
Abstract
The threat of new viral outbreaks has heightened the need for ready-to-use vaccines that are safe and effective. Here we show that a subcutaneous vaccine consisting of live Zika virus electrostatically entrapped in a self-adjuvanting hydrogel recruited immune cells at the injection site and provided mice with effective protection against a lethal viral challenge. The hydrogel prevented the escape of the viral particles and upregulated pattern recognition receptors that activated innate antiviral immunity. The local inflammatory niche facilitated the engulfment of the virus by immune cells infiltrating the hydrogel, the processing and cross-presentation of antigens and the expansion of germinal centre B cells and induced robust antigen-specific adaptive responses and immune memory. Inflammatory immune niches entrapping live viruses may facilitate the rapid development of safe and efficacious vaccines.
Similar content being viewed by others

Main
Vaccines are essential for protecting humans from the threat of longstanding and emerging infectious diseases1. However, because of lagged vaccine development, the latest and very recent outbreaks of emerging virus infection caused by Zika virus (ZIKV) and severe acute respiratory syndrome coronavirus 2 still pose a great challenge for global public health2,3,4,5. Typically, whole-virus vaccine strategies, such as attenuation or inactivation, can be applied to directly convert a virulent virus into vaccines6. However, the reduced immunity, safety concerns and time-consuming manufacturing processes of whole virus vaccines hamper their extensive applications6,7. There is a notable impetus to develop a next-generation vaccine technology to rapidly convert wild virus strains into vaccines with high safety and effectiveness8. Nanotechnology is promising for viral vaccine development9,10,11 due to its ability to control the loading, delivery and release of antigens and to enhance the potency and longevity of immune responses12,13,14,15,16. However, spatial and temporal control remains an unresolved obstacle for nanoparticle-based engineering due to off-target distributions, systemic delivery and limited modulatory effects17,18,19.
An interesting alternative for vaccine engineering is to manipulate virus-residence immune microenvironments that enable spatial and temporal manipulation of the interplay between viruses and immune cells. In this regard, antigen- and chemokine-loaded three-dimensional macroscale scaffolds that can recruit cells at the injection site20,21 are suggested due to their on-site immune modulation properties22,23. However, the key challenge in using material scaffolds to convert viruses into vaccines is to restrict the viral infection while augmenting the local innate responses, which can locally rally immune cells at the injection site, trigger antiviral responses and process the viral antigens. However, previous macroscale scaffolds with loading and release effects do not meet the design criteria for virus vaccines due to the lack of spatial and temporal control over innate immunity. As the first line of host defence against viral infection, the activation of a robust innate immune response plays a critical role in eliminating viruses and in the subsequent adaptive immune responses24. Accordingly, a new approach requires the ability to entrap the virus while limiting its release; in the meantime, it should recruit and regulate the immune cells at the injection site to control the timing and location of virus processing.
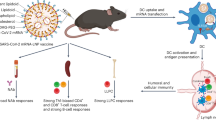
In this Article, we demonstrate a virus-entrapped hydrogel, named Vax, composed of chitosan oligomer hydrogel as a virus trapper and built-in adjuvant and calcium carbonate nanoparticles (nano-CaCO3) as a stabilizer and Ca2+ source. Chitosan scaffolds with positively charged side chains effectively entrap viruses via electrostatic interactions, while their self-adjuvanting properties25 enable the activation of innate immune responses and cell recruitment by the activation of pattern recognition receptors (PRRs). Thus, the hydrogel generates an inflammatory niche for virus uptake and antigen processing, which has a beneficial effect on antigen presentation in lymph nodes (Fig. 1), leading to effective humoral and cellular immunity due to the expanded germinal centre (GC) B cells and improved cross-presentation. Thus, a single-dose vaccination prepared by loading live pathogenic ZIKV into the scaffold without prior treatment can evoke effective immunity and protect mice against lethal infection, representing a promising strategy for protection against emerging infectious diseases.

Schematic showing that Vax is prepared by the presence of nano-CaCO3 and viruses. The administration of Vax forms a niche, where PRRs are activated to facilitate immune cell recruitment and the activation of innate immune responses, leading to antigen presentation, the elevation of GC B cells and cross-presentation in lymph nodes. Thus, Vax induces robust antigen-specific responses and memory responses.
Results
Vax locally entraps viruses and eliminates systemic infection
As the most commonly employed viral vector for gene therapy and vaccine development, recombinant adenovirus type-5 (Ad5) was used as a model virus26. For the preparation of Vax, we first mixed negatively charged Ad5 viral particles with positively charged low-molecular-weight chitosan oligosaccharide (COS), which is soluble over a wide pH range27. After the addition of calcium chloride, sodium bicarbonate and polyaspartic acid were added to the mixture of Ad5 and COS. During the reaction at 4 °C, the electrostatic repulsion between COS molecules was reduced by the alkaline environment, which allowed extensive hydrogen bonding and hydrophobic interactions between free –NH2 groups, resulting in the formation of hydrogel at 37 °C. The mechanical properties of Vax were studied to fully understand its rheological behaviours. The thermogelling behaviour of Vax was investigated by temperature sweeps, which showed that the loading of virus and CaCO3 shifted the gelation temperature of Vax higher from 15 °C to 20 °C (Fig. 2a). The reason is that negatively charged viruses and nano-CaCO3 combined with ammonium ions of chitosan and occupied cross-linking sites for gelation, decreasing the storage modulus (G′) of Vax compared with that of chitosan gel. Amplitude sweep and frequency sweep measurements of Vax at 37 °C showed that G′ was consistently higher than G″ across a wide frequency range, indicating that nano-CaCO3 and the virus did not affect gelation (Supplementary Fig. 1a,b). At 4 °C, the Vax precursor did not undergo a cross-linking reaction and remained in a sol state, indicating injectability (Supplementary Fig. 1c). When Vax was heated rapidly from 4 °C to 37 °C, gelation began at the 50th second, indicating gel formation at body temperature (Supplementary Fig. 1d). Therefore, the mechanical properties of Vax can be well controlled by varying the temperature (Supplementary Fig. 1e). The Ad5 encapsulation efficiency was more than 99% due to strong electrostatic interactions (Supplementary Fig. 1f).

a, Dynamic temperature sweep test of Vax loaded with virus and COS gel without adding CaCO3 and virus. b, Fluorescence microscopy observation of Vax sections. Green, calcein-labelled nano-CaCO3; red, AF555-labelled Ad5; blue, AF405-labelled chitosan. c, Representative cryo-SEM observation of Vax loaded with ZIKV. Scale bar, 1 μm. d, Virus confinement. Vax/Ad5, Vax/ZIKV and COS/ZIKV were immersed in various solutions to see the release of virus. Data represent the mean ± standard deviation (s.d.) (n = 3). For in vivo virus confinement experiment, IFNAR1−/− mice were subcutaneously injected with ZIKV-laden Vax, ZIKV or PBS. e, Viraemia of surviving IFNAR1−/− mice after infection (n = 6). f, Long-term detection of lingering ZIKV in the major organs of IFNAR1−/− mice after immunization (n = 5). LOD, limit of detection. g, Representative histopathological changes of the testes from PBS-, ZIKV- or Vax loaded with ZIKV-injected IFNAR1−/− mice at 5 and 15 d post-infection (d.p.i.). h, Detection of viral RNA at the injection site of IFNAR1−/− mice after injection (n = 5). i, Survival curves of suckling BALB/c mice after intracranial injection with ZIKV-embedded Vax or free ZIKV (n = 10). Data represent the mean ± s.d. In i, P values were determined by log-rank test.
The overall structure of Vax was examined through three-dimensional confocal laser scanning microscopy (CLSM), which illustrated the uniform distribution pattern of Alexa Fluor (AF) 555-labelled Ad5 and the calcein-labelled nano-CaCO3 inside the AF405-labelled chitosan hydrogel (Fig. 2b). The presence of nano-CaCO3 and viral proteins was also identified by the vibration of carbonate and proteins using a Fourier transform infra-red assay (Supplementary Fig. 2a) and the loss of carbon dioxide at approximately 645 °C using thermogravimetric analyses (Supplementary Fig. 2b). Observation of Vax using cryo-scanning electron microscopy (cryo-SEM)28 exhibited interconnected micrometre-scale void spaces 5–10 μm in diameter (Fig. 2c), and showed nano-CaCO3 particles approximately 200 nm in diameter and virus particles approximately 60 nm in diameter imbedded inside Vax (Supplementary Fig. 2c,d).
