
Abstract
Nucleic acid detection by isothermal amplification and the collateral cleavage of reporter molecules by CRISPR-associated enzymes is a promising alternative to quantitative PCR. Here, we report the clinical validation of the specific high-sensitivity enzymatic reporter unlocking (SHERLOCK) assay using the enzyme Cas13a from Leptotrichia wadei for the detection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)—the virus that causes coronavirus disease 2019 (COVID-19)—in 154 nasopharyngeal and throat swab samples collected at Siriraj Hospital, Thailand. Within a detection limit of 42 RNA copies per reaction, SHERLOCK was 100% specific and 100% sensitive with a fluorescence readout, and 100% specific and 97% sensitive with a lateral-flow readout. For the full range of viral load in the clinical samples, the fluorescence readout was 100% specific and 96% sensitive. For 380 SARS-CoV-2-negative pre-operative samples from patients undergoing surgery, SHERLOCK was in 100% agreement with quantitative PCR with reverse transcription. The assay, which we show is amenable to multiplexed detection in a single lateral-flow strip incorporating an internal control for ribonuclease contamination, should facilitate SARS-CoV-2 detection in settings with limited resources.
Similar content being viewed by others

Main
The COVID-19 pandemic caused by the spread of SARS-CoV-2 requires the deployment of rapid, sensitive and inexpensive diagnostic methods to facilitate the management and containment of the disease. Quantitative real-time PCR with reverse transcription (RT–qPCR), which measures the quantity of viral RNAs, remains the gold-standard technique for SARS-CoV-2 virus detection1. However, the lack of access to RT–qPCR instruments, reagents and trained instrument operators can limit the utility of this diagnostic tool, especially in resource-constrained developing countries. Quick point-of-care community-level testing for COVID-19—to be used as an initial screen before a subsequent confirmation by RT–qPCR at central laboratories—would greatly enhance diagnostic capacities, leading to improved quarantine and care procedures and ultimately helping to reduce strains on healthcare resources2. An accurate point-of-care testing platform for COVID-19 with a quick turnaround time is also needed to manage safe travel, school and work, and resumption of social and economic activities.
Several point-of-care RNA detection technologies that do not require special instruments exist, including reverse transcription–recombinase polymerase amplification3 (RT–RPA) and reverse transcription-loop-mediated isothermal amplification4,5 (RT-LAMP). RT–RPA and RT-LAMP are highly sensitive methods, but can suffer from nonspecific amplification under isothermal conditions, leading to false-positive results when used for viral RNA detections. The problem is exacerbated if non-sequence-specific probes, such as pH-sensitive dyes, are used for the detection6. Stringency of detection by these isothermal amplification methods can be improved by incorporating an additional sequence-specific detection module, such as hybridization-based fluorescent-oligonucleotide probes7,8.
Clustered regularly interspaced short palindromic repeats (CRISPR)-based diagnostic methods, which use collateral cleavage activity of bystander nucleic acid probes of RNA-guided CRISPR-associated 12/13 (Cas12/13) nucleases9,10,11,12, are typically used in combination with RT-LAMP or RT–RPA isothermal amplification methods. Primers for RT-LAMP or RT–RPA can be designed for specific viral RNA sequences, and the amplification methods are used before the Cas enzyme detection step, to multiply detection signals from the specific nucleic acid sequences. Since both the nucleic acid amplification step and the CRISPR–Cas detection step require sequence specificity to trigger signal amplification, CRISPR diagnostic methods are highly sensitive (attomolar level9,12) and highly specific (down to single-nucleotide level11). Readout of Cas-mediated nucleic acid probe cleavage can be done by fluorescence detection or using the lateral-flow strip method; the latter is advantageous as the strips are portable and the results can be read by eye13 and easily quantified by smartphones14. In contrast to hybridization-based detection methods, CRISPR–Cas-based detection can be performed in the same vessel, simultaneously with isothermal amplification15,16, greatly simplifying the operational procedure upon testing and reducing the risk of contamination. However, there is a trade-off between operational simplicity and the test performance: the easier-to-perform one-pot CRISPR diagnostic protocols are less sensitive than the two-step variant, where amplification and CRISPR-mediated detection steps are performed sequentially9.
During the present COVID-19 outbreak, the developers of CRISPR diagnostics have generously shared materials and protocols for the detection of SARS-CoV-216,17,18, which have been crucial in enabling researchers worldwide to rapidly evaluate and adapt these technologies for clinical use. Here, we report clinical validation of the two-step CRISPR–Cas13-based SHERLOCK system19 for sensitive and specific detection of SARS-CoV-2 viral RNA. We performed SHERLOCK detection on a total of 534 clinical samples. We first characterized the test characteristics on 154 clinical samples, 81 of which were positive for SARS-CoV-2, and then used the test in real diagnostic settings for pre-operation assessment for an additional 380 clinical samples. We challenged the testing procedures with clinical samples with diverse threshold cycle (Ct) values (11–37), as well as samples from asymptomatic cases. Overall, we found that the SHERLOCK detection system of SARS-CoV-2 RNA extracted from nasopharyngeal and throat swabs of infected patients in Thailand was 100% specific and 96% sensitive with the fluorescence readout, and 88% sensitive with the lateral-flow readout. These characteristics are comparable to the accuracy and the performances of other point-of-care genetic tests20,21 but have no requirement for specialized instruments. Within the characterized limit of detection (LoD) of the method (approximately 42 copies per reaction, corresponding to a Ct of 33.5), SHERLOCK has 100% specificity and 100% sensitivity with the fluorescence readout, and 97% sensitivity with the lateral-flow readout. To facilitate the potential use of SHERLOCK in testing settings with limited resources where there is an increased risk of ribonuclease (RNase) contamination, we designed the lateral-flow detection to contain an internal control for RNase contamination and demonstrated multiplex detection of SARS-CoV-2 RNA and RNase presence in a single lateral-flow strip.
