
Abstract
The emergence and re-emergence of highly virulent viral pathogens with the potential to cause a pandemic creates an urgent need for the accelerated discovery of antiviral therapeutics. Antiviral human monoclonal antibodies (mAbs) are promising candidates for the prevention and treatment of severe viral diseases, but their long development timeframes limit their rapid deployment and use. Here, we report the development of an integrated sequence of technologies, including single-cell mRNA-sequence analysis, bioinformatics, synthetic biology and high-throughput functional analysis, that enables the rapid discovery of highly potent antiviral human mAbs, the activity of which we validated in vivo. In a 78-d study modelling the deployment of a rapid response to an outbreak, we isolated more than 100 human mAbs that are specific to Zika virus, assessed their function, identified that 29 of these mAbs have broadly neutralizing activity, and verified the therapeutic potency of the lead candidates in mice and non-human primate models of infection through the delivery of an antibody-encoding mRNA formulation and of the respective IgG antibody. The pipeline provides a roadmap for rapid antibody-discovery programmes against viral pathogens of global concern.
Similar content being viewed by others


Main
Human monoclonal antibodies (mAbs) are increasingly being considered for use as therapeutic countermeasures against viral infectious diseases. In recent years, advances in the isolation of human B cells and antibody variable gene sequencing techniques have led to the identification of large numbers of therapeutic mAb candidates against many life-threatening viral pathogens. These targets include antigenically variable viruses, such as HIV1 and influenza virus2, newly emerging pathogens with high epidemic potential, including Ebola virus3, Marburg virus4, Zika virus (ZIKV)5,6,7, Lassa virus8, Middle East respiratory syndrome coronavirus (MERS-CoV)9, poxviruses10, Nipah virus11, and many other medically important viruses. More than 25 antiviral human mAbs are now being evaluated as therapeutics in clinical trials12,13.
Large human epidemics of zoonotic diseases are occurring on a regular basis. For example, Ebola virus reportedly caused 28,646 cases of disease and 11,323 deaths during the 2013–2016 epidemic in West Africa14, the 2015–2016 ZIKV epidemic resulted in millions of infections in new geographical areas15,16 and, recently, the novel highly transmissible coronavirus SARS-CoV-2 was first detected in China and caused a global pandemic with hundreds of thousands of deaths to date. These events highlight the potential of viral infections to cause global health emergencies and the need for rapid-response technologies to accelerate the development of medical countermeasures. Several obstacles impede progress in the widespread application of antiviral mAb therapies in outbreak scenarios. A principal impediment of deploying human antiviral mAbs is the difficulty in predicting which pathogen will cause an epidemic in the short term, and the long timeline needed for human mAb discovery and verification of therapeutic potency.
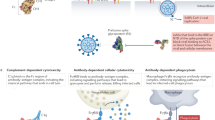
The goal of this study was to develop and demonstrate the ability of an integrated technology for accelerated discovery of potent human antiviral mAbs that are suitable for therapeutic development. As a model, we used the re-emerging pathogen ZIKV that is now endemic in multiple continents17 and could potentially be controlled with a neutralizing mAb treatment5,16,18,19. We designed an integrated workflow that could accomplish both discovery and validation of protective efficacy in small animals and non-human primates (NHP) in less than 90 d. The objectives were to rapidly execute a sequence of tasks, including virus stock production, target-specific antibody-gene rescue, bioinformatics analysis, mAb gene synthesis, mAb functional in vitro analysis, validation of mAb in vivo activity using IgG protein and RNA-encoded mAb delivery in a mouse model, and proof-of-principle NHP protection studies, as described in Fig. 1. Although clinically directed manufacturing was not within the scope of this timeline, both traditional protein antibody and experimental nucleic-acid-delivered mAb formulations were included within the discovery approach to accommodate for the diverse downstream development processes. The workflow implemented high-throughput and redundant features, with the goal of streamlining mAb discovery and building in orthogonal approaches to overcome or mitigate the failure of critical experimental processes.

A schematic of the proposed integrated technology workflow that incorporates pathogen production, target-specific B-cell isolation, single-cell V(D)J gene sequence analysis, bioinformatics analysis, mAb production, mAb in vitro validation, mAb-encoding RNA synthesis and mAb in vivo validation using protein and nucleic acid delivery technologies. The timeline for identifying and validating protective mAbs against ZIKV is indicated in the timeline chart, where day 0 was designated as the start point and day 90 was the projected end point of this study. The green star (day 78) shows the completion time. Fv, variable fragment.
Results
Virus stock production
We used a contemporary ZIKV strain of the Asian lineage (Brazil, Paraiba 2015) and a historical ZIKV strain of the African lineage (Dakar, Senegal 1984) to account for genetic diversity and, ultimately, breadth of protection. To model ZIKV infection in mice, we also used a mouse-adapted isolate of ZIKV Dakar (ZIKV Dakar MA), which has a single gain-of-function mutation in the viral NS4B protein that overcomes mouse innate immune restriction20. To model production of a high-titre virus stock for an unknown pathogen, we evaluated the growth of ZIKV in a panel of cell lines that are commonly used for virus propagation. High levels of virus replication were observed in Vero, U2OS, A549 and Huh7 cell lines, as determined using quantitative PCR with reverse transcription (RT–qPCR; Supplementary Fig. 1a), showing the ability of this approach to rapidly identify a permissive cell line(s). Identifying suitable conditions for large-scale production of the virus for subsequent in vitro and in vivo studies (Supplementary Fig. 1b,c) was accomplished in 21 d. Notably, the virus production was performed simultaneously and in parallel with initial mAb discovery steps and, therefore, the virus was obtained when it was needed for the neutralizing-activity screening step with purified mAbs, as described below.
Antibody variable gene sequence analysis and bioinformatics processing
To model a rapid response to a previously known pathogen, we used a mAb discovery approach, assuming that the envelope protein (ZIKV E protein) on the virion surface is a key protective antigen5,6,21,22. We used memory B (Bmem) cells from previously infected human individuals as the source for antibody discovery (Supplementary Table 1). The frequency of ZIKV-E-specific Bmem cells in most individuals with previous ZIKV infection is low23. We therefore pooled peripheral blood mononuclear cells (PBMCs) from a cohort of several individuals with previous exposure to ZIKV that were prescreened for E-specific Bmem-cell responses (Supplementary Fig. 2). B cells were enriched using antibody-coated magnetic beads and stained with phenotyping antibodies and soluble recombinant ZIKV E protein (see Methods). Antigen-labelled IgG-class-switched Bmem-cell–E complexes were isolated in bulk using fluorescence-activated cell sorting (FACS; Fig. 2a). To optimize the breadth of our mAb panel for diverse epitope specificities, we used several B-cell isolation approaches. The first approach (defined as direct) was based on the detection of bound biotinylated E protein using fluorescently labelled streptavidin. For the second approach (defined as indirect), B cells were labelled with E protein and then incubated with biotinylated non-neutralizing mAb ZIKV-88, which recognizes the fusion loop (FL) epitope5, and were detected using fluorescently labelled streptavidin. Furthermore, we sorted cells with high and low-to-intermediate intensity of E protein staining separately. Overall, more than 5,000 E-labelled B cells were sorted, and three independent sub-panels of mAbs were generated using the described B-cell isolation approaches (Supplementary Table 2).

a, Flow cytometric identification of target-specific Bmem cells after labelling magnetically enriched total B cells with ZIKV E antigen. Target-specific B cells were labelled with biotinylated recombinant E protein and detected using fluorochrome-conjugated streptavidin as indicated. The green arrow indicates FACS-sorted cells. Representative data of four experiments are shown. b, Bioinformatics filtering steps to select mAb sequences for synthesis. The timeline for identifying sequences of target mAbs and obtaining cDNA constructs for recombinant mAb expression is indicated by a blue arrow in the timeline chart.
To rescue the paired heavy- and light-chain antibody variable gene sequences from sorted B cells, we used a commercial single-cell encapsulation automated system (Chromium Technology; 10x Genomics). A feature of this sequencing approach is the large scale of paired antibody variable gene analysis, which is achieved by encapsulating single B cells using droplet microfluidics24. Approximately 800 sorted antigen-labelled B cells from the single specimen of pooled human cells were processed for library preparation and sequencing using the Chromium Controller device immediately after cell isolation using FACS. The remaining sorted cells were bulk-expanded in culture, and then the antibody variable genes for single cells were sequenced using Chromium Technology.
Using bioinformatics sieving, we downselected the total pool of possible sequences to choose 598 paired antibody heavy- and light-chain variable gene sequences for further evaluation (Fig. 2b). The filtered panel included mAbs of defined IgG isotypes, selecting the most somatically mutated individual clone in each antibody clonotype (that is, the clone with the most mutations compared with the inferred germline gene segments in the lineage of related sequences). Clonotypes were identified on the basis of the presence of identical inferred germline V and J gene assignments and identical amino acid sequences of the heavy-chain complementarity-determining region 3 (HCDR3; see Methods).
Synthetic complementary DNA constructs based on the sequences of all 598 selected ZIKV mAb candidates were synthesized using a rapid high-throughput cDNA synthesis technology (Twist Bioscience) and cloned into an IgG1 monocistronic expression vector for mammalian cell culture mAb secretion. This vector contains an enhanced 2A sequence and GSG linker that enables simultaneous expression of mAb heavy- and light-chain genes from a single construct after transfection25.
In summary, starting with samples from human participants, in 17 d, we produced recombinant IgG expression vectors for nearly 600 human mAbs from single cells, representing independent genetic clonotypes that were ready for expression and functional validation (Supplementary Table 2). These results demonstrate the ability to perform rapid target-specific B-cell selection and large-scale human antibody discovery, for a setting in which a known protective viral antigen is available.
