
Abstract
Although Fenton-like reactions have been extensively used to treat various organic contaminants in wastewaters (oily wastewaters, landfill leachate, denitrification with Fenton oxidized non-degradable large molecular organic pollutants, and removal of phosphorus, etc.), the difficulty in catalyst recovery and the instability of catalytic activity limit their practical application. Herein, iron atoms were inserted in a metal-organic framework (FeNC) with dual reaction sites as highly reactive and stable electro-Fenton-like catalysts for the catalytic oxidation of organic pollutants via the electro-Fenton-like activation reaction of peroxymonosulfate (PMS) with fractional leaching of metal ions. Experiments and density functional theory (DFT) calculations indicate that FeNC with FeN4 and Fe–Fe active sites can rapidly deliver electrons for PMS dissociation. Further, an electrolysis reactor was constructed for the on-site generation of reactive oxygen species, which can stably and continuously purify various organic wastewaters. The combined use of an electrolysis reactor and magnetic catalyst in the current study provides a direction for the long-term remediation of organic pollutants on an industrial scale.
Similar content being viewed by others


Introduction
Fenton-like reactions can efficiently degrade organic contaminants by generating highly reactive oxygen species (e.g., ROS, •OH, O2•–), and have been considered a promising method1,2,3 for eliminating the increasing environmental pollutants produced by rapid industrialization and human activity in groundwater or surface water as well as in wastewaters. The hydroxyl radical (•OH) released from the Fenton reaction, using hydrogen peroxide (H2O2) as an activator, can react rapidly and non-selectively with nearly all organic compounds and mineralize them from large molecules into small nontoxic molecules (e.g., H2O, CO2)4. However, the homogeneous Fenton reaction cannot be scaled up owning to the narrow pH range, low utilization of H2O2, excess consumption of Fe2+, rapid generation of iron-containing sludge, and poor stability5. Recently, the heterogeneous Fenton-like reaction caused by the activation of peroxymonosulfate (PMS) has received extensive research attention because of its ability to degrade and mineralize recalcitrant organic pollutants in water environments owing to its wide working pH range, high activity, and sustainability6,7. Although the heterogeneous Fenton-like reaction has been substantially studied owing to its promising performance in degrading various organic contaminants8,9, three critical challenges have substantially limited its practical application.
The first challenge is related to the leaching of metal ions in the reaction process. Although some single-atom catalysts can significantly reduce the release of metal ions10,11,12, the lower activity of 1O2 or O2•– than that of SO4•– and •OH is not beneficial to the rapid degradation of various recalcitrant organic pollutants13,14. Transition-metal-based catalysts (e.g. Fe–15, Co–16, Mn–17, Ni–18) on various supports (e.g. carbon nitride19, graphene20, carbon nanotubes21) can efficiently activate PMS to produce ROS (SO4•–and •OH) (Eq. 1). A traditional Fenton-like reaction based on the Mn+/PMS process (where M represents transition-metal atoms) proceeds according to the following equations:
Previous studies have demonstrated that the low transformation rate of M(n+1)+ back to Mn+ in a Fenton-like system can accumulate large amounts of Fe3+ ions, which is regarded as a rate-controlling and intrinsic shortcoming of this process22. The rapid reaction of Eq. 1 and the slow reaction of Eq. 2 not only reduces the efficiency of the M(n+1)+/Mn+ redox cycle, but also inactivates the catalyst23,24,25. In addition, the quenching reaction of ROS in acidic conditions by transition-metal atoms increases the leaching of metal ions (Eqs. (3) and (4))26. Consequently, the leaching of metal ions and the narrow working pH range further restrict Fenton-like reactions from comprehensive applications.
The second challenge with the Fenton-like reaction is catalyst recovery. As the core of efficient organic pollutant degradation, catalyst recovery and reuse significantly reduce operational costs. However, the continuous addition and recovery of catalysts is not feasible when Fenton-like technology is implemented in remote areas or in in-situ operational processes. A potential alternative method is using a catalyst with high activity and stability, which can be continuously activated and recovered via an in-situ method.
The third challenge with the Fenton-like process is the continuous generation of SO4•–and •OH. Previous studies have demonstrated that the catalysts are inactivated through a sustainable reaction owning to the active sites covered by adsorbed pollutants or the continued consumption of catalysts, thereby reducing the generation efficiency of ROS27,28,29. Although some studies recovered catalyst activity to generate more ROS via recalcination treatment30, complex operations and narrow working pH ranges are not beneficial for scaling up applications of the Fenton-like process.
Herein, we propose a wastewater purification technique for synthesizing effective catalysts that can successfully address the abovementioned challenges related to the electro-Fenton-like reaction. We explore the insertion of iron oxide into a metal-organic framework (MOF) to form iron-based catalysts with dual reaction sites (FeNC) and magnetism to solve the above challenges, and discuss the potential performance for PMS activation to generate SO4•–and •OH. In addition, we design and construct a reaction device that can use FeNC as the catalyst to continuously produce SO4•– and •OH to efficiently degrade organic pollutants without leaching metal ions or environmental pollutants. This device performs (i) an electro-Fenton-like process (to generate SO4•– and •OH to degrade organic pollutants), (ii) an ultrasonic recovering process (to restore the catalyst), and (iii) a solid-liquid separation process (to adsorb and separate the catalyst from the electrolyte). We also use the device to run six continuous cycles to evaluate the feasibility of the device and catalysts.