Results
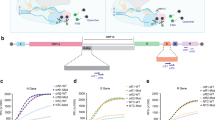
SHERLOCK detection of SARS-CoV-2 relies on RT–RPA to isothermally amplify viral gene segments of interest, followed by CRISPR–Cas-mediated detection of the amplified genes (in this case, using Cas13a from L. wadei (LwaCas13a), shown previously to confer the highest sensitivity in SHERLOCK-type detection9). The detection of the amplified gene sequences by CRISPR–Cas triggers collateral cleavage of reporter molecules for fluorescence or lateral-flow measurements (Fig. 1). We first designed and tested a total of four RPA-primer pairs and the corresponding CRISPR RNAs (crRNAs) targeting the spike (S), nucleoprotein (N) and replicase polyprotein 1ab (Orf1ab) genes of SARS-CoV-2 (Fig. 2a). Two of the primer pairs (and crRNAs), for S and Orf1ab genes, have previously been validated with synthetic RNAs17. The other two primer pairs and crRNAs, targeting the N gene and another region of Orf1ab, were designed to match gene regions used in the standard RT–qPCR assay for SARS-CoV-2 detection at Siriraj Hospital. All of the RPA primers and crRNA sequences were designed to be specific and selective towards SARS-CoV-2 viral RNA, and to minimize off-target affinity towards other common human coronaviruses (Supplementary Fig. 1).

A SARS-CoV-2 RNA region of interest is isothermally amplified to DNA by RT–RPA, then converted to RNA by T7 transcription. Cognate binding of Cas13a–crRNA complex to amplified RNA targets triggers collateral activity of Cas13a, which cleaves RNA reporters. Cleaved RNA reporters can be captured on a colorimetric lateral-flow strip (biotin–fluorescein RNA reporter, top path) or visualized by fluorescence signal (molecular beacon fluorescent reporter, bottom path). Fl, fluorescein.

a, SARS-CoV-2 genome map showing four regions—one in S, one in N and two in Orf1ab genes—selected for SHERLOCK detection. Selected regions are annotated with coloured rectangles and their nucleotide positions. UTR, untranslated region. b, LoD of SHERLOCK assays for detection of different SARS-CoV-2 gene regions, using lateral-flow readout. All lateral-flow strips contain a test (T) and control (C) band. c,d, LoD of SHERLOCK assays for detection of different SARS-CoV-2 gene regions, using fluorescence readout. c, Kinetics of fluorescence signal generation over 60 min of CRISPR–Cas13a reaction for each detected gene at different RNA serial dilutions. RFU, relative fluorescence units. d, Quantification of fluorescence signals generated after 20 min of CRISPR–Cas13a reaction at each dilution. Data are mean ± s.d. from triplicate measurements. Negative controls have RNase-free water as input instead of RNA dilutions.
Analysis of LoD and specificity
We first determined the LoD of SHERLOCK-based detection of SARS-CoV-2 upon using different combinations of RPA primers and crRNAs for the four selected gene regions. We used total RNA extracted from cultured SARS-CoV-2 in Vero cells (clinical isolate hCoV-19/Thailand/Siriraj_5/2020; GISAID accession ID: EPI_ISL_447908) as our LoD standards, performed serial dilutions of the extracted RNA and measured their corresponding Ct values with RT–qPCR for the N gene to ensure we obtained clinically relevant Ct ranges (Supplementary Table 1). We then verified that all four RPA primers and crRNA sets can be used effectively with the SHERLOCK assay to detect the presence of SARS-CoV-2 RNA (Fig. 2b–d). We found the detection of the S gene to be the most sensitive, under both lateral-flow strip (Fig. 2b) and fluorescence readouts (Fig. 2c,d), with reproducible detection down to 10−6 dilution, corresponding to Ct of approximately 33.5 in an RT–qPCR assay and about 42 copies per reaction, as quantified by digital-droplet PCR (Supplementary Table 1). We note that the low sensitivity in N gene detection is probably due to the longer N RPA amplicon we had designed; indeed, extending the RPA reaction time for the N gene to 1 h increased the sensitivity of detection to match that of the S gene (Supplementary Fig. 2). We further demonstrated that SHERLOCK-based detection of the SARS-CoV-2 S gene is specific to SARS-CoV-2, with no cross-reactivity towards other common human coronaviruses, including human coronavirus OC43 (hCoV-OC43), hCoV-NL63 and hCoV-229E (Fig. 3).

a, Alignment of the S gene region of SARS-CoV-2 with other common human coronaviruses, including hCoV-229E, hCoV-NL63, hCoV-HKU1 and hCoV-OC43. Nucleotides of RPA-primer- and crRNA-binding regions of the SARS-CoV-2 S gene (highlighted in pink) that are common between SARS-CoV-2 and the other viruses are highlighted in grey. b, Kinetics of fluorescence signal generation of the CRISPR–Cas13a reaction in RNA extracts from clinical samples tested positive with different coronaviruses, using SHERLOCK SARS-CoV-2 S gene detection. c, Fluorescence signals generated after 60 min of CRISPR–Cas13a reaction for each virus. Negative control is RNA extracted from a SARS-CoV-2-negative (by RT–qPCR) clinical sample.
Validation of SHERLOCK-based detection of SARS-CoV-2 in clinical samples
We empirically tested the amount of RNA added to the RT–RPA reaction and the amount of RPA reaction transferred to the CRISPR–Cas13a reaction, to optimize the detection signal from SARS-CoV-2 RNA in RNA extracts from nasopharyngeal swabs (Supplementary Fig. 3; see Methods for specific protocols). Subsequent to assessing the performance of SHERLOCK using the S gene as the targeted sequence, we performed the SHERLOCK-based detection of SARS-CoV-2 on COVID-19 clinical samples and directly benchmarked the SHERLOCK assay performance against the standard RT–qPCR assay. To minimize patient-selection bias and to ensure our validation study reflected the full distribution of Ct values among patients positive for COVID-19, we included all available positive samples obtained from a defined swab-collection time window, between 3 March and 10 April 2020, at Siriraj Hospital. To minimize information bias, positive and negative samples were randomized before being given to study staff, and the SHERLOCK results were interpreted without knowledge of the RT–qPCR results. As RT–qPCR was performed before SHERLOCK for all samples (we used RNA leftovers from RT–qPCR for SHERLOCK validation), we stored RNA samples at −80 °C to reduce degradation and always performed SHERLOCK validation experiments onsite at Siriraj Hospital, to eliminate the possibility of sample degradation during transport.