The virus confinement effect of Vax was determined both in vitro and in vivo by the incubation of virus-loaded Vax in solutions under different pH values and in conditions that mimic the physiological environment at 37 °C. No Ad5 was released from the scaffold within a week because of strong electrostatic adsorption by chitosan, indicating a virus confinement effect (Fig. 2d). Notably, nano-CaCO3 prevented the decomposition of the gel by scavenging the acid in the gel at pH 5; thus, no virus was leaked (Supplementary Fig. 3). Next, we endeavoured to determine whether a replication-competent virus could be effectively confined within Vax. We loaded ZIKV, a mosquito-borne single-stranded RNA virus belonging to the Flaviviridae family29, into the abovementioned Vax and incubated it in protein-rich Dulbecco’s modified Eagle medium (DMEM), and lysozyme-containing DMEM to mimic in vivo conditions. As expected, no ZIKV was detected in the soaking solutions containing foetal bovine serum (FBS), showing that a high concentration of negatively charged proteins had no effect on the electrostatic interaction between the virus and Vax (Fig. 2d). In addition, upon treatment with lysozyme, no ZIKV was released into the suspension, whereas virus from the COS gel leaked due to digestion (Fig. 1e and Supplementary Fig. 3). Confinement was maintained in the presence of lysozyme because cationic lysozyme tends to adsorb either calcium ions or calcium carbonate, resulting in the inhibition of enzymatic activity.
Given that ZIKV escape from Vax might lead to systemic infection in vivo, we evaluated the in vivo safety profile of live ZIKV-loaded Vax using IFNAR1−/− mice that lack the interferon receptor as a mouse model of ZIKV infection. IFNAR1−/− mice are highly vulnerable to ZIKV infection and can develop viraemia and mortality30. After subcutaneous (s.c.) implantation of 8 × 104 plaque-forming units (PFU) of wild-type (WT) ZIKV and ZIKV-loaded Vax, viraemia was monitored by quantifying viral RNA using quantitative reverse transcription polymerase chain reaction (qRT‒PCR). Owing to systemic distribution and replication, viraemia of free ZIKV peaked at 7.6 × 106 copies μl−1 by day 3 post-infection and lasted for 21 days (d), whereas viraemia was consistently undetectable in Vax-injected mouse sera for 56 d (Fig. 2e). In addition, the viral loads in the tested organs after s.c. inoculation with WT ZIKV, including the brain, heart, spleen, kidney and testes, remained at high levels even on day 52 after injection, indicating the accumulation and persistence of the virus (Fig. 2f). However, over the course of 2 months, no lingering viral RNA was detected in either the serum or organs of Vax-inoculated mice at any tested timepoint (Fig. 2f). Notably, ZIKV infection damaged the testis in mice and reduced the weight of the testis, whereas Vax did not induce weight loss or pathological changes (Fig. 2g and Extended Data Fig. 1). We next examined whether viral RNA persisted at the injection site and how long it persisted. Two weeks after Vax injection, viral RNA accumulation was observed only at the injection site, and that accumulation was completely cleared within 4–8 weeks (Fig. 2h). The survival rate of the ZIKV-in-Vax group was 100% because the confined ZIKV was unable to establish systemic infection, while the native ZIKV group had a 50% mortality rate (Extended Data Fig. 1).
We next examined the neurovirulence through intracranial injection of either ZIKV or ZIKV-laden Vax into postnatal day 1 BALB/c mice31. The newborn mice succumbed to ZIKV infection and showed marked neurological manifestations at a dosage of 2 × 104 PFU, leading to 100% mortality 23 d post inoculation (Fig. 2i). In contrast, all mice that were implanted with ZIKV-laden Vax survived and suffered minimal neurological symptoms, suggesting a good safety profile (Fig. 2i). We believe that the elimination of systemic infection and survival are partially attributable to the firm restriction effect of Vax. The immune modulation effect of Vax also plays a crucial role in eliminating viral infection.
Systemic toxicity of empty Vax
In addition to virus leakage, we also focused on whether the Vax induced long-term inflammation or systemic toxicity. BALB/C mice received s.c. injection with phosphate-buffered saline (PBS) or empty Vax. Haematoxylin and eosin (H&E) staining of the injection site of Vax-injected mice showed that a large number of inflammatory cells were infiltrated into the gel-containing dermis on day 7. However, the number of inflammatory cells was obviously reduced on day 14 and eventually reached a normal level compared with the PBS-injected mice (Supplementary Fig. 4). The acute inflammation at Vax-injected site did not augment the serum levels of inflammatory cytokines in mice, which also confirmed the safety profile of Vax (Supplementary Fig. 4). Furthermore, histopathological analysis of major organs at days 7, 14 and 28 after Vax injection exhibited no obvious morphological cytotoxic changes compared with the control (Supplementary Fig. 5 and Supplementary Table 1). Hepatic and renal functional biomarkers from the blood samples of Vax-injected mice showed that alanine transferase, total bile acids and uric acid were beyond the normal range only at day 7, which resolved to normal levels afterwards. Such abnormities might relate to the induction of restored liver toxicity. The haematological parameters in Vax-injected mice were identical to the PBS control group (Supplementary Fig. 6 and Supplementary Table 2). Together, the data suggested that the Vax efficiently restricted virus infection while showing minimized toxicity in vivo.
Vax stimulates local innate immune responses through the activation of PRRs
Since COS is an attractive adjuvant and can trigger host innate immunity, the scaffold made of COS is capable of driving innate immune responses of cells either inside or surrounding the scaffold. First, the impacts of Vax on differentially expressed genes (DEGs) from RAW 264.7 cells were investigated using RNA sequencing (RNA-seq). The top 15 Gene Ontology annotations demonstrated that most DEGs were mainly enriched in antiviral defence, lipopolysaccharide-induced cellular response, inflammatory responses, extracellular interaction, lysosome, redox signalling and cell metabolism (Fig. 3a and Supplementary Table 3), suggesting the activation of innate responses after Vax treatment.

a, Kyoto Encyclopedia of Genes and Genomes pathway analysis of DEGs of RAW 264.7 cells treated with Vax. b, Effects of Vax on the mRNA expression of CR3, CD14, TLR4 and Dectin-1 in BMDCs (n = 3). c, Secretion of TNF-α, IL-6 and IL-18 from BMDCs after treatment with Ad5, COS/Ad5, COS/CaCO3 or Vax (n = 3). d, Secretion of IFN-γ and IL-12p70 from BMDCs after treatment with Ad5, COS/Ad5, COS/CaCO3 or Vax. e, Secretion of chemokines from BMDCs after treatment (n = 3). f, Secretion of CXCL10 from BMDCs after treatment (n = 3). g, Effects of Vax on the mRNA expression of Ifna and Ifnb in BMDCs (n = 3). Data represent the mean ± standard deviation (s.d.), and P values were determined by two-tailed Student’s t-test. P < 0.05 was considered significant. NS, not significant.
We postulated that the Vax-induced PRR response may play an essential role in promoting the innate immune response. It is well known that a wide variety of PRRs can lead to the production of pro-inflammatory cytokines, chemotaxis, pathogen uptake and antigen presentation32. On the basis of the RNA-sequencing results, we found that the transcripts of some PRRs, such as integrin alpha M and CD14, were upregulated (Supplementary Table 3). To reveal whether Vax exerts an effect on PRRs, we investigated the messenger RNA levels of complement III receptor (CR3), CD14, Toll-like receptor 4 (TLR4) and dectin-1 after co-culture of bone marrow-derived dendritic cells (BMDCs) and RAW 264.7 cells with Vax. In line with a previous study33, we found that Vax drastically upregulated the expression of CR3, a PRR, by more than six-fold; CD14, a glycosylphosphatidylinositol-anchored protein as a co-receptor, by more than ten-fold; and TLR4, a PRR, by more than ten-fold. However, dectin-1, a C-type lectin-like carbohydrate recognition receptor, remained identical to the blank control (Fig. 3b), indicating a specific agonist effect of Vax on PRRs. Notably, compared with other control groups, Vax/virus showed a synergistic effect on PRR activation (Fig. 3b).