High-throughput mAb production and functional screening
We used high-throughput assays for rapid production, purification and functional analysis of mAbs from small sample volumes (designated here as microscale) to identify lead mAb candidates for in vivo studies. To produce mAbs, we performed transfection of Chinese hamster ovary (CHO) cell cultures using ~1 ml cell culture volume per antibody, and mAbs were subsequently purified using a microscale parallel purification approach. Of the 598 mAb cDNAs tested, 475 were produced successfully as recombinant IgG proteins, with a median yield of ~29 μg of purified antibodies from a single 1 ml culture (Fig. 3a; Supplementary Table 2).

a, Microscale-expressed and -purified mAbs. The dots indicate the average concentration of individual mAbs from assay duplicates, and the median mAb yield is shown by the horizontal line. LOD, limit of the detection. b, The relationships between the binding and neutralizing activities of individual mAbs of the panel shown as a heat map. Binding to E protein was determined using ELISA and neutralizing activity was measured using RTCA of mAbs that were purified as shown in a. c, ZIKV E protein binding by potently neutralizing mAbs representing three distinct epitope-binding groups. MAb rRSV-90, which is specific to the unrelated respiratory syncytial virus (RSV) fusion protein (F) antigen, was used as a control. OD450, optical density at 450 nm. d, Neutralization of ZIKV Brazil or Dakar strain viruses by representative cross-neutralizing mAbs from c, as determined using RTCA. Data are mean ± s.d. of assay triplicates in c and d, and represent at least two independent experiments. The timeline for identifying mAb candidates for in vivo testing is indicated by a blue arrow in the timeline chart.
Antibodies can mediate protection through a diverse range of mechanisms, including direct virus neutralization and/or engagement of innate immune cells through their crystallizable fragment (Fc) receptors26,27. Potent neutralization and Fc effector functions are desirable mAb features that can be used to prioritize candidate mAbs for in vivo testing. For rapid identification of neutralizing mAbs within the large panel that we expressed, we used a real-time cell analysis (RTCA) cellular impedance assay. RTCA assesses kinetic changes in cell physiology, including virus-induced cytopathic effect (CPE), which offers a generic screening approach for mAb neutralizing activity against cytopathic viruses28,29. Our comparative side-by-side study showed that the RTCA performed similarly to a conventional focus-reduction neutralization test to identify neutralizing mAbs against ZIKV (Supplementary Table 2). However, RTCA offered an advantage for the large mAb panel analysis owing to its speed and automation—24 h to 36 h for the initial qualitative screening of neutralizing activity, and ~48 h for determining half-maximal inhibitory concentration (IC50) values and ranking ZIKV mAbs by neutralizing potency (Supplementary Fig. 3). Approximately 15% (92 out of 598) of the mAbs bound strongly to recombinant ZIKV E protein, as measured by enzyme-linked immunosorbent assay (ELISA), and ~8% (48 out of 598) of the mAbs neutralized ZIKV (Fig. 3b–d; Supplementary Table 2). Fourteen additional ZIKV-specific mAbs were identified only by neutralization, as they did not show detectable binding to the recombinant E antigen in the solid-phase on the basis of an ELISA assay, even though their B cells were sorted with the same antigen in solution. Thus, using a combination of binding and neutralization assays, we validated that at least 106 out of the mAbs from the panel were ZIKV-specific (Supplementary Table 2).
The individuals that we studied had probably been infected with a strain of ZIKV of the Asian lineage, as they contracted the disease during the recent outbreak in South America (Supplementary Table 1). Twenty-nine mAbs of the panel possessed neutralizing activity against viruses that were antigenically homologous (Brazil strain; Asian lineage) or heterologous (Dakar strain; African lineage) to the infecting strain (Supplementary Table 2). Twenty mAbs fully neutralized at least one out of the two tested viruses, with estimated IC50 values on the basis of RTCA data ranging from about 7 ng ml−1 to 1,100 ng ml−1, and 15 mAbs completely neutralized both viruses. The three most potent mAbs by neutralization IC50 value (designated mAbs ZIKV-893, ZIKV-752 and ZIKV-940) neutralized both the Brazil and Dakar ZIKV strains with IC50 values of less than 100 ng ml−1 but exhibited differential binding to recombinant E protein. ZIKV-752 bound E strongly, ZIKV-940 bound weakly and binding of ZIKV-893 was not detected at the highest mAb concentration tested (10 μg ml−1; Fig. 3c,d). Competition-binding analysis with the previously characterized ZIKV-E-specific human mAb ZIKV-117 (which recognizes the E protein dimer–dimer interface in domain II of the viral particles)30, ZIKV-116 (which binds to the E protein domain III lateral ridge epitope)31 and ZIKV-88 (which is specific for the E protein FL)5 showed that the three identified neutralizing mAbs ZIKV-893, ZIKV-752 and ZIKV-940 recognized non-overlapping epitopes (Supplementary Table 2). This conclusion was supported by epitope mapping data from loss-of-binding analysis to a ZIKV prM-E protein alanine scanning mutagenesis library expressed in cells5. Notably, these mAbs were identified using three different E-antigen-sorting strategies (Supplementary Table 2). This finding highlights the importance of study design in the antibody-discovery workflow, which relied on concurrent independent strategies for B-cell isolation conducted in parallel to identify potently neutralizing mAbs of diverse epitope specificity.
We next demonstrated the ability to rapidly assess the Fc-mediated effector function of the mAb panel. Using high-throughput flow cytometry analysis, we measured mAb-mediated activation of human effector cells in vitro after binding individual mAbs to bead- or plate-immobilized recombinant ZIKV E. Most of the mAbs in the panel (all of the IgG1 sub-class), which were reactive to recombinant E by ELISA, also showed activation of the effector cells after mAb binding to bead- or plate-bound E (Supplementary Table 2). This demonstrates the ability to perform high-throughput Fc profiling, and that the approach can be used in cases in which further engineering of the function of the Fc fragment (for example, tuning up or down) of individual mAbs is necessary for improved protection in vivo.
In summary, these data demonstrate the high performance of a suite of microscale assays in an integrated workflow and accomplished accelerated production and functional analysis of large panels of recombinant human mAbs to identify lead candidates for in vivo studies—all of which was accomplished in 13 d.
IgG protein and RNA production for the lead therapeutic mAb candidates for in vivo studies
We selected 20 lead mAb candidates for in vivo testing. We administered the mAbs using two different delivery systems: (1) recombinant IgG protein expressed in mammalian cell culture; and (2) mRNA delivery of antibody genes to enable in vivo expression of antibodies from transient gene transfer. We initiated the production of 20 recombinant IgG proteins in cultures scaled to obtain 3–7 mg of purified IgG. In parallel, the same 20 antibody variable gene sequences were cloned into a reading frame for human IgG1 and sub-cloned into plasmids containing an alphavirus replicon32 for in vivo analysis using nucleic acid delivery. To deliver the replicon, we chose a cationic nanostructured lipid carrier (NLC) formulation, in which RNA was bound to the nanoparticle surface by electrostatic association, enabling rapid formulation32. Linearized DNAs were used as a template to transcribe and post-transcriptionally cap replicon RNA at a 500 μg scale. The IgG protein preparation and RNA production and formulation were both accomplished in parallel in 8 d.
Protective efficacy of identified neutralizing mAbs in a lethal ZIKV challenge mouse model
We next used an established lethal, immunocompromized challenge model in mice for ZIKV5,21 to evaluate the protective ability of the 20 lead candidate mAbs (Supplementary Tables 3 and 4). In prophylaxis experiments, C57BL6/J mice were treated by intraperitoneal (i.p.) injection with anti-IFN-alpha/beta receptor (Ifnar1) antibodies and individual ZIKV mAbs on day −1. Candidate mAbs were compared to a potently protective human mAb, ZIKV-117, that we described previously5. An irrelevant IgG1 isotype mAb (FLU-5J8, which is specific to the influenza A virus haemagglutinin protein33) or PBS was used as a negative control. On day 0, mice were challenged through the subcutaneous (s.c.) route with 103 focus-forming units (FFU) of the ZIKV Dakar MA and monitored for 21 d for survival. High-dose prophylaxis (about 70 μg of mAbs per mouse; ~5 mg kg−1 mAb dose) provided complete protection from mortality for three out of the five tested mAbs. The mAb ZIKV-668 offered partial protection, whereas the mAbs ZIKV-434 and FLU-5J8 or PBS treatment did not provide protection (Supplementary Table 4). As multiple individual mAbs conferred full protection with high-dose mAb prophylaxis, we could not discriminate which mAb was the most effective in vivo. We therefore next tested a larger panel of ten neutralizing mAbs using a low-dose prophylaxis approach (using about 9 μg of mAbs; 0.65 mg kg−1 mAb dose). Even at low doses, six out of the ten human mAbs were detected readily in mouse blood as assessed 2 d after infection (Fig. 4a). As an early-time-point correlate of mAb-mediated protection, we measured viraemia from individual mice of each treatment group using RT–qPCR. All of the groups treated with ZIKV mAbs showed substantially (500–1,000-fold) reduced levels of virus measured as genome equivalents per 1 ml of serum by 2 d after infection compared with the control group that was treated with the mAb FLU-5J8, which developed high viraemia in all of the animals (Fig. 4b). This finding demonstrates efficient inhibition of viraemia using a low dose of anti-ZIKV mAbs in a prophylaxis setting.