A validation study was conducted with a total of 154 total clinical samples, consisting of 81 RT–qPCR-verified COVID-19-positive samples (Ct of N gene ranging from 11–37) and 73 RT–qPCR-verified COVID-19-negative samples (Fig. 4). We envisioned that readouts from the SHERLOCK method could be performed with either fluorescence or lateral-flow readout, depending on the setting and the throughput, with the fluorescence readout being preferred for higher-throughput assessment, and the lateral-flow readouts intended for point-of-care usage. Both detection methods were assessed in parallel on clinical samples.

Positive COVID-19 clinical samples were binned according to their Ct values from RT–qPCR, with the total number of clinical samples in each Ct bin shown. Representative lateral-flow and fluorescence detection are shown for each Ct bin as well as for negative COVID-19 samples. The full dataset is shown in Supplementary Fig. 4. For the lateral-flow readout, the appearance of a coloured test band, often in conjunction with the disappearance of a coloured control band, indicated a positive COVID-19 result. Quantification of band intensities is shown in Supplementary Fig. 4. For the fluorescence readout, we set the threshold (blue line) of signal-to-noise (S/N) of fluorescence intensities (with noise being the fluorescence intensity from a negative sample with water as input performed in parallel) for a positive result to be 2. Bottom rows show the numbers of false-negative samples; rates of false-negative results are shown for each Ct bin and for the two SHERLOCK readout modes.
Among the 154 clinical samples, we were able to identify all 73 negative COVID-19 samples by both fluorescence and lateral-flow strip readouts, 78 out of 81 positive COVID-19 samples by fluorescence readouts, and 71 out of 81 positive COVID-19 samples by lateral-flow strip readouts (Fig. 4, Supplementary Fig. 4 and Table 1). Samples with Ct < 32 were all detectable by both fluorescence and lateral-flow readouts, closely matching the LoD Ct (33.5) determined from cultured viral RNA. Beyond this LoD, we observed better sensitivity of detection using fluorescence readouts, with sample Ct as high as 37 being detected, albeit at much lower signal-to-noise ratio. Detection of samples at higher Ct values (>32) with lateral flow was less robust, presumably due to a requirement for a critical concentration of the cleaved RNA reporter to accumulate sufficient gold nanoparticles to generate an observable colorimetric signal at the test line22. Since not all of the high-Ct samples can be detected by the SHERLOCK method, we suspected that at low levels of target RNA, amplification by RPA can become variable, and initial RNA input needs to be sufficiently high to ensure productive amplification and detection. Thus, we performed further empirical optimizations on the RPA step, and found that doubling components of the RPA reaction can boost the sensitivity of detection for samples with low viral load (Supplementary Fig. 5).
While almost all of our clinical samples were nasopharyngeal or throat swabs, we analysed three positive sputum samples (Ct for N of 26, 28 and 29), all of which could be detected by the SHERLOCK method under both readout modes. In addition, while 90% of samples were obtained from symptomatic patients (presenting with upper respiratory tract infection, diarrhoea, pneumonia, bronchitis and/or fever; see Supplementary Table 2 for patient characteristics), 10% (8 samples) of our 81 RT–qPCR-positive samples were from asymptomatic cases. We were able to detect SARS-CoV-2 in six out of eight asymptomatic cases using the SHERLOCK method. Indeed, two of the three patients who tested false negative with SHERLOCK with fluorescence readout were asymptomatic; all three false-negative samples did show detectable fluorescent signal over background but did not pass the threshold we set. Samples were obtained from patients who had not taken antimicrobial or antiviral treatments, except for two samples, one from a patient undergoing treatment with lopinavir, ritonavir and chloroquine, and the other was from a patient treated with lopinavir, ritonavir, chloroquine and favipiravir.
Compared with RT–qPCR, we found SHERLOCK-based detection of the SARS-CoV-2 S gene in clinical samples to be 96% sensitive and 100% specific using the fluorescence readout, and 88% sensitive and 100% specific using the lateral-flow readout (Table 2). These correspond to 100% positive predictive agreement (PPA) for either readout, 96% negative predictive agreement (NPA) for the fluorescence readout, and 88% NPA for the lateral-flow readout. Within the determined LoD, SHERLOCK is 100% sensitive and 100% specific using the fluorescence readout, and 97% sensitive and 100% specific using the lateral-flow readout. The diagnostic odds ratios (DORs) of SHERLOCK detection of SARS-CoV-2 are in the range of 6.91–9.94 (Table 2; values shown as ln(DOR)), among the highest when compared to DORs of other point-of-care genetic tests for SARS-CoV-223. The higher sensitivity and DOR when using fluorescence make this readout suitable for point-of-care and routine diagnostics, where a light source with appropriate filters and a smartphone camera can be used. The lateral-flow readout, with its ease of use and current sensitivity, may already be suitable for screening purposes—potentially outside of diagnostic laboratory settings—before diagnostic confirmation.
Deploying SHERLOCK as a pre-operative screen for COVID-19 in surgical patients
Protection of healthcare workers is a fundamental strategy in maintaining societal functions and limiting the spread of COVID-19. Among healthcare workers, those who perform aerosol-producing procedures—including otolaryngologists, dentists and intensive care unit staff—are considered the most susceptible to infection24. A rapid and accurate test to detect SARS-CoV-2 in incoming patients could help safeguard at-risk healthcare personnel while enabling greater accessibility to treatment for a higher number of patients.
