neutral pH but can phase-separate and form coacervates after conjugation of the sole Lys residue with a self-immolative moiety (HBpep-SR, middle left). In a reducing environment such as the GSH-rich cytosol, HBpep-SR is reduced, followed by self-catalytic cleavage of the SR moiety, resulting in HBpep-K again and the disassembly of the coacervates (bottom left). During coacervation of HBpep-SR near neutral pH (top right), macromolecular therapeutics are readily recruited within the coacervates. On incubation with cells, the therapeutics-loaded coacervates cross the cell membrane through an energy-independent endocytic pathway, possibly mediated by cholesterol-dependent lipid rafting, to migrate into the cytosol (bottom right), where they are reduced by GSH, resulting in the disassembly and release of the therapeutic.
By single amino acid level manipulation, the pH at which the HBpep phase-separates was dramatically altered. Specifically, the insertion of a single lysine (Lys) residue at position 16 (HBpep-K) shifted the phase separation from approximately pH 7.5 (ref. 22) to 9.0 (Fig. 2a and Supplementary Table 1). Next, a disulfide-containing moiety was conjugated to the amine group of the inserted Lys residue to neutralize the extra positive charge and increase the hydrophobicity of the peptide (Fig. 1). The conjugated moiety is self-immolative, eventually restoring the amine group of the Lys residue through a series of autocatalytic reactions, starting with the reduction of the disulfide bond and followed by a series of side-group rearrangements (Fig. 1)28,29. After the modification, both conjugated peptides with acetyl (HBpep-SA) and phenyl (HBpep-SP) groups at the extremity of the self-immolative moiety were able to phase-separate at the lower pH of 6.5, forming stable microdroplets (Fig. 2a,b and Supplementary Table 1). This design allowed the modified peptides (HBpep-SA and HBpep-SP, collectively referred to as HBpep-SR) to form coacervate microdroplets with a diameter of ~1 μm and a relatively narrow size distribution (Extended Data Fig. 1a,b). Critically, HBpep-SR peptides were able to recruit a wide range of macromolecules during the self-coacervation process at pH 6.5, including proteins, small peptides and mRNAs (Extended Data Fig. 1a–c), without a significant change in their size and zeta potential. The cargo-loaded coacervates were stable at near-physiological and serum conditions until internalization by the cells (Extended Data Fig. 2a,b). Owing to the self-immolative nature of the flanking moiety, reducing agents such as glutathione (GSH), which is abundant in the cytosol, could trigger the reduction and subsequent cleavage of the modified side chain, to convert HBpep-SR back to HBpep-K (Fig. 1). As verified by HPLC and matrix-assisted desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF) (Extended Data Fig. 3), most of the HBpep-SP coacervates were reduced in the presence of GSH for 24 h, producing a thiol-containing intermediate that was eventually converted into HBpep-K. Because HBpep-K reverts to the single phase at neutral pH (that is, monomeric peptide in solution, Fig. 2a), we postulated that GSH-triggered reduction would cause disassembly of coacervate microdroplets in the cytosol, thus releasing the cargo. Therefore, as shown in Fig. 1, by combining pH- and redox-responsivity of the peptides, it should be possible to design an intracellular delivery platform. It is noteworthy that a simple modification at the end of the flanking moiety of HBpep-SR (HBpep-SA versus HBpep-SP) resulted in significant variation in the rate of peptide reduction (Fig. 2c). The concentration decay of HBpep-SA and HBpep-SP in the early stage (2–10 h) could be well fitted by a first-order reaction (Supplementary Fig. 4), with the reaction rate of HBpep-SP about twofold higher than that of HBpep-SA, thus potentially providing a way to tune the kinetics of therapeutic release.

a, Turbidity measurements of HBpep-SA and HBpep-SP at various pHs and comparison with HBpep-K. Data are presented as the mean ± s.d. of n = 3 independent measurements. b, Optical micrograph of HBpep-SP coacervates at pH 6.5 and an ionic strength of 0.1 M (phosphate buffer). c, GSH-induced reduction of HBpep-SA and HBpep-SP coacervates. Data are presented as the mean ± s.d. of n = 3 independent experiments; two-sided Student’s t-test, *P = 0.00028 compared to HBpep-SA (1 mM GSH) at 24 h. d,e, Four-hour (d) and 24-h (e) fluorescence micrographs of HepG2 cells treated with EGFP-loaded HBpep-SA coacervates. f,g, Four-hour (f) and 24-h (g) fluorescence micrographs of HepG2 cells treated with EGFP-loaded HBpep-SP coacervates. h–j, Intracellular delivery of EGFP into A549 (h), NIH 3T3 (i) and HEK293 (j) cells mediated by HBpep-SP coacervates.
EGFP model protein delivery mediated by redox-responsive coacervates
To evaluate the intracellular delivery efficiency of our designed peptide coacervates, EGFP was first employed as a model protein and recruited inside both HBpep-SA and HBpep-SP coacervates, before being incubated with cells. As shown in Fig. 2d–g, both EGFP-loaded coacervates were internalized by liver cancer cells (HepG2) within 4 h, and subsequently released inside the cytoplasm within 24 h. As a control, we also verified that EGFP alone could not cross the cell membrane (Supplementary Fig. 5a). Another finding was that HBpep-SP exhibited a faster release rate than HBpep-SA and started to release its EGFP cargo after 4 h, which is consistent with the faster reduction rate of HBpep-SP shown in Fig. 2c and Supplementary Fig. 4. This further highlights the possibility of controlling the kinetics of cargo release by slight modifications of the conjugate moiety side group. To further investigate the versatility of this delivery system, EGFP-loaded HBpep-SP coacervates were tested on another cancerous cell line (A549) as well as two healthy cell lines, namely NIH 3T3 and HEK293. Based on the fluorescence signals observed inside the cells (Fig. 2h–j), the intracellular delivery and release ability of the HBpep-SP coacervates was verified for all cell lines. We emphasize that, for unmodified HBpep (that is, without the additional Lys residue and not conjugated with the self-immolative moieties), the coacervates remained as spherical puncta in the cytosol between three days to one week but did not release their payload (Supplementary Figs. 1–3 and Supplementary Videos 1 and 2), suggesting that disassembly and release is activated by the redox properties of the self-immolative groups. To further verify this mechanism, we measured the release profiles of EGFP from HBpep-SP coacervates in phosphate-buffered saline (PBS, pH 7.4, ionic strength of 0.15 M) with or without GSH. As shown in Extended Data Fig. 4a, the release of EGFP was negligible in PBS without GSH, but reached ~90% after 24 h in PBS containing 1 mM GSH. Furthermore, the release could be modulated between these two profiles at an intermediate concentration of 0.1 mM, and it is important to note that the change in GSH concentration was the only variable among the three samples. We also conducted cell uptake assays on HepG2 cells that were pre-incubated with the GSH-depleting reagent l-buthionine-sulfoximine (BSO)30,31. Compared to the uniform release of EGFP in normal HepG2 cells (Extended Data Fig. 4b), GSH-depleted cells only contained a few fluorescent dots, indicating that EGFP was not released in the cytosol in this case (Extended Data Fig. 4c). Together, these data support that coacervates disassembly and therapeutics release is activated by GSH.
Delivery and release of proteins with different molecular weights and IEPs by HBpep-SP coacervates
After the successful delivery and release of EGFP, we decided to check whether proteins with a wider range of molecular weights and IEPs could also be delivered into HepG2 cells using HBpep-SP coacervates. We first assessed lysozyme and bovine serum albumin (BSA), two common proteins with substantially different molecular weights and IEPs. As shown in Fig. 3a,b, both Alexa Fluor 488 (AF)-labelled lysozyme and BSA could be delivered into HepG2 cells and then released into the cytoplasm within 24 h. On the other hand, in their free form (not recruited into coacervates), neither of these proteins was internalized by cells (Supplementary Fig. 5b,c).

a–c, Fluorescence micrographs of AF-lysozyme delivery mediated by HBpep-SP coacervates (a), AF-BSA delivery mediated by HBpep-SP coacervates (b) and R-PE delivery mediated by HBpep-SP coacervates (c). d, Concentration-dependent cytotoxicity of free saporin and saporin-loaded HBpep-SP coacervates. Data are presented as the mean ± s.d. of n = 3 independent experiments; two-sided Student’s t-test, *P = 0.00010 compared to saporin at 500 ng ml−1. e, X-Gal staining of cells treated with β-Gal-loaded HBpep-SP coacervates after 24 h. f, X-Gal staining of cells treated with free β-Gal. g, Summary of proteins with a wide range of IEP and molecular weight demonstrated to be successfully delivered in the cytosol, including lysozyme (IEP, 10.7; Mw, 14 kDa), saporin (IEP, 9.4; Mw, 28.6 kDa), EGFP (IEP, 6.2; Mw, 32.7 kDa), BSA (IEP, 4.8; Mw, 66.4 kDa), R-PE (IEP, 4.1; Mw, 255 kDa) and β-Gal (IEP, 4.6; Mw, 465 KDa).
