
Abstract
SARS-CoV-2 infection is controlled by the opening of the spike protein receptor binding domain (RBD), which transitions from a glycan-shielded ‘down’ to an exposed ‘up’ state to bind the human angiotensin-converting enzyme 2 receptor and infect cells. While snapshots of the ‘up’ and ‘down’ states have been obtained by cryo-electron microscopy and cryo-electron tomagraphy, details of the RBD-opening transition evade experimental characterization. Here over 130 µs of weighted ensemble simulations of the fully glycosylated spike ectodomain allow us to characterize more than 300 continuous, kinetically unbiased RBD-opening pathways. Together with ManifoldEM analysis of cryo-electron microscopy data and biolayer interferometry experiments, we reveal a gating role for the N-glycan at position N343, which facilitates RBD opening. Residues D405, R408 and D427 also participate. The atomic-level characterization of the glycosylated spike activation mechanism provided herein represents a landmark study for ensemble pathway simulations and offers a foundation for understanding the fundamental mechanisms of SARS-CoV-2 viral entry and infection.

Similar content being viewed by others


Main
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is an enveloped RNA virus and the causative agent of coronavirus disease 2019 (COVID-19), a disease that has caused substantial morbidity and mortality worldwide1,2. The main infection machinery of the virus, the spike protein that sits on the outside of the virus, is the first point of contact that the virion makes with the host cell, and is a major viral antigen3. A substantial number of cryo-electron microscopy (cryo-EM) structures of the spike protein have been recently reported, collectively informing on structural states of the spike protein. The vast majority of resolved structures fall into either ‘down’ or ‘up’ states, as defined by the position of the receptor binding domain (RBD), which modulates interaction with the angiotensin-converting enzyme 2 (ACE2) receptor for cell entry4,5,6.
The RBDs must transition from a ‘down’ to an ‘up’ state for the receptor binding motif (RBM) to be accessible for ACE2 binding (Fig. 1), and therefore the activation mechanism is essential for cell entry. Lu et al.7 used single-molecule fluorescence (Förster) resonance energy transfer (smFRET) imaging to characterize spike dynamics in real time. Their work showed that the spike dynamically visits four distinct conformational states, the populations of which are modulated by the presence of the human ACE2 receptor and antibodies. However, smFRET, as well as conventional structural biology techniques, are unable to inform on the atomic-level mechanisms underpinning such dynamical transitions. Recently, all-atom molecular dynamics (MD) simulations of the spike protein, with experimentally accurate glycosylation together with corroborating experiments, indicated the extensive shielding by spike glycans, as well as a mechanical role for glycans at positions N165 and N234 in supporting the RBD in the ‘open’ conformation8. Conventional MD simulations as performed in Casalino et al.8 also revealed microsecond-timescale dynamics to better characterize the spike dynamics but were limited to sampling configurations that were similar in energy to the cryo-EM structures. Several enhanced sampling MD simulations have been performed to study this pathway; however, these simulations lacked glycosylation for the spike protein9 or involved the addition of an external force10 or did not provide mechanistic detail11.

a, The SARS-CoV-2 spike head (grey) with glycans (dark blue) as simulated, with the stalk domain and membrane (not simulated here, but shown as transparent for completeness). RBD shown in cyan, RBM in pink. b,c, Side view of the RBDdown (shielded, b) and RBDopen (exposed, c). d,e, Top view of the RBMclosed (shielded, d) and RBMopen (exposed, e). Composite image of glycans (dark blue lines) shows many overlapping snapshots of the glycans over the microsecond simulations.
