
Abstract
Stem cells need to be protected from genotoxic and proteotoxic stress to maintain a healthy pool throughout life1,2,3. Little is known about the proteostasis mechanism that safeguards stem cells. Here we report endoplasmic reticulum-associated degradation (ERAD) as a protein quality checkpoint that controls the haematopoietic stem cell (HSC)–niche interaction and determines the fate of HSCs. The SEL1L–HRD1 complex, the most conserved branch of ERAD4, is highly expressed in HSCs. Deletion of Sel1l led to niche displacement of HSCs and a complete loss of HSC identity, and allowed highly efficient donor-HSC engraftment without irradiation. Mechanistic studies identified MPL, the master regulator of HSC identity5, as a bona fide ERAD substrate that became aggregated in the endoplasmic reticulum following ERAD deficiency. Restoration of MPL signalling with an agonist partially rescued the number and reconstitution capacity of Sel1l-deficient HSCs. Our study defines ERAD as an essential proteostasis mechanism to safeguard a healthy stem cell pool by regulating the stem cell–niche interaction.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
Previously published sequencing data that were re-analysed here are available under accession codes GSE60101 for Fig. 1a (ref. 7) and GSE109125 for Extended Data Fig. 2b (ref. 6). All other data supporting the findings of this study are available from the corresponding author on reasonable request. Source data are provided with this paper.
References
Garcia-Prat, L., Sousa-Victor, P. & Munoz-Canoves, P. Proteostatic and metabolic control of stemness. Cell Stem Cell 20, 593–608 (2017).
Vilchez, D., Saez, I. & Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 5, 5659 (2014).
Bakker, S. T. & Passegue, E. Resilient and resourceful: genome maintenance strategies in hematopoietic stem cells. Exp. Hematol. 41, 915–923 (2013).
Brodsky, J. L. Cleaning up: ER-associated degradation to the rescue. Cell 151, 1163–1167 (2012).
Morrison, S. J. & Scadden, D. T. The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334 (2014).
Yoshida, H. et al. The cis-regulatory atlas of the mouse immune system. Cell 176, 897–912 (2019).
Lara-Astiaso, D. et al. Chromatin state dynamics during blood formation. Science 345, 943–949 (2014).
Schinzel, R. T. et al. The hyaluronidase, TMEM2, promotes ER homeostasis and longevity independent of the UPRER. Cell 179, 1306–1318 (2019).
Ehninger, A. et al. Loss of SPARC protects hematopoietic stem cells from chemotherapy toxicity by accelerating their return to quiescence. Blood 123, 4054–4063 (2014).
Sigurdsson, V. et al. Bile acids protect expanding hematopoietic stem cells from unfolded protein stress in fetal liver. Cell Stem Cell 18, 522–532 (2016).
van Galen, P. et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 510, 268–272 (2014).
Miharada, K., Sigurdsson, V. & Karlsson, S. Dppa5 improves hematopoietic stem cell activity by reducing endoplasmic reticulum stress. Cell Rep. 7, 1381–1392 (2014).
Karagoz, G. E., Acosta-Alvear, D. & Walter, P. The unfolded protein response: detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 11, a033886 (2019).
Sun, S. et al. IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 17, 1546–1555 (2015).
Qi, L., Tsai, B. & Arvan, P. New Insights into the physiological role of endoplasmic reticulum-associated degradation. Trends Cell Biol. 27, 430–440 (2017).
Cortez, L. & Sim, V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion 8, 197–202 (2014).
Zhao, N. et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Invest. 128, 1283–1299 (2018).
Sidrauski, C. et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife 2, e00498 (2013).
Pinho, S. & Frenette, P. S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 20, 303–320 (2019).
Walter, P. & Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 (2011).
Acar, M. et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature 526, 126–130 (2015).
Kunisaki, Y. et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643 (2013).
Ballmaier, M. & Germeshausen, M. Advances in the understanding of congenital amegakaryocytic thrombocytopenia. Br. J. Haematol. 146, 3–16 (2009).
Varghese, L. N., Defour, J. P., Pecquet, C. & Constantinescu, S. N. The thrombopoietin receptor: structural basis of traffic and activation by ligand, mutations, agonists, and mutated calreticulin. Front Endocrinol. 8, 59 (2017).
Fox, N. E., Lim, J., Chen, R. & Geddis, A. E. F104S c-Mpl responds to a transmembrane domain-binding thrombopoietin receptor agonist: proof of concept that selected receptor mutations in congenital amegakaryocytic thrombocytopenia can be stimulated with alternative thrombopoietic agents. Exp. Hematol. 38, 384–391 (2010).