To further challenge the molecular-weight ceiling of the cargo proteins, we recruited R-phycoerythrin, a larger red fluorescence protein (R-PE; Mw, 255 kDa), inside HBpep-SP coacervates, and incubated the loaded coacervates with HepG2 cells. After 4 h of uptake and another 20 h of release, a strong red fluorescence signal was detected inside the cytoplasm, confirming that R-PE was delivered and released inside HepG2 cells (Fig. 3c). We also attempted to co-deliver both EFGP and R-PE using HBpep-SP coacervates. As shown in Extended Data Fig. 5, both green and red fluorescence signals were observed in HepG2 cells treated with EGFP/R-PE co-loaded coacervates, demonstrating the ability of our HBpep-SP coacervate system to simultaneously deliver a combination of protein therapeutics.
Beside the successful delivery and release of cargo proteins, maintaining their bioactivity after delivery is critical for protein-based therapies. Saporin from Saponaria officinalis seeds is a well-known ribosome-inactivating protein32,33. However, due to its poor membrane permeability, a suitable delivery system is required for further applications of saporin in biomedicine32. Notably, the viability of HepG2 cells treated with saporin-loaded HBpep-SP coacervates significantly decreased compared to those treated with saporin alone (Fig. 3d). This demonstrates not only that saporin was delivered and released from HBpep-SP coacervates, but also that its bioactivity was preserved during the recruitment and delivery process.
To further confirm the versatility of our peptide coacervate delivery system, we selected β-galactosidase (β-Gal), a very high-molecular-weight enzyme (Mw, 430 kDa) whose intracellular delivery is challenging owing to its difficulty in forming complexes with common nanocarriers because of its high molecular weight34. Our system appeared to overcome these hurdles, as almost all of the HepG2 cells treated with β-Gal-loaded HBpep-SP coacervates turned blue due to the pigment generated by the β-Gal-catalysed hydrolysis of 5-bromo-4-chloro-3-indolyl-β-d-galactoside (X-Gal; Fig. 3e). By contrast, no blue pigment was formed in cells treated with β-Gal alone (Fig. 3f), further corroborating that HBpep-SP coacervates are capable of delivering large enzymes while maintaining their bioactivities.
We also compared the delivery efficiency of fluorescent EGFP- and R-PE-loaded HBpep-SP coacervates in HepG2 cells with commercial protein transfection reagents, including Pro-Ject and Xfect. For both proteins, HBpep-SP displayed better performance than Pro-Ject and Xfect (Fig. 4a–c,e–g), as evidenced by the stronger and much more homogeneous fluorescence signal within the cells. Further quantification by fluorescence-activated cell sorting (FACS) analyses (Fig. 4d,h) confirmed the extremely high delivery efficiency of HBpep-SP for both EGFP (99.3%) and R-PE (98.8%), which outperformed Pro-Ject (EGFP, 65.9%; R-PE, 33.3%) and Xfect (EGFP, 49.4%; R-PE, 71.0%). In addition to its stability and high delivery efficiency, our coacervate system also benefits from aqueous-based preparation and facile handleability, providing an advantage over lipid-based delivery reagents such as Pro-Ject, which requires organic solvents during sample preparation.

a–c, Fluorescence micrographs of EGFP delivery mediated by HBpep-SP coacervates (a), Pro-Ject (b) and Xfect (c). d, FACS analysis of HepG2 cells transfected with EGFP by HBpep-SP coacervates, Pro-Ject and Xfect. NC, negative control. e–g, Fluorescence micrographs of R-PE delivery mediated by HBpep-SP coacervates (e), Pro-Ject (f) and Xfect (g). h, FACS analysis of HepG2 cells transfected with R-PE by HBpep-SP coacervates, Pro-Ject and Xfect.
Taken together, these results show that HBpep-SR coacervates can efficiently recruit (Extended Data Fig. 6) and directly deliver proteins exhibiting a wide range of molecular weights and IEPs into the cytosol (Fig. 3g), at a very high delivery rate, and using a cargo-recruitment process that is fully aqueous, easy and rapid. These characteristics enable HBpep-SR coacervates to recruit both native and engineered proteins without further chemical modifications and in a manner that preserves their bioactivity, making this approach a promising and flexible platform for single- and multi-protein-based therapies.
Peptides delivery mediated by HBpep-SP coacervates
Compared to protein-based therapeutics, peptides display specific advantages such as a low immune response and scalability35. Therefore, two short peptides including the second mitochondria-derived activator (Smac, AVPIAQK) and the proapoptotic domain (PAD, KLAKLAK KLAKLAK) peptides were selected to be delivered into HepG2 cells using HBpep-SP coacervates. We note that both peptides were recruited within HPpep-SP coacervates with over 90% recruitment efficiency (Extended Data Fig. 6). Both peptides have previously demonstrated anticancer effects by promoting caspase activity or causing mitochondrial membrane disruption36,37. As shown in Fig. 5a, strong fluorescence signals were detected inside HepG2 cells treated with FITC-Smac-loaded coacervates. By contrast, FITC-Smac alone could not cross the cell membrane (Fig. 5b). Similar results were also obtained in the delivery of FITC-PAD peptide as shown in Fig. 5d,e. Furthermore, the anticancer activity of Smac- and PAD-loaded HBpep-SP coacervates was evaluated as shown in Fig. 5c,f. HepG2 cells treated with Smac-loaded and PAD-loaded coacervates showed 28% and 33% cell death, respectively, at 10 μg ml−1 peptide concentration. In comparison, there was negligible cytotoxicity for cells treated with Smac or PAD alone, and HBpep-SR coacervates without therapeutic peptide cargos did not exhibit cytotoxicity either.

a,b, FITC-Smac delivery mediated by HBpep-SP coacervates in HepG2 (a) and comparison with free FITC-Smac (b). c, Concentration-dependent cytotoxicity of Smac and Smac-loaded HBpep-SP coacervates. Data are presented as the mean ± s.d. of n = 3 independent experiments; two-sided Student’s t-test, *P = 0.0057 compared to Smac at 10 μg ml−1. d,e, FITC-PAD delivery mediated by HBpep-SP coacervates in HepG2 (d) and comparison with free FITC-PAD (e). f, Concentration-dependent cytotoxicity of PAD and PAD-loaded HBpep-SP coacervates. Data are presented as the mean ± s.d. of n = 3 independent experiments; two-sided Student’s t-test, *P = 0.0074 compared to PAD at 10 μg ml−1. g, Western blots of p53 and p21 in HCT116 p53+/+ cells treated with HBpep-SP coacervates loaded with four MDM2/4 inhibiting peptides (dPMI-δ, MP-189, MP-081 and MP-950) and one non-binding peptide (MP-202) as a negative control. Molecular-weight markers indicate 50 kDa and 37 kDa for p53, 20 kDa and 15 kDa for p21, and 50 kDa and 37 kDa for actin. The increased levels of p53 and p21 are particularly striking for the stapled peptides MP-081 and MP-950 (red dashed boxes), with activity comparable to the cell-permeable small molecule Nutlin-3a. h, Lactate dehydrogenase (LDH) release assay data for the same cells as treated in g, showing that HBpep-SP does not damage the plasma membrane. Data are presented individually (yellow dots) and as the mean ± s.d. (column with error bar) of n = 3 independent measurements.