Results
Synthesis and characterizations of FeNC catalyst
A magnetic FeNC catalyst can be synthesized based on the hydrothermal reaction and calcination process, which are outlined in the schematic shown in Fig. 1a. After a series of simple hydrothermal reactions at 200 °C, iron nanoparticles were successfully synthesized and denoted as FeNs. An MOF was prepared to form a nitrogen-doped carbon structure through high-temperature annealing. Through an additional hydrothermal reaction and calcination, an MOF structure with added iron nanoparticles (FeNs/MOF) was formed. The hydrothermal method has many benefits such as low reaction temperature, environmental friendliness, rich product morphology, and small particle size31,32. The quick reaction time of the calcination process allowed for the preparation of a pure product and control of product particle size and morphology33,34. Here, using the hydrothermal method, FeNs were successfully prepared and several active sites were exposed for inserting into supporting materials (MOF). FeNs/MOF was first dispersed in methanol through sonication. Subsequently, NH4OH and CTAB were added to the mixture and stirred to form a homogenous solution. After a series of processes, namely, centrifugation, washing, and drying, the samples were further heated via calcination. The mixing and evaporation of low-boiling-point Zn atoms could regulate the distance of adjacent Fe atoms and anchor them on the N–C support. Finally, a magnetic catalyst (FeNC) with dual reaction sites was obtained with functional modifications under the action of NH3•H2O and hexadecyltrimethylammonium bromide (CTAB).

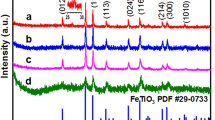
a Preparation procedure of the FeNC catalyst. b SEM and c TEM images of FeNC. d XRD pattern of different catalysts. e TEM and the corresponding (f) HRTRM images of FeNC. g HAADF image and elemental mappings of FeNC with Fe (green), C (blue), and N (red).
To explore the morphology of the FeNC catalyst, we performed scanning electron microscopy (SEM) to observe the microstructure of the FeNC catalyst. As illustrated in Fig. 1b and Supplementary Fig. 1, FeNC exhibits a fullerene-like uniform structure with various sizes ranging from 120 to 230 nm. Transmission electron microscopy (TEM) was performed to further observe the three-dimensional structure (Fig. 1c, e). The crystalline structures of MOF studied by X-ray diffraction (XRD) exhibit two diffraction peaks of graphene in the (101) and (002) planes. The diffraction peaks exhibit an obvious change with the introduction of Fe species, suggesting that the Fe species are successfully inserted into the MOF structure (Fig. 1d). The high-resolution TEM image in Fig. 1f exhibits a d-spacing of 0.38 nm, assigned to the (002) plane of FeNC, and the result is in accordance with the XRD patterns. The Fourier transform infrared (FTIR) spectra of FeNC are considerably smoother than those of the other catalysts (Supplementary Fig. 2), and the peak at 1387 cm–1 confirms the formation of the N–C structure35. The specific Brunauer–Emmett–Teller (BET) surface area and total pore volume (Supplementary Fig. 3) of FeNC measured using nitrogen sorption isotherms are 229.6 m2 g–1 and 0.484 cm3 g–1, respectively, which are higher than those of MOF (84.3 m2 g–1 and 0.225 cm3 g–1) and FeNs (22.2 m2 g−1 and 0.144 cm3 g−1), indicating an increase in the surface area of FeNC. HAADF image and energy-dispersive X-ray spectroscopy (EDX) elemental mappings of FeNC, as presented in Fig. 1g, suggest that the Fe, N, and C species are uniformly distributed in the catalyst, evidencing the formation of FeNC.
X-ray absorption fine structure measurements were performed to examine the chemical states and coordinated environments of FeNC. As displayed in Fig. 2a, the adsorption edge position of FeNC is higher than that of Fe foil and FeO and lower than that of Fe2O3, suggesting that the oxidation state of the Fe atom in FeNC is situated between +2 and +336. The Fourier-transformed (FT) k3-weighted EXAFS spectra of FeNC exhibit a peak at approximately 1.5 Å (Fig. 2b), ascribed to the Fe–N coordination37,38. A second peak located at 2.6 Å is observed; however, it differs from that of FeO and Fe foil39, indicating the presence of Fe–N and Fe–Fe species in FeNC. Compared with Fe foil, FeO, and Fe2O3, two intensity maximums at 4.7 Å−1 and 6.3 Å−1 can be observed from the wavelet transforms of the EXAFS plots for FeNC, which can be ascribed to Fe–N and Fe–Fe coordination, respectively (Fig. 2d–f, and Supplementary Fig. 4). As presented in Fig. 2c and Supplementary table 1, the FT-EXAFS of FeNC is fitted in R-space to further determine the coordination structure. The coordination properties of FeNC are 0.9 for Fe–Fe and 4.2 for Fe–N, respectively, indicating the presence of Fe–Fe coordination. These results indicate that Fe atoms with FeN4 and Fe–Fe configuration can be designated as Fe atomic clusters, as illustrated in the inset of Fig. 2c.

a Normalized Fe K-edge XANES spectra of FeNC and the reference spectra. b k3-weighted Fourier transforms spectra for Fe K-edge-extended X-ray absorption fine structure (EXAFS). c EXAFS fitting curves of FeNC in R-space. WT of (d) Fe foil, (e) FeO, and (f) FeNC.
Performance of the electro-Fenton-like reaction
We further utilized the electrochemical properties to evaluate the catalytic activity of these catalysts. As illustrated in Supplementary Fig. 5 and 3A, all catalysts exhibited multiple pairs of obvious redox peaks, and the peak current significantly increased with an increase in the scanning rate from 5 to 100 mV s−1. FeNC exhibited a substantially larger electrochemically active surface area of 247.41 m2g−1 than the other catalysts (Supplementary Fig. 6), which was beneficial to the electron transfer between the electrode and catalyst. The results of cyclic voltammetry (CV; Supplementary Fig. 7), linear sweep voltammetry (LSV; Supplementary Fig. 8), and Tafel curves (Supplementary Fig. 9) further confirmed the rapid electron transfer and excellent electrochemical activity of FeNC. Figure 3b shows that the current of FeNC exhibited a noticeable enhancement (39.8 mA) with the addition of acid orange (AO7), and this increase was significantly higher than that in other catalysts. In addition, the current response of FeNs/MOF and FeNC remained stable for 2 h, whereas that of the MOF and FeNs catalyst slightly decreased. This finding suggests that the presence of an electron donor increased the electron transfer to some extent; thus, FeNC exhibited better electrocatalytic activity for oxidizing AO7 than the other catalysts.