In this study, we characterized the spike RBD-opening pathway for the fully glycosylated SARS-CoV-2 spike protein to gain a detailed understanding of the activation mechanism. We used the weighted ensemble (WE) path-sampling strategy12,13 (Supplementary Fig. 1) to enable the simulation of atomistic pathways for the spike-opening process. As a path-sampling strategy, WE focuses computing power on the functional transitions between stable states rather than the stable states themselves14. This is achieved by running multiple trajectories in parallel and periodically replicating trajectories that have transitioned from previously visited to newly visited regions of configurational space15, thus minimizing the time spent waiting in the initial stable state for ‘lucky’ transitions over the free energy barrier. Given that these transitions are much faster than the waiting times16,17, the WE strategy can be orders of magnitude more efficient than conventional MD simulations in generating pathways for rare events such as protein folding and protein binding18,19. This efficiency is even higher for slower processes, increasing exponentially with the effective free energy barrier20. Not only are dynamics carried out without any biasing force or modifications to the free energy landscape, but suitable assignment of statistical weights to trajectories provides an unbiased characterization of the system’s time-dependent ensemble properties13. The WE strategy therefore generates continuous pathways with unbiased dynamics, yielding the most direct, atomistic views for analysing the mechanism of functional transitions, including elucidation of transient states that are too fleeting to be captured by laboratory experiments. Furthermore, while the strategy requires a progress coordinate towards the target state, the definition of this target state need not be fixed in advance when applied under equilibrium conditions21, enabling us to refine the definition of the target ‘open’ state of the spike protein on the basis of the probability distribution of protein conformations sampled by the simulation.
Our work characterizes a series of transition pathways of the spike opening, in agreement with conformations detected in the cryo-EM dataset by ManifoldEM22, and identifies key residues, including a glycan at position N343, that participate in the opening mechanism. Our simulation findings are corroborated by biolayer interferometry (BLI) experiments, which show a reduction in the ability of the spike to interact with ACE2 after mutation of these key residues.
Results and discussion
WE simulations of spike opening
As mentioned above, simulations of the spike-opening process require an enhanced sampling strategy as the process occurs beyond the microsecond timescale (that is, the seconds timescale7). We therefore used the WE path-sampling strategy, which enabled the generation of continuous, atomistic pathways for the spike-opening process with unbiased dynamics (Fig. 2a–e and Supplementary Video 1); these pathways were hundreds of nanoseconds long, excluding the waiting times in the initial ‘down’ state. The protein model was based on the head region (residues 16 to 1,140) of the glycosylated SARS-CoV-2 spike from Casalino et al.8 (Fig. 1), which in turn was built on the cryo-EM structure of the three-RBD-down spike (Protein Data Bank (PDB) ID, 6VXX (ref. 5)). The entire simulation system, including explicit water and salt ions, reaches almost half a million atoms. We focused sampling along a two-dimensional progress coordinate to track RBD opening: the difference in the centre of mass of the spike core to the RBD and the root-mean-square deviation of the RBD from the RBDup state (Fig. 2f,g). On the San Diego Supercomputer Center (SDSC) Comet and Texas Advanced Computing Center (TACC) Longhorn supercomputers, 100 graphics processing units (GPUs) ran the WE simulations in parallel for over a month, generating over 130 µs of glycosylated spike trajectories and more than 200 TB of trajectory data. We simulated a total of 310 independent pathways, including 204 pathways from the RBDdown conformation (PDB ID, 6VXX (ref. 5)) to the RBDup conformation (PDB ID, 6VSB (ref. 4)) and 106 pathways from the RBDdown to the RBDopen state, in which the RBD twists open beyond the 6VSB (ref. 4) cryo-EM structure. Remarkably, the RBDopen state that we sampled includes conformations that align closely with the ACE2-bound spike cryo-EM structure (PDB ID, 7A95 (ref. 6)) even though this structure was not a target state of our progress coordinate (Fig. 2f,g, Supplementary Video 1 and Supplementary Figs. 2 and 3). This result underscores the value of using (1) equilibrium WE simulations that do not require a fixed definition of the target state and (2) a two-dimensional progress coordinate that allows the simulations to sample unexpected conformational space along multiple degrees of freedom. The ACE2-bound spike conformation has also been sampled by the Folding@home-distributed computing project11, and RBD rotation has been detected in cryo-EM experiments6.