We next explored whether HBpep-SP could mediate the delivery of anticancer peptides that bind to MDM2 and MDM4, two key negative regulators of the tumour suppressor protein p53 but that have limited cellular permeabilities. These comprised a linear d-amino-acid peptide (dPMI-δ)38, a linear peptide carrying the α-helical inducing residue Aib (MP-189)39 and a chemically stapled peptide (MP-950)40 that was purposely engineered to be impermeable through its polyanionic nature. As a control, we also included MP-081, a stapled peptide with validated cell permeability on its own39,40 (the peptide sequences are provided in Supplementary Table 2). Inhibition of MDM2/4 by competitive antagonists manifests in increased levels of p53 and transcription of p53-dependent genes such as p2141. In all cases, HBpep-SP enhanced the delivery and intracellular activity as measured by elevated p53 and p21 levels in colorectal carcinoma HCT116 p53+/+ cells (Fig. 5g and Supplementary Fig. 6). This was particularly noticeable for the higher-affinity MP-081 (Kd = 14.1 ± 7.3 nM) and MP-950 (Kd = 2.3 ± 0.8 nM)40 stapled peptides, both showing comparable intracellular activity to the cell-permeable small-molecule MDM2 inhibitor Nutlin-3a42. This phenotype was not observed for their respective non-binding controls (MP-202 and MP-400). HBpep-SP coacervates did not compromise plasma membrane integrity compared to the controls, as measured by lactate dehydrogenase (LDH) release (Fig. 5h and Supplementary Fig. 6), ruling out non-specific p53 activation due to membrane toxicity. We emphasize that MP-081 was engineered to exhibit intrinsic cell permeability39,40, as can be seen by comparing lanes 8 (MP-081) and 13 (untreated control) in Fig. 5g. However, the cell permeability of MP-081 was further enhanced once its delivery was mediated by HBpep-SP coacervates: compare lanes 8 (MP-081 alone) and 9 (MP-081 recruited in HBpep-SP coacervates). Overall, the data indicate that our HBpep-SP coacervates can safely deliver and release a range of therapeutic peptides intracellularly, without compromising their activities.
The distribution of labelled peptides and proteins inside cells was also observed using confocal laser scanning microscopy (CLSM; Extended Data Fig. 7). Whereas the larger-molecular-weight R-PE protein was mostly found in the cytosol, the smaller EGFP and the FITC-Smac peptide were detected inside the nucleus, suggesting that nuclear permeability depends on the size of the cargo. On the other hand, HBpep-SP did not appear to facilitate transfer across the nuclear membrane, possibly because coacervates are already disassembled in the cytosol.
mRNA delivery mediated by HBpep-SP coacervates
Gene therapy has long been considered as a possible treatment or cure for serious diseases such as cancer, genetic disorders and infectious diseases43. Among these, mRNA-based therapy has recently attracted increasing interest because of its biosafety and the ability for mass production44,45. In its most successful and dramatic application so far, mRNA-based technology was used for the first vaccines approved for the COVID-19 pandemic5,6. These advances demonstrate that synthetic mRNA can indeed enter human cells to produce multiple copies of the protein of choice—opening a spectrum of new possibilities for treating human pathologies. However, lipid nanoparticles—the current mRNA delivery vehicle of choice—have a series of drawbacks, including toxicity and limited tissue distribution. Accordingly, we assessed whether our redox-responsive coacervate microdroplets could also be used to deliver mRNA. HBpep-SP transfection efficiency was evaluated using mRNA encoding a luciferase reporter protein in both HepG2 and HEK293 cell lines and their efficiencies were compared to commonly employed mRNA transfection reagents (polyethylenimine (PEI) and Lipofectamine 2000 and 3000). At the optimal peptide concentration, the transfection efficiencies of HBpep-SA and HBpep-SP coacervates were higher than for PEI and Lipofectamine 3000, but slightly lower than for Lipofectamine 2000 in HepG2 cells (Fig. 6a). Additionally, in HEK293 cells, HBpep-SP coacervates showed a transfection efficiency comparable to that of Lipofectamine 2000 (Fig. 6d). Importantly, neither HBpep-SA nor HBpep-SP coacervates caused cytotoxicity at their optimal concentration (Extended Data Fig. 8).

a, Luciferase-encoding mRNA transfection efficiency of HBpep-SA and HBpep-SP coacervates compared to common commercial transfection reagents in HepG2 cells. Data are presented individually (yellow dots) and as the mean ± s.d. (column with error bar) of n = 3 independent experiments. RLU, relative light units. b,c, Fluorescence micrograph (b) and FACS analysis (c) of HepG2 cells transfected with EGFP-encoding mRNA (Cy5-labelled) loaded in HBpep-SP coacervates. d, Luciferase-encoding mRNA transfection efficiency of HBpep-SA and HBpep-SP coacervates compared to common commercial transfection reagents in HEK293 cells. Data are presented individually (yellow dots) and as the mean ± s.d. (column with error bar) of n = 3 independent experiments. e,f, Fluorescence micrograph (e) and FACS analysis (f) of HEK293 cells transfected with EGFP-encoding mRNA (Cy5-labelled) loaded in HBpep-SP coacervates.
After the successful delivery of luciferase-encoding mRNA, the transfection efficiency of HBpep-SP coacervates was further investigated with EGFP-encoding mRNA (Cy5-labelled). Based on the fluorescence micrographs shown in Fig. 6b,e, the vast majority of HepG2 and HEK293 cells were successfully transfected with mRNA, as most cells exhibited intense green fluorescence. Using FACS analysis, we determined that the uptake efficiency of EGFP-encoding mRNA loaded in HBpep-SP coacervates reached ~98% in HepG2 cells (Fig. 6c and Supplementary Fig. 7a). Furthermore, 72% of HepG2 cells expressed EGFP after 24 h. For HEK293 cells, 94.8% of cells exhibited coacervate internalization and 81.6% expressed EGFP after 24 h (Fig. 6f and Supplementary Fig. 7b). Such a high mRNA transfection efficiency suggests that our redox-responsive HBpep-SP coacervates may represent an efficient vector for gene therapy. For example, we envision that other nucleic acids such as plasmid DNA, microRNA and small interfering RNA could in principle be delivered using this platform. In combination with their protein delivery ability and the high recruitment efficiency of biomacromolecules (Extended Data Fig. 6), HBpep-SP coacervates may also be employed as a tool for the delivery of protein/nucleic acid complex, which is a critical step in genome editing systems such as CRISPR/Cas946.
One limitation of mRNA-based therapies is their instability and rapid degradation by RNase47. Thus, we assessed whether HBpep-SP coacervates could improve the stability of mRNA by exposing mRNA-loaded coacervates to a high concentration (1 mg ml−1) of RNase A for 2 h. Electrophoresis experiments (Extended Data Fig. 9) showed that the molecular weight of mRNA recruited within the coacervates remained unchanged. By contrast, free mRNA was no longer detected in the gel, indicating rapid degradation in that case. These data therefore indicate that HBpep-SP coacervates can protect mRNA from premature degradation.
Internalization mechanism study of HBpep-SP coacervates
With a size of ~1 μm (Extended Data Fig. 1a)—substantially larger than typical nanocarriers—and with liquid-like characteristics, it is intriguing that coacervate microdroplets display such a high cell uptake efficiency, which suggests an internalization pathway distinct from regular endocytosis. To determine whether the HBpep-SP coacervates are trapped in endosomal compartments, LysoTracker dye was used to stain acidic organelles such as lysosomes. Based on confocal microscopy images (Fig. 7a and Extended Data Fig. 10), both EGFP-loaded and AF-BSA-loaded HBpep-SP coacervates showed no co-localization with lysosomes and maintained a high fluorescence intensity. Because the fluorescence of EGFP is quenched at low pH, no fluorescence signal would be expected if the microdroplets were entrapped in the acidic pH of lysosomal compartments, which further supports that the microdroplets are not entrapped in lysosomes although they may possibly be present in early endosomes.

a, Confocal microscopy image of HepG2 cells treated with EGFP-loaded HBpep-SP coacervates (green) for 2 h. The nucleus was stained with Hoechst (blue) and the lysosomes were stained with LysoTracker (red). Coacervates are not co-localized with lysosomes. b,c, FACS (b) and fluorescence micrographs (c) of HepG2 cells treated with various inhibitors before incubation with EGFP-loaded HBpep-SP coacervates for 4 h. Two groups were included as control: totally untreated cells (negative control, NC) and cells treated by EGFP-loaded coacervates without any inhibitors (blank). Only the cholesterol-depletion compound MβCD inhibits cell uptake, as well as low temperature (4 °C).
We then treated the cells with endocytosis inhibitors, including the clathrin-mediated endocytosis inhibitor chlorpromazine (CPM)48,49, the pinocytosis inhibitor amiloride (AM)48,50 and the energy-dependent endocytosis inhibitor sodium azide (NaN3)50,51. None of these inhibitors affected the uptake of EGFP-loaded HBpep-SP coacervates (Fig. 7b,c). However, the cells pretreated with methyl-β-cyclodextrin (MβCD) showed almost no uptake of HBpep-SP coacervates. The effect of MβCD is to deplete cholesterol13, which apparently blocked the internalization of HBpep-SP coacervates, suggesting that the mechanism of coacervates uptake is cholesterol-dependent lipid rafting48. These results suggest that the uptake mechanism does not follow classical endocytosis, especially clathrin-mediated endocytosis, the most common endocytic pathway of cellular uptake52. Instead, cellular uptake appears to be lipid-raft-mediated endocytosis. We emphasize that NaN3—which inhibits all energy-dependent pathways53—did not prevent the uptake of coacervates. This points towards a passive uptake mechanism that depends on membrane fluidity, because cholesterol depletion by MβCD as well as low temperature (Fig. 7b,c), which are well established to affect membrane fluidity54, both inhibited uptake. Although questions remain about the precise uptake mechanism, these results nevertheless indicate that HBpep-SP coacervates avoid endosomal escape such that the biomacromolecular cargo is directly delivered and released inside the cytosol, and in a manner that preserves bioactivity.