a Electrochemical active surface area of different catalysts. b I–t curves of different catalysts. c Degradation curves of AO7 in sole AO7, PMS, and electrooxidation processes with or without PMS. d AO7 degradation curves of the MOF, FeNs, FeNs/MOF, and FeNC catalyst during the electrooxidation PMS process. e UV–vis absorbance of AO7 at a different time interval. f AO7 degradation rate by a FeNC-activated PMS system under different scavengers. g Electron paramagnetic resonance (EPR) spectra of different radicals. h EPR spectra of ·OH and SO4− for various catalysts. (i) Quantitative determination of ·OH and SO4−. Conditions: current density=10 mA cm−2, AO7 = 50 mg L−1, PMS = 10 mM, pH=7.0, T = 28 °C. Error bars show the standard deviation (n = 3 independent replicates).
The catalytic performance of the electro-Fenton-like reaction of the FeNC catalysts was assessed for AO7 removal by PMS activation. As illustrated in Fig. 3c, FeNC adsorption and PMS reaction were negligible for AO7 removal; furthermore, the combination of electrolysis and PMS without a catalyst exhibited no evident oxidation. However, almost 100% AO7 was removed when the FeNC catalyst was added to this system, signifying that large amounts of ROS were generated during the electro-Fenton-like reaction. In addition, further results of AO7 degradation by MOF, FeNs, and FeNs/MOF were compared (Fig. 3d), and the corresponding removal rates were found to be 11.8%, 19.7%, and 72.8% within 60 min, respectively. FeNC achieved significant improvement in AO7 degradation owing to the activation reaction of Fe active sites on PMS. The kinetic degradation rate of AO7 was calculated by data fitting using a pseudo-first-order equation. The kinetic degradation rate of AO7 in the FeNC system reached 0.051 min−1, which was 26, 17, and 2 times higher than that of MOF (0.002 min−1), FeNs (0.003 min−1), and MOF/FeNs (0.021 min−1), respectively (Supplementary Fig. 10). Furthermore, the absorbance spectra of AO7-polluted solution samples at various time intervals were collected, and the absorption peak intensity was drastically decreased at 485 nm in the electro-Fenton-like reaction system, which finally completely disappeared within 60 min (Fig. 3e). Various Fenton-like catalysts were used to remove organic pollutants, as shown in Supplementary table 2. FeNC exhibited considerably higher degradation efficiency and kinetic rate than those recorded in many published studies. Although the kinetic rates of MIL101-(Fe)/g-C3N4 or FeS2@Mo2C achieved a higher kinetic rate than that of FeNC in the present investigation, these Fenton-like reactions with MIL101-(Fe)/g-C3N4 or FeS2@Mo2C required significantly higher catalysts and PMS dosages. Furthermore, these Fenton-like reactions with MIL101-(Fe)/g-C3N4 or FeS2@Mo2C exhibited weaker stability than FeNC in our study. The loss of removal efficiency was only 0.2% after six cycles in our study, and this system with MIL101-(Fe)/g-C3N4 and FeS2@Mo2C were 10.6% and 3.9%, respectively. The loss of removal efficiency in this study was significantly lower than that in other studies, further indicating the stability and effectiveness of FeNC in the present study.
In addition to investigating the catalytic behavior, we conducted a series of control experiments to achieve the maximum performance for AO7 removal. The removal efficiency increased from 68.4% to 100% with an increase in the current density from 3 to 20 mA cm−1 (Supplementary Fig.11), and the corresponding kinetic degradation rate improved from 0.020 to 0.067 min−1 (Supplementary Fig. 11). Considering that a high current would destroy the electrode, 10 mA cm−2 was chosen to effectively remove AO7. In addition, continuous addition of PMS did not improve the removal efficiency and kinetic degradation rate of AO7 (Supplementary Fig.11), which was mainly ascribed to the scavenging reaction between PMS and ROS8. These results demonstrate that the promising electro-Fenton-like reaction performance of FeNC may be due to the highly effective activation reaction of PMS.
Generation of reactive species
As illustrated in Supplementary Fig. 12, the consumption rate of PMS was faster than the degradation rate of AO7 in the FeNC-activated electro-Fenton-like system, suggesting that ROS can be abundantly and rapidly generated in this system. In addition, the PMS concentration remained at a stable level in the presence of lone PMS, further confirming the effectiveness of the electro-Fenton-like system. To further investigate the main contributions of crucial ROS to the AO7 degradation, different radical quenchers were utilized to explore their effects. Adding these quenchers, such as NaN3, furfuryl alcohol (FFA), L-histidine (LH), p-benzoquinone (BQ), KI, superoxide dismutase (SOD), and catalase (CAT), had no evident influence on the degradation rate of AO7 (Fig. 3f), indicating the absence of 1O2 and O2•–. When tert-butanol (TBA), ethanol (EtOH), and methanol (MeOH) were used to probe for •OH and SO4•–, the degradation rate of AO7 was sharply reduced, and the inhibiting effect was enhanced with an increase in the concentration of the quenchers (Supplementary Fig.13). Note that the inhibiting effect of MeOH was more evident than that of TBA, indicating that the SO4•–radical played a more important role than the •OH radical in AO7 degradation. Various gases (O2, Ar, and air) bubbled into the electrolyte did not obviously affect the degradation rate of AO7 (Supplementary Fig. 14), indicating that the O element for the SO4•–and •OH radicals was derived not from dissolved oxygen but from the PMS activation reaction.
To further confirm the ROS produced in the electro-Fenton-like system, EPR spectroscopy was performed to detect the ROS. The four- and six-line peak intensities for •OH and SO4•–were significantly increased with the reaction time (Fig. 3g). Notably, the FeNC catalyst exhibited a stronger EPR peak intensity (SO4•–and •OH radicals) than other catalysts under the same reaction time (Fig. 3h), whereas the peak intensities for 1O2 and O2•–were very weak (Supplementary Fig. 15). Moreover, the concentration of the ROS in this electro-Fenton-like system was determined with different catalysts. The proportion of SO4•–and •OH radicals in the ROS generated by the FeNC reaction system was 99.05% (Fig. 3i and Supplementary Fig.15), indicating that a radical process occurred in the electro-Fenton-like system. In addition, the proportions of SO4•–and •OH radicals in other reaction systems were substantially higher than that of 1O2 and O2•–. Furthermore, the concentration of the SO4•–radical was lower than that of the •OH radical in all reaction systems, mainly because of the reaction of SO4•– and H2O for producing the •OH radical40. The quenching experiment, EPR spectroscopy, and quantification experiments confirmed that SO4•– and •OH radicals were the dominant ROS for organic pollutant degradation in the electro-Fenton-like system.