Hidalgo San Jose, L. et al. Modest declines in proteome quality impair hematopoietic stem cell self-renewal. Cell Rep. 30, 69–80 (2020).
Signer, R. A., Magee, J. A., Salic, A. & Morrison, S. J. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54 (2014).
Iwawaki, T., Akai, R., Yamanaka, S. & Kohno, K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl Acad. Sci. USA 106, 16657–16662 (2009).
Lee, A. H., Scapa, E. F., Cohen, D. E. & Glimcher, L. H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320, 1492–1496 (2008).
Sun, S. et al. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl Acad. Sci. USA 111, E582–E591 (2014).
Murone, M., Carpenter, D. A. & de Sauvage, F. J. Hematopoietic deficiencies in c-mpl and TPO knockout mice. Stem Cells 16, 1–6 (1998).
Miller, C. L., Dykstra, B. & Eaves, C. J. Characterization of mouse hematopoietic stem and progenitor cells. Curr. Protoc. Immunol. 80, 22B.22.21–22B.22.31 (2008).
Kankaanpaa, P. et al. BioImageXD: an open, general-purpose and high-throughput image-processing platform. Nat. Methods 9, 683–689 (2012).
Kawamoto, T. Use of a new adhesive film for the preparation of multi-purpose fresh-frozen sections from hard tissues, whole-animals, insects and plants. Arch. Histol. Cytol. 66, 123–143 (2003).
Kumar, S. & Filippi, M. D. An alternative approach for sample preparation with low cell number for TEM analysis. J. Vis. Exp. 12, 54724 (2016).
Fellmann, C. et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 5, 1704–1713 (2013).
Nakada, D., Saunders, T. L. & Morrison, S. J. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature 468, 653–658 (2010).
Betts, S., Speed, M. & King, J. in Methods in Enzymology, Vol. 309 (ed. Wetzel, R.) 333–350 (Academic Press, 1999).
Acknowledgements
We thank M. Goodell and K. King for their advice, discussion and critical review of the manuscript. We thank to L. Glimcher for providing the Xbp1flox/flox mice. This work was supported by the National Institutes of Health (grant nos R01HL146642 and R37CA228304 to X.C.; R01DK107413 and R01CA193235 to D.N.; R01CA016303 to J.M.R.; R01CA228140 to W.L.; R01DK115454 to A.C.; R01 AI1143992 and K22CA 218467 to S.A.; R01AR072018 and R21AG064345 to D.P.; R01HL095675 and R01HL133828 to W.T.; and R35GM130292, R01DK120330 and R01DK120047 to L.Q.), US Department of Defense Congressionally Directed Medical Research Programs (grant no. W81XWH1910524 to X.C.), Cancer Prevention and Research Institute of Texas (grant no. RR150009 CPRIT Scholar in Cancer Research award to X.C., RR140038 CPRIT Scholar in Cancer Research award to A.C., and RP160283 Baylor College of Medicine Comprehensive Cancer Training Program award to F.P.), St. Baldrick’s Foundation and Alex’s Lemonade Stand Foundation (to W.T.). This work was supported by the Cytometry and Cell Sorting Core at the Baylor College of Medicine with funding from the CPRIT Core Facility Support Award (grant no. CPRIT-RP180672), the NIH (grant nos CA125123, S10OD025251 and RR024574) and the assistance of J. M. Sederstrom. Transmission electron microscopy for this project was supported by the High Resolution Electron Microscopy Facility at MD Anderson Cancer Center with funding from NIH (grant no. P30CA016672). Imaging for this work was supported by the Integrated Microscopy Core at the Baylor College of Medicine with funding from NIH (grant nos DK56338 and CA125123) and CPRIT (grant nos RP150578 and RP170719).
Author information
Authors and Affiliations
Contributions
L.X. and X.C. conceived the project and designed the research. L.X., X.L., F.P., W.Z., L.Z., Y.D., T.G., L.O., K.L., T.H., X.S., G.Shi, G.Shang, S.S. and Y.J. performed the experiments and analysed the data. J.W., T.I., W.L., J.M.R., X.H.-F.Z., D.P., S.A., A.C., W.T., D.N. and L.Q. contributed to discussions, experimental design and critical reagents. X.C. supervised the project. X.C. and L.X. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
J.W. is the co-founder of CoActigon Inc. and Chemical Biology Probes LLC. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 SEL1L–ERAD is highly expressed in HSCs compared with progenitors.