We then determined the induction of pro-inflammatory cytokines upon Vax injection. In comparison with the blank control and Ad5 groups, COS/CaCO3, COS/Ad5 and Vax enhanced the secretion of pro-inflammatory cytokines, suggesting the positive effects of chitosan on pro-inflammatory responses (Fig. 3c,d). Among them, IL-12p70 and IL-18, which are involved in the secretion of IFN-γ (ref. 34) and regulation of both innate and adaptive immunity, were improved (Fig. 3d). Vax also markedly promoted the secretion of IFN-γ, which has antiviral and immunoregulatory activities, by seven- to eight-fold (Fig. 3d). The stimulation of innate immune responses by Vax also facilitated the production of chemokines, including CCL2, CCL4, CCL5 and CXCL2, which are beneficial for the recruitment of immune cells such as monocytes, T cells and neutrophils (Fig. 3e)32,35. Importantly, CXCL10 (IP-10), which plays an essential role in the recruitment of T-helper-1 cells, natural killer (NK) cells, macrophages and dendritic cells (DCs) into sites of inflammation, was also elevated by Vax engagement (Fig. 3f)36. The mRNA levels of type I interferons, including Ifnb and Ifna, were also evaluated (Fig. 2g). Both the COS/Ad5 and Vax groups had increases of more than ten-fold in the mRNA level of IFNβ but not IFNα, indicating a powerful role in promoting type I interferon (Fig. 3g). Unlike the chitosan gel, the addition of CaCO3 into hydrogel showed very weak regulatory effect on pro-inflammatory responses.
Chitosan is known as a NLRP3 inflammasome activator. However, Vax hydrogel treatment impaired IL-1β since the level of caspase 1 and IL-1β depended less on NLRP3 inflammasome response (Supplementary Fig. 7). In terms of downstream signalling, Vax drove the nuclear factor kappa B (NF-κB) signalling and antiviral innate responses of RAW 264.7 cells by upregulating the mRNAs of key proteins (Supplementary Fig. 8). Vax activated the corresponding innate immune reactions, such as pro-inflammatory responses, type I interferon responses and antiviral responses, suggesting that the stimulation of PPRs is one of its mechanisms of activation of innate immune responses, which enable the formation of an inflammatory niche and antiviral defence at the Vax site.
Vax enhances the recruitment of immune cells
We used an in vitro transwell assay to assess whether Vax contributes to cell migration by counting the BMDCs that migrated across the filter over 12 hours (h). Increased naïve BMDCs in the upper chambers migrated through the filter in response to Vax-treated BMDCs in the lower chambers compared with cells treated with COS/Ad5, Ad5 and the blank control (Fig. 4a and Supplementary Fig. 8a), indicating that the chemokines enhanced cell recruitment. Nano-CaCO3 showed a cooperative impact on promoting cell migration.

a, The migration of BMDCs towards the blank control, Ad5, COS/Ad5 and Ad5-in-Vax groups (n = 3). b, A nodule formed up to 4 d after s.c. injection of Vax. c, The numbers of infiltrated cells from the retrieved nodules (n = 6). d, The size change of nodules after Vax administration in vivo (n = 3). Data represent the mean ± standard deviation (s.d.). e, Representative H&E staining of retrieved nodules (n = 5). f, Flow cytometry analysis of the viability of infiltrated cells in the retrieved nodules formed by COS, COS/CaCO3, COS/Ad5 and Vax using a Fixable Viability Kit (n = 5). g, The frequency of granulocytes, NK cells, macrophages, CD4+ T cells, dendritic cells, CD8+ T cells, monocytes, and B cells in recruited cells in vivo. h, The numbers of NK cells, macrophages, CD4+ T cells, dendritic cells, CD8+ T cells, monocytes, and B cells in the recruited cells in vivo by flow cytometry. Data represent the mean ± s.d. In a, c and f, P values were determined by two-tailed Student’s t-test. P < 0.05 was considered significant. NS, not significant.
In vivo cell recruitment was evaluated by s.c. injection of Vax into the right flanks of BALB/c mice. Upon injection, a nodule was formed at the site of administration and was gradually healed 28 d post administration (Fig. 4b and Supplementary Fig. 8b). Due to the cell attraction property, Vax recruited larger numbers of cells than COS or COS/ Ad5 (Fig. 4c). The nodule size peaked on day 5 and decreased afterwards, indicating increased cell recruitment and subsequent cell migration (Fig. 4d). According to H&E staining and SEM of retrieved nodules, most recruited cells at the Vax-injected site were embedded within the nodules (Fig. 4e and Supplementary Fig. 8c) and remained viable (Fig. 4f), while the recruited cells at the COS- and COS/Ad5-injected sites were mainly located peripherally (Fig. 4e). By analysing the types of infiltrated cells using flow cytometry, we found that 81% of the infiltrated cells were host immune cells, including granulocytes, macrophages, DCs, T cells, B cells and NK cells (Fig. 4g,h and Supplementary Figs. 8 and 9). Among the infiltrated cells, professional antigen-presenting cells (APCs) recruited by Vax were able to process the viruses embedded in the Vax.
Co-internalization of virus and Vax drives local activation and antigen presentation
Since the activation of PRRs could benefit the phagocytosis of viruses, we investigated the uptake of Ad5 by BMDCs following incubation of fluorescent-labelled Ad5-in-Vax with BMDCs for 2 h. The CLSM images showed the co-localization of Ad5 (green) and Vax (red) in the cytoplasm after incubation for 2 h, whereas in the control groups that expressed fewer PRRs, the uptake of Ad5 was less efficient (Fig. 5a,b and Supplementary Fig. 10a). Moreover, the uptake efficiency was enhanced by the presence of nano-CaCO3.

a, The BMDCs took up Ad5-Vax, where Ad5 was labelled with AF555 and chitosan was labelled with AF660. b, Mean fluorescence intensity (MFI) of Ad5-AF555 in BMDCs (n = 3). c,d, The corresponding quantification (c) and representative flow cytometric analysis images (d) of CD80- and CD86-positive BMDCs after treatment with COS, COS/Ad5, COS/CaCO3 or Vax (n = 3). e, Flow cytometry analysis of MHC-II-positive BMDCs after treatment with COS, COS/Ad5, COS/CaCO3 or Vax. f,g, The corresponding quantification (f) and representative flow cytometric analysis images (g) of MHC-I molecule-positive BMDCs that presented SIINFEKL on the surface after treatment with COS, COS/Ad5, COS/CaCO3 or Vax (n = 3). h, IHC of retrieved nodule sections from mice injected with COS, COS/Ad5, COS/CaCO3 or Vax at day 3 after s.c. injection were probed using an Ad5-specific anti-hexon antibody (n = 3). i, The bioluminescence images of BALB/c mice after injection with COS, COS/Ad5, COS/CaCO3 or Vax. j, Quantitative analysis of the bioluminescence images (n = 3). k, Quantification of the numbers of CD11c+ cells isolated from retrieved nodules at day 3 after s.c. injection (n = 5). l, Quantification of the numbers of CD80+ CD86+ CD11c+ cells isolated from retrieved nodules (n = 5). m, Comparative evaluation of MHC-I and MHC-II in CD11c+ cells isolated from retrieved nodules (n = 5). Data represent the mean ± standard deviation (s.d.), and P values were determined by two-tailed Student’s t-test. P < 0.05 was considered significant. NS, not significant.
Having observed a positive effect on virus uptake, we next investigated the effect of Vax on DC activation and antigen presentation in vitro. Upon loading BMDCs into Vax, the presence of Ad5 together with nano-CaCO3 showed a significant synergistic effect on the upregulation of co-stimulatory markers, including CD80, CD86, CD40 and MHC-II, in comparison with the control groups (Fig. 5c–e and Supplementary Fig. 10b). To test the local process of antigen generation in the context of Vax, we loaded Vax with ovalbumin (OVA) and analysed the presentation of antigen peptide on the MHC-I of BMDCs. As expected, Vax was capable of generating a substantial number of SIINFEKL-MHC-I+ CD11c+ BMDCs (Fig. 5f,g). These results imply localized DC activation and antigen presentation. Similar results were obtained using RAW 264.7 cells (Supplementary Fig. 10c).