Mechanism investigation
To investigate the mechanism of the electro-Fenton-like reaction for degrading organic contaminants, the change in the surface composition of FeNC was explored through XPS. Figure 4a demonstrates that the high-resolution XPS spectra of Fe 2p from fresh/used FeNC can be curve-fitted into six peak components. Two characteristic peaks at 726.2 and 712.9 eV were attributed to Fe3+, and the two peaks located at 723.9 and 710.4 eV were attributed to Fe2+. Following the activation reaction, the area ratio of the Fe 2p peak slightly changed, the area ratio of the Fe2+ peak located at 723.9 eV slightly decreased from 16.3% to 13.2%, and the ratio of the Fe2+ peak located at 710.4 eV gradually increased from 25.0% to 27.8%. Note that the ratio of the Fe3+ peak increased at 726.2 eV and decreased at 712.9 eV. The transformation of these ratios remained more stable after the reaction than before the reaction, indicating that an electron was transferred between Fe3+ and Fe2+ via the PMS activation reaction for ROS generation. A vibrating sample magnetometer (VSM) was used to explore the magnetic behavior of FeNC before and after the reaction (Fig. 4b). As can be observed, the saturation magnetization value of FeNC before the reaction was 23.6 emu g−1, which is similar to the value after the reaction (21.9 emu g−1). The above results suggest that the magnetic properties of FeNC underwent minor changes after the activation reaction, which benefitted to the magnetic separation and reuse of the FeNC catalyst by the electro-Fenton-like reaction device. EPR spectra were measured to further investigate the electronic properties of different catalysts (Fig. 4c). The EPR signal of FeNC was considerably stronger than that of FeNs and FeNs/MOF, indicating more unpaired electrons in the aromatic rings of FeNC.

a X-ray photoelectron spectroscopy (XPS) pattern of Fe 2p before and after the reaction. b EPR spectra of the catalysts. c Magnetic hysteresis loop of the catalysts before and after use. d Calculated free energy profile for PMS to SO4·–process in the FeNC-activated electro-Fenton-like reaction system. e PMS adsorption on the surface of FeNs/MOF and FeNC. f Calculated electron density difference diagrams of FeNs/MOF and FeNC with PMS adsorbed on the FeNC site.
To determine the critical active site, the FeNC catalyst was treated with strong acid and alkali solutions for one week to efficiently remove the Fe atom. The kinetic degradation rate of AO7 via treated FeNC was sharply reduced from 0.051 to 0.003 min−1 (Supplementary Fig. 16), indicating that the primary catalytic contribution originated from the anchor of the Fe atom. DFT calculations were used to assess the catalytic decomposition properties of PMS; the corresponding Gibbs free energy is presented in Fig. 4d. The occurrence and formation of SO4–* and HSO5–* in the FeNs/PMS and FeNs/MOF/PMS systems were both exothermic reactions, suggesting that both the adsorption and decomposition processes of PMS in the above system were spontaneous. In the PMS decomposition process, the Gibbs free energy of FeNC was −2.47 eV, which is lower than that of FeNC/MOF (−3.06 eV) because the electrons located near the iron atom helped transfer to the PMS adsorbed on the surface. Note that desorption was the rate-limiting process of the catalytic reaction, and the desorption energy of FeNC was considerably lower than that of FeNs/MOF. Thus, the desorption performance of PMS can be considerably enhanced by increasing the rate-limiting process, which can improve the catalytic reaction between ROS and pollutants in the following process and further accelerate organic pollutant degradation.
The electron density difference between the catalysts and PMS (Fig. 4e) showed the electron transfer from PMS to Fe active sites. Then, the electron was transferred to the Fe–Fe bond during PMS adsorption. In the reaction system (Fig. 4f), PMS was adsorbed on the Fe actives sites because of the strong positively charged Fe nanoparticles. In addition, the strong interaction between negatively charged oxygen and positively charged Fe caused the disassembly of PMS accompanied by the generation of SO4•–and •OH radicals. Notably, the dissociation reaction of PMS on the surface of FeNs/MOF and FeNC was thermodynamically favorable without energy barriers. In addition, the short Fe–O bond between FeNC and PMS was markedly beneficial to the electron transfer reaction compared with FeNs/MOF. Consequently, FeNC can serve as an electron donor to adsorb PMS to generate active radicals during electro-Fenton-like reactions.
Scalability and recyclability of the electro-Fenton-like reaction
As illustrated in Fig. 5a, the electro-Fenton-like reaction device for degrading organic pollutants is constructed with three reaction chambers. In the electro-Fenton-like reaction chamber, the FeNC catalyst activates PMS to generate SO4•– and •OH to remove organic pollutants under electrochemical conditions with 0.05 M Na2SO4 as the electrolyte. In the ultrasonic regeneration chamber, the activity of the used FeNC catalysts is recovered through ultrasonic treatment at 200 Hz for 15 min. In the magnetic force separation chamber, the recovered catalysts are separated by the adsorption force of the magnet. Here, ultrasonic treatment can restore the catalytic activity of the FeNC catalyst. Finally, the magnetic FeNC catalysts are separated from the solution by the adsorption force of the magnet. In particular, the separated magnetic catalysts are continuously used as activators to generate SO4•– and •OH to remove organic pollutants in the electro-Fenton-like reaction chamber. The continuous operation of the device is examined in the following sections, and its viability for the purification of organic wastewater is assessed.