a, Serum concentrations of human mAbs (2 d after infection) were determined by ELISA from mice that were treated prophylactically (day −1) with the indicated mAbs (9 μg per mouse) and challenged with ZIKV (day 0). The dots show measurements from individual mice. ZIKV-609, ZIKV-624, ZIKV-635, ZIKV-668, ZIKV-869, ZIKV-980, ZIKV-1006 and FLU5J8 were examined using n = 5 mice per group in one experiment; ZIKV-752 (n = 14 mice), ZIKV-893 (n = 11 mice), ZIKV-940 (n = 9 mice) and ZIKV-117 (n = 12 mice) were examined in three independent experiments of which cumulative data are shown. Data are mean ± s.d. NA, not available. b, Serum ZIKV titres (2 d after infection) from mice treated and challenged as shown in a. The dots show measurements from individual mice; n = 5 mice per group were analysed and data represent one experiment, except for the ZIKV-752 and ZIKV-117 treatment groups, for which a total of n = 14 mice per group were analysed in three independent experiments of which cumulative data are shown. Data are mean ± s.d. c, Survival of C57BL6/J mice that were treated and challenged as shown in a. The overall difference in survival between the groups was estimated using two-sided log-rank (Mantel–Cox) tests. P values are indicated. d, Low-dose (4.5 μg per mouse) prophylaxis of three lead candidate mAbs, ZIKV-752, ZIKV-893 and ZIKV-940, representing each of the three competition-binding groups; n = 5 mice per group were analysed in one experiment. Mice were treated and challenged as shown in a. The overall difference in survival between the groups was estimated using two-sided log-rank (Mantel–Cox) tests. P values are indicated. e, Low-dose (9 μg per mouse) post-exposure therapeutic efficacy (1 d after infection) of three lead candidate mAbs in mice challenged with ZIKV; n = 5 mice per group were analysed in one experiment. The overall difference in survival between the groups was estimated using two-sided log-rank (Mantel–Cox) tests. P values are indicated. The mAbs ZIKV-117 and FLU-5J8 and PBS were used as controls. The timeline for scaling-up mAb protein production and identifying the lead protective mAbs is shown by the blue arrows in the timeline chart.
Consistent with the reduced viral load in the plasma, each of the tested mAbs conferred protection against disease, as shown by reduced and/or delayed mortality compared with the control group that was treated with the mAb FLU-5J8. Three out of ten tested mAbs that neutralized both Brazil and Dakar strains (ZIKV-635, ZIKV-668 and ZIKV-893) conferred complete protection (100% survival by 21 d after infection); partial protection was conferred by the other two neutralizing mAbs, ZIKV-752 and ZIKV-940 (Fig. 4c; Supplementary Table 4). Protection with a 9 μg dose of ZIKV-893 or ZIKV-752 was confirmed in three independent experiments. Moreover, similar protection was observed using a twofold-to-fourfold lower dose of ZIKV-893, ZIKV-752 or ZIKV-940 (Fig. 4d; Supplementary Table 4). The above results demonstrate a high level of efficacy for several of these rapidly identified mAbs against ZIKV when given as prophylaxis.
We next tested the therapeutic efficacy after infection was established by administering a low dose of 9 μg of mAbs per mouse 1 d after infection. We tested the three mAbs ZIKV-893, ZIKV-752 and ZIKV-940 owing to their broad and potent neutralization of ZIKV, recognition of distinct non-overlapping epitopes on E and a high level of protection as prophylaxis (Fig. 4a–d; Supplementary Table 4). ZIKV-893 and ZIKV-940 conferred complete protection that was comparable to the control human mAb ZIKV-117, whereas ZIKV-752 was partially protective (Fig. 4e; Supplementary Table 4). A similar level of therapeutic protection was observed using a twofold lower dose of ZIKV-752, ZIKV-893 or ZIKV-940 mAbs (Supplementary Table 4).
In an outbreak scenario, an effective rapid public health response incorporating antibody treatments may be limited by the timeline required for protein mAb production in a format that can be safely administered to humans. Nucleic-acid-encoded antibody genes offer a potential route to antibody-mediated protection with shorter delivery timelines. As such, and in parallel, we tested the therapeutic potency of the 20 lead candidate mAbs discovered here with antibody-encoding RNA delivery in mice using a technology that enables in vivo mAb expression after intramuscular (i.m.) delivery of an NLC-formulated RNA34. Groups of C57BL6/J mice were treated with 40 μg of individual mAb-encoding RNA formulations on day −1 and then challenged with ZIKV Dakar MA on day 0. Groups treated with RNA encoding FLU-5J8 or ZIKV-117 were used as negative or positive controls, respectively. Human IgG protein was readily detected in the serum of individual mice from most of the treatment groups (Fig. 5a). Five mAbs that were identified to be highly protective with IgG protein treatment (ZIKV-635, ZIKV-668, ZIKV-752, ZIKV-893 and ZIKV-940) also mediated a high level of protection with this RNA antibody-gene delivery method, as judged by reduced viraemia on 2 d after infection and improved survival of the mice (Fig. 5b,c). These proof-of-concept experiments demonstrate the use of an antibody-encoding-RNA delivery approach for antiviral treatment against ZIKV. The activities of the tested 20 lead candidate mAbs are summarized in Table 1. Together, these results demonstrate the ability to rapidly identify several highly protective neutralizing mAbs against ZIKV and set the stage for evaluating monotherapy efficacy in the rhesus macaque NHP ZIKV challenge model.

a, The serum concentration of human mAbs (2 d after infection) in mice that were treated with the indicated mAb-encoding RNA formulations (40 μg per mouse) on day −1 and challenged with ZIKV on day 0 was determined using ELISA. The dots show measurements from individual mice; n = 5 mice per group were analysed and data represent one experiment, except for ZIKV-117, for which n = 10 mice per group were analysed. Data are mean ± s.d. b, Serum ZIKV titres (2 d after infection) from mice treated and challenged as shown in a were determined using RT–qPCR. The dots show measurements from individual mice; n = 5 mice per group were analysed in one experiment, except for ZIKV-117, for which n = 10 mice per group were analysed. Data are mean ± s.d. c, The survival of mice that were treated and challenged as shown in a; n = 5 mice per group were analysed in one experiment. The overall difference in survival between the groups was estimated using two-sided log-rank (Mantel–Cox) tests. P values are indicated. The mAbs ZIKV-117 and FLU-5J8 were used as controls. The timeline for scaling-up mAb-encoding RNA production and identifying the lead protective mAb-encoding RNA formulations is indicated by the blue arrows in the timeline chart.
Modelling a timeline for NHP protection study against ZIKV
At the conclusion of the murine protection studies, we immediately initiated a mAb treatment study of ZIKV infection in NHPs. We produced RNA formulations using a Good Manufacturing Practice (GMP)-compatible process for two lead therapeutic candidate mAbs (ZIKV-752 and ZIKV-893) in a 200 mg scaled-up process (Supplementary Fig. 4) that could be used in NHP studies to enable an Investigational New Drug (IND) application. In order to demonstrate protection in NHPs, we used our previously described potently neutralizing human mAb ZIKV-117 (ref. 5) for the challenge study. We decided to test this antibody in NHPs to maximize the use of the data, as ZIKV-117 is already on an accelerated clinical development path. Large-scale production and purification of the ZIKV-117 IgG protein used here was not a part of the timeline. The treatment group (n = 3 NHPs) received a single dose (10 mg kg−1) of ZIKV-117 intravenously on day −1 and was challenged s.c. with 103 plaque-forming units (p.f.u.) of ZIKV strain Brazil the next day. An additional group of NHPs (n = 3) that received IgG of irrelevant specificity (sham) was used as a control. We did not observe any detectable viral RNA in the plasma or the other collected specimens in any of the ZIKV-117-treated NHPs (limit of detection = 2 log10[genome equivalents per ml]), whereas we found that each of the control NHPs were positive for infection (Fig. 6a). ELISA analysis of serum from ZIKV-117 mAb-treated NHPs detected ~50 μg ml−1 of circulating ZIKV-117 the day after the treatment (Fig. 6b). Together, this timeline modelling study enabled us to define the efficacy of mAb monotherapy against ZIKV infection in NHPs in only 9 d.

Animals received a single dose (10 mg kg−1) of the mAb ZIKV-117 (n = 3 NHPs per group) or mAb PGT121 (n = 3 NHPs per group), which was used as a contemporaneous control, intravenously on day −1 and were then challenged s.c. with a target dose of 103 p.f.u. of ZIKV strain Brazil the next day. a, RT–qPCR measurement of viraemia in the plasma and other compartments at the indicated time points after virus challenge. b, The concentration of ZIKV-117 human mAbs that was determined in the serum of treated and control macaques (M1–M6) at the indicated time points after virus challenge. Data in a and b represent a single experiment.
Discussion
Here we described an integrated technology modelling deployment of the rapid response for discovering of antiviral antibodies. Recently, several laboratories have demonstrated the feasibility of rapidly identifying potent antiviral mAbs12. The shortest reported time frame of which we are aware to date is about 4 months, which was reported for isolation of the neutralizing mAb LCA60 against MERS-CoV, a process that included mAb identification, functional screening, a mouse protection study and the development of a stable cell line for mAb production35. Our study demonstrated that, using a pipeline of integrated technologies, human antiviral mAb discovery and therapeutic potency verification (including mAb RNA delivery and proof-of-principle NHP protection study) can be accomplished in 78 d, which is 12 d shorter than our 90 d goal. Note that current single-cell antibody-gene cloning techniques enable the rapid generation of antigen-reactive mAbs in 2 weeks or even less36,37. However, isolating mAbs that would be suitable for clinical development (for example, mAbs with exceptional in vitro and in vivo potency), remains a bottleneck in the antibody-discovery process12. Here we demonstrated high performance of an integrated technology. A comparison to historical studies with previously reported human mAbs against ZIKV6,7,18,23 qualifies the newly identified broadly neutralizing mAbs ZIKV-752, ZIKV-893 and ZIKV-940 as promising therapeutic candidates of high potency against ZIKV. Furthermore, these mAbs recognize a diverse range of epitopes, which has relevance for therapeutic cocktails with complementary specificity and activity. Although a conventional mouse challenge model to assess protection against ZIKV uses long timelines (typically 16–21 d per study), early examination of viraemia and a mAb dose-de-escalation approach could be used to identify highly protective mAbs in the short timeframe that is required by rapid response programmes. Together, these results suggest that the development and use of mAbs as alternative antiviral therapeutics could be practical compared with conventional antiviral countermeasures, such as vaccines or small-molecule antiviral drugs. Continuing improvement of instruments and technologies for high-throughput single-cell analysis24,38 warrants feasibility of rapid antibody discovery for emerging viruses in the future.
