
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the RNA virus responsible for the coronavirus disease 2019 (COVID-19) pandemic. Although SARS-CoV-2 was reported to alter several cellular pathways, its impact on DNA integrity and the mechanisms involved remain unknown. Here we show that SARS-CoV-2 causes DNA damage and elicits an altered DNA damage response. Mechanistically, SARS-CoV-2 proteins ORF6 and NSP13 cause degradation of the DNA damage response kinase CHK1 through proteasome and autophagy, respectively. CHK1 loss leads to deoxynucleoside triphosphate (dNTP) shortage, causing impaired S-phase progression, DNA damage, pro-inflammatory pathways activation and cellular senescence. Supplementation of deoxynucleosides reduces that. Furthermore, SARS-CoV-2 N-protein impairs 53BP1 focal recruitment by interfering with damage-induced long non-coding RNAs, thus reducing DNA repair. Key observations are recapitulated in SARS-CoV-2-infected mice and patients with COVID-19. We propose that SARS-CoV-2, by boosting ribonucleoside triphosphate levels to promote its replication at the expense of dNTPs and by hijacking damage-induced long non-coding RNAs’ biology, threatens genome integrity and causes altered DNA damage response activation, induction of inflammation and cellular senescence.
Similar content being viewed by others


Main
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is an RNA virus, responsible for the ongoing coronavirus disease 2019 (COVID-19) pandemic1. Its 30 kb genome encodes 26 polypeptides encompassing 16 non-structural proteins (NSPs), 4 structural proteins such as the nucleocapsid (N) protein, and 6 accessory ones2.
Viral infections can impact on several cellular pathways, including the autophagy pathway3, the ubiquitin–proteasome system (UPS)4 and the DNA damage response (DDR). While the interplay between some DNA viruses and DDR has been studied5, much less is known about RNA viruses6. Although SARS-CoV-2 infection has been suggested to engage components of the DDR machinery7,8,9,10, a thorough characterization and a mechanistic probing of the impact of SARS-CoV-2 on genome integrity and DDR engagement is lacking.
The DDR is a network of pathways that sense DNA lesions, signal their presence and coordinate their repair11. DNA single-strand and double-strand breaks (SSBs and DSBs) are detected by replication protein A (RPA) and by the MRE11–RAD50–NBS1 (MRN) complex12, respectively, which guide the recruitment of the apical DDR kinases ataxia telangiectasia and Rad3-related (ATR) or ataxia-telangiectasia mutated (ATM) at SSBs or DSBs12, respectively. ATR and ATM undergo autophosphorylation and phosphorylate several DDR factors, including the effector kinases CHK1 and CHK2, which contribute to enforce cell-cycle arrest12. DDR activation can cause cellular senescence13,14 and inflammation15 or cell death13.
We demonstrated that the induction of a DSB results in the recruitment of the RNA polymerase II complex, which transcribes a novel class of RNA molecules named damage-induced long non-coding RNAs (dilncRNAs)16,17,18. These RNAs, by interacting with DDR factors such as p53-binding protein 1 (53BP1), are necessary for their condensation into foci at DSBs16,17 by promoting liquid–liquid phase separation (LLPS)17,19. Inhibiting the synthesis or function of dilncRNAs disrupts DDR foci and impairs DNA repair16,17,20. Interestingly, also SARS-CoV-2 N-protein phase-separates in an RNA-dependent manner21,22.
In this Article, we demonstrate that SARS-CoV-2 infection causes DNA damage and activation of an altered DDR. DNA damage is the consequence of the degradation of CHK1 by ORF6 and NSP13 viral factors through the proteasome and the autophagy pathways, respectively. Depletion of CHK1 causes loss of RRM2, a component of the ribonucleotide reductase (RNR) complex23, which leads to deoxynucleoside triphosphate (dNTP) shortage that causes impaired S-phase progression, DNA damage accumulation, DDR activation, induction of inflammatory pathways and establishment of cellular senescence. The supplementation of deoxynucleosides (dNs) is sufficient to contrast this cascade of events. In addition to that, SARS-CoV-2 N-protein impairs 53BP1 recruitment at DSB by competing with dilncRNAs binding, ultimately hampering DNA repair. These events occur also in vivo in mice infected by SARS-CoV-2 and in patients with COVID-19.
Overall, our results reveal the impact of SARS-CoV-2 infection on genome integrity and its contribution to the inflammatory response observed in COVID-19 patients and the recently reported virus-induced cellular senescence24.
Results
SARS-CoV-2 causes DNA damage and an altered DDR activation
We studied the engagement of the DDR pathways at different timepoints upon infection by SARS-CoV-2 of Huh7 cells, a human cell line naturally permissive to SARS-CoV-2 (refs. 25,26), by immunoblotting of whole cell lysates. As negative control we used mock-infected cells; as positive control we exposed cells to hydroxyurea (HU), which induces DNA replication stress and activates the ATR–CHK1 axis27,28, or ionizing radiation (IR) that causes DSBs and activates the ATM–CHK2 pathway11 (Fig. 1a). We observed that SARS-CoV-2 infection triggered the autophosphorylation, and thus activation, of the master kinases DNA-PK (pDNA-PKS2056, involved in DNA repair12) and ATM (pATMS1981) but not ATR (pATRT1989) (Fig. 1a,b). CHK2, the direct downstream target of ATM, was not detectably phosphorylated on its activating site (T68); similarly, CHK1, a target of ATR, was not phosphorylated on S317. Also P53 was not significantly phosphorylated on S15, an ATM/ATR target site (Fig. 1a,b). Differently, KAP1 (also known as TRIM28), a chromatin-bound ATM target12, was strongly phosphorylated (pKAP1S824) together with phosphorylated H2AX (γH2AX) and RPA (pRPAS4/8), markers of DSB and SSB, respectively11 (Fig. 1a,b). Similar results were generated in infected human lung epithelial Calu-3 cells29,30 (Extended Data Fig. 1a,b).

a, Immunoblotting of whole cell lysates of Huh7 cells infected, or not, with SARS-CoV-2 analysed at different timepoints post-infection for markers of DDR activation. Lysates from Huh7 cells not treated (NT) or treated with 6 mM HU or exposed to 2 Gy IR and collected at different timepoints were used as positive controls. Viral infection was monitored by probing for SARS-CoV-2 N-protein. Where present, dashed lines indicate where the blot was cropped. b, Quantification of activated protein levels shown in a. Values are normalized to mock-infected samples. c, Representative immunofluorescence (IF) images of SARS-CoV-2-infected (V+) or mock-infected (V−) Huh7 cells fixed at 48 h post-infection and stained for DDR markers. SARS-CoV-2 N-protein was used to label infected cells. Nuclei were stained with DAPI. Scale bar, 10 μm. d, Quantification of DDR activation shown in c. Each dot is a nucleus. e, Images of comet assays of infected or mock-infected Huh7 cells. Scale bar, 100 μm. f, Quantification of comet tail moment shown in e. Horizontal bars represent the median values ± 95% confidence interval (CI) of three independent infections. Source numerical data and unprocessed blots are available in source data.
To confirm and extend at single-cell resolution the impact of SARS-CoV-2 infection on DDR, we performed quantitative immunofluorescence analyses of the conditions aforementioned. We observed increased numbers of pKAP1S824, pRPAS4/8 and γH2AX foci per cell in infected Huh7 compared with mock-infected cells (Fig. 1c,d). In addition, SARS-CoV-2 infection of human nasal epithelial primary cells (HNEpCs) confirmed DDR activation, as detected by pRPAS4/8 and γH2AX foci (Extended Data Fig. 1c,d).
To directly monitor the impact of the virus on physical DNA integrity, we performed comet assays in Huh7 and Calu-3 cells. We observed DNA fragmentation induction in both SARS-CoV-2-infected cell lines compared with control conditions, as measured by tail moment (Fig. 1e,f and Extended Data Fig. 1e,f).
Damaged DNA released in the cytoplasm can be sensed by the cGAS–STING pathway triggering an inflammatory response31. We therefore investigated cGAS–STING and other inflammatory pathways in cells upon SARS-CoV-2 infection and observed a higher number of micronuclei, which also stained positive for cGAS (Extended Data Fig. 1g,h) in Calu-3 cells, suggestive of the release of damaged nuclear DNA in the cytosol. In infected Huh7 cells, which do not express cGAS and STING32, P38 and STAT1, factors involved in the pro-inflammatory response7, were activated (Extended Data Fig. 1i).
To test the consequences of the activation of these pro-inflammatory pathways, we monitored by quantitative reverse transcription polymerase chain reaction (RT–qPCR) the transcriptional induction of IL6, IL8, CXCL9, CXCL10 and TNFα genes in Huh7 and Calu-3 upon SARS-CoV-2 infection. We detected their significant upregulation in both cell types, although generally stronger in Calu-3 (Extended Data Fig. 1j). Since increased expression of pro-inflammatory genes is consistent with the induction of cellular senescence by SARS-CoV-2, as recently reported33,34, we tested and confirmed the establishment of cellular senescence following SARS-CoV-2 infection in our settings, as demonstrated by increased senescence-associated β-galactosidase (SA-β-gal) activity (Extended Data Fig. 1l,m), augmented P21- and reduced KI67-positive cells (Extended Data Fig. 1n,o), and no significant induction of apoptosis (Extended Data Fig. 1k).
In sum, our results obtained by different techniques and in three independent cell types indicate that SARS-CoV-2 infection causes DNA damage and an altered DDR; this is associated with the induction of pro-inflammatory pathways and cytokines and cellular senescence.
SARS-CoV-2 causes dNTP shortage by decreasing CHK1 levels
While studying activation of individual DDR proteins, we noticed that total CHK1 protein levels progressively decreased in infected Huh7 and Calu-3 cells (Figs. 1a and 2a–d and Extended Data Fig. 2c,d), mainly post-transcriptionally (Extended Data Fig. 2e,f). CHK1 loss is reportedly sufficient to cause DNA replication stress and DNA damage accumulation35. CHK1 controls the expression of the ribonucleoside-diphosphate reductase subunit M2 (RRM2), the small subunit of the RNR enzyme that converts ribonucleoside triphosphates (rNTPs) into dNTPs, necessary for DNA synthesis23,36. By testing RRM2 messenger RNA and protein levels by RT–qPCR, immunoblotting and immunofluorescence, we consistently observed their progressive and significant decrease following SARS-CoV-2 infection (Fig. 2a–d and Extended Data Fig. 2a–f).

a, Immunoblotting of whole cell lysates of Huh7 infected, or not, with SARS-CoV-2 and analysed at different timepoints post-infection. b, Quantification of protein levels shown in a; values are shown as relative to mock-infected samples. c, Immunofluorescence (IF) images of infected (V+) or mock-infected (V−) Huh7 cells fixed 48 h post-infection; nuclei were stained with DAPI. Scale bar, 10 μm. d, Quantification of CHK1- or RRM2-positive cells shown in c; n = 3 independent experiments. e, dNTP concentration was measured in V− or V+ Huh7 and Calu-3; values are shown as relative to V−. f, Histograms show the percentage of cells in each phase of the cell cycle in V− or V+ Huh7 fixed 48 h post-infection. g, Fraction of V− or V+ Huh7 cells that did not incorporate BrdU (BrdU−) measured by flow cytometry 48 h post-infection. Source numerical data and unprocessed blots are available in source data.