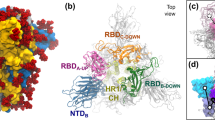
a–e, Snapshot configurations along the opening pathway with chain A shown in cyan, chain B in grey, chain C in pink and the glycan at position N343 in magenta. Each RBD and N-terminal domain (NTD) are subscripted with their chain ID (A, B or C). RBDs are also subscripted with their conformation from initial conformation with all three RBDs in the ‘down’ state (6VXX) (a), RBDA in a ‘transient’ state in between the ‘down’ and ‘up’ state (6VSB) (b) RBDA in the ‘up’ state (c), RBDA in the ‘open’ state (beyond 6VSB) (d) and RBDA in the furthest open state sampled (e). f, Scatter plot of data from the 310 continuous pathways with the Cα-root-mean-square deviation (RMSD) of the RBD from the RBDup state plotted against the RBD–core distance. Data points are coloured on the basis of the percentage RBD solvent-accessible surface area compared with the RBDdown state. The locations of the snapshots shown in a–e are labelled. g, Primary regions of spike defined for tracking progress of the opening transition. The spike core is composed of three central helices per trimer, coloured according to chains as in a–e. The RBD contains a structured pair of antiparallel beta-sheets, and an overlay of snapshots from a continuous WE simulation are shown coloured along a spectrum resembling the palette in f. Overlayed cryo-EM structures are highlighted and labelled including the initial RBDdown state (6VXX), the target RBDup state and the ACE2-bound RBDopen state (7A95).
Comparison with spike conformations detected by ManifoldEM
To validate our simulated RBDdown to RBDup pathway, the ManifoldEM framework22 was applied using the cryo-EM dataset of PDB 6VSB from McLellan and colleagues4. The ManifoldEM method allows characterization of conformational variations as obtained from a single-particle cryo-EM ensemble of a molecule in thermal equilibrium. Two conformational coordinates (that is, collective motion coordinates) CC1 and CC2 were discovered from this dataset, and observed from several exemplary projection directions (PDs) showing a (1) RBDdown to RBDup pathway and (2) RBD outward opening pathway (Supplementary Fig. 4 and Supplementary Videos 2 and 3).
These projections were next aligned to corresponding two-dimensional projections of coulomb potential maps generated with frames from the WE simulation (Supplementary Fig. 5 and Supplementary Videos 2 and 3). Overall, there was very good agreement between the ManifoldEM conformational coordinates and the WE trajectory, aside from two discrepancies. First, the CC2 observed in the ManifoldEM included concerted opening of all three RBDs, while the WE focused sampling on the opening of a single RBD (Supplementary Video 2). Second, the WE trajectory ultimately opens to an RBD–core distance 11 Å greater than the most open conformation in the ManifoldEM. This is probable because the simulations sample the S1 subunit en route to the postfusion conformation, whereas the experimental dataset does not.
The N343 glycan gates RBD opening
In the ‘down’ state, the RBD of the SARS-CoV-2 spike is shielded by glycans at positions N165, N234 and N343 (ref. 23). While glycan shielding had been investigated for the RBDdown and RBDup states8, our WE simulations allowed characterization of shielding during the opening process, revealing an abrupt decrease in glycan shielding when the RBD transitions from the ‘down’ to the ‘up’ state. The glycans at position N165 and N234 consistently shield the RBM, while shielding by the N343 glycan decreases with RBD opening (Supplementary Fig. 6). Beyond shielding, a structural role for glycans at positions N165 and N234 has been recently reported, stabilizing the RBD in the ‘up’ conformation through a ‘load and lock’ mechanism8.
Our WE simulations reveal an even more specific, critical role of a glycan in the opening mechanism of the spike: the N343 glycan acts as a ‘glycan gate’ pushing the RBD from the ‘down’ to the ‘up’ conformation by intercalating between residues F490, Y489, F456 and R457 of the ACE2 binding motif in a ‘hand-jive’ motion (Fig. 2a–e, 3 and Supplementary Video 4). Therefore, the N343 glycan plays an active role in initiating the transition, distinct from the stabilizing roles of glycans N165 and N234. This gating mechanism was initially visualized in several successful pathways of spike opening and then confirmed through analysis of all 310 successful pathways in which the N343 glycan was found to form contacts (within 3.5 Å) with each of the aforementioned residues in every successful pathway (Supplementary Fig. 7). The same mechanistic behaviour of the N343 glycan was observed in two fully independent WE simulations, suggesting the result is robust despite potentially incomplete sampling that can challenge WE and other enhanced sampling simulation methods15.