To clinically validate SHERLOCK as a potential rapid test method for such applications, we have integrated SHERLOCK detection of SARS-CoV-2 as a part of the pre-operative assessment for surgical patients at Siriraj Hospital, starting in early May 2020. The fluorescence readout of SHERLOCK was used, due to its excellent sensitivity, and the assay was performed alongside RT–qPCR for verification. We used a blue LED transilluminator and a smartphone camera to immediately visualize fluorescence of each sample in real time, and also captured the end-point fluorescence using a PCR with reverse transcription (RT–PCR) instrument (Supplementary Fig. 6). SHERLOCK detection of the SARS-CoV-2 S gene in nasopharyngeal or throat swab samples from 380 surgical patients showed 100% concordance with RT–qPCR results (Table 1), with an approximate test time of 70 min (and as fast as 35 min test time for strongly positive samples), compared with at least 120 min test time for RT–qPCR, excluding time for RNA extraction. All patient samples collected during this testing period (May 2020) were negative for COVID-19, as Thailand had been under lockdown since late March 2020 and had very few domestic cases nationwide in May.
Multiplexing SHERLOCK detection of SARS-CoV-2 RNA with RNase-contamination detection on a lateral-flow strip
As SHERLOCK is based on nucleic acid detection, the presence of active nucleases, especially RNases, can obscure testing results. RNases can lead to RNA-input degradation, leading to false negatives, or if carried over to the CRISPR–Cas detection step, can cleave RNA reporters and create false positives. The risk of RNase contamination is increased in resource-limited settings, where access to clean facilities and equipment to control RNases may be lacking. Current SHERLOCK protocols include RNase inhibitors and steps to inactivate nucleases13 and a negative control to ensure that there is no contamination leading to false-positive results (and a positive control to ensure functionality of the components). However, an in-strip confirmation could serve as an additional alert of the contamination.
Here we incorporated an internal RNase-contamination detection into the design of the SHERLOCK-based detection of RNAs. As LwaCas13a shows sequence preference for its collateral activity9, we designed an RNase-responsive RNA reporter that is resistant to Cas13a cleavage, but remains susceptible to RNase I-, RNase A- and RNase T1-mediated cleavage (Fig. 5a). The RNA reporter is functionalised with digoxigenin (DIG) and fluorescein, allowing capture on a lateral-flow strip with anti-DIG antibody and detection with anti-fluorescein conjugated to gold nanoparticles (anti-Fl–NP). Combining the DIG–fluorescein-functionalised RNase reporter, the biotin–fluorescein-functionalised LwaCas13a reporter, and a lateral-flow strip capable of detecting both DIG- and biotinylated analytes (for example a three-band HybriDetect 2 T strip) enables one-pot, simultaneous detection of both RNA target by SHERLOCK and RNase contamination. This multiplexed readout should enable easy differentiation of true-positive results from false results caused by RNase contamination.

a, Sequence of RNase reporter. Red lines indicate RNase A and RNase T1 cleavage sites. b, Predicted colorimetric outcomes for negative, positive and RNase-contaminated samples. c, Comparison of a conventional lateral-flow readout of SHERLOCK detection of SARS-CoV-2 RNA (left) and a lateral-flow strip with internal RNase-contamination detection (right), in the presence and absence of RNase A. d, Similar LoD obtained from conventional lateral-flow strips (left) and strips containing internal RNase detection (right), with SHERLOCK S gene detection of serially diluted SARS-CoV-2 RNA.
We first optimized the loadings of the biotin–fluorescein SHERLOCK reporter and the DIG–fluorescein RNase reporter to ensure near-complete capture at their respective bands (Supplementary Fig. 7). We further adjusted the amount of anti-FI–NP on the lateral-flow strip (see Methods) to minimize the spillover of anti-FI–NP beyond the first streptavidin band in the case where no Cas-mediated cleavage of the biotin–fluorescein SHERLOCK reporter occurs. In a true-negative control (no RNA input and no RNase), only one strong coloured band at the control (C) line is produced when all anti-FI–NP binds to intact biotin–fluorescein SHERLOCK reporter (Fig. 5b). Specific SHERLOCK detection of the RNA target would cleave the biotin–fluorescein SHERLOCK reporter, allowing production of two coloured bands at T2 (where anti-FI–NP binds to intact DIG–fluorescein RNase reporter) and T1 (where anti-rabbit IgG binds to excess anti-FI–NP). RNase-mediated cleavage cleaves both reporters regardless of specific RNA input, resulting in one strong band at the T1 line (where all anti-FI–NP binds).
We tested the multiplex detection of SARS-CoV-2 RNA with SHERLOCK and RNase contamination (Fig. 5c). True-positive samples (+SARS-CoV-2 RNA, Ct ≈ 27; −RNase) indeed produced two coloured bands at T1 and T2, readily distinguishable from true-negative controls (main band at C) and RNase-contaminated samples (one band at T1). Analysis of serial dilutions of SARS-CoV-2 RNA showed that the multiplex detection maintains the sensitivity of SARS-CoV-2 S gene detection when compared with the single-plex detection (Fig. 5d).
Discussion
The global scale of the COVID-19 pandemic highlights the need for diagnostic tests that are rapid, accurate, economical and can be deployed at point of care, which would inform decisions on disease-control strategies and help manage patients at healthcare facilities25. RT–qPCR, the current gold-standard test for genetic detection of SARS-CoV-2, has a long turnaround time and relies on materials and equipment that can be costly and subject to strains in the supply chain. These disadvantages are exacerbated in developing countries, which lack economic resources and bargaining power, transport infrastructure and local capability to manufacture testing components or devices.
Here we critically evaluated the performance of the SHERLOCK method, a CRISPR–Cas-based detection platform, for SARS-CoV-2 detection. We validated the SHERLOCK method on 154 clinical COVID-19 samples in total, and found it to be 100% specific and 96% sensitive with the fluorescence readout, and 88% sensitive with the lateral-flow readout. The method was able to detect SARS-CoV-2 in RNA extracts from nasopharyngeal and throat swab samples including sputum samples, without any cross-reactivity to other common human coronaviruses, and was able to detect the virus in asymptomatic cases. We demonstrated the viability of SHERLOCK as a rapid genetic test for COVID-19 by incorporating it as a part of the pre-operative assessment for incoming surgical patients. The utility of a rapid test such as SHERLOCK would be even greater outside of clinical laboratory settings where access to diagnostic equipment is limited, or where very quick turnaround times are needed.