Then, we measured individual dNTP concentrations in SARS-CoV-2 infected Huh7 and Calu-3 cells and observed reduced levels of cellular dNTPs compared with mock-infected conditions (Fig. 2e).
dNTP shortage can impair DNA synthesis, ultimately hampering S-phase progression23,27. To monitor cell-cycle progression, we measured DNA content in infected or mock-infected cells by propidium iodide (PI) staining followed by flow cytometry analysis. We observed a significant accumulation of infected cells in S-phase compared with control samples (Fig. 2f and Extended Data Figs. 3d and 8c). This was confirmed by strongly reduced levels of CDT1, a G1-phase marker37,38,39 (Extended Data Fig. 2g). By pulse labelling with 5-bromo-2′-deoxyuridine (BrdU) for 1 h before flow cytometry, we observed an increased percentage of BrdU-negative cells in S-phase in infected samples (Fig. 2g and Extended Data Figs. 3d and 8c). Altogether, these results indicate reduced dNTP levels and impaired S-phase progression following infection.
To determine the causal role of reduced dNTP levels, we tested the impact of dN supplementation to culture medium of infected cells. We observed that dN supplementation was sufficient to reduce DDR activation as shown by immunofluorescence and immunoblots of γH2AX, pRPAS4/8 and pKAP1S824 (Fig. 3a–d), DNA damage accumulation detected by comet assays (Fig. 3e,f) and transcription of several pro-inflammatory cytokines (Fig. 3g).

a, Immunofluorescence (IF) images of V− or V+ Huh7 cells, treated or not with dNs, fixed 48 h post-infection; nuclei were stained with DAPI. Scale bar, 10 μm. b, Quantification of DDR activation shown in a. Each dot is a nucleus. c, Immunoblots of Huh7 cells treated as in a. d, Quantification of protein levels shown in c. Values are normalized to untreated V+ cells. e, Images of comet assays of Huh7; conditions are as in a. Scale bar, 100 μm. f, Quantification of comet tail moment shown in e; horizontal bars represent the median values ± 95% CI of three independent infections. g, RT–qPCR of pro-inflammatory cytokine expression in V− or V+ Calu-3 cells, treated or not with dNs. Values are shown as relative to RPLP0 mRNA. Source numerical data and unprocessed blots are available in source data.
Overall, these results are consistent with a model in which SARS-CoV-2 gene products cause CHK1 loss, which reduces RRM2 levels and consequently the pool of available dNTPs, causing impaired DNA replication and S-phase progression, DNA damage accumulation and ultimately fuelling an inflammatory response. Supplementation of dNs is sufficient to tame these events.
CHK1 loss is sufficient to cause DNA damage and inflammation
To determine whether CHK1 loss is sufficient to recapitulate the events here described following SARS-CoV-2 infection, we studied the impact of CHK1 depletion by RNA interference. Consistent with previous reports23, we observed by flow cytometry that cells knocked down for CHK1 accumulate in S-phase (Extended Data Figs. 3a–d and 8c) and pulse labelling with BrdU for 1 h before flow cytometry analysis revealed a higher fraction of BrdU-negative S-phase cells compared with control samples (Extended Data Figs. 3d and 8c).
We also observed that CHK1 depletion was sufficient to reduce RRM2 levels and cause DNA damage, as shown by increased pRPAS4/8 and γH2AX signals (Extended Data Fig. 3e,f). In addition, CHK1 knockdown led to the activation of P38 and STAT1 (Extended Data Fig. 3e,f) and formation of γH2AX foci and micronuclei, often positive for cGAS (Extended Data Fig. 3g–j), indicating that CHK1 loss in infected cells probably contributes to the activation of pro-inflammatory pathways. Indeed, cells depleted for CHK1 displayed increased expression of most of the cytokine and chemokine genes tested (Extended Data Fig. 3k,l) and increased secretion of IL6, CXCL9 and CXCL10 in Calu-3 cells as monitored by immunoassays (Extended Data Fig. 3m).
In sum, CHK1 loss is sufficient to recapitulate several of the events observed in SARS-CoV-2 infected cells, namely, RRM2 reduction, S-phase progression impairment, DNA damage and secretion of inflammatory cytokines.
SARS-CoV-2 ORF6 and NSP13 trigger CHK1 protein degradation
To identify the viral gene products responsible for CHK1 downregulation, we individually expressed 24 of the 26 annotated SARS-CoV-2 proteins40 (SARS-CoV-2 reference genome, NC_045512.2) and analysed by immunoblotting their impact on CHK1 levels. Among the gene products tested, ORF6 and NSP13 were the factors with the strongest and most consistent impact on CHK1 protein levels (Fig. 4 and Extended Data Fig. 4a,b). Their sole expression was also sufficient to reduce RRM2 levels and increase γH2AX and RPA phosphorylation (S4/8) (Fig. 4a–d and Extended Data Fig. 4a,b).

a, Immunofluorescence (IF) images of Huh7 cells expressing Strep-tagged SARS-CoV-2 ORF6 or NSP13 fixed 48 h post-transfection and stained for DDR markers; GFP was used as control; staining with anti-Strep-tag was used to label transfected cells; nuclei were stained with DAPI. Scale bar, 10 μm. b, The histograms show the percentage of CHK1- or RRM2-expressing cells among the transfected ones (Strep-Tag+) as determined in a; n = 3 independent experiments (n = 4 for GFP-expressing cells in RRM2 analysis). The dot plots show the number of γH2AX or pRPAS4/8 foci in the samples described in a. c, Representative immunoblots of whole cell lysates from Huh7 cells transfected with plasmids encoding for HA-tagged SARS-CoV-2 ORF6, or Strep-tagged SARS-CoV-2 NSP13, or empty vector (EV) as a control. Where present, dashed lines indicate where the blot was cropped. d, Quantification of protein levels shown in c; values are the mean ± s.e.m. of four independent experiments and shown as relative to the control sample (EV). Source numerical data and unprocessed blots are available in source data.
ORF6 causes CHK1 degradation through the proteasome pathway
SARS-CoV-2 ORF6 has been shown to associate with the nuclear pore and to interfere with proteins’ nuclear–cytoplasmic trafficking41,42.
We observed that SARS-CoV-2-infected cells displayed cytoplasmic CHK1 localization compared with mock-infected cells in which CHK1 is mainly nuclear (Fig. 2c). It has been reported that accumulation of CHK1 in the cytoplasm leads to its degradation through the UPS43. To investigate the potential involvement of ORF6 in the cytoplasmic accumulation of CHK1, we took advantage of a point mutant form of ORF6 (ORF6M58R) unable to interact with the nuclear pore complex41. Interestingly, CHK1 protein levels did not decrease in Huh7 cells expressing the mutant ORF6, as detected both by immunofluorescence and immunoblots (Fig. 5a–d). Consistent with that, mutant ORF6 expression also had no impact on RRM2 protein levels and DNA damage accumulation as detected by pRPAS4/8 and γH2AX (Fig. 5c,d). This suggests that ORF6, by altering CHK1 nuclear–cytoplasm shuttling, may cause its degradation.

a, Images of Huh7 expressing GFP (negative control), ORF6 or its mutant form ORF6M58R. b, Quantification of the percentage of transfected cells expressing CHK1 shown in a. c, Immunoblotting of Huh7 treated as in a; EV-transfected cells were used as negative control. d, Quantification of the protein levels shown in c. Values are the mean ± s.e.m. of four independent experiments. e, Confocal images of GFP- or ORF6-expressing Huh7 ± MG132. f, Quantification of nuclear (n) and cytoplasmic (c) CHK1 levels in the cells described in e. g, Immunoblots of the samples described in e; EV-transfected cells were used as negative control. h, Quantification of the protein levels shown in g. i, Ubiquitination assay of CHK1 immunoprecipitated from ORF6- or ORF6M58R-expressing Huh7 ± MG132. j, Quantification of the samples shown in i. Values are shown as relative to immunoprecipitated CHK1 amounts (IP-CHK1); n = 3 independent experiments. k, Immunofluorescence (IF) images of GFP- or NSP13-expressing Huh7 ± BafA1 or CQ. l, CHK1 quantification in cells described in k. Values are the mean ± s.e.m. of four independent experiments, except for GFP and NSP13 not treated (NT) conditions (n = 6). m, IF images of NSP13-expressing Huh7 treated with BafA1 (1 h). Arrow points to CHK1 and P62 co-localization. n, Quantification of the percentage of cells displaying co-localizing CHK1 and P62 signals shown in m. o, IF images of CHK1 levels in Huh7 transfected with the indicated siRNAs before viral NSP13 overexpression. p, Quantification of the percentage of CHK1-expressing cells in the transfected samples represented in o. Values are the mean ± s.e.m. of four independent experiments. Scale bar, 10 μm and DAPI-stained nuclei in a,e,k,m and o. Source numerical data and unprocessed blots are available in source data.
We therefore probed the engagement of UPS in ORF6-dependent CHK1 loss. Treatment of ORF6-expressing Huh7 cells with MG132—a proteasome inhibitor—recovered CHK1 protein levels, detected mostly in the cytoplasm (Fig. 5e–h), consistent with impaired protein trafficking of ORF6-expressing cells.
To demonstrate that SARS-CoV-2 ORF6 expression causes CHK1 poly-ubiquitination, a prerequisite of proteosome-dependent degradation, we immunoprecipitated endogenous CHK1 in Huh7 cells expressing either ORF6 or ORF6M58R, treated or not with MG132, and probed for ubiquitinated CHK1. Proteasomal inhibition led to a higher accumulation of ubiquitinated CHK1 in ORF6-expressing cells compared with samples that overexpressed ORF6M58R (Fig. 5i,j).
These results indicate that SARS-CoV-2 ORF6 prevents CHK1 nuclear import, causing its accumulation in the cytoplasm and its consequent degradation through the UPS.
NSP13 causes CHK1 degradation through autophagy
It has been shown that NSP13 can promote protein degradation in an autophagy-dependent manner44. Therefore, to test whether CHK1 loss in NSP13-expressing cells was dependent on the autophagic route, we transiently expressed the viral NSP13 gene in Huh7 cells in the presence of either Bafilomycin A1 (BafA1) or chloroquine (CQ), two specific inhibitors of autophagy45; efficacy was confirmed by the observed accumulation of P62 cytoplasmic aggregates45 (Extended Data Fig. 5a,b). NSP13-mediated reduction of CHK1 protein levels was abolished by both treatments (Fig. 5k,l). A short BafA1 exposure highlighted a clear co-localization of CHK1 with P62 cytoplasmic aggregates, suggesting its accumulation in autophagosomes (Fig. 5m,n). To confirm and extend these results, we individually knocked down Beclin 1 (BECN1) and LC3B—two key regulators of autophagy46—and observed a significant restoration of CHK1 protein levels in NSP13-expressing cells (Fig. 5o,p and Extended Data Fig. 5c,d).
These results indicate that SARS-CoV-2 NSP13 causes the accumulation of CHK1 in the cytoplasm, where it co-localizes with P62, in this way promoting its degradation through autophagy.
N-protein impairs 53BP1 recruitment at DSB and hinders NHEJ
We noticed that γH2AX foci accumulation was not accompanied by co-localizing 53BP1 foci in SARS-CoV-2-infected Huh7, Calu-3 and HNEpC (Fig. 6a–d and Extended Data Fig. 6a,b), despite unaltered 53BP1 protein levels (Extended Data Fig. 6c).