The in vivo antigen uptake, activation and antigen presentation were assessed following the implantation of Vax. The antigen uptake inside the nodule was estimated by immunohistochemistry (IHC) of paraffin sections of the nodules. The localization of Ad5 was probed using an anti-hexon antibody, which clearly showed a larger number of Ad5-positive cells in the Vax-injected nodule than in the other groups (Fig. 5h). We next evaluated the transduction efficiency of luciferase expressing Ad5 vectors (Ad5-Luc) inside the nodule to determine whether a deficiency in cell infiltration influences its transduction activity. At the injection site, Luc expression in Ad5-injected mice rapidly declined, while Luc expression in Vax-injected mice increased by 9.5-fold and persisted for over 3 d (Fig. 5i,j and Supplementary Fig. 10d). Conversely, without nano-CaCO3 the Luc signal was completely undetectable, indicating impaired transduction activity due to low phagocytic efficiency and reduced cell infiltration.
Furthermore, the viral RNA at the Vax-injected site was identical to that in ZIKV-injected mice in the first 15 d, but by day 21, only one of the six mice remained positive, indicating in situ virus clearance (Extended Data Fig. 1). This long-term kinetics represented a depot effect. In addition, we noticed that larger numbers of DC cells infiltrated into the nodule at the Vax injection site (Fig. 5k), where the CD80+ CD86+, MHC-II+ and CD40+ DC numbers were notably elevated (Fig. 5l and Supplementary Fig. 10e). In the CD86+ CD11c+ DC population, the expression of MHC-I was 20-fold higher than that of MHC-II at the Vax injection site (Fig. 5m). Together, cellular uptake, intracellular processing and antigen presentation are crucial processes for eliciting an effective adaptive host response.
Potent adaptive immune responses
Having efficiently initiated antigen presentation in the Vax-injected microenvironments, we next evaluated the subsequent adaptive responses after a single vaccination. The Ad5-vectored vaccines containing the ZIKV pre-membrane-envelope proteins (Ad5-prM-E)37 were imbedded into the Vax, and ZIKV E protein-specific IgG in mouse sera was determined at 2, 4 and 6 weeks post-vaccination (Fig. 6a). Compared with the control group, Ad5-prM-E-Vax significantly mounted the E protein-specific IgG response in the 4–6 weeks, indicating an enhanced effect on humoral immunity (Fig. 6b). The efficacy of E-specific IgG was then evaluated by a 50% plaque reduction neutralization test (PRNT50), which is regarded as the gold standard for determining neutralizing antibody titres. The ZIKV E protein-specific IgG response and PRNT50 titre of Vax-vaccinated mice endured for 6 weeks (Fig. 6c), indicating the protective efficacy of Vax-based vaccination.

a, Overview of the immune schedule. b, Serum anti-ZIKV-Env IgG after immunization with free Ad5-prM-E, Vax, or untreated (n = 5). c, Neutralizing activity (n = 5). d, E protein-specific CD4+ T cells and e, CD8+ T cells secreting IL-2, IFN-γ or TNF-α (n = 6). Data represent the mean ± standard deviation (s.d.), and P values were determined by two-tailed Student’s t-test. P < 0.05 was considered significant. NS, not significant.
The ZIKV-Env-specific CD8+ and CD4+ T-cell responses were evaluated in mice immunized with Ad5-prM-E-Vax. The effector T cells that secreted T-helper-1 cytokines, including IFN-γ, IL-2 and TNF-α, were identified through multiparameter intracellular cytokine staining analysed by flow cytometry (Supplementary Fig. 11). We found that, after 2 weeks post-vaccination, the mice in the Vax group had enhanced IFN-γ-, TNF-α- and IL-2-secreting ZIKV-Env-specific CD4+ and CD8+ T cells compared with those in the Ad5-prM-E group (Fig. 6d,e). The robust cytokine-secreting ZIKV-Env-specific CD8+ T cells elicited by Vax denoted augmented cytotoxic T-cell responses. These results suggested the elevated production of both protective cellular responses and humoral immune responses by Vax, indicating the migration of antigen presenting cells from nodules to lymph nodes.
Vax enhanced immune responses in lymph nodes to exert systemic effects
The enhanced specific CD8+ T-cell responses and humoral responses of Vax may involve the APC and the T-cell responses in lymph nodes following the migration of APCs from nodules to draining lymph nodes (dLNs). Thus, after injection with PBS (BC), COS/CaCO3, Ad5, COS/Ad5 or Vax, the ability of Vax to initiate antigen presentation in the dLN was examined. The number of CD80+ CD86+ DCs in the dLNs was elevated after inoculation with Vax compared with that of other control groups (Fig. 7a). To test antigen presentation in the dLNs, we loaded Vax with OVA (Vax/OVA) as a model antigen and tested the numbers of DCs that presented SIINFEKL on MHC-I 7 d after injection. Vax/OVA immunization was capable of generating a significant number of SIINFEKL-MHC-I+ CD11c+ DCs in the dLNs compared with the OVA- and COS/OVA-injected groups, suggesting the effect of nano-CaCO3 on cross-presentation (Fig. 7b). The increased numbers of MHC-II+ and CD40+ DCs in the Vax group also confirmed the maturation of DCs, which is beneficial for subsequent T-cell activation (Fig. 7c and Supplementary Fig. 12). As previous studies demonstrated, upregulated CD40 was also involved in the migration of antigen-bearing DCs to the dLNs38,39. Moreover, the enhanced presentation of OVA peptide onto MHC-I by Vax could cross-present antigens by MHC-I to CD8+ T cells. Thus, we examined OVA-specific CD8+ T cells in the dLNs after vaccination using OVA-tetramer staining. Compared with other groups containing OVA, OVA-Vax drastically elevated the numbers of OVA tetramer + CD8+ T cells by 16-fold (Fig. 7d), contributing to the antigen-specific cytotoxic CD8+ T-cell response.

a, Quantification of the numbers of CD80+ CD86+ CD11c+ cells in the dLNs of C57BL/6 mice immunized with Ad5-, COS/Ad5-, COS/CaCO3- or Ad5-loaded Vax at 2 d post immunization. b, Number of DCs presenting SIINFEKL on the MHC-I molecule in the dLNs 7 d post immunization. c, Quantification of the numbers of MHC-II+ CD11c+ cells in the dLNs of mice at 2 d post immunization. d, Numbers of OVA Tetramer+ CD8+ cells in the dLNs 7 d post immunization. e, Representative flow cytometry plots to analyse Fas+ GL7+ B220+ cells in dLNs at 7 d post immunization. f, Number of Fas+ GL7+ B220+ GC B cells. g, Representative immunohistological images of GCs at 7 d post immunization. h, Sizes of GCs in representative immunofluorescent images. Throughout, data represent the mean ± standard deviation (s.d.) of five mice, and P values were determined by two-tailed Student’s t-test. P < 0.05 was considered significant. NS, not significant.
Since GC reactions are essential determinants of antibody responses and immune memory, we tested the ability of Vax to mediate the activation of GC B cells, which can undergo affinity maturation and somatic hypermutation to generate memory B cells and high-affinity antibodies in dLNs37. The GCs were identified by their expression of high levels of Fas and N-glycolylneuraminic acid (probed by GL7 antibody)40 using an immunofluorescence assay and flow cytometry. Analysis of dLNs in immunized mice revealed that ~9% of B220+ B cells were GL7+ Fas+ GC B cells in the Vax/Ad5 group (Fig. 7e), showing a three- to eight-fold increase in the number of GC-positive cells compared with the numbers in the Ad5 and COS/Ad5 groups (Fig. 7f). To explore the active B cells organized in bona fide GC structures, we examined the GC B cells (GL7 and B220) in dLN sections post-prime and observed highly organized GC structures, indicating downstream effects of Vax on the B cells in the dLN (Fig. 7g). In contrast, the GCs in mice that received Ad5 or no treatment showed smaller sizes and numbers than those in mice that received Vax (Fig. 7g,h). Taken together, these data suggest that Vax is capable of activating both T-cell responses and humoral responses by enhanced cross-presentation and GC activation.