Discussion
We have shown that HBpep-K conjugated with self-immolative moieties (HBpep-SR) exhibits LLPS, forming coacervate microdroplets within which a wide range of biomacromolecules (including proteins, peptides and mRNAs) can be efficiently recruited. The cargo-loaded coacervates are taken up by various cell lines and achieve redox-triggered cargo release in the cytosol. The versatility of cargo recruitment and subsequent release allows these redox-responsive coacervates to deliver a single or combination of macromolecular therapeutics, making this intracellular delivery platform a promising candidate for the treatment of a variety of human pathologies such as cancer and metabolic and infectious diseases. Another useful feature of HBpep-SR coacervates is their ability to inhibit RNase-induced premature degradation of mRNA, probably because RNase cannot diffuse through the microdroplets once the latter are formed. It is noteworthy that our approach does not involve either endosomal escape or cell membrane fusion (the two main mechanisms of intracellular delivery8) and that the coacervates are micrometre-sized carriers as opposed to the nanocarriers used in the vast majority of current intracellular delivery strategies. Presumably, the liquid-like properties of coacervates achieved via LLPS are critical to their ability to cross the cell membrane, resulting in cholesterol-dependent uptake, although the precise entry mechanism is still unclear and is currently under investigation.
The physicochemical strategy behind cargo recruitment and subsequent intracellular release combines LLPS of a pH-responsive peptide with self-immolative disulfide chemistry. Self-immolative linkers are promising molecular tools to conjugate thiol-free drugs to help trigger their intracellular release28. The present study expands this chemistry as a way to finely tune the phase behaviour of peptide coacervates and should be applicable to other types of phase-separating peptide that have shown translational potential in recent years, such as elastin-like polypeptides.
Future work will aim at elucidating the uptake and membrane trafficking pathways of the peptide microdroplets as well as evaluating their in vivo safety, efficacy and tissue distribution. We also envision that microdroplets disassembly and therapeutics release could be induced via other physicochemical triggers, which could further broaden their translational potential for intracellular drug delivery applications.
Methods
Materials
HBpep peptide, resins and Fmoc-protected amino acids used in solid-phase peptide synthesis were purchased from GL Biochem. N-hydroxysuccinimide, tetrahydrofuran, triphosgene, sodium azide, triphosgene and benzoic acid were purchased from Tokyo Chemical Industry (TCI). N,N′-diisopropylcarbodiimide, acetic acid, 2-hydroxyethyl disulfide, N,N-diisopropylethylamine, piperidine, trifluoroacetic acid, triisopropylsilane, 2,4,6-trinitrobenzenesulfonic acid, glutathione, bovine serum albumin, lysozyme, saporin, β-galactosidase, R-phycoerythrin, methylthiazolyldiphenyl-tetrazolium bromide, Hoechst 33342, methyl-β-cyclodextrin, chlorpromazine hydrochloride, amiloride chloride, monoclonal anti-β-actin-peroxidase antibody l-buthionine-sulfoximine, reduced l-glutathione (GSH) and the Pur-A-Lyzer Maxi Dialysis Kit Maxi 50000 were obtained from Sigma-Aldrich. Dichloromethane, N,N-dimethylformamide, LysoTracker Red DND-99, Opti-MEM, RNase A, Pierce protein transfection reagent (Pro-Ject), Ni-NTA His Bind resin and 5-bromo-4-chloro-3-indolyl β-d-galactopyranoside were purchased from Thermo Fisher Scientific. Xfect protein transfection reagent was purchased from Takara Bio. Organic solvents, including ethyl acetate, hexane and diethyl ether were purchased from Aik Moh Paints & Chemicals Pte Ltd. Dulbecco’s modified Eagle medium (DMEM), fetal bovine serum (FBS), phosphate-buffered saline (PBS) and antibiotic-antimycotic (100X) liquid were purchased from Gibco. The Nano-Glo Dual-Luciferase kit used for luciferase detection and the CytoTox 96 non-radioactive cytotoxicity assay kit were purchased from Promega. HyClone McCoy’s 5A medium was purchased from Cytiva. Trans-Blot Turbo 0.2-μm nitrocellulose transfer packs, 4–20% Criterion TGX stain-free protein gel and Clarity and Clarity Max Western ECL substrate were purchased from Bio-Rad. Mouse monoclonal p21 (F-5) and p53 (DO-1) horseradish peroxidase (HRP) antibodies were purchased from Santa Cruz Biotechnology. Polyclonal rabbit anti-mouse immunoglobulin HRP was purchased from Dako. EGFP was expressed in Escherichia coli BL21 strain and purified with Ni-NTA His Bind resin. Luciferase-encoding mRNA and EGFP-encoding mRNA used for mRNA transfection experiments were obtained from Trilink. HepG2, HEK293, A549, NIH 3T3, H1299 and HCT116 cell lines were obtained from ATCC. T22 and ARN8 cell lines were established by following previous works55,56.
Self-immolative moiety synthesis
The self-immolative moieties conjugated to HBpep-K peptide were designed based on the literature29 (the synthesis routes for the amine-reactive species are shown in Supplementary Fig. 8). First, for the synthesis of the side-blocked intermediate product, HO-SS-R, 2-hydroxyethyl disulfide (1 equiv., 10 mmol) was dissolved in 15 ml of tetrahydrofuran (THF), then another 15 ml of THF containing a carboxylic acid reactant such as acetic acid or benzoic acid (0.9 equiv., 9 mmol) was added. Then, under an ice bath, 15 mmol of N,N′-diisopropylcarbodiimide (DIC) was slowly added into the reaction mixture. The reaction was kept at 0 °C for another 0.5 h and then increased to room temperature. After overnight reaction, the mixture was filtered and the supernatant evaporated under reduced pressure. The raw products were purified using silica gel chromatography with ethyl acetate/hexane (1/4) as elute. The purified products were isolated by rotary evaporation (R-215 Rotavapor, BUCHI).
Intermediate products HO-SS-R and N-hydroxysuccinimide (NHS) were coupled using triphosgene. Specifically, HO-SS-R (1 equiv., 5 mmol) and 4-dimethylaminopyridine (DMAP, 0.1 equiv., 0.5 mmol) were dissolved in 10 ml of THF. Triphosgene (0.37 equiv., 1.85 mmol) in 10 ml of THF was added into the previous solution dropwise under an ice bath. After another 0.5 h on the ice bath, the reactions were continued at 40 °C for 4 h, followed by evaporation under reduced pressure to remove excess phosgene. NHS (1.5 equiv., 7.5 mmol) in 20 ml of THF, and N,N-diisopropylethylamine (DIPEA, 1.5 equiv., 7.5 mmol) were then pipetted into the mixtures. The reactions were kept at 40 °C for 24 h before evaporation. The raw products were purified using silica gel chromatography with ethyl acetate/hexane (1/3) as elute. The purified products were isolated by rotary evaporation. The amine-reactive products NHS-SS-Ac and NHS-SS-Ph were synthesized from acetic acid and benzoic acid. The chemical structures of the HO-SS-R and NHS-SS-R were verified by 1H NMR spectroscopy, as shown in Supplementary Fig. 9. The NMR spectra were collected on a Bruker Advance 400 spectrometer.
Peptide synthesis and purification
The peptides used in this study were synthesized by the classical Merrifield solid-phase peptide synthesis (SPPS) technique57. Wang resin (1.0 g, 0.56 mmol) was first swollen in 15 ml of dichloromethane (DCM) for 0.5 h with bubbling nitrogen flow. The DCM was drained with increased pressure, and the resin was washed three times with N,N-dimethylformamide (DMF).
For N-terminal protected amino acid (Fmoc-AA-OH) coupling, Fmoc-AA-OH (2 equiv., 1.12 mmol) was dissolved in 5 ml of DMF, then 5 ml of DMF with 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU, 1.9 equiv., 1.064 mmol) and DIPEA (5 equiv., 2.80 mmol) was added into the solution. The mixture was reacted for 2 min at room temperature before being added onto the resin for 1 h of coupling reaction with bubbling nitrogen flow. The resin was washed with DCM and then DMF three times each after the coupling reaction. The coupling efficiency was evaluated using 2,4,6-trinitrobenzenesulfonic acid (TNBS).