a, Immunofluorescence (IF) images of V+ or V− Huh7; nuclei were stained with DAPI. b, Quantification of 53BP1 foci shown in a. Each dot represents the number of 53BP1 foci per nucleus. c, IF images of infected HNEpC in which SARS-CoV-2 RNA was detected by FISH; nuclei were stained with Hoechst. d, Quantification of 53BP1 foci shown in c; the histograms show the percentage of nuclei with 53BP1 foci (>1) in cells expressing (+) or not (−) SARS-CoV-2 RNA; n = 3 independent infections. e, IF images of irradiated Huh7 transfected with N-protein or EV as control; nuclei were stained with DAPI. f, Quantification of DDR foci shown in e; the dot plots show the number of γH2AX and 53BP1 foci per nucleus in N-protein- or EV-expressing samples. Values are relative to irradiated cells transfected with EV; bars represent the mean ± 95% CI of three independent experiments. g, Quantification of 53BP1 foci per nucleus over time in irradiated cells injected with recombinant N-protein or BSA as control. Error bars represent s.e.m.; the experiment was repeated three times with similar results. h, NIH2/4 expressing (n = 3) or not (n = 2) I-SceI were transfected with N-protein. Cell lysates were incubated with anti-N-protein or normal rabbit IgG and co-precipitated RNA analysed by strand-specific RT–qPCR. H2AX mRNA was used as an unrelated transcript. Values are shown as percentage of input RNA. i, Endogenous 53BP1 was immunoprecipitated from I-SceI-expressing NIH2/4 transfected with N-protein or EV as control. 53BP1-bound transcripts were monitored as in h and shown as percentage of input RNA. Values are the average of two independent experiments. j, EJ5-GFP U2OS were transfected with N-protein or EV, ± I-SceI. DSB re-joining events were evaluated by qPCR on gDNA isolated at 72 h post-transfection. Values are relative to I-SceI-transfected cells not expressing N-protein. Scale bar, 10 μm (a, c and e). Source numerical data are available in source data.
SARS-CoV-2 N-protein is an RNA-binding protein capable to undergo RNA-dependent LLPS21,22,47,48,49. We previously reported that also 53BP1 phase-separates in an RNA-dependent manner17. To test the potential impact of N-protein on 53BP1 foci formation, we expressed the viral N gene40 in Huh7 cells and exposed them to IR. We observed that irradiated cells expressing N showed increased numbers of γH2AX foci per cell, but fewer 53BP1 foci compared with control cells (Fig. 6e,f). To reduce the possibility of an indirect effect mediated by altered gene expression, we micro-injected purified recombinant N-protein into the nuclei of irradiated cells stably expressing 53BP1-GFP50 and immediately studied the kinetics of 53BP1 foci by live imaging. We observed 53BP1 foci number decreasing with a faster (~8.5-fold) kinetic in cells injected with the N-protein compared with control cells (Fig. 6g, Extended Data Fig. 6d and Supplementary Video 1).
Next, we sought to elucidate the molecular mechanisms underlying N-protein impact on 53BP1 functions. In co-immunoprecipitation experiments, 53BP1 did not interact with N-protein (Extended Data Fig. 6e). We previously reported that dilncRNAs generated at DSB drive LLPS of 53BP1 (refs. 16,17). Intriguingly, both viral and cellular RNAs have been reported to associate with N-protein and promote its phase separation48, as we confirmed (Extended Data Fig. 6f,g).
Since N-protein, although mainly cytoplasmic, also localizes in the nucleus51,52,53 (Extended Data Fig. 6j,k), we tested whether N associates with cellular dilncRNAs by performing RNA immunoprecipitation (RIP) against the N-protein in NIH2/4 cells, which we previously characterized for the expression of dilncRNAs upon DSB induction by I-SceI endonuclease16. Therefore, following SARS-CoV-2 N gene expression into NIH2/4 cells and DSB induction by I-SceI, we immunoprecipitated N-protein and analysed the associated RNAs by RT–qPCR. We observed that N-protein was associated with dilncRNA upon DSB generation, but not with H2AX mRNA used as a negative control (Fig. 6h). Next, we immunoprecipitated endogenous 53BP1 in I-SceI-induced NIH2/4 cells expressing or not the viral N-protein, and monitored dilncRNA association with 53BP1. We observed that 53BP1 association with dilncRNAs was reduced in cells expressing N-protein (Fig. 6i), despite unaltered 53BP1 protein levels or immunoprecipitation (IP) efficiency following N-protein overexpression (Extended Data Fig. 6h).
Since 53BP1 plays important DNA repair functions through non-homologous end-joining (NHEJ)54, we tested the impact of N-protein on NHEJ. We took advantage of a cell line bearing an integrated GFP construct flanked by two I-SceI recognition sites (EJ5-GFP)55: following I-SceI expression, DSBs are generated and repair can be quantified by qPCR on genomic DNA (gDNA) with primers flanking the re-joined site56. EJ5-GFP U2OS were transfected with a plasmid expressing I-SceI together with N-protein or an EV. Seventy-two hours post-transfection, gDNA was collected and analysed. NHEJ efficiency in cells expressing N-protein was significantly decreased compared with control samples (Fig. 6j) to an extent comparable to that previously observed upon 53BP1 depletion17,56,57 while leaving I-SceI levels unchanged (Extended Data Fig. 6i).
In sum, our evidence indicates that SARS-CoV-2 N competes with 53BP1 for dilncRNAs binding and thus reduces 53BP1 focus formation at DSB, ultimately hampering DNA repair by NHEJ.
SARS-CoV-2 causes DNA damage in mice and patients with COVID-19
We next extended our analyses in in vivo settings of SARS-CoV-2 infection. Lung sections of mice expressing human ACE2 (hACE2) were stained for DDR markers following intranasal administration of SARS-CoV-2 (Fig. 7a,b and Extended Data Fig. 7a,b); mice exposed to IR were used as positive control (Extended Data Fig. 7c,d). Immunostaining of γH2AX and pRPAS4/8 together with N-protein demonstrated strong γH2AX and pRPAS4/8 signal induction in infected samples compared with mock-infected ones. Instead, 53BP1, CHK1 and RRM2 signals were reduced in infected murine lungs (Fig. 7a,b). Thus, consistent with our observations in cultured cells, SARS-CoV-2 infection in mice causes DNA damage accumulation, impairs 53BP1 activation and reduces CHK1 and RRM2 protein levels.

a, IHC images of lungs from SARS-CoV-2-infected hACE2-mice or mock-infected (PBS) wild-type mice that were stained for the indicated markers at 6 days following intranasal administration; the presence of the virus was assessed by staining for SARS-CoV-2 N or by probing the viral genome through RNA ISH; nuclei were stained with haematoxylin (light blue). Scale bar, 50 μm. b, Quantification of the percentage of cells positive for the indicated markers as shown in a. Values are the mean ± s.e.m.; at least three mice were studied for each condition; the precise number is indicated below each histogram. Source numerical data are available in source data.
As SARS-CoV-2 infection has been recently shown to induce cellular senescence and contribute to inflammation in vivo9,10,33,34, we probed the same tissues for p16 and p21 and observed an increase of both in infected lungs (Extended Data Fig. 7e,f), with p21 detected almost exclusively in pneumocytes and in bronchial epithelium of the infected lungs, while p16 also associated with inflammatory cells populating the lung parenchyma of infected mice (Extended Data Fig. 7g).
We next analysed lungs and nasal mucosa sections from patients who died with a clinical diagnosis of COVID-19, determined by a positive naso-pharyngeal swab or bronchoalveolar lavage during their last hospitalization58. In situ hybridization (ISH) and immunohistochemistry (IHC) were used to confirm the presence of SARS-CoV-2 RNA or N-protein, respectively. As negative control, we studied the lungs of patients not diagnosed for the pathology (non-COVID) but affected by viral pneumonia of different aetiologies58. Since for ethical reasons we were not able to collect nasal mucosa from non-COVID subjects, we compared samples scored for the presence or absence of SARS-CoV-2, as detected by fluorescence in situ hybridization (FISH).
We probed lung and nasal mucosa for γH2AX, SARS-CoV-2 genome and cytokeratin-18 (CK18), a marker of epithelial cells, and observed increase γH2AX foci in epithelial cells of infected specimens, compared with controls (Fig. 8a,c). Similarly, pRPAS4/8 levels were strongly induced in the lungs of patients with COVID-19 (COVID) compared with non-COVID subjects (Fig. 8e,f).

a, Immunofluorescence (IF) images of lungs of patients diagnosed (COVID, n = 17) or not (non-COVID, n = 9) with COVID-19, and nasal mucosa of patients with COVID-19 in which cells were detected as positive (FISH+, n = 18) and negative (FISH−, n = 11) for SARS-CoV-2. Tissues were stained for SARS-CoV-2 RNA, γH2AX and CK18 to label epithelial cells; nuclei were counter-stained with Hoechst. Scale bar, 10 μm. b, IF images of lung (n = 4) and nasal mucosa (n = 5) of patients with COVID-19 stained for SARS-CoV-2 N-protein, γH2AX and 53BP1. Sections (n = 6) of a lung treated ex vivo with 2 μM bleomycin were used as positive control for DDR activation. Nuclei were stained with Hoechst. Scale bar, 10 μm. c, Quantification of DDR determined in a. Histograms show the percentage of epithelial cells bearing γH2AX foci (>1). d, Quantification of 53BP1 recruitment at γH2AX sites determined in b. The histograms show the percentage of nuclei with 53BP1 foci (>1) among those positive for γH2AX. e, IHC images of tissues of patients with COVID-19 stained for the indicated markers. Virus presence was assessed by probing for N-protein. Nuclei were stained with haematoxylin (light blue). Conditions are as in a; scale bar, 50 μm. f, The histograms show the percentage of cells positive for the markers determined in e. pRPAS4/8: non-COVID (n = 5 patients); COVID (n = 10). CHK1: non-COVID (n = 7); COVID (n = 3); FISH− (n = 4); FISH+ (n = 4). Values are the mean ± s.e.m. g, IF images of tissues of patients with COVID-19 stained for RRM2; scale bar, 10 μm. h, The histograms show the average intensity of RRM2 signal as determined in g. Values are the mean ± s.e.m.; n = 3 individuals for lungs and n = 5 for nasal mucosa. i, Schematic model of the impact of SARS-CoV-2 infection on genome integrity and cellular senescence. Source numerical data are available in source data.
We also analysed 53BP1 in both lung and nasal mucosa. In lung samples of patients with COVID, γH2AX foci were significantly less associated with 53BP1 than ex vivo bleomycin-treated sections of human non-COVID lungs (non-COVID bleo+) used as positive control, indicating impaired 53BP1 recruitment in infected lungs (Fig. 8b,d). Similarly, in nasal mucosa, COVID FISH+ cells displayed a reduced recruitment of 53BP1 compared with COVID FISH− cells (Fig. 8b,d). Both CHK1 and RRM2 expression was invariably and significantly lower in infected tissues compared with not infected ones (Fig. 8e–h).
Staining of lung tissues for P16 and P21 together with SARS-CoV-2 markers revealed that both were increased in patients with COVID compared with non-COVID individuals (Extended Data Fig. 8a,b).
Overall, these results indicate that SARS-CoV-2 infection in vivo causes an altered DDR activation that is associated with increased levels of DNA damage and cellular senescence in mouse and patients with COVID-19.
Discussion
Viruses are known to hijack cellular activities, including DDR, as a strategy to promote their replication6,59. This may have deleterious effects on the cell, potentially leading to genome instability59. SARS-CoV-2 infection has been reported to alter different host pathways7,60,61 and to correlate with the activation of some DDR markers and senescence7,8,9,10,33,62,63,64. The observation that some DDR inhibitors reduce SARS-CoV-2 replication65 further hints at a mutual interplay. However, when studied, DNA damage was correlated mainly with reactive oxygen species10,33.
Here, we showed that SARS-CoV-2 infection causes DNA damage (Fig. 1e,f and Extended Data Fig. 1e,f), as observed in two immortal cell lines, in primary human cells and in vivo in mice and humans. We identified at least two mechanisms responsible for DNA damage accumulation: one impacting on cellular dNTP metabolism leading to DNA replication impairment; another impeding 53BP1 activation and reducing DNA repair (Fig. 8i). The DNA damage accumulated triggers DDR activation, but in an altered way (Fig. 1a,d and Extended Data Fig. 1a,d). For instance, CHK1, together with P53, decreases following SARS-CoV-2 infection (Figs. 1a and 2a–d and Extended Data Fig. 2c,d). Degradation of DDR factors is a strategy shared by different viruses to override host defences59,66,67,68.