a, Diagram showing the ERAD and UPR pathways. The ER is the major subcellular site for synthesis and maturation of all transmembrane and secreted proteins. To maintain protein homeostasis and normal cell function, cells have evolved highly sensitive and sophisticated quality control systems and/or stress response pathways to ensure the fidelity of protein structure. Two such systems conserved across different species are ERAD and UPR. Among the mammalian ERAD complexes, the SEL1L–HRD1 complex, consisting of the E3 ubiquitin ligase HRD1 and its adaptor protein SEL1L, is the most conserved branch that ubiquitinates and targets selective substrates for proteasomal degradation. Many physiological or pathological stresses cause the accumulation of unfolded or misfolded proteins in the ER and activate the ER stress response or UPR that is mediated by three ER transmembrane sensors IRE1α, ATF6 and PERK. IRE1α is the most ancient and conserved sensor of the UPR. Upon activation, IRE1α dimerizes and trans-autophosphorylates to activate its RNase domain to induce unconventional splicing of its substrate XBP1. Recent study reveals that IRE1α, rather than ATF6 and PERK, as a selective endogenous substrate that is degraded by SEL1L–HRD1 ERAD. b, Heatmap showing the expression of ERAD genes in mouse HSCs and progenitors. LT-HSC: long-term HSC (LSK CD150+CD48−CD135−); ST-HSC: short-term HSC (LSK CD150-CD48-CD135-); MPP: multipotent progenitors (LSK CD135+). Data extracted from GSE109125. c, Quantitative RT–PCR analysis of Hrd1 expression in mouse HSCs and progenitors. Data are presented relative to Gapdh. n = 3 biologically independent mice. One-way ANOVA was used to calculate P values. Results are shown as mean ± s.d. Statistical information is provided as source data.
Extended Data Fig. 2 Representative flow cytometry gating strategy used in this study.
a, Strategy used to isolate the hematopoietic stem and progenitor cell populations in mouse bone marrow presented on Figs. 1d,f,h,j, 2c,f,h,i, 3e,g, 4a, 5b,c,f,g and Extended Data Figs. 3c,d, 4e,g,i,n–q,s,t, 5c,f, 6b,g.i,l, 7b–e,i,m,p,r (right panel), 8a–f (right panel), 9e,g,k and 10a,f. LSK: Lin-Sca-1+c-Kit+; LT-HSC: long-term HSC (LSK CD34-Flk2-); ST-HSC: short-term HSC (LSK CD34+Flk2-); MPP: multipotent progenitors (LSK CD34+Flk2+); CLP: common lymphoid progenitors (Lin-Sca-1lowc-KitlowFlk2+IL7Rα+); CMP: common myeloid progenitors (LS-K CD34+FcγR-); GMP: granulocyte/macrophage progenitors (LS-K CD34+ FcγR+); MEP: megakaryocytic/erythroid progenitors (LS-K CD34-FcγR-). b, Strategy used to isolate myeloid cell (CD11b+Gr1+), B cell (B220+CD3ε-) and T cell (CD3ε+B220-) in mouse bone marrow presented on Figs. 2b,e, 3d, 5e and Extended Data Figs. 3e, 5b,e, 6h,k, 7h,l,o,r(left panel), 8b(left panel), 8d(left panel), 8f(left panel) and 9f.
Extended Data Fig. 3 Progressive loss of progenitor cells in Sel1l-knockout mice.
a, Western blot analysis of SEL1L expression in LSK cells from 2 control (Ctrl) and 2 Sel1lMx1 mice 1 week after last poly (I:C) injection. β-ACTIN was used as loading control. b, Bone marrow (2 femurs and 2 tibiae) cellularity of Ctrl and Sel1lMx1 mice at the indicated time points. -1 indicates 1 week before poly (I:C) injection. Age and gender matched littermates were used in each time points. Ctrl (12, 20): n = 5, Ctrl (5): n = 6 and the others: n = 4. c, d, Percentage (c) and number (d) of CMP, GMP, MEP and CLP in the BM of control (Ctrl) and Sel1lMx1 mice at the indicated time points. Ctrl (12, 20): n = 5, Ctrl (5): n = 6 and the others: n = 4. e, f, Percentage (e) and number (f) of B, T and myeloid cells in the BM of control (Ctrl) and Sel1lMx1 mice at the indicated time points. Ctrl and Sel1lMx1 (1, 12): n = 3, Ctrl (5): n = 6 and the others: n = 4. g, Representative image of femur bone from control and Sel1lMx1 mice 30 weeks after poly (I:C) injection. h, i, Quantification of Red blood cell (RBC, h) and haemoglobin (HGB, i) in peripheral blood of Ctrl and Sel1lMx1 mice 30 weeks after poly (I:C) injection. n = 4. j-l, Body weight (j), spleen weight (k) and thymus weight (l) in control and Sel1lMx1 mice as in h. n = 4. m, n, BM (m) and spleen (n) cellularity in mice as in h. n = 4. Results are shown as mean ± s.d. Two-way ANOVA (b-f) or two-tailed Student’s t-tests (h-n) was used to assess statistical significance. ns, not significant. Statistical information and unprocessed blots are provided as source data.