a–d, Snapshot configurations along the opening pathway with chain A shown in cyan, chain B in grey, chain C in pink and the glycan at position N343 in magenta. RBDA in the ‘down’ conformation is shielded by the glycan at position N343 of the adjacent RBDB (a). The N343 glycan intercalates between (b) and underneath (c) residues F490, Y489, F456 and F457 to push the RBD up and open (d).
To test the role of the N343 glycan as a key-gating residue, we performed BLI experiments. BLI experiments assess the binding level of the spike RBM (residues 438 to 508) to ACE2, acting as a proxy for the relative proportion of RBDs in the ‘up’ position for each spike variant. No residues directly involved in the binding were mutated (that is, at the RBM–ACE2 interface) to ensure controlled detection of the impact of RBD opening in response to mutations. Although previous results have shown reduced binding levels for N165A and N234A variants in the SARS-CoV-2 S-2P protein8, the N343A variant displayed an even greater decrease in ACE2 binding, reducing the spike binding level by ~56% (Fig. 4 and Supplementary Table 1). As a negative control, the S383C/D985C variant24, which is expected to be locked by disulfides into the three-RBD-down conformation, showed no association with the ACE2 receptor. These results support the hypothesis that the RBDup conformation is substantially affected by glycosylation at position N343.

a, BLI sensorgrams of HexaPro spike variants binding to ACE2. For clarity, only the traces from the first replicate are shown. b, Graph of the binding response for BLI data collected in triplicate with error bars representing the standard deviation from the mean.
Atomic details of the opening mechanism
The RBDdown state features a hydrogen bond between T415 of the RBDA and K986 of chain C, a salt bridge between R457 of RBDA and D364 of RBDB, and a salt bridge between K462 of RBDA and D198 of N-terminal domain C (NTDC) (Fig. 5a–c,e and Supplementary Fig. 8). The hydrogen bond T415A–K986C spends an average of 12% of the successful pathways to the ‘up’ state before K986C makes a short lived (2% average duration to the ‘up’ state) salt bridge with RBDA–D427. (Fig. 5b,e and Supplementary Fig. 8). Next, K986C forms salt bridges with E990C and E748C as the RBDA continues to open. These contacts are formed in all 310 successful pathways (Supplementary Fig. 8). Mutation of K986 to proline has been used to stabilize the prefusion spike25,26, including in vaccine development27, and these simulations provide molecular context to an additional role of this residue in RBD opening.

a–d, Salt-bridge or hydrogen-bond contacts made between RBDA, shown in blue, and RBDB, shown in grey, or RBDC, shown in pink, within the ‘down’ (a), ‘transient’ (b), ‘up’ (c) and ‘open’ (d) conformations. Nitrogen atoms are coloured blue and oxygen atoms are coloured red in the stick representation of amino acids to show hydrogen-bond and salt-bridge contacts. e, Histogram showing the frequency at which residues from a–d are within 3.5 Å of each other relative to the RBD–core distance. Frequencies are normalized to 1.
Subsequently, at an average of 16% of the way through the successful pathways to the ‘up’ state, the R457A–D364B salt bridge is broken, prompting the RBDA to twist upward, away from RBDB towards RBDC and forming a salt bridge between R408 of RBDA and D405 of RBDC (Fig. 5c,e and Supplementary Fig. 8). This salt bridge persists for 20% of the successful trajectories to the ‘up’ state and is present in all 310 successful pathways.