The current LoD and sensitivity of SHERLOCK SARS-CoV-2 detection are on par with the performance of approved point-of-care genetic diagnostic tests20,21 such as Abbott ID Now and Cepheid Xpert Xpress and other point-of-care tests under development23, but it does not require specific instrumentation. This potentially extends the utility of SHERLOCK beyond point-of-care to at-home tests, which can be performed more frequently and cheaply, pending the implementation of a safe sample-collection procedure. A recent study has suggested a relationship between measured Ct values of SARS-CoV-2 E gene in patient samples and their infectivity in in vitro cell culture, and that viral infectivity of patients with Ct > 24 may be low26. An at-home test with good coverage of infective Ct ranges could prove useful, and in our study, SHERLOCK was already able to detect SARS-CoV-2 in samples with Ct < 33.5 with 97–100% sensitivity and 100% specificity.
Outlook
Future work can further close the gap in sensitivity, especially for lateral-flow detection, and improve the user-friendliness of the protocol. The two-step SHERLOCK protocol evaluated here is best performed with physical separation of reagent preparation, amplification and detection steps to minimize contamination caused by aerosols; this is cumbersome to arrange outside of molecular diagnostic laboratories. One-pot CRISPR diagnostics protocols such as the SHERLOCK testing in one pot (STOP) COVID protocol16 and SHERLOCK and HUDSON integration to navigate epidemics15 (SHINE) bypass RNA extraction, combine genetic amplification and CRISPR-based detection into one step, and are much easier to perform. Continual, rapid advances in better isothermal amplification and CRISPR-mediated detection conditions (for instance, the addition of RNase H to improve RT–RPA efficiency8, the beneficial effect of which we have verified (Supplementary Fig. 8)), especially in the context of a simple one-pot reaction, will make variations of SHERLOCK and CRISPR diagnostics viable testing platforms for COVID-19 in limited-resource settings, at point of care or at home.
While CRISPR diagnostic technologies do not need extensive instrumentation, many specialized reagents needed are costly and proprietary. However, with its inherently scalable biochemical components, CRISPR diagnostics offers a unique opportunity for resource-constrained developing countries to self-assemble test components to further save the cost of operation. In this study, we produced Cas enzymes and RNAs in Thailand, and started to develop testing procedures that would be compatible with diverse testing scenarios, including the multiplexed detection of RNA target and RNase contamination using orthogonal RNA reporters. Beyond COVID-19, we hope that CRISPR diagnostics technology will have lasting utility in infectious disease monitoring and diagnosis, as well as other applications related to human health and biotechnology in Southeast Asia.
Methods
Expression and purification of His6–SUMO–LwaCas13a
Escherichia coli BL21 (DE3) cells harbouring PC013-His6-Twinstrep-SUMO-huLwCas13a (Addgene #90097) were used for the expression of His6–SUMO–LwaCas13a. The recombinant gene expression was induced by addition of 0.5 mM IPTG at 16 °C overnight in 4.5 l LB medium containing 100 µg ml−1 ampicillin. The culture was collected by centrifugation at 8,000 rpm for 20 min at 4 °C. The supernatant was discarded and the cell pellet was resuspended in extraction buffer (50 mM Tris-HCl, 500 mM NaCl, 1 mM DTT, 1× protease inhibitor cocktail, 0.25 mg ml−1 lysozyme and 5 mM imidazole; pH 7.5). The cell resuspension was lysed by sonication (Sonics Vibracell VCX750) using 40% pulse amplitude (on 5 s and off 10 s) until completely lysed. The lysate was centrifuged at 11,000 rpm for 30 min at 4 °C. Following clarification, the soluble fraction was filtered through a 0.45 mm polyethersulfone membrane and purified by HisTrap FF column connected to a fast protein liquid chromatography system. The column was pre-equilibrated with binding buffer (50 mM Tris-HCl, 500 mM NaCl, 5 mM imidazole pH 7.5). The soluble fraction was loaded at 5 ml min−1 and washed with 5 column volumes of binding buffer. The recombinant His6–SUMO–LwaCas13a was eluted in a linear gradient of elution buffer (50 mM Tris-HCl, 0.5 M NaCl and 0.5 M imidazole; pH 7.5). The eluted fractions were pooled and diluted with 20 mM Tris-HCl and 5% glycerol pH 8.0 to give a conductivity around 17–18 mS cm−1. Subsequently, the protein was applied to a HiTrap SP HP column, which was pre-equilibrated with binding buffer (20 mM Tris-HCl, 1 mM DTT, 5% glycerol, 150 mM NaCl pH 8.0). After washing with the binding buffer to remove nonspecific binders, the bound protein was eluted with a linear gradient of NaCl from 0.15 M–2 M NaCl in 20 mM Tris-HCl, 1 mM DTT, 5% glycerol pH 8.0. The purified His6–SUMO–LwaCas13a fractions were pooled and exchanged against SUMO cleavage buffer (20 mM Tris, 150 mM NaCl, 1 mM DTT, 10% glycerol pH 8.0) before concentration.
To obtain LwaCas13a without SUMO, His6–SUMO–LwaCas13a was digested with Ulp1 SUMO protease (Addgene #64697) to remove a fusion tag using 10:1 of SUMO substrate (SUMO protease in 20 mM Tris-HCl, 150 mM NaCl, 10% glycerol, 1 mM DTT and 0.2% Triton X-100 pH 8.0). The reaction mixture was incubated at 25 °C for 3 h with gentle shaking before loading onto Superdex200 Increase 10/300 GL followed by 20 mM Tris-HCl, 150 mM NaCl pH 7.5. Finally, the purified protein was exchanged against 40 mM Tris-HCl, 400 mM NaCl and stored at −20 °C in 20 mM Tris, 200 mM NaCl, 2 mM DTT, 40% glycerol pH 7.5.