Extended Data Fig. 4 Characterization of the Sel1lVav-knockout mice.
a, b, Expression of Sel1l mRNA (a) or protein (b) in primary control (Ctrl) or Sel1lVav-KO HSCs. n = 5 in a. c, Body weight of Ctrl and Sel1lVav-KO mice at indicated time points, n = 6. d, BM cellularity of Ctrl and Sel1lVav-KO mice at the indicated time points. n = 6. e, Frequency and absolute number of LT-HSCs from 8-week-old Ctrl and Sel1lVav-KO mice. n = 3. f, Colony formation in methylcellulose from equal number of Ctrl or Sel1lVav-KO HSCs. CFU-GM/G/M: colony forming unit granulocytic and monocytic (GM)/ granulocytic (G)/ monocytic (M); CFU-GEMM: colony forming unit granulocytic, erythrocytic, monocytic and megakaryocytic; BFU-E: blast forming unit erythrocytic. The colonies were counted after 12 days of culture. n = 3. g-j, Representative pseudocolor dot plots showing the gating strategy to identify CMP, GMP, MEP, CLP (g, i) and quantification (h, j) in the BM of Ctrl and Sel1lVav-KO mice. n = 6 for each group. k, RBC count in peripheral blood of Ctrl and Sel1lVav-KO mice at indicated age. 8-week-old Ctrl: n = 3; 8-week-old Sel1lVav: n = 7; 50-week-old Crl: n = 7; 50-week-old Sel1lVav: n = 5. l, Kaplan–Meier survival analysis of Ctrl and Sel1lVav-KO mice. n = 20. m, Schematic diagram of the Ki67 assay under BMT condition. n, Cell-cycle analysis of donor and competitor derived HSCs using Ki67 and 7-AAD. n = 3. o, p, Representative FACS plot for apoptosis analysis from freshly isolated control and Sel1lVav-KO HSCs. Sample without Annexin V stain served as negative control. Sample treated by heat shock served as positive control. q, Quantification of apoptotic HSCs in control and Sel1lMx1-KO mice at indicated time points, n = 3. r, Quantification of apoptosis-related genes expression in control and Sel1lVav-KO HSCs. n = 4. s, Quantification of phos-p38, phos-JNK and phos-ERK in control and Sel1lMx1 HSC at indicated time points. n = 3 for all groups except 5 weeks Ctrl (n = 2). t, Percentage of apoptotic HSCs in the BM of 8-week-old Ctrl and Sel1lVav-KO mice. Ctrl: n = 8; Sel1lVav: n = 7. Two-way ANOVA (c, d, h, j, k, q, s), two-tailed Student’s t-tests (a, e, f, n, r, t), or log-rank test (l) were used to calculate P values. Results are shown as mean ± s.d. ns, not significant. Statistical information and unprocessed blots are provided as source data.
Extended Data Fig. 5 SEL1L is required for HSC regenerative potential under haematological stress condition.
a, Schematic depiction of the competitive bone marrow transplantation scheme using whole bone marrow cells. CD45.2+ donor BM cells isolated from control (Ctrl) or Sel1lVav mice were mixed with CD45.1+ competitor BM cells at 3:1 ratio, transplanted into lethally irradiated CD45.1+ recipients respectively. Reconstitution levels were monitored for 16 weeks in peripheral blood (PBMC) including myeloid cells, B cells and T cells and HSCs were examined 16 weeks after transplantation. b, c, Percentage of donor-derived myeloid cells, B cells and T cells in peripheral blood at indicated time points (b) or HSC in the BM at 16 weeks after transplantation (c). Week4 in b: n = 8; n = 5 in all the other time points. n = 5 in c. d, Schematic depiction of the competitive bone marrow transplantation scheme using LT-HSCs (LSK CD34-Flk2-) as donor. 250 HSCs were mixed with 250,000 CD45.1+ BM cells for each recipient mice in this assay. e, Percentage of donor-derived myeloid cells, B cells and T cells in peripheral blood at indicated time points using LT-HSC as donor. n = 7. f, Quantification of HSCs number in the spleen of control (Ctrl) and Sel1lVav mice 15 days after 5-FU treatment. n = 5. Results are shown as mean ± s.d. Two-way ANOVA (b, e) or two-tailed Student’s t-tests (c, f) were used to assess statistical significance. ns, not significant. Statistical information is provided as source data.