Vax directly converted WT ZIKV into an effective vaccine
Finally, to demonstrate the possibility of Vax converting a WT virus into an effective vaccine, we used a pathogenic strain of ZIKV as an example and investigated the in vivo protective effect of ZIKV-loaded Vax (Vax/ZIKV) on lethal ZIKV challenge (Fig. 8a). True benefits of loading live ZIKV into Vax were verified by comparing its IgG responses and immune protection with clinically relevant controls, including Ad5-prM-E, inactivated ZIKV (iZIKV) with PolyI:C, and iZIKV with Alum, and material-related control groups, including empty Vax without virus, iZIKV mixed with nano-CaCO3 (iZIKV-CaCO3), iZIKV with COS gel (COS/iZIKV) and iZIKV with COS/CaCO3 gel (Vax/iZIKV). Four- to 6-week-old IFNAR1−/− mice received a single s.c. vaccination of abovementioned remedies. The Vax/ZIKV-immunized IFNAR1−/− mice developed faster and higher ZIKV E-specific serum IgG responses and neutralizing titres than the iZIKV-related groups (Fig. 8b, c).

Four- to 6-week-old IFNAR1−/− mice were immunized through the s.c. route with PBS, empty Vax, Ad5-prM-Env, inactivated ZIKV (iZIKV), iZIKV with Alum, iZIKV with PolyI:C, iZIKV with nano-CaCO3 (iZIKV+CaCO3), iZIKV loaded COS gel (COS/iZIKV), iZIKV-loaded Vax (Vax/iZIKV) or live ZIKV-loaded Vax (Vax/ZIKV) (n = 6). a, The schedule for immune protection evaluation after one-dose Vax-ZIKV vaccination in IFNAR1−/− mice. b, The production of anti-ZIKV-Env IgG by mice immunized for 2 and 4 weeks. c, Neutralizing activity of ZIKV-Vax-treated IFNAR1−/− mice before the challenge. d, Survival curves of immunized IFNAR1−/− mice after lethal challenge with ZIKV. P values were determined by log-rank test. e, Mouse weight after challenge. The T- and B-cell immune memory effect was evaluated using IFNAR1−/− mice. PBS, Vax/iZIKV or Vax/ZIKV was prime-boost administrated at days 0 and 14 (n = 6). f, Splenocytes were isolated at day 42 for analysis of central memory T cells (CD4+ CD44+ CD62L+ and CD8+ CD44+ CD62L+) and effector memory T cells (CD4+ CD44+ CD62L− and CD8+ CD44+ CD62L−). g, Memory B cells (MBCs) subsets were defined by CD80+ PDL2+ MBCs that related to antibody-forming cell (AFC) responses or CD80− PDL2− MBCs that seeded GCs. Data represent the mean ± standard deviation (s.d.), and P values were determined by two-tailed Student’s t-test (b, c, f and g) or two-way analysis of variance with a Tukey post-hoc test (e). P < 0.05 was considered significant. NS, not significant.
After s.c. immunization, mice from all the groups were infected with WT ZIKV on day 29 post immunization (Fig. 8a). The infected mice were evaluated for weight loss and survival. The Vax/ZIKV-administered mice showed some, albeit statistically non-significant, weight loss compared with Ad5-prM-E-vaccinated mice (Fig. 8e). Mice immunized with iZIKV-related formulations all exhibited a significant weight loss on days 7–12 compared with the Vax ZIKV-vaccinated groups. Neither morbidity nor mortality was observed in Vax/ZIKV- and Ad5-prM-E-vaccinated animals, whereas approximately 20–70% mortality was observed in animals from other groups (Fig. 8d). These data demonstrated that equivalent responses cannot be obtained by adding inactivated ZIKV into Vax or adding adjuvants to iZIKV, showing an advantage over the traditional vaccine adjuvant technology and the virus inactivation strategy. After the challenge, the PRNT50 titre of Vax-immunized mice was enhanced among surviving mice in all groups (Supplementary Fig. 13).
Since vaccination with Vax/ZIKV showed a long-lasting depot effect and robust dLN response, we investigated whether loading live ZIKV into Vax could effectively trigger long-term immune memory. To assess memory responses, mice were first subcutaneously immunized with 2 × 105 PFU of Vax/iZIKV and Vax/ZIKV and 100 μl of PBS for 2 weeks after secondary immunization with the same dosage of the above samples (Fig. 8a). The immune memory markers on T and B cells in the spleens of recipient mice were investigated 2 weeks after vaccination. The subpopulations of central memory (CD44+ and CD62L+) and effector memory (CD44+ and CD62L−) T cells among CD4+ and CD8+ T cells were significantly promoted in Vax/ZIKV-injected mice compared with those in Vax/iZIKV-vaccinated mice (Fig. 8f and Supplementary Fig. 14). The markers CD80 and programmed cell death 1 ligand 2 (PDL2) were used to define memory B-cell subsets. In Vax/ZIKV-immunized mice, CD80+ PDL2+ memory B cells that are related to plasmablasts and CD80− PDL2− memory B cells that indicate the GC reaction were elevated compared with PBS control (Fig. 8g). However, these memory B-cell responses were identical to Vax/iZIKV. Collectively, these results showed that loading living ZIKV into the Vax scaffold considerably enhanced the T-cell immune memory responses. Overall, Vax is a safe material-based strategy to enable a virus-to-vaccine conversion via eliminating infectivity while enhancing immune responses.
Discussion
This proof-of-concept study suggests that virulent virus strains can be directly transformed into vaccines by engineering virus-entrapped microenvironments. Compared with widely used nanovaccine technology, which is unable to efficiently recruit and regulate multiple immune cells41,42,43,44, the advantage of this strategy is the creation of a peripheral compartment for virus entrapment and the mass production of different types of activated immune cells. This immune-cell-rich environment can produce cytokine cascades and provide a positive feedback loop for immune activation, resulting in the spatial and temporal control of virus activation, processing, and local antigen presentation by APCs. Thus, instead of ZIKV infection throughout the body, the formation of an immune niche can directly eliminate the virus inside the gel scaffold while initiating antigen processing since the PRR-dependent innate immune responses of infiltrated cells are fully activated. Subsequently, the robust local innate immune responses enable the migration of the antigen-laden APCs to dLNs45, resulting in the activation of cross-presentation and GC responses in dLNs.
We used chitosan as the framework because it has excellent biocompatibility and has been widely used as a vaccine adjuvant due to its immunomodulatory and chemoattractive properties46,47. It has been documented that chitosan particles can be readily phagocytosed by immune cells48 and promote innate immune responses that can lead to pathogen clearance, pro-inflammatory responses and antigen presentation. Chitosan, regarded as a PAMP, can be recognized by some PRRs. However, the specific receptors that can recognize chitosan to induce an innate immune response have yet to be definitively elucidated. Some innate immune receptors, including TLR-2, TLR-4, dectin-1, the mannose receptor, CR3 and CD14, have been implicated in mediating the innate responses upon treatment with chitin or chitosan33,49,50. In our case, the RNA-sequencing results indicated the upregulation of the αM chain of integrin and CD14 (Fig. 3b). CR3 (CD11b/CD18-integrin, alpha M/beta 2-integrin, Mac-1) plays a central role in the immunological functions of phagocytes. Note that the αM chain of CR3 is a ‘lectin domain’ that has high β-glucan binding activity and binds to other microbial carbohydrates via non-opsonized phagocytosis51,52. Although CR3 alone is sufficient to mediate the phagocytosis of some bacteria, in some cases, CD14, as the glycosylphosphatidylinositol-anchored protein, also participates in the internalization of microorganisms mediated by integrin53. In line with previous studies, we found that the engagement of Vax activates CR3, CD14 and TLR-4, which results in the stimulation of local innate immune responses and the promotion of virus phagocytosis by DCs and macrophages, indicating pathogen clearance via a phagocytosis-related pathway. In addition, compared with the general understanding that chitosan can activate the NLRP3 inflammasome pathway and stimulate IL-1β release54, our results showed that the release of IL-1β in response to Vax and COS/CaCO3 was notably impaired compared with the chitosan gel. The NLRP3 ablation did not effectively inhibit the IL-1β release of Vax-treated BMDCs, indicating that the enhanced innate immune responses upon Vax immunization were not closely correlated with the NLRP3 pathway (Supplementary Fig. 7). Further efforts are needed to reveal the immune modulating effect of the Vax scaffold. To augment the immune modulation effect of COS hydrogel, we also added nano-CaCO3, known as an immunologic adjuvant55. We found that CaCO3 did not obviously increase the inflammatory responses but notably enhanced cell recruitment and promotes cross-presentation in the dLNs (Fig. 5m), probably because of immune modulation and the regulation of ion homeostasis (Supplementary Table 3).