CHK1 is known to control the expression of E2F transcription factors, important regulators of cell-cycle progression69, and consequently of the RRM2 gene, allowing DNA synthesis in S-phase23,36,70. We demonstrated that SARS-CoV-2 infection leads to CHK1 loss and consequent RRM2 decrease (Figs. 1a and 2a–d), causing dNTP shortage and prolonged S-phase (Fig. 2e–g), consistent with the generation of DNA replication stress and DNA damage. This cascade of events leads to the establishment of cellular senescence and activation of pro-inflammatory pathways (Extended Data Figs. 1g–o, 7e–g and 8).
CHK1 depletion is sufficient to recapitulate RRM2 reduction, DNA damage accumulation and cytokine expression (Extended Data Fig. 3). Importantly, the administration of dNs to SARS-CoV-2-infected cells reduced virally induced DNA damage, DDR activation and cytokine expression (Fig. 3), thus demonstrating the causative role of dNTP depletion in these events.
We propose that this is probably the unmeant consequence of the dire need for rNTPs of SARS-CoV-2. Staggering two-thirds of total RNA in SARS-CoV-2-infected cells is of viral origin71: thus infected cells need to triple their normal RNA synthesis capacity. Therefore, the virus has been under evolutionary pressure to boost rNTP levels. One way is to reduce CHK1 levels, causing decreased RRM2 activity and consequent accumulation of rNTPs at the expense of dNTPs. Interestingly, a similar yet opposite mechanism was observed in the DNA virus HPV31, which boosts RRM2 to increase dNTPs and favour its genome replication72.
At least two SARS-CoV-2 products cause CHK1 degradation. ORF6, by associating with the nuclear pore complex, interferes with CHK1 nuclear import, leading to CHK1 cytoplasmic mis-localization and consequent proteasomal degradation. Notably, a point mutation that disrupts ORF6 binding to the nuclear pore complex prevented CHK1 poly-ubiquitination, degradation and DNA damage accumulation (Figs. 5a–d,i,j). In addition, proteasome inhibition with MG132 in ORF6-expressing cells was sufficient to rescue CHK1 levels (Fig. 5e–h). Differently, NSP13 leads to CHK1 depletion through the autophagic route, as indicated by the recovery of CHK1 levels upon treatment with autophagy inhibitors or with RNAi against key autophagy factors (Fig. 5k–p).
In addition to induce DNA damage, SARS-CoV-2 inhibits its repair. We observed a strikingly reduced ability of 53BP1 to form DDR foci, despite unaltered protein levels, in infected cells (Fig. 6a–d and Extended Data Fig. 6a–c). We propose that SARS-CoV-2 N, an avid RNA-binding protein, impairs 53BP1 condensation at DSB by competing for dilncRNA binding. Indeed, both 53BP1 and N-protein undergo LLPS in an RNA-dependent manner, and we demonstrate that N-protein, just like 53BP1 (ref. 16), binds to dilncRNA (Fig. 6g–i and Supplementary Video 1).
These data suggest a nuclear role of SARS-CoV-2 N-protein. Although both SARS-CoV and SARS-CoV-2 N-proteins bear functional nuclear localization signals, they are only partly nuclear73,74,75 (for example, Extended Data Fig. 6j,k), but phylogenetic studies have correlated the enhancement of motifs that promote nuclear localization of viral N-proteins with coronavirus pathogenicity and virulence75.
Intriguingly, enoxacin, a molecule that we reported to boost RNA-mediated 53BP1 foci assembly and DNA repair56, has been predicted in silico to reduce SARS-CoV-2 replication76. It is tempting to speculate that enoxacin, by enhancing 53BP1 activities, counteracts SARS-CoV-2 N-protein competition with 53BP1 for dilncRNAs.
Hyperactivation of inflammatory pathways is responsible for fatal COVID-19 cases77. DNA damage accumulation and chronic DDR activation are potent inducers of inflammation78. Consistent with previous studies62,63, we observed that SARS-CoV-2 infection of cultured cells activates multiple pro-inflammatory signalling pathways, including cGAS/STING, STAT1 and p38/MAPK (Extended Data Fig. 1g–j), similar to CHK1 depletion (Extended Data Fig. 3e–j). Supported by reports that disruption of the CHK1–RRM2 pathway triggers cellular senescence79 and our own evidence (Extended Data Figs. 1l–o, 7e–g and 8), we propose that SARS-CoV-2-mediated CHK1 loss promotes a pro-inflammatory programme akin to the senescence-associated secretory phenotype (Extended Data Fig. 3k–m).
A role for SARS-CoV-2-induced senescence in promoting macrophage infiltration and inflammation in vivo has been proposed33. We observed that SARS-CoV-2 infection causes DNA damage accumulation that correlates with markers of cellular senescence, in primary cells and in vivo (Extended Data Figs. 1l–o, 7e–g and 8). In particular, infected pneumocytes express high p21 levels, while polymorphonuclear and monocytoid inflammatory elements have elevated p16 (Extended Data Fig. 7g), reminiscent of a two-wave model of inflammatory response: an initial cell-intrinsic one and a second one triggered by the immune system9,10.
Altogether, our results indicate that SARS-CoV-2-induced DNA damage triggers a cell-intrinsic pro-inflammatory programme that, in concert with the immune response, fuels the strong inflammatory response observed in patients with COVID-19. The observed ageing phenotypes recently reported in patients with severe COVID-19 (refs. 80,81) are consistent with our observations.
Finally, by proposing a mechanism for the generation of DNA damage and the activation of DDR pathways and of a pro-inflammatory programme, we provide a model to improve our understanding of SARS-CoV-2-induced cellular senescence9,33. In this regard, it will also be interesting to determine if persistent DNA damage and DDR activation, features of cellular senescence82,83, following SARS-CoV-2 infection, contribute to the chronic manifestations of the pathology known as long COVID84.
Methods
Mice and treatments
Experiments involving animals have been carried out in accordance with the Italian Laws (D.lgs. 26/2014), which enforce Directive 2010/63/EU (Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes). Accordingly, the project has been authorized by the Italian Competent Authority (Ministry of Health). B6.Cg-Tg(K18-ACE2)2Prlmn/J mice were purchased from The Jackson Laboratory. Mice were housed under specific-pathogen-free conditions as already described85. SARS-CoV-2 isolation and propagation for infection in mice has been carried out as shown before85. Virus infection of K18-hACE2 mice was performed via intranasal administration of 1 × 105 tissue culture infectious dose 50 per mouse under isoflurane 2% (# IsoVet250) anaesthesia, as described85. Mice were monitored to record body weight, clinical and respiratory parameters. Mice were killed by cervical dislocation. At the time of autopsy, mice were perfused through the right ventricle with phosphate-buffered saline (PBS). Lung tissues were collected in liquid nitrogen or in Zn-formalin and transferred into 70% ethanol 24 h later. For irradiation experiments, an 11-month-old C57BL/6J mouse was irradiated with 2 Gy TBI using GADGIL X-Ray irradiator. An age- and sex-matched mouse was used as control. Mice were killed by CO2 inhalation at 1 h post-IR, and lungs were collected for fixation in 10% neutral buffered formalin overnight, washed in water and paraffin-embedded for histological analysis. RNA from mouse lungs was extracted and analysed as described85.
Patients diagnosed with COVID-19 and subjects negative for COVID-19
Histological analysis of lungs of patients with COVID-19 and subjects not diagnosed with the pathology (non-COVID)—but affected by viral pneumonia (influenza virus infection)—was performed by expert technicians and pathologists at the Pathology Unit of Trieste University Hospital. The same pathologists analysed all samples considered in this study, excluding operator-dependent biases. Viral presence in nasal mucosa of patients with COVID-19 was assessed by FISH against SARS-CoV-2 RNA. This study was approved by the Joint Ethical Committee of the Regione Friuli Venezia Giulia, Italy (re. 0019072/P/GEN/ARCS). All patients provided written informed consent to the use of their samples for research purposes at the time of hospital admission.
Cell culture and treatments
Vero E6 (ATCC-1586), Huh7 (kindly provided by Ralf Bartenschlager, University of Heidelberg, Germany) and Calu-3 cell lines (ATCC HTB-55) were cultured in Dulbecco’s modified Eagle medium (DMEM, ThermoFisher) supplemented with 10% foetal bovine serum (FBS, ThermoFisher) and 50 µg ml−1 gentamicin. 53BP1-GFP U2OS, EJ5-GFP U2OS and NIH2/4 cell lines were cultured as already described16,17,56. Cell cultures were maintained at 37 °C under 5% CO2. Cells were routinely tested for mycoplasma contamination. DNA damage was induced in cultured cells by 6 mM HU treatment for 4 h, or by exposure to IR (2 Gy) and analysed 15 or 60 min post-IR. Proteasome inhibition was conducted by incubating Huh7 cells for 6 h in growth medium containing 10 μM MG132 (M7449, Sigma). To inhibit autophagy, Huh7 cells were incubated with medium supplemented with either 100 nM Bafilomycin A1 (S1413, Selleckchem) or 50 μM CQ (C6628, Sigma) for 6 h, unless indicated otherwise.
SARS-CoV-2 propagation and in vitro infection
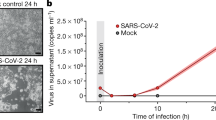
Working stocks of SARS-CoV-2 ICGEB-FVG_5 isolated and sequenced in Trieste, Italy86, were propagated on semiconfluent Vero E6 cells. Cultured Huh7 and Calu-3 cells were mock-infected or infected at a multiplicity of infection (MOI) of 0.1, in DMEM without supplements for 1 h at 37 °C and 5% CO2. Then, the non-bound virus was rinsed off with PBS 1× and fresh DMEM containing 2% of heat-inactivated FBS was added to the cells. Uniformity of viral infection in all experiments was confirmed by viral titration, RNA detection and immunofluorescence.
Isolation and SARS-CoV-2 infection of HNEpCs
HNEpCs were collected from healthy adult volunteers. Nasal cavities were anaesthetized using lidocaine, and nasal epithelial cells were collected by repeatedly scraping turbinates with a disposable bronchial cytology brush (CONMED). The tissue was resuspended in DMEM supplemented with 10% FBS and centrifuged at 300g for 5 min. The pellet was digested in a solution containing 1,000 U of Accutase (Sigma, A6964), 5,000 U of Dispase (Corning, 354235) and 1 mg ml−1 DNAse II (Sigma, D8764) for 8 min at 37 °C. The digestion was stopped by adding an equal volume of Pneumacult medium (Stem Cell Technologies, 5050) supplemented with 10% FBS and filtered through a 100 µm cell strainer. The cell suspension was centrifuged at 300g for 5 min, and the pellet was resuspended in red blood cell lysis solution (150 mM NH4Cl and 10 mM KHCO3) for 2 min. Cells were centrifuged at 300g for 5 min, resuspended in medium and seeded on eight-well chambers (Ibidi) at a concentration of 5 × 105 cells per well. After 48 h, cells were infected with SARS-CoV-2 (ref. 86) at 1 MOI as described above. Cells were cultured for 48 or 72 h and fixed in 4% paraformaldehyde for 15 min at room temperature for staining.
Immunofluorescence and FISH on HNEpCs
Fixed HNEpCs were permeabilized with 0.5% Triton X-100 (Sigma, T8787) in PBS for 15 min at room temperature. FISH to detect SARS-CoV-2 genome was performed using a kit by Molecular Instruments, following the manufacturer’s instructions. After FISH, samples were incubated with a blocking solution (0.5% Triton X-100, 10% horse serum in PBS) for 30 min and incubated for 16 h at 4 °C with primary antibodies (Supplementary Table 1) diluted in the blocking solution. Samples were stained with the appropriate secondary antibody (Supplementary Table 1). Nuclei were counter-stained using Hoechst 33342 trihydrochloride (Invitrogen, h3570) and samples mounted using Mowiol (Sigma, 81381).