One of the remaining challenges for the widespread application of antiviral antibody therapies is the potentially higher production costs that are associated with intravenous administration. An alternative to passive IgG protein administration is nucleic acid delivery of genes that encode antibodies using cDNA or mRNA. A previous study showed that DNA-encoded i.m. mAb delivery protected NHPs from ZIKV challenge after three sequential DNA administrations39. Another study reported that, after intravenous infusion of a lipid-nanoparticle- (LNP)-encapsulated mRNA encoding antiviral mAbs, physiologically relevant concentrations of human mAbs for viral protection were achieved in NHP serum40, and this formulation was used in a first-in-man clinical trial of mRNA-encoded antibodies in 2019 (ClinicalTrials.gov ID, NCT03829384). The NLC formulation used in this study may have an advantage over LNP-encapsulated RNA formulation41, in that the RNA is externally bound to the NLC in contrast to the internally placed RNA of the LNP formulation. This approach could enable stockpiling of the stable nanoparticles without cold-chain manufacturing. Thus, final formulation could be performed as needed once the desired mRNA encoding an antibody is identified and produced. Formulated RNA enables a high level of mAb expression after i.m. injection in vivo in mice34. The extent to which these findings translate to NHPs and humans remains unclear, and this aspect of the approach must be determined in future studies.
The timeline modelled in this study extends from acquisition of immune donor samples through in vivo mAb validation and does not consider downstream manufacturing-related activities. Other considerations that could not be predicted a priori include the timeline to obtain patient samples and the time required for antibody affinity maturation to occur in vivo. Studies of other primary infections of Ebola and yellow fever viruses have demonstrated that it takes about 3 months for the emergence of neutralizing mAbs in the Bmem-cell compartment, and 6–9 months for maximal affinity maturation42,43. In recent efforts to use the general approach that we describe here (for Zika virus) instead to isolate antibodies against SARS-CoV-2, we found a relatively low frequency of circulating Bmem cells for the virus at ~1 month after acute primary infection44. However, at ~2 months after infection, sufficient numbers of Bmem cells were present to rapidly isolate high-potency neutralizing antibodies, suggesting that the convalescence period that is required for this type of work may be shortened to 2 months from the more extended periods of 4–9 months previously investigated. For novel viruses causing an epidemic, such as the current SARS-CoV-2 pandemic, the development of recombinant antigens, functional assays and animal models also may extend the timelines for rapid antibody identification and testing. In an outbreak scenario, recombinant antigen may not readily available, and alternative discovery approaches—including target-specific B-cell isolation based on phenotypic markers, direct screening for neutralizing activity or use of phage display libraries12,36—should all be considered for parallel rapid-response approaches. However, even with these considerations, the integrated technology that we describe here provides a roadmap for large-scale accelerated antiviral antibody-discovery programmes and, in fact, was successfully used for the rapid discovery of potent anti-SARS-CoV-2 spike-antigen-specific human mAbs44,45.
Methods
Research participants
We studied 11 participants in the United States with previous or recent ZIKV infection and one uninfected control participant (Supplementary Table 1). The studies were approved by the Institutional Review Board of Vanderbilt University Medical Center; samples were collected after written informed consent was obtained by the Vanderbilt Clinical Trials Center. The participants were infected during the 2015–2016 outbreak of an Asian lineage strain, after exposure in Brazil, Nicaragua, Puerto Rico, Dominican Republic, Guatemala or Haiti.
Animals
The mouse challenge studies were approved by the Washington University School of Medicine (assurance number, A3381-01) Institutional Animal Care and Use Committee. The facility in which the animal studies were conducted is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International and follows guidelines set forth by the Guide for the Care and Use of Laboratory Animals, National Research Council, 2011. Blood was obtained as approved by submandibular vein bleeding. Virus inoculations were performed under anaesthesia that was induced and maintained with ketamine hydrochloride and xylazine, and all efforts were made to minimize animal suffering. Mice were distributed into the different groups non-specifically and in a blinded manner.
The NHP research studies were conducted in compliance with all relevant local, state and federal regulations and were approved by the Animal Care and Use Committee. The facility in which this research was conducted (Alpha Genesis) is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International and has an approved Office of Laboratory Animal Welfare Assurance (A3645-01).
Cell culture
Vero-E6 cells (American Type Culture Collection) were maintained at 37 °C in 5% CO2 in Dulbecco’s minimal essential medium (DMEM) containing 10% (v/v) heat-inactivated fetal bovine serum (FBS) and 1 mM sodium pyruvate. All of the cell lines were tested and found to be negative for mycoplasma contamination.
Viruses
Previously described human isolates of ZIKV Brazil, Paraiba 01/2015 (GenBank, KX280026.1)21,46 and ZIKV Dakar 41525, Senegal 1984 (GenBank, KU955591)20 were used for in vitro studies. A previously described mouse-adapted isolate of ZIKV Dakar 41525 (ref. 20) ZIKV Dakar MA (GenBank, MG758786.1) was used for both in vitro and in vivo studies. For NHP studies, a human ZIKV strain from Brazil (Brazil 2015) was used47,48.
Virus stock production
To identify cells suitable for the production of high-titre ZIKV stocks in an antigen-independent manner, we used RT–qPCR to rapidly detect ZIKV viral RNA in a panel of immortalized cell lines, including those that are commonly used for virus propagation: Vero-E6 (ATCC), BHK (ATCC), JEG-3 (ATCC), HeLa (ATCC), HEK-293T (ATCC), U2OS (ATCC), A549 (ATCC), Huh7, Huh7.5, EA.Hy926 (ATCC) and Hap-1 (Horizon). For subsequent ZIKV production studies, we chose Vero cells owing to their well-characterized use in virus production. To generate stocks of ZIKV for subsequent in vitro and in vivo studies, Vero cells plated in T175 flasks were inoculated with a seed stock and the cell culture supernatant was collected 66–72 h after inoculation and titrated. Virus stocks were then titrated by focus-forming assay (FFA), aliquoted and stored at −80 °C until use. To identify suitable conditions for large-scale production of ZIKV stocks in Vero cells grown in roller bottles, we examined ZIKV titres in various serum concentrations ranging from 0.5% to 5% of FBS in cell culture supernatant collected 24–72 h after inoculation. We observed that even 0.5% fetal bovine serum was sufficient to generate high-titre ZIKV stocks by 72 h after virus inoculation. To identify conditions for concentration and inactivation of virus stocks, we determined the infectivity of ZIKV stocks by FFA after ultracentrifugation as previously described49 and after inactivation with hydrogen peroxide in an attempt to preserve immunogenicity as described previously50. We observed increased ZIKV titres in the virus pellet after ultracentrifugation and could not detect infectious ZIKV after treatment with 3% hydrogen peroxide for 1 h at ambient temperature and 0.024 U μl−1 of catalase for 10 min at ambient temperature (to inactivate hydrogen peroxide).
Human participant selection and target-specific Bmem-cell isolation
B-cell responses to ZIKV in PBMCs from a cohort of 11 individuals with previous exposure to the ZIKV Asian lineage were assessed to identify individuals with the highest response. The frequency of ZIKV-specific B cells was enumerated from frozen PBMCs using an African ZIKV lineage soluble recombinant E protein (Meridian Bioscience), the antigen that was used previously to identify potent ZIKV mAbs5. In brief, B cells were purified magnetically (STEMCELL Technologies) and stained with anti-CD19 phycoerythrin-conjugated (1:10 dilution), anti-IgD fluorescein isothiocyanate (FITC)-conjugated (1:20 dilution) and anti-IgM FITC-conjugated (1:20 dilution) phenotyping antibodies (BD Biosciences) and biotinylated E protein. We used 4′,6-diamidino-2-phenylindole (DAPI) as a viability dye to identify dead cells. Antigen-labelled class-switched Bmem-cell–E complexes (CD19+IgM−IgD−ZIKV E+DAPI−) were detected with allophycocyanin (APC)-labelled streptavidin conjugate and quantified using an iQue Plus Screener flow cytometer (IntelliCyt). After identifying the seven individuals with the highest B-cell response against ZIKV, target-specific Bmem cells were isolated by FACS using an SH800 cell sorter (Sony) from pooled PBMCs of these individuals, after labelling B cells with biotinylated E protein (defined as the direct labelling approach that generated sub-panel 1).
Although sorting using a biotinylated E antigen was used successfully for isolating human neutralizing ZIKV mAbs, this approach can fail to identify mAbs of which the antigenic sites are altered by the biotin labelling of the target protein. To overcome this limitation, we also used alternate approaches. The second approach used binding of soluble intact E protein antigen to B cells, but included a labelled antibody detected at the E FL (defined as the indirect label method that generated sub-panel 2). This approach circumvented any binding issues stemming from biotin labelling and also excluded the subset of B cells that encode FL-specific mAbs, which dominate in the response to ZIKV but typically poorly neutralize the virus5. Third, all labelled B cells (that is, mAb sub-panel 2) and a subset of B cells with high fluorescence intensity for E labelling (mAb sub-panel 3) were sorted separately. Overall, from >5 × 108 PBMCs, >5,000 E-specific B cells were sorted and further analysed. Two methods were implemented to prepare sorted B cells for sequencing. Approximately 800 sorted cells were processed for direct sequencing immediately after FACS analysis. Although a direct-sequencing approach is the fastest approach, it could be less efficient owing to the low RNA copy numbers in Bmem cells; this approach may lead to misidentification of heavy- and light-chain pairs. The remaining sorted cells were expanded in culture for 8 d in the presence of irradiated 3T3 feeder cells that were engineered to express human CD40L, IL-21 and BAFF as described previously51. The expanded lymphoblastoid cell lines secreted high levels of E-specific mAbs, as confirmed by ELISA from culture supernatants. Approximately 40,000 expanded lymphoblastoid cell lines were sequenced using the Chromium sequencing method (10x Genomics).