a The electro-Fenton-like reaction device includes an FeNC catalyst and a separate reactor chamber. b AO7 degradation rate in the FeNC-activated electro-Fenton-like reaction system in the presence of different ions. c AO7 degradation rate in the reaction system with different initial pH values. d Degradation performance of various pollutants in the electro-Fenton-like reaction system. e AO7 degradation properties of the FeNC-activated electro-Fenton-like reaction system in different natural waters. f Cyclic stability of FeNC with AO7 as an indicator. g Leaching concentration of Fe ions under continuous run. Conditions: current density=10 mA cm−2; AO7 = 50 mg L−1, PMS = 10 mM, pH=7.0, T = 28 °C. Error bars show the standard deviation (n = 3 independent replicates).
As illustrated in Fig. 5b, none of the cations (Mg2+, Ca2+, K+, and Na+), anions (NO3–, SO42−, HCO3−, and Cl−), or natural organic matter (Supplementary Fig. 17) affected the degradation rate constant of AO7 at the different tested dosages, signifying that the electro-Fenton-like reaction with the FeNC catalyst experiences less disturbance from the usual background inorganic and organic substances that exist in water. In addition, FeNC demonstrated excellent pH adaptability, exhibiting similar degradation performance of AO7 in the pH range of 2.5–11.5 (Fig. 5c), indicating that FeNC catalysts with dual reaction sites had high tolerance to changes in pH. In addition, the electro-Fenton-like system can degrade various organic pollutants, including phenols (4-chlorophenol [4-CP], phenol, and bisphenol A [BPA]), antibiotics (tetracycline [TC], oxytetracycline [OTC], and caffeine [CAF]), and dyes (methyl blue [MB], rhodamine B [RhB], and AO7) (Fig. 5d). Consequently, the electro-Fenton-like system has great potential for the purification of chronic and emerging organic contaminants in water.
Figure 5e indicates that the electro-Fenton-like system can achieve similar degradation properties and kinetic rate constants as AO7 in deionized and natural water (i.e., ground water, sea water, and Pearl river water), indicating its high practical application potential. The stability of the system for AO7 degradation was tested by six continuous cycles. As illustrated in Fig. 5f, the electro-Fenton-like system with the FeNC catalyst exhibited a stable degradation level, as confirmed by the flat kinetic rate plateau (inset of Fig. 5f). No significant changes in the morphology (Supplementary Fig. 18) or peak shift of Fe 2p (Fig. 4a) were observed after the continuous reaction. TOC removal efficiency remained stable during the six testing cycles (Supplementary Fig. 19). This phenomenon was mainly attributed to the rapid recovery of catalyst activity after the introduction of sonication treatment when AO7 molecules adsorbed on the surface captured the active center of FeNC. The sonication treatment was an effective approach for removing carbon deposition on the surface of catalysts41 and prevent the poisoning of the catalyst, further recovering the ability of the catalysts. As illustrated in Fig. 5g, the leaching amount of Fe ions is less than 70 µg L−1 after continuous five-cycle operations, which is below the standard for drinking water specified by the World Health Organization.
The highly effective and continuous catalytic ability of FeNC in the electro-Fenton-like system reveals considerable potential for remediating real industrial wastewater. In addition, benefiting from the ubiquitous existence of dual reaction sites, various catalysts with similar properties can be used to degrade organic wastewater. These catalysts are typically low-cost and commercially available MOF materials.
Discussion
In summary, we synthesized a magnetic catalyst and designed an engineering device to address three challenges of the Fenton-like reaction in treating organic wastewater: (i) the leaching of metal ions; (ii) the recovery of catalysts; and (iii) the continuous generation of ROS. First, we comprehensively investigated the effectiveness and stability of the magnetic catalyst (FeNC) in removing organic pollutants via the electro-Fenton-like reaction. The FeNC catalysts synthesized herein provide dual reaction sites for efficient PMS activation to generate SO4•– and •OH radicals. Additionally, this structure can significantly reduce the leaching of metal ions (70 µg L−1). Subsequently, we designed and constructed an electrolysis reaction device containing an electro-Fenton-like reaction chamber, an ultrasonic regeneration chamber, and a magnetic force separation chamber. The FeNC catalysts used in this device can be regenerated, separated, and reused to continuously generate ROS through the electro-Fenton-like reaction for the effective degradation of organic pollutants. We also used this device to treat various organic pollutants containing phenols, antibiotics, and dyes, and found that the kinetic degradation rate of phenols (0.077–0.126 min−1) was substantially higher than that of antibiotics (0.036–0.049 min−1) and dyes (0.051–0.099 min−1). The proposed technology is a promising alternative for large-scale applications in biological and chemical engineering as well as areas related to electro-Fenton-like reactions for the remediation and purification of wastewater.
Methods
Materials
Zinc nitrate (Zn(NO3)2·6H2O, 99%), 1,4-Benzene dicarboxylic acid (H2BDC, 99%), acetic acid (HAc), N, N-Dimethylformamide (DMF, 99%), ferric chloride hexahydrate (FeCl3·6H2O), Iron(III) nitrate (Fe(NO3)3·H2O, 99%), urea (99%), polyethylene glycol, sodium acetate trihydrate (NaAc, 98.5%), CTAB, ammonia solution (NH4OH, 99.99%), sodium sulfate (Na2SO4), oxone (KHSO5·0.5KHSO4·0.5K2SO4, PMS, 99%), 4-CP, CAF, MB, TC, OTC), BPA, RhB, AO7, phenol, magnesium chloride (MgCl2), calcium chloride (CaCl2), potassium chloride (KCl), sodium chloride (NaCl), sodium nitrate (NaNO3), sodium sulfate (Na2SO4), sodium bicarbonate (NaHCO3), MeOH, TBA, EtOH, NaN3, FFA, LH, BQ, KI, SOD, CAT, dimethyl pyridine N-oxide (DMPO), and 2,2,6,6-tetramethylpiperidine (TEMP) were purchased from Aladdin.
Preparation of MOF
BPDC (0.3 mM) and Zn(NO3)2·6H2O (0.3 mM) were added subsequently to the DMF solution (20 mL) via sonication for 30 min. Then, HAc (0.5 mL) and HCl (0.1 mL) were added to the mixture solution through sonication for 30 min. Next, the above solution was transferred into a 100 mL Teflon-lined hydrothermal reaction kettle at 120 °C for 24 h. Finally, the sample (MOF) was obtained via further calcination at 900 °C for 2 h under argon gas protection.