RPA primers, crRNAs, and RNA reporters
Information on nucleic acid components of the detection are given in Supplementary Table 3.
S gene RNA fragment production
The in vitro transcription of S gene was adapted from a previous protocol19. The T7–3G oligonucleotide was annealed with the DNA template by denaturation at 95 °C for 30 s and cooling down to 25 °C with a ramp rate 0.1 °C s−1. The annealed oligonucleotide was diluted with DEPC-treated water. Four picomoles of the annealed product was incubated with T7 RNA polymerase with RiboMAX Large Scale RNA Production System–T7 (ProMega) at 37 °C overnight. The product RNA was then purified with VAHTS RNA Clean Beads (Vazyme Biotech) and eluted with DEPC-treated water.
RT–RPA
One lyophilized RPA pellet (TwistAmp Basic kit, TwistDx) is enough to provide an RPA mix for five reactions, and the protocol here is for preparation of five RPA reactions at a time. First, one RPA pellet was resuspended with 29.5 µl of the rehydration buffer supplied in the TwistAmp Basic kit. RPA forward primer (2.5 µl, 10 µM stock), 2.5 µl RPA reverse primer (10 µM stock) and 1 µl of EpiScript reverse transcriptase (200 U µl−1 stock; Lucigen) were then added to the resuspended RPA mixture. RPA-primer–reverse transcriptase mix (7.1 µl) was aliquoted into five precooled 1.5 ml Eppendorf tubes, to each of which 6.5 µl of RNA extracts from nasopharyngeal swabs was added. Concentrations of RNA extracted from clinical samples can be variable, and we have had success with samples containing 0.1 pg µl−1 total RNA. Once RNA was added to the RPA-primer–reverse transcriptase mix, 0.7 µl of magnesium acetate (280 mM stock) was added as the last component to initiate the amplification reaction. The reactions were incubated at 42 °C for 25 min. Thereafter, each RPA reaction tube was placed back on ice before proceeding with Cas13-based detection19.
For positive and negative controls, the reactions were set up with RNA extracts from cultured SARS-CoV-2 virus and DEPC-treated water, respectively.
Colorimetric-based lateral-flow detection assay
Each Cas13-based detection reaction was prepared in a precooled 1.5 ml microcentrifuge tube with the following components: 7.5 μl DEPC-treated water, 2.0 μl Tris (400 mM stock, pH 7.4), 1.0 μl MgCl2 (120 mM stock), 0.8 μl ribonucleoside tri-phosphate mix (25 mM stock; NEB), 2 µl LwaCas13a in storage buffer (63.3 µg ml−1 enzyme stock concentration), 1 µl RiboGuard RNase inhibitor (40 U μl−1 stock; Lucigen), 0.6 µl T7 RNA polymerase (50 U µl−1 stock; Lucigen), 1 µl crRNA (10 ng µl−1 stock) and 1 µl of the biotin–fluorescein ssRNA reporter (20 µM stock; IDT). Two microlitres of RPA products from the RT–RPA amplification step were transferred to the Cas reaction (for multiple Cas reactions, a master mix containing the above components can be prepared before aliquoting and addition of individual RPA reactions). The reaction tubes were incubated at 37 °C for 30 min.
To analyse the Cas reaction via lateral-flow strip, 80 µl of HybriDetect assay buffer was added to each Cas reaction and mixed thoroughly. A HybriDetect lateral-flow strip (Milenia Biotech) was placed into each reaction tube, allowing the reaction to flow into the strips and provide readout. The band intensities were quantified using gel analyser tool in Fiji ImageJ software27.
Fluorescence-based detection assay
Each Cas13-based detection reaction was prepared in a precooled 1.5 ml microcentrifuge tube with the following components: 9.5 µl DEPC-treated water, 0.4 µl HEPES (1 M stock pH 6.8), 1 µl MgCl2 (120 mM stock), 0.8 µl ribonucleoside tri-phosphate mix (25 mM stock), 2 µl LwaCas13a enzyme in storage buffer (63.3 µg ml−1 enzyme stock concentration), 1 µl RiboGuard RNase inhibitor (40 U µl stock; Lucigen), T7 RNA polymerase (0.6 µl, 50 U µl−1 stock; Lucigen), 1 µl of crRNA (10 ng µl−1 stock) and 1.25 µl RNase Alert v1 (2 µM stock; IDT). After mixing, 18 µl of the reaction mixture was transferred to a 384-well plate. Two microlitres of RPA products from the RT–RPA amplification step was then added. The results were collected by monitoring fluorescence (fluorescein excitation/emission filters) on a Varioskan microplate reader over 1 h at 37 °C with a 1-min interval between each data collection.
Alternatively, the Cas-based detection reactions could be visualized using an LED transilluminator. Reactions were performed as above, with minor modifications: 0.4 µl HEPES (1 M stock, pH 6.8) and 1.25 µl of RNase Alert v1 (2 µM stock; IDT) were replaced with 2.0 µl Tris (400 mM stock, pH 7.4) and 1.25 µl of FAM-Iowa Black polyU reporter (4 µM stock; IDT) and a volume of DEPC-treated water was reduced to 7.9 µl. After 30 min incubation at 37 °C using a smartphone camera and a BluPAD Dual LED Blue/White Light Transilluminator with 460 nm blue light with 580 nm amber filter (Bio-Helix). Ten microlitres of samples was transferred to a PCR strip tube, and the samples were incubated for 1 min at 37 °C in CFX96 Touch Real-Time PCR System (Bio-Rad). The end-point fluorescence intensity was detected in a FAM channel.
Multiplexed detection of SARS-CoV-2 RNA and RNase contamination on a lateral-flow strip
The RNase reporter was designed corresponding to LwaCas13a- and RNase-cleavage preferences. In particular, Cas13a-cleavable dinucleotide preferences of UU, AU, GU and CU were taken into account9. Cleavage preferences of mammalian RNase A and Aspergillus oryzae RNase T1 are rY/rN28 and rG/rN29, respectively. Our reporter should also detect activity of non-sequence-specific RNases such as E. coli RNase I30.