Generation of single-cell antibody variable gene profiling libraries
The Chromium Single Cell V(D)J workflow with the B-cell-only enrichment option was chosen for generating linked heavy-chain and light-chain antibody profiling libraries. Approximately 800 directly sorted E-specific B cells were split evenly into two replicates and separately added to 50 μl of RT reagent mix, 5.9 μl of Poly-dt RT primer, 2.4 μl of additive A and 10 μl of RT enzyme mix B to complete the reaction mix according to the vendor’s protocol; the samples were then was loaded directly onto a Chromium chip (10x Genomics). Similarly, for the remaining sorted cells that were bulk-expanded, approximately 40,000 cells from two separate sorting approaches were split evenly across six reactions and separately processed as described above before loading onto a Chromium chip. The libraries were prepared according to the user guide for Chromium Single Cell V(D)J Reagents kits (CG000086_REV C).
Next-generation sequencing
Chromium single-cell V(D)J B-cell enriched libraries were quantified, normalized and sequenced according to the user guide for Chromium Single Cell V(D)J Reagents kits (CG000086_REV C). The two enriched libraries from direct flow cytometry cell sorting were combined into one library pool and sequenced using a NovaSeq sequencer (Illumina) with a NovaSeq 6000 S1 Reagent Kit (300 cycles; Illumina). The six enriched libraries from bulk expansion were combined into one library pool and sequenced on a NovaSeq sequencer with a NovaSeq 6000 S4 Reagent Kit (300 cycles; Illumina). All of the enriched V(D)J libraries were targeted for a sequencing depth of at least 5,000 raw read pairs per cell. After sequencing, all of the samples were demultiplexed and processed using 10x Genomics Cell Ranger (v.2.1.1) as described below.
Bioinformatics analysis
Down-selection to identify lead candidates for expression was performed in two phases. In the first phase, all paired antibody heavy- and light-chain variable gene cDNA nucleotide sequences obtained that contained a single heavy- and light-chain sequence were processed using our Python-based antibody variable gene analysis tool (PyIR; https://github.com/crowelab/PyIR). We considered heavy- and light-chain encoding gene pairs to be productive and retained them for additional downstream processing if they met the following criteria: (1) they did not contain a stop codon; (2) they had an intact CDR3; and (3) they contained an in-frame junctional region. The second phase of processing grouped all productive heavy chain nucleotide sequences on the basis of their V3J clonotype (defined by identical inferred V and J gene assignments and identical amino acid sequences of the CDR3)52. All heavy-chain nucleotide sequences from individual sub-panels were grouped by their heavy-chain V3J clonotype. The somatic variants belonging to each heavy chain V3J clonotype grouping were then rank-ordered on the basis of the degree of somatic mutation, and only the most mutated heavy-chain sequence was retained for downstream expression and characterization. Any somatic variant that was not designated as an IgG isotype (on the basis of the sequence and assignment using 10x Genomics Cell Ranger V(D)J (v.2.1.1)) was removed from consideration. All heavy- and light-chain nucleotide sequences were translated, and redundant entries were removed to avoid expressing identical mAbs. The identities of antibody variable gene segments, CDRs and mutations from inferred germline gene segments were determined by alignment using the ImMunoGeneTics database53.
Antibody-gene synthesis
Sequences of selected mAbs were synthesized using a rapid high-throughput cDNA synthesis platform (Twist Bioscience) and subsequently cloned into an IgG1 monocistronic expression vector (designated as pTwist) for mammalian cell culture mAb secretion. This vector contains an enhanced 2A sequence and GSG linker that enables simultaneous expression of mAb heavy- and light-chain genes from a single construct after transfection25.
MAb production and purification
For high-throughput production of recombinant mAbs, we used approaches that are designated as microscale. For mAb expression, we performed microscale transfection (~1 ml per antibody) of CHO cell cultures using the Gibco ExpiCHO Expression System and a protocol for deep 96-well blocks (Thermo Fisher Scientific). In brief, synthesized antibody-encoding lyophilized DNA (~2 μg per transfection) was reconstituted in OptiPro serum free medium (OptiPro SFM), incubated with ExpiFectamine CHO Reagent and added to 800 µl of ExpiCHO cell cultures into 96-deep-well blocks using a ViaFlo 384 liquid handler (Integra Biosciences). The plates were incubated on an orbital shaker at 1,000 r.p.m. with an orbital diameter of 3 mm at 37 °C in 8% CO2. The next day after transfection, ExpiFectamine CHO Enhancer and ExpiCHO Feed reagents (Thermo Fisher Scientific) were added to the cells, followed by 4 d incubation for a total of 5 d at 37 °C in 8% CO2. Culture supernatants were collected after centrifuging the blocks at 450g for 5 min and were stored at 4 °C until use. For high-throughput microscale mAb purification, we used fritted deep-well plates containing 25 μl of settled protein G resin (GE Healthcare Life Sciences) per well. Clarified culture supernatants were incubated with protein G resin for mAb capturing, washed with PBS using a 96-well plate manifold base (Qiagen) connected to the vacuum and eluted into 96-well PCR plates using 86 μl of 0.1 M glycine-HCL buffer pH 2.7. After neutralization with 14 μl of 1 M Tris-HCl pH 8.0, purified mAbs were buffer-exchanged into PBS using Zeba Spin Desalting Plates (Thermo Fisher Scientific) and stored at 4 °C until use.
To scale up mAb production for the in vivo mouse studies, the transfection was performed in 125 ml Erlenmeyer vented-cap flasks (Corning) containing 35 ml of ExpiCHO cells according to the manufacturer’s protocol. Antibodies were purified from filtered culture supernatants by fast protein liquid chromatography using an ÄKTA Pure instrument with a HiTrap MabSelect Sure column (GE Healthcare Life Sciences). Purified mAbs were buffer-exchanged into PBS, filtered using sterile filter devices (pore size, 0.45 μm; Millipore), concentrated and stored in aliquots at 4 °C until use. To quantify purified mAbs, absorption at 280 nm (A280) was measured using a NanoDrop (Thermo Fisher Scientific), and mAb concentration was calculated using the IgG sample-type setting on the NanoDrop that uses a molar extinction coefficient that is typical for IgG, equal to 210,000 M−1 cm−1.
Production of antibody-encoding RNA
cDNAs encoding the 20 lead mAb candidates were amplified from the mammalian expression pTwist vector using universal primer sets and cloned into plasmids encoding Venezuelan equine encephalitis virus replicon (strain TC-83) under the control of a T7 promoter (pT7-VEE-Rep)32. To generate linear templates for RNA transcription for RNA in vitro transcription and capping at midi scale (500 μg), plasmid DNA was cleaved using NotI or BspQI restriction enzymes (New England Biolabs), respectively, and purified using phenol–chloroform extraction and sodium acetate precipitation. RNA was transcribed using T7 polymerase, RNase inhibitor, pyrophosphatase enzymes (Aldevron) and reaction buffer. RNA transcripts were capped with vaccinia virus capping enzyme using GTP and S-adenosyl-methionine (Aldevron) as substrates to create cap-0 structures. RNA was purified using lithium chloride precipitation. Large-scale demonstration of RNA-production ability was performed using a 200 mg scaled-up process and purified using a column on a Capto Core 700 instrument (GE Healthcare Life Sciences) and tangential flow filtration.
RNA formulation production
The NLC nanoparticle formulation was manufactured as previously described32. The oil phase is composed of squalene (the liquid-phase of the oil core), glyceryl trimyristate (Dynasan 114; the solid-phase of the oil core), a non-ionic sorbitan ester surfactant (sorbitan monostearate; Span 60) and the cationic lipid DOTAP (N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium chloride). The aqueous phase is a 10 mM sodium citrate trihydrate buffer containing the non-ionic PEGylated surfactant Tween-80. Separately, the two phases were heated and equilibrated to 60 °C in a bath sonicator. After complete dissolution of the solid components, the oil and aqueous phases were mixed at 5,000 r.p.m. in a high-speed laboratory emulsifier (Silverson Machines) to produce a crude mixture containing micrometre-sized oil droplets. Further particle-size reduction was achieved by high-shear homogenization in a M-110P microfluidizer (Microfluidics). The colloid mixtures were processed at 30,000 p.s.i. (~207 MPa) for five discrete microfluidization passes. The final pH was adjusted to between 6.5 and 6.8. Formulations were filtered using a 0.2 µm polyethersulfone membrane syringe filter and stored at 2–8 °C. RNA was complexed with NLC at a nitrogen-to-phosphate ratio of 5, diluting NLC in 10 mM citrate buffer and RNA in a 20% sucrose solution and complexing on ice for 30 min before use.
ELISA binding screening assays
Wells of 96-well microtitre plates were coated with purified recombinant ZIKV E protein (Meridian Bioscience) at 4 °C overnight. The plates were blocked with 2% non-fat dry milk and 2% normal goat serum in DPBS containing 0.05% Tween-20 (DPBS-T) for 1 h. For mAb screening assays, CHO cell culture supernatants or purified mAbs were diluted 1:20 in blocking buffer, added to the wells, and incubated for 1 h at ambient temperature. The bound antibodies were detected using goat anti-human IgG conjugated with horseradish peroxidase (HRP; Southern Biotech) at 1:5,000 dilution and 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (Thermo Fisher Scientific). Colour development was monitored, 1 N hydrochloric acid was added to stop the reaction and the absorbance was measured at 450 nm using a spectrophotometer (Biotek). An optical density at 450 nm of 0.3 was set as the threshold for mAb reactivity. This screening approach could underestimate the total number of target-specific mAbs in the panel but enabled the elimination of poorly produced mAbs. For dose–response assays, serial dilutions of purified mAbs were applied to the wells in triplicate or quadruplicate, and mAb binding was detected as described above.