Compared with the current vaccine engineering strategies, such as live-attenuated and inactivated vaccines that still suffer from safety concerns and long development cycles, our virus-confined immune-modulating microenvironment provides a safe and efficient strategy for the spatial–temporal modulation of immune responses. Vax gathers immune cells to form an immune niche and changes the immune patterns of immune cells, leading to the generation of pro-inflammatory factors and antiviral responses, which contribute to eliminating virus infection (Fig. 3c–g). Moreover, the niche enables further immune activation in the dLN due to the elevation of GC B cells and cross-presentation to CD8+ T cells, resulting in enhanced adaptive immune responses. This microenvironment enables complete protection against ZIKV infection and ablates the need for complicated biological engineering of viruses. We believe that the concept that materials can convert viruses into vaccines will provide a vaccine-development strategy for protection against newly emerging viruses for which currently there is no vaccine available.
Methods
Materials
COS lactate, polyaspartic acid, sodium bicarbonate (NaHCO3) and calcium chloride (CaCl2) were purchased from Sigma-Aldrich and used without further purification. The cell lines, including RAW 264.7 cells and Vero cells, were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). C57BL/6 mice (4–6 weeks) and female BALB/c mice (4–6 weeks) were purchased from Charles River, China. IFNAR1−/− mice (4–8 weeks old) were purchased from Shanghai Model Organisms Centre and Institute of Laboratory Animal Sciences, CAMS&PUMC. Ad5-GFP and Ad5-null were purchased from Hanbio Biotechnology (Shanghai, China). Ad5-Luc and Ad5-prM-E were stored in the laboratory. The ZIKV strain ZIKA-SMGC-1, GenBank accession number KX266255, was a laboratory-stored virus.
All animal experiments were carried out according to the Guidelines on the Care and Use of Animals for Scientific Purposes issued by Zhejiang University and approved by the Animal Care and Use Committee of the Beijing Institute of Biotechnology.
Preparation of Vax
Briefly, calcium chloride solution was added to 25 mg ml−1 chitosan solutions containing viruses (Ad5 or ZIKV) with stirring. Then, a mixed sodium bicarbonate and polyaspartic acid solution was added to the virus–chitosan mixture dropwise to initiate the formation of nano calcium carbonate. Carboxyl-group-rich polyaspartic acid was added to stabilize the nano-CaCO3 produced during gelation. The mixture was then stirred at 4 °C for 15 min. The alkaline environment reduced the electrostatic repulsion and allowed extensive hydrogen bonding and hydrophobic interactions between free –NH2 groups. The pre-hydrogel liquid was then brought to 37 °C for gelation. Empty Vax (eVax) was prepared using the same method without adding virus. The conventional chitosan gel (COS) was prepared by directly adding sodium bicarbonate to chitosan or chitosan–virus solution under stirring, followed by a temperature change to 37 °C for gelation. To prepare the nano-CaCO3 control, 100 μl of 1 M sodium bicarbonate solution was mixed with 100 μl of 1 mg ml−1 polyaspartic acid and added to 1 ml of 0.1 M calcium chloride solution with stirring to initiate the formation of nano-CaCO3. For immunological evaluation, the endotoxin in all the used solutions was removed using endotoxin removal filter (PALL) before use (Supplementary Fig. 2e).
In vitro confinement of virus in Vax
The in vitro release of Ad5 or ZIKV from Ad5-loaded Vax (Vax/Ad5) or ZIKV-loaded Vax (Vax/ZIKV) was studied after incubation of Vax/Ad5 or Vax/ZIKV in various solutions. Vax/Ad5 was soaked in sodium acetate buffer (pH 5.0), Tris–HCl buffer (pH 7.4) and Tris–HCl buffer (pH 8.0) at 37 °C. Vax/ZIKV was immersed in complete DMEM medium supplemented with 10% FBS and a lysozyme (10 μg ml−1)-containing buffer at 37 °C to mimic physiological environments. Released viruses in the supernatant were collected over time and measured using real-time qPCR or qRT‒PCR.
Total DNA was isolated with a DNeasy Blood & Tissue Kit (QIAGEN). The obtained DNA was quantified by one-step quantitative real-time RT‒PCR using TB Green Premix (Takara) and Ad5 primers (forward primer: 5′-CACCACCTCCCGGTACCATA-3′; reverse primer: 5′-CCGCACCTGGTTTTGCTT-3′).
ZIKV RNA in supernatant was isolated using a QIAamp Viral RNA Mini Kit (QIAGEN). The obtained RNA was quantified by real-time RT‒PCR using a QuantiNova Probe RT‒PCR Kit (QIAGEN), ZIKV primers (forward primer: CGYTGCCCAACACAAGG; reverse primer: CCACYAAYGTTCTTTTGCABACA) and probe (FAM AGCCTACCTTGAYAAGCARTCAGACACTC-BHQ-1) (ref. 37).
In vivo confinement of virus in Vax
In vivo systemic release of ZIKV was tested by detecting ZIKV RNA in mouse sera, at the injection site and in major organs. IFNAR1−/− mice were subcutaneously injected with PBS, ZIKV or ZIKV-loaded Vax containing 3.33 mg of chitosan per mouse, 0.31 mg of CaCO3 per mouse, and 8 × 104 PFU of ZIKV per mouse. Sera, organs and the skins from the injection site were collected at days 5, 10, 15, 28 and 52 for RNA extraction. ZIKV RNA in sera or tissues was isolated using a QIAamp Viral RNA Mini Kit (QIAGEN). The obtained RNA was quantified by real-time RT‒PCR. When ≥20% weight loss occurred, the mice were killed. To evaluate ZIKV confinement in developing mouse brains, 1-d-old suckling BALB/c mice were intracranially injected with ZIKV-embedded Vax or free ZIKV, and survival was monitored; herein, the injected dose was reduced to 0.333 mg of chitosan, 0.031 mg of nano-CaCO3 and 2 × 104 PFU of ZIKV per mouse (n = 10). The mice were killed at 28 d post inoculation or when ≥20% weight loss occurred.
BMDC isolation and culture
BMDCs were derived using standard techniques56. In brief, bone marrow cells were isolated from C57BL/6 mice and cultured in RPMI 1640 medium (Gibco) supplemented with 10% heat-inactivated FBS (Gibco), 1% penicillin–streptomycin, 10 ng ml−1 IL-4 (Peprotech) and 20 ng ml−1 GM-CSF (Peprotech). DCs were collected and used for experiments after 7 d.
In vitro cell migration
BMDCs or RAW 264.7 cells were starved for 24 h the day before the assay. An attraction well without Transwell inserts was prepared by adding blank control, Ad5, COS/Ad5 and Ad5-loaded Vax to the lower wells with BMDCs or RAW 264.7 cells and immersing in 600 μl of RPMI 1640 medium (Gibco) plus 10% FBS (Gibco). Then, 1 × 104 cells were dispensed into Transwell (Costar) inserts, which were then gently transferred into the attraction wells. The 24-well plate was gently placed in a 37 °C, 5% CO2 incubator and allowed to incubate undisturbed for 9–12 h. To count the cells that migrated from the upper to the lower wells, the Transwell inserts were removed, and the cells attached to the bottom membranes were stained with crystal violet for cell counting.
Detection of chemokine and cytokine secretion
BMDCs were cultured within Ad5-loaded Vax and with control samples. The cell culture supernatants were collected at 24 h to examine using a ProcartaPlex kit (Invitrogen).