Immunoblotting
Whole cell extracts were obtained by lysing Huh7 and Calu-3 in 1× Laemmli buffer (2% SDS, 10% glycerol and 60 mM Tris pH 6.8). Before fractionation on 4–12% gradient SDS–PAGE (ThermoFisher), whole extracts were boiled and sonicated for 15 s at low intensity using Bioruptor Next Gen (Diagenode) in a water bath at 4 °C. Proteins were then transferred onto nitrocellulose membrane and analysed as described before56 with the antibodies listed in Supplementary Table 1. Bio-Rad Image Lab 6.1 was used for immunoblot data collection.
Quantitative immunofluorescence analysis in cultured cells
Quantitative immunofluorescence assays were carried out in cultured cells as described56, with minor modifications. Specifically, Calu-3 were fixed with 4% paraformaldehyde and permeabilized first with methanol/acetone (1:1) for 2 min at room temperature and then with Triton 0.2% in PBS 1× for 10 min at room temperature. The antibodies used are listed in Supplementary Table 1. Images at widefield microscope were acquired with MetaVue software or with Zen 2.0 Software (Zeiss); confocal microscope images were collected with Leica Application Suite X; data analyses were carried out by using CellProfiler 3.1.9 or ImageJ 1.53a.
RNA extraction and RT–qPCR analysis
SARS-CoV-2-infected or mock-infected Huh7 and Calu-3 were collected in TriFast (EMR507100, Euroclone), and total RNA was extracted using RNeasy Kit (Qiagen). RNA from short interfering RNA (siRNA)-transfected Calu-3 and Huh7 was purified with Maxwell RSC simplyRNA Tissue Kit (Promega). DNase I was added during RNA purification, following the manufacturers’ protocols. Purified total RNA was reverse transcribed with SuperScript VILO cDNA Synthesis Kit (ThermoFisher), and complementary DNA was analysed by qPCR using SYBR Green I Master Mix (Roche) with the primers listed in Supplementary Table 2.
Comet assay
Alkaline comet assay was performed on 48 h SARS-CoV-2-infected or mock-infected Huh7, or on 24 h infected or mock-infected Calu-3 using CometAssay Reagent Kit for Single Cell Gel Electrophoresis Assay (Trevigen, 4250-050-K), following the manufacturer’s instructions. Tail moment was measured using CometScore 2.0 software.
SA-β-gal assay
SA-β-gal activity was measured in 48 h SARS-CoV-2-infected or mock-infected Huh7 or in 72 h infected or mock-infected HNEpC as described87. Cells treated with 6 mM HU treatment for 4 h were used as positive controls of senescence induction.
dNTP quantification
dNTP pools were extracted from 48 h SARS-CoV-2-infected or mock-infected Huh7 or from 24 h infected or mock-infected Calu-3. Cell plates were carefully washed free of medium with cold PBS and extracted with ice-cold 60% methanol. Methanolic extract was centrifuged, boiled for 3 min, brought to dryness by centrifugal evaporation (Savant, SC100 SpeedVac Concentrator and RT100A Refrigerated Condensation Trap) and stored at −80 °C until use. Cells left on the plate were dried and dissolved on 0.3 M NaOH, and the absorbance at 260 nm of the lysates was used as an index of cell mass, in turn an approximation for cell number88. The dry residue was dissolved in water and used to determine the size of dNTP pools by the DNA polymerase-based assay as described89. Two different aliquots of each pool extract were analysed, and pool sizes were normalized by the A260nm of the NaOH lysates.
BrdU staining and flow cytometry analysis
For cell-cycle analysis, SARS-CoV-2-infected or mock-infected Huh7 were pulsed with 10 µM BrdU (Sigma-Aldrich) for 1 h. Then, cells were collected and fixed first in formaldehyde 2% and then in 75% ethanol. Cells were probed with anti-BrdU primary antibody (Supplementary Table 1) diluted in PBS 1% bovine serum albumin (BSA) at room temperature for 1 h. After washing, cells were incubated with the secondary antibody, diluted 1:400, at room temperature for 1 h in the same buffer. Finally, cells were stained with PI (Sigma-Aldrich, 50 µg ml−1) in PBS 1% BSA and RNase A (Sigma-Aldrich, 250 µg ml−1). Samples were acquired with Attune NxT (ThermoFisher) using a 561 nm laser and 695/40 filter for PI; 488 nm laser and 530/30 filter for BrdU. Analysis was carried out using FlowJo 10.7.1 (BD Biosciences). At least 104 events were analysed for each sample.
Plasmid and siRNA transfection
pLVX-EF1α-2xStrep-IRES-Puro vectors encoding for SARS-CoV-2 proteins are a kind gift from Professor Nevan J. Krogan40. pCAGGS-ORF6 and -ORF6M58R vectors were gently provided by Lisa Miorin41. pcDNA3.1 (+) Mammalian Expression Vector (ThermoFisher) was used as a control EV where indicated. Plasmids were transfected with Lipofectamine 2000 Transfection Reagent (ThermoFisher) in Opti-MEM (ThermoFisher). siRNAs were purchased from Dharmacon (Horizon) and transfected into Huh7 or Calu-3 with Lipofectamine RNAiMAX Transfection Reagent (ThermoFisher) in Opti-MEM. Cells were collected for analyses at 48 or 72 h post-transfection.
Multiplex immunoassay
Bio-Plex Pro-Human Cytokine Immunoassay kits (Bio-Rad) were used to measure the concentration of pro-inflammatory cytokines and chemokines in cell supernatants, according to the manufacturer’s guidelines. The magnetic-bead-based antibody detection kit allows simultaneous quantification of the analytes of interest. Each sample was tested undiluted and in duplicate. Positive and negative controls were included in the plate. The plate was read on the Bio-Plex 200 system (Bio-Rad), powered by the Luminex xMAP technology. The concentration of analyte bound to each bead was proportional to the median fluorescence intensity of the reporter signal, and was corrected by the standards provided in the kit (Bio-Rad). Data were expressed in pg ml−1.
dN supplementation
Following exposure to SARS-CoV-2 (0.1 MOI) for 1 h, Huh7 were washed once with PBS 1× and then incubated with fresh DMEM containing 2% of heat-inactivated FBS supplemented with 5 μM deoxy-adenosine (dA), deoxy-cytidine (dC) and deoxy-guanidine (dG)90 (D8668, D0776 and D0901, Sigma) for 48 h before being collected for downstream analyses. dN supplementation in Calu-3 was carried out by incubating cells with medium containing 50 μM dA and dC, 5 μM dG and deoxy-thymidine (dT, T1895, Sigma) for 24 h.
Ubiquitination assay
Forty-eight hours post plasmid transfection, Huh7 were lysed with IP buffer (150 mM KCl, 25 mM Tris–HCl pH 7.4, 5% glycerol, 0.5% NP40, 10 mM MgCl2 and 1 mM CaCl2), supplemented with 1× protease inhibitors (Roche) and 250 U ml−1 Benzonase (E1014-25KU, Sigma) for 45 min at 4 °C. Lysates were cleared by centrifugation and supernatants incubated overnight at 4 °C with 1:200 of anti-CHK1 (Supplementary Table 1), previously coupled with Protein G Dynabeads (10004D, ThermoFisher). After five washes with IP buffer, proteins were eluted with 1× LDS buffer (B0007, ThermoFisher), separated by 4–12% SDS–PAGE (ThermoFisher) and transferred onto nitrocellulose membrane (Amersham). Membrane was autoclaved before overnight blocking at 4 °C in 5% BSA TBS-T. After blocking, membrane was incubated with anti-Ubiquitin antibody (Supplementary Table 1) 1:1,000 in 5% BSA TBS-T for 1 h at room temperature.
SARS-CoV-2 N-protein expression and purification
pET28 plasmid bearing SARS-CoV-2 N-protein fused to a C-terminal His6-tag (a kind gift from S. Pasqualato, Human Technopole, Milan, Italy) was expressed in Escherichia coli BL21-CodonPlus (DE3)-RP (Agilent) upon induction with 500 µM isopropyl-β-d-1-thiogalactopyranoside for 16 h at 18 °C. Cells were resuspended in resuspension buffer (25 mM Tris pH 8, 500 mM NaCl, 5% glycerol, 2 mM β-mercaptoethanol and 10 mM imidazole), supplemented with protease inhibitors and TurboNuclease and sonicated. After polyethyleneimine addition and centrifugation, the supernatant was applied onto Ni-NTA agarose beads (Qiagen) and the His6-nucleocapsid protein was eluted in elution buffers containing 250 mM and 500 mM imidazole. The eluted protein was applied on a HiTrap Heparin HP column (Cytiva) and loaded on a Superdex 200 Increase 10/300 GL (Cytiva) pre-equilibrated in SEC buffer (25 mM Tris pH 8, 500 mM NaCl and 2 mM β-mercaptoethanol). Protein purity was assessed by Comassie blue SDS–PAGE as >90% pure.
Micro-injection and live imaging analysis
Before micro-injection experiments, both purified N-protein and acetylated BSA (ThermoFisher) were dialysed overnight against micro-injection buffer (25 mM Tris pH 8 and 150 mM NaCl). Micro-injection experiments were performed as already described17, with minor changes. An UltraVIEW VoX spinning-disk confocal system (PerkinElmer) with a motorized Luigs & Neumann SM7 micromanipulator was used. Glass borosilicate capillaries were pulled to a diameter of 0.7 μm using a Flaming/Brown micropipette puller Model P-1000 (Sutter Instrument) and loaded with 6 mg ml−1 N-protein or BSA solution as control, in presence of Rhodamine B Dextran (ThermoFisher) as marker for micro-injected cells. Samples were injected into cell nuclei 30 min after irradiation (2 Gy) using a FemtoJet pump (Eppendorf) at a constant pressure of 20 hPa. A z-stack was acquired every minute for 100 min total using a 60× oil-immersion objective (Nikon Plan Apo VC, 1.4 numerical aperture). To excite GFP and Rhodamine B, 488 nm and 561 nm lasers were used, respectively. Images in micro-injection experiments were acquired with Volocity 6.4.0. Quantification of 53BP1-GFP foci per nucleus per single frame was performed using the software CellProfiler 3.1.9. Foci number per nucleus was plotted along time, and the corresponding curve was fitted to an exponential function to determine the decay rate (k) using Prism 9.3.0 software.
Turbidity assay
Purified recombinant N-protein (5 mM) in micro-injection buffer (25 mM Tris pH 8, 150 mM NaCl) was mixed with increasing concentrations of RNA extracted from SARS-CoV-2-infected (48 h) or mock-infected Huh7. Turbidity of the solutions was measured at 350 nm using a NanoVue Plus Spectrophotometer (Cytiva) after 30 min, 2 h and 24 h of incubation at room temperature.
SARS-CoV-2 N IP
Huh7 were transfected with the plasmid encoding for N-protein40 or with an EV as control. Forty-eight hours post-transfection, cells were irradiated or not. One hour post-IR, cells were collected by trypsinization and washed twice in ice-cold 1× PBS. Cell pellets were lysed in IP buffer supplemented with 1× protease inhibitors (Roche), 0.5 mM dithiothreitol, 40 U ml−1 RNaseOUT and 1,000 U ml−1 DNase I (Roche), and incubated at room temperature for 15 min and at 4 °C for an extra 15 min with gentle rotation. Lysates were cleared by centrifugation at maximum speed for 20 min at 4 °C. After addition of 5 mM EDTA (pH 8), lysates were incubated overnight at 4 °C with 5 μg of anti-N-protein (40588-T62 Sino Biological) or with normal rabbit IgGs (Cell Signaling), which were previously bound to Dynabeads Protein G (ThermoFisher). After five washes with 1 ml of IP buffer, immunoprecipitated proteins were analysed by immunoblotting.