A salt bridge between R466 of RBDA and E132 from NTDB is present in 189 out of 204 successful pathways to the ‘up’ state, and all 106 pathways to the ‘open’ state. This contact is most prevalent during the transition between the ‘up’ and ‘open’ state. Finally, the salt bridge between D428 of RBDA and R454 of RBDC is present only in all 106 pathways from the ‘up’ to the ‘open’ state and is the last salt bridge between the RBD and the spike in the ‘open’ state before the S1 subunit begins to peel off (Fig. 5d,e and Supplementary Fig. 8), at which point the last remaining contact to the RBDA is the glycan at position N165 of NTBB.
Additional BLI experiments of the key identified spike residues R408A, D405A and D427A corroborate the pathways observed in our simulations. Each of these reduces the binding interactions of the spike with ACE2 by ~13%, ~27% and ~52%, respectively (Fig. 4 and Supplementary Table 1). We also note that identified residues D198, N343, D364, D405, R408, T415, D427, D428, R454, R457, R466, E748, K986 and E990 are conserved between SARS-CoV and SARS-CoV-2 spikes, supporting their importance in coordinating the primary spike function of RBD opening. The emerging mutant SARS-CoV-2 strains, B.1 (D614G), B.1.1.7 (H69–V70 deletion and Y144–Y145 deletions, N501Y, A570D, D614G, P681H, T716I, S982A and D1118H), B.1.351 (L18F, D80A, D215G, R246I, K417N, E484K, N501Y, D614G and A701V), P.1 (L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y and T1027I), and CAL.20 C (L452R and D614G)28, do not contain mutants in the residues we identified here to facilitate RBD opening. Analysis of neighbouring residues and glycans to those mutated in the emerging strains along the opening pathway is detailed in Supplementary Table 2, and distances between each residue and glycan to RBDA is summarized in Supplementary Video 5.
Conclusions
We report extensive WE MD simulations of the glycosylated SARS-CoV-2 spike head characterizing the transition from the ‘down’ to ‘up’ conformation of the RBD. Over 130 µs of simulation provide more than 300 independent RBD-opening transition pathways. The simulated opening pathways align very well to conformations detected from cryo-EM with the ManifoldEM method. Analysis of these pathways from independent WE simulations indicates a clear gating role for the glycan at N343, which lifts and stabilizes the RBD throughout the opening transition. We also characterize an ‘open’ state of the spike RBD, in which the N165 glycan of chain B is the last remaining contact with the RBD en route to further opening of S1. BLI experiments of residues identified as key in the opening transitions, including N343, D405, R408 and D427, broadly supported our computational findings. Notably, a 56% decrease in ACE2 binding of the N343A mutant, compared with a 40% decrease in N234A mutant and a 10% decrease in the N165A mutant reported previously8, evidenced the key role of N343 in gating and assisting the RBD-opening process, highlighting the importance of sampling functional transitions to fully understand mechanistic detail. None of the individual mutations fully abolished ACE2 binding, indicating that the virus has evolved a mechanism involving multiple residues to coordinate spike opening. Our work indicates a critical gating role of the N343 glycan in spike opening and provides new insights into mechanisms of viral infection for this important pathogen.
Data availability
Data supporting the findings of this study are included in the article and its Supplementary Information files. We endorse the community principles around open sharing of COVID-19 simulation data29. All simulation input files and data are available at the NSF MolSSI COVID-19 Molecular Structure and Therapeutics Hub at https://covid.molssi.org and the Amaro Lab website http://amarolab.ucsd.edu. Source data are provided with this paper.
Code availability
This study utilized the standard builds of the simulation software WESTPA 2020.02 (https://github.com/westpa/westpa) and AMBER 18 (https://ambermd.org) according to best practices for running WE simulations30 with no special modifications.
References
Chan, J. F.-W. et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 395, 514–523 (2020).
Lu, R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574 (2020).
Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 3, 237–261 (2016).
Wrapp, D. et al. Cryo-EM structure of the 2019-NCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292.e6 (2020).
Benton, D. J. et al. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 588, 327–330 (2020).
Lu, M. et al. Real-time conformational dynamics of SARS-CoV-2 spikes on virus particles. Cell Host Microbe 28, 880–891.e8 (2020).