Extended Data Fig. 6 Characterization of ER stress and UPR activation in Sel1l-KO HSCs.
a, Transmission electron microscopic analysis of the ER and mitochondria (M) in HSCs from Control (Ctrl) or Sel1lVav-KO mice. Scale bar, 1 μm. b, ER-tracker staining in Ctrl and Sel1lMx1 HSCs. n = 3. c, WB analysis of HSCs from 8-week-old control and Sel1lVav-KO mice. d, RT-PCR analysis of UPR and ERAD in 8-week-old control or Sel1lVav-KO HSCs. Ctrl: n = 4; Sel1lVav: n = 3. e, f, RT-PCR analysis of Xbp1s (e) and Bip (f) expression in c-Kit-enriched BM cells from Ctrl and Sel1lVav-KO mice treated with vehicle or TUDCA (TUDCA-2: 200 mg/kg, TUDCA-5: 500 mg/kg). Vehicle: n = 2; TUDCA: n = 3. g, Frequency of HSCs in vehicle- or TUDCA (200 mg/kg)-treated Ctrl and Sel1lVav mice. n = 2 for vehicle-treated control group and n = 3 for the other 3 groups. h, i, Percentage of control and Sel1lMx1 donor-derived PBMC (h) and HSCs (i) in recipient mice at indicated time after vehicle or TUDCA treatment (daily i.p. injection) starting from the first poly(I:C) injection (4 weeks after donor-reconstitution). HSCs were analysed at week 5. Ctrl: n = 2; Sel1lMx1: n = 3. The Vehicle groups and donor chimerism of n=3 randomly sampled mice from control or Sel1lMx1 donor group before poly(I:C) injection (left panel of h) is the same as in Extended Data Figs. 6k,l, 7 and 8. j, RT-PCR analysis of Xbp1s expression in LSK cells from 4-PBA treated mice. n = 2. k, l, Percentage of Ctrl and Sel1lMx1 donor-derived PBMC (k) and HSCs (l) in recipient mice at indicated time after vehicle or 4-PBA treatment (in drinking water) starting from the first poly(I:C) injection. Ctrl: n = 2; Sel1lMx1: n = 3. Results are shown as mean ± s.d. Two-tailed Student’s t-tests (b, d, j) or two-way ANOVA with Bonferroni test (e-i, k, l) were used to calculate P values. ns, not significant. Statistical information and unprocessed blots are provided as source data.
Extended Data Fig. 7 Regulation of HSC identity by SEL1L ERAD is independent of ER stress and UPR.
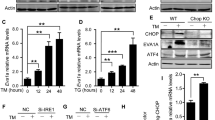
a, Quantification of Xbp1s (left) and Bip (right) expression in control (Ctrl) or Sel1lVav-KO HSCs treated with tunicamycin (0.1mg/kg) or thapsigargin (0.05 mg/kg) for 4 weeks (i.p. injection, 3 times/week). n = 3. b, c, Number of HSCs (b) and quantification of apoptotic HSCs (c) from Ctrl and Sel1lVav-KO mice as in a. n = 3. d, Frequency of HSCs in the BM of 8-week-old mice with indicated genotypes. Sel1lf/f; Ire1αf/f (Ctrl): n = 5; Sel1lVav (Sel1l-KO): n = 3; Ire1αVav (Ire1α-KO): n = 6; Sel1l; Ire1α double-knockout (dKO): n = 5; Vav-iCre: Ire1αhet, n = 3; Vav-iCre; Sel1lf/f; Ire1αhet, n = 5, data are combined from 2 independent experiments. e, Frequency of HSCs in the BM of 8-week-old mice with indicated genotypes. Sel1lf/f; XBP1f/f (Ctrl): n = 11, Sel1lVav (Sel1l-KO): n = 4, Xbp1Vav(Xbp1-KO): n = 8, Sel1l; Xbp1 double-knockout (dKO): n = 6, data are combined from 3 independent experiments. f, Schematic of BMT assay using retroviral transduced HSPCs. g, Quantification of Xbp1s expression in Ctrl and Sel1lMx1-KO HSPCs transduced by empty vector (EV) or Xbp1s. n = 3 independent samples. h, i, Percentage of Ctrl and Sel1lMx1-KO donor-derived PBMC (h) and HSCs (i) transduced with indicated construct in recipient mice. n = 3. j. Schematic of treatment strategy for competitive BMT assay. k, Quantification of Xbp1s expression in vehicle or IRE1α inhibitor MKC8866 (IRE1αi) treated HSCs. HSCs were sorted from Ctrl + Vehicle or Ctrl + IRE1αi group at the end of the rescue experiments. n = 2. l, m, Percentage of Ctrl and Sel1lMx1 donor-derived PBMC (l) and HSCs in the BM (m) of recipient mice at the indicated time points. HSCs were analysed at week 5. Ctrl: n = 2; Sel1lMx1: n = 3. n, WB analysis of p-PERK and CHOP in WBM cells from mice treated with PERK inhibitor GSK2656157 (PERKi) or eIF2α inhibitor ISRIB respectively as in j. o, p, Percentage of Ctrl and Sel1lMx1 donor-derived PBMC (o) and HSCs (p) in recipient mice treated with vehicle, PERKi or ISRIB at the indicated time points. HSCs were analysed at week 5. Ctrl: n = 2; Sel1lMx1: n = 3. q, Quantification of Atf6 expression in Ctrl and Sel1lMx1 HSPCs transduced with control shRNA (shRen) or shRNA targeting Atf6 (shAtf6). n = 3 independent samples. r, Percentage of Ctrl and Sel1lMx1 donor-derived PBMC (left) and HSCs (right) in recipient mice transduced with indicated construct. n = 3. The same vehicle group was used in l, m, o, p. Two-way ANOVA (a-c, g-i, l, m, o-r), one-way ANOVA (d, e) or two-tailed Student’s t-tests (k) were used to calculate P values. Results are shown as mean ± s.d or mean ± s.e.m (c). Statistical information and unprocessed blots are provided as source data.
Extended Data Fig. 8 Regulation of HSC identity by SEL1L ERAD is independent of MAPK signalling.
a, Analysis of p-p38 levels in whole bone marrow (WBM) by western blot (left) or in HSCs by flow cytometry from mice treated with vehicle or p38 inhibitor SB202190 (p38i). For flow analysis, n = 3 (3 of the 5 vehicle or p38i treated mice were randomly picked and donor-competitor HSCs were not distinguished). b, Percentage of control (Ctrl) and Sel1lMx1 donor-derived PBMC (left) and HSCs in the BM (right) of recipient mice treated with vehicle or p38i at the indicated time points. HSCs were analysed at week 5. Ctrl: n = 2; Sel1lMx1: n = 3. c, Analysis of p-JNK levels in WBM by western blot (left) or in HSCs by flow cytometry from mice treated by vehicle or JNK inhibitor SP600125 (JNKi). n = 3. d, Percentage of control (Ctrl) and Sel1lMx1 donor-derived PBMC (left) and HSCs in the BM (right) of recipient mice treated with vehicle or JNKi at the indicated time points. HSCs were analysed at week 5. Ctrl: n = 2; Sel1lMx1: n = 3. e, Analysis of p-ERK1/2 levels in WBM by western blot (left) or in HSCs by flow cytometry from mice treated with vehicle or ERK1/2 inhibitor SCH772984 (ERKi). n = 3. f, Percentage of control (Ctrl) and Sel1lMx1 donor-derived PBMC (left) and HSCs in the BM (right) of recipient mice treated with vehicle or ERKi at the indicated time points. HSCs were analysed at week 5. Ctrl: n = 2; Sel1lMx1: n = 3. The same vehicle group was used in b, d and f. Two-way ANOVA (b, d and f), or two-tailed Student’s t-tests (a, c and e) were used to calculate P values. Results are shown as mean ± s.d. Statistical information and unprocessed blots are provided as source data.
Extended Data Fig. 9 SEL1L regulates MPL maturation.