Focus reduction neutralization test
Serial dilutions of mAbs were incubated with 102 FFU of different ZIKV strains (Dakar or Brazil) for 1 h at 37 °C. The mAb–virus complexes were added to Vero cell monolayers in 96-well plates for 90 min at 37 °C. Subsequently, cells were overlaid with 1% (w/v) methylcellulose in minimum essential medium supplemented with 4% heat-inactivated FBS. Plates were fixed 40 h later with 1% paraformaldehyde (PFA) in PBS for 1 h at room temperature. The plates were incubated sequentially with 500 ng ml−1 previously described mouse anti-ZIKV mAb ZV-16 (ref. 5) and 1:5,000 dilution of HRP-conjugated goat anti-mouse IgG (Sigma-Aldrich) in PBS supplemented with 0.1% (w/v) saponin and 0.1% bovine serum albumin (BSA). ZIKV-infected cell foci were visualized using TrueBlue peroxidase substrate (KPL) and quantified on an ImmunoSpot 5.0.37 Macro Analyzer (Cellular Technologies).
High-throughput quantification of mAbs
High-throughput quantification of microscale-produced mAbs was performed from CHO culture supernatants or microscale-purified mAbs in a 96-well plate format using the Cy-Clone Plus Kit and an iQue Plus Screener flow cytometer (IntelliCyt) according to the vendor’s protocol. Purified mAbs were assessed at a single dilution (1:10 final, using 2 μl of purified mAbs per reaction), and a control human IgG solution with known concentration was used to generate a calibration curve. Data were analysed using ForeCyt v.6.2 (IntelliCyt).
RTCA
To screen for neutralizing activity in the panel of recombinantly expressed mAbs, we used a high-throughput and quantitative RTCA assay and xCelligence Analyzer (ACEA Biosciences) that assesses kinetic changes in cell physiology, including virus-induced CPE. Cell culture medium (50 μl; DMEM supplemented with 2% FBS) was added to each well of a 96-well E-plate using a ViaFlo384 liquid handler (Integra Biosciences) to obtain a background reading. Vero-E6 cells (18,000) in 50 μl of cell culture medium were seeded in each well, and the plate was placed on the analyzer. Measurements were taken automatically every 15 min, and the sensograms were visualized using RTCA v.2.1.0 (ACEA Biosciences). For the screening neutralization assay, the virus (multiplicity of infection, 0.5; ~9,000 FFU per well) was mixed with a 1:10 dilution of CHO supernatants obtained from the microscale expression experiments, or a 1:25 dilution of microscale-purified antibodies in a total volume of 50 μl using DMEM supplemented with 2% FBS as a diluent and then incubated for 1 h at 37 °C in 5% CO2. At ~12 h after seeding the cells, the virus–mAb mixtures were added in replicates to the cells in 96-well E-plates. Wells containing virus only (in the absence of mAbs) and wells containing only Vero cells in medium were included as controls. Plates were measured continuously (every 15 min) for 48–72 h to assess virus neutralization. A mAb was considered to be neutralizing if it partially or completely inhibited ZIKV-induced CPE. For mAb potency ranking experiments, individual mAbs identified as fully neutralizing from the screening study were assessed at six fivefold dilutions starting from 1 μg ml−1 in replicates after normalization of mAb concentrations. IC50 values were estimated as the change in cellular index over time, using nonlinear fit with variable slope analysis performed in the RTCA v.2.1.0 (ACEA Biosciences). The potency ranking study was repeated using midi-scale-purified mAbs to confirm the activity and to ensure the quality of antibody preparations before performing in vivo protection studies in mice.
Antibody-mediated cellular phagocytosis by human monocytes
Recombinant soluble ZIKV E protein was biotinylated and coupled to Alexa-Fluor-488-dye-coupled Neutravidin beads (Life Technologies). Microscale-purified mAbs were tested at a single 1:10 dilution; concentrations were not normalized. Antibodies were diluted in cell culture medium and incubated with beads for 2 h at 37 °C. THP-1 cells (ATCC) were added at 2.5 × 104 cells per well and incubated for 18 h at 37 °C. Cells were fixed with 4% PFA and analysed on a BD LSRII flow cytometer, and a phagocytic score was determined using the percentage of Alexa Fluor 488+ cells and the median fluorescence intensity (MFI) of the Alexa Fluor 488+ cells. Data are represented as Z score, where Z = (x − μ)/σ, where x is a phagocytic score, μ is the mean of the population and σ is the s.d. of the population. The human mAb ZIKV-117 was used as a positive control, and the human mAb FLU-5J8 was used as a negative control.
Antibody-mediated neutrophil phagocytosis
Recombinant soluble ZIKV E protein was biotinylated and coupled to Alexa-Fluor-dye-coupled Neutravidin beads (Life Technologies). Microscale-purified mAbs were tested at a single 1:10 dilution; concentrations were not normalized. Antibodies were diluted in cell culture medium and incubated with beads for 2 h at 37 °C. White blood cells were isolated from the peripheral blood of participants by lysis of red blood cells, followed by three washes with PBS. Cells were added at a concentration of 5.0 × 104 cells per well and incubated for 1 h at 37 °C. Cells were stained with anti-human-CD66b (Pacific Blue, Clone G10F5; BioLegend), anti-human-CD3 (AF700, Clone UCHT1; BD Biosciences) and anti-human-CD14 (APC-Cy7, Clone MφP9; BD Biosciences) antibodies (1:100 dilution of each), then fixed with 4% PFA and analysed using flow cytometry with a BD LSR II flow cytometer. Neutrophils were defined as SSC-AhighCD66b+CD3−CD14−. A phagocytic score was determined using the percentage of Alexa Fluor 488+ cells and the MFI of the Alexa Fluor 488+ cells. Z-score values were calculated as described above. The previously identified mAb ZIKV-117 was used as a positive control, and the mAb FLU-5J8 was used as a negative control.
Antibody-mediated complement deposition
Recombinant soluble ZIKV E protein was biotinylated and coupled to red fluorescent Neutravidin beads (Life Technologies). Antibodies were diluted to 5 μg ml−1 in RPMI-1640, and incubated with GP-coated beads for 2 h at 37 °C. Freshly reconstituted guinea pig complement (Cedarlane Labs) was diluted in veronal buffer with 0.1% fish gelatin (Boston Bioproducts), added to the antibody-bead complexes and incubated for 20 min at 37 °C. Beads were washed twice with PBS containing 15 mM EDTA, and stained with 1:100 dilution of an anti-guinea-pig-C3 antibodies conjugated to FITC (MP Biomedicals) for 15 min at ambient temperature. Beads were washed twice more with PBS, and C3 deposition onto beads was analysed using a BD LSRII flow cytometer and the MFI of the FITC+ of all beads was measured. Z-score values were calculated as described above.
Competition-binding analysis and epitope mapping
For the competition-binding assay, wells of 384-well microtitre plates were coated with purified recombinant ZIKV E protein at 4 °C overnight. Plates were blocked with 2% BSA in DPBS containing 0.05% Tween-20 (DPBS-T) for 1 h. Purified unlabelled mAbs were diluted in blocking buffer to 5 μg ml−1, added to the wells (25 μl per well) and incubated for 1 h at ambient temperature. Biotinylated previously characterized ZIKV E-specific human mAbs, ZIKV-117, ZIKV-116 and ZIKV-88, were added to the indicated wells at 5 μg ml−1 in a 2.5 μl per well volume (final concentration of biotinylated mAbs, ~0.5 μg ml−1) without washing of the unlabelled antibodies and then incubated for 1 h at ambient temperature. Plates were washed, and the bound antibodies were detected using HRP-conjugated avidin (Sigma) and TMB substrate. The signal obtained for binding of the biotin-labelled reference antibody in the presence of the unlabelled tested antibody was expressed as the percentage of the binding of the reference antibody alone after subtracting the background signal. Tested mAbs were considered to be competing if their presence reduced the reference antibody binding to less than 41% of its maximal binding and non-competing if the signal was greater than 71%. A level of 40–70% was considered to be intermediate competition.
Epitope mapping was performed using shotgun mutagenesis essentially as described previously54. A ZIKV prM/E protein expression construct (based on ZIKV strain SPH2015) was processed for high-throughput alanine scanning mutagenesis to generate a comprehensive mutation library. Each residue within prM/E was changed to alanine, with alanine codons mutated to serine. In total, 672 ZIKV prM/E mutants were generated (100% coverage), sequence confirmed and arrayed into 384-well plates. Each ZIKV prM/E mutant was transfected into HEK-293T cells and was allowed to express for 22 h. Cells were fixed in 4% (v/v) paraformaldehyde (Electron Microscopy Sciences), and then permeabilized with 0.1% (w/v) saponin (Sigma-Aldrich) in PBS plus calcium and magnesium (PBS++). Cells were incubated with purified mAbs diluted in PBS++, 10% normal goat serum (Sigma) and 0.1% saponin. Primary antibody screening concentrations were determined using an independent immunofluorescence titration curve against wild-type ZIKV prM/E to ensure that the signals were within the linear range of detection. Antibodies were detected using 3.75 μg ml−1 (final antibody concentration) of Alexa-Fluor-488-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) in 10% normal goat serum with 0.1% saponin. Cells were washed three times with PBS++/0.1% saponin followed by two washes in PBS. Mean cellular fluorescence was detected using a high-throughput flow cytometer (HTFC, Intellicyt). Antibody reactivity against each mutant prM/E clone was calculated relative to the wild-type prM/E protein reactivity by subtracting the signal from mock-transfected controls and normalizing to the signal of the wild-type prM/E-transfected controls. Mutations within clones were identified to be critical to the mAb epitope if they did not support reactivity of the test MAbs, but supported reactivity of other ZIKV antibodies. This counter-screen strategy facilitates the exclusion of prM/E mutants that are locally misfolded or have an expression defect.