Casalino, L. et al. Beyond shielding: the roles of glycans in the SARS-CoV-2 spike protein. ACS Cent. Sci. 6, 1722–1734 (2020).
Gur, M. et al. Conformational transition of SARS-CoV-2 spike glycoprotein between its closed and open states. J. Chem. Phys. 153, 075101 (2020).
Fallon, L. et al. Free energy landscapes for RBD opening in SARS-CoV-2 spike glycoprotein simulations suggest key interactions and a potentially druggable allosteric pocket. Preprint at ChemRxiv https://doi.org/10.26434/chemrxiv.13502646.v1 (2020).
Zimmerman, M. I. et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 13, 651–659 (2021).
Huber, G. A. & Kim, S. Weighted-ensemble Brownian dynamics simulations for protein association reactions. Biophys. J. 70, 97–110 (1996).
Zhang, B. W., Jasnow, D. & Zuckerman, D. M. The “weighted ensemble” path sampling method is statistically exact for a broad class of stochastic processes and binning procedures. J. Chem. Phys. 132, 054107 (2010).
Chong, L. T., Saglam, A. S. & Zuckerman, D. M. Path-sampling strategies for simulating rare events in biomolecular systems. Curr. Opin. Struct. Biol. 43, 88–94 (2017).
Zuckerman, D. M. & Chong, L. T. Weighted ensemble simulation: review of methodology, applications, and software. Annu. Rev. Biophys. 46, 43–57 (2017).
Pratt, L. R. A statistical method for identifying transition states in high dimensional problems. J. Chem. Phys. 85, 5045–5048 (1986).
Zuckerman, D. M. & Woolf, T. B. Transition events in butane simulations: similarities across models. J. Chem. Phys. 116, 2586–2591 (2002).
Adhikari, U. et al. Computational estimation of microsecond to second atomistic folding times. J. Am. Chem. Soc. 141, 6519–6526 (2019).
Saglam, A. S. & Chong, L. T. Protein–protein binding pathways and calculations of rate constants using fully-continuous, explicit-solvent simulations. Chem. Sci. 10, 2360–2372 (2019).
DeGrave, A. J., Ha, J.-H., Loh, S. N. & Chong, L. T. Large enhancement of response times of a protein conformational switch by computational design. Nat. Commun. 9, 1013 (2018).
Suárez, E. et al. Simultaneous computation of dynamical and equilibrium information using a weighted ensemble of trajectories. J. Chem. Theory Comput. 10, 2658–2667 (2014).
Dashti, A. et al. Trajectories of the ribosome as a Brownian nanomachine. Proc. Natl Acad. Sci. USA 111, 17492–17497 (2014).
Watanabe, Y., Allen, J. D., Wrapp, D., McLellan, J. S. & Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 369, 330–333 (2020).
Henderson, R. et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 27, 925–933 (2020).
Hsieh, C.-L. et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 369, 1501–1505 (2020).
Pallesen, J. et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl Acad. Sci. USA 114, E7348–E7357 (2017).
Cross, R. The tiny tweak behind COVID-19 vaccines. Chem. Eng. News 98, 18–20 (2020).
Corum, J. & Zimmer, C. Coronavirus variants and mutations. New York Times (10 February 2021).
Amaro, R. E. & Mulholland, A. J. A community letter regarding sharing biomolecular simulation data for COVID-19. J. Chem. Inf. Model. 60, 2653–2656 (2020).
Bogetti, A. T. et al. A suite of tutorials for the WESTPA rare events sampling software. Living J. Comput. Mol. Sci. 1, 10607 (2019).