a, Schematic of HSC (LSK CD150+CD48-)-vasculature distance measurement in the BM. b, c, Representative 2D images (b) and quantification (c) of the distances between CFSE-labelled HSCs (green) and vascular structure (CD31+ and/or CD144+, magenta). 177 control (Ctrl) and 147 Sel1lVav-KO HSCs were analysed over n=3 independent experiments. Scale bar, 20 μm. d, Schematic for non-conditioned transplantation of donor cells into Ctrl or Sel1lMx1-KO congenic mice. e, The HSCs numbers in Ctrl and Sel1lMx1-KO mice 5 weeks after poly (I:C) injection. n = 5. f, Percentage of donor-derived (CD45.1+) cells in the PBMC of non-irradiated Ctrl or Sel1lMx1-KO recipient mice 16 weeks after transplantation. n = 4. g, h, Flow analysis of surface MPL expression in HSCs at different cell-cycle states. Representative flow cytometry plots (g) and quantification (h) are shown. n = 4. i, Quantification of surface c-Kit expression from 8-week-old Ctrl and Sel1lVav-ko HSCs. n = 5. j, RT-PCR analysis of Mpl mRNA in 8-week-old Ctrl or Sel1lVav-KO HSCs. Data are presented relative to Actb. Ctrl: n = 8; Sel1lVav: n = 7. k, Representative histogram and quantification of phospho-STAT5 (Tyr694) level from 30-week-old Ctrl or Sel1lVav-KO HSCs. n = 4. l, Quantification of p57 mRNA by qPCR in 8-week-old Ctrl or Sel1lVav-KO HSCs. n = 4. m, WB analysis of total MPL in HSCs from 8-week-old Ctrl and Sel1lVav-KO mice under native or denature conditions. n, Quantification of total MPL signal in Ctrl and Sel1lVav-KO HSCs from immunostaining experiment in Fig. 4f. Ctrl HSCs: n = 100; Sel1lVav HSCs: n = 250. o, Sequence alignment of MPL from indicated species with R257 (Human) highlighted in red. p, 293T cells were transfected with Myc-tagged wild-type MPL (WT-Myc), FLAG-tagged mutant MPL (Mut-FLAG) or 1:1-mixed WT and Mut MPL (WT-Myc + Mut-FLAG) for 48h. The expression of WT or Mutant MPL was quantified by qPCR primers specific to Myc or FLAG tag. n = 3 independent samples. q, WT or SEL1LCRISPR-KO 293T cells were transfected with HA-tagged MPL and surface MPL expression was determined by flow cytometry 48h later. n = 3. r, WB analysis of HRD1 expression in WT, SEL1LCRISPR-KO or HRD1CRISPR-KO 293T cells. s, t, WT or HRD1CRISPR-KO 293T cells were transfected with HA-tagged MPL and surface MPL or C-KIT expression were determined by flow cytometry 48h after transfection. n = 3. u-x, Expression of aggregation-prone mutant proAVP (G57S) (u) or POMC (C28F) (v) forms aggregates in WT 293T cells, but is not sufficient to inhibit ERAD activity (u, v, w) and thus does not reduce surface MPL expression (x). HRD1 autoubiquitination (u, v) and OS9 (OS9-1 and OS9-2) accumulation (u, v, w) were used as validated indicators for ERAD activity. The experiments for q and u-x are performed together, the MPL level in WT (r) and Mock (x) was from the same experiment. n=3. Results are shown as mean ± s.d. Two-tailed Student’s t-test (e, f, h-l, n, p, q, s, t, x), or two-sample Kolmogorov–Smirnov test (c) were used to assess statistical significance. ns, not significant. Statistical information and unprocessed blots are provided as source data.
Extended Data Fig. 10 MPL–TPO is important for HSC–niche interaction.
a, Frequency of HSCs in 20-week-old control (Ctrl) and Mpl-KO mice. n = 3. b, Schematic of HSC (LSK CD150+CD48-)-vasculature distance measurement in the BM. Donor HSCs were analysed by whole-mount confocal microscope in femurs and tibiae. c, d, Representative 2D images (c) and quantification (d) of the distances between CFSE-labelled HSCs (green) and vascular structure (CD31+ and/or CD144+, magenta). A total of n=71 Ctrl and n=57 Mpl-KO HSCs were analysed over 3 independent experiments. Scale bar, 20 μm. e, Mice were treated by indicated antibodies daily for 7 days before examining the HSC-vasculature distance in the BM. f, g, Both MPL and TPO antibodies blocked TPO-induced phospho-STAT5 in HSCs in vitro. n = 3 independent samples. h, i, Representative 2D images (h) and quantification (i) of the distances between indicated HSCs (CD150+Lin-CD48-CD41-, Red) and the closest vascular structure (CD31+ and/or CD144+, Blue). HSCs from mice treated with IgG control (combined, total of 71 HSCs), anti-TPO antibody (total of 83 HSCs) or anti-MPL antibody (total of 73 HSCs) were analysed in n = 3 independent experiments for all groups. Scale bar, 20 μm. j, k, Representative 2D images (j) and quantification (k) of the distance between indicated HSCs (Red) and the closest vascular structure (Blue). HSCs from mice treated with vehicle (total = 123), tunicamycin (total = 101) or thapsigargin (total = 98) were analysed. n = 3 independent samples. Scale bar, 10 μm. l, Diagram showing the structure of MPL agonist and its binding to the juxtamembrane end of MPL. m, Schema depicting the regulation of HSC identity by SEL1L–HRD1 ERAD. Results are shown as mean ± s.d. Two-tailed Student’s t-test (a, d, g), or two-sample Kolmogorov–Smirnov test (i, k) were used to assess statistical significance. ns, not significant. Statistical information and unprocessed blots are provided as source data.