RIP and analysis
RIP and dilncRNA analysis was carried out in NIH2/4 as previously described16, with minor modifications. Briefly, I-SceI-GR-expressing NIH2/4 cells were transfected with the plasmid encoding for N-protein or with an EV as a control at 24 h before triamcinolone acetonide 0.1 µM (Sigma-Aldrich) administration. IP was performed using 5 μg of anti-N-protein, or 10 μg of anti-53BP1 (Supplementary Table 1), or with normal rabbit IgGs (Cell Signaling) as a mock IP.
NHEJ repair assay
Evaluation of repair efficiency by NHEJ following viral N-protein expression was conducted as shown previously56, with minor modification: HA-I-SceI expressing plasmid was transfected along with the plasmid encoding for SARS-CoV-2 N-protein.
Immunolocalization, in situ RNA hybridization and analyses in mouse and human paraffin-embedded tissues
IHC staining was performed as previously reported91. Double IHC staining was conducted by applying SignalStainBoost IHC Detection (#18653; #31926, Cell Signaling) and Vulcan Fast Red as substrate chromogen.
For combined ISH and IHC staining, SARS-CoV-2 RNA probe hybridization (RNAscope Probe - V-nCoV2019-S; 848561; ACD) was performed using RNAscope 2.5 HD Detection Reagent-BROWN (ACD), following the manufacturer’s protocol. After blocking, samples were incubated with primary antibodies (Supplementary Table 1) following by SignalStainBoost IHC Detection and Vulcan Fast Red. Nuclei were counter-stained with Gill’s Hematoxylin n.1 (Bio-Optica). Quantitative analyses of IHC experiments were performed by calculating the average percentage of positive signals in five non-overlapping fields at medium-power magnification (×200) using the Nuclear v9 or Positive Pixel Count v9 ImageScope software (v12.3.2.8013, Leica Biosystems). Combined ISH/IHC images were analysed through the use of a segmentation-based software (HALO v3.5.3577.140, Indica Labs), quantifying the percentage of positive cells.
Double-marker immunofluorescence was conducted as previously described92.
Statistics and reproducibility
All the data are shown as mean ± standard error of the mean (s.e.m.) of three independent experiments, unless stated otherwise in the figure legends. We did not use any criteria to determine the sample size. As much data as possible was collected depending on the nature of the experiments or to allow statistical analysis. Where single-cell analysis was performed, we scored at least 30 cells per condition in each biological replicate. Throughout the manuscript, no data were excluded. Only in rare occasions were individual data points removed following unbiased criteria of outlier identification using Prism 9.3.0 software. In addition, in the case of IHC staining in infected mouse lungs, we excluded the tissue sections that show low rates of SARS-CoV-2 infection. The number of animals and replicates are indicated in each figure legend. Animals were randomized to the experimental groups. For in vitro experiments, wells were randomly assigned into each group and all cells were analysed equally. No blinding method was applied. No statistical method was used to pre-determine sample sizes. Ordinary one-way analysis of variance (ANOVA) with Dunnet’s post hoc test was applied in Figs. 1b and 2b and Extended Data Figs. 1b, 2d–f, 4d and 5c. Unpaired two-tailed t-test was applied in Figs. 1d,f, 2d, 6b,d,g, 7b and 8b,d,f,h and Extended Data Figs. 1d,f,h,o, 2b,n,o,p,r, 5b, 6d,f and 7b. Multiple paired two-tailed t-test was applied in Extended Data Fig. 1j. Ordinary one-way ANOVA with Fisher’s least significant difference test was applied in Figs. 2j,l,n,o, 3d, 4h and 5b,f and Extended Data Figs. 1m and 2q. Ordinary one-way ANOVA with Šidák’s post hoc test was applied in Figs. 2e, 4j and 6f,i,j. Ordinary two-way ANOVA with Šidák’s post hoc test was applied in Fig. 2g and Extended Data Fig. 5d. Ratio paired two-tailed t-test was applied in Figs. 2h and 6h. Ordinary two-way ANOVA with Bonferroni’s post hoc test was applied in Extended Data Fig. 2k. Repeated measures one-way ANOVA with Fisher’s least significant difference test was applied in Extended Data Fig. 2s. Ordinary one-way ANOVA with Tukey’s post hoc test was applied in Figs. 3b, 4b,d,f and 5d and Extended Data Figs. 4b and 5k.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper, available online for Figs. 1–8 and Extended Data Figs. 1–8. All other data that support the findings of this study are available from the corresponding author on reasonable request.
References
V’kovski, P., Kratzel, A., Steiner, S., Stalder, H. & Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 19, 155–170 (2021).
Yang, H. & Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 19, 685–700 (2021).
Choi, Y., Bowman, J. W. & Jung, J. U. Autophagy during viral infection—a double-edged sword. Nat. Rev. Microbiol. 16, 341–354 (2018).
Luo, H. Interplay between the virus and the ubiquitin–proteasome system: molecular mechanism of viral pathogenesis. Curr. Opin. Virol. 17, 1–10 (2016).
Lilley, C. E., Schwartz, R. A. & Weitzman, M. D. Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol. 15, 119–126 (2007).
Ryan, E. L., Hollingworth, R. & Grand, R. J. Activation of the DNA damage response by RNA viruses. Biomolecules 6, 2–24 (2016).
Bouhaddou, M. et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 182, 685–712.e19 (2020).
Victor, J. et al. SARS-CoV-2 triggers DNA damage response in Vero E6 cells. Biochem. Biophys. Res. Commun. 579, 141–145 (2021).
Lipskaia, L. et al. Evidence that SARS-CoV-2 induces lung-cell senescence: potential impact on COVID-19 lung disease. Am. J. Respir. Cell Mol. Biol. https://doi.org/10.1165/rcmb.2021-0205le (2021).
D’Agnillo, F. et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci. Transl. Med. 13, 1–18 (2021).
Jackson, S. P. & Bartek, J. The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (2009).
Polo, S. E. & Jackson, S. P. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 25, 409–433 (2011).
d’Adda di Fagagna, F. Living on a break: cellular senescence as a DNA-damage response. Nat. Rev. Cancer 8, 512–522 (2008).
Di Micco, R., Krizhanovsky, V., Baker, D. & d’Adda di Fagagna, F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 22, 75–95 (2021).
Rodier, F. et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 (2009).
Michelini, F. et al. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat. Cell Biol. 19, 1400–1411 (2017).
Pessina, F. et al. Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat. Cell Biol. 21, 1286–1299 (2019).
Francia, S. et al. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature https://doi.org/10.1038/nature11179 (2012).
Kilic, S. et al. Phase separation of 53 BP 1 determines liquid-like behavior of DNA repair compartments. EMBO J. https://doi.org/10.15252/embj.2018101379 (2019).
D’Alessandro, G. et al. BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nat. Commun. 9, 5376 (2018).
Savastano, A., de Opakua, A. I., Rankovic, M. & Zweckstetter, M. Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat. Commun. https://doi.org/10.1101/2020.06.18.160648 (2020).
Perdikari, T. M. et al. SARS-CoV-2 nucleocapsid protein phase-separates with RNA and with human hnRNPs. EMBO J. https://doi.org/10.15252/embj.2020106478 (2020).
Naruyama, H. et al. Essential role of Chk1 in S phase progression through regulation of RNR2 expression. Biochem. Biophys. Res. Commun. 374, 79–83 (2008).
Schmitt, C. A. et al. COVID-19 and cellular senescence. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-022-00785-2 (2022).
Schmidt, N. et al. The SARS-CoV-2 RNA–protein interactome in infected human cells. Nat. Microbiol. https://doi.org/10.1038/s41564-020-00846-z (2020).
Kratzel, A. et al. A genome-wide CRISPR screen identifies interactors of the autophagy pathway as conserved coronavirus targets. PLoS Biol. 19, e300149 (2021).
Koç, A., Wheeler, L. J., Mathews, C. K. & Merrill, G. F. Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J. Biol. Chem. 279, 223–230 (2004).
Shechter, D., Costanzo, V. & Gautier, J. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair 3, 901–908 (2004).
Bestle, D. et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 3, e202000786 (2020).
Li, Y. et al. SARS-CoV-2 induces double-stranded RNA-mediated innate immune responses in respiratory epithelial-derived cells and cardiomyocytes. Proc. Natl Acad. Sci. USA 118, e2022643118 (2021).
Harding, S. M. et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 (2017).
Thomsen, M. K. et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology 64, 746–759 (2016).
Lee, S. et al. Virus-induced senescence is driver and therapeutic target in COVID-19. Nature 599, 283–289 (2021).
Evangelou, K. et al. Pulmonary infection by SARS-CoV-2 induces senescence accompanied by an inflammatory phenotype in severe COVID-19: possible implications for viral mutagenesis. Eur. Respir. J. https://doi.org/10.1183/13993003.02951-2021 (2022).
González Besteiro, M. A. et al. Chk1 loss creates replication barriers that compromise cell survival independently of excess origin firing. EMBO J. 38, 1–16 (2019).
Zhang, Y. W., Jones, T. L., Martin, S. E., Caplen, N. J. & Pommier, Y. Implication of checkpoint kinase-dependent up-regulation of ribonucleotide reductase R2 in DNA damage response. J. Biol. Chem. 284, 18085–18095 (2009).
Sansam, C. L. et al. DTL/CDT2 is essential for both CDT1 regulation and the early G2/M checkpoint. Genes Dev. 20, 3117–3129 (2006).
Higa, L. A. A., Mihaylov, I. S., Banks, D. P., Zheng, J. & Zhang, H. Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat. Cell Biol. 5, 1008–1015 (2003).
Hu, J., McCall, C. M., Ohta, T. & Xiong, Y. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat. Cell Biol. 6, 1003–1009 (2004).
Gordon, D. E. et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020).
Miorin, L. et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl Acad. Sci. USA 117, 28344–28354 (2020).
Addetia, A. et al. SARS-CoV-2 orf6 disrupts bidirectional nucleocytoplasmic transport through interactions with rae1 and nup98. mBio 12, e00065-21 (2021).
Wang, J., Han, X., Feng, X., Wang, Z. & Zhang, Y. Coupling cellular localization and function of checkpoint kinase 1 (Chk1) in checkpoints and cell viability. J. Biol. Chem. 287, 25501–25509 (2012).
Sui, C. et al. SARS-CoV-2 NSP13 inhibits type I IFN production by degradation of TBK1 via p62-dependent selective autophagy. J. Immunol. 208, 753–761 (2022).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 17, 1–382 (2021).
Green, D. R. & Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 157, 65–75 (2014).
Chen, H. et al. Liquid–liquid phase separation by SARS-CoV-2 nucleocapsid protein and RNA. Cell Res. https://doi.org/10.1038/s41422-020-00408-2 (2020).
Wang, J., Shi, C., Xu, Q. & Yin, H. SARS-CoV-2 nucleocapsid protein undergoes liquid–liquid phase separation into stress granules through its N-terminal intrinsically disordered region. Cell Discov. 7, 3–7 (2021).
Wang, S. et al. Targeting liquid–liquid phase separation of SARS-CoV-2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat. Cell Biol. 23, 718–732 (2021).
Bekker-Jensen, S., Lukas, C., Melander, F., Bartek, J. & Lukas, J. Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. J. Cell Biol. 170, 201–211 (2005).
Tsuji, S. et al. SARS-CoV-2 infection triggers paracrine senescence and leads to a sustained senescence-associated inflammatory response. Nat. Aging 2, 115–124 (2022).