Acknowledgements
We are grateful for the efforts of the Texas Advanced Computing Center (TACC) Longhorn team and for the computing time made available through a Director’s Discretionary Allocation (made possible by National Science Foundation (NSF) award OAC-1818253). We thank Z. Gaieb for helpful discussions around system construction. We thank M. Tatineni for help with computing on SDSC Comet, as well as a COVID-19 HPC Consortium Award for computing time. We also thank C. Simmerling and his research group (SUNY Stony Brook), and A. Mulholland and his research group (University of Bristol), for helpful discussions related to the spike protein, as well as D. Zuckerman, J. Copperman, M. Zwier and A. Saglam for helpful methodological discussions. T.S. is funded by an NSF GRFP grant (DGE-1650112). This work was supported by: a National Institutes of Health (NIH) grant (GM132826); an NSF RAPID grant (MCB-2032054); an award from the RCSA Research Corp. and a UC San Diego Moores Cancer Center 2020 SARS-CoV-2 seed grant to R.E.A.; an NIH grant (R01-GM31749) to J.A.M.; an NIH grant (R01-AI127521) to J.S.M.; an NIH grant (R01 GM115805) and an NSF grant (CHE-1807301) to L.T.C.; and NIGMS grants (R01 GM29169 and R35 GM139453) to J.F. A.O. and G.M. acknowledge support by the US Department of Energy, Office of Science, Basic Energy Sciences under award DE-SC0002164 (underlying dynamical algorithms), and by the US National Science Foundation under awards STC 1231306 (underlying data-analytical techniques) and DBI-2029533 (underlying data-analytical models).
Author information
Authors and Affiliations
Contributions
T.S. and S.-H.A. contributed equally to this work. R.E.A. and L.T.C. oversaw the project. T.S. and L.C. prepared the simulation model. T.S. and S.-H.A. performed WE simulations and A.T.B. provided WESTPA scripts. S.-H.A., A.T.B., T.S. and L.T.C. carried out WE analysis. T.S. and F.L.K. performed simulation analyses. L.C., T.S. and F.L.K. created figures and videos. J.S.M. designed and oversaw BLI experiments. R.S.M. and J.A.G. performed BLI experiments and wrote the corresponding parts in Results and Methods. J.F. and A.O. directed the ManifoldEM study, and E.S., F.A.-R., S.M. and G.M. performed the ManifoldEM study. E.S. and F.A.-R. analysed the results and compared them with WE simulations. E.S. and J.F. described the ManifoldEM methods and results. T.S., S.H.-A., L.T.C. and R.E.A. wrote the manuscript with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Chemistry thanks Syma Khalid and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Methods, Figs. 1–16, Table 1 and legends for Supplementary Videos.
Supplementary Table 2
Evaluation of neighbouring residues of key altered residues in circulating SARS-CoV-2 spike variants.
Supplementary Video 1
Continuous pathway of RBD opening.
Supplementary Video 2
A comparison of the WE trajectory and ManifoldEM CC1 and CC2 from a side view (PD 1386).
Supplementary Video 3
A comparison of the WE trajectory and ManifoldEM CC2 from a top-down view (PD 112).
Supplementary Video 4
Glycan gate at position N343 intercalates with residues to facilitate RBD opening.
Supplementary Video 5
Mapping of residue contacts to RBD throughout opening pathway.
Source data
Source Data Fig. 2
Raw values for plot in Fig. 2f.
Source Data Fig. 4
Raw values from BLI experiments.
Source Data Fig. 5
Raw values for histogram in Fig. 5e.
Rights and permissions
About this article

Cite this article
Sztain, T., Ahn, SH., Bogetti, A.T. et al. A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat. Chem. 13, 963–968 (2021). https://doi.org/10.1038/s41557-021-00758-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41557-021-00758-3
This article is cited by
-
Targeting the receptor binding domain and heparan sulfate binding for antiviral drug development against SARS-CoV-2 variants
Scientific Reports (2024)
-
Structural insights into the modulation of coronavirus spike tilting and infectivity by hinge glycans
Nature Communications (2023)
-
Enhanced sampling in molecular dynamics simulations and their latest applications—A review
Nano Research (2023)
-
Analysis of the N-glycosylation profiles of the spike proteins from the Alpha, Beta, Gamma, and Delta variants of SARS-CoV-2
Analytical and Bioanalytical Chemistry (2023)
-
Molecular recognition of SARS-CoV-2 spike protein with three essential partners: exploring possible immune escape mechanisms of viral mutants
Journal of Molecular Modeling (2023)