Detection of circulating human mAbs in mouse serum
The amount of human mAbs in serum was detected using a capture ELISA with a standard curve of recombinant anti-ZIKV mAb ZIKV-117 or an IgG1 isotype-matched control anti-influenza mAb, FLU-5J8. In brief, plates were coated with 2 µg ml−1 (1:500 dilution) of goat anti-human-kappa or goat anti-human-lambda antibodies cross-absorbed against mouse IgG (Southern Biotech) at 4 °C overnight. The plates were blocked using 2% BSA in PBS for 1 h at 37 °C and then incubated for 1 h at 4 °C with serial dilutions of heat-inactivated mouse serum in parallel with a serial dilution of a known quantity of ZIKV-117 IgG1 protein. After washing, bound antibodies were detected using a 1:5,000 dilution of HRP-conjugated goat anti-human-Fc multiple-species cross-absorbed antibodies (Southern Biotech) in blocking buffer for 1 h at 4 °C. Plates were developed using TMB substrate (Thermo Fisher Scientific), and the reaction was stopped with H2SO4. ELISA plates were read using a TriBar LB941 plate reader (Berthold Technologies). The optical density values from the known quantity of ZIKV-117 were fitted to a standard curve and compared with the optical density values of serum to determine the concentration of ZIKV-117. The antibodies ZIKV-609, ZIKV-869 and ZIKV-980 failed to perform in this detection assay, even in cases in which they were used as purified IgG. Serum concentration measurements for these mAbs are not available.
ZIKV titre measurements
Blood was collected from ZIKV-infected mice at various time points, allowed to clot at ambient temperature and serum was separated using centrifugation. Viral RNA was isolated using the 96-well Viral RNA kit (Epigenetics), as described by the manufacturer. ZIKV RNA levels were determined using TaqMan one-step RT–qPCR as described previously55. Alternatively, ZIKV Dakar MA and Brazil strain titres were determined using a focus-forming assay on Vero cell monolayer cultures, as previously described56.
Mouse challenge experiments
Wild-type male C57BL/6J mice (aged 4 weeks) were purchased from Jackson Laboratory and housed in groups of up to 5 mice per cage at 18–24 °C ambient temperatures and 40–60% humidity. Mice were fed a 20% protein diet (PicoLab 5053, Purina) and maintained in a 12 h–12 h light–dark cycle (06:00 to 18:00). For studies involving ZIKV challenge, mice were treated with 2 mg of Ifnar1-blocking antibodies (MAR1-5A3, Leinco Technologies) by i.p. injection 1 d before virus inoculation. For the prophylaxis experiments, mice were treated i.p. 1 d before virus challenge with purified individual mAbs and monitored for 21 d for survival. For the therapeutic protection experiments, mice were treated i.p. 1 d after virus challenge with varying defined doses of purified individual antibodies and monitored for 21 d for survival. For the antibody-encoding-RNA delivery studies, mice were inoculated with 40 µg of individual RNA through the i.m. route (into each quadricep and hamstring muscle groups) 1 d before virus challenge, with four injections of 50 μl of RNA/NLC complex per injection. For ZIKV infections, mice were inoculated by a s.c. (by the footpad) route with 103 FFU of ZIKV Dakar MA in a volume of 30 μl of PBS. The antibody ZIKV-117 was used as a positive control, and the mAb FLU-5J8 or PBS was used as a negative control.
NHP challenge study
Six healthy adult rhesus macaques (Macaca mulatta) of Chinese origin (body weight, 5–15 kg) were studied. The rhesus macaques were aged 5–7 years and were mixed male and female. The animals were allocated randomly to two treatment groups (n = 3 per group) and two control sham-treated groups (n = 3 per group). All of the animals were inoculated by the s.c. route with a target dose of 106 viral particles (~103 p.f.u.) of ZIKV Brazil before mAb infusions. The macaques in the treatment group received 10 mg kg−1 of ZIKV-117 mAbs by intravenous injection 1 d before ZIKV challenge. The control sham-treated animals received 10 mg kg−1 of the mAb PGT121, which is specific to gp120 protein antigen of HIV-1, 1 d before ZIKV challenge. All of the animals were given physical exams, and blood was collected at the time of ZIKV inoculation and at indicated times after ZIKV inoculation. Furthermore, all of the animals were monitored daily with an internal scoring protocol approved by the Institutional Animal Care and Use Committee. These studies were not performed blinded.
Detection of virus load in NHPs by RT–qPCR analysis
Titration of virus in the indicated specimens was performed using RT–qPCR analysis as previously described47,48. Viral RNA was isolated from plasma and other tested specimens using a QIAcube HT (QIAGEN) system. A QIAcube 96 Cador Pathogen kit or RNeasy 96 QIAcube HT kit was used for RNA extraction. cDNA of the wild-type BeH815744 Cap gene was cloned into the pcDNA3.1 expression plasmid, and then transcribed in vitro to RNA using the AmpliCap-Max T7 High Yield Message Maker Kit (Cellscript). RNA quality was assessed by the Beth Israel Deaconess Medical Center molecular core facility. Tenfold dilutions of the RNA were prepared for standards and reverse transcribed to cDNA. Primers were synthesized by Integrated DNA Technologies (Coralville), and probes were obtained from Biosearch Technologies (Petaluma). Viral loads were calculated as virus particles per ml, and the assay sensitivity was 100 copies per ml.
Detection of circulating human mAbs in NHP serum
A modified protocol using the commercially available human anti-ZIKV-Env IgG kit (Alpha Diagnostics International) was used to quantify ZIKV-117 mAb levels in NHP serum samples. In brief, plate wells were equilibrated several times with the kit NSB wash buffer and 100 µl of standard calibrators, and NHP serum dilutions were prepared in the kit sample diluent buffer plated in duplicate and incubated for 1 h at ambient temperature. The plates were washed, and NHP IgG preadsorbed HRP-conjugated anti-human secondary antibodies (Novus Biological), which were diluted to 1:2,000 in kit sample diluent buffer, were added at 100 µl per well and incubated for 1 h. The plates were washed and developed for 2 min using the kit TMB solution, stopped by adding the kit stop solution and analysed at 450 nm/550 nm on a VersaMax microplate plate reader (Molecular Devices) using Softmax Pro v.6.5.1. A four-parameter logistic standard curve was generated using Prism v.8.0 (GraphPad) for the standard calibrators, and the concentrations of the unknown samples were interpolated from the linear portion of the curve.
Quantification and statistical analysis
The descriptive statistics mean ± s.e.m. or mean ± s.d. were determined for continuous variables as noted. Survival curves were estimated using the Kaplan–Meier method, and an overall difference between groups was estimated using the two-sided log-rank (Mantel–Cox) test. In the neutralization assays using focus-reduction neutralization tests, IC50 values were calculated after log transformation of antibody concentrations using a three-parameter nonlinear fit analysis. In the RTCA neutralization assays, IC50 values were estimated as cellular index change over time using nonlinear fit with variable slope analysis determined in the RTCA v.2.1.0 (ACEA Biosciences). Technical and biological replicates are described in the figure legends. Statistical analyses were performed using Prism v.8.0 (GraphPad).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. The ImMunoGeneTics database is available from http://www.imgt.org/. The raw and analysed datasets generated during the study are too large to be publicly shared, yet they are available for research purposes from the corresponding authors on reasonable request.
Code availability
The 10x Genomics Cell Ranger (v.2.1.1) bioinformatics processing pipeline is available from https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome. Our PyIR (v.1.0) processing pipeline is freely available at GitHub (https://github.com/crowelab/PyIR). Prism v.8.0 (GraphPad) is commercially available from https://www.graphpad.com.
References
Sok, D. & Burton, D. R. Recent progress in broadly neutralizing antibodies to HIV. Nat. Immunol. 19, 1179–1188 (2018).
Laursen, N. S. & Wilson, I. A. Broadly neutralizing antibodies against influenza viruses. Antiviral Res. 98, 476–483 (2013).
Bornholdt, Z. A. et al. Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science 351, 1078–1083 (2016).
Flyak, A. I. et al. Mechanism of human antibody-mediated neutralization of Marburg virus. Cell 160, 893–903 (2015).
Sapparapu, G. et al. Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 540, 443–447 (2016).
Stettler, K. et al. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 353, 823–826 (2016).
Wang, Q. et al. Molecular determinants of human neutralizing antibodies isolated from a patient infected with Zika virus. Sci. Transl. Med. 8, 369ra179 (2016).
Robinson, J. E. et al. Most neutralizing human monoclonal antibodies target novel epitopes requiring both Lassa virus glycoprotein subunits. Nat. Commun. 7, 11544 (2016).
Corti, D., Passini, N., Lanzavecchia, A. & Zambon, M. Rapid generation of a human monoclonal antibody to combat Middle East respiratory syndrome. J. Infect. Publ. Health 9, 231–235 (2016).
Gilchuk, I. et al. Cross-neutralizing and protective human antibody specificities to poxvirus infections. Cell 167, 684–694 (2016).
Geisbert, T. W. et al. Therapeutic treatment of Nipah virus infection in nonhuman primates with a neutralizing human monoclonal antibody. Sci. Transl. Med. 6, 242ra282 (2014).
Walker, L. M. & Burton, D. R. Passive immunotherapy of viral infections: ‘super-antibodies’ enter the fray. Nat. Rev. Immunol. 18, 297–308 (2018).
Salazar, G., Zhang, N., Fu, T. M. & An, Z. Antibody therapies for the prevention and treatment of viral infections. npj Vaccines 2, 19 (2017).
Coltart, C. E., Lindsey, B., Ghinai, I., Johnson, A. M. & Heymann, D. L. The Ebola outbreak, 2013-2016: old lessons for new epidemics. Philos. Trans. R. Soc. B 372, 20160297 (2017).
Sikka, V. et al. The emergence of Zika virus as a global health security threat: a review and a consensus statement of the INDUSEM Joint Working Group (JWG). J. Glob. Infect. Dis. 8, 3–15 (2016).