For deprotection of the N-terminal amine, 15 ml of 20% piperidine in DMF (volume ratio) was added onto the resin. The deprotection continued for 0.5 h at room temperature with bubbling nitrogen flow, then the resin was washed with DCM then DMF three times, and the deprotection efficiency was evaluated using TNBS.
After all amino acids in the peptide sequence were coupled onto the resin by performing coupling/deprotection cycles in the C-terminal to N-terminal direction, peptides were cleaved from the resins using a cocktail containing 95% of trifluoroacetic acid (TFA), 2.5% of H2O and 2.5% of triisopropylsilane (TIPS). After 2 h of cleavage, the reaction mixtures were filtered. The supernatants were concentrated using a nitrogen flow and precipitated into 50 ml of cold diethyl ether. After centrifugation, the pellets were dried under vacuum and re-dissolved using 90% of 10 mM acetic acid and 10% acetonitrile for purification by HPLC (1260 Infinity, Agilent Technologies) equipped with a C8 column (Zorbax 300SB-C8, Agilent Technologies). The purified peptides were isolated by lyophilization (FreeZone 4.5 Plus, Labconco) from HPLC elutes.
Peptide modification
The redox-responsive peptides were synthesized by reacting the ε-amine of the single Lys residue of the N-terminal protected peptide (Fmoc-HBpep-K, Fmoc-GHGVY-GHGVY-GHGPY-K-GHGPY-GHGLYW) with the amine-reactive species NHS-SS-R, followed by deprotection. First, the Fmoc-HBpep-K peptide (1 equiv., 15 μmol) was dissolved in 5 ml of DMF containing DIPEA (15 equiv., 225 μmol). After 30 min of deprotonation, NHS-SS-R (1.5 equiv., 22.5 μmol) in 0.5 ml of DMF was added into the solution. The mixture solutions were allowed to react at room temperature for 24 h before precipitation by adding 50 ml of cold diethyl ether. The raw products were collected from the pellets by centrifugation and dried under reduced pressure. The purification of modified peptides was conducted on an HPLC system equipped with a C8 column. The purified Fmoc-protected peptides were isolated by lyophilization from the HPLC fractions.
The purified Fmoc-protected peptides were dissolved in 5 ml of DMF containing 20% piperidine. The mixture was stirred at room temperature for 2 h of N-terminal deprotection. The raw products were collected from the precipitates after adding 50 ml of cold diethyl ether into the reaction mixtures and purified by HPLC. The final products were isolated by lyophilization as white solids. Two modified peptides were synthesized, HBpep-SA from NHS-SS-Ac and HBpep-SP from NHS-SS-Ph. The molecular weights of Fmoc-HBpep-K and the modified peptides were verified by MALDI-TOF mass spectrometry using α-cyano-4-hydroxycinnamic acid (CHCA) as the matrix (Supplementary Fig. 10). Both molecular weights were consistent with the expected molecular weights of the peptides. The MALDI-TOF spectra were collected on an AXIMA Performance spectrometer (Shimadzu Corporation). The modified peptides HBpep-SA and HBpep-SP were dissolved in 10 mM acetic acid solution at 10 mg ml−1 as stock solution.
Coacervation of peptide and therapeutic recruitment
The phase separation behaviour of the HBpep-K and HBpep-SR peptides at various values of pH was monitored by turbidity measurements using a UV–vis spectrometer (UV-2501PC, Shimadzu). The absorbance at 600 nm (A600) was used to calculate the relative turbidity as22
The recruitment of the macromolecules within the peptide coacervates was conducted during the coacervation process at the optimal pH (7.5 for HBpep and 6.5 for HBpep-SR). The therapeutics were dissolved or diluted in 10 mM phosphate buffers (pH 7.5 or 6.5, ionic strength of 100 mM) to achieve the target concentrations. The peptide stock solutions were then mixed with the therapeutics containing the buffer at a 1:9 volume ratio to induce coacervation and recruitment of the therapeutics. As shown in Extended Data Fig. 6, the recruitment efficiency of HBpep-SP coacervates was calculated by comparing the supernatant fluorescence in the buffer solution before and after coacervation and centrifugation using microplate reader (Infinite M200 Pro, Tecan). The fluorescence of EGFP (or FITC), R-PE and Cy5 was detected using 488 nm/519 nm, 532 nm/584 nm and 640 nm/680 nm for the excitation/emission wavelengths, respectively.
Confocal microscopy of EGFP delivery mediated by HBpep coacervates
For a three-dimensional (3D) view of coacervate-treated cells, cells treated overnight with EGFP-loaded coacervates (0.01 mg ml−1 of EGFP, 0.2 mg ml−1 of HBpep) were first stained with a plasma membrane stain and fixed before image acquisition. Briefly, cells were rinsed with HBSS buffer and stained with either 1× CellTracker CM-DiI (C7000; Thermo Fisher) for 5 min at 37 °C followed by 15 min at 4 °C or 1× CellMask Deep Red Plasma membrane stain (Thermo Fisher) for 10 min at 37 °C. Cells stained with membrane dyes were rinsed once with PBS and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. After fixing, the cells were washed three times with PBS and finally resuspended in PBS. Confocal Z-stack images were collected on an Olympus FV1000 inverted scanning confocal microscope using a ×40 oil immersion objective (NA 1.3). The Z-stacks were reconstructed into 3D images or animations with the aid of Imaris software 3D View and Animation modes.
For live-cell imaging, T22 cells treated with EGFP-loaded DgHBP-2 coacervates were split and seeded on days 3 and 7 to achieve 50–60% confluency, and images were acquired after cells adhered ~4 h after seeding. Z-stack images (differential interference contrast or fluorescence) were acquired on a Nikon Eclipse Ti inverted fluorescence microscope, using a ×40 oil immersion objective (NA 1.3) and sum slices projection was applied to all the stack images using ImageJ software.
Characterization of redox-responsive peptide coacervates
Optical and fluorescence microscopy images of HBpep-SP coacervates and fluorescence images of macromolecules-loaded HBpep-SP coacervates were taken using an inverted fluorescence microscope (AxioObserver.Z1, Zeiss). A dynamic light scattering (DLS, ZetaPALS) system was used to measure the size and zeta potential of pristine HBpep-SR coacervates and macromolecules-loaded HBpep-SR coacervates. The freshly prepared pristine or macromolecules-loaded coacervates (with or without 0.1 mg ml−1 macromolecules, 1 mg ml−1 modified peptides) were diluted into PBS or PBS containing various percentages of FBS with a volume ratio of 1:9 before the DLS test. The redox-responsivities of HBpep-SA and HBpep-SP were evaluated by measuring the decrease in concentration in the presence of GSH. The freshly prepared HBpep-SA or HBpep-SP coacervates (50 μl, 1 mg ml−1 peptide) were diluted in 450 μl of PBS containing 1 mM of GSH. The mixtures were incubated at 37 °C before adding 25 μl of acetic acid to dissolve all the unreacted peptides, and their concentrations were measured by HPLC. The HBpep-SP coacervates incubated in PBS containing various concentrations of GSH (0, 1 and 10 mM) at 37 °C for 24 h were injected into an HPLC system equipped with a C8 column. Fractions were collected and measured by MALDI-TOF for their molecular weights using CHCA as matrix. The redox-triggered EGFP release was conducted using a dialysis tube (molecular weight cutoff of 50 kDa). The freshly prepared EGFP-loaded HBpep-SP coacervates (200 μl, 1 mg ml−1 HBpep-SP, 0.1 mg ml−1 EGFP) were diluted in 1.8 ml of PBS (pH 7.4, ionic strength of 0.15 M) and dialysed against 20 ml of PBS (pH 7.4, ionic strength of 0.15 M) containing various concentrations of GSH (0, 0.1 and 1 mM). A 0.3 ml sample was collected from each group and replaced with 0.3 ml of fresh PBS every 2 h to measure the release of EGFP by using a microplate reader using 488 nm/519 nm for the excitation/emission wavelengths.