Preparation of Fe nanoparticles (FeNs)
FeCl3·6H2O (5 mM) and Fe(NO3)3·H2O (5 mM) were dissolved in ethylene glycol (40 mL) to form a saffron yellow solution, followed by the addition of NaAc (20 mM) and polyethylene glycol (1.0 g). Then, the sample was heated to 200 °C for 12 h in an autoclave. Fe nanoparticles (FeNs) were obtained by washing and drying.
Preparation of FeNs/MOF
During FeNs preparation, MOF (500 mg) and urea (20 mM) were introduced into the reaction solution through sonication for 60 min. Following the hydrothermal reaction, the sample was further heated to 550 °C for 4 h under argon atmosphere at 2 °C min−1. The synthesized sample is denoted as FeNs/MOF.
Preparation of FeNC
FeNs/MOF (100 mg) was dispersed in 20 mL methanol via sonication for 30 min. Subsequently, NH4OH (3 mL) and CTAB (60 mL) were added to the mixture and stirred for 6 h to form a homogenous solution. After a series of processes, namely, centrifugation, washing, and drying, the samples were further heated to 900 °C for 4 h under argon atmosphere at a rate of 2 °C min−1. The final sample is denoted as FeNC.
Material characterization and analysis methods
Surface morphology and structure of the samples were observed using SEM (ZEISS Gemini 300) and high-resolution TEM (FEI Talos F200s) equipped with EDX. The pattern of the samples was measured via powder XRD (Rigaku Ultima IV, Japan) with Cu Kα radiation. The elemental composition of the samples was determined via XPS (Thermo Scientific K-Alpha) with Al Kα radiation. The sample surface area and pore size distribution were measured using the BET method based on the nitrogen adsorption/desorption isotherm by an analyzer (ASAP2460). The surface functional groups of the samples were determined through FTIR (Vertex 70, Germany), and the magnetic properties of the samples were explored through VSM (ATM 2900). The concentrations of AO7, RhB, and MB were determined using a UV–vis spectrophotometer (UV-3600 Plus) in the wavelength range of 200–800 nm. The concentrations of contaminants (4-CP, CAF, TC, OTC, BPA, phenol, and PMS) were determined using a high-performance liquid chromatographer (HPLC, Agilent 1260). The amount of Fe ions in the solution was determined using an inductively coupled plasma atomic emission spectrometer (ICAP 7200). The XAS of the samples was collected at the Singapore Synchrotron Light Source center, where a pair of channel-cut Si (111) crystals was used in the monochromator. The storage ring operated at an energy of 2.5 GeV with an average electron current below 200 mA. The TOC was detected using a TOC analyzer (VCPH, Japan). The EPR spectra were measured using a Bruker EMXnano spectrometer with DMPO or TEMP as a spin-trapping agent to detect free radicals (such as SO4–, ·OH, and O2–) and singlet oxygen (1O2), respectively. SO4–, ·OH, and O2– were qualitatively analyzed via a rapid spectrophotometric method42, the terephthalic acid photoluminescence method43, and the nitroblue tetrazolium transformation method44, respectively.
Activity evaluation
The degradation experiments were conducted in a 250 mL reactor with an anode and a cathode with a constant stirring speed (300 rpm). Ti/RuO2–IrO2 and Ti plate electrodes were used as anode and cathode electrodes, respectively. In a typical experiment, 50 mg L−1 AO7 was introduced into the reactor, and the current density was set to 10 mA cm−2. In addition, PMS was added into the solution at a concentration of 10 mM. The electrolyte contained 0.05 M Na2SO4 solution. In addition, the catalyst was added to the electrolyte to maintain a concentration of 50 mg L−1. The reaction solution at an interval time (2.5, 5, 10, 20, 40, 60 min) was detected through HPLC. The degradation process of AO7 in this study was consistent with pseudo-first-order kinetics, and the kinetic degradation rate constant (k) of AO7 degradation was further explored. The corresponding equation is given as follows:
where Ct and C0 denote the concentration of AO7 at time t and zero min, respectively.
Electrochemical measurements
The electrochemical performance of the samples was measured using an electrochemical workstation (CorrTest, 310) with a traditional three-electrode cell. A Pt wire electrode and Ag/AgCl electrode were used as the counter and reference electrodes, respectively. A glass carbon (GC) electrode with an area of 0.196 cm2 was used as the working electrode. Ink was prepared by dispersing 5 mg catalysts and 1 mL ethanol into 20 µL Nafion with continuous sonication. Then, the slurry was pipetted onto the GC surface, and the loading amount was 0.25 mg cm−2. The electrolyte was 0.5 M Na2SO4 solution. LSV was performed in the range of 0.4 to 2.0 V at a scanning rate of 2 mV s−1. CV was performed in the range of −0.2 to 0.6 V at a scanning rate of 2 mV s−1. Quenchers such as EtOH, MeOH, TBA, NaN3, FFA, LH, and BQ were used to distribute the contributions of ·OH and SO4·–, O2·–, and 1O2. KI, SOD, and CAT were used to quench surface hydroxyl radicals, O2−, and H2O245, respectively.
Density functional theory (DFT) calculations
Calculations were performed in the framework of DFT using the Castep module in the Material Studio software46 using the Perdew–Burke–Ernzerhof generalized gradient approximation47. The kinetic energy cutoff was set to 450 eV, and the k-point for the bulk was 2 × 2 × 7. The force on each atom was set to less than 0.03 eV Å−1 based on the convergence criterion of geometry relaxation. The adsorption energy of PMS (Ead) on FeNC was calculated based on the following equation:
where Etotal is the total energy of the adsorbed system, and Epms and Esub are the energies of the free PMS and substrate, respectively.
Data availability
The data analyzed or generated during this study are included in this published article.
References
Dong, C. et al. Self-cycled photo-Fenton-like system based on an artificial leaf with a solar-to-H2O2 conversion efficiency of 1.46. Nat. Commun. 13, 4982 (2022).