Pierson, T. C. & Diamond, M. S. The emergence of Zika virus and its new clinical syndromes. Nature 560, 573–581 (2018).
Musso, D. & Gubler, D. J. Zika virus. Clin. Microbiol. Rev. 29, 487–524 (2016).
Robbiani, D. F. et al. Recurrent potent human neutralizing antibodies to Zika virus in Brazil and Mexico. Cell 169, 597–609 (2017).
Magnani, D. M. et al. Neutralizing human monoclonal antibodies prevent Zika virus infection in macaques. Sci. Transl. Med. 9, eaan8184 (2017).
Gorman, M. J. et al. An immunocompetent mouse model of Zika virus infection. Cell Host Microbe 23, 672–685 (2018).
Zhao, H. et al. Structural basis of Zika virus-specific antibody protection. Cell 166, 1016–1027 (2016).
Slon-Campos, J. L. et al. A protective Zika virus E-dimer-based subunit vaccine engineered to abrogate antibody-dependent enhancement of dengue infection. Nat. Immunol. 20, 1291–1298 (2019).
Rogers, T. F. et al. Zika virus activates de novo and cross-reactive memory B cell responses in dengue-experienced donors. Sci. Immunol. 2, eaan6809 (2017).
Setliff, I. et al. High-throughput mapping of B cell receptor sequences to antigen specificity. Cell 179, 1636–1646 (2019).
Chng, J. et al. Cleavage efficient 2A peptides for high level monoclonal antibody expression in CHO cells. mAbs 7, 403–412 (2015).
Crowe, J. E. Jr. Principles of broad and potent antiviral human antibodies: insights for vaccine design. Cell Host Microbe 22, 193–206 (2017).
Lu, L. L., Suscovich, T. J., Fortune, S. M. & Alter, G. Beyond binding: antibody effector functions in infectious diseases. Nat. Rev. Immunol. 18, 46–61 (2018).
Charretier, C. et al. Robust real-time cell analysis method for determining viral infectious titers during development of a viral vaccine production process. J. Virol. Methods 252, 57–64 (2018).
Tian, D. et al. Novel, real-time cell analysis for measuring viral cytopathogenesis and the efficacy of neutralizing antibodies to the 2009 influenza A (H1N1) virus. PLoS ONE 7, e31965 (2012).
Long, F. et al. Structural basis of a potent human monoclonal antibody against Zika virus targeting a quaternary epitope. Proc. Natl Acad. Sci. USA 116, 1591–1596 (2019).
Zhao, H. et al. Mechanism of differential Zika and dengue virus neutralization by a public antibody lineage targeting the DIII lateral ridge. J. Exp. Med. 217, e20191792 (2020).
Erasmus, J. H. et al. A nanostructured lipid carrier for delivery of a replicating viral RNA provides single, low-dose protection against Zika. Mol. Ther. 26, 2507–2522 (2018).
Hong, M. et al. Antibody recognition of the pandemic H1N1 influenza virus hemagglutinin receptor binding site. J. Virol. 87, 12471–12480 (2013).
Erasmus, J. H. et al. Intramuscular delivery of replicon RNA encoding ZIKV-117 human monoclonal antibody protects against Zika virus infection. Mol. Ther. Methods Clin. Dev. 18, 402–414 (2020).
Corti, D. et al. Prophylactic and postexposure efficacy of a potent human monoclonal antibody against MERS coronavirus. Proc. Natl Acad. Sci. USA 112, 10473–10478 (2015).
Huang, J. et al. Isolation of human monoclonal antibodies from peripheral blood B cells. Nat. Protoc. 8, 1907–1915 (2013).
Guthmiller, J. J., Dugan, H. L., Neu, K. E., Lan, L. Y. & Wilson, P. C. An efficient method to generate monoclonal antibodies from human B cells. Methods Mol. Biol. 1904, 109–145 (2019).
DeKosky, B. J. et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat. Biotechnol. 31, 166–169 (2013).
Esquivel, R. N. et al. In vivo delivery of a DNA-encoded monoclonal antibody protects non-human primates against Zika virus. Mol. Ther. 27, 974–985 (2019).
Kose, N. et al. A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against chikungunya infection. Sci. Immunol. 4, eaaw6647 (2019).
Kowalski, P. S., Rudra, A., Miao, L. & Anderson, D. G. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol. Ther. 27, 710–728 (2019).
Davis, C. W. et al. Longitudinal analysis of the human B cell response to Ebola virus infection. Cell 177, 1566–1582 (2019).
Wec, A. Z. et al. Longitudinal dynamics of the human B cell response to the yellow fever 17D vaccine. Proc. Natl Acad. Sci. USA 117, 6675–6685 (2020).
Zost, S. J. et al. Rapid isolation and profiling of a diverse panel of human monoclonal antibodies targeting the SARS-CoV-2 spike protein. Nat. Med. https://doi.org/10.1038/s41591-020-0998-x (2020).
Zost, S. J. et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature. https://doi.org/10.1038/s41586-020-2548-6 (2020).
Tsetsarkin, K. A. et al. A full-length infectious cDNA clone of Zika virus from the 2015 epidemic in Brazil as a genetic platform for studies of virus-host interactions and vaccine development. mBio 7, e01114–16 (2016).
Abbink, P. et al. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science 353, 1129–1132 (2016).
Larocca, R. A. et al. Vaccine protection against Zika virus from Brazil. Nature 536, 474–478 (2016).
Brien, J. D., Lazear, H. M. & Diamond, M. S. Propagation, quantification, detection, and storage of West Nile virus. Curr. Protoc. Microbiol. 31, 15D.3.1–15D.3.18 (2013).
Amanna, I. J., Raue, H. P. & Slifka, M. K. Development of a new hydrogen peroxide-based vaccine platform. Nat. Med. 18, 974–979 (2012).
Gilchuk, P. et al. Analysis of a therapeutic antibody cocktail reveals determinants for cooperative and broad Ebolavirus neutralization. Immunity 52, 388–403 (2020).
Soto, C. et al. High frequency of shared clonotypes in human B cell receptor repertoires. Nature 566, 398–402 (2019).
Giudicelli, V. & Lefranc, M. P. IMGT/junctionanalysis: IMGT standardized analysis of the V-J and V-D-J junctions of the rearranged immunoglobulins (IG) and T cell receptors (TR). Cold Spring Harb. Protoc. 6, 716–725 (2011).
Davidson, E. & Doranz, B. J. A high-throughput shotgun mutagenesis approach to mapping B-cell antibody epitopes. Immunology 143, 13–20 (2014).
Govero, J. et al. Zika virus infection damages the testes in mice. Nature 540, 438–442 (2016).
Lazear, H. M. et al. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19, 720–730 (2016).
Acknowledgements
We thank M. Mayo for assistance with acquisition of the human survivor samples and coordination across study sites; J. Govero for assistance with RNA protection experiments; A. Jones and K. Beeri for assistance and coordination of NGS sequencing timelines; J. C. Slaughter, M. Goff and R. Troseth for assistance with data analysis; and STEMCELL Technologies and ACEA Biosciences for providing resources. This study was supported by Defense Advanced Research Projects Agency (DARPA) grant HR0011-18-2-0001 and HHS contract HHSN272201400058C (to J.E.C. and B.J.D.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the DARPA.
Author information
Authors and Affiliations
Contributions
P.G., R.G.B., J.H.E., R.N., C.S., T.J.S., E.D., B.J.D., L.T., G.A., S.G.R., N.V.H., D.H.B., M.S.D., J.E.C., L.B.T. and R.C. planned the studies. P.G., R.G.B., J.H.E., Q.T., R.N., C.S., T.J., L.A.D., A.K., J.A., J.L., M.E.F., P.A., E.L., S.E., B.G., J.F.-S., V.R., T.B., T.C.L., C.H.L. and J.N. conducted experiments. P.G., E.D., R.G.B., J.H.E., C.S., T.J.S., M.J.G., N.V.H., M.S.D., J.E.C., L.B.T. and R.C. interpreted the studies. P.G., J.E.C. and R.C. wrote the first draft of the paper. B.J.D., G.A., S.G.R., D.H.B., M.S.D. and J.E.C. obtained funding. All of the authors reviewed, edited and approved the paper.
Corresponding authors
Ethics declarations
Competing interests
J.L., E.D., M.E.F. and B.J.D. are employees of Integral Molecular. B.J.D. is a shareholder of Integral Molecular. G.A. has a financial interest in SeromYx, a company developing technology that describes the antibody immune response. G.A. interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. M.S.D. is a consultant for Inbios and Emergent BioSolutions and on the Scientific Advisory Board of Moderna. J.E.C. has served as a consultant for Sanofi and is on the Scientific Advisory Boards of CompuVax and Meissa Vaccines, is a recipient of previous unrelated research grants from Moderna and Sanofi and is the founder of IDBiologics. J.H.E., A.K. and N.V.H. are listed as inventors on a patent application describing the NLC formulation. Vanderbilt University has applied for a patent (US Provisional Patent no. 62/937,603) concerning ZIKV antibodies that is related to this research. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary figures, tables and references.
Supplementary Table 2
Summary of antibody discovery approaches and characterization assays.
Rights and permissions
About this article

Cite this article
Gilchuk, P., Bombardi, R.G., Erasmus, J.H. et al. Integrated pipeline for the accelerated discovery of antiviral antibody therapeutics. Nat Biomed Eng 4, 1030–1043 (2020). https://doi.org/10.1038/s41551-020-0594-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41551-020-0594-x
This article is cited by
-
Multifunctional human monoclonal antibody combination mediates protection against Rift Valley fever virus at low doses
Nature Communications (2023)
-
Efficient discovery of SARS-CoV-2-neutralizing antibodies via B cell receptor sequencing and ligand blocking
Nature Biotechnology (2022)
-
Lipid nanoparticle-encapsulated mRNA antibody provides long-term protection against SARS-CoV-2 in mice and hamsters
Cell Research (2022)