Delivery of proteins and peptides
For protein delivery into cells, 1 × 105 cells were suspended in 1 ml of DMEM supplemented with 10% FBS, 100 U ml−1 of penicillin and 100 μg ml−1 of streptomycin, and then transferred into 35 cm2 culture dishes. After 24 h of incubation at 37 °C with 5% CO2, the medium was replaced with 900 μl of Opti-MEM, then 100 μl of freshly prepared peptide or protein-loaded HBpep-SR coacervate suspensions (0.05 mg ml−1 peptide or 0.1 mg ml−1 protein, 1 mg ml−1 HBpep-SR) were prepared by adding the HBpep-SR peptide stocks into cargos containing buffer, then added into the medium. After 4 h of incubation, the coacervates-containing medium was removed and the cells were washed with PBS twice before adding 1 ml of fresh medium (DMEM, 10% FBS, antibiotics). The cells were incubated for another 20 h and then washed twice at pH 5.0 in phosphate buffer to remove any coacervates that had not entered the cells. The release of proteins was determined by the distribution of fluorescence signals inside the cells using fluorescence microscopy (AxioObserver.Z1, Zeiss) or confocal microscopy (LSM 780, Zeiss). For comparison with commercially available protein transfection reagents, Pro-Ject (Thermo Fisher Scientific) and Xfect (Takara Bio) were used according to protocols from the manufacturers to evaluate the delivery efficiency. The EGFP- and R-PE-transfected cells were imaged under a fluorescence microscope (AxioObserver.Z1, Zeiss) and analysed by FACS (LSR Fortessa X20, BD Biosciences).
Delivery of p53-activating peptides
HCT116 p53(+/+) cells suspended in HyClone McCoy’s 5A medium supplemented with 10% FBS, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin were seeded at a density of 8.0 × 104 cells per well in 24-well culture dishes. After 24 h of incubation at 37 °C with 5% CO2, the medium was replaced with 450 μl of Opti-MEM, then 50 μl of freshly prepared free peptide (32.81 μM), peptide-loaded HBpep-SP coacervate suspensions (32.81 μM cargo peptide, 1 mg ml−1 HBpep-SP) or HPpep-SP-only coacervate suspensions (1 mg ml−1) was added into the medium. After 4 h of incubation, the medium was removed and replaced with 0.5 ml of fresh medium. For Nutlin-3a treatment, 10 μM Nutlin-3a was added to the medium. After an additional 20 h of incubation, 50 μl of medium was removed for lactate dehydrogenase release assay and cells collected for western blots. Western blots were performed by loading equal amounts of total protein (30 μg) from whole-cell lysates on 4–20% Criterion TGX stain-free protein gel (Bio-Rad). Proteins were transferred to nitrocellulose membranes and blotted with p53 (D0-1) HRP, anti-β-actin-peroxidase or p21(F-5) antibody followed with anti-mouse immunoglobulin HRP antibodies. Immunoblots were developed using Clarity Western ECL substrate or Clarity Max Western ECL substrate for weaker signals and detected with a Bio-Rad ChemiDoc imaging system. For the LDH release assay, 50 μl of culture medium from treated cells was transferred to 96-well microplates and assayed using the CytoTox 96 Non-Radioactive Cytotoxicity Assay kit. Lysis solution was added to lytic control wells for maximum LDH release 45 min before the culture medium was transferred for the assay.
EGFP delivery into the cell with GSH depletion
To verify that the cargo release of HBpep-SR coacervates was triggered by the endogenous reducing agent GSH, HepG2 cells were pretreated with 0.5 mM l-buthionine-sulfoximine (BSO) for 12 h (ref. 31). The medium was then replaced with 900 μl of Opti-MEM and 100 μl of freshly prepared EGFP-loaded HBpep-SP coacervate suspensions (0.1 mg ml−1 EGFP, 1 mg ml−1 HBpep-SP). After 4 h of uptake, the medium was removed and the cells were washed with PBS twice before adding 1 ml of BSO containing full medium (DMEM, 10% FBS, antibiotics, 0.5 mM BSO). The cells were incubated for another 20 h, then washed twice at pH 5.0 in phosphate buffer to remove any coacervates that had not entered the cells, before being imaged under the fluorescence microscope (AxioObserver.Z1, Zeiss).
Delivery and transfection of mRNA
Two reporter genes including luciferase and EGFP were used to evaluate the mRNA transfection efficiency of the HBpep-SR coacervates. Before transfection, HepG2 or HEK293 cells were incubated in 96-well plates with a density of 1 × 104 cells per well for 24 h. The medium was replaced with 90 μl of Opti-MEM, followed by the addition of 10 μl of freshly prepared mRNA-loaded coacervate suspensions (1 or 2 mg ml−1 of modified peptides). The final concentration of luciferase-encoding mRNA used in the transfection was 3.3 μg ml−1. After 4 h of incubation, the medium was removed and the cells were washed with PBS twice before adding 100 μl of medium (DMEM, 10% FBS, antibiotics). Transfection was then continued for another 20 h before testing the luminescence using the Nano-Glo Dual-Luciferase kit and a microplate reader. For EGFP-encoding mRNA (Cy5-labelled) transfection, the cultures were conducted in 35-cm2 dishes into which 100 μl of mRNA-loaded HBpep-SP coacervates (1 mg ml−1 HBpep-SP) was added to achieve the final mRNA concentration of 1 μg ml−1. The transfection was conducted for 4 h of uptake and 20 h of expression before imaging the cells under a fluorescence microscope and testing the transfection efficiency by FACS (LSR Fortessa X20, BD Biosciences).
Protection of mRNA from RNase A
To test whether the coacervates could protect recruited mRNA from enzymatic degradation, 10 μl of the freshly prepared mRNA (luciferase)-loaded HBpep-SP coacervate suspension (1 mg ml−1 of HBpep-SP, 0.1 μg ml−1 mRNA) was diluted into 30 μl of PBS before adding 4 μl of RNase A (10 mg ml−1). The mixture was incubated at 37 °C for 2 h, then 2 μl of 2-mercaptoethanol was added into the mixture and the temperature was raised to 70 °C for 30 min to deactivate the RNase A and release mRNA from the coacervates. Two control groups, including untreated mRNA and free mRNA treated with RNase A, were also used. The integrity of the mRNA in the three groups was determined by 1% agarose gel electrophoresis.
Cytotoxicity study
The cytotoxicity of the therapeutics-loaded or pristine peptide coacervates was evaluated using the methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay. Following literature protocols58, 1 × 104 HepG2 or HEK293 cells in 100 μl of medium (DMEM, 10% FBS, antibiotics) were transferred into 96-well plates and incubated for 24 h. The medium was then replaced with 100 μl of Opti-MEM containing therapeutics-loaded coacervates (various concentration of therapeutics, 0.1 mg ml−1 HBpep-SP) or various concentrations of pristine coacervate suspensions. After 4 h of uptake, the medium was removed and the cells were washed by PBS twice before adding 100 μl of medium (DMEM, 10% FBS, antibiotics). The cells were incubated for another 20 h before 10 μl of 5 mg ml−1 MTT dissolved in PBS was added. The medium was removed after 4 h of incubation with MTT, and the cells were washed by PBS twice. Next, 100 μl of DMSO was added per well for absorbance measurements at 570 nm using a microplate reader (Infinite M200 Pro, Tecan). The relative cell viability was calculated as
where At, Ab and Ac represent the absorbance of tested cells, no cells and untreated cells, respectively.
Internalization mechanism study
LysoTracker staining was conducted by following the manual from the manufacturer. Similar to protein delivery, 1 × 105 of HepG2 cells were incubated in 35-cm2 dishes with DMEM for 24 h, then the medium was replaced with 900 μl of Opti-MEM and 100 μl of EGFP-loaded or AF-BSA-loaded HBpep-SP coacervates (0.1 mg ml−1 EGFP, 1 mg ml−1 HBpep-SP). The cells were cultured for another 2 h before being washed twice with phosphate buffer (pH 5.0) to remove any coacervates that had not entered the cells. After that, 1 ml of Opti-MEM containing 50 nM LysoTracker was added for 30 min of staining under cell culture conditions. The treated HepG2 cells were washed by PBS twice and fixed with 4% formaldehyde solution. Before being imaged by confocal microscopy (LSM 780, Zeiss), the cells were treated with 1 μg ml−1 of Hoechst 33342 for 10 min to stain the nucleus.
Based on the literature13,50,51, various inhibitors were used to study the pathway of coacervates internalization. HepG2 cells were treated with chlorpromazine (CPM, 30 μM), amiloride chloride (AM, 20 μM), sodium azide (NaN3, 100 mM) or methyl-β-cyclodextrin (MβCD, 2.5 mM) separately for 1 h, then 100 μl of EGFP-loaded HBpep-SP coacervates (0.1 mg ml−1 EGFP, 1 mg ml−1 HBpep-SP) was added. After another 4 h of incubation, the cells were washed twice with pH 5.0 phosphate buffer followed by PBS twice. The treated cells were imaged by fluorescence microscopy or dissociated by trypsin for FACS. For the 4 °C treated group, the HepG2 cells were pre-incubated for 1 h and kept at low temperature during the 4 h uptake process. Two control groups—totally untreated cells (negative control, NC) and cells treated by EGFP-loaded coacervates without any inhibitors (blank)—were also examined.