Supplementary information
Supplementary Table 1
Supplementary Table 1. Reagents and kits used in this study. Supplementary Table 2. Antibodies used in this study. Supplementary Table 3. Primers used in this study.
41556_2020_581_MOESM3_ESM.avi
Supplementary Video 1. Interaction of control HSCs with the perivascular niche. CD45.2+ control HSCs (CD150+CD48− LSK from Sel1lflox/flox mice) were purified, labelled with CFSE (green) and transplanted into irradiated CD45.1+ recipient mice. The recipient mice were euthanized after 24 h. The femurs and tibias were collected and perfusion fixed. The bones were embedded and sectioned for immunostaining with anti-CD31 and anti-CD144 antibodies (magenta). Three-dimensional images were captured using a Zeiss LSM780 confocal microscope and videos were produced by the 3D viewer plugins
41556_2020_581_MOESM4_ESM.avi
Supplementary Video 2. Interaction of Sel1l-KO HSCs with the perivascular niche. CD45.2+ Sel1l-KO HSCs (CD150+CD48− LSK from Sel1lflox/flox; Vav-iCre mice) were purified, labelled with CFSE (green) and transplanted into irradiated CD45.1+ recipient mice. The recipient mice were euthanized after 24 h. The femurs and tibias were collected and perfusion fixed. The bones were embedded and sectioned for immunostaining with anti-CD31 and anti-CD144 antibodies (magenta). Three-dimensional images were captured using a Zeiss LSM780 confocal microscope and videos were produced by the 3D viewer plugins
Source data
Source Data Fig. 1
Statistical source data
Source Data Fig. 2
Statistical source data
Source Data Fig. 3
Statistical source data
Source Data Fig. 4
Statistical source data
Source Data Fig. 4
Unprocessed western blots
Source Data Fig. 5
Statistical source data
Source Data Extended Data Fig. 1
Statistical source data
Source Data Extended Data Fig. 3
Statistical source data
Source Data Extended Data Fig. 3
Unprocessed western blots
Source Data Extended Data Fig. 4
Statistical source data
Source Data Extended Data Fig. 4
Unprocessed western blots
Source Data Extended Data Fig. 5
Statistical source data
Source Data Extended Data Fig. 6
Statistical source data
Source Data Extended Data Fig. 6
Unprocessed western blots
Source Data Extended Data Fig. 7
Statistical source data
Source Data Extended Data Fig. 7
Unprocessed western blots
Source Data Extended Data Fig. 8
Statistical source data
Source Data Extended Data Fig. 8
Unprocessed western blots
Source Data Extended Data Fig. 9
Statistical source data
Source Data Extended Data Fig. 9
Unprocessed western blots
Source Data Extended Data Fig. 10
Statistical source data
Rights and permissions
About this article

Cite this article
Xu, L., Liu, X., Peng, F. et al. Protein quality control through endoplasmic reticulum-associated degradation maintains haematopoietic stem cell identity and niche interactions. Nat Cell Biol 22, 1162–1169 (2020). https://doi.org/10.1038/s41556-020-00581-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41556-020-00581-x
This article is cited by
-
SEL1L-HRD1 interaction is required to form a functional HRD1 ERAD complex
Nature Communications (2024)
-
Proteomic screens of SEL1L-HRD1 ER-associated degradation substrates reveal its role in glycosylphosphatidylinositol-anchored protein biogenesis
Nature Communications (2024)
-
SEL1L preserves CD8+ T-cell survival and homeostasis by fine-tuning PERK signaling and the IL-15 receptor-mediated mTORC1 axis
Cellular & Molecular Immunology (2023)
-
SEL1L–HRD1 endoplasmic reticulum-associated degradation controls STING-mediated innate immunity by limiting the size of the activable STING pool
Nature Cell Biology (2023)
-
Mitochondrial oxidative phosphorylation is dispensable for survival of CD34+ chronic myeloid leukemia stem and progenitor cells
Cell Death & Disease (2022)