Wu, Y., Zhou, S., Qin, F., Ye, X. & Zheng, K. Mathematical Model Analysis of Fenton Oxidation of Landfill Leachate. Waste Manag. 31, 468–474 (2011).
Li, H. S., Zhou, S. Q., Sun, Y. B. & Lv, J. Nitrogen and carbon removal from Fenton-treated leachate by denitrification and biofiltration. Bioresour. Technol. 101, 7736–7743 (2010).
Yang, X. J., Xu, X. M., Xu, J. & Han, Y. F. Iron oxychloride (FeOCl): an efficient Fenton-like catalyst for producing hydroxyl radicals in degradation of organic contaminants. J. Am. Chem. Soc. 135, 16058–16061 (2013).
Su, L., Wang, P., Ma, X., Wang, J. & Zhan, S. Regulating Local Electron Density of Iron Single Sites by Introducing Nitrogen Vacancies for Efficient Photo-Fenton Process. Angew. Chem. Int. Ed. 60, 21261–21266 (2021).
Duan, X., Sun, H. & Wang, S. Metal-Free Carbocatalysis in Advanced Oxidation Reactions. Acc. Chem. Res. 51, 678–687 (2018).
Li, M. et al. FeN4-doped carbon nanotubes derived from metal organic frameworks for effective degradation of organic dyes by peroxymonosulfate: Impacts of FeN4 spin states. Chem. Eng. J. 431, 133339 (2022).
Li, X. et al. Single Cobalt Atoms Anchored on Porous N-Doped Graphene with Dual Reaction Sites for Efficient Fenton-like Catalysis. J. Am. Chem. Soc. 140, 12469–12475 (2018).
Forooshani, P. K. et al. Hydroxyl Radical Generation Through the Fenton-Like Reaction of Hematin- and Catechol-Functionalized Microgels. Chem. Mater. 32, 8182–8194 (2020).
Yan, Q. et al. Constructing an Acidic Microenvironment by MoS2 in Heterogeneous Fenton Reaction for Pollutant Control. Angew. Chem. Int. Ed. 60, 17155–17163 (2021).
Xu, H. et al. Improving PMS oxidation of organic pollutants by single cobalt atom catalyst through hybrid radical and non-radical pathways. Appl. Catal. B Environ. 263, 118350 (2020).
Gao, Y. et al. Unraveling the High-Activity Origin of Single-Atom Iron Catalysts for Organic Pollutant Oxidation via Peroxymonosulfate Activation. Environ. Sci. Technol. 55, 8318–8328 (2021).
Wang, J. & Wang, S. Reactive species in advanced oxidation processes: Formation, identification and reaction mechanism. Chem. Eng. J. 401, 126158 (2020).
Guo, Z. Y. et al. Mn-O Covalency Governs the Intrinsic Activity of Co-Mn Spinel Oxides for Boosted Peroxymonosulfate Activation. Angew. Chem. Int. Ed. 60, 274–280 (2021).
Wang, P. et al. Catalytic degradation of micropollutant by peroxymonosulfate activation through Fe(III)/Fe(II) cycle confined in the nanoscale interlayer of Fe(III)-saturated montmorillonite. Water Res. 182, 116030 (2020).
Cai, C., Kang, S., Xie, X. & Liao, C. Ultrasound-assisted heterogeneous peroxymonosulfate activation with Co/SBA-15 for the efficient degradation of organic contaminant in water. J. Hazard. Mater. 385, 121519 (2020).
Gao, B. et al. Superoxide radical mediated Mn(III) formation is the key process in the activation of peroxymonosulfate (PMS) by Mn-incorporated bacterial-derived biochar. J. Hazard. Mater. 431, 128549 (2022).
Yu, D. et al. Peroxymonosulfate activation using a composite of copper and nickel oxide coated on SBA-15 for the removal of sulfonamide antibiotics. Environ. Res. 206, 112301 (2022).
Lv, Y. et al. Fabrication of magnetically recyclable yolk-shell Fe3O4@TiO2 nanosheet/Ag/g-C3N4 microspheres for enhanced photocatalytic degradation of organic pollutants. Nano Res. 14, 2363–2371 (2021).
Sun, P. et al. Strategic combination of N-doped graphene and g-C3N4: Efficient catalytic peroxymonosulfate-based oxidation of organic pollutants by non-radical-dominated processes. Appl. Catal. B Environ. 272, 119005 (2020).
Duan, X., Sun, H., Wang, Y., Kang, J. & Wang, S. N-doping-induced nonradical reaction on single-walled carbon nanotubes for catalytic phenol oxidation. ACS Catal. 5, 553–559 (2015).
Zou, J. et al. Rapid acceleration of ferrous iron/peroxymonosulfate oxidation of organic pollutants by promoting Fe(III)/Fe(II) cycle with hydroxylamine. Environ. Sci. Technol. 47, 11685–11691 (2013).
Li, Y. et al. Constructing Solid-Gas-Interfacial Fenton Reaction over Alkalinized-C3N4 Photocatalyst To Achieve Apparent Quantum Yield of 49% at 420 nm. J. Am. Chem. Soc. 138, 13289–13297 (2016).
Koo, S. et al. Enhanced Chemodynamic Therapy by Cu-Fe Peroxide Nanoparticles: Tumor Microenvironment-Mediated Synergistic Fenton Reaction. ACS Nano. 16, 2535–2545 (2022).
Li, M. et al. Exploring key reaction sites and deep degradation mechanism of perfluorooctane sulfonate via peroxymonosulfate activation under electrocoagulation process. Water Res. 207, 117849 (2021).
Dong, C., Ji, J., Shen, B., Xing, M. & Zhang, J. Enhancement of H2O2 Decomposition by the Co-catalytic Effect of WS2 on the Fenton Reaction for the Synchronous Reduction of Cr(VI) and Remediation of Phenol. Environ. Sci. Technol. 52, 11297–11308 (2018).
Li, X. et al. Fine-Tuning Radical/Nonradical Pathways on Graphene by Porous Engineering and Doping Strategies. ACS Catal. 11, 4848–4861 (2021).