Statistics and reproducibility
All experiments were repeated three times. The data are presented as mean ± standard deviation (s.d.). Statistical significance (P < 0.01) was evaluated using a two-sided Student’s t-test when only two groups were compared. All microscopy experiments were repeated independently three times and showed no differences.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All relevant data supporting the findings of this study are available within the paper, the Extended Data files and the Supplementary Information. Source data are provided with this paper. Data are available from the corresponding author on request.
References
Zou, P. et al. Recent advances: peptides and self-assembled peptide-nanosystems for antimicrobial therapy and diagnosis. Biomater. Sci. 8, 4975–4996 (2020).
Fu, A., Tang, R., Hardie, J., Farkas, M. E. & Rotello, V. M. Promises and pitfalls of intracellular delivery of proteins. Bioconjug. Chem. 25, 1602–1608 (2014).
Nelson, A. L., Dhimolea, E. & Reichert, J. M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 9, 767–774 (2010).
Dowdy, S. F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 35, 222–229 (2017).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403–416 (2021).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Du, S., Liew, S. S., Li, L. & Yao, S. Q. Bypassing endocytosis: direct cytosolic delivery of proteins. J. Am. Chem. Soc. 140, 15986–15996 (2018).
Goswami, R., Jeon, T., Nagaraj, H., Zhai, S. & Rotello, V. M. Accessing intracellular targets through nanocarrier-mediated cytosolic protein delivery. Trends Pharmacol. Sci. 41, 743–754 (2020).
Li, M. et al. Discovery and characterization of a peptide that enhances endosomal escape of delivered proteins in vitro and in vivo. J. Am. Chem. Soc. 137, 14084–14093 (2015).
Ping, Y. et al. Supramolecular β-sheets stabilized protein nanocarriers for drug delivery and gene transfection. ACS Nano 11, 4528–4541 (2017).
Scaletti, F. et al. Protein delivery into cells using inorganic nanoparticle-protein supramolecular assemblies. Chem. Soc. Rev. 47, 3421–3432 (2018).
Liu, C. et al. A boronic acid-rich dendrimer with robust and unprecedented efficiency for cytosolic protein delivery and CRISPR-Cas9 gene editing. Sci. Adv. 5, eaaw8922 (2019).
Mout, R. et al. Direct cytosolic delivery of CRISPR/Cas9-ribonucleoprotein for efficient gene editing. ACS Nano 11, 2452–2458 (2017).
Lee, S. et al. Transmembrane epitope delivery by passive protein threading through the pores of the OmpF porin trimer. J. Am. Chem. Soc. 142, 12157–12166 (2020).
Akishiba, M. et al. Cytosolic antibody delivery by lipid-sensitive endosomolytic peptide. Nat. Chem. 9, 751–761 (2017).
Hu, Y., Mignani, S., Majoral, J.-P., Shen, M. & Shi, X. Construction of iron oxide nanoparticle-based hybrid platforms for tumor imaging and therapy. Chem. Soc. Rev. 47, 1874–1900 (2018).
Buse, J. & El-Aneed, A. Properties, engineering and applications of lipid-based nanoparticle drug-delivery systems: current research and advances. Nanomedicine 5, 1237–1260 (2010).
Yang, J., Tu, J., Lamers, G. E. M., Olsthoorn, R. C. L. & Kros, A. Membrane fusion mediated intracellular delivery of lipid bilayer coated mesoporous silica nanoparticles. Adv. Healthcare Mater. 6, 1700759 (2017).
Tai, W., Zhao, P. & Gao, X. Cytosolic delivery of proteins by cholesterol tagging. Sci. Adv. 6, eabb0310 (2020).
Khlebtsov, N. & Dykman, L. Biodistribution and toxicity of engineered gold nanoparticles: a review of in vitro and in vivo studies. Chem. Soc. Rev. 40, 1647–1671 (2011).
Fadeel, B. & Garcia-Bennett, A. E. Better safe than sorry: understanding the toxicological properties of inorganic nanoparticles manufactured for biomedical applications. Adv. Drug Deliv. Rev. 62, 362–374 (2010).
Lim, Z. W., Ping, Y. & Miserez, A. Glucose-responsive peptide coacervates with high encapsulation efficiency for controlled release of insulin. Bioconjug. Chem. 29, 2176–2180 (2018).
Lim, Z. W., Varma, V. B., Ramanujan, R. V. & Miserez, A. Magnetically responsive peptide coacervates for dual hyperthermia and chemotherapy treatments of liver cancer. Acta Biomater. 110, 221–230 (2020).
Blocher McTigue, W. C. & Perry, S. L. Protein encapsulation using complex coacervates: what Nature has to teach us. Small 16, 1907671 (2020).
Blocher, W. C. & Perry, S. L. Complex coacervate-based materials for biomedicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 9, e1442 (2017).
Gabryelczyk, B. et al. Hydrogen bond guidance and aromatic stacking drive liquid–liquid phase separation of intrinsically disordered histidine-rich peptides. Nat. Commun. 10, 5465 (2019).
Lampel, A. Biology-inspired supramolecular peptide systems. Chem 6, 1222–1236 (2020).
Riber, C. F., Smith, A. A. A. & Zelikin, A. N. Self-immolative linkers literally bridge disulfide chemistry and the realm of thiol-free drugs. Adv. Healthcare Mater. 4, 1887–1890 (2015).
Tang, L. et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol. 36, 707–716 (2018).
Zhou, Z. et al. GSH depletion liposome adjuvant for augmenting the photothermal immunotherapy of breast cancer. Sci. Adv. 6, eabc4373 (2020).
Wang, Y.-M. et al. Depletion of intracellular glutathione mediates butenolide-induced cytotoxicity in HepG2 cells. Toxicol. Lett. 164, 231–238 (2006).
Lv, J. et al. Fluoropolymers for intracellular and in vivo protein delivery. Biomaterials 182, 167–175 (2018).
Wang, M., Alberti, K., Sun, S., Arellano, C. L. & Xu, Q. Combinatorially designed lipid-like nanoparticles for intracellular delivery of cytotoxic protein for cancer therapy. Angew. Chem. Int. Ed. 53, 2893–2898 (2014).
Mitragotri, S., Burke, P. A. & Langer, R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat. Rev. Drug Discov. 13, 655–672 (2014).
Fosgerau, K. & Hoffmann, T. Peptide therapeutics: current status and future directions. Drug Discov. Today 20, 122–128 (2015).
Li, M. et al. Smac therapeutic peptide nanoparticles inducing apoptosis of cancer cells for combination chemotherapy with doxorubicin. ACS Appl. Mater. Interfaces 7, 8005–8012 (2015).
Toyama, K., Nomura, W., Kobayakawa, T. & Tamamura, H. Delivery of a proapoptotic peptide to EGFR-positive cancer cells by a cyclic peptide mimicking the dimerization arm structure of EGFR. Bioconjug. Chem. 29, 2050–2057 (2018).
Zhan, C. et al. An ultrahigh affinity d-peptide antagonist of MDM2. J. Med. Chem. 55, 6237–6241 (2012).
Partridge, A. W. et al. Incorporation of putative helix-breaking amino acids in the design of novel stapled peptides: exploring biophysical and cellular permeability properties. Molecules 24, 2292 (2019).
Peier, A. et al. NanoClick: a high throughput, target-agnostic peptide cell permeability assay. ACS Chem. Biol. 16, 293–309 (2021).
Momand, J., Zambetti, G. P., Olson, D. C., George, D. & Levine, A. J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69, 1237–1245 (1992).
Vassilev, L. T. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 (2004).
Naldini, L. Gene therapy returns to centre stage. Nature 526, 351–360 (2015).
Pardi, N. et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543, 248–251 (2017).
Pardi, N. et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 8, 14630 (2017).
Liu, C., Zhang, L., Liu, H. & Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control. Release 266, 17–26 (2017).
Yoshinaga, N. et al. Bundling mRNA strands to prepare nano-assemblies with enhanced stability towards RNase for in vivo delivery. Angew. Chem. Int. Ed. 58, 11360–11363 (2019).
Panja, P. & Jana, N. R. Lipid-raft-mediated direct cytosolic delivery of polymer-coated soft nanoparticles. The. J. Phys. Chem. B 124, 5323–5333 (2020).
Sangsuwan, R., Tachachartvanich, P. & Francis, M. B. Cytosolic delivery of proteins using amphiphilic polymers with 2-pyridinecarboxaldehyde groups for site-selective attachment. J. Am. Chem. Soc. 141, 2376–2383 (2019).