Each Cas13-based detection reaction was performed in a similar manner to the single-plex assay previously described with 2 µl of the DIG–fluorescein RNA reporter (1 µM stock, GenScript) added. A total reaction volume of 20 μl was adjusted with DEPC-treated water. For samples spiked with RNase, 1 µl of FAPD1 buffer with RNase A (FavorPrep plasmid DNA extraction mini kit, Favorgen) was added to the tubes before the incubation.
To analyse the Cas reaction via lateral-flow strip, 80 µl of HybriDetect 2T assay buffer was added to the tube, followed by placing the strip as previously described. We cut out 9 mm of the sample pad of the HybriDetect 2T strip to empirically reduce the amount of anti-FI–NP to match the amount of RNA reporters used.
Sample collection, RNA preparation, RT–qPCR and ethical approval
Nasopharyngeal and throat swabs from patients with suspected SARS-CoV-2 infection were processed at the Diagnostic Molecular Laboratory, Department of Microbiology, Faculty of Medicine, Siriraj Hospital. RNA was extracted from 200 µl viral transport medium (Gibco) used to store the swab using a MagLEAD 12gC automated extraction platform (Precision System Science) according to the manufacturer’s instructions, and eluted in 100 µl. RNA extracts were initially used for molecular diagnosis of SARS-CoV-2 by RT–qPCR targeting RdRp, E and N genes using Allplex 2019-nCoV assay (Seegene). Excess RNA extracts from these samples were then used, without any personally identifiable information being collected, for validation of CRISPR diagnostics detection of SARS-CoV-2. Eighty-one samples with known positive results from RT–qPCR for SARS-CoV-2, 73 samples with known negative results from RT–qPCR for SARS-CoV-2, and three known samples with positive results for hCoV-OC43, hCoV-NL63 and hCoV-229E were included for the method validation. Samples were randomized when given to study staff, who were kept blinded to the SARS-CoV-2 RT–qPCR diagnostics results of the samples when they performed validation experiments of CRISPR diagnostics.
Ethical approval of the study was given by the Siriraj Institutional Review Board (COA: Si 339/2020 and Si 424/2020).
Sample-size calculation for clinical validation study
Minimal sample size was determined before experiments on the basis of sensitivity (≥95%) and specificity (≥95%) requirements of diagnostic test characteristics by the Department of Medical Sciences, Ministry of Public Health, Thailand, according to the formula:
where Z is the critical value of the normal distribution at the required confidence level, P is the percentage picking a choice of sensitivity/specificity, expressed as decimal, and d is margin of error, expressed as decimal.
In our study, n = 73 when Z = 1.96 for a 95% confidence level, P = 0.95 and d = 0.05. We therefore proceeded by using at least 73 SARS-CoV-2-positive (by RT–PCR) samples and 73 SARS-CoV-2-negative samples for the CRISPR diagnostics validation study.
Statistical analysis
Data were analysed using GraphPad Prism 8. Two-sided confidence intervals of sensitivity, specificity, PPA and NPA were calculated using the Clopper–Pearson method. Two-sided confidence intervals of ln(DOR) were calculated as described for odds ratio31.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The main data supporting the results in this study are available within the paper and its supplementary information. Raw datasets generated and analysed during the study are available from the corresponding authors on reasonable request.
References
Vogels, C. B. F. et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT–qPCR primer–probe sets. Nat. Microbiol. https://doi.org/10.1038/s41564-020-0761-6 (2020).
Kelly-Cirino, C. D. et al. Importance of diagnostics in epidemic and pandemic preparedness. BMJ Glob. Health 4, e001179 (2019).
Piepenburg, O., Williams, C. H., Stemple, D. L. & Armes, N. A. DNA detection using recombination proteins. PLoS Biol. 4, e204 (2006).
Notomi, T. et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, e63 (2000).
Rabe, B. A. & Cepko, C. SARS-CoV-2 detection using an isothermal amplification reaction and a rapid, inexpensive protocol for sample inactivation and purification. Preprint at https://doi.org/10.1101/2020.04.23.20076877 (2020).
Becherer, L. et al. Loop-mediated isothermal amplification (LAMP)—review and classification of methods for sequence-specific detection. Anal. Methods 12, 717–746 (2020).
Bhadra, S., Riedel, T. E., Lakhotia, S., Tran, N. D. & Ellington, A. D. High-surety isothermal amplification and detection of SARS-CoV-2, including with crude enzymes. Preprint at https://doi.org/10.1101/2020.04.13.039941 (2020).
Qian, J. et al. An enhanced isothermal amplification assay for viral detection. Preprint at https://doi.org/10.1101/2020.05.28.118059 (2020).
Gootenberg, J. S. et al. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444 (2018).
Chen, J. S. et al. CRISPR–Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439 (2018).
Harrington, L. B. et al. Programmed DNA destruction by miniature CRISPR–Cas14 enzymes. Science 362, 839–842 (2018).
Gootenberg, J. S. et al. Nucleic acid detection with CRISPR–Cas13a/C2c2. Science 356, 438–442 (2017).
Myhrvold, C. et al. Field-deployable viral diagnostics using CRISPR–Cas13. Science 360, 444–448 (2018).
Xiao, W. et al. A simple and compact smartphone-based device for the quantitative readout of colloidal gold lateral flow immunoassay strips. Sens. Actuators B 266, 63–70 (2018).
Arizti-Sanz, J. et al. Integrated sample inactivation, amplification, and Cas13-based detection of SARS-CoV-2. Preprint at https://doi.org/10.1101/2020.05.28.119131 (2020).
Joung, J. et al. Point-of-care testing for COVID-19 using SHERLOCK diagnostics. Preprint at https://doi.org/10.1101/2020.05.04.20091231 (2020).
Zhang, F., Abudayyeh, O. O. & Gootenberg, J. S. A protocol for detection of COVID-19 using CRISPR diagnostics. https://static1.squarespace.com/static/5b7c640be2ccd1703a3da4d3/t/5e7773f40df723159b16f262/1584886772519/COVID-19+detection+%28v.20200321%29.pdf (2020).