In vitro antigen processing
To detect whether cell uptake and antigen expression were affected by Vax, BMDCs were cultured inside Vax with AF660-labelled chitosan and AF55-labelled Ad5 for 2 h. The cells were then observed by CLSM (Olympus). Stimulation, activation and maturation were studied by detecting the upregulation of co-stimulatory markers on the APC. The Ad5-loaded Vax and other control samples were incubated with BMDCs. After incubation for 24 h, the cells were analysed by flow cytometry using a CytoFLEX LX Flow cytometer (Beckman Coulter). Before the tests, the isolated cells were first blocked with anti-mouse CD16/CD32 and then incubated with PerCP anti-mouse CD11c (BioLegend), BV650 anti-mouse CD80 (BioLegend), APC-cy7 anti-mouse CD86 (BioLegend), PE anti-mouse CD40 (BioLegend), BV510 anti-mouse I-A/I-E (BioLegend) or APC anti-mouse H-2Kb bound to SIINFEKL antibodies diluted in Staining Buffer. All cells were gated on the basis of forward and side scatter characteristics to limit debris, including dead cells. Antibodies were diluted according to the manufacturer’s suggestions. Cells were gated on the basis of fluorescence-minus-one controls, and the frequencies of cells staining positive for each marker were recorded.
In vivo antigen presentation in dLNs
Four- to 6-week-old C57BL/6 mice were immunized with PBS, COS/CaCO3, OVA, COS/OVA and OVA-loaded Vax. The lymph nodes from immunized mice were collected on day 14 for intracellular cytokine staining and flow cytometric analysis. Cell suspensions from dLNs were prepared by mechanical disruption and pressing of the tissue through 40 μm cell strainers and examined by flow cytometry. Cells were blocked with anti-mouse CD16/CD32 and then incubated with PerCP anti-mouse CD11c (BioLegend), BV650 anti-mouse CD80 (BioLegend), APC-cy7 anti-mouse CD86 (BioLegend), PE anti-mouse CD40 (BioLegend), BV510 anti-mouse I-A/I-E (BioLegend), or APC anti-mouse H-2Kb bound to SIINFEKL antibodies diluted in Staining Buffer. The cells were also incubated with APC-Cy7 anti-mouse CD3 (BioLegend), PerCP-Cy5.5 anti-mouse CD4 (BioLegend), PE anti-mouse CD8 (BioLegend) and T-Select H-2Kb OVA Tetramer-SIINFEKL-APC (MBL). The OVA-derived peptide SIINFEKL bound to H-2Kb of MHC class I CD11c+ DCs were probed by SIINFEKL monoclonal antibody that specifically reacts with the MHC class I-binding 257–264 epitope from OVA. Then, antigen-specific CD8+ T cells were analysed by the recognition of T-Select H-2Kb OVA Tetramer-SIINFEKL-APC upon binding to cognate TCRs.
Detection of GC formation
To analyse GC formation, dLNs were isolated on day 7 after vaccination. Cells were enumerated and stained with BV421 anti-mouse B220 (BioLegend), PE anti-mouse GL7 (eBioscience) and APC anti-mouse CD95 (Fas) (BioLegend) antibodies for 15 min on ice. Cells were washed and assessed by flow cytometry. To assess GC formation, dLNs were isolated, flash-frozen, cryosectioned, stained with anti-B220 and anti-GL7 (BioLegend), and observed by OLYMPUS SLIDEVIEW VS200. The GC area was obtained by analysing the images using OlyVIA software.
Cellular responses
Four- to 6-week-old BALB/c mice were randomly divided into three groups to receive Ad5-prM-E (1 × 108 PFU per mouse), Ad5-prM-E-loaded Vax or PBS. Immunized mice were killed on day 14 for intracellular cytokine staining and flow cytometric analysis. The isolated spleen lymphocytes were stimulated for 2 h with a pool of 15-mers with five-offset overlapping peptides covering the prM or Env proteins (Glbiochem)37 dissolved in DMSO (2.5 μg ml−1 each) and then incubated with BD GolgiStop for 4 h. The cells were washed with PBS, blocked with Mouse BD Fc Block and then incubated with BV421 anti-mouse CD3e (BD), BV605 anti-mouse CD4 (BD), BV510 anti-mouse CD8a (BD) and APC anti-mouse CD107a (BD) antibodies diluted in Cell Staining Buffer (BD). After simultaneous fixation and permeabilization with Cytofix/Cytoperm Fixation and Permeabilization Solution (BD), cells were incubated with FITC anti-mouse IFN-γ (BD), PerCP-Cy5.5 anti-mouse TNF-α (BD) and PE anti-mouse IL-2 (BD) antibodies diluted in 1× Perm/Wash Buffer. Anti-rat/hamster Ig and κ/negative control compensation particle sets (BD) were used for fluorescence compensation. Flow cytometry was performed using a CytoFLEX LX Flow cytometer (Beckman Coulter).
Protection efficacy
The protection efficacy after vaccination with ZIKV-loaded Vax was examined using the IFNAR1−/− mouse model. Briefly, 4- to 6-week-old IFNAR1−/− mice (six animals per group) were immunized with 8 × 104 PFU of iZIKV, iZIKV with alum, iZIKV with polyI:C, iZIKV with CaCO3, iZIKV with COS gel, iZIKV with empty Vax and ZIKV-loaded Vax (dose was the same as the above IFNAR1−/− mouse experiments) or 10 mM PBS, empty Vax and Ad5-PreM-E as controls. At 4 weeks post-vaccination, mice were subcutaneously challenged with 2 × 105 PFU of WT ZIKV. Body weight was monitored for 18 d. Survival rate as monitored for 2 months post inoculation. However, after infection for 28 d, none of the mice died.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. All data generated in this study, including source data for the figures, are available from figshare with the identifier https://doi.org/10.6084/m9.figshare.22126691.
References
Plotkin, S. A. Vaccines: past, present and future. Nat. Med. 11, S5–S11 (2005).
Larocca, R. A. et al. Vaccine protection against Zika virus from Brazil. Nature 536, 474–478 (2016).
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Wang, C., Horby, P. W., Hayden, F. G. & Gao, G. F. A novel coronavirus outbreak of global health concern. Lancet 395, 470–473 (2020).
Amanat, F. & Krammer, F. SARS-CoV-2 vaccines: status report. Immunity 52, 583–589 (2020).
Gao, Q. et al. Development of an inactivated vaccine candidate for SARS-CoV-2. Science 369, 77 (2020).
Rappuoli, R., Pizza, M., Del Giudice, G. & De Gregorio, E. Vaccines, new opportunities for a new society. Proc. Natl Acad. Sci. USA 111, 12288 (2014).
Graham, B. S. Rapid COVID-19 vaccine development. Science 368, 945–946 (2020).
Irvine, D. J., Swartz, M. A. & Szeto, G. L. Engineering synthetic vaccines using cues from natural immunity. Nat. Mater. 12, 978–990 (2013).
Kim, J. et al. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat. Biotechnol. 33, 64–72 (2015).
Wilson, D. S. et al. Antigens reversibly conjugated to a polymeric glyco-adjuvant induce protective humoral and cellular immunity. Nat. Mater. 18, 175–185 (2019).
Kasturi, S. P. et al. Programming the magnitude and persistence of antibody responses with innate immunity. Nature 470, 543–U136 (2011).
Liu, H. et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522 (2014).
Wang, X. et al. Biomineralization-based virus shell-engineering: towards neutralization escape and tropism expansion. Adv. Healthc. Mater. 1, 443–449 (2012).
Wang, G. et al. Rational design of thermostable vaccines by engineered peptide-induced virus self-biomineralization under physiological conditions. Proc. Natl Acad. Sci. USA 110, 7619 (2013).
Wang, X. et al. Vaccine engineering with dual-functional mineral shell: a promising strategy to overcome preexisting immunity. Adv. Mater. 28, 694–700 (2016).
Fries, C. N. et al. Advances in nanomaterial vaccine strategies to address infectious diseases impacting global health. Nat. Nanotechnol. 16, 385–398 (2020).
Wen, A. M. & Steinmetz, N. F. Design of virus-based nanomaterials for medicine, biotechnology, and energy. Chem. Soc. Rev. 45, 4074–4126 (2016).
Singh, A. Eliciting B cell immunity against infectious diseases using nanovaccines. Nat. Nanotechnol. 16, 16–24 (2021).
Seliktar, D. Designing cell-compatible hydrogels for biomedical applications. Science 336, 1124 (2012).