Liu, X. et al. SARS‐CoV‐2–host proteome interactions for antiviral drug discovery. Mol. Syst. Biol. 17, 1–26 (2021).
V’kovski, P. et al. Disparate temperature-dependent virus–host dynamics for SARS-CoV-2 and SARS-CoV in the human respiratory epithelium. PLoS Biol. 19, e3001158 (2021).
Zimmermann, M. & De Lange, T. 53BP1: pro choice in DNA repair. Trends Cell Biol. 24, 108–117 (2014).
Gunn, A. & Stark, J. M. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol. Biol. 531, 588 (2012).
Gioia, U. et al. Pharmacological boost of DNA damage response and repair by enhanced biogenesis of DNA damage response RNAs. Sci. Rep. 9, 6460 (2019).
Shamanna, R. A. et al. WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat. Commun. 7, 1–12 (2016).
Bussani, R. et al. Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced COVID-19 pathology. EBioMedicine 61, 103104 (2020).
Weitzman, M. D., Lilley, C. E. & Chaurushiya, M. S. Genomes in conflict: maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 64, 61–81 (2010).
Banerjee, A. K. et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183, 1325–1339.e21 (2020).
Blanco-Melo, D. et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181, 1036–1045.e9 (2020).
Ren, H. et al. Micronucleus production, activation of DNA damage response and cGAS-STING signaling in syncytia induced by SARS-CoV-2 infection. Biol. Direct 16, 1–10 (2021).
Zhou, Z. et al. Sensing of cytoplasmic chromatin by cGAS activates innate immune response in SARS-CoV-2 infection. Signal Transduct. Target. Ther. https://doi.org/10.1038/s41392-021-00800-3 (2021).
Jiang, H. & Mei, Y. F. SARS-CoV -2 spike impairs DNA damage repair and inhibits V(D)J recombination in vitro. Viruses https://doi.org/10.3390/v13102056 (2021).
Garcia, G. et al. Antiviral drug screen identifies DNA-damage response inhibitor as potent blocker of SARS-CoV-2 replication. Cell Rep. 35, 108940 (2021).
Turnell, A. S. & Grand, R. J. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 93, 2076–2097 (2012).
Ma-Lauer, Y. et al. P53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc. Natl Acad. Sci. USA 113, E5192–E5201 (2016).
Lilley, C. E. et al. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 29, 943–955 (2010).
Trimarchi, J. M. & Lees, J. A. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 3, 11–20 (2002).
Gong, C. et al. ATR-CHK1-E2F3 signaling transactivates human ribonucleotide reductase small subunit M2 for DNA repair induced by the chemical carcinogen MNNG. Biochim. Biophys. Acta Gene Regul. Mech. 1859, 612–626 (2016).
Kim, D. et al. The architecture of SARS-CoV-2 transcriptome. Cell 181, 914–921.e10 (2020).
Anacker, D. C. et al. HPV31 utilizes the ATR-Chk1 pathway to maintain elevated RRM2 levels and a replication-competent environment in differentiating keratinocytes. Virology 499, 383–396 (2016).
Timani, K. A. et al. Nuclear/nucleolar localization properties of C-terminal nucleocapsid protein of SARS coronavirus. Virus Res. 114, 23–34 (2005).
Rowland, R. R. R. et al. Intracellular localization of the severe acute respiratory syndrome coronavirus nucleocapsid protein: absence of nucleolar accumulation during infection and after expression as a recombinant protein in Vero cells. J. Virol. 79, 11507–11512 (2005).
Gussow, A. B. et al. Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. Proc. Natl Acad. Sci. USA 117, 15193–15199 (2020).
Ahmadi, A. & Moradi, S. In silico analysis suggests the RNAi-enhancing antibiotic enoxacin as a potential inhibitor of SARS-CoV-2 infection. Sci. Rep. 11, 1–14 (2021).
Mehta, P. et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395, 1033–1034 (2020).
Li, T. & Chen, Z. J. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 215, 1287–1299 (2018).
Aird, K. M. et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 3, 1252–1265 (2013).
Mavrikaki, M., Lee, J. D., Solomon, I. H. & Slack, F. J. Severe COVID-19 is associated with molecular signatures of aging in the human brain. Nat. Aging 2, 1130–1137 (2022).
Cao, X. et al. Accelerated biological aging in COVID-19 patients. Nat. Commun. 13, 1–7 (2022).
Fumagalli, M. et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365 (2012).
Rossiello, F., Herbig, U., Longhese, M. P. & Fumagalli, M. & d’Adda di Fagagna, F. Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr. Opin. Genet. Dev. 26, 89–95 (2014).
Nalbandian, A. et al. Post-acute COVID-19 syndrome. Nat. Med. 27, 601–615 (2021).
Fumagalli, V. et al. Administration of aerosolized SARS-CoV-2 to K18-hACE2 mice uncouples respiratory infection from fatal neuroinvasion. Sci. Immunol. 7, 1–11 (2022).
Licastro, D. et al. Isolation and full-length genome characterization of SARS-CoV- 2 from COVID-19 cases in Northern Italy. J. Virol. 94, 9–12 (2020).
Dimri, G. P. et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA 92, 9363–9367 (1995).
Frangini, M. et al. Synthesis of mitochondrial DNA precursors during myogenesis, an analysis in purified C2C12 myotubes. J. Biol. Chem. 288, 5624–5635 (2013).
Ferraro, P., Franzolin, E., Pontarin, G., Reichard, P. & Bianchi, V. Quantitation of cellular deoxynucleoside triphosphates. Nucleic Acids Res. 38, e85–e85 (2010).
Pontarin, G., Ferraro, P., Bee, L., Reichard, P. & Bianchi, V. Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc. Natl Acad. Sci. USA 109, 13302–13307 (2012).
Morello, G. et al. T cells expressing receptor recombination/revision machinery are detected in the tumor microenvironment and expanded in genomically over-unstable models. Cancer Immunol. Res. 9, 825–837 (2021).
Sepe, S. et al. DNA damage response at telomeres boosts the transcription of SARS-CoV-2 receptor ACE2 during aging. EMBO Rep. https://doi.org/10.15252/embr.202153658 (2021).
Acknowledgements
We thank N. J. Krogan (UCSC, San Francisco, CA, USA), S. Pasqualato (Human Technopole, Milan, Italy) and L. Miorin (Icahn School of Medicine at Mount Sinai, New York, NY, USA) for reagents; S. Martone and M. G. Totaro (IFOM, Milan, Italy) for technical support with flow cytometry analyses; S. Magni, E. Martini and D. Parazzoli (IFOM, Milan, Italy) for support with imaging analyses; L. Falbo (IFOM, Milan, Italy) for providing antibodies; E. Maspero and S. Polo (IFOM, Milan, Italy) for suggestions for ubiquitination analyses; S. Martin and S. Santaguida (IEO, Milan, Italy) for support with autophagy studies and reagents; V. Cianfanelli, V. Nazio and F. Cecconi for support with autophagy studies; F. Giavazzi (Università degli Studi di Milano, Segrate, Italy) for helpful advice with micro-injection analyses. M.I. is supported by the European Research Council (ERC) Consolidator Grant (725038), ERC Proof of Concept Grant (957502), Italian Association for Cancer Research (AIRC) Grants (19891, 22737), Italian Ministry of Health Grant (RF-2018-12365801) and Funded Research Agreements from Gilead Sciences, Asher Bio, Takis Biotech and Vir Biotechnology. V.F. is supported by a donation from FONDAZIONE PROSSIMO MIO and from AIRC Fellowship for Italy (26813-2021). C.T. laboratory is supported by AIRC 5 × 1000 2019 (22759). The work in A.M. laboratory related to SARS-CoV-2 is supported by grants from SNAM Foundation, Generali SpA, Beneficentia Stiftung, CARIPLO (INNATE-CoV) and the #FarmaCovid crowdfunding initiative. S.Z. laboratory is supported by AIRC IG (24529). F.d’A.d.F laboratory is supported by: ERC advanced grant (TELORNAGING—835103); AIRC IG (21762); Telethon (GGP17111); AIRC 5 × 1000 (21091); ERC PoC grant (FIREQUENCER—875139); Progetti di Ricerca di Interesse Nazionale (PRIN) 2015 ‘ATR and ATM-mediated control of chromosome integrity and cell plasticity’; Progetti di Ricerca di Interesse Nazionale (PRIN) 2017 ‘RNA and genome Instability’; Progetto AriSLA 2021 ‘DDR & ALS’; POR FESR 2014–2020 Regione Lombardia (InterSLA project); FRRB—Fondazione Regionale per la Ricerca Biomedica—under the frame of EJP RD, the European Joint Programme on Rare Diseases with funding from the European Union’s Horizon 2020 research and innovation programme under the EJP RD COFUND-EJP NO 825575. S.T. was a PhD student within the European School of Molecular Medicine (SEMM).
Author information
Authors and Affiliations
Contributions
U.G. conceived, conducted and analysed RIP, co-IP, comet, ubiquitination and NHEJ repair assays; performed immunoblot analyses; expressed individual SARS-CoV-2 proteins in cultured cells and analysed their impact on DDR activation by immunoblot; designed, carried out and analysed the experiments upon CHK1 knockdown by RNAi; analysed all the remaining data; wrote and edited the manuscript. S.T. performed immunofluorescence staining of infected and transfected cultured cells, carried out the immunostaining for flow-cytometry analyses, generated cDNA from infected cultured cells, conceived the autophagy experiments, contributed to design the multiplex immunoassays and the dN supplementation experiments, and wrote and edited the manuscript. P.M.-O. and S.R. performed all the SARS-CoV-2 infection assays, measured virus by immunoblot, immunofluorescence, plaque assay and RT–PCR and prepared samples for analyses. P.M.-O. conducted SA-β-gal assay. G.C. performed IHC analyses on mouse and human specimens. A.C. and P.M.-O. performed the analyses of DDR activation in infected HNEpC. A.C. and A.P. carried out FISH in infected HNEpC, IHC and immunofluorescence analyses in human lung and nasal mucosa tissues. M.Ce. produced recombinant SARS-CoV-2 N and performed micro-injection experiments and turbidity assays. M.Ca. purified and analysed by RT–qPCR the RNA of infected cultured cells and CHK1-depleted cells. A.C.H. provided technical support with immunoblots and immunofluorescence. V.F. propagated SARS-CoV-2, infected hACE2 mice and generated lung tissues from such animals. E.P. performed the analysis of dNTP levels in infected cultured cells. N.I. performed Bio-Plex multiplex immunoassays. F.P. contributed to set up IHC in mouse specimens. V.M. contributed to the analyses of RNA of infected cultured cells by RT–qPCR. S.S. generated lung samples from irradiated or control mice. M.I.C. analysed flow cytometry experiments. S.B. and Z.L. assisted M.Ce. in conducting and analysing micro-injection experiments. T.C. assisted in the management of the BSL3 facility and virus stock preparation. M.C.V. prepared and characterized nasal primary cells. P.C. designed Bio-Plex multiplex immunoassays, supervised N.I. and edited the manuscript. M.I. supervised V.F. C.R. designed the quantification of dNTP levels, the dN supplementation experiments, supervised E.P. and edited the manuscript. R.B. performed post-mortem pathology on human lungs. C.T. supervised G.C. and edited the manuscript. S.Z. coordinated the analysis of human tissues, supervised A.C., A.P. and M.C.V., and edited the manuscript. A.M. coordinated the virology aspects of the work, supervised P.M.-O., S.R. and T.C., and edited the manuscript. F.d’A.d.F. conceived the study, coordinated experimental activities, and assembled and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.I. participates in advisory boards/consultancies for Gilead Sciences, Roche, Third Rock Ventures, Antios Therapeutics, Asher Bio, Amgen, Allovir. All the other authors declare no competing interests.