Lin, Q. et al. Imaging the cytosolic drug delivery mechanism of HDL-like nanoparticles. Pharm. Res. 31, 1438–1449 (2014).
Xu, C. et al. Self-assembled nanoparticles from hyaluronic acid-paclitaxel prodrugs for direct cytosolic delivery and enhanced antitumor activity. Int. J. Pharm. 493, 172–181 (2015).
Mayor, S. & Pagano, R. E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 8, 603–612 (2007).
Rennick, J. J., Johnston, A. P. R. & Parton, R. G. Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat. Nanotechnol. 16, 266–276 (2021).
Murata, N. & Los, D. A. Membrane fluidity and temperature perception. Plant Physiol. 115, 875–879 (1997).
Lu, X. et al. Discordance between accumulated p53 protein level and its transcriptional activity in response to UV radiation. Oncogene 13, 413–418 (1996).
Blaydes, J. P. & Hupp, T. R. DNA damage triggers DRB-resistant phosphorylation of human p53 at the CK2 site. Oncogene 17, 1045–1052 (1998).
Merrifield, R. B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 85, 2149–2154 (1963).
Chang, H. et al. Rational design of a polymer with robust efficacy for intracellular protein and peptide delivery. Nano Lett. 17, 1678–1684 (2017).
Acknowledgements
This research was funded by the Singapore Ministry of Education (MOE) through an Academic Research Fund (AcRF) Tier 3 grant (grant no. MOE 2019-T3-1-012). S.C.C. acknowledges support from the Merck Research Laboratories (MRL) Postdoctoral Research Program.
Author information
Authors and Affiliations
Contributions
Y.S. designed, performed and analysed experiments and wrote the manuscript. S.Y.L. and Z.W.L. designed and performed the preliminary study of EGFP delivery mediated by HBpep coacervates. S.Y.L. conducted the stapled peptide delivery study. S.C.C. helped with designing mRNA delivery and provided mRNA and Cy5-mRNA samples. F.G. suggested protein delivery studies mediated by coacervates, designed the stapled peptides delivery study and commented on the manuscript. A.P. suggested and helped with mRNA delivery and commented on the manuscript. A.M. designed the project, supervised the work and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
Y.S and A.M. have filed a PCT application on the peptide coacervates described in this study (PCT application no. PCT/SG2021/050309).
Peer review
Peer review information
Nature Chemistry thanks Yuan Ping and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Characterization of pristine and biomacromolecules-loaded HBpep-SP coacervates.
(a, b) Particle sizes (a) and distributions (b) of pristine, EGFP-loaded, Smac peptide-loaded and mRNA-loaded coacervates. Data are presented individually (yellow dots) and as the mean ± SD (column with error bar) of n = 3 independent measurements. (c) Zeta potentials of pristine, EGFP-loaded, Smac peptide loaded and mRNA-loaded coacervates. Data are presented individually (yellow dots) and as the mean ± SD (column with error bar) of n = 3 independent measurements. Fluorescence micrograph of EGFP-loaded HBpep-SP coacervates. (d-f) Fluorescence micrograph of EGFP-loaded (d), FITC-Smac-loaded (e) and Cy5-mRNA-loaded (f) HBpep-SP coacervates.
Extended Data Fig. 2 Serum stability of HBpep-SP coacervates.
(a) Size distributions and (b) zeta potentials of HBpep-SP coacervates in PBS containing various percentages of FBS. Data are presented individually (yellow dots) and as the mean ± SD (column with error bar) of n = 3 independent measurements.
Extended Data Fig. 3 Reduction of the self-immolative moiety of HBpep-SP.
(a) HPLC chromatograms of HBpep-SP coacervates incubated in PBS containing different concentrations of GSH. (b-d) MALDI-TOF of the products collected from HPLC. The measured MWs are consistent with the indicated chemical structures.
Extended Data Fig. 4 EGFP release from HBpep-SP coacervates triggered by GSH.
(a) Cumulative release of EGFP from coacervates in PBS buffer (pH = 7.4, ionic strength = 0.15 M) containing no (0 mM, PBS only), low (0.1 mM), and medium (1 mM) concentration of GSH. Data are presented as the mean ± SD of n = 3 independent measurements. (b-c) Fluorescence micrographs of normal HepG2 cells (b) and GSH-depleted HepG2 cells (c) treated with EGFP loaded coacervates.
Extended Data Fig. 5 Co-delivery of EGFP and R-PE by HBpep-SP coacervates.
(a) EGFP channel; (b) R-PE channel; and (c) merged micrographs of HepG2 cells treated with EGFP/R-PE co-loaded HBpep-SP coacervates for 24 hours.
Extended Data Fig. 6 Recruitment efficiency of biomacromolecules by HBpep-SP coacervates (1 mg/mL).
Cargos including proteins (EGFP, AF-lysozyme, AF-BSA and R-PE) (0.1 mg/mL), peptides (FITC-Smac and FITC-PAD) (0.1 mg/mL), and Cy5-mRNA (10 μg/mL). Data are presented individually (yellow dots) and as the mean ± SD (column with error bar) of n = 3 independent measurements.
Extended Data Fig. 7 Confocal images of HepG2 cells treated with cargo-loaded HBpep-SP coacervates.
Cargos including (a) FITC-Smac peptide; (b) EGFP; and (c) R-PE.
Extended Data Fig. 8 Cytotoxicity of empty redox-responsive HBpep-SR coacervates.
(a,b) Relative cell viability of HepG2 (a) and HEK293 (b) treated with HBpep-SA and HBpep-SP coacervates and comparison with commercial transfection reagents including PEI and lipofectamine 2000 and 3000. All experiments in the present study were conducted at a concentration of 0.1 mg/mL. Data are presented individually (yellow dots) and as the mean ± SD (column with error bar) of n = 3 independent measurements.
Extended Data Fig. 9 Gel electrophoresis of mRNAs showing protection from RNase A enzyme for mRNA recruited in HBpep-SP coacervates.
(C+R): mRNA recruited in HBpep-SP coacervates after exposure to RNase A for 2 hrs. (R): free mRNA after exposure to RNase A for 2 hrs. (NC): untreated free mRNA used as a negative control.
Extended Data Fig. 10 Confocal microscopy images of HepG2 cells treated with AF-BSA loaded HBpep-SP coacervates (green) for 2 hours.
The nucleus was stained with Hoechst (blue) and the lysosomes were stained with LysoTracker (red). Coacervates are not co-localized with lysosomes.
Supplementary information
Supplementary Information
Supplementary Tables 1 and 2 and Figs. 1–11.
Supplementary Video 1
3D reconstruction of scanning confocal microscopy of T22 cells treated with EGFP-loaded HBpep coacervates.
Supplementary Video 2
3D reconstruction of scanning confocal microscopy of H1299 cells treated with EGFP-loaded HBpep coacervates.
Source data
Source Data Fig. 2
Statistical and microscopy source data.
Source Data Fig. 3
Statistical and microscopy source data.
Source Data Fig. 4
Statistical and microscopy source data.
Source Data Fig. 5
Statistical and microscopy source data, unprocessed western blots.
Source Data Fig. 6
Statistical and microscopy source data.
Source Data Fig. 7
Statistical and microscopy source data.
Source Data Extended Data Fig. 1
Statistical and microscopy source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical and microscopy source data.
Source Data Extended Data Fig. 5
Statistical and microscopy source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Microscopy source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Unprocessed gel.
Source Data Extended Data Fig. 10
Microscopy source data.
Rights and permissions
About this article

Cite this article
Sun, Y., Lau, S.Y., Lim, Z.W. et al. Phase-separating peptides for direct cytosolic delivery and redox-activated release of macromolecular therapeutics. Nat. Chem. 14, 274–283 (2022). https://doi.org/10.1038/s41557-021-00854-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41557-021-00854-4
This article is cited by
-
Sequestration within peptide coacervates improves the fluorescence intensity, kinetics, and limits of detection of dye-based DNA biosensors
Communications Chemistry (2024)
-
Tuning the viscoelastic properties of peptide coacervates by single amino acid mutations and salt kosmotropicity
Communications Chemistry (2024)
-
Self-assembly of stabilized droplets from liquid–liquid phase separation for higher-order structures and functions
Communications Chemistry (2024)
-
Dipeptide coacervates as artificial membraneless organelles for bioorthogonal catalysis
Nature Communications (2024)
-
Polyelectrolyte Complexes and Coacervates Formed by De novo-Designed Peptides and Oligonucleotide
Chinese Journal of Polymer Science (2024)