Broughton, J. P. et al. CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 38, 870–874 (2020).
Kellner, M. J., Koob, J. G., Gootenberg, J. S., Abudayyeh, O. O. & Zhang, F. SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 14, 2986–3012 (2019).
Basu, A. et al. Performance of Abbott ID Now COVID-19 rapid nucleic acid amplification test using nasopharyngeal swabs transported in viral transport media and dry nasal swabs in a New York City academic institution. J. Clin. Microbiol. 58, e01136-20 (2020).
Smithgall, M. C., Scherberkova, I., Whittier, S. & Green, D. A. Comparison of Cepheid Xpert Xpress and Abbott ID Now to Roche cobas for the rapid detection of SARS-CoV-2. J. Clin. Virol. 128, 104428 (2020).
Khlebtsov, B. N., Tumskiy, R. S., Burov, A. M., Pylaev, T. E. & Khlebtsov, N. G. Quantifying the numbers of gold nanoparticles in the test zone of lateral flow immunoassay strips. ACS Appl. Nano Mater. 2, 5020–5028 (2019).
Subsoontorn, P., Lohitnavy, M. & Kongkaew, C. The diagnostic accuracy of nucleic acid point-of-care tests for human coronavirus: a systematic review and meta-analysis. Preprint at https://doi.org/10.1101/2020.07.09.20150235 (2020).
Kowalski, L. P. et al. COVID-19 pandemic: effects and evidence-based recommendations for otolaryngology and head and neck surgery practice. Head Neck 42, 1259–1267 (2020).
Esbin, M. N. et al. Overcoming the bottleneck to widespread testing: a rapid review of nucleic acid testing approaches for COVID-19 detection. RNA 26, 771–783 (2020).
Bullard, J. et al. Predicting infectious SARS-CoV-2 from diagnostic samples. Clinical Infectious Diseases https://doi.org/10.1093/cid/ciaa638 (2020).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Raines, R. T. Ribonuclease A. Chem. Rev. 98, 1045–1066 (1998).
Steyaert, J., Wyns, L. & Stanssens, P. Subsite interactions of ribonuclease T1: viscosity effects indicate that the rate-limiting step of GpN transesterification depends on the nature of N. Biochemistry 30, 8661–8665 (1991).
Zhu, L. & Deutscher, M. P. The Escherichia coli RNA gene encoding RNase I: sequence and unusual promoter structure. Gene 119, 101–106 (1992).
Glas, A. S. et al. The diagnostic odds ratio: a single indicator of test performance. J. Clin. Epidemiol. 56, 1129–1135 (2003).
Acknowledgements
We thank P. Chuchottaworn, J. Limtrakul and P. Watanapa for generous support and advice on the project; M. Fuangthong for Bioanalyzer chips and P. Pannopard and the PTT Expresso Team for management and logistical support; A. Ting for the UltraTEV protease expression plasmid and protein; M. Howarth for the streptavidin variant expression plasmid and protein; C. Gilbert for recombinase and polymerase expression plasmids; and M. Sanchez Lopez and S. Han for helpful advice. Several pieces of equipment used for the project were generously provided on loan by Bang Trading, Eppendorf Thailand, PCL Holding and PTT Innovation Institute. We thank Baan Wanglang Riverside for facilitating the team’s work at Siriraj Hospital. The project is financially supported by PTT, PTTEP, Kasikorn Bank, Siam Commercial Bank, GPSC, VISTEC and Siriraj Hospital.
Author information
Authors and Affiliations
Contributions
C.U., N.H. and F.Z. designed the study. C.U. and N.H. supervised the study. M.P., K.J., A.P., K.A., S. Suraritdechachai, N.L., P.M., K.S., T. Wongsatit, N.A., B.E., B.L., T.P. and T.R. designed and performed experiments. F.Z., J. Joung, A.L., J.S.G., O.O.A., D.P., S.C., A.H., R.T., P.K., K.P., S. Soithongcharoen, C.K., V.P., D.T., J. Jaroensuk, T. Wongnate, S.M. and P.C. provided critical materials and reagents for the experiments. N.H., S.K., J.S., S.C., Y.S., C.C., R.S. and W.K. provided and facilitated testing of clinical samples. N.N. and X.J. contributed to the design, analysis and management of experiments. All authors contributed to the writing of the manuscript and interpretation of data.
Corresponding authors
Ethics declarations
Competing interests
O.O.A., J.S.G. and F.Z. are co-founders and scientific advisors for and hold equity interests in Sherlock Biosciences, Inc. F.Z. is also a co-founder of Editas Medicine, Beam Therapeutics, Pairwise Plants and Arbor Biotechnologies. C.U., S. Suraritdechachai, K.A., N.L., T. Wongsatit, M.P., K.J., P.M., K.S. and T.R. are inventors on a patent application (Thai patent application no. 2001004005) related to lateral-flow detection of nuclease activity filed by VISTEC. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary figures and tables.
Rights and permissions
About this article

Cite this article
Patchsung, M., Jantarug, K., Pattama, A. et al. Clinical validation of a Cas13-based assay for the detection of SARS-CoV-2 RNA. Nat Biomed Eng 4, 1140–1149 (2020). https://doi.org/10.1038/s41551-020-00603-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41551-020-00603-x
This article is cited by
-
Polydopamine-assisted aptamer-carrying tetrahedral DNA microelectrode sensor for ultrasensitive electrochemical detection of exosomes
Journal of Nanobiotechnology (2024)
-
Micrometer-thick and porous nanocomposite coating for electrochemical sensors with exceptional antifouling and electroconducting properties
Nature Communications (2024)
-
Transcript-specific induction of stop codon readthrough using a CRISPR-dCas13 system
EMBO Reports (2024)
-
Nanotechnology’s frontier in combatting infectious and inflammatory diseases: prevention and treatment
Signal Transduction and Targeted Therapy (2024)
-
Fast and sensitive CRISPR detection by minimized interference of target amplification
Nature Chemical Biology (2024)