Dellacherie, M. O., Seo, B. R. & Mooney, D. J. Macroscale biomaterials strategies for local immunomodulation. Nat. Rev. Mater. 4, 379–397 (2019).
Veiseh, O. et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nat. Mater. 14, 643–651 (2015).
Vegas, A. J. et al. Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nat. Biotechnol. 34, 345–352 (2016).
Thompson, M. R., Kaminski, J. J., Kurt-Jones, E. A. & Fitzgerald, K. A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 3, 920–940 (2011).
Gong, N. et al. Proton-driven transformable nanovaccine for cancer immunotherapy. Nat. Nanotechnol. 15, 1053–1064 (2020).
Le Gal La Salle, G. et al. An adenovirus vector for gene transfer into neurons and glia in the brain. Science 259, 988 (1993).
Ahmed, T. A. & Aljaeid, B. M. Preparation, characterization, and potential application of chitosan, chitosan derivatives, and chitosan metal nanoparticles in pharmaceutical drug delivery. Drug Des. Dev. Ther. 10, 483–507 (2016).
Chen, Q. et al. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat. Nanotechnol. 14, 89–97 (2019).
Abbink, P. et al. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science 353, 1129 (2016).
Lazear, H. M. et al. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19, 720–730 (2016).
Shan, C. et al. A live-attenuated Zika virus vaccine candidate induces sterilizing immunity in mouse models. Nat. Med. 23, 763–767 (2017).
Newton, K. & Dixit, V. M. Signaling in innate immunity and inflammation. Cold Spring Harbor Perspect. Biol. 4, a006049 (2012).
Chang, S.-H., Lin, Y.-Y., Wu, G.-J., Huang, C.-H. & Tsai, G. J. Effect of chitosan molecular weight on anti-inflammatory activity in the RAW 264.7 macrophage model. Int. J. Biol. Macromol. 131, 167–175 (2019).
Hamza, T., Barnett, J. B. & Li, B. Y. Interleukin 12 a key immunoregulatory cytokine in infection applications. Int. J. Mol. Sci. 11, 789–806 (2010).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Yoneyama, H. et al. Pivotal role of dendritic cell-derived CXCL10 in the retention of T helper cell 1 lymphocytes in secondary lymph nodes. J. Exp. Med. 195, 1257–1266 (2002).
Guo, Q. et al. Immunization with a novel human type 5 adenovirus-vectored vaccine expressing the premembrane and envelope proteins of zika virus provides consistent and sterilizing protection in multiple immunocompetent and immunocompromised animal models. J. Infect. Dis. 218, 365–377 (2018).
Moodycliffe, A. M. et al. Cd40–Cd40 ligand interactions in vivo regulate migration of antigen-bearing dendritic cells from the skin to draining lymph nodes. J. Exp. Med. 191, 2011–2020 (1999).
Bennett, S. R. M. et al. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 393, 478–480 (1998).
Shlomchik, M. J. & Weisel, F. Germinal centers. Immunol. Rev. 247, 5–10 (2012).
Tang, L. et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol. 36, 707–716 (2018).
Chaudhuri, O. et al. Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat. Mater. 15, 326–334 (2016).
Wang, C., Wang, J., Zhang, X., Yu, S. & Gu, Z. In situ formed reactive oxygen species–responsive scaffold with gemcitabine and checkpoint inhibitor for combination therapy. Sci. Transl. Med. 10, eaan3682 (2018).
Conde, J., Oliva, N., Atilano, M., Song, H. S. & Artzi, N. Self-assembled RNA-triple-helix hydrogel scaffold for microRNA modulation in the tumour microenvironment. Nat. Mater. 15, 353–363 (2016).
Hori, Y., Winans, A. M., Huang, C. C., Horrigan, E. M. & Irvine, D. J. Injectable dendritic cell-carrying alginate gels for immunization and immunotherapy. Biomaterials 29, 3671–3682 (2008).
Li, X. M. et al. Immunostimulatory effect of chitosan and quaternary chitosan: a review of potential vaccine adjuvants. Carbohydr. Polym. 264, 118050 (2021).
Noh, K. H. et al. GM-CSF-loaded chitosan hydrogel as an immunoadjuvant enhances antigen-specific immune responses with reduced toxicity. BMC Immunol. 15, 48 (2014).
Bueter, C. L. et al. Chitosan but not chitin activates the inflammasome by a mechanism dependent upon phagocytosis. J. Biol. Chem. 286, 35447–35455 (2011).
Mora-Montes, H. M. et al. Recognition and blocking of innate immunity cells by candida albicans chitin. Infect. Immun. 79, 1961–1970 (2011).
Dasgupta, P. & Keegan, A. D. Contribution of alternatively activated macrophages to allergic lung inflammation: a tale of mice and men. J. Innate Immun. 4, 478–488 (2012).
Goodridge, H. S., Wolf, A. J. & Underhill, D. M. Beta-glucan recognition by the innate immune system. Immunol. Rev. 230, 38–50 (2009).
Linehan, S. A., Martinez-Pomares, L. & Gordon, S. Macrophage lectins in host defence. Microbes Infect. 2, 279–288 (2000).
Da Silva, C. A. et al. Chitin is a size-dependent regulator of macrophage TNF and IL-10 production. J. Immunol. 182, 3573–3582 (2009).
Jia, J., Liu, Q., Yang, T., Wang, L. & Ma, G. Facile fabrication of varisized calcium carbonate microspheres as vaccine adjuvants. J. Mat. Chem. B 5, 1611–1623 (2017).
Hawley, K. L. et al. CD14 cooperates with complement receptor 3 to mediate MyD88-independent phagocytosis of Borrelia burgdorferi. Proc. Natl Acad. Sci. USA 109, 1228–1232 (2012).
Song, H. et al. Injectable polypeptide hydrogel for dual-delivery of antigen and TLR3 agonist to modulate dendritic cells in vivo and enhance potent cytotoxic T-lymphocyte response against melanoma. Biomaterials 159, 119–129 (2018).
Acknowledgements
We thank J. Xuan for the assistance with CLSM data analysis, and Q. Huang and J. Chen for their help with histological analysis. This work was supported by The National Natural Science Foundation of China (22037005 and 21625105) and National Science & Technology Major Project (2016ZX10004001).
Author information
Authors and Affiliations
Contributions
R.T. and X.W. initiated this study. X.W., R.T. and L.H. supervised and supported the research. H.H. performed most experiments including physicochemical characterization, cell experiments and animal experiments. Y.Z., F.T., X.W. and X.X. participated in performing the physicochemical characterization. J.L., Z.T., Y.M.Z., F.T., M.Z. and Y.C. participated in performing the animal experiments. H.H., X.W., Q.G. and S.W. analysed the data. The manuscript was written by H.H., X.W. and R.T. All authors provided critical feedback and helped shape the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Biomedical Engineering thanks Michael Mitchell and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Vax hydrogel restricted systemic infection in vivo.
IFNAR1−/− mice were injected with PBS, ZIKV, and Vax; n = 5 for each group at days 5–28, and n = 3 for each group at day 52. a, Viral RNA in the spleens, kidneys, liver, hearts, brains and testes of PBS-injected mice was determined by real-time quantitative RT-PCR. L.O.D., limit of detection. b, The weight of testes from IFNAR1−/− mice after injected with PBS, ZIKV, or Vax. c, Representative H&E staining of the testes from IFNAR1−/− mice after injected with PBS, ZIKV, and Vax. d, Survival curves of IFNAR1−/− mice after injection with PBS, ZIKV or ZIKV loaded Vax; n = 6 for each group. All the mice in the Vax injected group survived compared with the 50% survival rate of the WT ZIKV group. Statistical analyses were performed by using log-rank test.
Supplementary information
Supplementary Information
Supplementary methods, figures, tables, and references.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hao, H., Wu, S., Lin, J. et al. Immunization against Zika by entrapping live virus in a subcutaneous self-adjuvanting hydrogel. Nat. Biomed. Eng 7, 928–942 (2023). https://doi.org/10.1038/s41551-023-01014-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41551-023-01014-4
This article is cited by
-
Inorganic ionic polymerization: From biomineralization to materials manufacturing
Nano Research (2024)
-
A hydrogel-entrapped live virus immunization
Nature Biomedical Engineering (2023)