Peer review
Peer review information
Nature Cell Biology thanks Fangfang Zhou and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 SARS-CoV-2 induces DNA damage, inflammation and cellular senescence.
A) Immunoblotting of Calu-3, infected (V+ ) or not (V−) with SARS-CoV-2. Calu-3 exposed or not (NT) to HU or IR were used as controls. Infection was monitored by probing for N-protein. B) Quantification of protein levels shown in A; values are relative to V−. C) IF images of HNEpC; nuclei were stained with Hoechst. D) Quantification of the percentage of γH2AX or pRPAS4/8 positive cells shown in C. E) Images of comet assays of Calu-3. Scale bar, 50 μm. F) Quantification of comet tail moment shown in E. Horizontal bars represent the median values ± 95% CI of three independent infections. G) IF images of Calu-3; nuclei were stained with DAPI; arrows point to cGAS+ micronuclei. H) Percentage of cells positive for cGAS dots, micronuclei and cGAS+ micronuclei shown in G. Values are the means ± s.e.m. I) Immunoblots from samples described in Fig. 1A. The experiment was repeated three times with similar results. J) RT–qPCR for pro-inflammatory cytokines expression in Calu-3 and Huh7, 24 h and 48 h after infection, respectively. Values are the means ± s.e.m. of four independent experiments, except for IL6 and IL8 in Huh7 (n = 3) and shown as relative to V−. K) Immunoblotting for cleaved Caspase-3 (cCASP3) from Huh7 lysates. Valproic acid (VPA)-treated HCT116 were used as positive control. Tubulin was used as loading control. The experiment was repeated three times with similar results. L) Images of Huh7 and HNEpC stained for SA-β-gal; HU-treated cells were used as positive control. M) Quantification of positive cells shown in L. N) IF images of HNEpC. O) Percentage of P21- or KI67-positive cells. Scale bar, 10 μm for panels C, G, L, N. Source numerical data and unprocessed blots are available in source data.
Extended Data Fig. 2 SARS-CoV-2 causes CHK1 and RRM2 reduction leading to cell cycle progression impairment.
A) IF images of infected HNEpC; nuclei were stained with Hoechst. Scale bar, 10 μm. B) Quantification of the percentage of RRM2-expressing cells in infected (SARS-CoV-2 N + ) or not (SARS-CoV-2 N-) HNEpC. C) Immunoblotting of whole cell lysates of Calu-3 infected, or not, with SARS-CoV-2 and analyzed at different time points post-infection. D) Quantification of protein levels shown in C; values are shown as relative to mock-infected samples. E,F) RT–qPCR of CHK1 and RRM2 mRNA expression in infected (V+ ) or mock-infected (V−) Huh7 and Calu-3 cells, respectively. G) Immunoblotting of CDT1 in Huh7 and Calu-3 cells treated as indicated. The experiment was repeated three times with similar results. Source numerical data and unprocessed blots are available in source data.
Extended Data Fig. 3 CHK1 depletion is sufficient to recapitulate the effects of SARS-CoV-2 infection.
A) Huh7 transfected with siRNAs against CHK1 mRNA (siCHK1) or siCTRL were stained for CHK1 and propidium iodide (PI) prior to flow cytometry. B) DNA content analysis of V− or V+ Huh7 fixed 48 h post-infection and siCHK1- or siCTRL-transfected Huh7. C) Histograms show the percentage of cells in each phase of the cell cycle upon siCHK1 or siCTRL treatment. D) Bivariate plot showing DNA content (PI) and BrdU incorporation measured by flow cytometry of V− or V+ Huh7 fixed 48 h post-infection and siCHK1- or siCTRL-transfected Huh7. E) Immunoblots of siCHK1- or siCTRL-transfected Huh7. F) Quantification of protein levels shown in E; values are the means ± s.e.m. of two independent experiments and shown as relative to the siCTRL-transfected sample. G) IF images of Calu-3 transfected with the indicated siRNAs; nuclei were stained with DAPI. Scale bar, 10 μm. H) Quantification of γH2AX foci per cell shown in G. I) IF images of cGAS staining in samples as in G; nuclei were stained with DAPI. Scale bar, 10 μm. J) Micronuclei and cGAS+ micronuclei quantifications on total cell number; at least 300 nuclei were scored for each sample. K,L) RT–qPCR for pro-inflammatory cytokines and CHK1 mRNA expression in siCHK1-treated Calu-3 and Huh7 cells, respectively. Values are shown as relative to siCTRL-transfected samples. M) Quantification of the amounts of secreted cytokines and chemokines from siCHK1- or siCTRL-transfected Calu-3 by Bio-Plex multiplex immunoassays. Source numerical data and unprocessed blots are available in source data.
Extended Data Fig. 4 Impact of SARS-CoV-2 proteins on CHK1 and RRM2 expression.
A) Immunoblotting of whole cell lysates from Huh7 cells transfected with plasmids encoding for the indicated SARS-CoV-2 proteins and stained for CHK1 and RRM2. Predominant bands at the expected molecular weights are indicated by red arrows. B) Quantification of CHK1 and RRM2 protein levels shown in A; values are the means of two independent experiments, except for NSP13 and ORF6 (n = 4); protein levels are normalized to either vinculin or tubulin protein amounts (house-keeper, HK) and shown as relative to the control-sample expressing GFP. Arrows indicate the viral proteins that most affected both CHK1 and RRM2 levels. Huh7 cells did not express detectable levels of the following viral proteins: NSP4, NSP11, ORF9c, ORF10, M and therefore CHK1 and RRM2 protein levels where not quantified. Source numerical data and unprocessed blots are available in source data.
Extended Data Fig. 5 Controls for autophagy experiments.
A) Representative IF images of autophagosome accumulation in Huh7 cells following GFP or viral NSP13 expression and treatment with the autophagy inhibitors BafA1 and CQ. Autophagosomes were visualized by probing for P62; nuclei were stained with DAPI. Scale bar, 10 μm. B) Quantification of the area (μm2) of cytoplasmic P62+ aggregates shown in A. At least 100 cells were scored for each sample. C) Representative immunoblots showing the efficiency of BECN1 and LC3B knock-down in Huh7 cells expressing NSP13; as control, cells were transfected with an EV. D) Quantification of LC3 and BECN1 protein levels shown in C. Values are shown as relative to samples transfected with a non-targeting siRNA and with the EV siCTRL. Vinculin was used as a loading control. Source numerical data and unprocessed blots are available in source data.
Extended Data Fig. 6 SARS-CoV-2 N-protein impairs 53BP1 foci formation in a RNA-dependent manner.
A) IF images of Calu-3 cells fixed 24 h post-infection; nuclei were stained with DAPI. Scale bar, 10 μm. B) Quantification of DDR foci shown in A; the dot-plots show the number of γH2AX or 53BP1 foci per nucleus; horizontal bars are the means ± s.e.m. of four independent infections. C) Immunoblot of infected Huh7 shown in Fig. 1A; tubulin was used as loading control. The histograms show the quantification of total 53BP1 levels; values are shown as relative to mock-infected samples. D) Quantification of 53BP1 foci per nucleus in irradiated 53BP1-GFP after micro-injection of purified recombinant N-protein or BSA as control (n = 6). E) Protein lysates of Huh7 cells transfected with N-protein or EV, irradiated (IR) or not (NI), were incubated with an anti-N-protein antibody (IP: N) or with normal rabbit IgGs (IgG) and analyzed by immunoblotting for the presence of 53BP1. The experiment was repeated three times with similar results. F) Representative pictures of turbidity assays. G) Analysis of turbidity represented in F; n = 4 independent experiments. H) Immunoprecipitation efficiency of the RIP experiments shown in Fig. 6H,I. Asterisk marks unspecific signal. The experiment was repeated three times with similar results. I) IF images of samples described in Fig. 6J. The experiment was repeated three times with similar results. Scale bar, 50 μm. J) IF images of Huh7 fixed 48 h post-infection and stained for N-protein. Arrows indicate cells with nuclear N-protein signal. Scale bar, 10 μm. K) Quantification of N-protein levels in the nucleus (n) and cytoplasm (c) of the samples described in J. Horizontal bars represent the means ± s.e.m. of a representative experiment of infection. Source numerical data and unprocessed blots are available in source data.
Extended Data Fig. 7 SARS-CoV-2 infection induces senescence in hACE2-mouse lungs.
A) Mouse body weight was monitored daily for 7 days and expressed as the percentage relative to day -1. Single values of PBS-control mice (black dots) and infected mice (red dots) are represented. B) Quantification of SARS-CoV-2 RNA in the lung of PBS-treated control (n = 3) and SARS-CoV-2 infected mice (n = 3) measured at 6 days post-infection. Values are expressed as copy number per ng of total RNA. C) IHC images of sections (n = 3) of the lungs from irradiated (IR, n = 1) or not irradiated (NI, n = 1) wild-type mice analyzed as controls of DDR activation. Scale bar, 20 μm. D) Quantification of DDR activation represented in C, each dot corresponds to a section. E) IHC images of lungs from SARS-CoV-2 infected hACE2-mice or mock-infected (PBS) wild-type mice probed for p16 or p21. Scale bar, 50 μm. F) Quantification of the percentage of cells positive for the markers shown in E, each dot corresponds to a mouse. G) IF images of a representative experiment of lungs, from SARS-CoV-2 infected hACE2-mice probed for the indicated proteins. CD68 and prosurfactant protein C (proSP-C) markers were used to label macrophages and pulmonary alveolar type II cells, respectively. Scale bar, 50 μm. The experiment was repeated three times with similar results. Source numerical data are available in source data.
Extended Data Fig. 8 SARS-CoV-2 infection induces senescence in COVID-19 patients’ lungs.
A) Representative IHC images of lung tissues stained for the senescence markers P16 (n = 5) or P21 (non-COVID individuals: n = 5; COVID-19 patients: n = 10); the presence of the virus was assessed by staining for SARS-CoV-2 N or by probing the viral genome through RNA ISH; nuclei were stained with hematoxylin (light blue); scale bar, 50 μm. Conditions are as in Fig. 8A. B) Quantification of the expression of the senescence markers determined in A. Values are the means ± s.e.m.; each dot corresponds to one individual. Source numerical data are available in source data. C) Gating strategy for flow cytometry.
Supplementary information
Supplementary Table 1
Tables listing the antibodies and the RT–qPCR primers used in this work.
Supplementary Video 1
Movie showing 53BP1-GFP foci over time upon micro-injection of 53BP1-GFP U2OS nuclei with recombinant SARS-CoV-2 N-protein or BSA as control. Arrows mark micro-injected cells. Images relative to pre-injected samples are also shown. The video was recorded at one frame per minute for 100 min total. The experiment was repeated three times with similar results.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 1
Unprocessed western blots.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 2
Unprocessed western blots.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 4
Unprocessed western blots.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 5
Unprocessed western blots.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 7
Statistical source data.
Source Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 1
Unprocessed western blots.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 2
Unprocessed western blots.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 3
Unprocessed western blots.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 4
Unprocessed western blots.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed western blots.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 6
Unprocessed western blots.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gioia, U., Tavella, S., Martínez-Orellana, P. et al. SARS-CoV-2 infection induces DNA damage, through CHK1 degradation and impaired 53BP1 recruitment, and cellular senescence. Nat Cell Biol 25, 550–564 (2023). https://doi.org/10.1038/s41556-023-01096-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41556-023-01096-x
This article is cited by
-
The interplay of aging, adipose tissue, and COVID-19: a potent alliance with implications for health
GeroScience (2024)
-
GrimAge is elevated in older adults with mild COVID-19 an exploratory analysis
GeroScience (2024)
-
Senolytic therapy alleviates physiological human brain aging and COVID-19 neuropathology
Nature Aging (2023)