Li, M. et al. Exploration of perfluorooctane sulfonate degradation properties and mechanism via electron-transfer dominated radical process. Water Res. 215, 118259 (2022).
Li, M. et al. Efficient decomposition of perfluorooctane sulfonate by electrochemical activation of peroxymonosulfate in aqueous solution: Efficacy, reaction mechanism, active sites, and application potential. Water Res. 221, 118778 (2022).
Yang, L. et al. Iron active sites encapsulated in N-doped graphite for efficiently selective degradation of emerging contaminants via peroxymonosulfate (PMS) activation: Inherent roles of adsorption and electron-transfer dominated nonradical mechanisms. Chem. Eng. J. 444, 136623 (2022).
Nayem, S. M. A., Islam, S., Aziz, M. A. & Ahammad, A. J. S. Mechanistic insight into hydrothermally prepared molybdenum-based electrocatalyst for overall water splitting. Electrochim. Acta. 445, 142050 (2023).
Del Rio, P. G. et al. Current breakthroughs in the hardwood biorefineries: Hydrothermal processing for the co-production of xylooligosaccharides and bioethanol. Bioresour. Technol. 343, 126100 (2022).
Yv, L., Wang, J., Shi, Z., Shi, J. & Wang, X. Preparation of high-entropy ceramic oxide powder at different calcination temperatures and electrochemical applications. Ceram. Int. 48, 26370–26377 (2022).
Wijesinghe, W. P. et al. Facile synthesis of both needle-like and spherical hydroxyapatite nanoparticles: effect of synthetic temperature and calcination on morphology, crystallite size and crystallinity. Mater. Sci. Eng. C. 42, 83–90 (2014).
Yan, K. et al. Biomass waste-derived porous carbon efficient for simultaneous removal of chlortetracycline and hexavalent chromium. iScience 24, 102421 (2021).
Zhu, Y. et al. Environment of Metal-O-Fe Bonds Enabling High Activity in CO2 Reduction on Single Metal Atoms and on Supported Nanoparticles. J. Am. Chem. Soc. 143, 5540–5549 (2021).
Zhang, L. S. et al. Carbon Nitride Supported High-Loading Fe Single-Atom Catalyst for Activation of Peroxymonosulfate to Generate 1O2 with 100 % Selectivity. Angew. Chem. Int. Ed. 60, 21751–21755 (2021).
Pan, Y. et al. A Bimetallic Zn/Fe Polyphthalocyanine-Derived Single-Atom Fe-N4 Catalytic Site:A Superior Trifunctional Catalyst for Overall Water Splitting and Zn-Air Batteries. Angew. Chem. Int. Ed. 57, 8614–8618 (2018).
Peng, L., Duan, X., Shang, Y., Gao, B. & Xu, X. Engineered carbon supported single iron atom sites and iron clusters from Fe-rich Enteromorpha for Fenton-like reactions via nonradical pathways. Appl. Catal. B: Environ. 287, 119963 (2021).
Mi, X. et al. Almost 100 % Peroxymonosulfate Conversion to Singlet Oxygen on Single-Atom CoN2+2 Sites. Angew. Chem. Int. Ed. 60, 4588–4593 (2021).
Ji, J. et al. Metallic Active Sites on MoO2(110) Surface to Catalyze Advanced Oxidation Processes for Efficient Pollutant Removal. iScience 23, 100861 (2020).
Liang, C., Huang, C. F., Mohanty, N. & Kurakalva, R. M. A rapid spectrophotometric determination of persulfate anion in ISCO. Chemosphere 73, 1540–1543 (2008).
Xing, M. et al. Metal Sulfides as Excellent Co-catalysts for H2O2 Decomposition in Advanced Oxidation Processes. Chem 4, 1359–1372 (2018).
Jiang, X.-H. et al. Simultaneous photoreduction of Uranium(VI) and photooxidation of Arsenic(III) in aqueous solution over g-C3N4/TiO2 heterostructured catalysts under simulated sunlight irradiation. Appl. Catal. B Environ. 228, 29–38 (2018).
Chen, N. et al. Visible Light Driven Organic Pollutants Degradation with Hydrothermally Carbonized Sewage Sludge and Oxalate Via Molecular Oxygen Activation. Environ. Sci. Technol. 52, 12656–12666 (2018).
Segall, M. D. et al. First-principles simulation: ideas, illustrations and the CASTEP code. J. Phys.: Condens. Matter. 14, 2717–2744 (2002).
Yao, Y. et al. First principles calculation of anomalous Hall conductivity in ferromagnetic bcc Fe. Phys. Rev. Lett. 92, 037204 (2004).
Acknowledgements
We gratefully thank the financial support from Project Funded by Key Program of National Natural Science Foundation of China (42030713), National Natural Science Foundation of China (42007358), Guangdong Basic and Applied Basic Research Foundation (2020A1515110518), the Hong Kong Scholarship Program (XJ2020059), and the China Postdoctoral Science Foundation (2019M663382). K.Y. acknowledges the Project of Hetao Shenzhen-Hong Kong Science and Technology Innovation Cooperation Zone (HZQB-KCZYB-2020083). S.Z. thanks the project of the State Key Research & Development Project of China (2022YFC3705003) and the Project funded by the Department of Science & Technology of Guizhou Province (2023-115). The authors would like to thank Shiyanjia Lab (www.shiyanjia.com) for the XAS measurements.
Author information
Authors and Affiliations
Contributions
M.L. designed and conducted all the preparation, characterization, and measurements. M.L. wrote and revised the whole manuscript. K.Y., S.Z., and C.M. brought a clear study pathway and guided the project. W.H. and J.S. help analyze the data.
Corresponding authors
Ethics declarations
Competing interests
These authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, M., Song, J., Han, W. et al. Iron-organic frameworks as effective fenton-like catalysts for peroxymonosulfate decomposition in advanced oxidation processes. npj Clean Water 6, 37 (2023). https://doi.org/10.1038/s41545-023-00251-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41545-023-00251-z
